

Keywords: extracellular histones, heparin, lung endothelial permeability, Toll-like receptors, two-hit model
Abstract
Extracellular histones released into the circulation following trauma, sepsis, and ARDS may act as potent damage-associated molecular pattern signals leading to multiple organ failure. Endothelial cell (EC) dysfunction caused by extracellular histones has been demonstrated in vitro and in vivo; however, precise mechanistic details of histone-induced EC dysfunction and exacerbation of ongoing inflammation remain poorly understood. This study investigated the role of extracellular histones in exacerbating preexisting endothelial dysfunction and acute lung injury. Histone subunits H3 and H4, but not H1, H2A, or H2B, induced permeability in human pulmonary EC. H3 and H4 at concentrations above 30 µg/mL caused EC inflammation reflected by activation of the NF-κB pathway, transcriptional activation, and release of cytokines and chemokines including IL-6 and IL-8, and increased mRNA and protein expression of EC adhesion molecules VCAM-1 and ICAM-1. Pharmacological inhibitors targeting Toll-like receptor TLR4 but not TLR2/6, blocked histone-induced EC dysfunction. H3 and H4 also strongly augmented EC permeability and inflammation caused by Gram-negative and Gram-positive bacterial particles, endotoxin, and TNFα. Heparin blocked histone-induced augmentation of EC inflammation caused by endotoxin and TNFα. Injection of histone in mouse models of lung injury caused by bacterial wall lipopolysaccharide (LPS) and heat-killed Staphylococcus aureus (HKSA) augmented ALI parameters: increased protein content, cell count, and inflammatory cytokine secretion in bronchoalveolar lavage fluid. Important clinical significance of these findings is in the demonstration that even a modest increase in extracellular histone levels can act as a severe exacerbating factor in conjunction with other EC barrier disruptive or proinflammatory agents.
INTRODUCTION
Histones are highly conserved, alkaline, positively charged DNA-binding proteins that in complex with genomic DNA form chromatin located in cell nucleus. Following post-translational modifications such as acetylation, phosphorylation, methylation, ubiquitylation, and sumoylation, histones play a key role in epigenetic regulation of gene expression by altering chromatin structure or recruiting histone modifiers (1). Besides these conventional functions, histones act as potent damage-associated molecular pattern (DAMP) molecules with cytotoxic and proinflammatory activities once released into the extracellular space by damaged or activated cells (2, 3). Indeed, patients with sepsis and trauma, affected tissues reveal high rates of apoptotic or necroptotic programmed cell death and histone is one of the major DAMPs released by injured cells into blood circulation (4). Furthermore, patients with severe trauma, ARDS, and sepsis exhibit elevated levels of circulating histones that directly correlated with incidence and severity of acute lung injury (ALI) (5–8). Conversely, lipopolysaccharide (LPS), hydrochloric acid, or complement factor-induced ALI in animal models showed the release of histone into bronchoalveolar lavage (6, 9).
Endothelial damage accompanied by lung dysfunction with the occurrence of pulmonary edema, preferential influx of inflammatory cells into the lung and hemorrhage was the earliest pathological event observed in rodents receiving intravenous injection of histone mix (5), suggesting that lung dysfunction is the major target of histone action.
In the lung, vascular endothelial cells (EC) by virtue of their direct exposure to circulating factors represent a primary target of histone toxicity. It has been shown that histone concentrations exceeding 50 µg/mL directly caused cell death in cultured endothelial cells (EC) (5, 10, 11). Multiple animal studies have demonstrated histone-induced endothelial damage, inflammatory cytokines elevation, platelet aggregation, and coagulation activation [reviewed in Karki et al. (12)]. For instance, blocking of histone activities with activated protein C or anti-histone antibody protected mice from lethality (11).
Although activation of inflammation, coagulation, and thrombosis have been described as major factors contributing to histone-induced organ injury, precise mechanisms mediating EC damage and ensuing lung injury remain incompletely understood. Activation of Toll-like receptors (TLR) 4 and 2 has been reported to be involved in histone-induced liver and renal injury (13, 14). Similar mechanisms of TLR2 and TLR4 activation mediate histone-induced tissue factor expression in ECs (15). Other studies have reported an induction of cytokine storm and activation of NLRP3 inflammasome during histone-induced toxicity (8, 16). In endothelial cells, histone-induced barrier breach has been linked to elevation of intracellular calcium levels (11), disassembly of EC adherens junctions, and cytoskeletal reorganization with actin stress fiber formation (17).
Although the pathological role of high histones concentrations in triggering endothelial dysfunction is now well recognized, the role of more pathologically relevant submaximal histone concentrations in exacerbating the preexisting lung injury syndromes and augmenting EC barrier disruption and inflammation caused by proinflammatory agonists is not known. This study investigated the synergistic effects of various histone subtypes on barrier disruption and inflammation in cultured human lung EC and mouse models of ALI caused by heat-killed bacteria, LPS, and tumor necrosis factor-α (TNFα).
MATERIALS AND METHODS
Reagents
Human pulmonary artery endothelial cells (HPAECs) and growth media kit were obtained from Lonza (Allendale, NJ). Purified recombinant human histone subunits H1, H2A, H2B, H3, and H4 were purchased from Cayman Chemical (Ann Arbor, MI). Calf thymus histone mixture was obtained from Sigma (St. Louis, MO). TLR4 inhibitor CLI-095, synthetic TLR4 activator CRX-527, and heat-killed bacteria (Staphylococcus aureus, Escherichia coli, Pseudomonas aeruginosa, and Streptococcus pneumoniae) were from Invivogen (San Diego, CA). Antibodies to phospho-NF-κB (1:1,000), HRP-linked anti-mouse (1:5,000), and anti-rabbit IgG (1:5,000) were from Cell Signaling (Beverly, MA); ZO-1 (1:1,000) and p120-catenin (1:1,000) were from BD Transduction Laboratories (San Diego, CA); antibodies to VE-cadherin (1:1,000), VCAM-1 (1:1,000), and ICAM-1 (1:1,000) were from Santa Cruz Biotechnology (Santa Cruz, CA). LPS (E. coli 0111: B4) and β-tubulin (1:5,000) antibodies were from Sigma (St. Louis, MO). TNFα and TLR2 inhibitor CU-CPT22 were from R&D systems (Minneapolis, MN). Texas Red-conjugated phalloidin and Alexa Fluor 488-labeled secondary antibodies were purchased form Molecular Probes (Eugene, OR). All antibodies have been validated in the previous studies (18). All other reagents were obtained from Sigma unless otherwise indicated.
EC Permeability Assays
The cellular barrier properties were analyzed by measurements of transendothelial electrical resistance (TER) across confluent HPAEC monolayers using an electrical cell-substrate impedance sensing system (Applied Biophysics, Troy, NY) as previously described (19). Endothelial permeability to macromolecules was determined by express permeability testing (XPerT) assay developed by our group (20). This assay is based on high-affinity binding of cell impermeable avidin-conjugated FITC-labeled tracer to the biotinylated extracellular matrix proteins immobilized on the surface covered with EC monolayers. Briefly, after desired periods of EC stimulation with agonists, FITC-avidin solution was added directly to the culture medium for 3 min before termination of the experiment. Unbound FITC-avidin was washed out with PBS (pH 7.4, 37°C), cells were fixed with 3.7% formaldehyde in PBS (10 min, room temperature), and visualization of FITC-avidin on the bottoms of coverslips was performed using Nikon imaging system Eclipse TE 300 (Nikon, Tokyo, Japan) equipped with a digital camera (DKC 5000, Sony, Tokyo, Japan); ×10 objective lenses were used. Images were processed with Adobe Photoshop 7.0 software (Adobe Systems, San Jose, CA), and green fluorescence signal was quantitated using ImageJ software (NIH, Bethesda, MD).
Coimmunoprecipitation and Western Blotting
At the end of agonist stimulation, cells were washed in cold phosphate-buffered saline (PBS) and lysed on ice with cold TBS-NP40 lysis buffer (20 mM Tris pH 7.4, 150 mM NaCl, 1% NP40) supplemented with protease and phosphatase inhibitor cocktails (Roche, Indianapolis, IN). Clarified lysates were then incubated with antibodies to p120-catenin (1:100) (BD Transduction Laboratories, San Diego, CA) or VE-cadherin (1:100) (Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4°C, washed three to four times with TBS-NP40 lysis buffer, and the complexes were analyzed by Western blotting. For Western blotting, protein samples were subjected to 8% SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose membrane (17 V for 1 h, semi-dry transfer system, Bio-Rad), and incubated with desired primary antibodies (1:1,000) overnight at 4°C. After washing three times with TBS-Tween buffer (0.1% Tween-20), HRP-conjugated anti-mouse or anti-rabbit secondary antibodies (1:5,000) were added for 1 h at room temperature. Immunoreactive proteins were visualized by enhanced chemiluminescence (ECL) according to the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, MA). The relative intensities of the protein bands were quantified by scanning densitometry using ImageJ software.
Quantitative Real-Time PCR
One microgram of total RNA isolated using the RNeasy plus mini kit (Qiagen, Germantown, MD) was reverse transcribed to cDNA using iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). The quantitative real-time PCR was carried out with PerfeCTa SYBR Green Fastmix (Quantabio, Beverly, MA) on a Bio-Rad CFX96 real-time PCR system. To analyze gene expression changes, Ct values were normalized to GAPDH and fold changes in expression were calculated using ΔΔCt method.
Measurement of Cytokines
The concentrations of sICAM, IL-6, and IL-8 were measured using commercially available ELISA kits (R&D systems, Minneapolis, MN). Briefly, conditioned media supernatants from treated EC were collected and centrifuged to remove debris and ELISA was performed following manufacturer’s protocol. Absorbance was read at 450 nm within 30 min in microplate reader (Molecular Devices, Menlo Park, CA). Standard curves were generated with expected minimum detectable concentration of 8 pg/mL and cytokines levels were quantified.
