

Abstract
Bile acid‐CoA: amino acid N‐acyltransferase (BAAT) catalyzes bile acid conjugation, the last step in bile acid synthesis. BAAT gene mutation in humans results in hypercholanemia, growth retardation, and fat‐soluble vitamin insufficiency. The current study investigated the physiological function of BAAT in bile acid and lipid metabolism using Baat −/− mice. The bile acid composition and hepatic gene expression were analyzed in 10‐week‐old Baat −/− mice. They were also challenged with a westernized diet (WD) for additional 15 weeks to assess the role of BAAT in bile acid, lipid, and glucose metabolism. Comprehensive lab animal monitoring system and cecal 16S ribosomal RNA gene sequencing were used to evaluate the energy metabolism and microbiome structure of the mice, respectively. In Baat −/− mice, hepatic bile acids were mostly unconjugated and their levels were significantly increased compared with wild‐type mice. Bile acid polyhydroxylation was markedly up‐regulated to detoxify unconjugated bile acid accumulated in Baat −/− mice. Although the level of serum marker of bile acid synthesis, 7α‐hydroxy‐4‐cholesten‐3‐one, was higher in Baat −/− mice, their bile acid pool size was smaller. When fed a WD, the Baat −/− mice showed a compromised body weight gain and impaired insulin secretion. The gut microbiome of Baat −/− mice showed a low level of sulfidogenic bacteria Bilophila. Conclusion: Mouse BAAT is the major taurine‐conjugating enzyme. Its deletion protected the animals from diet‐induced obesity, but caused glucose intolerance. The gut microbiome of the Baat −/− mice was altered to accommodate the unconjugated bile acid pool.
BaatKO mice exhibited almost complete absence of taurine conjugated bile acids in the liver. The mutant mice thrived on this hydrophobic bile acid condition via induction of BA hydroxylation mechanism with compromised body weight gaining.

INTRODUCTION
The conjugation of bile acids (BAs) with glycine or taurine is the last step in hepatic BA synthesis before their release into the gallbladder. The step is catalyzed by bile acid CoA: amino acid N‐acyltransferase (BAAT) and lowers the pKa of BAs to prevent passive absorption in the duodenum for proper emulsification of dietary lipids. In the liver, the classic BA synthesis pathway is initiated by the rate‐limiting enzyme, cholesterol 7α‐hydroxylase (CYP7A1), to synthesize two predominant primary BAs, cholic acid (CA) and chenodeoxycholic acid (CDCA), in humans.[ 1 ] Sterol 12α‐hydroxylase (CYP8B1) catalyzes 12α‐hydroxylation and determines the ratio of CA to CDCA. 7α‐hydroxy‐4‐cholesten‐3‐one (C4) is the common precursor for CA and CDCA synthesis; thus, the serum C4 level is a surrogate marker for the rate of BA synthesis.[ 2 ] Mitochondrial sterol 27‐hydroxylase (CYP27A1) catalyzes the sterol side‐chain oxidation. CYP27A1 initiated the alternative BA synthesis pathway, which requires oxysterol 7α‐hydroxylase (CYP7B1). BA intermediates are activated by a peroxisomal CoA ligase, very‐long‐chain acyl‐CoA synthase (fatty acid transport protein 2 [FATP2]; SLC27A2) to produce CoA esters of 27 carbon (C27) BAs.[ 3 ] They undergo β‐oxidation to produce C24 BA‐CoA esters, which are conjugated with glycine and taurine by BAAT and are then secreted to the bile through the bile salt export pump (BSEP; ABCB11), a canalicular membrane transporter for conjugated BAs.[ 4 ] In contrast, deconjugated BAs circulated from the intestine to liver are directly activated by BA‐CoA ligase (FATP5; SLC27A5) before conjugation in hepatocytes (Figure S1).[ 3 ]
Inborn errors of BA conjugation cause malabsorption of dietary fats and lipid‐soluble vitamins. A single homozygous BAAT mutation (226G/226G) was first reported in Amish individuals with familial hypercholanemia, who presented an almost complete absence of conjugated BAs in the serum.[ 5 ] Most of the patients failed to thrive at the time of diagnosis, but 1 patient was asymptomatic from birth. In 2013, Setchell et al. reported detailed clinical and biochemical features manifested by conjugated BA deficiency caused by homozygous BAAT mutations. Most of the patients developed rickets due to low serum fat soluble vitamin levels.[ 6 ] General clinical features identified in human BA conjugation deficiency were very similar to what Dr. Hofmann predicted from the physicochemical properties of amino acid conjugation.[ 7 ]
There have been several mouse studies with BA conjugation deficiency. One noticeable study was carried out by targeting deletion of Fatp5, which led to a significant BA conjugation deficiency in the mutant mouse.[ 8 ] However, the mice failed to present any significant pathophysiological features manifested by human BA conjugation deficiency by BAAT mutations, such as hypercholanemia and overt fat malabsorption. Therefore, a proper mouse model of BA conjugation deficiency is needed to understand the pathophysiology presented by human BA conjugation deficiency further and to assess potential effects of unconjugated BAs on whole‐body metabolic homeostasis.
In the present study, we used Baat −/− mice to gain insights into the role of mouse BAAT in lipid and BA metabolism and to evaluate any pathophysiology unidentified from rare human cases of BA conjugation deficiency. The Baat −/− mice exhibited strong BA conjugation deficiency along with higher plasma BA levels as observed in humans, and gained less weight but consumed more food than their wild‐type (WT) littermates on a Western diet (WD) regimen. In addition, the gut microbiome comprised a distinctive structure to accommodate the BA conjugation deficiency.
EXPERIMENTAL PROCEDURES
Animals and diets
A male mouse and a female mouse with heterozygous Baat deletion (Baat[em1(IMPC)Mbp], MGI:6148072, the Mutant Mouse Resource & Research Centers at the University of California, Davis) were bred to generate experimental Baat −/− mice and their WT littermates (Figure S2A–C). Throughout this study, only male mice were used (different phenotypes manifested by female Baat −/− mice will be subjects of a future study). At 10 weeks old, the offspring were either euthanized for tissue and blood collection or started on a WD regimen (Envigo; 21.2% [wt/wt] total fat [42% Kcal from fat], 0.2% [wt/wt] cholesterol [4.5 kcal/g]) for 15 weeks. All tissue and blood samples were collected after overnight fasting, except 5 h fasting for BA pool‐size measurements. The regular chow (6.7% [wt/wt] total fat [16.7% Kcal from fat], 280 ppm cholesterol) was purchased from Formulab Diet. All mice were housed in the accredited pathogen‐free facility at Northeast Ohio Medical University on a 12‐h light/dark cycle. All animal care and use protocols were approved by the Institutional Animal Care and Use Committee of Northeast Ohio Medical University.
Determination of bile acids by mass spectroscopy
Samples of liver (25 mg), ileum (25 mg), plasma (25 μl), or gallbladder (1 μl) were processed and their BA compositions were determined by liquid chromatography–mass spectrometry (LC/MS).[ 9 ]
For polyhydroxylated BA quantification, liver homogenates (100 mg) or plasma (50 μl) were analyzed by a liquid chromatography–tandem mass spectrometry (LC/MS‐MS) system.[ 10 ]
C4 quantification
A total of 20 μl of plasma was mixed with 80 μl internal standard containing 50 ng/ml of C4‐d7 (Cayman Chem, Ann Arbor, MI, USA) in methanol. The mixture was centrifuged at 17,000 g for 15 min at 4°C after a thorough vortex. A total of 10 μl of the supernatant was injected to a Vanquish ultrahigh‐performance liquid chromatography system coupled with a Q‐Exactive Orbitrap mass spectrometry (Thermo Fisher). After separation by an Accucore Vanquish C18 column (Thermo Fisher), all product ions of 401.3414 (C4) and 408.3853 (C4‐d7) were monitored on parallel reaction monitoring mode at the collision energy of 60 eV. The representative m/z transitions for C4 and C4‐d7 were analyzed using skyline software (University of Washington, Seattle, WA), and the C4 concentrations were determined with calibrators raging from 3 ng/ml to 1 μg/ml.
RNA isolation and quantitative polymerase chain reaction
Tissues were homogenized in TRIzol reagent (Thermo Fisher) to extract total RNA. PrimeScript RT Master Mix (Takara Bio, Inc.) was used to synthesize complementary DNA. Quantitative polymerase chain reaction (PCR) was performed and analyzed on a CFX96 (Bio‐Rad Laboratories).
