

Abstract
Chronic cholestatic liver diseases are characterized by injury of the bile ducts and hepatocytes caused by accumulated bile acids (BAs) and inflammation. Wnt/β‐catenin signaling is implicated in organ fibrosis; however, its role in cholestatic liver fibrosis remains unclear. Therefore, we explored the effect of a selective cAMP response element‐binding protein‐binding protein (CBP)/β‐catenin inhibitor, PRI‐724, on murine cholestatic liver fibrosis. PRI‐724 suppressed liver fibrosis induced by multidrug resistance protein 2 knockout (KO), bile duct ligation, or a 3.5‐diethoxycarbonyl‐1.4‐dihydrocollidine (DDC) diet; it also suppressed BA synthesis and macrophage infiltration. The expression of early growth response‐1 (Egr‐1), which plays a key role in BA synthesis, was increased in the hepatocytes of patients with cholestatic liver disease. PRI‐724 inhibited Egr‐1 expression induced by cholestasis, and adenoviral shEgr‐1‐mediated Egr‐1 knockdown suppressed BA synthesis and fibrosis in DDC diet–fed mice, suggesting that PRI‐724 exerts its effects, at least in part, by suppressing Egr‐1 expression in hepatocytes. Hepatocyte‐specific CBP KO in mice suppressed BA synthesis, liver injury, and fibrosis, whereas hepatocyte‐specific KO of P300, a CBP homolog, exacerbated DDC‐induced fibrosis. Intrahepatic Egr‐1 expression was also decreased in hepatocyte‐specific CBP‐KO mice and increased in P300‐KO mice, indicating that Egr‐1 is located downstream of CBP/β‐catenin signaling. Conclusion: PRI‐724 inhibits cholestatic liver injury and fibrosis by inhibiting BA synthesis in hepatocytes. These results highlight the therapeutic effect of CBP/β‐catenin inhibition in cholestatic liver diseases.
CBP/β‐catenin signaling could be new therapeutic target for cholestatic liver fibrosis.

INTRODUCTION
Primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC) are chronic progressive cholestatic liver diseases caused by autoimmune disorders of unknown etiology,[ 1 ] and there is currently no effective treatment for these conditions aside from liver transplantation. Cholestatic liver injury results from liver parenchymal cell destruction because of the retention of hydrophobic bile acids (BAs). The initial hepatocyte damage triggers subsequent inflammatory responses, exacerbating hepatocyte and intralobular bile duct injury, resulting in fibrosis and, eventually, liver failure from cirrhosis.[ 1 ] Ursodeoxycholic acid and bezafibrate have been recognized as therapeutic agents for PBC[ 2 , 3 ]; however, antifibrotic drugs for liver cirrhosis associated with PBC and PSC are yet to be put into practical use.[ 2 , 4 ] BA receptors such as farnesoid X receptor (FXR) and its transporter system, and fibroblast growth factor 19, are attracting attention as therapeutic targets for cholestatic liver diseases and liver fibrosis.[ 5 , 6 ] However, there is currently no therapeutic drug that lowers BAs.
Several studies have reported that abnormal Wnt/β‐catenin signaling is involved in fibrosis.[ 7 , 8 ] Transforming growth factor‐β (TGF‐β)/mothers against decapentaplegic homolog 3 (Smad3) signaling, a key mediator of abnormal extracellular matrix production, cross‐talks with the Wnt/β‐catenin pathway.[ 9 ] Studies using hepatocyte‐specific β‐catenin gene KO in mice revealed dual roles of β‐catenin signaling: Deletion of β‐catenin increased liver injury and fibrosis induced by cholestasis in Mdr2‐KO mice, whereas it suppressed liver injury and fibrosis due to cholestasis in bile duct–ligated (BDL) mice.[ 10 , 11 ]
The transcription factor early growth response‐1 (Egr‐1) is a mediator Smad, independent of TGF‐β signaling.[ 12 ] TGF‐β enhances collagen type I alpha 2 (COL1A2) transcription induced by Egr‐1 in vitro and in vivo.[ 13 ] Egr‐1 also regulates Wnt/β‐catenin‐mediated signals.[ 14 ] Studies have revealed the involvement of Egr‐1 in cholestatic liver fibrosis.[ 15 ] BAs increase Egr‐1 expression, and inversely, Egr‐1 induces BA accumulation through the up‐regulation of Cyp7A1 and Cyp8B1.[ 15 ] However, the relationship between Egr‐1 and Wnt/β‐catenin signaling remains unclear.
β‐Catenin recruits cAMP response element‐binding protein‐binding protein (CBP) or P300, a homolog of CBP,[ 16 ] as a coactivator to induce target gene transcription, with CBP and P300 playing distinct roles. Recently, we showed that PRI‐724, a low‐molecular‐weight compound that selectively inhibits the interaction between CBP and β‐catenin, exerts antifibrotic effects in liver fibrosis models.[ 17 ] PRI‐724 is highly tolerated as it does not inhibit P300/β‐catenin signaling,[ 18 ] and phase 1/2a clinical trials have confirmed that PRI‐724 does not have harmful effects on humans.[ 19 ] In this study, the efficacy of PRI‐724 in suppressing cholestatic liver fibrosis was evaluated in mice. Mouse models mimicking human PSC were established using three different methods: Mdr2 KO, BDL, and a 3.5‐diethoxycarbonyl‐1.4‐dihydrocollidine (DDC) diet.[ 20 ] In addition, to investigate the distinct roles of CBP and P300 in the progression of cholestatic liver disease, hepatocyte‐specific CBP‐KO and P300‐KO mice were used.