Immunofluorescent Staining and Image Analysis
Following agonists stimulation, cells were fixed in 3.7% formaldehyde solution in PBS for 10 min at 4°C, washed with PBS, permeabilized with 0.1% Triton X-100 in PBS for 30 min at room temperature, and blocked with 2% BSA in PBS for 30 min. Then, cells were incubated with VE-cadherin antibody (1:1,000) for 1 h at room temperature followed by staining with Alexa 488-conjugated secondary antibody (1:500). Actin filaments were stained with Texas Red-conjugated phalloidin (1:500) diluted in the blocking solution. After immunostaining, the slides were analyzed using an inverted microscope Nikon Eclipse TE300 connected to SPOT RT monochrome digital camera and image processor (Diagnostic Instruments, Sterling Heights, MI). The images were processed with Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA). For each experimental condition, at least 10 microscopic fields in each independent experiment were analyzed.
Animal Studies
All the protocols involving animal care and treatment procedures were approved by the Institutional Animal Care and Use Committee of University of Maryland. Adult male and female C57BL/6J mice, 8–12 wk old, with an average weight of 20–25 g (Jackson Laboratories, Bar Harbor, ME) were used at equal proportions in this study. Sterile saline solution was used as a vehicle control, LPS (0.75 mg/kg) was given intratracheally (it), HKSA (2 × 108 bacterial particle/mouse) was administered intranasally (in), and calf thymus histone (Cat. No. 10223565001, Sigma) was administered intravenously (iv) into jugular vein at the dose of 25 mg/kg. After 18–24 h, animals were anesthetized with 0.03-mL intraperitoneal injection of ketamine (12 mg/kg) and euthanized. Parameters of lung injury were evaluated by measurements of BAL cell count and protein concentration and evaluation of the Evans blue accumulation in the lung tissue, as previously described (21, 22).
Statistical Analysis
Results are expressed as means ± SD of three to eight independent experiments. Agonist-stimulated samples were compared with controls by unpaired Student’s t test. One-way analysis of variance (ANOVA) followed by the post hoc Fisher’s test was used for multiple comparisons with P < 0.05 considered as statistically significant.
RESULTS
H3 and H4, but Not H1, H2A, and H2B Histone Subunits Cause EC Permeability
Effects of histone subunits on EC permeability were evaluated using measurements of transendothelial electrical resistance (TER) in human pulmonary artery EC monolayers grown on golden electrodes, as described in materials and methods. Treatment of EC monolayers with 50 µg/mL of H1, H2A, H2B, H3, or H4 subunits showed that only H3 and H4 histones caused pronounced EC permeability, whereas other subunits were without effect (Fig. 1A, top). Both H3 and H4 histones caused dose-dependent TER decline in the 10–50 µg/mL concentration range with maximal effect at 50 µg/mL which developed within 3–4 h after histone administration (Fig. 1A, bottom).
Figure 1.
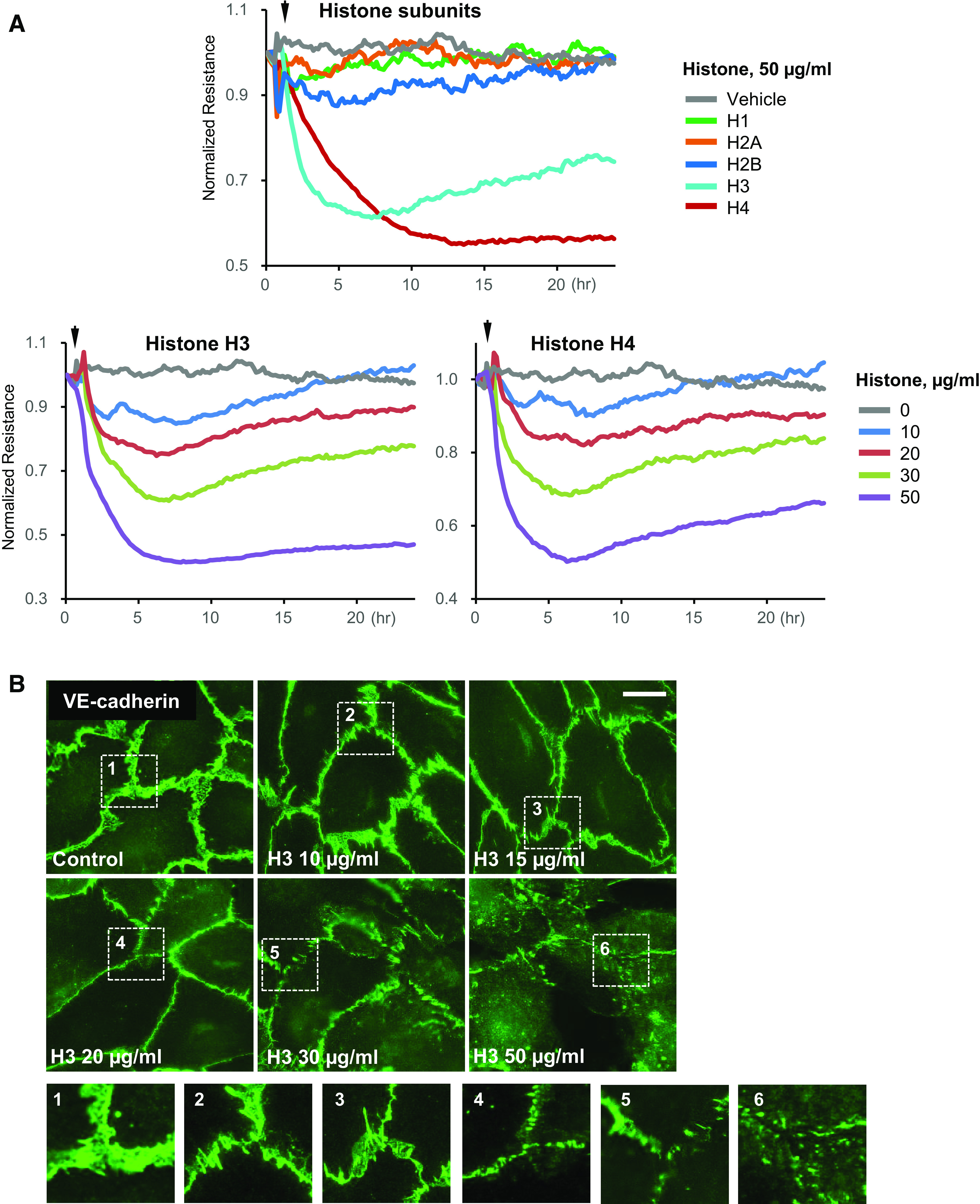
Selectivity of histone subunits in causing endothelial dysfunction. A: HPAECs were stimulated with indicated histone subunits, and endothelial permeability was determined by monitoring TER over time (top). The dose dependence of H3 and H4 on endothelial barrier disruption is also presented (bottom). Representative TER traces are shown for n = 8–10. B: HPAECs were treated with indicated concentrations of histone H3 for 4 h, and VE-cadherin immunostaining was performed. Insets: higher magnification images from the marked areas to illustrate the loss of VE-cadherin from cell membrane. Shown are representative images captured from 10 microscopic fields/condition (n = 3); bar = 5 µm. HPAECs, human pulmonary artery endothelial cells; TER, transendothelial electrical resistance.
Barrier disruptive effects of H3 histone were accompanied by dose-dependent gradual reduction of VE-cadherin positive areas at cell–cell interface reflecting weakening of the EC adhesive structures (Fig. 1B). Higher magnification insets depict the details of cell junction disassembly caused by increasing doses of histone H3.
H3 and H4 Exacerbate Agonist-Induced EC Inflammation
Testing histone subunits for their effects on EC inflammatory signaling showed that even at high doses (50 µg/mL), H1, H2A, and H2B did not induce phosphorylation of NF-κB subunit and expression of EC adhesion molecules ICAM-1 and VCAM-1, the hallmarks of EC inflammatory activation. In contrast, H3 and H4 at this concentration induced pronounced NF-κB phosphorylation and ICAM-1 and VCAM-1 protein expression 6 h after stimulation, indicating strong EC inflammatory response (Fig. 2A, left). It is important to note that direct activation of EC inflammatory response by H3 was observed only at higher concentrations (30–50 µg/mL), whereas lower concentrations (10–20 µg/mL) increased EC permeability (Fig. 1) but did not activate inflammatory signaling (Fig. 2A, right). Similar effects were caused by H4 subunit (data not shown).
Figure 2.