Body weight, body composition, and comprehensive lab animal monitoring system analysis
Mouse body weight was measured daily during the first 2 weeks and every other week thereafter. During 15 weeks of WD regimen, their lean and fat masses were measured using magnetic resonance imaging (EchoMRI) 2 days before euthanasia for tissue collection. A comprehensive lab animal monitoring system (CLAMS; Columbus Instruments) was used to measure VO2, VCO2, food intake, and ambulatory activity at the Comparative Medicine Unit at Northeast Ohio Medical University 2 weeks before tissue collection. All parameters were normalized to lean mass.
Hepatic and fecal lipid extraction
Hepatic lipids were extracted using chloroform, methanol, and a phosphate‐buffered saline mixture (2:1:0.75). Fecal lipids were extracted using H2O, chloroform and a methanol mixture (1:3.3:4).[ 8 ] Extracted lipids in the chloroform layer were dried overnight and dissolved in a solvent consisting of 95% isopropanol and 5% Triton X‐100.
Triglyceride, cholesterol, alanine aminotransferase, and aspartate aminotransferase measurement
Details are provided in the Supporting Methods.
Hepatic, intestinal, and fecal BA extraction and quantification
Approximately 200 mg of liver, intestinal content, or overnight dried feces were homogenized in 1 ml of 75% ethanol. The BA levels in the supernatant of the homogenized liver, feces, and intestinal contents were measured using a colorimetric assay (Cell Biolabs Inc.).
Glucose and insulin tolerance tests
Details are provided in the Supporting Methods.
Fibroblast growth factor 15, insulin, glucagon‐like peptide 1, vitamin A, and vitamin D enzyme‐linked immunosorbent assay
Details are provided in the Supporting Methods.
Islet isolation
Pancreas was perfused with collagenase via the common bile duct as described.[ 11 ] Islets were hand‐picked under a microscope. Each data point contains islets collected from one or two mice.
N‐acyl taurine quantification
Plasma was prepared from 10‐week‐old mice and processed for N‐acyl taurine measurement by LC/MS‐MS.[ 12 ]
Cecum DNA content and microbiome analysis
Bacterial DNA in cecum was extracted using an E.Z.N.A. stool DNA kit (Omega Bio‐tek). 16S ribosomal RNA gene sequencing of the cecal content was performed by EzBiome Inc.
Statistical analysis
The data are presented as means ± SEM. Statistical significance was determined by Student's t test or two‐way analysis of variance (ANOVA). Analyses were performed using Microsoft Excel or GraphPad Prism software, respectively.
RESULTS
BA conjugation deficiency is evident in Baat −/− mice
Baat‐deficient mice were created by CRISPR‐mediated technology to delete a 324 bp of Baat genomic DNA, including exon 3 (Figure S2A, left). The heterozygous mice were in a C57BL/6NCrl background and were crossed to generate mice with null alleles. We confirmed homozygotic deletion of the allele among newborn pups using PCR analysis with three recommended primers (Figure S2A, right). We extracted RNA from several tissues of Baat −/− mice and WT littermates to assess Baat expression. Figure S2B showed that liver is the major tissue Baat expresses and the expression of Baat messenger RNA (mRNA) is almost completely absent in the null mice. Western blot analysis shown in Figure S2C was in strong agreement with the observed mRNA expression.
Analysis of hepatic BAs in Baat −/− mice showed almost complete absence of taurine‐conjugated BAs in the liver (1.5 ± 0.48% vs. 86.7 ± 4.57% in WT controls) without affecting glyco‐BA levels (data not shown), suggesting that BAAT is the major taurine‐conjugating enzyme in mice (Figure 1A,C). BA levels were increased significantly with notable increases of overall CA and α‐muricholic acid (α‐MCA) species in mutant mouse liver compared with WT mouse. We also analyzed BA composition in the gallbladder. As shown in Figure 1B,C and Figure S2D, most (96.7%) BAs were tauro‐conjugated in the WT gallbladder. In contrast, only 14.2% of BAs were tauro‐conjugated in the Baat −/− gallbladder. In contrast to the compositions in liver, levels of almost all unconjugated BAs in the Baat −/− mice were lower than those of corresponding conjugated BA species in WT mice. Therefore, overall BA concentration in the Baat −/− gallbladder was significantly lower than in WT (Figure S2E). In contrast, Baat −/− mice had significantly higher plasma and urine BA concentrations than WT mice, which is consistent with the hypercholanemia observed in humans with BAAT mutations (Figure 1C, Figure S2D,E). Unconjugated BA levels in Baat −/− ileum were significantly decreased compared with those in other tissues, probably due to the suggested passive absorption of unconjugated BAs in duodenum area (Figure 1C).
FIGURE 1.
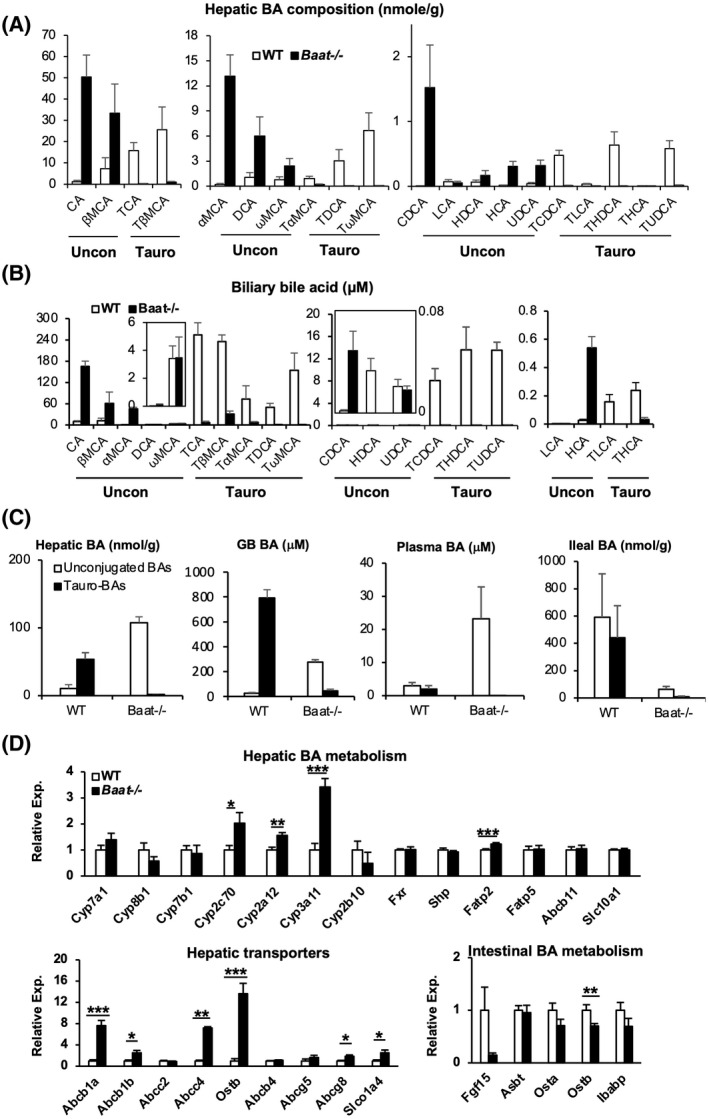
Bile acid (BA) conjugation deficiency and altered BA metabolism in bile acid‐CoA: amino acid N‐acyltransferase (Baat −/− ) mice. Ten‐week‐old mice fed chow were sacrificed after overnight fasting for the following BA and gene‐expression analysis. (A) Hepatic BA composition was determined using liquid chromatography–mass spectrometry (LC/MS). Among identified 25 BAs, 20 unconjugated BAs and their tauro‐conjugated counterparts were presented with average ± SEM (n = 5). Glycoconjugated BAs[ 5 ] were omitted because of their extremely low levels. Each panel contains unconjugated BAs (left) and their corresponding tauro‐conjugated BAs (right). (B) Biliary BA composition was determined using LC/MS as described in (A). (C) Unconjugated and tauro‐conjugated BA levels of liver, gallbladder (GB), plasma, and ileum (from left to right) calculated from the LC/MS analysis are plotted as absolute values. (D) Gene expression was quantified using quantitative polymerase chain reaction (PCR) analysis for hepatic BA homeostasis (top panel), hepatobiliary transporters (bottom left panel), and intestinal BA metabolism (bottom right panel). *p < 0.05, **p < 0.01, ***p < 0.005. CA, cholic acid; Cyp7a1, cholesterol 7α‐hydroxylase; Cyp8b1, sterol 12α‐hydroxylase; DCA, deoxycholic acid; Fatp2, fatty acid transport protein 2; Fgf15, fibroblast growth factor 15; FXR, farnesoid X receptor; HCA, hyocholic acid; HDCA, hyodeoxycholic acid; Ibabp, ileal bile acid‐binding protein; LCA, lithocholic acid; MCA, muricholic acid; Ostb, organic solute transporter β; Shp, small heterodimer partner; TCA, trichloroacetic acid; TCDCA, taurochenodeoxycholic acid; TDCA, taurodeoxycholic acid; TDHCA, taurodehydrocholic acid; THCA, tetrahydrocannabinolic acid; TLCA, taurolithocholic acid; TUDCA, tauroursodeoxycholic acid; UDCA, ursodeoxycholic acid; WT, wild type.