Briefly, we investigated the effectiveness of PRI‐724 in three cholestasis fibrosis models and focused on hepatocyte CBP/β‐catenin‐Egr‐1 signaling with regard to its potential antifibrotic effects. Although many points regarding the therapeutic potential of PRI‐724 remain unclear, this study reports on the importance of hepatocyte CBP signal and bile acid metabolism mechanism.
MATERIALS AND METHODS
Animals and treatments
All mice were maintained in ventilated cages under 12‐h light/dark cycles with free access to enrichment, water, and feed.
Mdr2‐KO model
Mdr2‐KO (FVB.129P2‐Abcb4tm1Bor/J, #002539) mice were purchased from the Jackson Laboratory, and wild‐type littermates in the friend leukemia virus B (FVB)/NJ background were used as a control. Male Mdr2‐KO mice and littermate controls (8–10 weeks or 10–11 months of age) were intraperitoneally injected with 20 mg/kg PRI‐724 (Prism BioLab) dissolved in phosphate‐buffered saline (PBS) or with PBS as a control 3 times a week for 10 weeks.
BDL model
Male wild‐type (C57BL/6J) mice aged 8–10 weeks were obtained from Japan SLC and were subjected to BDL by dissecting the common bile duct above the pancreas as described previously.[ 17 ] The mice were intraperitoneally injected with 20 mg/kg PRI‐724 dissolved in PBS 3 times a week and sacrificed on day 14 after BDL.
DDC model
The following mouse strains were obtained from the Jackson Laboratory: albumin promoter–driven Cre recombinase (Alb/Cre) mice (B6.FVB[129]‐Tg[Alb1‐cre]1Dlr/J, #016832), Lyz2 promoter–driven Cre recombinase (LysM/Cre) mice (B6.129P2‐Lyz2 tm1[cre]Ifo /J, #004781), CBPflox mice (B6.Cg‐Crebbp tm1Jvd /J, #025178), and P300flox mice (B6.129P2‐Ep300 tm2Pkb /J, #025168). CBPflox mice were crossed with Alb/Cre and LysM/Cre mice to delete CBP in hepatocytes and macrophages. P300flox mice were crossed with Alb/Cre mice to delete P300 in hepatocytes. These male mice and C57BL/6J wild‐type male mice that were 8 weeks old were fed a 0.1% DDC‐enriched (Sigma‐Aldrich) diet for 18 days. Some wild‐type mice were injected with the adeno‐associated virus (AAV) construct on day 4 of DDC administration.
The animals were euthanized by exsanguination. The livers were immediately removed, frozen in liquid nitrogen, and stored until analysis. A portion of the dissected liver tissues was fixed with 10% formalin for histological analysis.
AAV transfection
AAVs (serotype 8) were custom‐packed at SignaGen Laboratories. Egr‐1 short hairpin RNA (shRNA; AAV8/shEgr‐1) (Cat # SL100810) or control shRNA (AAV8/control) (Cat # SL100862) was delivered by tail vein injection (2 × 1012 copies/mouse) (Figure S5).
Serum cytokines, chemokines, and parameters
Levels of serum cytokines and chemokines were measured using Bio‐Plex Cytokine Assay Kits (Bio‐Rad Laboratories) according to the manufacturer's instructions. Specifically, the Bio‐Plex mouse Cytokine 40‐Plex Panel, Chemokine Panel, and matrix metalloproteinase (MMP) panel were used. The samples were analyzed in a 96‐well plate reader using a Bio‐Plex Suspension Array System and Bio‐Plex Manager software (Bio‐Rad Laboratories). Serum alanine aminotransferase (ALT) and alkaline phosphatase (ALP) levels were analyzed using the Transaminase CII test and LabAssay ALP (Fuji Firm Wako). Serum bilirubin was measured using the QuantiChrom Bilirubin Assay Kit (BioAssay Systems).
BA analysis
Serum and liver total BA (TBA), cholic acid (CA), and chenodeoxycholic acid (CDCA) levels were analyzed using the Total Bile Acid Assay Kit, Cholic Acid ELISA Kit, and Chenodeoxycholic Acid ELISA Kit, respectively (Cell Biolabs), according to the manufacturer's instructions.
Hydroxyproline measurement
Liver tissues were homogenized and hydrolyzed in 6 N HCl at 110°C for 24 h. The samples were oxidized with chloramine‐T (Sigma‐Aldrich) and incubated in Ehrlich's perchloric acid solution as previously reported.[ 21 ] The absorbance of the samples at 558 nm was measured by a GloMax explorer multimode microplate reader (Promega). Purified hydroxyproline (Sigma‐Aldrich) was used as the standard. Hydroxyproline content was expressed as micrograms of hydroxyproline per gram of liver.