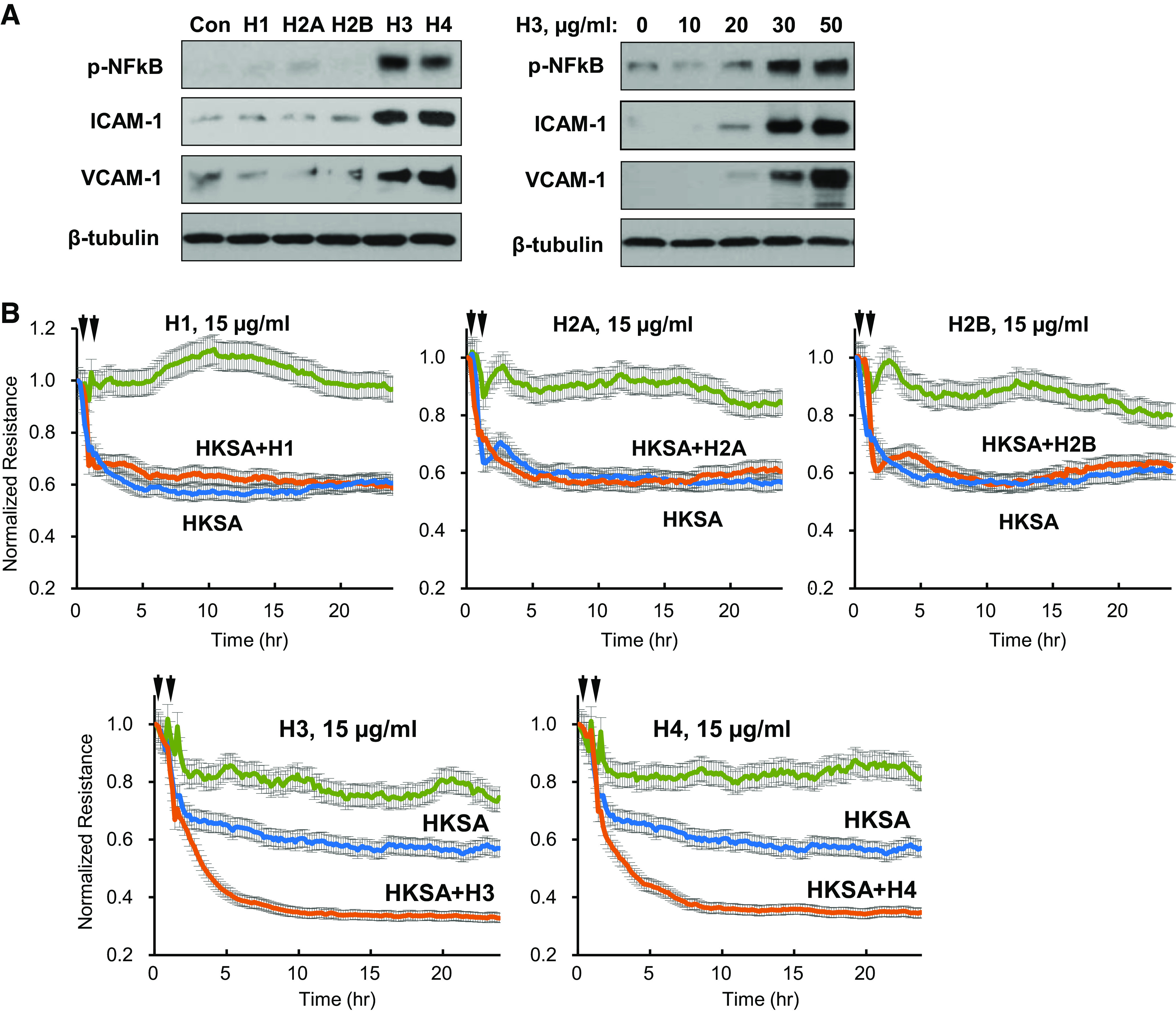
Histones H3 and H4 augment agonist-induced endothelial barrier disruption. A: HPAECs were treated with 50 µg/mL of each histone subunit. Expression of indicated inflammatory proteins was detected by Western blotting (left). Dose-dependent effect of histone H3 on expression of inflammatory markers (right). Shown are representative blots from four independent experiments. B: TER measurements in HPAEC monolayers treated with HKSA alone (5 × 108 particles/mL) or with addition of indicated histone subtypes (15 µg/mL, added 30 min after HKSA stimulation). Single stimulation with HKSA or each histone subunit was used as comparison controls. Shown are averaged TER traces from 8 to 10 individual runs. C: at the time points marked by first arrows, EC was treated with vehicle, heat-killed bacteria (HKSA, HKEC, HKSP, HKPA; 5 × 108 particles/mL), FSL1 (200 ng/mL), CRX-527 (100 ng/mL), TNFα (5 ng/mL), or LPS (100 ng/mL) followed by stimulation with H3 subunit (20 µg/mL, second arrows). TER was monitored over 20–24 h. Bar graph summarizes TER changes in HPAEC monolayers 10 h after agonist stimulation; *P < 0.05, n = 5–8. D: cells were exposed to LPS, TNFα, or heat-killed bacteria: HKSA, HKSP, HKEC, or HKPA alone or in combination with H3 (20 µg/mL). Visualization of permeability for FITC avidin was performed as described in materials and methods. FITC fluorescence images depict FITC-avidin tracer added to culture medium and bound to biotin-coated matrix. Counterstaining of cell nuclei was performed by DAPI. Images were randomly selected from 10 to 12 microscopic fields/condition from three independent experiments: bar = 10 µm. Bar graph depicts quantitative analysis of tracer fluorescence intensity; *P < 0.05. HKEC, heat-killed E. coli; HKPA, heat-killed P. aeruginosa; HKSA, heat-killed S. aureus; HPAEC, human pulmonary artery endothelial cell; LPS, lipopolysaccharide; TER, transendothelial electrical resistance; TNF-α, tumor necrosis factor-α.
The next experiments tested whether submaximal doses of histone subunits can modulate EC barrier dysfunction caused by heat-killed S. aureus bacterial particles (HKSA). HKSA caused abrupt TER decline within 15 min after addition and sustained EC barrier dysfunction reaching maximal levels by 5–6 h. Addition of submaximal doses (15 µg/mL) of H1, H2A, or H2B subunits to EC monolayers after 30-min incubation with HKSA did not affect barrier-disruptive effects of HKSA. In contrast, H3 and H4 at 15 µg/mL strongly exacerbated HKSA-induced EC barrier compromise (Fig. 2B). Because H3 and H4 subunits demonstrated similar direct effects on EC barrier function and augmentation of EC permeability caused by other inflammatory agonists, further experiments focused on H3 as a representative subunit.
We investigated in more detail potential mechanisms of H3-induced exacerbation of EC permeability caused by inflammatory agents. H3 augmented TER decline caused by Gram-negative (heat-killed E. coli, HKEC, and heat-killed P. aeruginosa, HKPA) and Gram-positive (HKSA, and heat-killed S. pneumoniae, HKSP) bacteria known to engage TLR4 and TLR2/6 receptors, respectively (Fig. 2C, top). EC treatment with selective activators of TLR4 and TLR2/6 receptors: CRX527 and FSL1, respectively, or another TLR4 ligand, LPS, showed similar permeability augmenting effect by low doses of H3 (Fig. 2C). Besides effects on TLR-mediated pathways, H3 also augmented EC barrier dysfunction caused by inflammatory cytokine TNFα acting via distinct receptor (Fig. 2C, bottom).
Alternative analysis of EC monolayer permeability using visualization of fluorescent tracer penetration through intercellular gaps by XPerT assay further supported TER data and demonstrated enhanced EC barrier disruption in EC monolayers treated with LPS, TNFα, HKSA, HKEC, HKSP, or HKPA in the presence of submaximal H3 concentrations (Fig. 2D).
H3 Augments Agonist-Induced Disintegration of EC Adhesive Complexes and Cytoskeletal Remodeling
VE-cadherin is a transmembrane protein that forms adhesive contacts between neighboring EC and serves as a key structural element of endothelial cell adherens junctions (AJ) essential for maintaining EC monolayer barrier. Immunofluorescence staining of EC monolayers showed that EC incubation with HKSA, LPS, or TNFα caused disruption of continuous VE-cadherin line at cell–cell junctions and led to formation of intercellular gaps (shown by arrows) indicating compromised EC barrier integrity (Fig. 3A). Cotreatment of EC exposed to LPS, HKSA, or TNFα with submaximal doses of H3 (Fig. 3A) or H4 (data not shown) further exacerbated EC barrier disruption. EC barrier disruption caused by HKSA, LPS, and TNFα was accompanied by F-actin remodeling characterized by weakening of peripheral actin cytoskeletal rim and formation of central F-actin bundles. Addition of H3 or H4 at submaximal concentrations alone to EC monolayers did not cause dramatic cytoskeletal remodeling. However, cell cotreatment with H3 or H4 and HKSA caused collapse of actin cytoskeleton with formation of perinuclear patches (Fig. 3A) and further promoted gap formation in EC treated with LPS and TNFα (Fig. 3B). Analysis of AJ protein complexes assembly state was performed using coimmunoprecipitation assays with VE-cadherin antibody. The results show that HKSA decreased association of AJ component, p120-catenin, and integral cell junction protein, ZO-1, with VE-cadherin. This effect was further augmented by addition of H3 (Fig. 3C, left). Reciprocal coimmunoprecipitation assays with p120-catenin antibody confirmed previous result and demonstrated exacerbation of HKSA-induced dissociation of ZO-1 and VE-cadherin protein complex with p120-catenin caused by additional introduction of H3 (Fig. 3C, right).
Figure 3.

Histone H3 exacerbates disassembly of adherens junctions caused by inflammatory agents. A: HPAECs were stimulated with H3 or H4 (20 µg/mL) alone or in combination with LPS (100 ng/mL) or TNFα (5 ng/mL) for 6 h followed by immunofluorescence staining with VE-cadherin antibody. Actin stress fibers were visualized by staining with Texas Red phalloidin. Intercellular gaps are marked by arrows; bar = 10 µm. Images are representative of three independent experiments, 10–12 microscopic fields/condition. B: quantitative analysis of gap formation. Data represent means ± SD, n = 4, *P < 0.05. C: HPAECs were treated with HKSA (5 × 108 particles/mL) alone or in combination with 20 µg/mL histone H3 followed by coimmunoprecipitation assays with VE-cadherin (left) or p120-catenin (right) antibodies. Western blot analysis of immunocomplexes was performed. Blots were reprobed with the antibodies used for immunoprecipitation as internal controls. Shown are representative blots of four independent experiments. Bar graph depicts quantitative densitometry analysis of Western blot data; *P < 0.05. HPAEC, human pulmonary artery endothelial cell; HKSA, heat-killed S. aureus; LPS, lipopolysaccharide; TNF-α, tumor necrosis factor-α.