Altered expression of BA metabolism genes in Baat −/− mice
To investigate the effect of the conjugation deficiency on BA metabolism, we assessed mRNA expression of the genes in BA metabolism in the livers of 10‐week‐old male Baat −/− mice by real‐time quantitative PCR. We observed no significant changes in the expression of Cyp7a1, Cyp8b1, Cyp7b1, farnesoid X receptor (Fxr), Abcb11 (Bsep), and Slc10a1 (Na+ −taurocholate co‐transporting polypeptide peptide, Ntcp). However, mRNA expression of the genes encoding enzymes involved in BA hydroxylation at the 6β (Cyp2c70),[ 9 ] 1β and 6α (Cyp3a11) positions,[ 13 ] and 7α‐rehydroxylation (Cyp2a12)[ 14 ] were significantly increased in Baat −/− mice compared with WT mice, but no difference for CYP2B10 involved in hydroxylation of drugs (Figure 1D). These might explain higher α‐MCA (6β‐BA) and hyocholic acid (6α‐BA) levels in the Baat‐deleted liver. The genes encoding phase II detoxification (sulfation and glucuronidation) enzymes showed diverse responses. The mRNA expression of the genes (uridine diphosphate glucuronosyltransferase 1 family, polypeptide A1 [Ugt1a1] and Ugt1a2) encoding uridine diphosphoglucuronate–glucuronosyltransferases for BA glucuronidation was not altered (Figure S3A). In male Baat −/− mice, the female‐dominant sulfotransferase 2a1 (Sult2a1) gene expression was unaltered, whereas the male‐dominant Sult2a8 gene expression was down‐regulated without any significant changes in the mRNA expression of 3′‐phosphoadenosine 5′‐phosphosulfate synthase 1&2 (Papss1&2), responsible for production of 3′‐phosphoadenosine 5′‐phosphosulfate, a universal sulfate donor. Consistent with these gene‐expression patterns, a significant amount of polyhydroxylated (tetra and penta) BAs were detected in Baat −/− serum (8.1% of serum BA, calculated from the CA level) and liver (10.2% of hepatic BA), while they were almost absent in their WT counterparts (Figure 2A–C). Meanwhile, although the sulfated CA level was slightly higher in Baat −/− mice (probably due to higher availability of CA), the level difference between the two genotypes was not statistically significant (Figure S3B). These results agreed with the earlier reports that the phase I detoxification by hydroxylation is the major elimination process for toxic BAs in rodent models of cholestasis.[ 15 , 16 ] In addition, increased hydroxylation of secondary BAs was also anticipated with higher Cyp2a12 gene expression in the mutant mice. The lack of recirculating conjugated BAs in Baat −/− mice induced expression of the Fatp2 gene (encoding a BA CoA ligase) expression but not the Fatp5 gene expression (Figure 1D). The mRNA expression levels of Abcb1a, Abcb1b, Abcc4 (Mrp4), organic solute transporter β (Ostb), and solute carrier organic anion transporter 1a4 (Slco1a4, also known as Oatp1a4) were increased in Baat −/− mice, which is a manifestation of hepatic BA overload or cholestasis.[ 17 ] In contrast, mRNA expression of the major transporter for biliary phospholipid excretion (Abcb4, also known as Mdr2) was not altered in the mutant mice. However, the genes for the apical cholesterol transporters ABCG5 (no significance) and ABCG8 were up‐regulated in the mutant mice. The mRNA expression levels of intestinal FXR target genes, fibroblast growth factor 15 (Fgf15), and Ostb were down‐regulated in Baat −/− mice, indicating that the effect of reduced intestinal BA level is dominant over the antagonizing effect of TβMCA for FXR transcriptional activity in Baat −/− intestine (TβMCA, 6.19 ± 2.63 nmol/g in Baat −/− vs. 211.47 ± 114.33 nmol/g in WT mice).[ 18 ]
FIGURE 2.
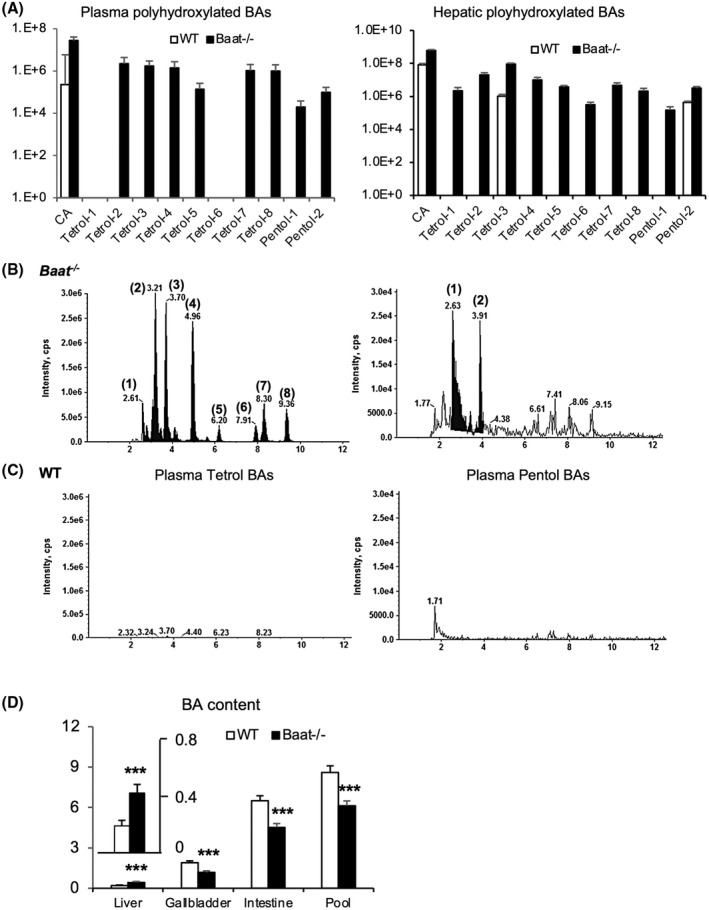
Hydroxylation of unconjugated BAs and BA pool size in Baat −/− mice. (A) Tetra‐hydroxylated and penta‐hydroxylated BAs from plasma (left panel) and liver (right panel) of 10‐week‐old mice were quantified using liquid chromatography tandem mass spectrometry (LC/MS‐MS) as described. The means of the area under each representative peak was plotted to logarithmic scale (log10) graph along with SEM (n = 6 per group). All of the detected hydroxylated BAs were unconjugated, and the differences between the two genotypes were all statistically significant. (B,C) Their representative chromatograms of tertrahyhdroxylated (left) and pentahydroxylated (right) BAs from plasma samples are presented (top panel, Baat −/− mice; bottom panel, WT mice). All of the values are significantly different between the two genotypes (p < 0.005). (D) BA pool size was determined by combining BAs from liver, gallbladder, and whole small intestine of 10‐week‐old animals (n = 8–9). Total BA contents were quantified by a commercial kit. ***p < 0.005.
To determine the effect of the dramatic changes in BA composition and BA‐related gene expression on total circulating BA pool size, we measured total BA content in liver, gallbladder, and whole intestine. Hepatic BA content was significantly increased, while biliary and intestinal BA contents were significantly decreased, in Baat −/− mice compared with those levels in WT controls. Thus, the BA pool size was significantly decreased in Baat −/− mice compared with WT littermates (Figure 2D).
Baat −/− mice are protected from diet‐induced obesity
Although 10‐week‐old Baat −/− mice had similar body weight to their WT counterparts, at weaning they appeared to be smaller than WT littermates. To assess body weight change during early age, newborn pups from heterozygous breeding cages were weighed every day during lactation period and then every week from weaning to 10 weeks of age (Figure 3A). The Baat −/− mice had significantly lower body weights during the first 7 weeks. Thereafter, they gained extra weight to reach that of their WT counterparts at 10 weeks. This observation corroborated the growth failure observed in human BA conjugation deficiency at a young age.
FIGURE 3.
