

Abstract
HIV-1-specific CD4+ T cells (TCD4+) play a critical role in controlling HIV-1 infection. Canonically, TCD4+ are activated by peptides derived from extracellular (“exogenous”) antigens displayed in complex with major histocompatibility complex class II (MHCII) molecules on the surfaces of “professional” antigen presenting cells (APC) such as dendritic cells (DCs). In contrast, activated human TCD4+, which express MHCII, are not typically considered for their APC potential due to their low endocytic capacity and the exogenous antigen systems historically used for assessment. Using primary TCD4+ and monocyte-derived DCs from healthy donors, we show that activated human TCD4+ are highly effective at MHCII-restricted presentation of an immunodominant HIV-1-derived epitope following infection and subsequent noncanonical processing and presentation of endogenously produced antigen. Our results indicate that, in addition to marshalling HIV-1-specific immune responses during infection, TCD4+ also act as APCs, leading to the activation of HIV-1-specific TCD4+
INTRODUCTION
Despite over forty years of intensive research, human immunodeficiency virus 1 (HIV-1) continues to pose a significant global health threat. HIV-1 attacks the human immune system, eventually leading to a state of profound immune deficiency known as acquired immunodeficiency syndrome (AIDS). In 2020, an estimated 37.7 million people were living with HIV, 1.5 million were newly infected, and 680,000 died of AIDS-related illnesses (1). HIV-1 mainly infects activated CD4+ T cells (TCD4+), macrophages, and microglia, and to a lesser extent dendritic cells and osteoclasts (2–4). Infection causes profound immune dysregulation and TCD4+ death, leaving infected individuals vulnerable to opportunistic infections and death in the absence of therapeutic intervention (5).
TCD4+ play a critical role in the immune response against a wide variety of viral pathogens (6–8). Previous studies indicate that vigorous, HIV-1-specific TCD4+ responses play an important role in controlling viremia (9) and are also correlated with broader neutralizing antibody responses in infected individuals (10). Data from chronically infected individuals indicate a link between strong HIV-1-specific TCD4+ responses and effective HIV-1-specific CD8+ T cell (TCD8+) responses (11), and IL-21-producing TCD4+ have been shown to be important for maintaining HIV-1-specific TCD8+ (12). Additionally, HIV-1-specific cytotoxic TCD4+ work cooperatively with HIV-1-specific TCD8+ to control HIV-1 viremia (13) and expansion of cytolytic TCD4+ during acute HIV-1 infection predicts disease outcome (14). T follicular helper cells (Tfh) also play a key role in the anti-HIV-1 immune response. Work by Cohen et al. demonstrated an association between the frequency of Tfh cells during early, untreated infection and the future development of broadly neutralizing antibodies (BNAbs) (15). Additionally, a longitudinal study in rhesus macaques found that continuous HIV-1 envelope antigen production is required for driving Tfh activation, leading to more effective broadly neutralizing antibody responses (16).
Given the importance of TCD4+ in host responses to HIV-1, it is crucial to understand the basis for HIV-1-specific TCD4+ activation. By convention, TCD4+ activation is mediated by professional antigen-presenting cells (APCs) -dendritic cells (DCs), macrophages, and B cells- that that: 1) internalize extracellular antigen, such as whole virions, 2) proteolyze internalized material into within the endocytic network, 3) load the resulting peptides onto major histocompatibility complex class II (MHCII) molecules in the late endosome, and 4) transport resulting peptide:MHCII complexes to the APC plasma membrane, where they are recognized by T cell receptors (TCR) on the surfaces of antigen-specific TCD4+ (17). Peptide:MHCII-TCR contact, along with costimulatory signals, leads to TCD4+ activation.
Several studies have shown that MHCII-restricted processing and presentation is more complex than this conventional presentation pathway. (18). For example, endogenous processing has been described as a robust alternative to classical processing (19–24), coming into play when an APC becomes productively infected, the nascent viral proteins within the APC providing antigen processing substrates for presentation via MHCII. Endogenous processing has been demonstrated in several viral systems, including measles (25), influenza (24), and, notably, HIV-1 (26). Indeed, we have reported that endogenous processing and presentation is the primary driver of the TCD4+ response to influenza infection (24).
The cell types capable of MHCII-restricted presentation may also be more extensive than originally appreciated. Many cell types besides professional APCs constitutively express MHCII (23). In addition, many other cell types express MHCII in response to inflammatory signals such as interferon gamma (IFNγ) (27, 28). This includes human TCD4+, which, unlike their murine counterparts, express MHCII as well as the costimulatory molecule CD86 upon activation (29). Historically, these cells have not been considered as APCs, likely due to their limited ability to internalize extracellular material (a prerequisite for effective classical processing) and their non-constitutive expression of MHCII. However, the function of MHCII in these cells has been the subject of scrutiny for some time. In addition to their induced MHCII expression, activated human TCD4+ are a main cellular target of HIV-1, providing a large intracellular depot of newly synthesized viral antigen that could be accessed for endogenous MHCII processing and presentation. A limited number of studies have shown that, under the right circumstances, activated TCD4+ can present HIV-1-derived antigen on MHCII (30, 31), though TCD4+ were determined to be inferior to professional APC types, such as B cell lines and monocytes, at stimulating TCD4+ responses. However, it is worth noting that these studies were performed using 1) inert purified proteins, not infectious HIV-1, and 2) TCD4+ clones as APCs instead of primary ex vivo TCD4+, potentially limiting their relevance to the study of endogenous processing in primary TCD4+.
Despite the key role that HIV-1-specific TCD4+ responses play, there are very few studies dedicated to the details of MHCII-restricted presentation of HIV-1 epitopes in any cell type. We were intrigued by the possibility of endogenous MHCII-restricted processing of HIV-1 antigens by activated human TCD4+. Infection would obviate the low internalization capacity of TCD4+, and endogenous processing of abundantly produced antigen might lead to potent presentation of HIV-1-derived epitopes. In this study, we asked if activated human TCD4+ could present antigen derived from infectious HIV-1 to HIV-1-specific TCD4+, using DCs, the prototypical professional APC, as a comparator.
MATERIALS AND METHODS
Cell Procurement:
Human peripheral blood mononuclear cells (PBMCs) were isolated from healthy HLA-DR1, DR11, and DR15+ donors by the University of Pennsylvania Human Immunology Core (HIC) by density gradient centrifugation using Lymphoprep (Stemcell Technologies, Vancouver, Canada). CD4+ T cell (TCD4+) and monocytes were further isolated by the HIC using the RosetteSep Human CD4+ T Cell Enrichment Cocktail and EasySep Human Monocyte Enrichment kits (Stemcell Technologies). All studies were conducted with the permission of the University of Pennsylvania Institutional Review Board.
TCD4+ and Dendritic Cell Culture:
TCD4+ were cultured in RPMI 1640 (Invitrogen, Waltham, MA) supplemented with 100 units/mL penicillin (Invitrogen), 100 μg/mL streptomycin (Invitrogen), 2 mM L-glutamine (Corning Inc., Corning, NY), 10% heat-inactivated fetal bovine serum (R&D Systems, Minneapolis, MN), and 10 mM HEPES (Invitrogen). Media was further supplemented with 100 IU/mL recombinant human IL-2 (Biolegend, San Diego, CA). TCD4+ were activated using K562 based artificial APCs (aAPCs) expressing CD64 and CD86 (32) that were additionally engineered to express a chimeric antigen receptor based on OKT3 antibody irradiated at 100 Gys. TCD4+ and aAPCs were plated at a 2:1 ratio at a concentration of 1 × 106 cells/mL in a 24 well plate. On day 2, the media volume was quadrupled, and cells were moved to a T25 cell culture flask. Cells were counted at days 4 and 6 and fed with fresh media + 100 IU/mL IL-2 to maintain a cell concentration of 2.5 × 105 cells/mL. We confirmed that K562-activated TCD4+ expressed typical activation markers using flow cytometry (Fig. S1A). To generate DCs, 5 × 106 monocytes were cultured in 3 mLs of RPMI 1640 (Invitrogen) supplemented with 100 units/mL penicillin (Invitrogen), 100 μg/mL streptomycin (Invitrogen), 2 mM L-glutamine (Corning Inc.), 10% fetal bovine serum (R&D Systems) and 0.05 mM 2-mercaptoethanol (Sigma-Aldrich, St. Louis, MO). Media was further supplemented with 100 ng/mL recombinant human GM-CSF (Biolegend) and 50 ng/mL recombinant human IL-4 (Biolegend). On days 3 and 6, fresh media supplemented with the same concentration of GM-CSF and IL-4 was added to DC cultures. Unless otherwise indicated, TCD4+ and DCs were used on Day 7.
Viruses and Infection:
NL4-R3A (+Nef) has been previously described (33). Nef/Vpu KO NL4-R3A, and K574D NL4-R3A viruses were also generated by the laboratory of Dr. James Hoxie. To make the K574D fusion deficient mutant, HIV-1 R3A gp120/41 Env was mutated at position K574 (34) with QuikChange || XL Site-Directed Mutagenesis (Aligent Technologies) with primers CTCACAGTCTGGGGCATCGATCAGCTCCAGGCAAGAGTC (Fwd) and GACTCTTGCCTGGAGCTGATCGATGCCCCAGACTGTGAG (Rev). Mutations were confirmed by sequencing. The Env was then cloned into the pNL4–3 HIV-1 provirus. This virus was used to inoculate TCD4+ cells in vitro and no infected cells were detected by microscopy using an anti-p24 antibody. To make the Nef-/Vpu double KO R3A, the R3A Env was cloned into the pNL4–3 vector (which is Nef- by design) then the Vpu gene was mutated with QuickChange XL (Agilent Technologies) at the amino acid L12 with the primer introducing a premature stop codon Fwd 5’-GCAATAGTAGCATAAGTAGTAGCAGCAATAATAGCAATAGTAG-3’. For clarity, the amino acid is underlined: MQSLQILAIVALVVAAIIAIVVWSIALIEYRKILRQRKIDRIINRIIERAEDSGNESEGDQEELSALVEMGHHAPWDINDL*. Aldrithiol-2 (AT2) inactivated NL4-R3A (+Nef) was obtained from the University of Pennsylvania Center for AIDS Research (CFAR) Viral and Molecular Core. All viruses were made in HEK 293T cells transfected with Lipofectamine 2000 (Invitrogen) and were titered for p24 content by the University of Pennsylvania CFAR Viral and Molecular Core using Alliance HIV-1 p24 Antigen ELISA kits (PerkinElmer). Viruses were aliquoted and stored at −80°C until use.