Selectivity of Histone Subunits in Exacerbation of Agonist-Induced EC Inflammation
Analysis of histone subunits effects on EC barrier function in earlier experiments aforementioned (Fig. 1) showed exclusive potency of H3 and H4 to induce EC permeability. The next experiments evaluated the potential of different histone subunits to exacerbate EC inflammation caused by other pathogenic agonists. Pulmonary EC challenged with submaximal concentrations of TNFα or LPS for 30 min were coincubated with low doses of histone subunits: H1, H2A, H2B, H3, or H4 for 6 h, and expression of ICAM1 and VCAM1 was evaluated by Western blotting (Fig. 4A). Although both, TNFα and LPS upregulated ICAM1 and VCAM1 protein expression, cotreatment with H1, H2A, or H2B did not cause further increase in ICAM1 and VCAM1 protein levels. In contrast, addition of H3 and H4 further increased ICAM1 and VCAM1 expression primed by TNFα and LPS. These findings were further supported by ELISA assays detecting secretion of soluble ICAM1, IL-6, and IL-8 into culture medium by pulmonary EC stimulated with TNFα or LPS and cotreated with individual histone subunits. Although the levels of soluble ICAM1, IL-6, and IL-8 were elevated by TNFα and LPS, additional cotreatment with H1, H2A, or H2B did not cause further increase in secreted products. In contrast, cotreatment with the same concentrations of H3 and H4 significantly elevated soluble ICAM1 and cytokine production induced by TNFα or LPS (Fig. 4B).
Figure 4.

H3 and H4 augment LPS- and TNF-α-induced inflammatory response in human pulmonary endothelium. HPAECs were stimulated with LPS (25 ng/mL) or TNFα (0.1 ng/mL) alone or combined with 20 µg/mL of indicated histone subunit. A: ICAM-1 and VCAM-1 protein expression was evaluated by Western blotting. B: the levels of secreted sICAM-1, IL-6, and IL-8 were determined by ELISA. Data represent means ± SD, n = 5, *P < 0.05 vs. LPS or TNFα alone. HPAEC, human pulmonary artery endothelial cell; LPS, lipopolysaccharide; TNF-α, tumor necrosis factor-α.
H3 Augments Expression of Inflammatory Markers by EC Exposed to Various Bacterial Strains and Cytokines
The augmenting effect of H3 on endothelial dysfunction was further investigated by analysis of expression levels of adhesion molecules and cytokines in EC challenged with a broad range of inflammatory agonists. HPAEC treatment with heat-inactivated E. coli, P. aeruginosa, S. aureus, and S. pneumoniae upregulated transcriptional activation of VCAM1, ICAM1, IL-6, IL-8, IL-1b, CXCL5, and TNFα, although the absolute levels of gene activation depended on particular stimulation (Fig. 5A, top). Most importantly, H3 at low dose, while causing modest proinflammatory effect on its own, dramatically enhanced inflammatory gene expression in EC challenged with any bacterial strain that we tested. Similar effects were observed in human pulmonary EC challenged with TLR4 ligands LPS and CRX527, TLR2/6 agonists lipoteichoic acid (LTA), peptidoglycan-G (PepG), FSL1, and inflammatory cytokine TNFα. Inflammatory gene expression caused by each agonist was augmented by cotreatment with low-dose H3 histone (Fig. 5A, bottom).
Figure 5.
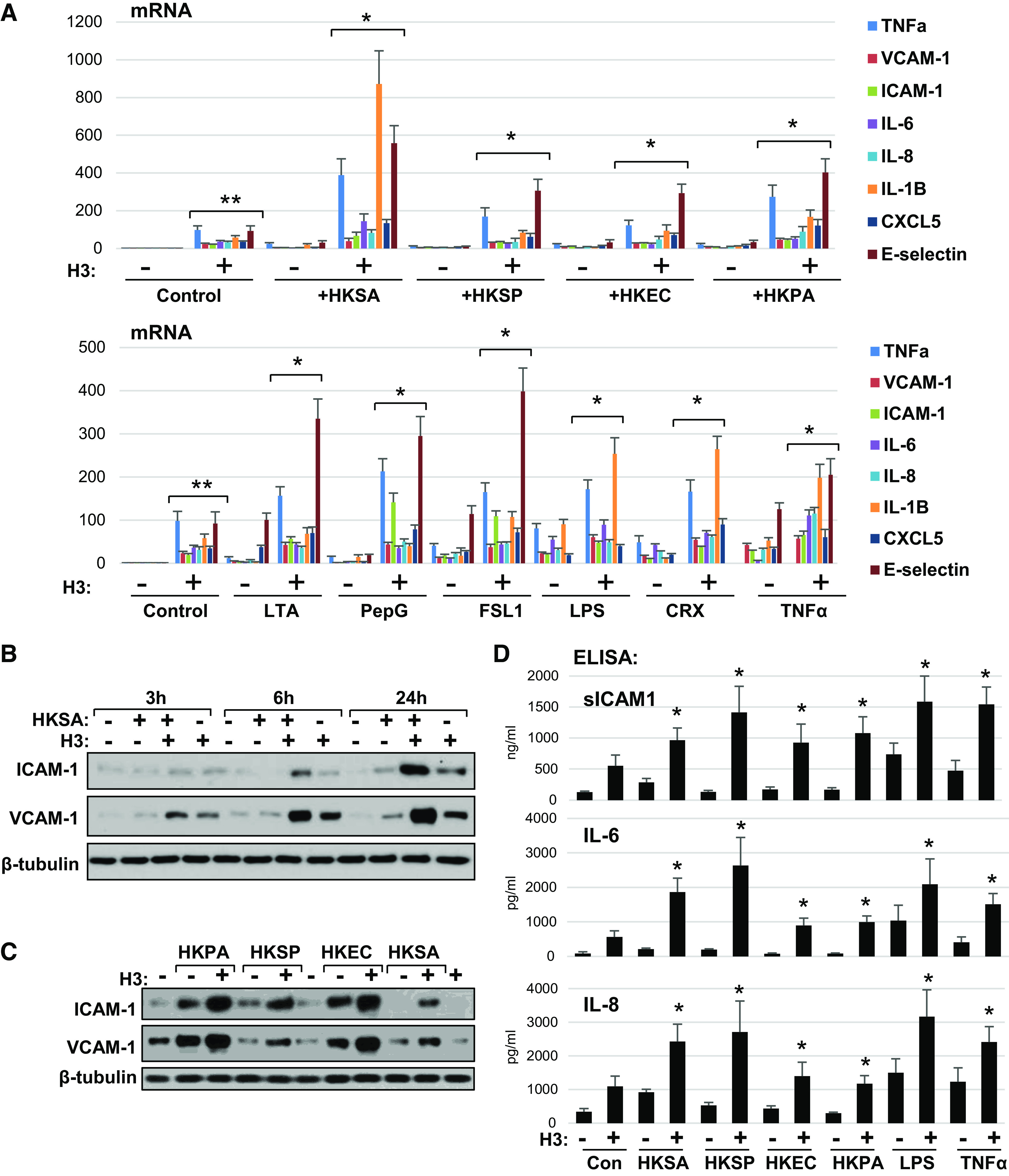
Universal augmenting effect of H3 histone on endothelial inflammation caused by various pathogens. A: HPAEC exposed to indicated heat-killed bacteria (5 × 108 particles/mL) with or without H3 (20 µg/mL) for 3 h were subjected for qPCR analysis. Bar graph depicts changes in expression of mRNA transcripts of various proinflammatory cytokines, chemokines, and adhesion molecules. Data represent means ± SD, n = 5, **P < 0.01 vs. vehicle; *P < 0.01 vs. agonist alone. Bottom: similar qPCR analysis of EC stimulated with bacterial cell wall components: LTA (200 ng/mL), PepG (200 ng/mL), LPS (25 ng/mL); specific TLR2/6 (FSL1, 200 ng/mL) and TLR4 (CRX-527, 25 ng/mL) activators, or TNFα (0.1 ng/mL) alone or in combination with 20 µg/mL H3. B: HKSA and histone H3 were added to HPAEC either alone or in combination and incubated for indicated times followed by Western blot analysis of ICAM-1 and VCAM-1 protein expression levels. C: HPAECs were treated with indicated heat-killed bacteria alone or in combination with histone H3, and immunoblotting was performed to detect ICAM-1 and VCAM-1 protein levels. Shown (B and C) are Western blot results representative of three independent experiments. D: secretion of sICAM-1, IL-6, and IL-8 was determined by ELISA of conditioned media obtained from cells treated with H3 in combination with other agonists as in A. Data are means ± SD, n = 3, *P < 0.05 vs. stimulation with indicated agonist alone. HPAEC, human pulmonary artery endothelial cell; HKSA, heat-killed S. aureus; LPS, lipopolysaccharide; LTA, lipoteichoic acid; PepG, peptidoglycan-G; TNF-α, tumor necrosis factor-α.
Western blot analysis showed time-dependent activation of ICAM1 and VCAM1 protein expression by HKSA-stimulated EC (Fig. 5B) which became detectable after 6 h and reached maximal levels by 24 h after HKSA challenge. EC cotreatment with low H3 dose (20 µg/mL) unable to activate inflammatory response per se, showed significant enhancement of HKSA-induced expression of ICAM1 and VCAM1. Ability of H3 to augment inflammatory activation by other agonists was further investigated in EC challenged with different bacterial pathogens: heat-inactivated P. aeruginosa, S. pneumoniae, and E. coli. (Fig. 5C). Introduction of H3 significantly increased ICAM1 and VCAM1 expression levels in EC stimulated with each bacterial agonist. Additional analysis of cytokine and soluble ICAM1 release by activated EC was performed using ELISA assays (Fig. 5D). The results show robust enhancement of soluble ICAM1, IL-6, and IL-8 release into culture medium by H3 costimulation of human pulmonary EC challenged with heat-inactivated bacteria, LPS, and TNFα.
