

Abstract
Sixty years ago, the geneticist James Neel proposed that the epidemics of obesity and diabetes today may have evolutionary roots. Specifically, he suggested that our ancestors may have accumulated mutations during periods of famine that provided a survival advantage at that time. However, the presence of this “thrifty genotype” in today’s world where food is plentiful would predispose us to obesity and diabetes. The ‘thrifty gene’ hypothesis, attractive to some, has been challenged over the years. We have previously postulated that the loss of the uricase gene, resulting in a rise in serum and intracellular uric acid levels, satisfies the criteria of a thrifty genotype mutation. Here we review and bring up-to-date the evidence supporting the hypothesis as well as discuss the current arguments that challenge this hypothesis. While further studies are needed to test the hypothesis, the evidence supporting a loss of uricase as a ‘thrifty gene’ is substantial and supports a role for evolutionary biology in the pathogenesis of the current obesity and diabetes epidemics.
Keywords: Evolution, uric acid, gout, obesity, diabetes, uricase, thrifty gene hypothesis
Introduction
In 1962, James Neel proposed that the remarkable rise in obesity and diabetes in western civilization might have evolutionary origins (1). Specifically, he suggested that there were likely times in our past where food and water availability may have been tenuous, and that spontaneously occurring mutations that could provide survival and reproductive advantage during these harsh times would have been “naturally selected” and integrated into future generations. While this “thrifty genotype” would have been advantageous during a time of food scarcity, the persistence of these genotypes into times when food is plentiful may be poorly adapted and lead to obesity or diabetes (1). Accordingly, the thrifty gene hypothesis could not only explain the remarkable rise during the twentieth century of not only obesity and diabetes, but potentially the rise of other conditions, including high blood pressure, fatty liver disease, chronic kidney disease and heart disease.
While the thrifty gene hypothesis has been attractive, there have been very few gene candidates. This has led to alternative hypotheses, such as our ability to avoid predation in the last million years has allowed obesity to develop in our society (the “drifty gene” hypothesis) (2). We have suggested that, rather than the gain of a new undefined gene, the loss of the uricase gene meets the criteria of a thrifty genotype (3) (Table 1). Here we review the hypothesis that the uricase mutation, by raising serum and intracellular uric acid levels, aided the survival of our ancestors during a prolonged period of seasonal famine, but today appears to increase our susceptibility to obesity and diabetes in response to western diet. We also discuss the challenges and limitations to the hypothesis.
Table 1.
Arguments for Uricase as a Thrifty Gene
| • The uricase mutation occurred during periods of climate change over millions of years leading to loss of food resources and the near extinction of our ancestors |
| • The uricase mutation triggered biologic changes that improved survival in the setting of starvation or threatened starvation. |
| • The introduction of western diet set the stage for the uricase mutation to switch from a mechanism to prevent starvation to one driving obesity and metabolic syndrome. |
| • Evidence supports the premise that reducing uric acid levels or fructose intake can prevent or treat obesity and metabolic syndrome. |
Evidence that the Uricase Mutation May have acted as a “Thrifty Gene”
Uricase and the Regulation of Uric acid
Uric acid is a nitrogenous waste product that is generated during the breakdown of RNA, DNA and ATP. In most mammals circulating uric acid levels are around 1 mg/dL, as blood levels are tightly regulatable by an enzyme, uricase, that degrades uric acid, eventually generating allantoin (4). However, unlike most mammals, uricase activity progressively weakened during primate evolution, and became completely silenced in ancestors of humans prior to the emergence of lesser apes, millions of years ago (5).
A consequence of the uricase mutation is that serum uric acid levels are higher and less easily regulatable. While animals expressing uricase typically maintain serum uric acid in the 1 to 2 mg/dL range, studies of apes that lack uricase document a doubling of serum uric acid to levels of 3 or 4 mg/dL (4). Furthermore, a study of the Yanomamö Indians that live on their traditional diets in southern Venezuela also found serum uric acid levels in the 3 to 4 mg/dl range (4). In contrast, serum uric acid levels tend to be higher in western culture, with levels typically in the 3 to 6 mg/dL range, and with over 30 million people in the USA having serum uric acid levels of 6.8 mg/dL or higher (defined as hyperuricemic). A consequence of higher serum uric acid levels is the predisposition to gout, in which uric acid crystallizes into the joints to cause a severe and painful arthritis. Indeed, gout is extremely rare in indigenous populations, and its frequency increases in modern societies (4).
The Uricase Mutation Occurred During Seasonal Starvation and Extinction
Molecular biology studies involving comparisons among extant primates have demonstrated that there was a stepwise reduction in uricase activity in primates coinciding with a decline in global temperatures from the Eocene climatic optimum through the Oligocene and early Miocene (5, 6). Uricase gene activity became silenced in a common ancestor of the ape/human lineage following a single amino acid replacement (mutation F222S) (6). The timing of this latter mutation occurred before the split of the lesser apes from the great apes. As the fossil evidence of the earliest apes date to 20 to 24 million years ago, the timing of the mutation likely occurred between that time and the split of the orangutan from the African great apes around 12 million years ago (5, 7).
Studies suggest that the earliest apes lived in East Africa, and were frugivorous and lived in tropical rain forests and deciduous woodlands (reviewed in (8)). They were initially quite successful and within a few million years had expanded to a half-dozen or more species. However, global temperatures which had been declining from the Eocene climate optimum continued to fall, resulting in the accumulation of ice in the Antarctic and a fall in sea level. By 17 to 18 million years ago, land bridges developed that allowed early apes to migrate out of Africa to Eurasia. Temperatures continued to fall, resulting in the Middle Miocene Climate Transition between 13 to 15 million years ago and the Middle Miocene Disruption (14.8 to 14.1 million years ago) in which nearly 30 percent of species became extinct.
Initially the apes that arrived in Eurasia continued to consume fruits all year, but as the climate cooled, there was a progressive change from subtropical forest to open woodlands and deciduous forests interspersed with open country. The changes in habitat resulted in greater seasonality, leading to decreases in fruit availability during the cooler seasonal months. Evidence for seasonal starvation is apparent from the presence of recurrent rings of enamel hypoplasia on the incisors of growing teeth in ancestral apes found at sites in Spain and eastern Europe (reviewed in (8)).
A consequence of the loss of fruit availability and seasonal starvation was the necessity for all fruit-eaters to find other foods. During this period the ancestral apes became more terrestrial, foraging for ‘fallback’ foods in the ground such as tubers and roots. This led to a selective advantage for apes that were better able to do this, and there was rapid evolution of apes in Europe with fossil evidence of changes in the axial skeleton, the development of a more robust (stronger) jaw, and for thicker enameled teeth. Nevertheless, as global cooling continued, the ape colonies declined, retreating to small isolated colonies (refugia) and eventually the last European ape became extinct around 8 to 9 million years ago (reviewed in (8)).
While the apes in Europe became extinct, the fossil record suggests that some of these apes may have migrated back to Africa or other sites to later become modern apes and humans. This ‘back to Africa’ hypothesis is based on evidence that the morphological and genetic change during the middle Miocene of Europe over a period of 15 to 8 million years ago in response to climate change were shared and retained in both the lineage leading to the orangutan in Asia and to the African apes and humans in Africa (reviewed in (8)). Indeed, fossil evidence suggests that some skeletal and dental adaptations that were acquired in Europe show up later in our primate lineage in Africa. For example, Kenyapithecus kizili identified in Paşalar, Turkey is a candidate ancestor of great apes and humans, and had a unique tooth structure that does not appear until two million years later in Kenyapithecus wickeri from Fort Ternan, Kenya (9). Other fossil apes may also have migrated to Asia where they evolved to become the lesser apes or gibbons while others founded the orangutan lineage (reviewed in (8)).
Thus, there is fossil evidence that a European ape may have returned to Africa to become the ancestor of the African great apes and humans (reviewed in (8)). Consistent with the fossil evidence, molecular biology studies confirm that the Middle Miocene Climatic Transition and extinctions coincided with a marked increase in mutations (noted by genetic duplications) that were incorporated into our genome (10). Thus, relevant to the thrifty gene hypothesis, the uricase mutation, which is shared by all apes and humans, may have occurred during this period of food shortage and starvation and must have carried a selection advantage as no apes survived that lacked this mutation (6).