Real‐time quantitative polymerase chain reaction
RNeasy and DNase Kits (Qiagen) and the High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems) were used for RNA extraction from liver tissues and cultured cells, DNA removal, and reverse transcription. Quantitative polymerase chain reactions (PCRs) were run in triplicate using probe and primer sets (Table S1) purchased from Thermo Fisher Scientific and TaqPath qPCR Master Mix, CG (Applied Biosystems) in a LightCycler 480 (Roche Applied Science). Target gene‐expression levels were normalized to that of GAPDH (glyceraldehyde 3‐phosphate dehydrogenase) in each sample.
PCR array
The RT2 Profiler™ PCR Array (Qiagen Sciences) was used for messenger RNA (mRNA) expression analysis of WNT/β‐catenin‐related genes (mouse WNT signaling pathway [PAMM‐043Z] and mouse WNT signaling targets [PAMM‐243Z]) in the liver. The assay was performed using RT2 SYBR Green qPCR Master Mix and the RT2 First Strand Kit (Qiagen). The target genes on the PCR arrays can be obtained from the Qiagen website.
Human PBC samples
Normal liver tissues (non‐cancerous tissues from patients with metastatic liver tumors) (n = 5) and PBC liver tissues (n = 8) were obtained by needle biopsy. PBC diagnoses were made according to clinical guidelines and typical histological findings. Written informed consent to participate in this study was obtained from all patients.
Histological analysis
Mouse liver tissues were fixed in 10% formalin, sectioned, and stained with hematoxylin and eosin (HE). Collagen deposits were stained with sirius red (saturated picric acid containing 0.1% direct red 80 and 0.1% Fast Green FCF). In addition, the samples were immunohistochemically stained using antibodies against S100A4, GS, cytokeratin 19 (Abcam), Egr‐1 (Santa Cruz Biotechnology), F4/80 (Invitrogen), CBP (Cell Signaling Technology), P300 (GeneTex), and Ki‐67 (DAKO) using the VECTASTAIN Elite ABC Kit or the M.O.M Immunodetection Kit (Vector Lab). For human liver‐tissue staining, antibodies against CBP (Cell Signaling Technology), P300 (GeneTex), and Egr‐1 (Cell Signaling Technology) were used. Diaminobenzidine tetrahydrochloride was used as a peroxidase substrate, and sections were counterstained with hematoxylin.
To quantify sirius red–positive areas, standardized computer‐assisted image analysis was performed.[ 22 ] An independent pathologist blindly selected five sirius red–stained parenchyma spots in all biopsy samples and automatically measured the sirius red–positive areas using the HistoQuant software (3DHISTECH).
Statistical analysis
Data are expressed as the mean ± SD of data collected from at least three independent experiments. Means of two groups were compared using a two‐tailed Student's t test, and means of multiple groups were compared by one‐way analysis of variance followed by Bonferroni's post‐hoc tests using GraphPad Prism 8.0 (GraphPad Prism Inc.). Statistical significance was set at *p < 0.05, **p < 0.01, ***p < 0.005, and ****p < 0.0001.
RESULTS
PRI‐724 suppresses cholestatic liver fibrosis in Mdr2‐KO mice
To investigate whether PRI‐724 has a therapeutic effect on liver fibrosis due to cholestasis, Mdr2‐KO mice, which fail to secrete phospholipids from the liver into the bile, resulting in cholestasis and subsequent portal inflammation followed by liver fibrosis, were used.[ 23 ] The treatment protocol is shown in Figure 1A. Sirius red–positive areas, indicating collagen deposition, were observed in the livers of Mdr2‐KO mice in the control group at 18 weeks, whereas the fibrotic areas were reduced in the livers of mice with intraperitoneally administered PRI‐724 (20 mg/kg/mouse) (Figure 1B,C). In addition, collagen deposition, as measured by hydroxyproline quantification, decreased following PRI‐724 treatment in Mdr2‐KO mice (Figure 1D). Gene‐expression analysis of the livers revealed that the mRNA levels of Col1a1, Col1a2, and tissue inhibitor of metalloproteinase 1 (TIMP1; an endogenous MMP inhibitor) were significantly reduced in the PRI‐724‐administered group compared with those in the control group (Figure 1F), whereas MMP‐9 expression was increased. In addition, the serum levels of MMP‐8 and proMMP‐9 were increased by PRI‐724 (Figure 1E). These results suggest that PRI‐724 suppresses fibrosis induced by Mdr2‐KO. HE staining and the bile duct injury experiment showed that PRI‐724 suppressed the infiltration of inflammatory cells around the portal vein. After PRI‐724 administration, we collected leukocytes from the livers of Mdr2‐KO mice and confirmed a decrease in the numbers of inflammatory cells (e.g., CD8 T cells, macrophages, monocytes) (Figure S1).
FIGURE 1.