Inhibition of H3-Induced Exacerbation of Endothelial Inflammation by Heparin
Heparin binds to histone subunits in an electrostatic-dependent manner and has been shown to neutralize deleterious effects of high-dose extracellular histones on cells (23, 24). However, anticoagulant activity of heparin may represent an obstacle for its clinical application as neutralizer of circulating histones in injury settings. We investigated effects of heparin and its modification lacking anticoagulant activity, N-acetylated heparin, on H3-induced exacerbation of EC inflammation caused by TNFα. Cotreatment of TNF-stimulated EC with low-dose H3 histone (20 µg/mL) which, if added alone, did not induce inflammatory response, augmented activation of ICAM1 and VCAM1 protein expression caused by TNFα (Fig. 6A). Addition of acetylated heparin (30 U/mL and 60 U/mL) completely abolished histone-mediated synergistic effect, but retained initial level of ICAM1 and VCAM1 upregulation induced by TNFα.
Figure 6.
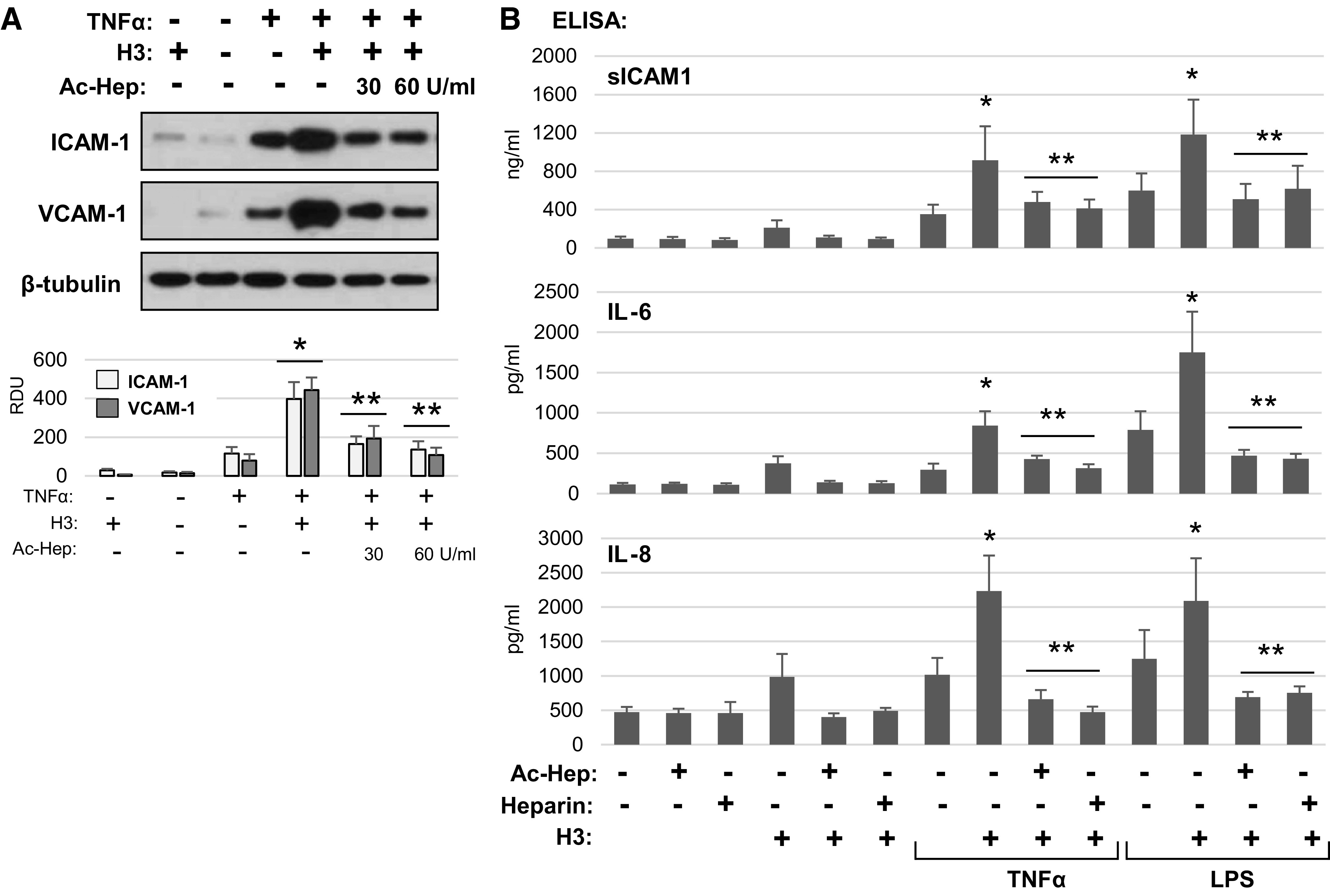
Heparin ameliorates histone-induced endothelial inflammation. A: HPAECs were pretreated with acetylated heparin (30 or 60 U/mL) for 30 min followed by stimulation with histone H3 (20 µg/mL) alone or in combination with TNFα (0.1 ng/mL). ICAM-1 and VCAM-1 protein levels in cell lysates were determined by Western blotting. Probing for α-tubulin was used as a loading control. Bar graphs depict quantitative densitometry analysis of Western blot data; *P < 0.01 vs. H3 or TNFα alone; **P < 0.01 vs. combined treatment with H3 and TNFα; n = 3. B: sICAM-1, IL-6, and IL-8 were determined by ELISA in conditioned media from cells treated with H3 alone or combined with LPS or TNFα in the presence or absence of heparin or acetylated heparin. Data are means ± SD, n = 3, *P < 0.05 vs. LPS or TNFα alone; **P < 0.05 vs. combined agonist/H3 treatment without heparin. HPAEC, human pulmonary artery endothelial cell; LPS, lipopolysaccharide; TNF-α, tumor necrosis factor-α.
Effects of native and N-acetylated heparin on EC inflammation caused by TNFα and LPS with or without H3 cotreatment were further investigated. EC inflammatory activation was evaluated by ELISA assays of ICAM1, IL-6, and IL-8 secretion by activated cells (Fig. 6B). Addition of native and N-acetylated heparin was without effect, whereas H3 alone caused slight elevation of ICAM1, IL-6, and IL-8 secretion by pulmonary EC. This effect of H3 was completely abolished by native and N-acetylated heparin. More importantly, although H3 significantly enhanced ICAM1 and cytokine production by TNFα- and LPS-stimulated EC, this effect was abolished by both, native and N-acetylated heparin. Again, it is important to note that heparin did not completely inhibit proinflammatory effects of TNFα- and LPS when added alone.
Inhibition of TLR4 Signaling Rescues Histone H3-Induced Inflammation in Lung EC
To gather further information about mechanisms of H3 response, we examined the effects of TLR4 (CLI-095) and TLR2/6 (CU-CPT-22) inhibitors on EC inflammation caused by H3 at the dose 30 µg/mL shown to induce prominent EC dysfunction (Figs. 1A and 2A), and in two-hit models of EC failure caused by LPS or HKSA and lower H3 dose (20 µg/mL), which leads to modest inflammation when added alone.
The results showed that NF-κB activation and increased ICAM1 and VCAM1 protein expression induced by H3 alone were abolished by CLI-095, but not by CU-CPT22 (Fig. 7A). In agreement with our previous findings (Figs. 4 and 5), H3 augmented ICAM1 and VCAM1 expression triggered by LPS. This effect was completely abolished by TLR4 inhibitor CLI-095, but was not affected by TLR2/6 inhibitor CU-CPT-22 (Fig. 7B). Increased ICAM1 and VCAM1 expression caused by TLR2/6 ligand HKSA was also augmented by low dose of H3. TLR2/6 inhibitor did not significantly affect HKSA-induced ICAM1 and VCAM1 expression, whereas TLR4 inhibitor strongly attenuated synergistic effect of HKSA and H3 (Fig. 7C).
Figure 7.