On Day 4 of cell culture, TCD4+ and DCs were infected in their original media with NL4-R3A or Nef/Vpu KO NL4-R3A (100 ng of p24) per 1 × 106 cells in a 48 well plate. Infection was allowed to proceed for 3 days before cell staining or antigen presentation assays were performed. 100 ng p24 AT2 treated NL4-R3A and 69 ng p24 K574D NL4-R3A per 1 × 106 cells were added to TCD4+ and DCs 6 days post culture, approximately 12 hours before antigen presentation assays were performed.
Flow Cytometry:
Expression of antigen processing and presentation machinery was assessed as follows: DCs were stained extracellularly with anti-HLA-DR (1:100 dilution, G46-6, BD), anti-CD86 (1:100 dilution, FUN-1, BD). Activated TCD4+ were stained extracellularly with anti-CD3 (1:200 dilution, UCHT1, Biolegend) and anti-CD4 (1:100 dilution, OKT4, Biolegend), anti-HLA-DR (G46-6, BD), anti-CD86 (1:100 dilution, FUN-1, BD) for 20 minutes at 4°C. For intracellular staining, both DCs and TCD4+ permeabilized with fixation/permeabilization solution (BD Biosciences) for 15 minutes at room temperature and were then stained with anti-CD74 (1:100 dilution, Pin.1, Biolegend) and anti-HLA-DM (1:100 dilution, MaP.DM1, Biolegend) for 25 minutes at room temperature. To assess the activation state of aAPC-activated TCD4+ as compared to anti-CD3/CD28 stimulated TCD4+, cells were stained extracellularly with anti-CD4 (OKT4, Biolegend), anti-CD69 (1:100 dilution, FN50, Biolegend), anti-HLA-DR (G46-;6, BD), anti-CD25 (1:100 dilution, BC96, Biolegend), anti-CD62L (1:100 dilution, DREG-56, Biolegend) for 20 minutes at 4°C. Cells were then stained intracellularly with anti-CD3 (UCHT1, Biolegend) and anti-TNFα (1:100 dilution, MAb1, Biolegend). For DQ-OVA internalization and processing assays, DCs were stained extracellularly with anti-HLA-DR (G46-6, Biolegend), and activated TCD4+ were stained with anti-HLA-DR (G46-6, Biolegend), anti-CD4 (OKT4, Biolegend) and anti-CD3 (UCHT1, Biolegend). To validate the HIV-specific TCR expression in transduced TCD4+, cells were stained extracellularly with anti-CD4 (OKT4, Biolegend) and anti-TCR Vβ22 (1:5 dilution, IMMU 546, Beckman Coulter, Brea, CA) for 20 minutes at 4°C. For antigen presentation and viral spread assays, cocultures were stained extracellularly with anti-CD4 (OKT4, Biolegend) and anti-CD69 (FN50, Biolegend) for 20 minutes at 4°C. Cocultures were stained intracellularly with anti-CD3 (UCHT1, Biolegend), anti-TNFα (MAb1, Biolegend), anti-IL2 (1:75 dilution, MQ1–17H12, BD), anti-IFNγ (1:100 dilution, B27, BD Biosciences), and anti-p24 (1:500 dilution, Kc57, Beckman Coulter). All samples were incubated with LIVE/DEAD Fixable Near-IR (Invitrogen) to exclude dead cells. Cells were fixed with and 4% paraformaldehyde prior to analysis. Cells were acquired on a BD LSR Fortessa (BD Biosciences) and data were analyzed using FlowJo software (Tree Star, Ashland, OR).
Endocytosis Experiments:
To assess the ability of DCs and activated TCD4+ to internalize and process the fluorescent DQ-OVA substrate, 200K DCs or TCD4+ were plated in 200 μLs R10 in a 96 well U-bottom plate. DQ-OVA (Invitrogen) was then added at a concentration of 10 μg/mL. After 2 hours at 37°C, cells were washed 3 times in sterile phosphate-buffered saline (PBS) and stained for flow cytometry as described. To assess the ability of DCs and activated TCD4+ to internalize FITC-Dextran, 200K DCs or TCD4+ were plated in 200 μLs R10 in a 96 well U-bottom plate. FITC-Dextran (Sigma-Aldrich) was then added at a concentration of 1 mg/mL. After 2 hours at either 4°C or 37°C, cells were washed 3 times in sterile phosphate-buffered saline (PBS) and stained for flow cytometry as described.
Cathepsin Activity Assays:
To determine the cathepsin D, L, and S activities of DCs and activated TCD4+, DCs and TCD4+ were cultured for 7 days as described above. DCs and TCD4+ lysates were then made according to the instructions in the SensoLyte 520 Cathepsin D Assay Kit, SensoLyte 520 Cathepsin L Assay Kit, and SensoLyte 520 Cathepsin S Assay Kit (all by Anaspec). Lysates were then assessed for their cathepsin D and L activities according to the manufacturer’s instructions. Lysate from 100K cells was assayed per well in a 96 well plate. Fluorescence was measured after 30 minutes using an Infinite M200 Pro plate reader (Tecan).
Generation of HIV-specific TCR-Encoding Lentiviruses:
Gag293-specific TCR (F5) sequences have been previously described (35, 36) and were synthesized by Genscript (Piscataway, NJ). These TCR sequences were cloned into pTRPE transfer plasmid (37) with the TCRβ and TCRα sequences separated by a furin domain (serine-glycine-serine-glycine) + T2A (38). 27 μg pTRPE transfer plasmid was mixed with 3 μg codon-optimized Cocal-g plasmid (ATUM, Newark, California) (39). 18 μg HIV gag-pol packaging plasmid, 18 μg HIVREV expression plasmid (pTRP Rev) and transfected using Lipofectamine 2000 (Invitrogen) into 293T cells. At 24 and 48 hours, supernatants were collected and ultracentrifuged for 2.5 hours at 25000 rpm at 4°C, split into four aliquots, and stored at −80°C until use. A ZnT8-specific TCR was obtained from Roberto Mallone (INSERM, FR) (40), used as a control TCR, and was made according to the protocol above.
Generation of HIV-specific TCD4+:
To generate HIV-specific TCD4+, TCD4+ from healthy donors were stimulated with irradiated aAPCs and IL-2 (Biolegend) as described above. 24 hours after stimulation, TCD4+ were transduced with lentiviruses encoding either a ZnT8 TCR or the Gag293-specific TCR. Cells were expanded and fed as described above, but were allowed to expand for 10 days, with an additional feeding on Day 8. Successful transduction was validated by flow cytometric analysis for TCR Vβ22 (Fig. S1B) and by measuring TCD4+ responses to Gag293 peptide pulsed DCs (Fig. S1C). Transduced TCD4+ were then frozen in freezing media consisting of 10% DMSO and 90% FBS and stored at −80°C until needed.
Antigen Presentation Assays:
To assess the ability of DCs and activated TCD4+ to present HIV-1-derived, MHCII-restricted epitope, cells were cultured and infected as described above. On the day of the assay, both uninfected and infected DCs and TCD4+ were collected, washed with PBS, and incubated with 12.5 μg/mL BFA (Biolegend) in fresh R10 media for a total of 3.5 hours. After an initial 1.5 hours of BFA treatment, 0.5 mg/mL purified Gag293 peptide (FRDYVDRFYKTLRAEQASQE) (Genscript) was added to appropriate wells of uninfected DCs and TCD4+. Cells were then washed with BFA-containing PBS and replated in fresh R10 with 12.5 μg/mL BFA. Lentivirus-transduced TCD4+ expressing either a ZnT8 TCR or a Gag293-specific TCR were labeled with CellTrace Violet (Invitrogen) according to manufacturer’s recommendations. 1 × 105 transduced TCD4+ and 5 × 104 candidate 50K potential antigen presenting cells were then cocultured for 8 hours at 37°C. Cells were then stained for flow cytometry as described above. To confirm MHCII-restriction, DCs and TCD4+ were collected, incubated with BFA, and treated with purified Gag293 peptide as described above. However, after 1 hour of peptide incubation, 50 μg/mL MHCII blocking antibody (Tü39, BD Biosciences) or CD71 blocking antibody (CY1G4, Biolegend) was added to relevant wells. After 1 hour, cells were washed with BFA-containing PBS and cocultured with transduced TCD4+ as described above. To test the ability of DCs and TCD4+ to present epitope derived from NL4-R3A (+Nef)-infected TCD4+ cultures, supernatant was collected from WT HIV-1-infected TCD4+ cultures 48 hours post infection. This supernatant was either treated with ultraviolet (UV) light for 1 minute to inactivate any infectious virions (validated by a lack of syncytia formation in Sup R5 cells) or was filtered through a 100 kDa Ultra-0.5 mL Centrifugal Filter (MilliporeSigma, Burlington, MA) to remove any infectious virions and large subcellular particles. This treated supernatant was then mixed at a 1:1 ratio with fresh media supplemented with IL-2 and added to uninfected DCs and TCD4+ 12 hours prior to antigen presentation assays.
Statistics:
Fold Change Calculations:
For all experiments, except where noted, significance was evaluated by two-tailed paired t test or one-way analysis of variance (ANOVA) test using PRISM software (GraphPad, San Diego, CA). The Tukey method was used to correct for multiple comparisons. A value of p<0.05 was considered significant.
Statistical Tests:
To convert antigen presentation data from percent cytokine secretion to fold induction, the data were loaded into R (41). Data were grouped by cell type, cytokine, and experiment replicate and summary statistics were calculated. Within each summary statistic group, the mean response rate of the negative condition (DMSO) was determined. Fold changes were calculated as the response to experimental conditions divided by the response to DMSO. The mean and standard deviation of the fold changes were calculated in R.
RESULTS
Activated human TCD4+ express antigen processing machinery components
Expression of proteins associated with antigen processing and presentation, such as HLA-DR, CD86, and invariant chain (Ii), have been previously reported in activated human TCD4+ (42–44). To verify that activated human TCD4+ express these proteins in our experimental system, and to compare their expression in TCD4+ vs. DCs from the same donor, activated human TCD4+ and DCs were analyzed by flow cytometry. DCs express high levels of HLA-DR, CD86, and HLA-DM (both by percent positive and median fluorescence intensity (MFI)) (Fig. 1A and B, gating strategies shown in Fig. S4A–C), an unsurprising finding given their role as professional APCs. Activated TCD4+ also express all these proteins, although at lower levels, supporting the potential for APC function. Since HIV-1 infection has been previously shown to downmodulate antigen processing and presentation machinery in myeloid-derived cells (45, 46), we were also interested to determine whether expression levels of these proteins are maintained in HIV-1 infected TCD4+. To identify HIV-1-infected cells, we gated on CD3+ cells with downregulated levels of surface CD4, as HIV-1 infection induces CD4 downregulation on the cell surface (47). When uninfected and infected TCD4+ were compared for levels expression of these proteins, a greater percentage of uninfected TCD4+ expressed HLA-DR and CD86, but there were no significant differences in the percentage of cells expressing HLA-DM and invariant chain (Fig. 1C). Furthermore, wildtype (WT) HIV-1 infection did not lead to decreased expression levels of these markers as measured by MFI. Indeed, infection slightly increased HLA-DM expression as assessed by MFI (Fig. 1D). We further confirmed that expression of these markers is upregulated upon activation, as a far lower proportion of freshly thawed, nonactivated TCD4+ express these markers (Fig. 1E) when compared to activated TCD4+, though MFI was high in the few cells that did express these proteins (Fig. 1F). Based on these data, we conclude that activated TCD4+, both uninfected and HIV-1-infected, express well known components of the antigen processing and presentation machinery.