Benefits of the Uricase Mutation in Circumstances of Seasonal Starvation
Uric acid was originally viewed as a nitrogen-rich biologic waste product. It is known, for example, that many fish and amphibians excrete nitrogenous wastes as ammonia, while mammals excrete mostly urea, and reptiles and birds (who also independently lost uricase in the past) excrete nitrogen as uric acid. It was thought that the loss of uricase in reptiles and birds provided a survival advantage as this allowed the precipitation of uric acid in the cloaca so it could be excreted as guano with minimal water loss. However, this did not explain why humans lack uricase, as we utilize urea as our means for excreting nitrogenous wastes.
One hypothesis to explain the reason for the loss of uricase was that increased serum uric acid, could function as an anti-oxidant, since uric acid can react with oxidants (11). Hyperuricemia could thereby counteract the mutation in L-gulono-lactone oxidase that led to the inability to make ascorbate (vitamin C) millions of years earlier(11). It was further suggested that the anti-oxidant benefits of uric acid might help improve longevity and also reduce mortality, especially from cancer (11).
Evidence supporting a role for uric acid as an anti-oxidant has been primarily been reported in the neurological literature, suggesting benefits of uric acid in multiple sclerosis and Parkinson’s disease. However, clinical trials were negative (12, 13). In addition, while uric acid blocks peroxynitrite, it also generates other reactive oxygen species and does not reduce overall oxidant levels released by inflammatory cells (14, 15). Indeed, lowering serum uric acid with pegylated uricase also does not increase oxidative stress in humans (16), while raising uric acid by blocking uricase actually increases oxidative stress, especially inside the cell (17, 18). Furthermore, the protective effect of raising uric acid in models of multiple sclerosis cannot be reproduced by ascorbate and appears to be due to an acute blockade of the blood-brain barrier (19, 20). This might be explained by the known effects of uric acid to acutely reduce endothelial nitric oxide (21) and could explain why acute administration of uric acid may provide some protection in acute ischemic stroke, especially in women (22). Finally, studies using inosine as a means for raising uric acid are difficult to interpret since the anti-inflammatory effects of inosine (23) may be due to its ability to bind adenosine or generate ATP via the salvage pathway as opposed to raising uric acid (23, 24).
It is thus not surprising that a higher serum uric acid does not protect against cancer and aging, but rather is associated with increased total and cardiovascular mortality (25, 26) as well as risk for cancer (27, 28). Indeed, knocking out or inhibiting uricase increases tumor growth and metastases (29).
The most compelling evidence of beneficial effects of the loss of uricase are related to the ability of fructose to aid survival (Table 2) (30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45). As mentioned, the main nutrient of ancestral apes was fruit, which is rich in fructose. Fructose has been shown to be relatively unique from other nutrients in that it causes rapid utilization of ATP followed by persistent maintenance of low intracellular levels due to suppression of mitochondrial function (18, 38, 46, 47) and the inhibition of ATP regeneration (such as by blocking AMP-activated protein kinase)(39, 48). This mechanism involves the production of uric acid from the deamination of AMP followed by its ability to stimulate the translocation of NADPH oxidase to the mitochondria where it causes mitochondrial oxidative stress (38, 46, 49, 50). Thus, while uric acid in the plasma may act as an antioxidant, the rise in intracellular uric acid paradoxically causes oxidative stress. The mitochondrial oxidative stress inhibits enzymes such as aconitase and enoyl CoA hydratase that act to preferentially shunt calories to fat as opposed to ATP, with the preferential stimulation of glycolysis and fat synthesis, while at the same time the beta oxidation of fatty acids is inhibited (39, 44, 47, 51, 52, 53).
Table 2.
Effect of Fructose on the Survival Response
| Stimulates Hunger |
| Acutely does not stimulate insulin or leptin, while reducing ghrelin (30) |
| Acute reduction in hepatic ATP (31) which stimulates food intake (32) |
| Induction of leptin resistance (shown in rats) (33) |
| Stimulates Thirst |
| Increases serum osmolarity by shifting fluid to intracellular compartment (in rats) (34) |
| Stimulates vasopressin production via osmolar and nonosmolar pathways (35) |
| Oral fructose increases serum vasopressin (copeptin) in humans (36) |
| Stimulates Foraging Responses |
| Stimulates foraging-like behavior (reviewed in(37)) |
| Stimulates Fat Accumulation |
| Stimulates hepatic lipogenesis (38) |
| Blocks beta fatty acid oxidation (39) |
| Blocks lipolysis in fat by causing hyperinsulinemia from insulin resistance |
| Increases Blood glucose levels |
| Induces Insulin Resistance (40) |
| Impairs Insulin Release by Islets (41) |
| Stimulates Gluconeogenesis (42) |
| Fructose Raises Blood Pressure (43) |
| Fructose Reduces Oxygen Needs by Stimulating Glycolysis (44) |
| Fructose Stimulates Innate Immune Pathways (45) |
A role for uric acid in the biological actions of fructose was shown by the observation that lowering uric acid could protect laboratory animals from developing fructose-induced metabolic syndrome (54, 55, 56). Moreover, if uric acid levels are raised, such as with a uricase inhibitor, then the effects of fructose to increase features of metabolic syndrome are enhanced (57). For example, the inhibition of uricase in laboratory rats receiving a fructose-glucose drink can significantly increase hepatic fat accumulation, serum triglycerides, serum glucose and blood pressure as well as induce worse oxidative stress than either giving fructose-glucose or uricase inhibition alone (57).
To further evaluate the function of the uricase gene in survival, we resurrected the ancient uricase prior to its final silencing and expressed it in a human hepatocyte cell line (HepG cells)(58). In cells expressing the ancient uricase, the rise in uric acid in response to fructose was less, as was the increase in triglyceride content, when compared to human hepatocyte cells that lacked uricase (5). Thus, the loss of uricase augmented the fat response to fructose. The loss of the ancient uricase also amplified the gluconeogenic response to fructose which suggests a role for uric acid as a means to enhance glucose production to aid the brain and muscle during periods of starvation (48).
While these studies document how a loss of uricase can amplify the effects of fructose to stimulate fat stores and insulin resistance, the uricase mutation may also enhance the ability to survive independently of fructose (Table 3) (17, 21, 48, 57, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74). For example, the inhibition of uricase would be expected to increase the effect of umami and purine foods (such as bone marrow) to raise uric acid levels (75). Indeed, the administration of monosodium glutamate and/or AMP and IMP (i.e., umami tastants) in drinking water stimulates weight gain, fat accumulation, hepatic steatosis, and hyperinsulinemia that is prevented by lowering uric acid. Directly adding uric acid to drinking water also enhances the ability of monosodium glutamate to stimulate weight gain (75).
Table 3.
In vivo evidence that Inhibiting Uricase is Involved in the Biological Survival Response
| Survival Response | Mechanism |
|---|---|
|
| |
| Foraging | Increases locomotor activity and hyperactivity (59) |
| Increased endurance (60) | |
| Increases Impulsivity (61) | |
| Decrease anxiety in stressful situations (62) | |
| Decrease Recent Memory (hippocampal)(63) | |
| Fat Accumulation | Increases Fat in the Liver (57, 64) |
| Hypertriglyceridemia (65) | |
| Impaired Glucose Tolerance | Stimulates Insulin Resistance (66) |
| Stimulates Gluconeogenesis (48) | |
| Reduces Insulin Secretion (67) | |
| Raises Blood Pressure | Reduces nitric oxide (21) |
| Stimulates renal and local RAS | |
| Stimulates aldosterone (68) | |
| Stimulates endothelin (69) | |
| Stimulates oxidative stress (17) | |
| Stimulates COX-2 associated thromboxane (70) | |
| Increases Renal Sodium Reabsorption (71) | |
| Inflammation | Systemic Inflammatory Markers (72) |
| Inflammasome activation (73) | |
| Alterations in Microbiome/Leaky Gut (74) | |
There is evidence that the inhibition of uricase with an increase in uric acid may assist other survival responses (Table 3). For example, blocking uricase raises blood pressure and increases salt sensitivity in rats (76). Blocking uricase also stimulates behaviors involved in the foraging response, including locomotor activity and impulsivity (59, 60, 61). Uric acid also appears to have an important role in maintaining glucose levels in the setting of starvation, by increasing gluconeogenesis, blocking insulin secretion, and stimulating insulin resistance (48, 66, 67). Injecting uric acid into starving animals can provide survival advantage similar to injecting glucose, while injection of the uricase product, allantoin, had no effect (77).