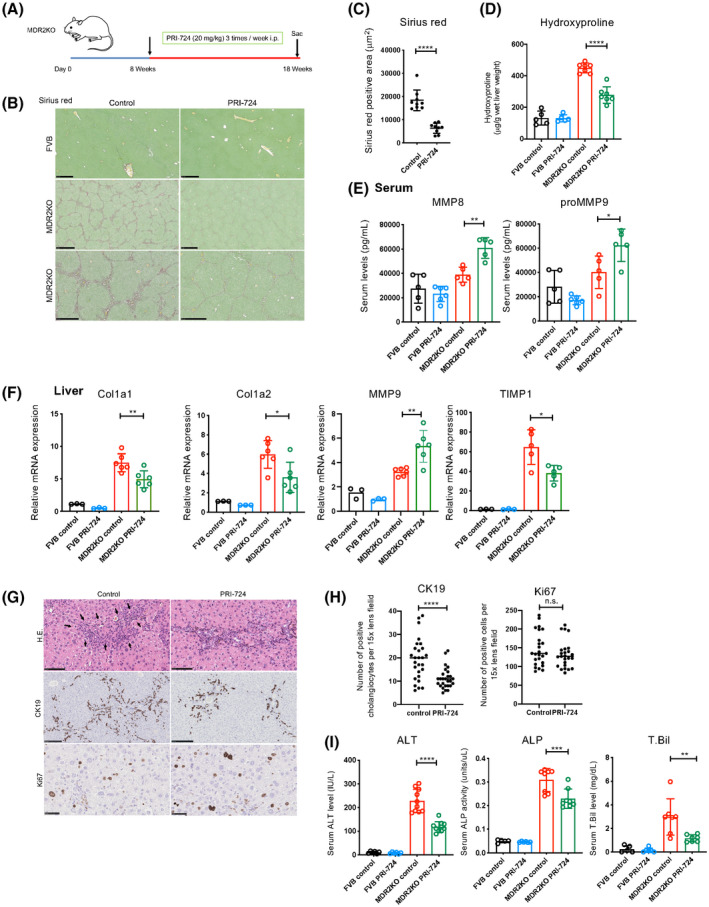
PRI‐724 suppresses liver injury and fibrosis in multidrug resistance protein 2 (Mdr2)–knockout (KO) mice. Male Mdr2‐KO mice (8–10 weeks old, n = 12) and friend leukemia virus B (FVB) background control mice (n = 6) were treated or not with PRI‐724 (20 mg/kg, 3 times a week) for 10 weeks. (A) Scheme of the treatment protocol. (B–D) Collagen deposition as assessed by sirius red staining (B; scale bars, 500 μm, 1 mm, and 500 μm, respectively, from the top figure) and graph (C) and by measuring hydroxyproline contents (D). (E) Serum matrix metalloproteinase (MMP) levels as analyzed using a Bio‐Plex assay. (F) Messenger RNA (mRNA) expression of the indicated genes in the livers as determined by real‐time quantitative polymerase chain reaction (PCR). (G,H) Hematoxylin and eosin (HE) staining and immunohistochemical staining using anti‐CK19 and Ki67 antibodies (scale bars, 100 μm, 250 μm, and 50 μm, respectively, from the top figure) (G) and quantification of the positive cells (H). (I) Serum alanine aminotransferase (ALT), alkaline phosphatase (ALP), and total bilirubin (T.Bil) levels (n = 5–7 per group). The results shown are representative of at least three independent experiments. Data represent the mean ± SD; *p < 0.05, **p < 0.01, ***p < 0.005, and ****p < 0.0001. Abbreviations: Col1A1, collagen type I alpha 1; i.p., intraperitoneally; n.s., not significant versus control‐treated Mdr2‐KO mice by one‐way analysis of variance; TIMP1, tissue inhibitor of metalloproteinase 1.
This is reflected by a decreased number of CK19‐positive proliferating bile ductules (Figures 1G,H) and a reduction in liver damage, as indicated by decreased serum alanine transaminase (ALT), alkaline phosphatase (ALP), and total bilirubin (T.Bil) levels (Figure 1I). The number of Ki67‐positive cells did not differ between the two groups (Figure 1G,H), indicating that PRI‐724 does not affect hepatocyte proliferation, although β‐catenin signaling has been reported to be implicated in liver regeneration.