Figure 1. Activated TCD4+ and DCs both express antigen presentation machinery components and have endocytic protease activities but TCD4+ have weak internalization capabilities.
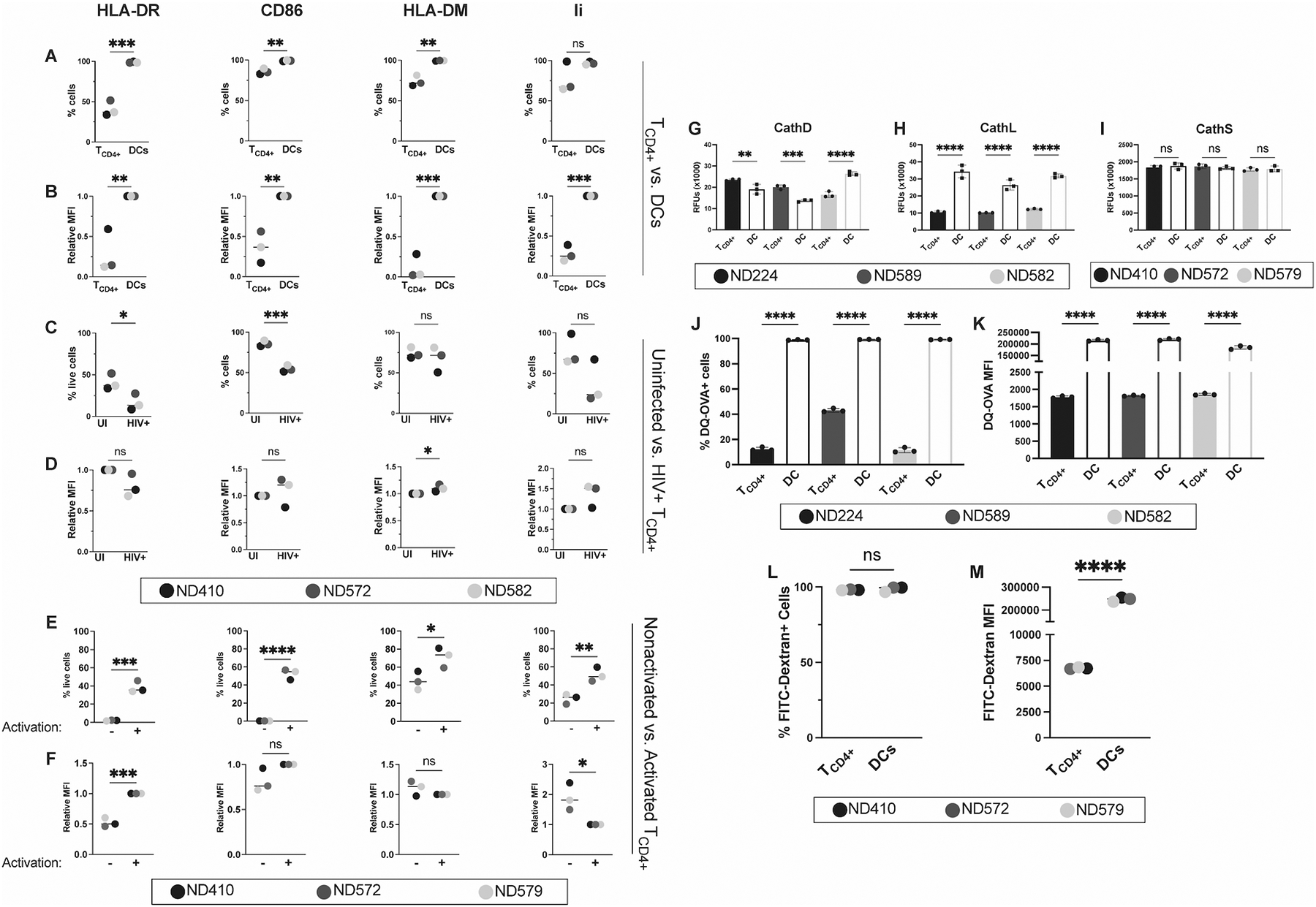
(A) percent positive and (B) relative mean fluorescence intensity (MFI) of HLA-DR, CD86, HLA-DM, and invariant chain in uninfected TCD4+ and DCs. TCD4+ and DCs were cultured as described. Cells were then stained for flow cytometry. HLA-DR and CD86 positive populations were gated on live singlets. HLA-DM and invariant chain expression was determined by pre-gating HLA-DR and CD86 double positive cells. (C) Percent positive and (D) relative MFI of these markers in uninfected (UI) and WT HIV-1 infected (HIV+) TCD4+. Where indicated, activated TCD4+ were infected with 100 ng p24 HIV-1 3 days prior to staining. Activated TCD4+ and DCs lysates were then analyzed for (E) cathepsin D, (F) cathepsin L, and (G) cathepsin S activity using fluorometric cathepsin substrates. Lysates from 1 × 105 cells were analyzed per replicate. Measurements were taken after 30 mins at 37°C. Activated TCD4+ and DCs were then tested for their ability to internalize and proteolyze DQ-OVA. All values are background subtracted. (H) Percent DQ-OVA positive cells and (I) MFI in DQ-OVA+ cells. TCD4+ and DCs were cultured as previously described and incubated with DQ-OVA fluorescent substrate for 2 hours at 37°C. Uptake and proteolysis were measured by flow cytometry on live singlets. Gates were drawn based on no-substrate controls. Percent FITC-Dextran positive cells (J) and (K) MFI in FITC-Dextran+ cells. TCD4+ and DCs were cultured as previously described and incubated with FITC-Dextran fluorescent substrate for 2 hours at 37°C. Uptake and proteolysis were measured by flow cytometry on live singlets. Gates were drawn based on no-substrate controls. Each color represents a unique donor, as indicated in the figure legend. For G-K, each dot represents a technical replicate. Representative of 3 independent experiments with the exception of L and M, which are representative of 2 independent experiments. For A-F and L-M, data were analyzed with an unpaired t test. For G-K, data were analyzed with a One-way ANOVA. Bars represent mean ± SD. *p<0.05, **p<0.01, ***p<0.005, ****p<0.001.
Activated TCD4+ have endocytic protease activities but limited internalization capabilities
Endocytic proteolysis is integral to classical antigen processing (48). Therefore, we investigated the activities of representative endosomal proteases, cathepsin D, L, and S in the lysates of DC and activated TCD4+ (Fig. 1E–G). Cathepsin D, a lysosomal aspartyl protease, and cathepsin L, a lysosomal endopeptidase, are necessary for intracellular protein breakdown and turnover and have been shown to play a role in antigen presentation (49–51). Cathepsin S is responsible for the degradation of invariant chain associated with mature MHCII molecules, allowing for the binding of antigenic peptides (52). While DCs had significantly greater Cathepsin L activity across 3 different donors, in 2/3 of donors (Fig. 1H), activated TCD4+ had greater Cathepsin D activity than did DCs (Fig.1G). DCs and TCD4+ had nearly indistinguishable Cathepsin S activities (Figure 1I). These activities, coupled with the expression of antigen processing and presentation components in Fig. 1A through D, suggests that activated human TCD4+ have the potential to be effective APCs during an HIV-1 infection.
We next investigated the ability of DCs and activated TCD4+ to internalize and proteolyze the fluorescent protease substrate DQ-OVA and the endocytic tracer FITC-Dextran, as internalization of extracellular antigen has been functionally linked to conventional MHCII-restricted processing and presentation (17, 53–56). DQ-OVA is internalized through receptor-mediated endocytosis (57) and fluid-phase micropinocytosis (58), and subsequent endocytic hydrolysis results in a fluorescent signal. Both DCs and activated TCD4+ internalize and proteolyze DQ-OVA (Fig. 1J and K). However, DCs internalize and proteolyze DQ-OVA at far greater levels than activated TCD4+, an unsurprising result given the well-documented ability of DCs to sample the external environment at a high rate (17, 53, 55, 56). Given that the fluorescent signal from DQ-OVA requires both internalization and proteolysis, and we can detect protease activity in activated TCD4+, the low signal likely reflects the modest internalization capabilities of this cell type. We confirmed these findings by examining the internalization of FITC-Dextran, another fluorescent substrate (59), by these two cell types. Similar to our DQ-OVA results, we found that while both DCs and activated TCD4+ are capable of internalizing FITC-Dextran (Fig. 1L), DCs do so at a much greater level (Fig. 1M). Taken together, these data suggest that activated TCD4+ have a limited ability to internalize and process exogenous antigens.
Activated TCD4+ present HIV-1-derived antigen to Gag293-specific TCD4+
Results thus far suggest that TCD4+ are limited in conventional processing of exogenous antigens due to their modest ability to internalize exogenous antigen. However, we reasoned that, because activated TCD4+ are highly susceptible to HIV-1 infection, they might be effective at endogenous processing following synthesis of viral proteins (19–24). To explore this possibility, we needed to establish a reliable system to read out epitope production. In initial experiments, we attempted to use activation of HIV-1-specific TCD4+ clones as the readout; however, we were unable to generate the cell numbers needed for sufficient biological and technical replicates and the experimental variations outlined below. We therefore turned to a lentiviral transduction system similar to that described by Benati et al. (35), in which we attained HIV-1-specificity by transducing TCD4+ from HIV-1-negative donors with an HIV-1-specific TCR. Extensive searches yielded only three full-length, MHCII-restricted HIV-1-specific TCR sequences, all specific for the immunodominant Gag293 epitope located within the HIV-1 capsid protein. The HIV-1-specific public TCR used in these studies, termed F5 (35), recognizes the Gag293 epitope in complex with HLA-DR1, DR11, and DR15 molecules. HLA-DR1+, DR11+, or DR15+ DCs and activated TCD4+ were infected with WT HIV-1 3 days prior to the assay. To minimize any nonspecific cytokine secretion from uninfected and HIV-1-infected TCD4+ over the course of infection, these cells were treated with brefeldin A (BFA) for 4 hours prior to coculture with HIV-1-specific TCD4+ and subsequently washed to remove any secreted cytokines. Synthetic Gag293 peptide was added to DCs and activated TCD4+ 2 hours prior to coculture (2 hours into the BFA pretreatment) and was washed out prior to coculture. HIV-1-specific TCD4+ were labeled with Cell Trace Violet dye to differentiate between the two TCD4+ populations. Candidate APCs (DCs and activated TCD4+) and HIV-1-specific TCD4+ were cocultured for 8 hours in the presence of BFA. Cocultures were then stained for flow cytometric analysis to detect production of interleukin-2 (IL-2), tumor necrosis factor alpha (TNFα) and interferon gamma (IFNγ) by responding TCD4+. Notably, we did not observe any differences in presentation patterns depending on identity of the presenting molecule (HLA-DR1, 11 or 15), though we did note occasional variability in the magnitude of responses depending on the donor and experiment. We measured three different indicators of activation (IL-2, TNFα, and IFNγ) to maximize the chances of detecting TCD4+ activation, as it has been observed that TCD4+ cytokine profiles may differ depending on the activating conditions used (60).