Thus, the mutation of uricase likely aided survival for starving apes, by stimulating foraging, enhancing fat stores and maintaining glucose levels from the fructose present in the little fruit available (Figure 1).
Figure 1. Hypothesis for How the Uricase Mutation may have Aided Survival to Hominids Facing Seasonal Starvation.
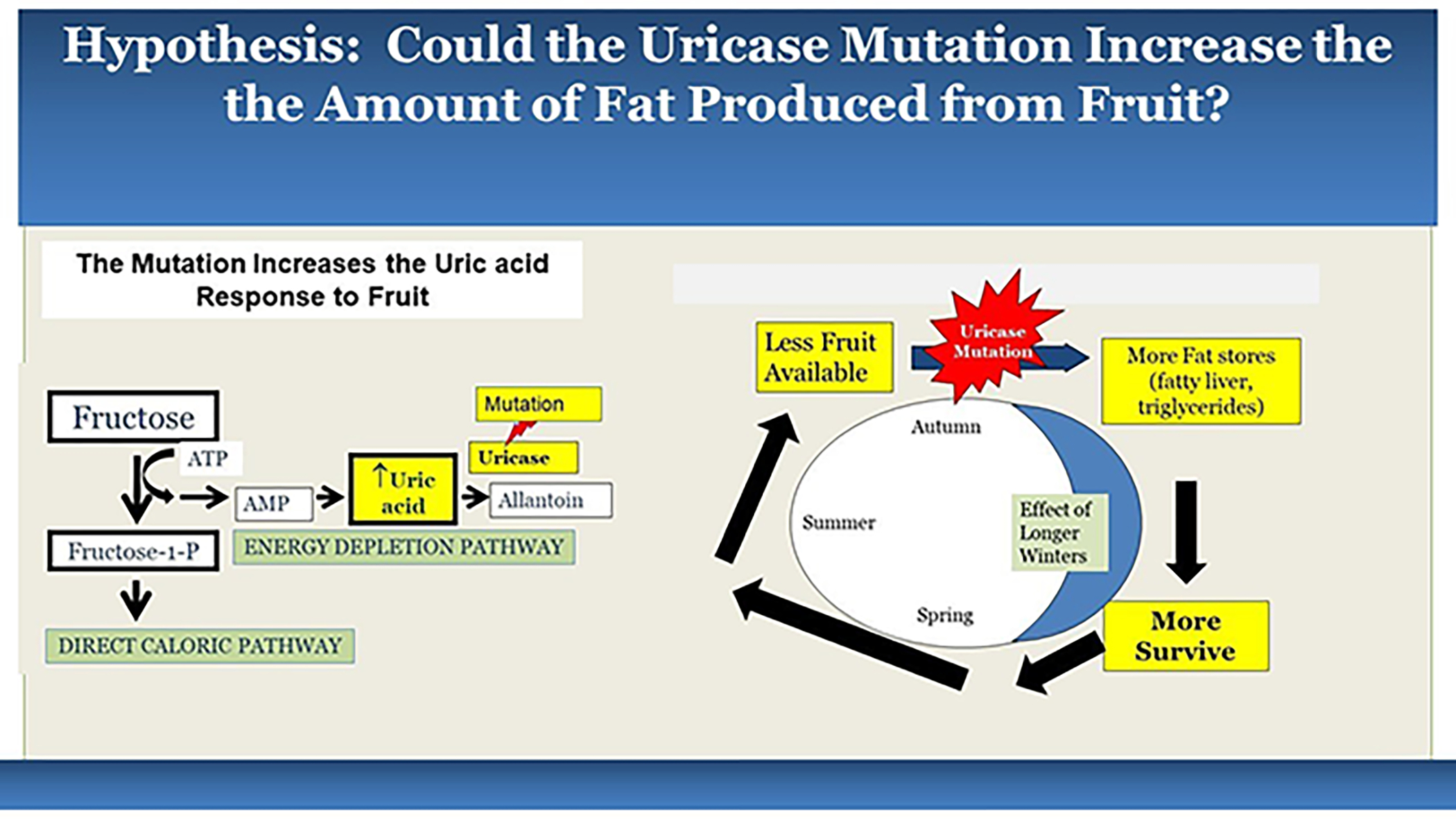
According to the hypothesis, the loss of uricase would have amplified the uric acid response to fructose, and this would have led to a greater activation of the biologic switch in response to dwindling fruit supplies, leading to increased fat stores that would allow survival through the cooler seasons.
Western Diet and the Switch from Preventing Starvation to Causing Obesity
The mutation in uricase was protective in situations of starvation and food scarcity and hence it rapidly took over the ancestral population, likely during the tumultuous times in the mid-Miocene. Today all humans are effectively uricase knockouts. However, the doubling of uric acid resulting from the mutation (4) still kept the uric acid at levels not expected to cause obesity. However, the loss of uricase resulted in a key inability to regulate uric acid levels effectively. As a consequence, obesity resulted with the introduction of western diet that contained large amounts of foods that could raise uric acid further.
One of the major foods is fructose itself, which historically was limited to fruits and honey. However, approximately 2500 years ago sugarcane was discovered in the Ganges River Valley, leading to the eventual commercialization of sugar (sucrose), which contains both glucose and fructose. Sushruta, a physician from that region, was one of the first to observe obesity, diabetes and heart disease, and he linked their presence to intake of the sweet liquid obtained from the cane. Later, sugarcane was brought to China, Persia and Egypt, and then to Europe via Venice. Initially sugar was expensive and obesity was primarily observed among royalty and the wealthy. With the expansion of the sugarcane industry to the Americas, along with the introduction of the sugar beet, sugar became more affordable by the general population.
The total intake of added sugars underwent another sharp rise in the 1970s following the introduction of high fructose corn syrup, which is a mixture of fructose and glucose, often with a relatively higher proportion of fructose than glucose. Intake of added sugars (i.e., table sugar and HFCS) peaked in 2005, and has been slowly decreasing since. However, average sugar intake remains about 14 to 16 percent of total calories, and adolescents, young adults, disadvantaged individuals, and ethnic minorities are ingesting higher amounts, with some individuals ingesting 25 percent of total caloric intake from added sugars (78).
Much has been written about the parallel rise in sugar and HFCS intake with the rise in serum uric acid, gout and the epidemics of obesity, diabetes and cardiovascular disease. Not only is the intake of added sugars, especially in the form of sugar-sweetened beverages, strongly associated with the development of obesity, diabetes, and metabolic syndrome (79), but also hyperuricemia is a strong predictor even in individuals who do not have manifestations of metabolic syndrome other than asymptomatic hyperuricemia (80). As the level of serum uric acid increases, there is a stepwise increase in the frequency of obesity, metabolic syndrome and related diseases (Figure 2) (81).
Figure 2. Relationship of Serum Uric acid Levels with Obesity and Diabetes.