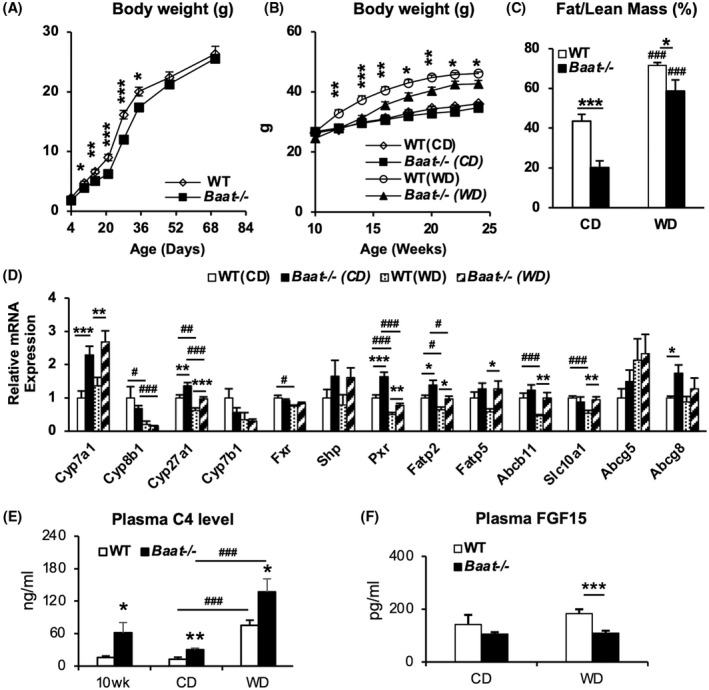
Compromised body weight gain in Baat −/− mice on high‐fat feedings. Table 1 is also associated with the Figure 3. (A) Newborn pups from heterozygous mating were monitored for their body weight gain from 4 days to 10 weeks. Average body weights were presented with SEM (n = 9–10). (B) Ten‐week‐old WT and Baat −/− mice were fed a Western diet (WD) containing high cholesterol for 15 weeks. Their biweekly body weights were plotted with means ± SEM (n = 7–11). (C) Body fat and lean mass were measured at 2 days before euthanasia for tissue collection without fasting. The ratio of fat to lean mass in each group was plotted with means ± SEM (6 WT chow [WTC], 5 knockout chow diet [KOC], 11 WT WD [WTWD], and 11 knockout WD [KOWD]). (D) Total RNA was extracted from liver and the indicated messenger RNA (mRNA) levels were quantified by real‐time quantitative PCR analysis after normalization to glyceraldehyde 3‐phosphate dehydrogenase (Gapdh) level. Average level relative to WTC was plotted with ±SEM (n = 7–9). (E) Plasma 7α‐hydroxy‐4‐cholesten‐3‐one (C4) levels of experimental animals (n = 6 per group; 10‐week‐old fed chow groups and 25‐week‐old WD regimen groups) were quantified using ultrahigh‐performance liquid chromatography–tandem mass spectrometry (UPLC‐MS/MS). Their means ± SEM were presented as a bar graph; *between genotypes; #between chow diet (CD) and WD. (F) Plasma FGF15 concentrations in the WT and Baat −/− mice on the WD regimen were measured using a commercial enzyme‐linked immunosorbent assay kit (n = 6 WTC, 6 KOC, 12 WTWD, and 11 KOWD).
As BA conjugation deficiency was suggested to diminish dietary fat absorption, Baat −/− mice were challenged with a WD containing high cholesterol at 10 weeks old for 15 weeks to investigate how BA conjugation deficiency affects fat and cholesterol/BA metabolism in detail at older age. As observed during lactation period, the body weights of Baat −/− mice were noticeably lower when challenged with WD, but showed no difference from WT controls under chow‐fed conditions (Figure 3B). EchoMRI analysis confirmed that the body fat content was significantly lower in Baat −/− mice than in WT mice, regardless of the diet condition (Table 1 and Figure 3C). Other important physiological and biochemical values from this diet regimen are presented in Table 1. Plasma aminotransferase values, which were higher in chow‐fed condition, were significantly lower in Baat −/− mice than WT counterparts after WD feeding. We also analyzed the hepatic expression of major genes involved in BA synthesis and metabolism, such as Cyp7a1, Cyp8b1, Cyp27a1, Fxr, pregnane X receptor (Pxr), Fatp2&5, Abcb11, Slc10a1, and Abcg5&8 (Figure 3D and Figure S4). In contrast to chow‐fed condition at 10 weeks, the 6‐month‐old Baat −/− mice showed higher expression of these genes (except Cyp8b1 and Fxr) than their WT counterparts. Interestingly, mRNA levels of nuclear hormone receptor PXR (Figure 3D) could explain higher expression of Cyp3a11 and higher BA hydroxylation in Baat −/− mice.[ 19 , 20 ] The plasma levels of C4, a biomarker for BA synthesis rate, were significantly increased in the Baat −/− mice in all diet and age conditions compared with WT mice and were further increased in WD‐fed mice (Figure 3E). It is known that WD containing cholesterol induces Cyp7a1 and BA synthesis in rodents. FXR is known to induce FGF15 in the intestine. Figure 3F shows that Baat −/− mice had lower serum FGF15 in both chow‐fed and WD‐fed condition and may contribute to increased Cyp7a1 mRNA expression. Because BA pool size was reduced in Baat −/− mice, the increased serum C4 is probably an indication of accumulation of BA intermediates due to BA synthesis defect in their liver.
TABLE 1.
Physiological and biochemical parameters of the experimental mice
| WT/CD | KO/CD | WT/WD | KO/WD | |
|---|---|---|---|---|
| Body weight (g) | 33.35 ± 0.82 | 29.94 ± 0.56* | 44.06 ± 0.93### | 39.72 ± 1.38*,### |
| Body lean (g) | 24.54 ± 0.55 | 26.16 ± 0.78 | 26.73 ± 0.44 | 26.31 ± 0.55 |
| Body fat (g) | 10.13 ± 0.83 | 5.04 ± 0.73* | 19.14 ± 0.35### | 14.55 ± 1.09*,### |
| Epididymal fat (g) | 1.48 ± 0.13 | 0.66 ± 0.10* | 1.84 ± 0.13 | 1.75 ± 0.16### |
| Brown adipose tissue (g) | 0.10 ± 0.00 | 0.07 ± 0.00* | 0.21 ± 0.02### | 0.18 ± 0.01### |
| Blood glucose (mg/dl) | 94.66 ± 2.58 | 110.36 ± 5.60* | 124.33 ± 7.61### | 144.58 ± 5.64*,### |
| Plasma ALT (U/L) | 42.66 ± 6.82 | 73.97 ± 15.88 | 194.91 ± 30.83### | 88.15 ± 14.18* |
| Plasma AST(U/L) | 75.09 ± 5.07 | 120.08 ± 33.20 | 203.31 ± 43.86## | 112.79 ± 16.06* |
| Plasma GLP‐1 (pM) | 12.26 ± 3.86 | 9.00 ± 1.69 | 17.58 ± 1.33 | 9.55 ± 1.38* |
| Plasma vitamin A (ng/ml) | 139.21 ± 4.09 | 111.01 ± 5.92* | 130.15 ± 6.85 | 118.03 ± 3.30 |
| Plasma vitamin D (ng/ml) | 1.03 ± 0.12 | 0.59 ± 0.14* | 3.68 ± 0.44### | 2.79 ± 0.17### |
| Fecal cholesterol (mg/g) | 3.23 ± 0.42 | 5.93 ± 0.16* | 8.47 ± 0.79### | 11.42 ± 0.29*,### |
| Fecal total lipid (mg/g) | 138.73 ± 7.36 | 210.50 ± 24.36* | 72.67 ± 3.40### | 98.86 ± 9.24*,### |
Note: All measurements were made from fasted mice after 15 weeks of the diet regimen (n = 6–15). For fecal measurements, feces were collected for 48 h during CLAMS analysis. Data are shown as means ± SEM. *p < 0.05 compared with WT counterparts; ## p < 0.01, ### p < 0.005 compared with chow‐fed counterparts.
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; KO, knockout.
While on the WD regimen, the mice were housed in CLAMS to assess their energy expenditure and food intake. The Baat −/− mice had a normal O2 consumption rate and a higher respiratory exchange ratio than the WT controls, indicating that Baat −/− mice used more carbohydrates for fuel than the WT controls (Figure 4A,B). There was no difference in the energy expenditure between the two genotypes (data not shown). Although the Baat −/− mice gained less weight on a WD compared with the controls, we observed an increased food intake of the Baat −/− mice on both chow and WD without significant changes in ambulatory activity (Figure 4C,D). The CLAMS data showed that Baat −/− mice had a shorter post‐meal interval time and a higher meal number than the controls, which indicated that Baat −/− mice had less satiety. The satiation appeared to be normal because their meal size was smaller, even with a longer meal duration (Figure S5A–D).
FIGURE 4.