Results reproducibly showed that DCs present synthetic Gag293 peptide but are unable to present the epitope derived from infectious HIV-1 (Fig. 2A). Conversely, activated TCD4+ present the Gag293 epitope derived from infectious HIV-1 but are unable to present synthetic Gag293 peptide (Fig. 2B, representative flow plots shown in Fig. S4D), even at much higher concentrations of synthetic peptide (Fig. S3C) despite high levels of MHCII expression. This phenomenon has been previously observed in other antigen systems (23). Two potential explanations are limited peptide exchange at the cell surface (61–63), or the need for peptide to be internalized by the cell for successful presentation (64), a mechanism that is likely severely compromised in TCD4+ considering the results shown in Fig. 1.
Figure 2. Activated TCD4+, but not DCs, can present epitope derived from infectious HIV-1 in an MHCII-dependent manner.
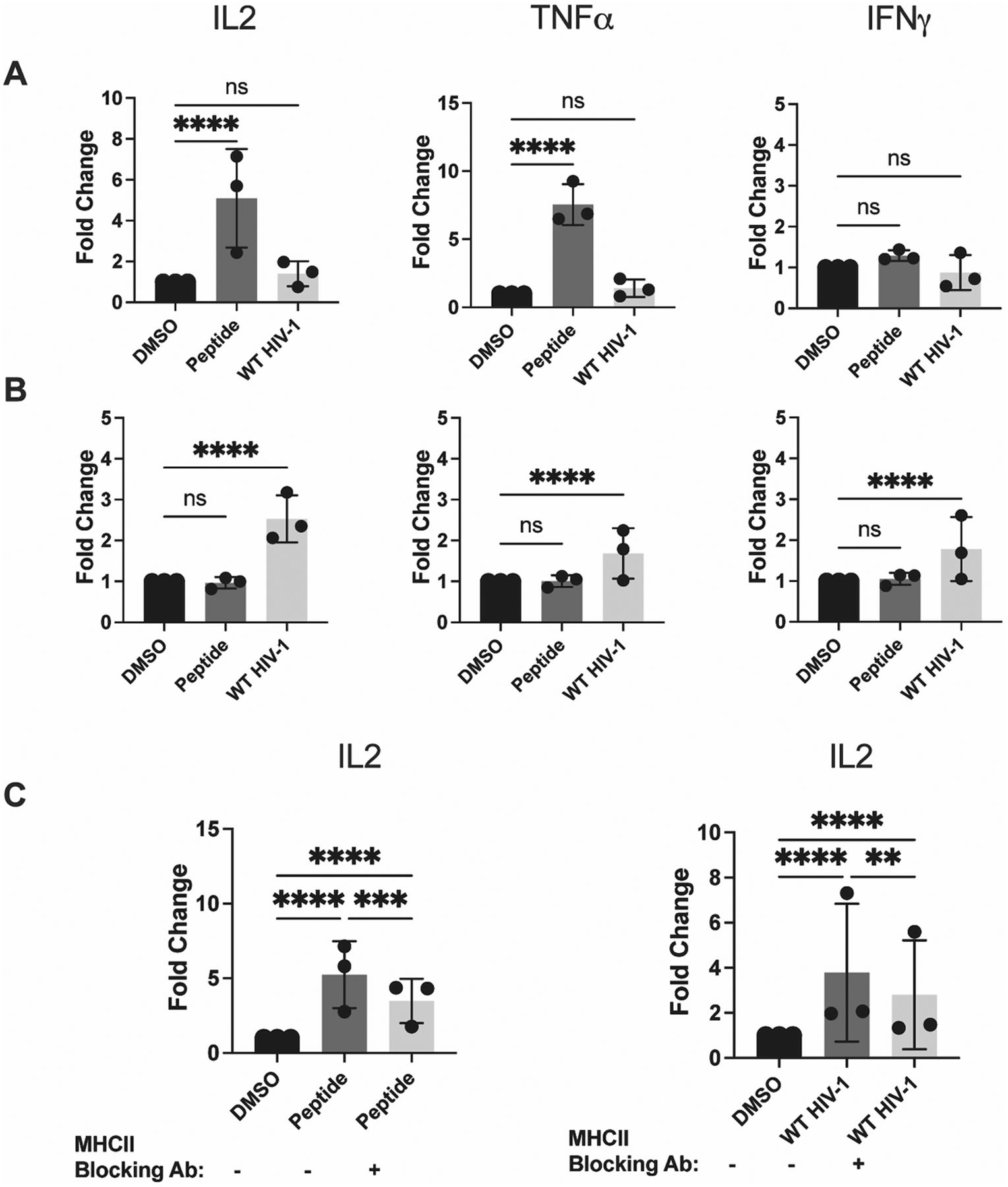
DCs and activated TCD4+ were cultured and infected as described in Materials and Methods. (A) DCs and (B) TCD4+ were assessed for their ability to present peptide and WT HIV-1 after 8 hours of coculture with HIV-1-specific TCD4+. IL-2, TNFα, and IFNγ expression was evaluated by flow cytometry. (C) DCs (left) and activated TCD4+ (right) were treated with 50 μg/mL MHCII blocking antibody 2 hours prior to the beginning of the assay. IL-2 expression was assessed by flow cytometry. Fold induction of each cytokine is shown, using DMSO as a baseline. Each dot represents an independent experiment. Bars represent mean ± SD. One way ANOVA, **p<0.01, ****p<0.001.
We confirmed that the activation of TCD4+ was epitope specific, as TCD4+ transduced with a TCR specific for ZnT8, a zinc transporter implicated in Type 1 diabetes (40) demonstrated either marginal or undetectable activation in response to infected TCD4+ (Fig. S2A–B). Additionally, we used an MHCII blocking antibody to confirm that presentation to HIV-1-specific TCD4+ is MHCII-dependent (Fig. 2C). Treatment with a control antibody against the transferrin receptor CD71 did not block presentation by infected TCD4+ (Fig. S2C). Notably, HIV-1 infection leads to CD4 downregulation at the infected TCD4+ surface, preventing superinfection (47) and likely preventing infected TCD4+ from presenting material derived from newly internalized virions. These results indicate that TCD4+, but not DCs, are capable of presenting Gag293 from infectious HIV-1 in an MHCII-restricted manner, leading to the activation of HIV-1-specific TCD4+. These data strongly suggest that activation is a direct consequence of MHCII-restricted epitope presentation.
Activated TCD4+ do not present inactivated or fusion-deficient HIV-1
Given the weak internalization capabilities of activated TCD4+ (Fig. 1G and H) and their susceptibility to productive HIV-1 infection, presentation seemed most attributable to endogenous processing, in which nascent viral antigen, not input virions, constitutes the major processing substrate (24, 26, 65). Thus, we hypothesized that activated TCD4+ would be unable to present epitope derived from structurally intact, chemically inactivated HIV-1. The same experimental setup as in Fig. 2 was used in these experiments, but a new experimental condition was introduced: aldrithiol-2 (AT2)-inactivated HIV-1. AT2-treated HIV-1 was added to uninfected DCs and activated TCD4+ 12 hours prior to the start of the assay. AT2-treated HIV-1 is still capable of fusing to its receptor, CD4, and its coreceptor, CCR5 or CXCR4, at the cell surface and inserting its nucleocapsid into the cytoplasm (66). However, the virus is unable to uncoat and initiate productive infection. If input virus was responsible for the presentation observed in Figure 2, then activated TCD4+ should be able to present this inactivated virus. As shown in Fig. 3A and B (representative flow plots shown in Fig. S4F–G), neither DCs nor activated TCD4+ can detectably present the Gag293 epitope derived from AT2-treated HIV-1, even at a much higher dose (Fig. S3A–B), supporting the notion that activated HIV-1-infected TCD4+ employ endogenous presentation to activate HIV-1-specific TCD4+.
Figure 3. DCs and activated TCD4+ are unable to present epitope derived from AT2-inactivated and K574D fusion-deficient HIV-1.