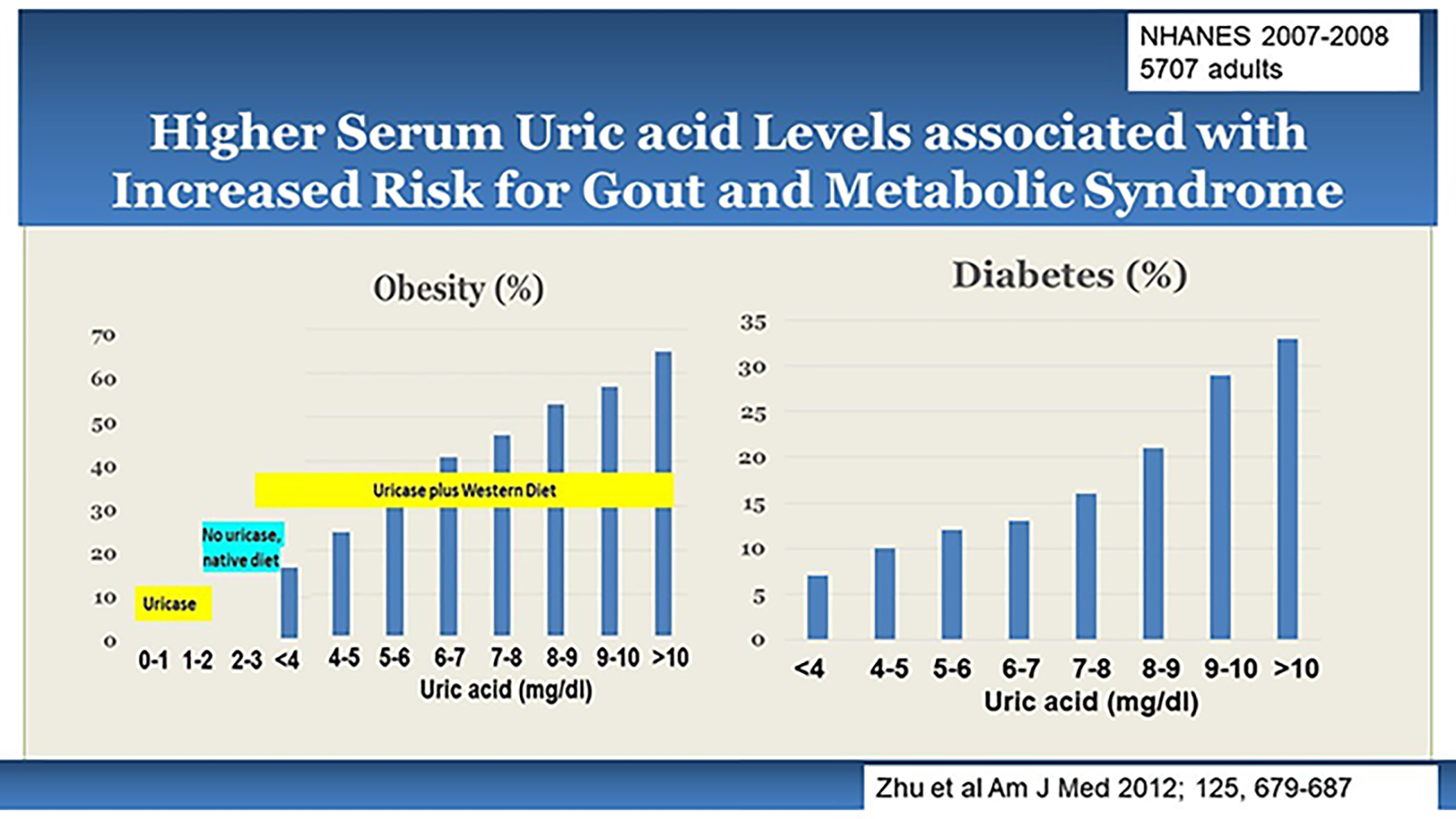
An analysis of 5707 adults from the NHANES population survey (2007 to 2008) demonstrated a strong relationship of serum uric acid levels with presence of obesity or diabetes(81). One can observed that the risk for obesity with uric acid levels < 4 mg/dl is relatively low, suggesting that the uricase mutation alone did not cause much obesity. It was the interaction of the uricase mutation with western diet that led to the dramatic rise in obesity and diabetes.
Strengths, Challenges and Limitations to the Uricase Thrifty Gene Hypothesis
If there is a thrifty gene, then why is not everyone obese?
A common criticism of the thrifty gene hypothesis is that famines are too short to allow a thrifty gene mutation to take over the entire population, and if it did, that it would drive obesity in everyone. It has also been stated that famines preferentially kill the young and old, and and the potential benefit of having obesity is not always evident.
However, the mutation that involved uricase occurred during a prolonged period of seasonal starvation that occurred over 7 million years (from around 15 million years ago to 8 million years ago). It was severe enough to lead to contraction of the population to refugia and eventually to the extinction of the apes living in Europe. Many mutations occurred during this time in our ancestral genome, and the loss of uricase could understandably have provided a survival advantage that would have rapidly spread to the entire population. Likely the key was the effect of the prolonged seasonal starvation on reproduction, which is severely affected by a loss of fat stores (82). For example, the Dutch Famine of the winter of 1944 found that only a few months of caloric restriction could markedly affect fertility, fecundity, and reproductive outcomes (83). In the case of the mid-Miocene, the fossil records suggest that not all the apes in Eurasia died, but that there were some that returned to Africa and southeast Asia prior to their extinction in Europe. Those apes that carried the mutation and returned to Africa may have carried a survival advantage over the endogenous apes as the climate in East Africa also subsequently became drier and more seasonal (8). Indeed, a mutation in alcohol dehydrogenase occurred during that time that allowed the apes to eat ripe fruit that had fallen to the ground and was fermenting, which allowed an additional food source but unwittingly allowed modern humans to consume alcohol (84). Thus, the ‘duration argument’ is unlikely to be correct and does not apply to the uricase hypothesis.
An important aspect of the uricase thrifty gene hypothesis is that it does not imply that the mutation causes obesity, but rather that it predisposes to obesity. The mutation acted to protect against starvation, but the uric acid levels only doubled, and this is not at a level that would cause obesity. However, the mutation affected our ability to regulate uric acid levels, and so in the setting of western diet rich in sugary foods, purine-rich foods and alcohol, the serum uric acid levels have been increasing in the population, and as this happens, the risk for obesity and diabetes also increase (Figure 2) (81). Hence, the loss of uricase is fully penetrant, but the manifestation of obesity requires ingesting the right foods or additional genetic mechanisms that enhance the biological survival response to fructose.
If high uric acid levels result from a thrifty gene, should not genetic polymorphisms that increase uric acid also increase the risk for obesity and diabetes?
If uric acid has a role in driving metabolic syndrome, then one might predict that genetic polymorphisms that raise serum uric acid should increase the risk for obesity and diabetes. For example, the Polynesian people tend to have higher uric acid levels that may be related to genetic polymorphisms acquired in the past. Gout, for example, was present in old Maori skeletons found prior to the introduction of western culture (85). As such, this group may be more prone to metabolic syndrome even in the absence of western diet.
Nevertheless, most Mendelian randomization studies do not find a relationship between genetic polymorphisms affecting serum uric acid levels with obesity, diabetes, hypertension, or chronic kidney disease even though elevated serum uric acid consistently predicts increased risk for obesity and diabetes (86, 87). In contrast, other genetic studies including Mendelian analyses have found associations of genetic polymorphisms of uric acid with obesity, diabetes, cardiovascular events and mortality (26, 88).
While the negative Mendelian randomization studies challenge the biological role of uric acid in obesity, interpretation of Mendelian randomization studies is complicated because these studies do not typically consider interactions of genetic polymorphisms with diet which we know is important when considering uric acid.
In addition, the uricase mutation increased both intrahepatic uric acid levels and serum uric acid levels, and it is the intrahepatic uric acid levels that drive the development of the metabolic syndrome (89). Normally serum uric acid reflects intracellular uric acid levels, but some genetic polymorphisms in urate transport that are used to calculate genetic scores for Mendelian randomization can have opposing effects on serum versus portal vein levels (90, 91). Hence, one cannot assume that a genetic score that predicts serum uric acid levels will also predict intrahepatic uric acid levels.
Indeed, one way serum and intrahepatic uric acid levels can be dissociated is when fructose is produced endogenously in the liver. Recently it was shown that fructose-induced obesity does not have to result from dietary intake of fructose, but rather the liver can also make fructose in response to foods that activate the polyol (aldose reductase) pathway, such as from high glycemic carbohydrates and salty foods (92, 93). Studies in humans suggest endogenous fructose production may be equivalent to the ingestion of a soft drink a day (94). Of interest, hepatic uric acid levels are high despite serum uric acid levels being normal in obesity induced by high salt intake (93).