PRI‐724 reduced BA synthesis and Egr‐1 expression in the liver of Mdr2‐KO mice
To assess the target cells of PRI‐724 in Mdr2‐KO mice, liver tissues were subjected to immunostaining. Staining of S100A4, which is downstream of CBP/β‐catenin signaling, was observed primarily in inflammatory cells in the control group, and S100A4‐positive cells were reduced in the PRI‐724‐administered group (Figure 2A,B). Similarly, the number of cells staining positively for F4/80, a macrophage marker, was reduced by PRI‐724 treatment (Figure 2A,B), suggesting that PRI‐724 affects inflammatory cells and especially reduces macrophage infiltration. Immunohistochemical analysis of glutamine synthetase (GS), a target of Wnt/β‐catenin signaling, revealed that the number of GS‐positive hepatocytes was significantly decreased in the PRI‐724‐administered group (Figure 2A,B), suggesting that PRI‐724 also affects hepatocytes. Wnt/β‐catenin‐related gene expression in the livers of Mdr2‐KO mice was analyzed using quantitative reverse‐transcription PCR arrays for Wnt/β‐catenin and their targets (Figure 2C). Seventeen genes showed increased expression in Mdr2‐KO mice, which was inhibited by PRI‐724. Among these, we focused on Egr‐1 because it is implicated in BA metabolism, TGF‐β signaling, and liver fibrosis due to cholestasis.[ 24 ] The decrease in Egr‐1 mRNA expression was confirmed by real‐time quantitative PCR in multiple specimens (Figure 2D). Egr‐1 protein expression was observed in the nuclei of hepatocytes, and the numbers of Egr‐1‐positive cells in PRI‐724‐administered Mdr2‐KO mice were decreased compared with those in the control group (Figure 2E,F). As Egr‐1 is implicated in BA metabolism, we also measured BA levels (TBA, CA, and CDCA). Hepatic TBA and CA levels were not affected by PRI‐724, but the CDCA level was decreased (Figure 2G). Serum TBA, CA, and CDCA levels were decreased by PRI‐724 (Figure 2G). In in vitro experiments using cultured hepatocytes, PRI‐724 treatment did not inhibit cell damage caused by BA administration (Figure S2). These results suggest that PRI‐724 inhibits liver injury by suppressing BA secretion from hepatocytes, Egr‐1 expression, and macrophage infiltration.
FIGURE 2.
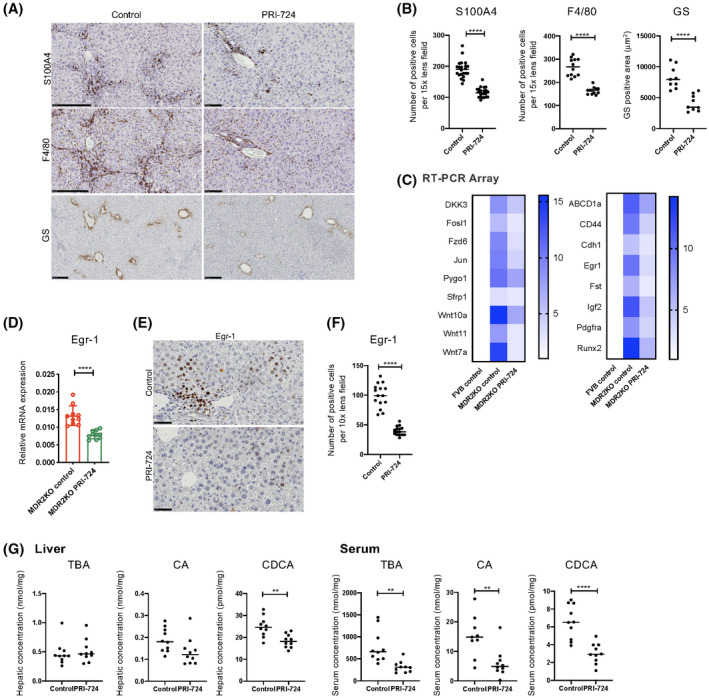
PRI‐724 reduces bile acid (BA) synthesis in Mdr2‐KO mice. Male Mdr2‐KO mice (8–10 weeks old, n = 12) and FVB background control mice (n = 6) were treated or not with PRI‐724 (20 mg/kg, 3 times a week) for 10 weeks. (A) Immunohistochemical staining using anti‐S100A4, F4/80, and Ki67 antibodies (scale bars, 250 μm). (B) Quantification of S100A4‐positive and F4/80‐positive cells and GS‐positive areas in the livers. (C) Clustergrams of PCR array analyses of WNT/β‐catenin‐related genes (left panel, WNT/β‐catenin signals; right panel, WNT/β‐catenin targets). (D) Early growth response‐1 (Egr‐1) mRNA expression in the livers as determined by real‐time quantitative PCR (n = 10 per group). (E) Immunohistochemical staining using anti‐Egr‐1 antibodies (scale bar, 100 μm). (F) Quantification of Egr‐1‐positive cells and glutamine synthetase (GS)–positive areas in the livers. (G) Hepatic and serum total BA (TBA), cholic acid (CA), and chenodeoxycholic acid (CDCA) levels (n = 10 per group). The results shown are representative of at least three independent experiments. Data represent the mean ± SD; *p < 0.05, **p < 0.01, ***p < 0.005, and ****p < 0.0001, by Student's t test.
Finally, the therapeutic effect of PRI‐724 in aged Mdr2‐KO mice with established fibrosis was examined to evaluate its clinical usefulness. Male Mdr2‐KO mice aged 10–11 months were divided into control (n = 3) and PRI‐724‐administered groups (n = 4). PBS and PRI‐724 (20 mg/kg/mouse) were intraperitoneally administered 3 times a week for 2 weeks (Figure S3A). After PRI‐724 administration in Mdr2‐KO mice, a decrease in the sirius red–positive area was observed in the liver (Figure S3B). Similarly, Col1a1, Col1a2, Col3a1, and smooth muscle actin mRNA levels in the liver were significantly reduced, indicating that PRI‐724 has a therapeutic effect on established liver fibrosis (Figure S3C).