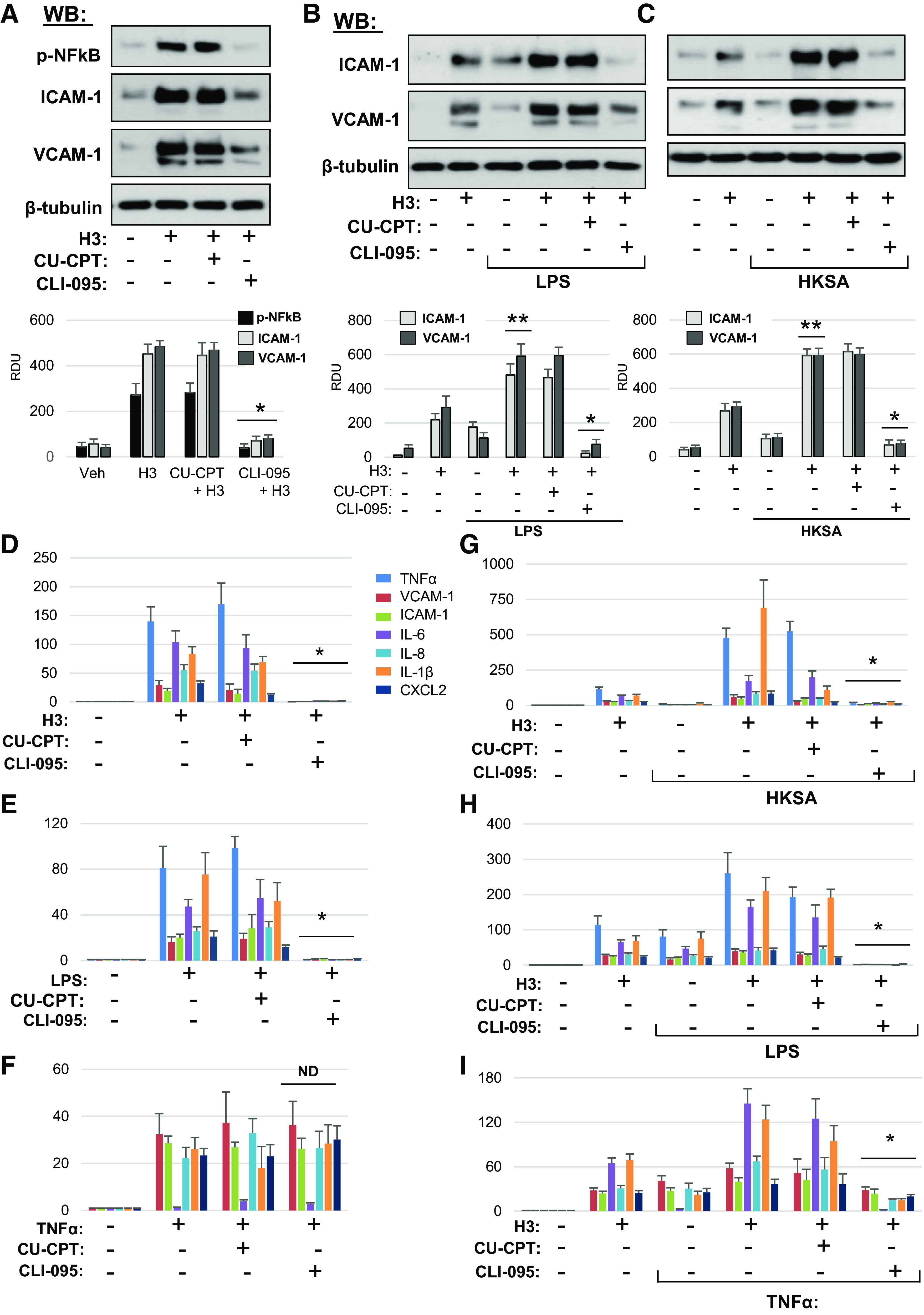
Inhibition of TLR4 signaling rescues histone H3-induced inflammation in lung EC. A: HPAEC pretreated with CU-CPT22 (5 µM) or CLI-095 (1 µM) for 30 min were stimulated with histone H3 (30 µg/mL, 5 h). NF-κB phosphorylation, ICAM-1, and VCAM1 protein levels in cell lysates were determined by Western blotting. Probing for β-tubulin was used as normalization control. Bar graphs depict quantitative densitometry analysis of Western blot data; *P < 0.01 vs. H3 alone; n = 4. Cells preincubated with CLI-095 or CU-CPT22 were treated with LPS (25 ng/mL) (B) or HKSA (5 × 108 particles/mL) (C) for 1 h followed by addition of 15 µg/mL H3 for 5 h. ICAM-1 and VCAM1 protein expression was measured by Western blotting. Probing for β-tubulin was used as normalization control. Bar graphs depict quantitative densitometry analysis of Western blot data; *P < 0.01 vs. HKSA+H3 or LPS+H3; **P < 0.01 vs. H3, LPS, or HKSA alone; n = 4. Effects of EC pretreatment with CU-CPT22 and CLI-095 on mRNA expression of inflammatory markers caused by H3 (D), LPS (E), and TNFα (F). HPAEC preincubated with vehicle, CLI-095, or CU-CPT22 were stimulated with H3 in combination with HKSA (G), LPS (H), or TNFα (I). Expression of markers of inflammation was analyzed by qRT-PCR. Data represent means ± SD, n = 4–6, *P < 0.01 vs. treatment without inhibitors. HPAEC, human pulmonary artery endothelial cell; HKSA, heat-killed S. aureus; LPS, lipopolysaccharide; ND, no difference; TNF-α, tumor necrosis factor-α; LPS, lipopolysaccharide.
Next, to assess the TLR4 and TLR2/6 involvement in the H3-augmenting effect of EC activation caused by bacterial pathogens and TNFα, we evaluated transcriptional activation of inflammatory markers. First, we investigated the effects of TLR4 and TLR2/6 inhibitors on EC inflammation caused by H3 alone (30 µg/mL). Although TLR2/6 inhibitor was without effect, pretreatment with TLR4 inhibitor CLI-095 abolished the H3-induced mRNA expression of inflammation markers TNFα, VCAM1, ICAM1, IL-6, IL-8, IL-1β, and CXCL2 (Fig. 7D). Control experiments were performed with TLR4 ligand LPS and inflammatory cytokine TNFα acting via distinct receptors. As expected, TLR4 but not TLR2/6 inhibitor suppressed LPS inflammatory response (Fig. 7E). Neither TLR2/6 nor TLR4 inhibitors affected the elevation of inflammatory markers caused by TNFα (Fig. 7F).
The mechanism of H3 synergistic effect on agonist-induced EC inflammation was further investigated in two-hit models. HPAEC stimulation with submaximal dose of HKSA did not cause pronounced inflammatory cell activation on its own, but the response was dramatically augmented by H3 subunit. TLR2/6 inhibitor CU-CPT22 partially attenuated this effect, and TLR4 inhibitor CLI-095 almost completely abolished EC inflammation caused by HKSA-H3 combination (Fig. 7G). In experiments with LPS, expression of inflammatory markers caused by LPS or H3 alone was augmented by combined treatment with both agonists. Pretreatment with TLR2/6 inhibitor did not have statically significant effect, whereas inhibition of TLR4 abolished inflammation caused by combined H3 + LPS treatment (Fig. 7H). Finally, EC inflammation caused by submaximal dose of TNFα was augmented by H3 but was not affected by TLR2/6 inhibition and was partially attenuated by inhibitor of TLR4 (Fig. 7I). Collectively, these results suggest that synergistic effects of histone H3 (as well as histone H4, data not shown) on pulmonary EC inflammation and increased permeability caused by proinflammatory agonists acting via TLR2/6, TLR4, and non-TLR receptors are mediated at least in part by TLR4-dependent mechanism.
Circulating Histones Augment Acute Lung Injury Caused by LPS and HKSA
Effects of circulating histones on lung injury caused by bacterial pathogens were evaluated. Histones isolated from calf thymus were injected intravenously in mice with LPS solution administered intratracheally, as detailed in methods and materials. Pilot studies determined safe histone dose range of 10–25 mg/kg which was well tolerated by mice, whereas increased histone doses caused animal death. Intravenously injected histone complex caused slight but statistically significant elevation of cell count in BAL (Fig. 8A, left) which was also accompanied by increased protein concentration in BAL samples (Fig. 8A, right) reflecting mild lung vascular leak. Administration of LPS caused robust increase in BAL cell count and protein concentrations, which was augmented by coadministration of histones. Exacerbation of LPS-induced lung barrier dysfunction by circulating histones was further demonstrated by increased Evans blue infiltration in the lung parenchyma (Fig. 8B). It was also noted that in addition to increased accumulation of Evans blue, the lungs of mice cotreated with LPS and histones also exhibited signs of intravascular coagulation manifested by increased red color intensity of lung preparations. The augmenting effect of histones on LPS-induced lung inflammation was further evaluated by RT-PCR analysis of IL-6, IL-1β, KC, CXCL2, and TNFα transcripts in lung tissue samples (Fig. 8C). Although histone administration alone caused low-grade activation of inflammatory marker transcripts, LPS stimulation caused more pronounced inflammatory response, which was further boosted by cotreatment with LPS and histones.
Figure 8.