Thus, the uricase mutation is likely driving the metabolic syndrome via its effects on intrahepatic uric acid level, and the serum uric acid is more of a biomarker and not the critical factor that drives the obesity response from the uricase mutation. Nevertheless, since serum uric acid tends to reflect intracellular levels, serum uric acid still carries valuable benefit in predicting the development of obesity and metabolic syndrome.
If a high uric acid has a role in obesity and diabetes, then should not the inhibition of uric acid production lead to a decreased frequency of these conditions?
There have only been a few pilot studies investigating the role of lowering uric acid on metabolic outcomes, and the studies have shown mild benefit on weight loss (95, 96) with improvement in glucose tolerance (97, 98, 99, 100), although one short, two-week study did not show a benefit of lowering uric acid on insulin resistance (43). However, this may be because experimental studies suggest uric acid also blocks insulin secretion (67) in addition to causing insulin resistance (66), so these two effects could potentially cancel each other out in the short-term. Likewise, pilot studies suggest lowering uric acid can reduce blood pressure in adolescents with hypertension, but the benefits in subjects with longstanding hypertension is less evident (101, 102).
The metabolic effects driven by the absence of uricase in affluent societies feeding on a western diet may have two phases, with an initiation phase and a persistent phase. For example, while fructose induces a metabolic phenotype through its ability to cause mitochondrial oxidative stress, over time this may lead to persistent injury with mitochondrial loss that may in itself predispose to obesity and thwart the ability to restore weight to baseline levels (103). We observed that chronic administration of sucrose (which contains fructose) initially caused insulin resistance, but over time there was a reduction in insulin secretion due to progressive injury to the islets (41). Interestingly, this latter effect was associated with increased expression of the urate transporter on the islets, and uric acid was found to induce islet injury in studies using islet cells (41). Furthermore, we found that raising uric acid levels with a uricase inhibitor caused acute hypertension, but over time the hypertension transitioned from being uric acid-dependent to one that was driven by inflammation in the kidney that caused persistent renal vasoconstriction (76). It is also possible that other factors besides uric acid itself may be driving some metabolic manifestations. For example, NAD+ is consumed during the production and metabolism of fructose as well as from the synthesis of uric acid, and this may result in a decrease in sirtuin activity that could amplify the development of obesity and metabolic syndrome (104). However, this would not easily explain why raising uric acid levels by blocking uricase has so many metabolic effects (Table 3).
Based on these observations, the lowering of uric acid (and fructose) might be most likely protective before mitochondrial injury, islet dysfunction, and kidney damage becomes permanent. This might explain why lowering fructose intake is so beneficial in children with metabolic syndrome (105) and why obesity and diabetes are harder to reverse in the adult who has had these conditions for a long time (76, 106, 107).
Conclusion
In summary, there is strong evidence that the uricase mutation likely represented a thrifty gene, and that it provided a survival advantage to our ancestors during a prolonged period of seasonal starvation during the Miocene, but that today it may be increasing our risk to become obese and diabetic. Nevertheless, more studies need to be performed, especially to understand the benefit of lowering uric acid on obesity and metabolic syndrome. Most studies suggest that the benefit is most likely greatest early in the course of metabolic syndrome, partly because with time other mechanisms may occur that maintain the phenotype besides the uric acid.
Study Importance
A classic hypothesis for the epidemic of obesity is that we acquired genetic mutations to aid survival from famine in the past may predispose us to obesity today (‘Thrifty Gene’ Hypothesis).
This hypothesis is controversial primarily because of the lack of a gene candidate.
Here we review arguments for and against the uricase mutation as being a thrifty gene which provides insights not only on uricase and obesity but also on evolutionary biology of obesity.
Main Findings
The uricase mutation occurred during a prolonged period of seasonal starvation and near extinction of our ancestors.
The uricase mutation likely had a role in survival by promoting the ability of fructose to store fat and cause insulin resistance.
Epidemiologic and clinical trials suggest elevated serum and intracellular uric acid levels play a role in the epidemics of obesity and diabetes.
How will this Affect Research Directions and Clinical Practice
The evidence supports the loss of uricase as a true thrifty gene
This is likely to stimulate more research on the role of fructose and uric acid in obesity and diabetes
It highlights the importance of incorporating evolutionary biology as a way to better understand the pathogenesis of diseases today
Acknowledgments.
We thank our many collaborators who assisted in these studies.
Funding:
Funded in part by NIH R01 DK121496 (RJJ and MAL)
Footnotes
Disclosure: RJJ, DT, LGL and MAL have equity with Colorado Research Partners LLC, RJJ also has stock with XORTX therapeutics, and RJJ has also received honoraria from Horizon Pharma. All others disclose no conflicts of interest.
Additional Notes: Additional references are also available if requested.
Authorship Roles: RJJ wrote the first draft. All other authors helped edit the manuscript.
References
- 1.Neel JV. Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? Am J Hum Genet 1962;14: 353–362. [PMC free article] [PubMed] [Google Scholar]
- 2.Speakman JR. The evolution of body fatness: trading off disease and predation risk. J Exp Biol 2018;221. [DOI] [PubMed] [Google Scholar]
- 3.Johnson RJ, Andrews P. Fructose, Uricase, and the Back-to-Africa Hypothesis. Evol Anthropol 2010;19 250–257. [Google Scholar]
- 4.Johnson RJ, Titte S, Cade JR, Rideout BA, Oliver WJ. Uric acid, evolution and primitive cultures. Semin Nephrol 2005;25: 3–8. [DOI] [PubMed] [Google Scholar]
- 5.Kratzer JT, Lanaspa MA, Murphy MN, Cicerchi C, Graves CL, Tipton PA, et al. Evolutionary history and metabolic insights of ancient mammalian uricases. Proc Natl Acad Sci U S A 2014;111: 3763–3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Z, Hoshino Y, Tran L, Gaucher EA. Phylogenetic Articulation of Uric Acid Evolution in Mammals and How It Informs a Therapeutic Uricase. Mol Biol Evol 2022;39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oda M, Satta Y, Takenaka O, Takahata N. Loss of urate oxidase activity in hominoids and its evolutionary implications. Mol Biol Evol 2002;19: 640–653. [DOI] [PubMed] [Google Scholar]
- 8.Andrews P An Apes’s View of Human Evolution. Cambridge University Press: Cambridge, 2015. [Google Scholar]
- 9.Kelley J, Andrews P, Alpagut B. A new hominoid species from the middle Miocene site of Pasalar, Turkey. J Hum Evol 2008;54: 455–479. [DOI] [PubMed] [Google Scholar]
- 10.Samonte RV, Eichler EE. Segmental duplications and the evolution of the primate genome. Nat Rev Genet 2002;3: 65–72. [DOI] [PubMed] [Google Scholar]