PRI‐724 suppresses cholestatic liver fibrosis in the BDL model
Next, the antifibrotic effects of PRI‐724 were investigated in the BDL model (Figure 3A). As observed in Mdr2‐KO mice, PRI‐724 suppressed inflammatory cell infiltration, liver damage, liver fibrosis, and bile duct reaction induced by BDL (Figure 3B–F). Moreover, serum levels of interleukin (IL)‐6, CC chemokine ligand (CCL)3, CCL4, and chemokine (C‐X‐C motif) ligand (CXCL)2 were decreased by PRI‐724 (Figure S4A), which was accompanied by reduced numbers of S100A4‐positive cells and F4/80‐positive macrophages (Figure S4B,C), suggesting that PRI‐724 suppressed immune responses in the BDL mice. Real‐time quantitative PCR arrays were used to analyze gene expression in the BDL livers (Figure 3G). Like in the Mdr2‐KO mice, Egr‐1 expression was increased at the mRNA and protein levels by BDL, and the increases were suppressed by PRI‐724 (Figure 3H–J). In addition to its effect on the immune responses, PRI‐724 decreased hepatic TBA, CA, and CDCA levels, as observed in Mdr2‐KO mice (Figure 3K). Thus, similar to the findings in Mdr2‐KO mice, PRI‐724 inhibited liver injury by reducing BA production by hepatocytes and suppressing Egr‐1 expression and macrophage infiltration in BDL mice.
FIGURE 3.
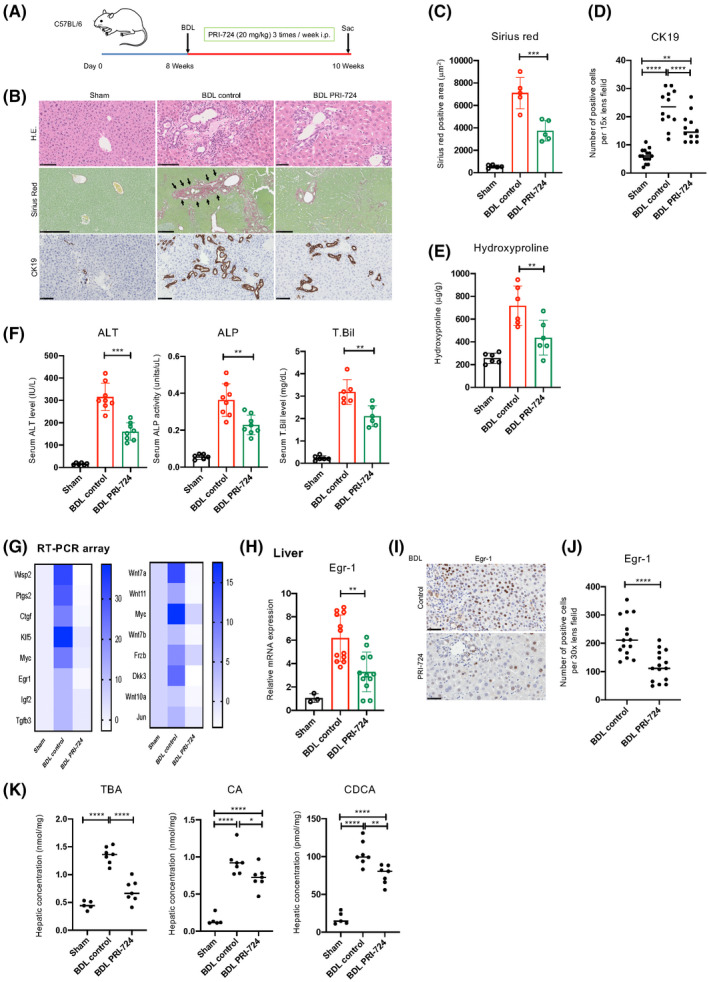
PRI‐724 suppresses liver injury and fibrosis accompanied by reduced hepatic and serum BA levels. Male wild‐type C57BL/6 male 8–10‐week‐old mice were subjected to bile duct ligation (BDL) or sham operation and treated with PRI‐724 (20 mg/kg, 3 times a week) or phosphate‐buffered saline (PBS). The animals were killed on day 14 after surgery. (A) Scheme of the treatment protocol. (B–D) HE, sirius red staining, and immunohistochemical staining for CK19 (B; scale bars, 100, 500, and 250 μm, respectively, from the top figure). Quantification of the sirius red–positive area (C) and CK19‐positive cells (D). (E) Collagen deposition as assessed by measuring hydroxyproline contents. (F) Serum ALT, ALP, and T.Bil levels. (G) Clustergrams of PCR array analyses of WNT/β‐catenin‐related genes (left panel, WNT/β‐catenin signals; right panel, WNT/β‐catenin targets). (H) Egr‐1 mRNA expression in the livers as determined by real‐time quantitative PCR (n = 3–12 per group). (I) Immunohistochemical staining using anti‐Egr‐1 antibodies (scale bar, 50 μm). (J) Quantification of Egr‐1‐positive cells in the livers. (K) Hepatic TBA, CA, and CDCA levels (n = 6–9 per group). The results shown are representative of at least three independent experiments. Data represent mean ± SD; *p < 0.05, **p < 0.01, ***p < 0.005, and ****p < 0.0001 by one‐way ANOVA (C–F,H,K) or unpaired Student's t test (J).