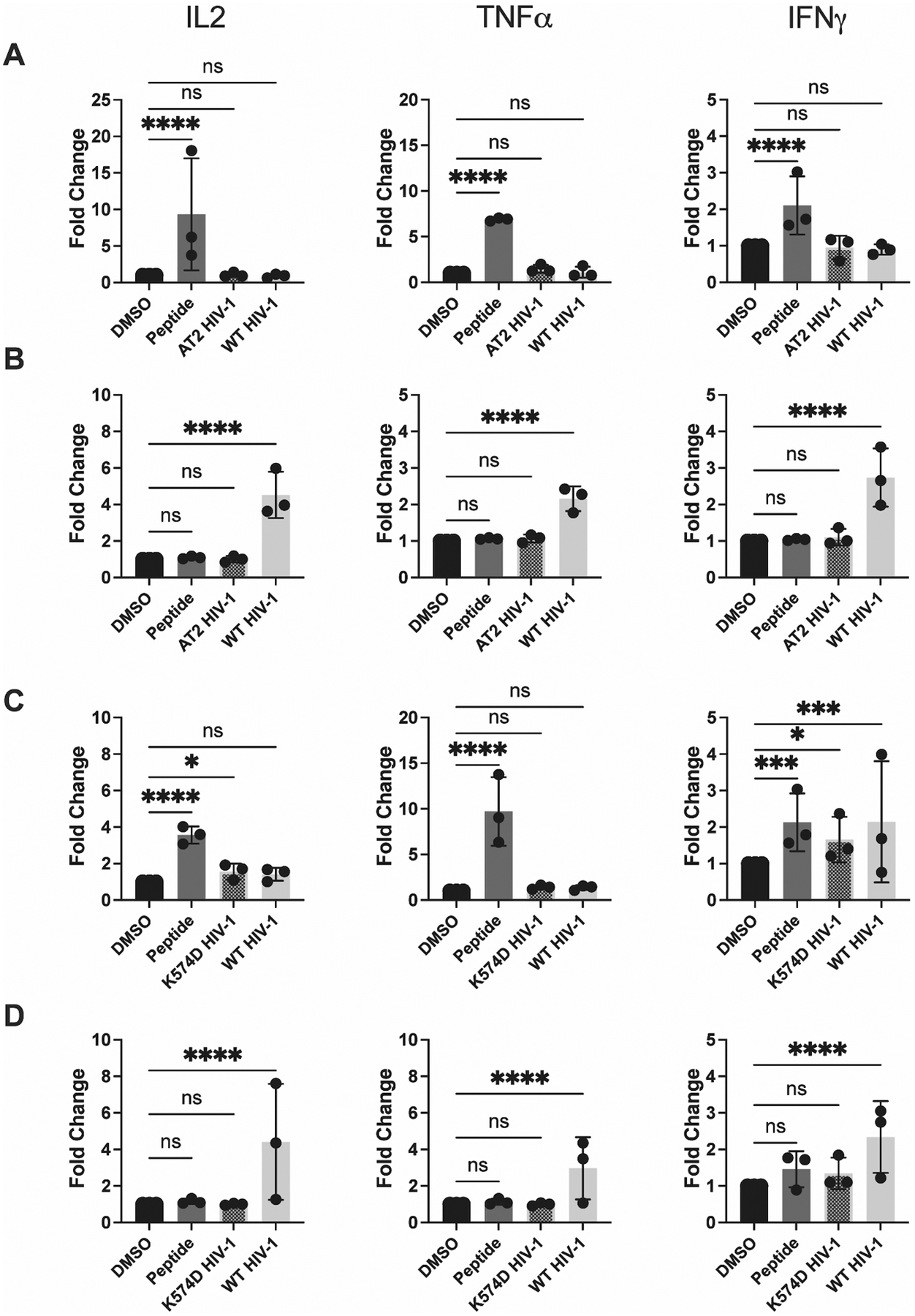
DCs and activated TCD4+ were cultured and infected with WT HIV-1 as described in Materials and Methods. AT2-treated HIV-1 and K574D fusion-deficient HIV-1 were added to DCs and activated TCD4+ 12 hours prior to the beginning of the assay. (A) DCs and (B) TCD4+ were assessed for their ability to present AT2-treated HIV-1 after 8 hours of coculture with HIV-1-specific TCD4+. IL-2, TNFα, and IFNγ expression was assessed by flow cytometry. (C) DCs and (D) activated TCD4+ were assessed for their ability to present fusion-deficient HIV-1 in a similar manner. Fold induction of each cytokine is shown, using DMSO as a baseline. Each dot represents an independent experiment. Bars represent mean ± SD. One way ANOVA, *p<0.05, **p<0.01, ****p<0.001.
To reinforce this finding, we tested the abilities of DCs and activated TCD4+ to generate epitope from infectious, but fusion-deficient, HIV-1 (K574D HIV-1). This virus was tested for two main reasons. First, chemical inactivation might have negatively impacted virion processibility, and thus use of a fusion-deficient virus provided an orthogonal approach to reinforce the finding. Second, since HIV-1 fuses at the target cell surface and injects its viral material directly into the cytoplasm (67), HIV-1 proteins may have limited opportunity to access endocytic compartments where conventional MHCII antigen processing occurs (17). Thus, absence of fusion at the cell surface may drive more input virions into endocytic compartments. Again, the same experimental setup was utilized in these experiments as in Fig. 2, except that fusion deficient HIV-1, quantified by p24 content, was compared to fusion-competent HIV-1, both being added to DCs and activated TCD4+ 12 hours prior to addition of Gag293-specific TCD4+. As indicated in Fig. 3C and D (representative flow plots shown in Fig. S4H–I), activated TCD4+ are unable to present fusion deficient HIV-1. DCs are able to present fusion deficient HIV-1, though the resulting HIV-1-specific TCD4+ responses are not robust. These data, combined with the lack of presentation of AT2-inactivated HIV-1, strongly suggest that HIV-1 must 1) fuse at the cell surface and 2) initiate productive infection in order to be processed and presented by activated TCD4+. However, in our hands, DCs are reproducibly unable to present the Gag293 epitope from infectious or chemically inactivated HIV-1 and are capable of only modest presentation of fusion deficient virus.
DCs, but not activated TCD4+, indirectly present subviral material
We have so far observed presentation by activated TCD4+ only when infectious HIV-1 was used. This could be due to direct, endogenous processing and presentation by the infected TCD4+ or via “indirect presentation” (68, 69), in which infected TCD4+ release viral material that is taken up and presented by uninfected, activated TCD4+. We subjected supernatant from HIV-1 infected TCD4+ to either 1) UV light to render the supernatant non-infectious or 2) 100 kDa filtration to remove whole virions and larger subcellular material. DCs and activated TCD4+ were incubated with treated supernatant for 12 hours prior to the assay. We found that DCs were able to present epitope from UV-treated supernatant, indicating that they are capable of MHCII-restricted indirect presentation (Fig. 4A, representative flow plots shown in Fig. S4J–K). This result is consistent with previous studies showing DCs are capable of indirect presentation (68, 69). Interestingly, DCs were unable to present filtered supernatant (Fig. 4A), and thus may require relatively large subcellular material, such as exosomes (69) or apoptotic bodies (70) for successful indirect presentation of the Gag293 epitope. In contrast, activated TCD4+ are capable of only modest presentation of UV-treated supernatant (Fig. 4B), indicating that indirect presentation does not play a major role in the presentation of HIV-1-derived epitope by activated TCD4+.
Figure 4. DCs, but not activated TCD4+, indirectly present HIV-1-derived epitope.
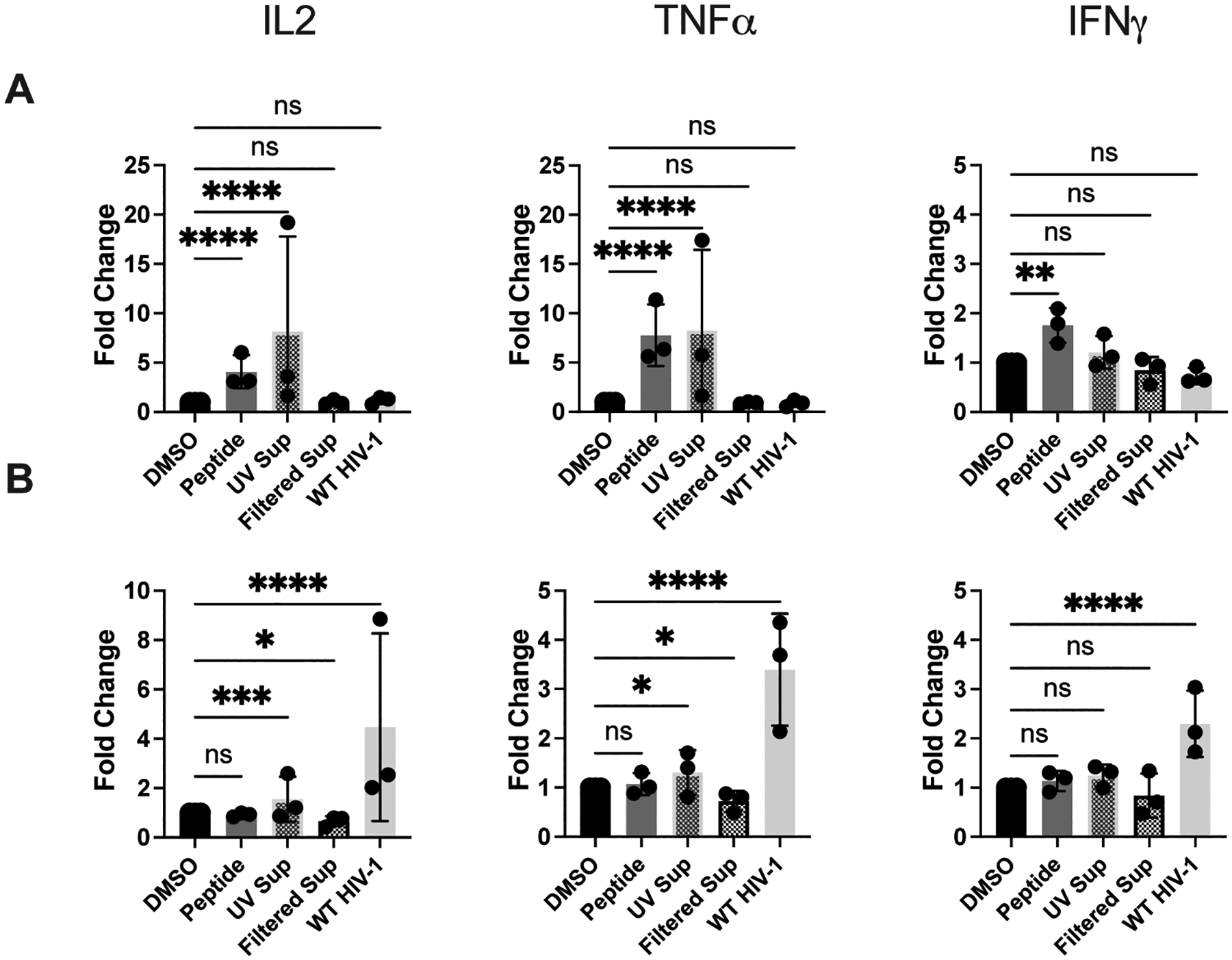
Presentation of UV-treated and filtered HIV-1+ supernatant by DCs and activated TCD4+ to HIV-1-specific TCD4+. DCs and activated TCD4+ were cultured and infected with WT HIV-1 as described in Materials and Methods. Supernatant from WT HIV-1-infected TCD4+ was collected and treated with UV light or filtered through a 100 kDa filter and then added to DCs and activated TCD4+ 12 hours prior to the beginning of the assay. (A) DCs and (B) TCD4+ were cocultured with HIV-1-specific TCD4+ for 8 hours and IL-2, TNFα, and IFNγ expression was assessed by flow cytometry. Fold induction of each cytokine is shown, using DMSO as a baseline. Each dot represents an independent experiment. Bars represent mean ± SD. One way ANOVA, *p<0.05, **p<0.01, ***p<0.005, ****p<0.001.