- 11.Ames BN, Cathcart R, Schwiers E, Hochstein P. Uric acid provides an antioxidant defense in humans against oxidant- and radical-caused aging and cancer: a hypothesis. Proc Natl Acad Sci U S A 1981;78: 6858–6862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parkinson Study Group S-PDI, Schwarzschild MA, Ascherio A, Casaceli C, Curhan GC, Fitzgerald R, et al. Effect of Urate-Elevating Inosine on Early Parkinson Disease Progression: The SURE-PD3 Randomized Clinical Trial. JAMA 2021;326: 926–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Munoz Garcia D, Midaglia L, Martinez Vilela J, Marin Sanchez M, Lopez Gonzalez FJ, Arias Gomez M, et al. Associated Inosine to interferon: results of a clinical trial in multiple sclerosis. Acta Neurol Scand 2015;131: 405–410. [DOI] [PubMed] [Google Scholar]
- 14.Carvalho LAC, Lopes J, Kaihami GH, Silva RP, Bruni-Cardoso A, Baldini RL, et al. Uric acid disrupts hypochlorous acid production and the bactericidal activity of HL-60 cells. Redox Biol 2018;16: 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Imaram W, Gersch C, Kim KM, Johnson RJ, Henderson GN, Angerhofer A. Radicals in the reaction between peroxynitrite and uric acid identified by electron spin resonance spectroscopy and liquid chromatography mass spectrometry. Free Radic Biol Med 2010;49: 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hershfield MS, Roberts LJ 2nd, Ganson NJ, Kelly SJ, Santisteban I, Scarlett E, et al. Treating gout with pegloticase, a PEGylated urate oxidase, provides insight into the importance of uric acid as an antioxidant in vivo. Proc Natl Acad Sci U S A 2010;107: 14351–14356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cristobal-Garcia M, Garcia-Arroyo FE, Tapia E, Osorio H, Arellano-Buendia AS, Madero M, et al. Renal oxidative stress induced by long-term hyperuricemia alters mitochondrial function and maintains systemic hypertension. Oxid Med Cell Longev 2015;2015: 535686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanchez-Lozada LG, Lanaspa MA, Cristobal-Garcia M, Garcia-Arroyo F, Soto V, Cruz-Robles D, et al. Uric Acid-Induced Endothelial Dysfunction Is Associated with Mitochondrial Alterations and Decreased Intracellular ATP Concentrations. Nephron Exp Nephrol 2012;121: e71–e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kean RB, Spitsin SV, Mikheeva T, Scott GS, Hooper DC. The peroxynitrite scavenger uric acid prevents inflammatory cell invasion into the central nervous system in experimental allergic encephalomyelitis through maintenance of blood-central nervous system barrier integrity. J Immunol 2000;165: 6511–6518. [DOI] [PubMed] [Google Scholar]
- 20.Spitsin SV, Scott GS, Mikheeva T, Zborek A, Kean RB, Brimer CM, et al. Comparison of uric acid and ascorbic acid in protection against EAE. Free Radic Biol Med 2002;33: 1363–1371. [DOI] [PubMed] [Google Scholar]
- 21.Khosla UM, Zharikov S, Finch JL, Nakagawa T, Roncal C, Mu W, et al. Hyperuricemia induces endothelial dysfunction. Kidney Int 2005;67: 1739–1742. [DOI] [PubMed] [Google Scholar]
- 22.Amaro S, Jimenez-Altayo F, Chamorro A. Uric acid therapy for vasculoprotection in acute ischemic stroke. Brain Circ 2019;5: 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hasko G, Sitkovsky MV, Szabo C. Immunomodulatory and neuroprotective effects of inosine. Trends Pharmacol Sci 2004;25: 152–157. [DOI] [PubMed] [Google Scholar]
- 24.Johnson TA, Jinnah HA, Kamatani N. Shortage of Cellular ATP as a Cause of Diseases and Strategies to Enhance ATP. Front Pharmacol 2019;10: 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuo CF, See LC, Yu KH, Chou IJ, Chiou MJ, Luo SF. Significance of serum uric acid levels on the risk of all-cause and cardiovascular mortality. Rheumatology (Oxford) 2012. [DOI] [PubMed] [Google Scholar]
- 26.Kleber ME, Delgado G, Grammer TB, Silbernagel G, Huang J, Kramer BK, et al. Uric Acid and Cardiovascular Events: A Mendelian Randomization Study. J Am Soc Nephrol 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yan S, Zhang P, Xu W, Liu Y, Wang B, Jiang T, et al. Serum Uric Acid Increases Risk of Cancer Incidence and Mortality: A Systematic Review and Meta-Analysis. Mediators Inflamm 2015;2015: 764250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kobylecki CJ, Afzal S, Nordestgaard BG. Plasma Urate, Cancer Incidence, and All-Cause Mortality: A Mendelian Randomization Study. Clin Chem 2017;63: 1151–1160. [DOI] [PubMed] [Google Scholar]
- 29.Fini MA, Lanaspa MA, Gaucher EA, Boutwell B, Nakagawa T, Wright RM, et al. Brief report: The uricase mutation in humans increases our risk for cancer growth. Cancer Metab 2021;9: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teff KL, Elliott SS, Tschop M, Kieffer TJ, Rader D, Heiman M, et al. Dietary fructose reduces circulating insulin and leptin, attenuates postprandial suppression of ghrelin, and increases triglycerides in women. J Clin Endocrinol Metab 2004;89: 2963–2972. [DOI] [PubMed] [Google Scholar]
- 31.Bawden SJ, Stephenson MC, Marciani L, Aithal GP, Macdonald IA, Gowland PA, et al. Investigating Alterations in Hepatic ATP levels following Fructose and Fructose+Glucose Ingestion: A Simple Non-invasive Technique to Assess Liver Function Using 31P MRS. Proc Intl Soc Mag Reson Med 2012;20: 1369. [Google Scholar]
- 32.Ji H, Graczyk-Milbrandt G, Friedman MI. Metabolic inhibitors synergistically decrease hepatic energy status and increase food intake. Am J Physiol Regul Integr Comp Physiol 2000;278: R1579–1582. [DOI] [PubMed] [Google Scholar]
- 33.Shapiro A, Mu W, Roncal C, Cheng KY, Johnson RJ, Scarpace PJ. Fructose-induced leptin resistance exacerbates weight gain in response to subsequent high-fat feeding. Am J Physiol Regul Integr Comp Physiol 2008;295: R1370–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson RJ, Stenvinkel P, Andrews P, Sanchez-Lozada LG, Nakagawa T, Gaucher E, et al. Fructose metabolism as a common evolutionary pathway of survival associated with climate change, food shortage and droughts. J Intern Med 2020;287: 252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song Z, Roncal-Jimenez CA, Lanaspa-Garcia MA, Oppelt SA, Kuwabara M, Jensen T, et al. Role of fructose and fructokinase in acute dehydration-induced vasopressin gene expression and secretion in mice. J Neurophysiol 2017;117: 646–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanbay M, Guler B, Ertuglu LA, Dagel T, Afsar B, Incir S, et al. The Speed of Ingestion of a Sugary Beverage Has an Effect on the Acute Metabolic Response to Fructose. Nutrients 2021;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson RJ, Wilson WL, Bland ST, Lanaspa MA. Fructose and Uric Acid as Drivers of a Hyperactive Foraging Response: A Clue to Behavioral Disorders Associated with Impulsivity or Mania? Evol Hum Behav 2021;42: 194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lanaspa MA, Sanchez-Lozada LG, Choi YJ, Cicerchi C, Kanbay M, Roncal-Jimenez CA, et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and -independent fatty liver. J Biol Chem 2012;287: 40732–40744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lanaspa MA, Cicerchi C, Garcia G, Li N, Roncal-Jimenez CA, Rivard CJ, et al. Counteracting Roles of AMP Deaminase and AMP Kinase in the Development of Fatty Liver. PLoS ONE 2012;7: e48801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagai Y, Yonemitsu S, Erion DM, Iwasaki T, Stark R, Weismann D, et al. The role of peroxisome proliferator-activated receptor gamma coactivator-1 beta in the pathogenesis of fructose-induced insulin resistance. Cell Metab 2009;9: 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roncal-Jimenez CA, Lanaspa MA, Rivard CJ, Nakagawa T, Sanchez-Lozada LG, Jalal D, et al. Sucrose induces fatty liver and pancreatic inflammation in male breeder rats independent of excess energy intake. Metabolism 2011;60: 1259–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caton PW, Nayuni NK, Khan NQ, Wood EG, Corder R. Fructose induces gluconeogenesis and lipogenesis through a SIRT1-dependent mechanism. J Endocrinol 2011;208: 273–283. [DOI] [PubMed] [Google Scholar]