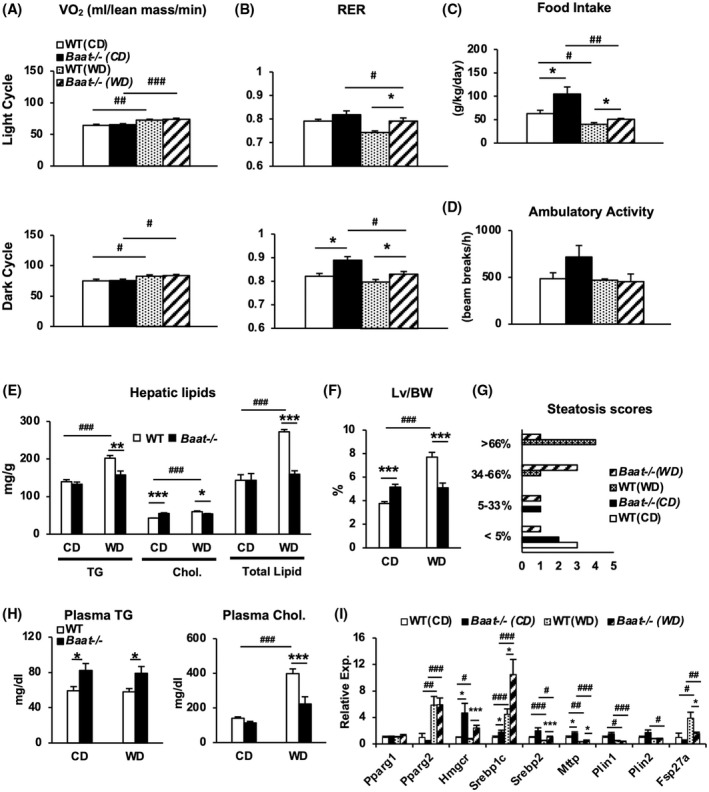
Comprehensive lab animal monitoring system (CLAMS) analysis and liver assessment on Baat −/− mice fed WD. (A–D) Oxygen consumption (VO2) (A), respiratory exchange ratio (RER) (B), food intake (C), and ambulatory activity (D) were measured using CLAMS. Average values of dark or night cycle from each group are presented with ±SEM for VO2 and RER. All measurements were from 23–24‐week‐old mice on CD (n = 7 and 9) or WD (n = 5 and 7). (E) Triglycerides (TG), cholesterol, and total lipid were measured from livers of mice fed CD or WD. Their average values are presented with ±SEM. (F) Liver (Lv)–to–body weight (BW) ratios from each group. (G) Steatosis scores were blindly evaluated by a pathologist (Dr. Novak) from hematoxylin and eosin–stained liver sections. The x‐axis represents the number of liver sections obtained from WT and Baat −/− mice, and y‐axis represents steatosis percentages (n = 5 per each group). (H) Plasma TG and plasma cholesterol levels were quantified in the experimental animals and are plotted as means ± SEM. (I) Expression of hepatic genes in lipid and cholesterol synthesis was quantified using quantitative PCR analysis. Relative expression to WTCD was is as means ± SEM (n = 13–15; Student's t test; *between genotypes; #between CD and WD). Fsp27a, fat‐specific protein 27a; Hmgcr, 3‐hydroxy‐3‐methyl‐glutaryl‐coenzyme A reductase; Mttp, mitochondria triglyceride transport protein gene; Pparg, peroxisome proliferator‐activated receptor gamma; Srebp, sterol regulatory element binding protein.
To verify reduced intestinal fat absorption in Baat −/− mice, we determined the total lipid, free fatty acid, and cholesterol levels in fecal samples from the animals. The Baat −/− mice had significantly increased total lipid and cholesterol levels in the feces. In addition, the plasma levels of lipid‐soluble vitamins (A and D) were significantly lower in Baat −/− mice than in WT mice, especially in the WD‐fed condition (Table 1). These data—along with ileal BA concentration—indicate that Baat −/− mice exhibit fat malabsorption due to reduced level of intestinal BA.
The fat malabsorption also protected the mutant mice from development of hepatic steatosis by WD challenge, as evidenced by lower hepatic triglycerides (TG), cholesterol, and total lipids compared with the WT counterparts (Figure 4E). The liver sections with hematoxylin and eosin staining further confirmed the protection of Baat −/− livers against WD‐induced fat accumulation (Figure S6A). The blinded histologic evaluation of the liver sections corroborated the protection against diet‐induced hepatic steatosis in Baat −/− mice, while demonstrating an opposite tendency in the chow‐fed condition, which was reflected not only by liver size but also by slightly higher steatosis score in the chow‐fed Baat −/− mice (Figure 4F,G and Figure S6B).
These changes in BA and lipid homeostasis decreased the plasma cholesterol level but unexpectedly increased the TG level on both diet conditions (Figure 4H). To understand the mechanism behind unexpected higher plasma TG level in the mutant mice, expression of hepatic genes involved in lipid metabolism was quantified by quantitative PCR analysis (Figure 4I). Baat −/− mice presented overexpression of important lipogenic genes (sterol regulatory element binding protein 1c [Srebp‐1c], Srebp‐2, and 3‐hydroxy‐3‐methyl‐glutaryl‐coenzyme A reductase [LXR]), which may have been caused by compensatory up‐regulation via liver X receptor activation due to the intestinal fat malabsorption. The mutant mice also exhibited higher expression of the mitochondria triglyceride transport protein gene (Mttp), encoding an essential protein for very‐low density lipoprotein assembly and secretion, and lower expression of fat‐specific protein 27a (Fsp27a, also known as Cidec) gene, encoding a lipid droplet binding protein for fat storage.[ 21 ] These data may explain that newly synthesized lipids in the liver are released into blood circulation rather than stored.
Glucose‐stimulated insulin secretion in Baat −/− mice is attenuated
Because we observed that the blood glucose levels in Baat −/− mice were significantly higher than in controls (Table 1), the glucose tolerance test (GTT) and insulin tolerance test were performed in the mice fed a WD for 9 to 11 weeks. The GTT showed that, compared with WT mice, the Baat −/− mice were glucose‐intolerant (Figure 5A). However, changes in the blood glucose levels were almost identical between the two genotypes after an insulin injection (Figure 5B). These results indicated that the glucose intolerance manifested in Baat −/− mice was likely caused by pancreatic islet dysfunction. Therefore, the plasma insulin levels were quantified after glucose challenge. The insulin levels of Baat −/− mice were substantially lower than those of WT mice at 15 min after glucose injection (Figure 5C). We next assessed the expression of a few major genes involved in insulin secretion in isolated islets. The genes encoding glucose transporter 2 (Glut2) and glucokinase (Gk) were down‐regulated in the pancreatic islets of Baat −/− mice, suggesting that impaired glucose use is one of the mechanisms under the disrupted insulin secretion in Baat −/− mice (Figure 5D).
FIGURE 5.
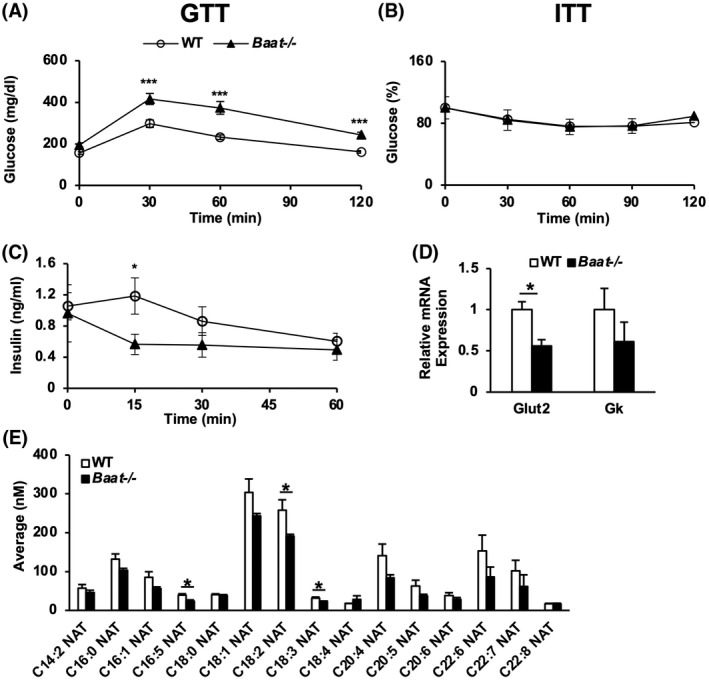
Disrupted glucose homeostasis of Baat −/− mice. (A) Glucose tolerance test (GTT) was performed on 19‐week‐old mice fed WD with intraperitoneal (i.p.) injection of 1 g/kg glucose after 6‐h fasting (n = 11–12). (B) Insulin tolerance test (ITT) was performed on 21‐week‐old male mice fed WD with i.p. injection of 1 U/kg Humulin after 6‐h fasting. Percentage values relative to zero time points re plotted with means ± SEM (n = 6–9). (C) Plasma insulin level was quantified with blood samples collected at 0 min, 15 min, 30 min, and 60 min during the GTT. Average raw values are plotted with SEM. (D) Expression of genes involved in the insulin secretion was quantified from the pancreatic islets of the animals on WD. (E) N‐acyl taurines were quantified in plasma of 10‐week‐old Baat −/− and WT mice using LC/MS‐MS (n = 6–7).