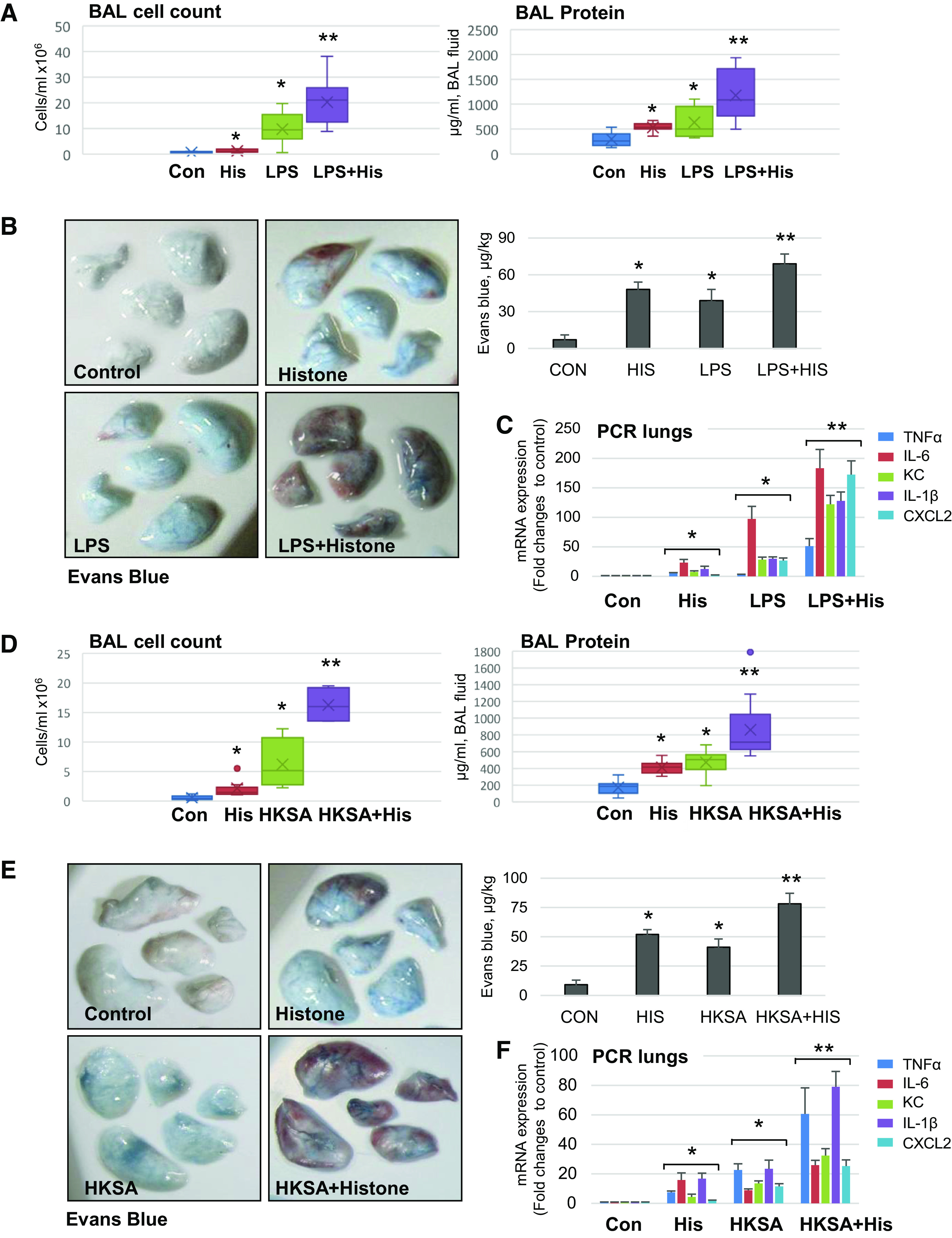
Circulating histone exacerbates LPS- and HKSA-induced ALI in mice. C57BL/6 mice were treated with calf thymus histone (25 mg/kg iv), LPS (0.75 mg/kg it), or HKSA (2 × 108 particles/mouse, in) alone, or with combination of histone with LPS (A–C) or HKSA (D–F) for 18 h. Lung injury was evaluated by analysis of BAL samples for total cell count and protein content (A and D). Shown data are means ± SD, n = 8–12, *P < 0.05 vs. control; **P < 0.05 vs. histone or agonist alone by ANOVA. B and E: images of Evans blue incorporation in the lungs. Bar graphs depict quantitative analysis of Evans blue-labeled albumin extravasation performed by spectrophotometric analysis of Evans blue extracted from the lung tissue samples; *P < 0.05 vs. control; **P < 0.05 vs. H3, LPS, or HKSA alone, n = 4. C and F: real-time PCR was performed from mice lung samples to determine mRNA transcript levels of indicated proinflammatory cytokines and chemokines. Shown data are means ± SD, n = 6, *P < 0.05 vs. control; **P < 0.05 vs. histone or agonist alone. HKSA, heat-killed S. aureus; LPS, lipopolysaccharide.
Synergistic effect of circulating histones on parameters of lung injury and inflammatory marker expression was observed in another model of ALI caused by intranasal administration of HKSA. Injection of submaximal dose of histone in jugular vein exacerbated HKSA-induced lung injury reflected by increased protein content and inflammatory cell counts in BAL (Fig. 8D). Experiments with Evans blue injection showed increased accumulation of Evans blue dye in the lungs of HKSA and histone cotreated animals versus single administration of HKSA or histones. These data indicate prominent vascular leak in HKSA-histone model (Fig. 8E). Histone injection also exacerbated HKSA-induced expression of IL-6, IL-1β, KC, CXCL2, and TNFα transcripts in the lungs (Fig. 8F).
DISCUSSION
Extracellular histones have been viewed as a sole factor causing cell death and endothelial barrier dysfunction. Indeed, high histone concentrations cause severe endothelial damage. However, in most pathological conditions, the concentrations of circulating histones are below the threshold levels causing direct EC damage. This study reveals a critical role of specific histone subunits in exacerbating the preexisting endothelial dysfunction and lung injury caused by bacterial pathogens or cytokine storm.
Our data support previous reports that identified histone subunits H3 and H4 as the major contributors to histone-induced injury (6, 11). Comprehensive analysis of various histone subunits on endothelial dysfunction performed in this study emphasizes pathological importance of histone H3 and H4, whereas histones H1, H2A, H2B were neither capable of inducing endothelial damage on their own, nor they affected the magnitude of EC barrier disfunction and inflammation caused by other agonists. Interestingly, histone H1 has been reported to possess neurotoxic properties (25). However, our data rule out the involvement of H1 and H2 subunits in endothelial dysfunction. It is intriguing that only a subtle structural difference exists between histone subunits exemplified by H3/H4 being arginine-rich versus lysine-rich histones H1, H2A, and H2B. The physicochemical nature of such dramatic differences in biological activities of arginine-rich and lysine-rich histones: the former being potent cytotoxic and latter being largely inert at least in regulating vascular endothelial function, awaits further investigation. It is also possible that H3 and H4 histones may be more prone to post-translational modifications facilitating their interactions with cell death receptors.
Several mechanisms have been suggested to mediate histone-induced endothelial dysfunction including elevation of intracellular calcium (11), apoptotic or autophagic EC death (26), and disruption of adherens junction (17). Our results show that breakdown of EC junctional assembly and reduced interaction between the components of adherens and tight junction complexes is the primary mechanism of histone-induced EC barrier breach. In contrast, our inhibitory analysis showed that calcium chelator BAPTA-AM and pan-caspase inhibitors failed to offer protection against histone-caused permeability and inflammatory gene expression (data not shown).
Activation of TLR receptor-mediated signaling, especially TLR4 and TLR2 pathways, has been described to mediate toxic effects of histones (14, 27). In line with the published data (8), histone stimulation of EC induced cytokine storm, as illustrated by the upregulation of mRNA expression and secretory levels of inflammatory cytokines TNF-α, IL-6, and IL-8. Our data also support activation of TLR4 as a trigger of histone-induced endothelial damage. Inhibition of TLR4 signaling by CLI-095 blocked H3-induced EC dysfunction. On the contrary, TLR2/6 antagonist CU-CPT-22 did not exhibit protective effects against histone H3-induced endothelial permeability and inflammation. Nevertheless, we cannot rule out additional mechanisms of histone-induced endothelial dysfunction.
The other major finding of this study is the ability of histone to act in synergy with other agonists to exacerbate ALI and augment agonist-induced endothelial dysfunction. The synergistic effects were again limited to histone subunits H3 and H4. Low levels of H3 and H4 that otherwise do not cause endothelial permeability or inflammation, induced severe endothelial damage when combined with submaximal doses of inflammatory agonists which, when added alone, only caused mild EC dysfunction. These findings reflect a clinical scenario and indicate that only a slight increase in the circulating histone levels in asymptomatic or moderately infected individuals with bacterial or viral infections may lead to serious illness. The concentrations of histones used in this two-hit cell culture model of ALI were higher than base levels of circulating histones reported in healthy subjects, but they were significantly lower than histone levels found in patients with ARDS and patients with sepsis (5, 7). More expanded screening showed that synergistic effect of H3 and H4 histones on agonist-induced EC dysfunction was observed with a broad range of agonists including TLR4 ligands (LPS and CRX-527), TLR2 activator (FSL1), heat-killed bacteria (S. aureus, E. coli, S. pneumoniae, P. aeruginosa), and inflammatory cytokine TNF-α. These results suggest H3 and H4 histones as a universal exacerbating factor in a wide variety of pathological settings causing endothelial permeability and inflammation. Synergistic effect of histones in exacerbating lung injury was also confirmed in animal models which showed that lung injury parameters in mice with intratracheal injection of LPS or HKSA were significantly worsened by intravenous coinjection of submaximal dose of calf thymus histone mixture.
Neutralization of histone activities with antihistone antibodies, activated protein C or heparin has been demonstrated to be effective in attenuating histone toxicity both in vitro and in vivo (7, 11, 24, 28). Among these, heparin appears to be the most promising agent owing to its efficient binding to histones. Moreover, to avoid undesired side effects of heparin such as bleeding, nonanticoagulant modified heparins and small polyanions have been developed that retain effective protection against histone toxicity (23, 24). Acetylated heparin without anticoagulant activity present in original heparin has been tested as a treatment of choice due to lack of hemorrhagic side effects observed with higher dosage of heparin and was shown to have better cytoprotective effects than native heparin (29). Our data show that acetylated heparin not only prevented histone-induced inflammation by interfering with histone-induced TLR4 activation, but also attenuated synergistic effects of histone with TNFα and LPS on endothelial dysfunction in EC culture and rescued histone-induced ALI in mouse model.
In summary, this study reveals a universal role of circulating histones in exacerbation of vascular endothelial dysfunction and enhancing the severity of mild inflammatory syndromes of different origin. These findings may have important clinical significance as they demonstrate that even a modest increase in extracellular histone levels can act as a severe exacerbating factor in conjunction with other EC barrier disruptive or proinflammatory agents. This notion may be true in various clinical scenarios such as mild bacterial or viral infection, including the current global COVID-19 pandemic, that would otherwise be noninjurious but may lead to severe lung injury and multiple organ failure. Thus, therapeutic approaches aimed at neutralization of circulating DAMPs and uncoupling DAMP-pathogen synergistic signaling hold promise in mitigation of severe illness and organ damage in patients with sepsis, trauma, or acute lung infection.
DATA AVAILABILITY
The data that support the findings of this study will be made available upon reasonable request from the corresponding author.