- 43.Perez-Pozo SE, Schold J, Nakagawa T, Sanchez-Lozada LG, Johnson RJ, Lillo JL. Excessive fructose intake induces the features of metabolic syndrome in healthy adult men: role of uric acid in the hypertensive response. Int J Obes (Lond) 2010;34: 454–461. [DOI] [PubMed] [Google Scholar]
- 44.Park TJ, Reznick J, Peterson BL, Blass G, Omerbasic D, Bennett NC, et al. Fructose-driven glycolysis supports anoxia resistance in the naked mole-rat. Science 2017;356: 307–311. [DOI] [PubMed] [Google Scholar]
- 45.Jones N, Blagih J, Zani F, Rees A, Hill DG, Jenkins BJ, et al. Fructose reprogrammes glutamine-dependent oxidative metabolism to support LPS-induced inflammation. Nat Commun 2021;12: 1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanchez-Lozada LG, Soto V, Tapia E, Avila-Casado C, Sautin YY, Nakagawa T, et al. Role of oxidative stress in the renal abnormalities induced by experimental hyperuricemia. Am J Physiol Renal Physiol 2008;295: F1134–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lanaspa MA, Sanchez-Lozada LG, Cicerchi C, Li N, Roncal-Jimenez CA, Ishimoto T, et al. Uric acid stimulates fructokinase and accelerates fructose metabolism in the development of fatty liver. PLoS One 2012;7: e47948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cicerchi C, Li N, Kratzer J, Garcia G, Roncal-Jimenez CA, Tanabe K, et al. Uric acid-dependent inhibition of AMP kinase induces hepatic glucose production in diabetes and starvation: evolutionary implications of the uricase loss in hominids. FASEB J 2014;28: 3339–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choi YJ, Shin HS, Choi HS, Park JW, Jo I, Oh ES, et al. Uric acid induces fat accumulation via generation of endoplasmic reticulum stress and SREBP-1c activation in hepatocytes. Lab Invest 2014;94: 1114–1125. [DOI] [PubMed] [Google Scholar]
- 50.Sautin YY, Nakagawa T, Zharikov S, Johnson RJ. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am J Physiol Cell Physiol 2007;293: C584–596. [DOI] [PubMed] [Google Scholar]
- 51.Mirtschink P, Krek W. Hypoxia-driven glycolytic and fructolytic metabolic programs: Pivotal to hypertrophic heart disease. Biochim Biophys Acta 2016;1863: 1822–1828. [DOI] [PubMed] [Google Scholar]
- 52.Softic S, Meyer JG, Wang GX, Gupta MK, Batista TM, Lauritzen H, et al. Dietary Sugars Alter Hepatic Fatty Acid Oxidation via Transcriptional and Post-translational Modifications of Mitochondrial Proteins. Cell Metab 2019;30: 735–753 e734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Herman MA, Birnbaum MJ. Molecular aspects of fructose metabolism and metabolic disease. Cell Metab 2021;33: 2329–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakagawa T, Hu H, Zharikov S, Tuttle KR, Short RA, Glushakova O, et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am J Physiol Renal Physiol 2006;290: F625–631. [DOI] [PubMed] [Google Scholar]
- 55.Baldwin W, McRae S, Marek G, Wymer D, Pannu V, Baylis C, et al. Hyperuricemia as a mediator of the proinflammatory endocrine imbalance in the adipose tissue in a murine model of the metabolic syndrome. Diabetes 2011;60: 1258–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sanchez-Lozada LG, Tapia E, Bautista-Garcia P, Soto V, Avila-Casado C, Vega-Campos IP, et al. Effects of febuxostat on metabolic and renal alterations in rats with fructose-induced metabolic syndrome. Am J Physiol Renal Physiol 2008;294: F710–718. [DOI] [PubMed] [Google Scholar]
- 57.Tapia E, Cristobal M, Garcia-Arroyo FE, Soto V, Monroy-Sanchez F, Pacheco U, et al. Synergistic effect of uricase blockade plus physiological amounts of fructose-glucose on glomerular hypertension and oxidative stress in rats. Am J Physiol Renal Physiol 2013;304: F727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.de Lima Balico L, Gaucher EA. CRISPR-Cas9-mediated reactivation of the uricase pseudogene in human cells prevents acute hyperuricemia. Mol Ther Nucleic Acids 2021;25: 578–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barrera CM, Hunter RE, Dunlap WP. Hyperuricemia and locomotor activity in developing rats. Pharmacol Biochem Behav 1989;33: 367–369. [DOI] [PubMed] [Google Scholar]
- 60.Cutler RG, Camandola S, Feldman NH, Yoon JS, Haran JB, Arguelles S, et al. Uric acid enhances longevity and endurance and protects the brain against ischemia. Neurobiol Aging 2019;75: 159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sutin AR, Cutler RG, Camandola S, Uda M, Feldman NH, Cucca F, et al. Impulsivity is associated with uric acid: evidence from humans and mice. Biol Psychiatry 2014;75: 31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tovchiga O, Shtrygol S. The influence of oxonate-induced hyperuricemia and allopurinol on behavioral reactions of random-bred mice. J Basic Clin Physiol Pharmacol 2012;23: 147–151. [DOI] [PubMed] [Google Scholar]
- 63.Shao X, Lu W, Gao F, Li D, Hu J, Li Y, et al. Uric Acid Induces Cognitive Dysfunction through Hippocampal Inflammation in Rodents and Humans. J Neurosci 2016;36: 10990–11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xie, Zhao H, Lu J, He F, Liu W, Yu W, et al. High uric acid induces liver fat accumulation via ROS/JNK/AP-1 signaling. Am J Physiol Endocrinol Metab 2021;320: E1032–E1043. [DOI] [PubMed] [Google Scholar]
- 65.Wexler BC, Greenberg BP. Effect of increased serum urate levels on virgin rats with no arteriosclerosis versus breeder rats with preexistent arteriosclerosis. Metabolism 1977;26: 1309–1320. [DOI] [PubMed] [Google Scholar]
- 66.Zhu Y, Hu Y, Huang T, Zhang Y, Li Z, Luo C, et al. High uric acid directly inhibits insulin signalling and induces insulin resistance. Biochem Biophys Res Commun 2014;447: 707–714. [DOI] [PubMed] [Google Scholar]
- 67.Lu J, Hou X, Yuan X, Cui L, Liu Z, Li X, et al. Knockout of the urate oxidase gene provides a stable mouse model of hyperuricemia associated with metabolic disorders. Kidney Int 2018;93: 69–80. [DOI] [PubMed] [Google Scholar]
- 68.Eraranta A, Kurra V, Tahvanainen AM, Vehmas TI, Koobi P, Lakkisto P, et al. Oxonic acid-induced hyperuricemia elevates plasma aldosterone in experimental renal insufficiency. J Hypertens 2008;26: 1661–1668. [DOI] [PubMed] [Google Scholar]
- 69.Long CL, Qin XC, Pan ZY, Chen K, Zhang YF, Cui WY, et al. Activation of ATP-sensitive potassium channels protects vascular endothelial cells from hypertension and renal injury induced by hyperuricemia. J Hypertens 2008;26: 2326–2338. [DOI] [PubMed] [Google Scholar]
- 70.Kang DH, Nakagawa T, Feng L, Watanabe S, Han L, Mazzali M, et al. A role for uric acid in the progression of renal disease. J Am Soc Nephrol 2002;13: 2888–2897. [DOI] [PubMed] [Google Scholar]
- 71.Xu W, Huang Y, Li L, Sun Z, Shen Y, Xing J, et al. Hyperuricemia induces hypertension through activation of renal epithelial sodium channel (ENaC). Metabolism 2016;65: 73–83. [DOI] [PubMed] [Google Scholar]
- 72.Lu J, Sun M, Wu X, Yuan X, Liu Z, Qu X, et al. Urate-lowering therapy alleviates atherosclerosis inflammatory response factors and neointimal lesions in a mouse model of induced carotid atherosclerosis. FEBS J 2019;286: 1346–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu M, Ma Y, Chen X, Liang N, Qu S, Chen H. Hyperuricemia causes kidney damage by promoting autophagy and NLRP3-mediated inflammation in rats with urate oxidase deficiency. Dis Model Mech 2021;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lv Q, Xu D, Zhang X, Yang X, Zhao P, Cui X, et al. Association of Hyperuricemia With Immune Disorders and Intestinal Barrier Dysfunction. Front Physiol 2020;11: 524236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Andres-Hernando A, Cicerchi C, Kuwabara M, Orlicky DJ, Sanchez Lozada LG, Nakagawa T, et al. Umami-Induced Obesity and Metabolic Syndrome is Mediated by Nucleotide Degradation and Uric acid Generation. Nature Metabolism 2021;in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Watanabe S, Kang DH, Feng L, Nakagawa T, Kanellis J, Lan H, et al. Uric acid, hominoid evolution, and the pathogenesis of salt-sensitivity. Hypertension 2002;40: 355–360. [DOI] [PubMed] [Google Scholar]