In an earlier report, taurine‐conjugated fatty acids, N‐acyl taurines (NATs), were shown to stimulate insulin secretion from pancreatic β cell lines by increasing calcium flux.[ 22 ] Our recent study also showed that a specific NAT (C18:1) enhanced glucose tolerance, increased glucagon‐like peptide 1 (GLP‐1) secretion, and reduced food intake in mice.[ 12 ] Because BAAT was suggested to be responsible for NAT production along with peroxisomal acyl‐CoA: amino acid N‐acyltransferase 1 (ACNAT1),[ 23 ] we assessed blood NAT level in Baat −/− mice. The N‐oleoyl taurine (C18:1) concentration was low (although not significantly), and other C18 NATs such as C18:2 and C18:3 were significantly decreased in the plasma of Baat −/− mice compared with their WT littermates. The mRNA levels of Acnat1 and Acnat2 were also lower in the Baat −/− mice (Figure 5E, Figure S3C). We also quantified plasma GLP‐1 levels: The GLP‐1 level in Baat −/− mice was reduced compared with the controls (Table 1), which may explain both the impaired insulin secretion and the hyperphagia in the mutant animals.
Effect of BA conjugation deficiency on gut microbiota
To examine whether BA conjugation deficiency alters gut microbiome, we collected ceca from 6‐month‐old Baat −/− mice and their WT littermates fed chow or 4 months of a WD. Bacterial DNA concentrations were measured to assess relative gut bacterial population changes in the Baat −/− mice with BA conjugation deficiency.[ 24 ] Due to a lower intestinal BA concentration, the Baat −/− mice showed a significantly higher bacterial DNA content than WT littermates when fed chow (Figure 6A). The bacterial population difference between the two genotypes was more significant when two‐way ANOVA was used. The operational taxonomic units obtained from the 16S rDNA sequencing were used to define the microbiota composition and to statistically analyze the microbiomes of the two groups. The Shannon diversity index is a statistical value that is used widely to characterize species diversity in a community. The Shannon index for microbial diversity was significantly higher in the Baat −/− mice than in the WT mice (Figure 6B). WD feeding significantly reduced the bacterial diversity in both genotypes, as has been previously reported.[ 25 , 26 ] Moreover, principal coordinate analysis showed significant distances between species‐level taxa in terms of the host's genotype in both diet regimens (Figure 6C,D and Figure S7A). The notable changes at the phylum level were significant increases in Actinobacteria and Firmicutes and significant decreases in Verrucomicrobia, Bacteroidetes, and Saccharibacteria_TM7 in Baat −/− mice that were fed chow. In the 4‐month WD feeding group, the phylogenetic changes were not as dramatic as in the chow feeding group. For the WD group, significant changes were found in three phyla: an increase in Firmicutes and decreases in Proteobacteria and TM7 (Figure S7B).
FIGURE 6.
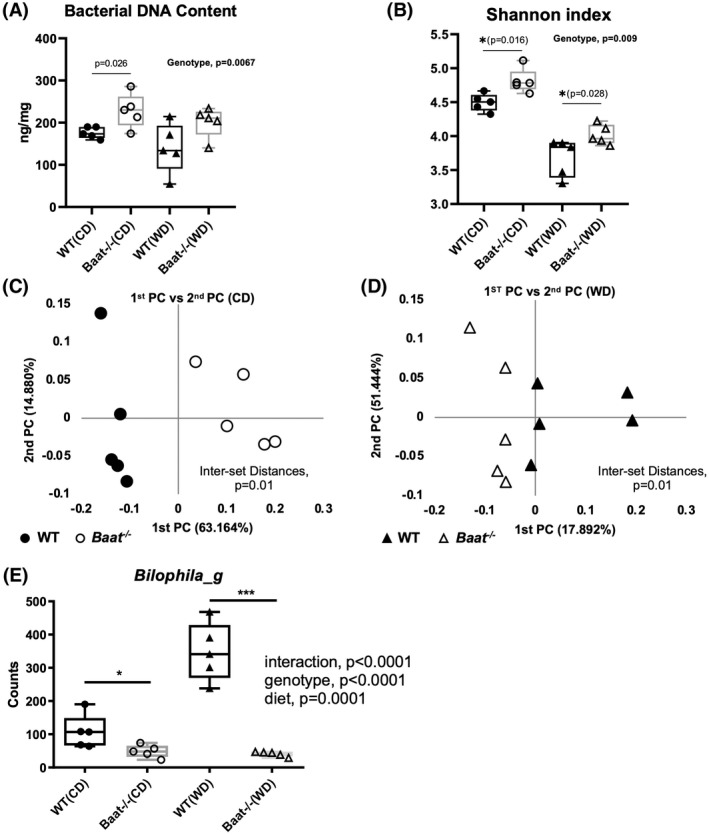
Altered gut microbiome in Baat −/− mice. (A) Total bacterial DNA content in the cecum was measured in Baat −/− mice and WT on both diets. The DNA content normalized by cecal weight is presented as a box‐and‐whisker graph using GraphPad Prism. (B) Alpha diversity of the gut microbiota from CD and WD groups are presented with Shannon indices. (C,D) Principle coordinate (PC) analysis of species‐level taxonomy was performed and plotted for CD (C) and for WD (D) groups using Unifrac. (E) Abundance of the genus Bilophila, a sulfidogenic bacterium, is presented as normalized by 16S RNA copy numbers. Two‐way analysis of variance was used to obtain the statistics.
We compared WT and Baat −/− taxonomic data obtained from both diet conditions and selected taxonomic biomarkers based on their genotypic differences between the conditions. As given in Table S1, two phyla (Firmicutes and Proteobacteria), three classes, three orders, seven families, 20 genera, and 64 species were identified as taxonomic biomarkers for Baat −/− mice. For the genus‐level biomarkers, 13 were enriched and seven were reduced in Baat −/− male mice compared with the WT counterparts (Figure S8). One of those identified, the genus Bilophila, a sulfidogenic pathobiont, has been known to expand on high‐fat diet feeding and uses the sulfite in taurine of the conjugated BAs, which are increased due to high fat consumption.[ 25 ] As shown in Figure 6E, the BA conjugation deficiency significantly reduced the Bilophila population in both diet conditions, probably due to a lack of taurine substrates in the gut. WD feeding did not increase the Bilophila population in Baat −/− mice, which was significantly increased in WT littermates.[ 25 ] Bile salt deconjugation is carried out by bile salt hydrolase (BSH) to produce unamidated BAs in mice. The BSH gene is widely found in normal microbiota, including the genera Clostridium, Enterococcus, Bifidobacterium, Lactobacillus, and Bacteroides.[ 27 ] However, none of these genera were significantly affected by a lack of conjugated BAs in Baat −/− mice (Table S1 and Figure S8).
Other notable changes in BA‐related biotransformation were the genera Alistipes and Muribaculum, which were significantly increased in mice fed CA and CDCA.[ 28 ] These genera were also significantly higher in Baat −/− mice than in WT controls, but only in the chow condition, which may have been due to a higher CA amount in the intestine of the mutant mice (Figure S9A,B). In the WD feeding, Baat −/− mice showed significant increases in the relative abundances of the genus Acetatifactor and the species Flintibacter butyricus; these bacteria were previously found to be increased after feeding a high‐fat diet that included CA and CDCA (Figure S9C,D).[ 28 ] Considering dietary fat malabsorption and the higher concentration of unconjugated CA in Baat −/− mice, this finding strongly supports an association between the dietary components and the abundance of Acetatifactor and Flintibacter butyricus.
We also assessed functional biomarkers using EzBioCloud, which provides up‐to‐date prediction of Kyoto Encyclopedia of Genes and Genomes (KEGG)–based orthology, modules, and pathways; EzBioCloud is based on the same algorithm as PICRUSt.[ 29 ] Notably, the prediction analysis selected BAAT KEGG orthology as a significant positive biomarker in Baat −/− mice (red highlight in Table S2). As novel microbial BA conjugation activities were detected in the gastrointestinal tracts of humans and mice in a recent study,[ 30 ] the predicted functional biomarker suggests that a lack of conjugated BAs might drive gut microbiota to an enrichment of the anabolic (amidation) pathway in the mutant mice.