In addition to addressing the question of indirect presentation, the results provide additional support for our conclusion that activated TCD4+ carry out endogenous processing. Supernatants were collected from activated TCD4+ 72 hours post-infection, the same duration of infection prior to addition of responding TCD4+. Lack of a robust level of presentation of the UV treated supernatant by activated TCD4+ (Fig. 4) argues against the presentation of exogenous material in the form of extracellular virions or subviral material and in favor of bona fide endogenous processing and presentation.
Nef and Vpu do not impact TCD4+-mediated presentation of HIV-1 antigen
The viral accessory proteins Nef and Vpu have been reported to disrupt MHCII-restricted antigen presentation in myeloid-derived cells (45, 71, 72). We investigated whether these proteins contribute to the lack of detectable presentation of live HIV-1 by DCs. To test this, and to determine the impact of these proteins on MHCII-restricted presentation by activated TCD4+, we utilized a Nef/Vpu double knockout (KO) HIV-1. Infection with the Nef/Vpu double KO virus was performed using the same method as in Fig. 2. Using this virus, we could not detect any impact of Nef or Vpu on presentation of the Gag293 epitope by either DCs or activated TCD4+ (Fig. 5A and B, representative flow plots shown in Fig. S4L–M). DCs were still unable to present the Gag293 epitope from infectious Nef/Vpu KO virus (Fig. 5A), and there were no significant differences in the presentation of WT and double KO HIV-1 by activated TCD4+ (Fig. 5B).
Figure 5. The viral accessory proteins Nef and Vpu do not impact the presentation of the Gag293 epitope by DCs and activated TCD4+.
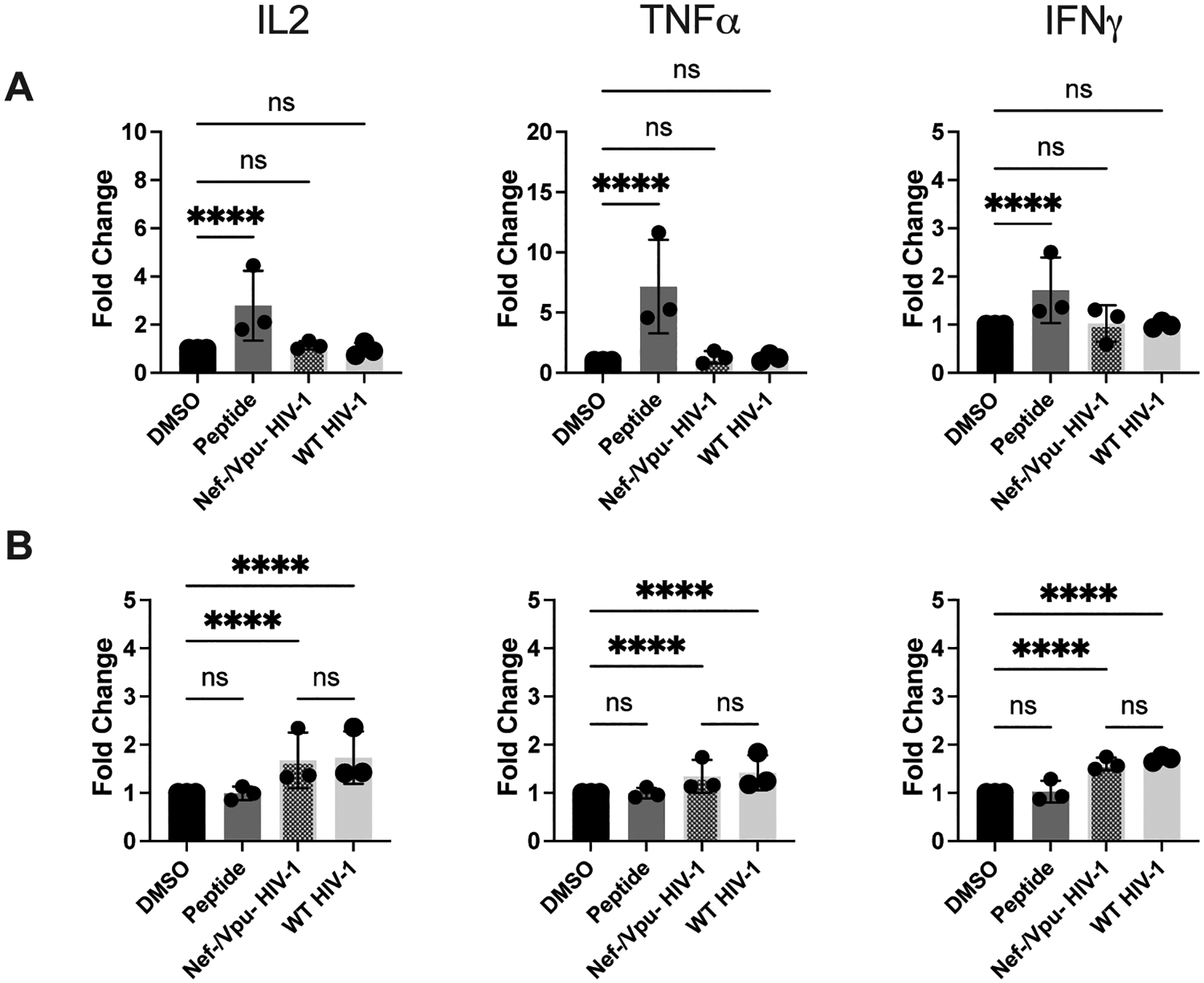
DCs and activated TCD4+ were cultured and infected with WT HIV-1 and Nef-/Vpu- HIV-1 as described in Materials and Methods. (A) DCs and (B) TCD4+ were cocultured with HIV-1-specific TCD4+ for 8 hours and IL-2, TNFα, and IFNγ expression was assessed by flow cytometry. Fold induction of each cytokine is shown, using DMSO as a baseline. Each dot represents an independent experiment. Bars represent mean ± SD. One way ANOVA, ****p<0.001.
DISCUSSION
Our study reveals that activated TCD4+ express several key components of the antigen processing and presentation machinery (Fig. 1A and B), and this expression remains intact in HIV-1-infected cells (Fig. 1C and D). We also show that activated TCD4+ express functional endosomal proteases (Fig. 1E and F), though their ability to internalize exogenous antigen is restricted, as demonstrated by their limited ability to unquench DQ-OVA (Fig. 1G and H) and internalize FITC-Dextran (Fig. 1L–M). Taken together, these data suggest that activated TCD4+ have the ability to process and present antigen on MHCII but are limited with respect to classical processing and presentation by their modest ability to internalize extracellular antigen.
Importantly, we observed that activated TCD4+ are able to present antigen derived from infectious HIV-1 to HIV-1-specific TCD4+, eliciting a strong cytokine response (Fig. 2A and B). This presentation is MHCII-dependent, as the stimulation of IL-2 secretion by HIV-1-specific TCD4+ can be inhibited with MHCII blocking antibody (Fig. 2C), and is antigen-specific, as ZnT8 TCR-transduced TCD4+ experienced either far weaker or undetectable activation in response to HIV-1-infected TCD4+ (Fig. S2A–B). We further confirmed that TCD4+ can present HIV-1-derived epitope only when they are productively infected, as these cells are unable to present chemically inactivated virus (Fig. 3B) or fusion-deficient virus (Fig. 3D) and are unable to indirectly present epitope from supernatants of HIV-1-infected cells (Fig. 4B). We also note that activated TCD4+ are unable to present synthetic peptide, even at high concentrations (Fig. S3C). This phenomenon has been previously observed in other antigen systems (23). Two potential explanations are limited peptide exchange at the cell surface (61–63), or the need for peptide to be internalized by the cell for successful presentation (64), which TCD4+ appear to be incapable of performing. These data lead us to conclude that activated TCD4+ employ endogenous processing to stimulate HIV-1-specific TCD4+. Thus, cell types with only a marginal capacity for classical processing and presentation can nevertheless be highly functional APCs via viral infection and endogenous processing and presentation. How far this paradigm extends beyond HIV-1 and TCD4+ remains to be seen. To date, the vast majority of endogenous processing studies in primary cells focus on professional APCs. However, Toulmin et al. recently demonstrated that type II alveolar cells, epithelial cells located in the distal lung, express MHCII and utilize endogenous processing to present influenza proteins and the endogenous E protein (23). These findings, along with ours, suggest that non-professional APCs may play key roles in MHCII-restricted presentation via endogenous antigen processing during viral infections.
Previous studies have reported that activated TCD4+ are capable of presenting HIV-1 gp120-derived epitope (30, 31). However, ours appears to be the first demonstration of MHCII-restricted presentation by activated TCD4+ of epitope derived from viable HIV-1 virions. These results suggest a novel function for TCD4+ during the immune response to HIV-1, though these findings will need to be confirmed in vivo. In addition to orchestrating the adaptive immune response to HIV-1, our data suggest that TCD4+ may also act as APCs, leading to the activation of HIV- 1-specific TCD4+, though this presentation could potentially come at the price of enhanced viral spread to HIV-1-specific TCD4+ (73).
Previous studies have shown that monocyte-derived DCs are capable of presenting the Gag293 epitope from infectious HIV-1 (36); however, our results are not necessarily in conflict with this work. Galperin et al. focused on TCD4+-mediated killing of infected DCs as a readout of TCD4+ activation, while we focused on cytokine secretion. It is possible that the presentation of infectious HIV-1 by DCs leads to cytotoxicity and not cytokine production in responding TCD4+, which could explain the differences in our results. Of note, DCs can present the Gag293 epitope when exposed to UV-treated, HIV-1-infected supernatant (Fig. 4A), indicating that indirect presentation is a viable production route for this particular epitope in DCs. We focused on the Gag293 epitope due to the limited number of available full length, MHCII-restricted HIV-1-specific TCR sequences. It will be interesting to determine whether our observations extend to other MHCII-restricted HIV-1 epitopes. Based on our previous work with influenza (24) (in which 6 out of 6 MHCII-restricted epitopes are endogenously presented), we anticipate that Gag293 is representative of other epitopes, though the processing pathways could be quite different.
We also showed that the HIV-1 accessory proteins Nef and Vpu do not impact presentation of the Gag293 epitope by either DCs or activated TCD4+ (Fig. 5A and B). Again, the different processing requirements for different epitopes (24, 65, 74, 75) may explain the apparent contradiction with previous studies, which showed that Nef and Vpu disrupt MHCII-restricted presentation in DCs (45, 71, 72). Additionally, while we can conclude that Nef and Vpu do not interfere with the presentation of the Gag293 epitope in DCs and activated TCD4+, we are unable to speculate on the effects of these proteins on the presentation by other APC types.