GRANTS
This study was supported by NIH Grants HL152761, HL155051, HL076259, and HL146829 from the National Heart, Lung, and Blood Institute and an Institute for Clinical & Translational Research (ICTR) grant from University of Maryland Baltimore.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.G.B. and A.A.B. conceived and designed research; J.K., R.B., Y.L., C.-O.Z., Y.K., and P.K. performed experiments; J.K., R.B., Y.L., C.-O.Z., Y.K., P.K., K.G.B., and A.A.B. analyzed data; J.K., R.B., Y.L., C.-O.Z., Y.K., P.K., K.G.B., and A.A.B. interpreted results of experiments; J.K., C.-O.Z., Y.K., and P.K. prepared figures; P.K. and A.A.B. drafted manuscript; K.G.B. and A.A.B. edited and revised manuscript; J.K., R.B., Y.L., C.-O.Z., Y.K., P.K., K.G.B., and A.A.B. approved final version of manuscript.
ACKNOWLEDGMENTS
Present address of J. Kim: Dept. of Pulmonology, Allergy, and Critical Care Medicine, CHA Bundang Medical Center, CHA University, Republic of South Korea.
REFERENCES
- 1. Berger SL. Histone modifications in transcriptional regulation. Curr Opin Genet Dev 12: 142–148, 2002. doi: 10.1016/S0959-437X(02)00279-4. [DOI] [PubMed] [Google Scholar]
- 2. Allam R, Kumar SV, Darisipudi MN, Anders HJ. Extracellular histones in tissue injury and inflammation. J Mol Med (Berl) 92: 465–472, 2014. doi: 10.1007/s00109-014-1148-z. [DOI] [PubMed] [Google Scholar]
- 3. Kawai C, Kotani H, Miyao M, Ishida T, Jemail L, Abiru H, Tamaki K. Circulating extracellular histones are clinically relevant mediators of multiple organ injury. Am J Pathol 186: 829–843, 2016. doi: 10.1016/j.ajpath.2015.11.025. [DOI] [PubMed] [Google Scholar]
- 4. Silk E, Zhao H, Weng H, Ma D. The role of extracellular histone in organ injury. Cell Death Dis 8: e2812, 2017. doi: 10.1038/cddis.2017.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abrams ST, Zhang N, Manson J, Liu T, Dart C, Baluwa F, Wang SS, Brohi K, Kipar A, Yu W, Wang G, Toh CH. Circulating histones are mediators of trauma-associated lung injury. Am J Respir Crit Care Med 187: 160–169, 2013. doi: 10.1164/rccm.201206-1037OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bosmann M, Grailer JJ, Ruemmler R, Russkamp NF, Zetoune FS, Sarma JV, Standiford TJ, Ward PA. Extracellular histones are essential effectors of C5aR- and C5L2-mediated tissue damage and inflammation in acute lung injury. FASEB J 27: 5010–5021, 2013. doi: 10.1096/fj.13-236380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lv X, Wen T, Song J, Xie D, Wu L, Jiang X, Jiang P, Wen Z. Extracellular histones are clinically relevant mediators in the pathogenesis of acute respiratory distress syndrome. Respir Res 18: 165, 2017. doi: 10.1186/s12931-017-0651-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ekaney ML, Otto GP, Sossdorf M, Sponholz C, Boehringer M, Loesche W, Rittirsch D, Wilharm A, Kurzai O, Bauer M, Claus RA. Impact of plasma histones in human sepsis and their contribution to cellular injury and inflammation. Crit Care 18: 543, 2014. doi: 10.1186/s13054-014-0543-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang Y, Wen Z, Guan L, Jiang P, Gu T, Zhao J, Lv X, Wen T. Extracellular histones play an inflammatory role in acid aspiration-induced acute respiratory distress syndrome. Anesthesiology 122: 127–139, 2015. doi: 10.1097/ALN.0000000000000429. [DOI] [PubMed] [Google Scholar]
- 10. Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, Lohmeyer J, Preissner KT. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One 7: e32366, 2012. doi: 10.1371/journal.pone.0032366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, Taylor FB, Esmon NL, Lupu F, Esmon CT. Extracellular histones are major mediators of death in sepsis. Nat Med 15: 1318–1321, 2009. doi: 10.1038/nm.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karki P, Birukov KG, Birukova AA. Extracellular histones in lung dysfunction: a new biomarker and therapeutic target? Pulm Circ 10: 2045894020965357, 2020. doi: 10.1177/2045894020965357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Allam R, Scherbaum CR, Darisipudi MN, Mulay SR, Hägele H, Lichtnekert J, Hagemann JH, Rupanagudi KV, Ryu M, Schwarzenberger C, Hohenstein B, Hugo C, Uhl B, Reichel CA, Krombach F, Monestier M, Liapis H, Moreth K, Schaefer L, Anders HJ. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol 23: 1375–1388, 2012. doi: 10.1681/ASN.2011111077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol 187: 2626–2631, 2011. doi: 10.4049/jimmunol.1003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang X, Li L, Liu J, Lv B, Chen F. Extracellular histones induce tissue factor expression in vascular endothelial cells via TLR and activation of NF-κB and AP-1. Thromb Res 137: 211–218, 2016. doi: 10.1016/j.thromres.2015.10.012. [DOI] [PubMed] [Google Scholar]
- 16. Allam R, Darisipudi MN, Tschopp J, Anders HJ. Histones trigger sterile inflammation by activating the NLRP3 inflammasome. Eur J Immunol 43: 3336–3342, 2013. doi: 10.1002/eji.201243224. [DOI] [PubMed] [Google Scholar]
- 17. Meegan JE, Yang X, Beard RS, Jannaway M, Chatterjee V, Taylor-Clark TE, Yuan SY. Citrullinated histone 3 causes endothelial barrier dysfunction. Biochem Biophys Res Commun 503: 1498–1502, 2018. doi: 10.1016/j.bbrc.2018.07.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Karki P, Ke Y, Tian Y, Ohmura T, Sitikov A, Sarich N, Montgomery CP, Birukova AA. Staphylococcus aureus-induced endothelial permeability and inflammation are mediated by microtubule destabilization. J Biol Chem 294: 3369–3384, 2019. doi: 10.1074/jbc.RA118.004030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Birukova AA, Smurova K, Birukov KG, Kaibuchi K, Garcia JG, Verin AD. Role of Rho GTPases in thrombin-induced lung vascular endothelial cells barrier dysfunction. Microvasc Res 67: 64–77, 2004. doi: 10.1016/j.mvr.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 20. Dubrovskyi O, Birukova AA, Birukov KG. Measurement of local permeability at subcellular level in cell models of agonist- and ventilator-induced lung injury. Lab Invest 93: 254–263, 2013. doi: 10.1038/labinvest.2012.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Birukova AA, Fu P, Xing J, Cokic I, Birukov KG. Lung endothelial barrier protection by iloprost in the 2-hit models of ventilator-induced lung injury (VILI) involves inhibition of Rho signaling. Transl Res 155: 44–54, 2010. doi: 10.1016/j.trsl.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Birukova AA, Xing J, Fu P, Yakubov B, Dubrovskyi O, Fortune JA, Klibanov AM, Birukov KG. Atrial natriuretic peptide attenuates LPS-induced lung vascular leak: role of PAK1. Am J Physiol Lung Cell Mol Physiol 299: L652–L663, 2010. doi: 10.1152/ajplung.00202.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Meara CHO, Coupland LA, Kordbacheh F, Quah BJC, Chang C-W, Simon Davis DA, Bezos A, Browne AM, Freeman C, Hammill DJ, Chopra P, Pipa G, Madge PD, Gallant E, Segovis C, Dulhunty AF, Arnolda LF, Mitchell I, Khachigian LM, Stephens RW, von Itzstein M, Parish CR. Neutralizing the pathological effects of extracellular histones with small polyanions. Nat Commun 11: 6408, 2020. doi: 10.1038/s41467-020-20231-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wildhagen KCAA, García de Frutos P, Reutelingsperger CP, Schrijver R, Aresté C, Ortega-Gómez A, Deckers NM, Hemker HC, Soehnlein O, Nicolaes GAF. Nonanticoagulant heparin prevents histone-mediated cytotoxicity in vitro and improves survival in sepsis. Blood 123: 1098–1101, 2014. doi: 10.1182/blood-2013-07-514984. [DOI] [PubMed] [Google Scholar]
- 25. Gilthorpe JD, Oozeer F, Nash J, Calvo M, Bennett DL, Lumsden A, Pini A. Extracellular histone H1 is neurotoxic and drives a pro-inflammatory response in microglia. F1000Res 2: 148, 2013. doi: 10.12688/f1000research.2-148.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ibañez-Cabellos JS, Aguado C, Pérez-Cremades D, García-Giménez JL, Bueno-Betí C, García-López EM, Romá-Mateo C, Novella S, Hermenegildo C, Pallardó FV. Extracellular histones activate autophagy and apoptosis via mTOR signaling in human endothelial cells. Biochim Biophys Acta Mol Basis Dis 1864: 3234–3246, 2018. doi: 10.1016/j.bbadis.2018.07.010. [DOI] [PubMed] [Google Scholar]
- 27. cSemeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, Esmon CT. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood 118: 1952–1961, 2011. doi: 10.1182/blood-2011-03-343061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li L, Yu S, Fu S, Ma X, Li X. Unfractionated heparin inhibits histone-mediated coagulation activation and thrombosis in mice. Thromb Res 193: 122–129, 2020. doi: 10.1016/j.thromres.2020.06.007. [DOI] [PubMed] [Google Scholar]
- 29. Zhang Y, Zhao Z, Guan L, Mao L, Li S, Guan X, Chen M, Guo L, Ding L, Cong C, Wen T, Zhao J. N-acetyl-heparin attenuates acute lung injury caused by acid aspiration mainly by antagonizing histones in mice. PLoS One 9: e97074, 2014. doi: 10.1371/journal.pone.0097074. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study will be made available upon reasonable request from the corresponding author.