DISCUSSION
Many features presented by Baat −/− mice, such as their apparent smaller body size at weaning, higher fecal lipid content, lower intestinal BA level, elevated plasma transaminase level, and lower plasma level of lipid‐soluble vitamins, were very similar to those reported in human BAAT mutations. However, the severity of phenotypes appeared to be weaker than that in humans, probably due to the presence of small amounts of conjugated BAs in the mutant mice. Additionally, BA polyhydroxylation was strongly induced to alleviate the toxicity of unconjugated BAs in the mutant animals. Enhanced BA hydroxylation was also observed in Bsep null mice, in which bile excretion of BAs was markedly decreased.[ 31 ] Increased BA polyhydroxylation was also seen in bile duct–ligated Fxr −/− mice, in which BSEP cannot be up‐regulated in the cholestatic condition.[ 16 ] An enzyme responsible for BA hydroxylation for detoxification is CYP3A11. An in vitro assay showed that unconjugated BAs activated the Cyp3a promoter more strongly than conjugated BAs.[ 32 ] As an extra note, Baat −/− mice had higher cholesterol saturation index in bile with lower BA concentration and higher cholesterol output under normal bile volume (data not shown; Figure S6H).
Interestingly, the detected percent values of conjugated and unconjugated BAs in the Baat −/− gallbladder (Figure S2D) were similar to those reported from the Fatp5 −/− mouse study (95% conjugated BAs in WT mice vs. 17% conjugated in Fatp5 −/− mice), in which a strong BA conjugation deficiency was evident by deletion of the BA‐CoA ligase (encoded by Fatp5) specific for reconjugation of deconjugated BAs from the enterohepatic recirculation.[ 8 ] This suggests that murine BAAT may be specific for the conjugation of recirculating BAs. Determination of subcellular localization of mouse BAAT would help us envision the speculation more correctly.
There were several interesting metabolic phenotypes of Baat −/− mice in WD feeding. The significantly higher food intake for Baat −/− mice was in contrast to a lower food intake and lower body weight gain in BA conjugation–deficient Fatp5 −/− mice.[ 8 ] Another interesting metabolic feature in Baat −/− mice was glucose intolerance. This phenotype was caused primarily by impaired glucose‐stimulated insulin secretion from pancreatic β cells, probably due to defective glucose uptake and use in β cells. NATs have been shown to play a role in glucose metabolism by stimulating insulin and GLP‐1 secretion and improving insulin sensitivity.[ 12 , 22 , 33 ] As N‐oleoyl taurine (C18:1 NAT) was also shown to reduce food intake in mice, lower levels of unsaturated NATs (including C18:1) may explain the glucose intolerance and hyperphagia in Baat −/− mice. Another possible scenario for impaired GLP‐1 secretion in the mutant mice is that intestinal Takeda G protein‐coupled receptor 5 (TGR5), an upstream regulator for GLP‐1 secretion, is not activated due to lower intestinal tauro–lithocholic acid level, a strong TGR5 agonist.[ 34 ]
The association of BA with gut microbiota has focused primarily on microbial effects on BA deconjugation and secondary BA formation, even though the traditional antimicrobial action of BAs is well recognized.[ 27 ] Importantly, recent studies have shown that changes in the BA composition by gut bacteria signal through the intestinal FXR to contribute to impaired host metabolism.[ 18 ] BA composition changes induced gut dysbiosis, which was one of the mechanisms protecting Fxr −/− mice from diet‐induced obesity.[ 35 ] These studies strongly suggest that BA composition plays a critical role in shaping the microbiome structure and maintaining host metabolic homeostasis. BA conjugation deficiency, along with lower gut BA concentrations (Figure S2F,G), clearly modified both cecal microbial structure and abundance. Genotype separation was more prominent in the chow‐fed condition than in the WD‐fed condition, and the effect of diet was less prominent in the Baat −/− group than in the WT group (Figure S7). Despite the reduced body weight gain in the WD challenge, Baat −/− mice showed a higher abundance of Firmicutes without any significant changes in Bacteroidetes, which is contrary to the established dogma related to the role of the two phyla in the development of obesity. We did not assess the effects of dysbiosis on the presented phenotypic manifestations. Nonetheless, a significant decrease of the genus Bilophila indicated a lack of taurine in the gut of Baat −/− mice. The identification of a predicted KEGG ortholog BAAT as a biomarker in the Baat −/− gut microbiome provides insight into the milder fat absorption defect. The overall effects of Baat deletion on BA/glucose metabolism and gut microbiota are summarized in Figure 7.
FIGURE 7.
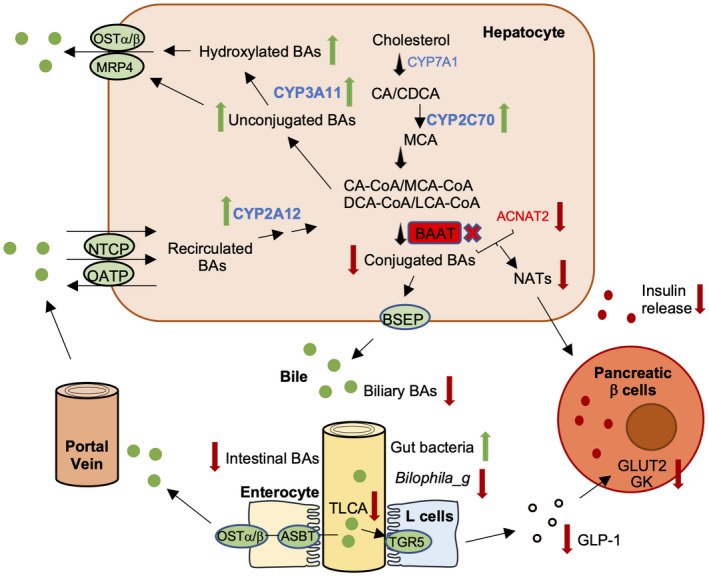
BA and glucose metabolism and gut dysbiosis in Baat −/− mice. Baat gene deletion reduces conjugated bile acids and increases unconjugated BAs dramatically in hepatocytes. Although BA excretion to the gallbladder is reduced due to higher affinity for conjugated BAs of bile salt export pump (BSEP), genes encoding basolateral BA export pumps (multidrug resistance‐associated protein 4 [MRP4] and OSTα/β) and hydroxylating enzymes such as CYP3A11, CYP2C70, and Cyp2A12 are increased in hepatocytes, which helps reduce accumulated unconjugated bile acid toxicity. Due to lower intestinal BA concentration, gut microbiota population is higher, but genus Bilophila, a sulfidogenic bacteria, is significantly less populated. Almost complete absence of TLCA fails to activate the Takeda G protein‐coupled receptor 5 (TGR5) signaling pathway and results in lower glucagon‐like peptide 1 (GLP‐1) level in serum. In addition, lower hepatic N‐acyl taurine levels due to absence of BAAT and lower synthesis of acyl‐CoA: amino acid N‐acyltransferases (ACNATs) lead to impaired glucose‐stimulated insulin secretion along with lower blood GLP‐1. ASBT, apical sodium–bile acid transporter; CDCA, chenodeoxycholic acid; GK, glucokinase; GLUT2, glucose transporter 2; NATs, N‐acyl taurines; NTCP, Na + −taurocholate co‐transporter peptide; OATP, organic anion transporting polypeptide.
AUTHOR CONTRIBUTIONS
Research: Bandar D. Alrehaili, Trisha J. Grevengoed, Yazen Alnouti, Xinwen Wang, Andrew D. Patterson, John Y. L. Chiang, Frank J. Gonzalez, and Yoon‐Kwang Lee designed research, Bandar D. Alrehaili, Mikang Lee, Shogo Takahashi, Bipin Rimal, Shannon Boehme, Samuel A. J. Trammell, Devendra Kumar, and Katya Chiti performed research. Data analysis: Bandar D. Alrehaili, Robert Novak, Trisha J. Grevengoed, and Yoon‐Kwang Lee. Manuscript editing: John Y. L. Chiang. Manuscript draft: Bandar D. Alrehaili and Yoon‐Kwang Lee.
FUNDING INFORMATION
Supported by the National Institutes of Health (R01DK093774, R01AA013623, R01DK044442, DK58379, and U01DK119702); a bridge fund from NEOMED; the Novo Nordisk Foundation (NNF18CC003490); and the Intramural Research Program, National Institutes of Health, National Cancer Institute, Center for Cancer Research (ZIABC005562).
CONFLICT OF INTEREST
Nothing to report.
Supporting information
Figure S1.
Figure S2.
Figure S3.
Figure S4.
Figure S5.
Figure S6.
Figure S7.
Figure S8.
Figure S9.
Appendix S1.
Appendix S2.