- 77.Kawaguchi T, Shimode M, Matsushita H, Nagase S. Frequent administration of uric acid extends survival of fasting analbuminemic rats under cold environment. Jpn J Physiol 1986;36: 295–303. [DOI] [PubMed] [Google Scholar]
- 78.Yang Q, Zhang Z, Gregg EW, Flanders WD, Merritt R, Hu FB. Added Sugar Intake and Cardiovascular Diseases Mortality Among US Adults. JAMA Intern Med 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Malik VS, Hu FB. The role of sugar-sweetened beverages in the global epidemics of obesity and chronic diseases. Nat Rev Endocrinol 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kuwabara M, Niwa K, Hisatome I, Nakagawa T, Roncal-Jimenez CA, Andres-Hernando A, et al. Asymptomatic Hyperuricemia Without Comorbidities Predicts Cardiometabolic Diseases: Five-Year Japanese Cohort Study. Hypertension 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhu Y, Pandya BJ, Choi HK. Comorbidities of gout and hyperuricemia in the US general population: NHANES 2007–2008. Am J Med 2012;125: 679–687 e671. [DOI] [PubMed] [Google Scholar]
- 82.Frisch RE. Critical fatness hypothesis. Am J Physiol 1997;273: E231–232. [DOI] [PubMed] [Google Scholar]
- 83.Stein Z, Susser M. Fertility, fecundity, famine: food rations in the dutch famine 1944/5 have a causal relation to fertility, and probably to fecundity. Hum Biol 1975;47: 131–154. [PubMed] [Google Scholar]
- 84.Carn D, Lanaspa MA, Benner SA, Andrews P, Dudley R, Andres-Hernando A, et al. The role of thrifty genes in the origin of alcoholism: A narrative review and hypothesis. Alcohol Clin Exp Res 2021;45: 1519–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gosling AL, Matisoo-Smith E, Merriman TR. Gout in Maori. Rheumatology (Oxford) 2013. [DOI] [PubMed] [Google Scholar]
- 86.Tin A, Marten J, Halperin Kuhns VL, Li Y, Wuttke M, Kirsten H, et al. Target genes, variants, tissues and transcriptional pathways influencing human serum urate levels. Nat Genet 2019;51: 1459–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Palmer TM, Nordestgaard BG, Benn M, Tybjaerg-Hansen A, Davey Smith G, Lawlor DA, et al. Association of plasma uric acid with ischaemic heart disease and blood pressure: mendelian randomisation analysis of two large cohorts. BMJ 2013;347: f4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gill D, Cameron AC, Burgess S, Li X, Doherty DJ, Karhunen V, et al. Urate, Blood Pressure, and Cardiovascular Disease: Evidence From Mendelian Randomization and Meta-Analysis of Clinical Trials. Hypertension 2021;77: 383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Andres-Hernando A, Orlicky DJ, Kuwabara M, Ishimoto T, Nakagawa T, Johnson RJ, et al. Deletion of Fructokinase in the Liver or in the Intestine Reveals Differential Effects on Sugar-Induced Metabolic Dysfunction. Cell Metab 2020;32: 117–127 e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dinour D, Gray NK, Campbell S, Shu X, Sawyer L, Richardson W, et al. Homozygous SLC2A9 mutations cause severe renal hypouricemia. J Am Soc Nephrol 2010;21: 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.DeBosch BJ, Kluth O, Fujiwara H, Schurmann A, Moley K. Early-onset metabolic syndrome in mice lacking the intestinal uric acid transporter SLC2A9. Nat Commun 2014;5: 4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lanaspa MA, Ishimoto T, Li N, Cicerchi C, Orlicky DJ, Ruzycki P, et al. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat Commun 2013;4: 2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lanaspa MA, Kuwabara M, Andres-Hernando A, Li N, Cicerchi C, Jensen T, et al. High salt intake causes leptin resistance and obesity in mice by stimulating endogenous fructose production and metabolism. Proc Natl Acad Sci U S A 2018;115: 3138–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Francey C, Cros J, Rosset R, Creze C, Rey V, Stefanoni N, et al. The extra-splanchnic fructose escape after ingestion of a fructose-glucose drink: An exploratory study in healthy humans using a dual fructose isotope method. Clin Nutr ESPEN 2019;29: 125–132. [DOI] [PubMed] [Google Scholar]
- 95.Madero M, Rodriguez Castellanos FE, Jalal D, Villalobos-Martin M, Salazar J, Vazquez-Rangel A, et al. A pilot study on the impact of a low fructose diet and allopurinol on clinic blood pressure among overweight and prehypertensive subjects: a randomized placebo controlled trial. J Am Soc Hypertens 2015;9: 837–844. [DOI] [PubMed] [Google Scholar]
- 96.Soletsky B, Feig DI. Uric acid reduction rectifies prehypertension in obese adolescents. Hypertension 2012;60: 1148–1156. [DOI] [PubMed] [Google Scholar]
- 97.Takir M, Kostek O, Ozkok A, Elcioglu OC, Bakan A, Erek A, et al. Lowering Uric Acid With Allopurinol Improves Insulin Resistance and Systemic Inflammation in Asymptomatic Hyperuricemia. J Investig Med 2015;63: 924–929. [DOI] [PubMed] [Google Scholar]
- 98.Liu P, Chen Y, Wang B, Zhang F, Wang D, Wang Y. Allopurinol treatment improves renal function in patients with type 2 diabetes and asymptomatic hyperuricemia: 3-year randomized parallel-controlled study. Clin Endocrinol (Oxf) 2015;83: 475–482. [DOI] [PubMed] [Google Scholar]
- 99.Meng J, Li Y, Yuan X, Lu Y. Effects of febuxostat on insulin resistance and expression of high-sensitivity C-reactive protein in patients with primary gout. Rheumatol Int 2017;37: 299–303. [DOI] [PubMed] [Google Scholar]
- 100.Ogino K, Kato M, Furuse Y, Kinugasa Y, Ishida K, Osaki S, et al. Uric acid-lowering treatment with benzbromarone in patients with heart failure: a double-blind placebo-controlled crossover preliminary study. Circ Heart Fail 2010;3: 73–81. [DOI] [PubMed] [Google Scholar]
- 101.Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA 2008;300: 924–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Higgins P, Walters MR, Murray HM, McArthur K, McConnachie A, Lees KR, et al. Allopurinol reduces brachial and central blood pressure, and carotid intima-media thickness progression after ischaemic stroke and transient ischaemic attack: a randomised controlled trial. Heart 2014;100: 1085–1092. [DOI] [PubMed] [Google Scholar]
- 103.Heo JW, No MH, Park DH, Kang JH, Seo DY, Han J, et al. Effects of exercise on obesity-induced mitochondrial dysfunction in skeletal muscle. Korean J Physiol Pharmacol 2017;21: 567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rodriguez-Iturbe B RJ J, Lanaspa-Garcia MA, Nakagawa T, Garcia Arroyo FE, Sanchez Lozada LG. Sirtuin deficiency and the adverse effects of fructose and uric acid synthesis. Am J Physiol Regul Integr Comp Physiol 2022;in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lustig RH, Mulligan K, Noworolski SM, Tai VW, Wen MJ, Erkin-Cakmak A, et al. Isocaloric fructose restriction and metabolic improvement in children with obesity and metabolic syndrome. Obesity (Silver Spring) 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes. J Clin Invest 2006;116: 1802–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gonzalez-Franquesa A, Gama-Perez P, Kulis M, Dahdah N, Moreno-Gomez S, Ana Latorre-Pellicer A, et al. Obesity causes irreversible mitochondria failure in visceral adipose tissue despite successful anti-obesogenic lifestyle-based interventions. BioRxiv 2020;registered. [Google Scholar]