In summary, our results indicate that activated, HIV-1 infected TCD4+ are highly effective at MHCII-restricted presentation of HIV-1-derived epitopes due to the tropism of HIV-1 for TCD4+ and the potency of endogenous MHCII-restricted processing and presentation. Given the critical importance of HIV-1-specific TCD4+, these results could significantly alter our understanding of the MHCII-restricted processing and presentation landscape that develops during HIV-1 infection.
Supplementary Material
KEY POINTS.
Activated TCD4+ can present MHCII-restricted antigen derived from infectious HIV-1.
Activated TCD4+ use endogenous processing to process and present HIV-1 antigen.
ACKNOWLEDGEMENTS
We are grateful to the following groups for their assistance with this project: 1) the University of Pennsylvania Human Immunology Core (supported by AI-045008 and CA-016520) for their invaluable assistance in cell procurement and processing, 2) the University of Pennsylvania Center for AIDS Research (CFAR) Viral and Reservoirs Core (supported by P30 AI 045008) for their help with HIV-1 titering and HIV-1 inactivation, 3) Roberto Mallone (INSERM, Fr) for supplying the ZnT8-specific TCR sequence, and 4) the Children’s Hospital of Philadelphia Flow Cytometry Core for their expertise and patience. The authors are also grateful to John Paul Bisciotti for his assistance and expertise with data transformation and statistical analysis. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
COMPETING INTERESTS
JLR is an equity holder of and receives sponsored research funding from Tmunity Therapeutics. The rest of the authors declare that they have no competing interests.
REFERENCES
- 1.UNAIDS. 2021. Global HIV & AIDS statistics — Fact sheet.
- 2.Wilen CB, Tilton JC, and Doms RW. 2012. HIV: cell binding and entry. Cold Spring Harb Perspect Med 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wallet C, De Rovere M, Van Assche J, Daouad F, De Wit S, Gautier V, Mallon PWG, Marcello A, Van Lint C, Rohr O, and Schwartz C. 2019. Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front Cell Infect Microbiol 9: 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raynaud-Messina B, Bracq L, Dupont M, Souriant S, Usmani SM, Proag A, Pingris K, Soldan V, Thibault C, Capilla F, Al Saati T, Gennero I, Jurdic P, Jolicoeur P, Davignon JL, Mempel TR, Benichou S, Maridonneau-Parini I, and Verollet C. 2018. Bone degradation machinery of osteoclasts: An HIV-1 target that contributes to bone loss. Proc Natl Acad Sci U S A 115: E2556–E2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moir S, Chun TW, and Fauci AS. 2011. Pathogenic mechanisms of HIV disease. Annu Rev Pathol 6: 223–248. [DOI] [PubMed] [Google Scholar]
- 6.Brown DM, Lee S, Garcia-Hernandez Mde L, and Swain SL. 2012. Multifunctional CD4 cells expressing gamma interferon and perforin mediate protection against lethal influenza virus infection. J Virol 86: 6792–6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swain SL, McKinstry KK, and Strutt TM. 2012. Expanding roles for CD4(+) T cells in immunity to viruses. Nat Rev Immunol 12: 136–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sant AJ, and McMichael A. 2012. Revealing the role of CD4(+) T cells in viral immunity. J Exp Med 209: 1391–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenberg ES, Billingsley JM, Caliendo AM, Boswell SL, Sax PE, Kalams SA, and Walker BD. 1997. Vigorous HIV-1-specific CD4+ T cell responses associated with control of viremia. Science 278: 1447–1450. [DOI] [PubMed] [Google Scholar]
- 10.Ranasinghe S, Soghoian DZ, Lindqvist M, Ghebremichael M, Donaghey F, Carrington M, Seaman MS, Kaufmann DE, Walker BD, and Porichis F. 2015. HIV-1 Antibody Neutralization Breadth Is Associated with Enhanced HIV-Specific CD4+ T Cell Responses. J Virol 90: 2208–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalams SA, Buchbinder SP, Rosenberg ES, Billingsley JM, Colbert DS, Jones NG, Shea AK, Trocha AK, and Walker BD. 1999. Association between virus-specific cytotoxic T-lymphocyte and helper responses in human immunodeficiency virus type 1 infection. J Virol 73: 6715–6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chevalier MF, Julg B, Pyo A, Flanders M, Ranasinghe S, Soghoian DZ, Kwon DS, Rychert J, Lian J, Muller MI, Cutler S, McAndrew E, Jessen H, Pereyra F, Rosenberg ES, Altfeld M, Walker BD, and Streeck H. 2011. HIV-1-specific interleukin-21+ CD4+ T cell responses contribute to durable viral control through the modulation of HIV-specific CD8+ T cell function. J Virol 85: 733–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson S, Eller M, Teigler JE, Maloveste SM, Schultz BT, Soghoian DZ, Lu R, Oster AF, Chenine AL, Alter G, Dittmer U, Marovich M, Robb ML, Michael NL, Bolton D, and Streeck H. 2015. Cooperativity of HIV-Specific Cytolytic CD4 T Cells and CD8 T Cells in Control of HIV Viremia. J Virol 89: 7494–7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soghoian DZ, Jessen H, Flanders M, Sierra-Davidson K, Cutler S, Pertel T, Ranasinghe S, Lindqvist M, Davis I, Lane K, Rychert J, Rosenberg ES, Piechocka-Trocha A, Brass AL, Brenchley JM, Walker BD, and Streeck H. 2012. HIV-specific cytolytic CD4 T cell responses during acute HIV infection predict disease outcome. Sci Transl Med 4: 123ra125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cohen K, Altfeld M, Alter G, and Stamatatos L. 2014. Early preservation of CXCR5+ PD-1+ helper T cells and B cell activation predict the breadth of neutralizing antibody responses in chronic HIV-1 infection. J Virol 88: 13310–13321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamamoto T, Lynch RM, Gautam R, Matus-Nicodemos R, Schmidt SD, Boswell KL, Darko S, Wong P, Sheng Z, Petrovas C, McDermott AB, Seder RA, Keele BF, Shapiro L, Douek DC, Nishimura Y, Mascola JR, Martin MA, and Koup RA. 2015. Quality and quantity of TFH cells are critical for broad antibody development in SHIVAD8 infection. Sci Transl Med 7: 298ra120. [DOI] [PubMed] [Google Scholar]
- 17.Roche PA, and Furuta K. 2015. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol 15: 203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller MA, Ganesan AP, and Eisenlohr LC. 2013. Toward a Network Model of MHC Class II-Restricted Antigen Processing. Front Immunol 4: 464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duraes FV, Niven J, Dubrot J, Hugues S, and Gannage M. 2015. Macroautophagy in Endogenous Processing of Self- and Pathogen-Derived Antigens for MHC Class II Presentation. Front Immunol 6: 459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thiele F, Tao S, Zhang Y, Muschaweckh A, Zollmann T, Protzer U, Abele R, and Drexler I. 2015. Modified vaccinia virus Ankara-infected dendritic cells present CD4+ T-cell epitopes by endogenous major histocompatibility complex class II presentation pathways. J Virol 89: 2698–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eisenlohr LC 2013. Alternative generation of MHC class II-restricted epitopes: not so exceptional? Mol Immunol 55: 169–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eisenlohr LC, Luckashenak N, Apcher S, Miller MA, and Sinnathamby G. 2011. Beyond the classical: influenza virus and the elucidation of alternative MHC class II-restricted antigen processing pathways. Immunol Res 51: 237–248. [DOI] [PubMed] [Google Scholar]
- 23.Toulmin SA, Bhadiadra C, Paris AJ, Lin JH, Katzen J, Basil MC, Morrisey EE, Worthen GS, and Eisenlohr LC. 2021. Type II alveolar cell MHCII improves respiratory viral disease outcomes while exhibiting limited antigen presentation. Nat Commun 12: 3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller MA, Ganesan AP, Luckashenak N, Mendonca M, and Eisenlohr LC. 2015. Endogenous antigen processing drives the primary CD4+ T cell response to influenza. Nat Med 21: 1216–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jacobson S, Sekaly RP, Jacobson CL, McFarland HF, and Long EO. 1989. HLA class II-restricted presentation of cytoplasmic measles virus antigens to cytotoxic T cells. J Virol 63: 1756–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coulon PG, Richetta C, Rouers A, Blanchet FP, Urrutia A, Guerbois M, Piguet V, Theodorou I, Bet A, Schwartz O, Tangy F, Graff-Dubois S, Cardinaud S, and Moris A. 2016. HIV-Infected Dendritic Cells Present Endogenous MHC Class II-Restricted Antigens to HIV-Specific CD4+ T Cells. J Immunol 197: 517–532. [DOI] [PubMed] [Google Scholar]
- 27.Shrikant P, and Benveniste EN. 1996. The central nervous system as an immunocompetent organ: role of glial cells in antigen presentation. J Immunol 157: 1819–1822. [PubMed] [Google Scholar]
- 28.Giroux M, Schmidt M, and Descoteaux A. 2003. IFN-gamma-induced MHC class II expression: transactivation of class II transactivator promoter IV by IFN regulatory factor-1 is regulated by protein kinase C-alpha. J Immunol 171: 4187–4194. [DOI] [PubMed] [Google Scholar]
- 29.June CH, Bluestone JA, Nadler LM, and Thompson CB. 1994. The B7 and CD28 receptor families. Immunol Today 15: 321–331. [DOI] [PubMed] [Google Scholar]
- 30.Lanzavecchia A, Roosnek E, Gregory T, Berman P, and Abrignani S. 1988. T cells can present antigens such as HIV gp120 targeted to their own surface molecules. Nature 334: 530–532. [DOI] [PubMed] [Google Scholar]
- 31.Siliciano RF, Lawton T, Knall C, Karr RW, Berman P, Gregory T, and Reinherz EL. 1988. Analysis of host-virus interactions in AIDS with anti-gp120 T cell clones: effect of HIV sequence variation and a mechanism for CD4+ cell depletion. Cell 54: 561–575. [DOI] [PubMed] [Google Scholar]