PRI‐724 suppresses liver fibrosis in the DDC diet model
Finally, we investigated the antifibrotic effect of PRI‐724 in cholestatic mice consuming a DDC diet. Eight‐week‐old male C57BL/6 mice were given a 0.1% DDC diet for 18 days. Simultaneously, PRI‐724 (n = 12) and PBS (n = 12) were intraperitoneally administered 3 times a week (Figure 4A). As shown in Figure 4B, the sirius red–positive region increased in the control group, and fibrosis was observed in the liver after DDC diet administration. In contrast, the fibrotic region decreased in the PRI‐724‐administered group (Figure 4B). The same findings were also reflected in the quantitative results of hydroxyproline in the liver (Figure 4C). We also found a significant reduction in Col1a1, Col1a2, and Col3a1 mRNA levels in the liver following PRI‐724 administration, indicating that PRI‐724 has a therapeutic effect on DDC diet–induced liver fibrosis. In addition, a significant increase in MMP‐8 and MMP‐9 mRNA levels and a significant decrease in TIMP‐1 mRNA were observed in the liver, indicating that PRI‐724 played a role in inducing the expression of these MMPs and in fibrosis suppression (Figure 4D). Similar to that seen in the two cholestatic fibrosis models, hepatocellular injury (ALT) and cholestasis (ALP and T.Bil) were alleviated by PRI‐724 administration (Figure 4E).
FIGURE 4.
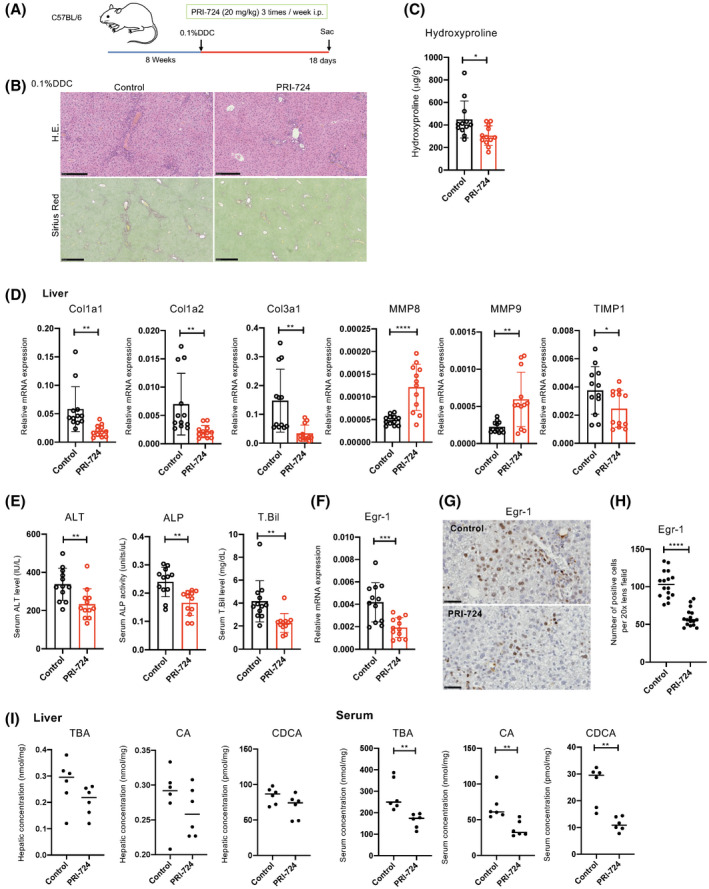
PRI‐724 suppresses liver injury and 3.5‐diethoxycarbonyl‐1.4‐dihydrocollidine (DDC) diet–induced fibrosis. Eight‐week‐old wild‐type C57BL/6 male mice were fed a 0.1% DDC diet for 18 days and administered PRI‐724 (20 mg/kg, 3 times a week) or PBS. (A) HE and sirius red staining (scale bars, 250 and 500 μm, respectively, from the top figure). (B) Collagen deposition as assessed by hydroxyproline measurement (n = 12 per group). (C,E) Messenger RNA (mRNA) expression of the indicated genes in the livers as determined by quantitative reverse‐transcription PCR (n = 12 per group). (D,F) Immunohistochemical staining for Egr‐1 (scale bars, 50 μm). (G) Quantification of Egr‐1‐positive cells in the livers. (H) Hepatic and serum TBA, CA, and CDCA levels (n = 6 per group). The results shown are representative of at least three independent experiments. Data represent the mean ± SD; *p < 0.05, **p < 0.01, and ***p < 0.005 by unpaired Student's t test.