Table S1.
Table S2.
ACKNOWLEDGMENT
The authors thank Drs. Kwang‐Hoon Song and Jessica Ferrell for the valuable advice and technical support.
Alrehaili BD, Lee M, Takahashi S, Novak R, Rimal B, Boehme S, Bile acid conjugation deficiency causes hypercholanemia, hyperphagia, islet dysfunction, and gut dysbiosis in mice. Hepatol Commun. 2022;6:2765–2780. 10.1002/hep4.2041
REFERENCES
- 1. Chiang JY. Bile acids: regulation of synthesis. J Lipid Res. 2009;50:1955–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Axelson M, Aly A, Sjovall J. Levels of 7 alpha‐hydroxy‐4‐cholesten‐3‐one in plasma reflect rates of bile acid synthesis in man. FEBS Lett. 1988;239:324–8. [DOI] [PubMed] [Google Scholar]
- 3. Mihalik SJ, Steinberg SJ, Pei Z, Park J, Kim DG, Heinzer AK, et al. Participation of two members of the very long‐chain acyl‐CoA synthetase family in bile acid synthesis and recycling. J Biol Chem. 2002;277:24771–9. [DOI] [PubMed] [Google Scholar]
- 4. Gerloff T, Stieger B, Hagenbuch B, Madon J, Landmann L, Roth J, et al. The sister of P‐glycoprotein represents the canalicular bile salt export pump of mammalian liver. J Biol Chem. 1998;273:10046–50. [DOI] [PubMed] [Google Scholar]
- 5. Carlton VE, Harris BZ, Puffenberger EG, Batta AK, Knisely AS, Robinson DL, et al. Complex inheritance of familial hypercholanemia with associated mutations in TJP2 and BAAT. Nat Genet. 2003;34:91–6. [DOI] [PubMed] [Google Scholar]
- 6. Setchell KD, Heubi JE, Shah S, Lavine JE, Suskind D, Al‐Edreesi M, et al. Genetic defects in bile acid conjugation cause fat‐soluble vitamin deficiency. Gastroenterology. 2013;144:945–55 e946; quiz e914–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hofmann AF, Strandvik B. Defective bile acid amidation: predicted features of a new inborn error of metabolism. Lancet. 1988;2:311–3. [DOI] [PubMed] [Google Scholar]
- 8. Hubbard B, Doege H, Punreddy S, Wu H, Huang X, Kaushik VK, et al. Mice deleted for fatty acid transport protein 5 have defective bile acid conjugation and are protected from obesity. Gastroenterology. 2006;130:1259–69. [DOI] [PubMed] [Google Scholar]
- 9. Takahashi S, Fukami T, Masuo Y, Brocker CN, Xie C, Krausz KW, et al. Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J Lipid Res. 2016;57:2130–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang J, Bathena SP, Csanaky IL, Alnouti Y. Simultaneous characterization of bile acids and their sulfate metabolites in mouse liver, plasma, bile, and urine using LC‐MS/MS. J Pharm Biomed Anal. 2011;55:1111–9. [DOI] [PubMed] [Google Scholar]
- 11. Park YJ, Kim SC, Kim J, Anakk S, Lee JM, Tseng HT, et al. Dissociation of diabetes and obesity in mice lacking orphan nuclear receptor small heterodimer partner. J Lipid Res. 2011;52:2234–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grevengoed TJ, Trammell SAJ, McKinney MK, Petersen N, Cardone RL, Svenningsen JS, et al. N‐acyl taurines are endogenous lipid messengers that improve glucose homeostasis. Proc Natl Acad Sci U S A. 2019;116:24770–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bodin K, Lindbom U, Diczfalusy U. Novel pathways of bile acid metabolism involving CYP3A4. Biochim Biophys Acta. 2005;1687:84–93. [DOI] [PubMed] [Google Scholar]
- 14. Honda A, Miyazaki T, Iwamoto J, Hirayama T, Morishita Y, Monma T, et al. Regulation of bile acid metabolism in mouse models with hydrophobic bile acid composition. J Lipid Res. 2020;61:54–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wagner M, Halilbasic E, Marschall HU, Zollner G, Fickert P, Langner C, et al. CAR and PXR agonists stimulate hepatic bile acid and bilirubin detoxification and elimination pathways in mice. Hepatology. 2005;42:420–30. [DOI] [PubMed] [Google Scholar]
- 16. Marschall HU, Wagner M, Bodin K, Zollner G, Fickert P, Gumhold J, et al. Fxr(−/−) mice adapt to biliary obstruction by enhanced phase I detoxification and renal elimination of bile acids. J Lipid Res. 2006;47:582–92. [DOI] [PubMed] [Google Scholar]
- 17. Wagner M, Zollner G, Trauner M. New molecular insights into the mechanisms of cholestasis. J Hepatol. 2009;51:565–80. [DOI] [PubMed] [Google Scholar]
- 18. Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro‐beta‐muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013;17:225–35. [DOI] [PubMed] [Google Scholar]
- 19. Staudinger JL, Goodwin B, Jones SA, Hawkins‐Brown D, MacKenzie KI, LaTour A, et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci U S A. 2001;98:3369–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xie W, Radominska‐Pandya A, Shi Y, Simon CM, Nelson MC, Ong ES, et al. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc Natl Acad Sci U S A. 2001;98:3375–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim SC, Kim CK, Axe D, Cook A, Lee M, Li T, et al. All‐trans‐retinoic acid ameliorates hepatic steatosis in mice by a novel transcriptional cascade. Hepatology. 2014;59:1750–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Waluk DP, Vielfort K, Derakhshan S, Aro H, Hunt MC. N‐Acyl taurines trigger insulin secretion by increasing calcium flux in pancreatic beta‐cells. Biochem Biophys Res Commun. 2013;430:54–9. [DOI] [PubMed] [Google Scholar]
- 23. Hunt MC, Siponen MI, Alexson SE. The emerging role of acyl‐CoA thioesterases and acyltransferases in regulating peroxisomal lipid metabolism. Biochim Biophys Acta. 2012;1822:1397–410. [DOI] [PubMed] [Google Scholar]
- 24. Ang QY, Alexander M, Newman JC, Tian Y, Cai J, Upadhyay V, et al. Ketogenic diets alter the gut microbiome resulting in decreased intestinal Th17 cells. Cell. 2020;181:1263–75.e1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Devkota S, Wang Y, Musch MW, Leone V, Fehlner‐Peach H, Nadimpalli A, et al. Dietary‐fat‐induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature. 2012;487:104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roytio H, Mokkala K, Vahlberg T, Laitinen K. Dietary intake of fat and fibre according to reference values relates to higher gut microbiota richness in overweight pregnant women. Br J Nutr. 2017;118:343–52. [DOI] [PubMed] [Google Scholar]
- 27. Ridlon JM, Harris SC, Bhowmik S, Kang DJ, Hylemon PB. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes. 2016;7:22–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Just S, Mondot S, Ecker J, Wegner K, Rath E, Gau L, et al. The gut microbiota drives the impact of bile acids and fat source in diet on mouse metabolism. Microbiome. 2018;6:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Quinn RA, Melnik AV, Vrbanac A, Fu T, Patras KA, Christy MP, et al. Global chemical effects of the microbiome include new bile‐acid conjugations. Nature. 2020;579:123–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang R, Salem M, Yousef IM, Tuchweber B, Lam P, Childs SJ, et al. Targeted inactivation of sister of P‐glycoprotein gene (spgp) in mice results in nonprogressive but persistent intrahepatic cholestasis. Proc Natl Acad Sci U S A. 2001;98:2011–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schuetz EG, Strom S, Yasuda K, Lecureur V, Assem M, Brimer C, et al. Disrupted bile acid homeostasis reveals an unexpected interaction among nuclear hormone receptors, transporters, and cytochrome P450. J Biol Chem. 2001;276:39411–8. [DOI] [PubMed] [Google Scholar]
- 33. Aichler M, Borgmann D, Krumsiek J, Buck A, MacDonald PE, Fox JEM, et al. N‐acyl taurines and acylcarnitines cause an imbalance in insulin synthesis and secretion provoking beta cell dysfunction in type 2 diabetes. Cell Metab. 2017;25:1334–47.e1334. [DOI] [PubMed] [Google Scholar]
- 34. Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, et al. TGR5‐mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10:167–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Parseus A, Sommer N, Sommer F, Caesar R, Molinaro A, Stahlman M, et al. Microbiota‐induced obesity requires farnesoid X receptor. Gut. 2017;66:429–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Figure S2.
Figure S3.
Figure S4.
Figure S5.
Figure S6.
Figure S7.
Figure S8.
Figure S9.
Appendix S1.
Appendix S2.
Table S1.
Table S2.