- 32.Golovina TN, Mikheeva T, Suhoski MM, Aqui NA, Tai VC, Shan X, Liu R, Balcarcel RR, Fisher N, Levine BL, Carroll RG, Warner N, Blazar BR, June CH, and Riley JL. 2008. CD28 costimulation is essential for human T regulatory expansion and function. J Immunol 181: 2855–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meissner EG, Duus KM, Gao F, Yu XF, and Su L. 2004. Characterization of a thymus-tropic HIV-1 isolate from a rapid progressor: role of the envelope. Virology 328: 74–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pacheco B, Alsahafi N, Debbeche O, Prevost J, Ding S, Chapleau JP, Herschhorn A, Madani N, Princiotto A, Melillo B, Gu C, Zeng X, Mao Y, Smith AB 3rd, Sodroski J, and Finzi A. 2017. Residues in the gp41 Ectodomain Regulate HIV-1 Envelope Glycoprotein Conformational Transitions Induced by gp120-Directed Inhibitors. J Virol 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benati D, Galperin M, Lambotte O, Gras S, Lim A, Mukhopadhyay M, Nouel A, Campbell KA, Lemercier B, Claireaux M, Hendou S, Lechat P, de Truchis P, Boufassa F, Rossjohn J, Delfraissy JF, Arenzana-Seisdedos F, and Chakrabarti LA. 2016. Public T cell receptors confer high-avidity CD4 responses to HIV controllers. J Clin Invest 126: 2093–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galperin M, Farenc C, Mukhopadhyay M, Jayasinghe D, Decroos A, Benati D, Tan LL, Ciacchi L, Reid HH, Rossjohn J, Chakrabarti LA, and Gras S. 2018. CD4(+) T cell-mediated HLA class II cross-restriction in HIV controllers. Sci Immunol 3. [DOI] [PubMed] [Google Scholar]
- 37.Richardson MW, Guo L, Xin F, Yang X, and Riley JL. 2014. Stabilized human TRIM5alpha protects human T cells from HIV-1 infection. Mol Ther 22: 1084–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang S, Cohen CJ, Peng PD, Zhao Y, Cassard L, Yu Z, Zheng Z, Jones S, Restifo NP, Rosenberg SA, and Morgan RA. 2008. Development of optimal bicistronic lentiviral vectors facilitates high-level TCR gene expression and robust tumor cell recognition. Gene Ther 15: 1411–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Humbert O, Gisch DW, Wohlfahrt ME, Adams AB, Greenberg PD, Schmitt TM, Trobridge GD, and Kiem HP. 2016. Development of Third-generation Cocal Envelope Producer Cell Lines for Robust Lentiviral Gene Transfer into Hematopoietic Stem Cells and T-cells. Mol Ther 24: 1237–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Culina S, Lalanne AI, Afonso G, Cerosaletti K, Pinto S, Sebastiani G, Kuranda K, Nigi L, Eugster A, Osterbye T, Maugein A, McLaren JE, Ladell K, Larger E, Beressi JP, Lissina A, Appay V, Davidson HW, Buus S, Price DA, Kuhn M, Bonifacio E, Battaglia M, Caillat-Zucman S, Dotta F, Scharfmann R, Kyewski B, Mallone R, and G. ImMaDiab Study. 2018. Islet-reactive CD8(+) T cell frequencies in the pancreas, but not in blood, distinguish type 1 diabetic patients from healthy donors. Sci Immunol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Team R. C. 2021. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 42.Levacher M, Tallet S, Dazza MC, Dournon E, Rouveix B, and Pocidalo JJ. 1990. T activation marker evaluation in ARC patients treated with AZT. Comparison with CD4+ lymphocyte count in non-progressors and progressors towards AIDS. Clin Exp Immunol 81: 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hathcock KS, and Hodes RJ. 1996. Role of the CD28-B7 costimulatory pathways in T cell-dependent B cell responses. Adv Immunol 62: 131–166. [DOI] [PubMed] [Google Scholar]
- 44.Zola H, Swart B, Nicholson I, and Voss E. 2007. Leukocyte and Stromal Cell Molecules: The CD Markers. John Wiley and Sons, Inc. [Google Scholar]
- 45.Schindler M, Wurfl S, Benaroch P, Greenough TC, Daniels R, Easterbrook P, Brenner M, Munch J, and Kirchhoff F. 2003. Down-modulation of mature major histocompatibility complex class II and up-regulation of invariant chain cell surface expression are well-conserved functions of human and simian immunodeficiency virus nef alleles. J Virol 77: 10548–10556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stumptner-Cuvelette P, Morchoisne S, Dugast M, Le Gall S, Raposo G, Schwartz O, and Benaroch P. 2001. HIV-1 Nef impairs MHC class II antigen presentation and surface expression. Proc Natl Acad Sci U S A 98: 12144–12149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kwon Y, Kaake RM, Echeverria I, Suarez M, Karimian Shamsabadi M, Stoneham C, Ramirez PW, Kress J, Singh R, Sali A, Krogan N, Guatelli J, and Jia X. 2020. Structural basis of CD4 downregulation by HIV-1 Nef. Nat Struct Mol Biol 27: 822–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bryant PW, Lennon-Dumenil AM, Fiebiger E, Lagaudriere-Gesbert C, and Ploegh HL. 2002. Proteolysis and antigen presentation by MHC class II molecules. Adv Immunol 80: 71–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hsieh CS, deRoos P, Honey K, Beers C, and Rudensky AY. 2002. A role for cathepsin L and cathepsin S in peptide generation for MHC class II presentation. J Immunol 168: 2618–2625. [DOI] [PubMed] [Google Scholar]
- 50.Rodriguez GM, and Diment S. 1992. Role of cathepsin D in antigen presentation of ovalbumin. J Immunol 149: 2894–2898. [PubMed] [Google Scholar]
- 51.Sadegh-Nasseri S, and Kim A. 2015. MHC Class II Auto-Antigen Presentation is Unconventional. Front Immunol 6: 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bania J, Gatti E, Lelouard H, David A, Cappello F, Weber E, Camosseto V, and Pierre P. 2003. Human cathepsin S, but not cathepsin L, degrades efficiently MHC class II-associated invariant chain in nonprofessional APCs. Proc Natl Acad Sci U S A 100: 6664–6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu Z, and Roche PA. 2015. Macropinocytosis in phagocytes: regulation of MHC class-II-restricted antigen presentation in dendritic cells. Front Physiol 6: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lim JP, and Gleeson PA. 2011. Macropinocytosis: an endocytic pathway for internalising large gulps. Immunol Cell Biol 89: 836–843. [DOI] [PubMed] [Google Scholar]
- 55.Stuart LM, and Ezekowitz RA. 2005. Phagocytosis: elegant complexity. Immunity 22: 539–550. [DOI] [PubMed] [Google Scholar]
- 56.Platt CD, Ma JK, Chalouni C, Ebersold M, Bou-Reslan H, Carano RA, Mellman I, and Delamarre L. 2010. Mature dendritic cells use endocytic receptors to capture and present antigens. Proc. Natl. Acad. Sci. U. S. A 107: 4287–4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wahid R, Cannon MJ, and Chow M. 2005. Dendritic cells and macrophages are productively infected by poliovirus. J Virol 79: 401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Park J, Wu CT, and Bryers JD. 2013. Chemokine programming dendritic cell antigen response: part I - select chemokine programming of antigen uptake even after maturation. Immunology 139: 72–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mee CJ, Grove J, Harris HJ, Hu K, Balfe P, and McKeating JA. 2008. Effect of cell polarization on hepatitis C virus entry. J Virol 82: 461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Olsen I, and Sollid LM. 2013. Pitfalls in determining the cytokine profile of human T cells. J Immunol Methods 390: 106–112. [DOI] [PubMed] [Google Scholar]
- 61.Jensen PE, Weber DA, Thayer WP, Westerman LE, and Dao CT. 1999. Peptide exchange in MHC molecules. Immunol Rev 172: 229–238. [DOI] [PubMed] [Google Scholar]
- 62.Freund C, and Hofer T. 2019. A Missing Switch in Peptide Exchange for MHC Class II Molecules. Front Immunol 10: 2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wieczorek M, Sticht J, Stolzenberg S, Gunther S, Wehmeyer C, El Habre Z, Alvaro-Benito M, Noe F, and Freund C. 2016. MHC class II complexes sample intermediate states along the peptide exchange pathway. Nat Commun 7: 13224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pathak SS, and Blum JS. 2000. Endocytic recycling is required for the presentation of an exogenous peptide via MHC class II molecules. Traffic 1: 561–569. [DOI] [PubMed] [Google Scholar]
- 65.Eisenlohr LC, and Hackett CJ. 1989. Class II major histocompatibility complex-restricted T cells specific for a virion structural protein that do not recognize exogenous influenza virus. Evidence that presentation of labile T cell determinants is favored by endogenous antigen synthesis. J Exp Med 169: 921–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rossio JL, Esser MT, Suryanarayana K, Schneider DK, Bess JW Jr., Vasquez GM, Wiltrout TA, Chertova E, Grimes MK, Sattentau Q, Arthur LO, Henderson LE, and Lifson JD. 1998. Inactivation of human immunodeficiency virus type 1 infectivity with preservation of conformational and functional integrity of virion surface proteins. J Virol 72: 7992–8001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Research, N. s. O. o. A. 2021. The HIV Life Cycle.
- 68.Forsyth KS, Roy NH, Peauroi E, DeHaven BC, Wold ED, Hersperger AR, Burkhardt JK, and Eisenlohr LC. 2020. Ectromelia-encoded virulence factor C15 specifically inhibits antigen presentation to CD4+ T cells post peptide loading. PLoS Pathog 16: e1008685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Testa JS, Apcher GS, Comber JD, and Eisenlohr LC. 2010. Exosome-driven antigen transfer for MHC class II presentation facilitated by the receptor binding activity of influenza hemagglutinin. J Immunol 185: 6608–6616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Munz C 2012. Antigen Processing for MHC Class II Presentation via Autophagy. Front Immunol 3: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Toussaint H, Gobert FX, Schindler M, Banning C, Kozik P, Jouve M, Kirchhoff F, and Benaroch P. 2008. Human immunodeficiency virus type 1 nef expression prevents AP-2-mediated internalization of the major histocompatibility complex class II-associated invariant chain. J Virol 82: 8373–8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chaudhry A, Verghese DA, Das SR, Jameel S, George A, Bal V, Mayor S, and Rath S. 2009. HIV-1 Nef promotes endocytosis of cell surface MHC class II molecules via a constitutive pathway. J Immunol 183: 2415–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Douek DC, Brenchley JM, Betts MR, Ambrozak DR, Hill BJ, Okamoto Y, Casazza JP, Kuruppu J, Kunstman K, Wolinsky S, Grossman Z, Dybul M, Oxenius A, Price DA, Connors M, and Koup RA. 2002. HIV preferentially infects HIV-specific CD4+ T cells. Nature 417: 95–98. [DOI] [PubMed] [Google Scholar]
- 74.Tewari MK, Sinnathamby G, Rajagopal D, and Eisenlohr LC. 2005. A cytosolic pathway for MHC class II-restricted antigen processing that is proteasome and TAP dependent. Nat Immunol 6: 287–294. [DOI] [PubMed] [Google Scholar]
- 75.Eisenlohr LC, Gerhard W, and Hackett CJ. 1988. Individual class II-restricted antigenic determinants of the same protein exhibit distinct kinetics of appearance and persistence on antigen-presenting cells. J Immunol 141: 2581–2584. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.