Next, to examine liver Egr‐1 expression, which was detected in Mdr2‐KO mice and the BDL model, Egr‐1 mRNA levels were analyzed using real‐time quantitative PCR in liver tissue and were found to be significantly decreased following PRI‐724 administration (Figure 4F). Egr‐1 immunostaining of liver tissue showed that the number of positive hepatocytes significantly decreased in the PRI‐724‐administered group (Figure 4G,H). Finally, we measured serum and liver tissue BA concentrations after PRI‐724 administration. As shown in Figure 4I, serum TBA, CA, and CDCA concentrations were significantly decreased in the PRI‐724‐administered group, but no change was observed in the liver tissue.
Cholestatic liver fibrosis outcomes differ between CBP and P300‐Alb/Cre KO mice on a DDC diet
As PRI‐724 affected BA metabolism and macrophage infiltration, we hypothesized that the CBP/β‐catenin inhibitor targets hepatocytes and macrophages. To confirm the effect of CBP on hepatocytes, Alb/Cre‐CBP‐KO and Alb/Cre‐P300‐KO mice, which lack CBP and its homolog P300, respectively, specifically in hepatocytes, were generated.[ 21 ] Because the generation of Mdr2‐KO/Alb/Cre/CBP‐flox mice is complicated and time‐consuming, a DDC‐induced cholestatic liver injury and fibrosis model was used instead (Figure 5A).[ 25 ] In Alb/Cre‐CBP‐KO mice, inflammatory cell infiltration, liver injury, ductal reaction, and fibrosis by DDC were reduced, whereas in Alb/Cre‐P300‐KO mice, these features were exacerbated compared with those in Alb/Cre control mice (Figure 5B–F). Of note, serum T.Bil levels in Alb/Cre‐CBP‐KO mice were higher than those in control mice, although the underlying mechanism is unknown. The number of Ki67‐positive cells was increased in Alb/Cre‐p300‐KO mice (Figure 5C), suggesting that hepatocyte regeneration may be activated by exacerbated liver injury. TIMP‐1 expression in the liver was decreased by PRI‐724, whereas it was increased in Alb/Cre‐CBP‐KO mice. As hepatocytes do not produce TIMP‐1, PRI‐724 may affect other TIMP‐1‐producing cells, such as fibroblasts.
FIGURE 5.
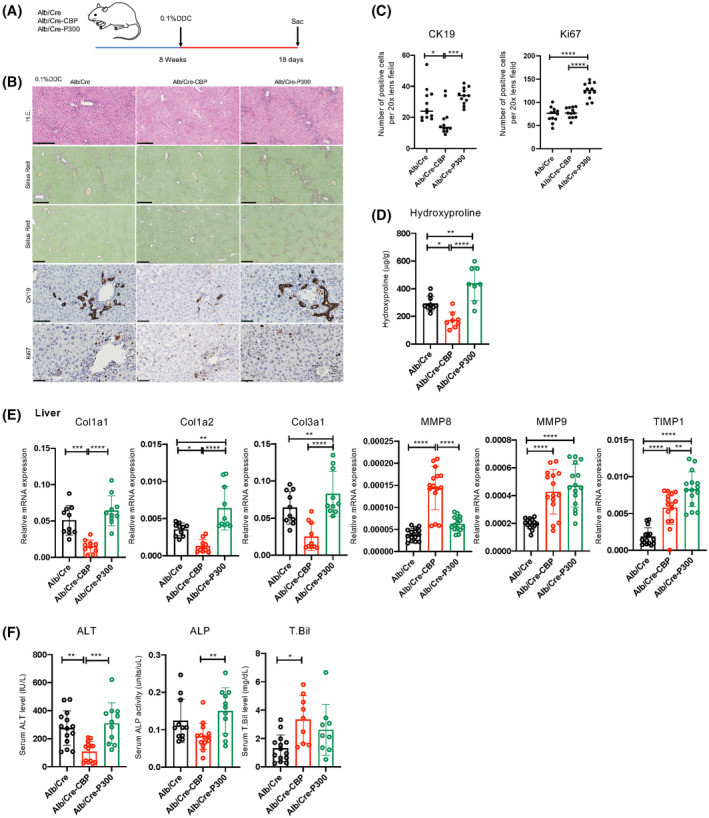
Liver fibrosis outcomes differ between cAMP response element‐binding protein‐binding protein (CBP) and P300‐Alb/Cre KO mice on a DDC diet. Alb/Cre‐CBP‐KO, Alb/Cre‐P300KO, and control Alb/Cre male mice (8 weeks old, n = 15 per group) were fed a 0.1% DDC diet for 18 days. (A) Scheme of the treatment protocol. (B) HE, sirius red, and immunohistochemical staining for CK19 and Ki67 (scale bars, 250, 250, 500, 100, and 50 μm, respectively, from the top figure). (C) Quantification of CK19‐positive and Ki67‐positive in the liver. (D) Collagen deposition as assessed by hydroxyproline measurement. (E) mRNA expression of the indicated genes in the livers as determined by real‐time quantitative PCR (n = 10 per group). (F) Serum ALT, ALP, and T.Bil levels (n = 10–15 per group). The results shown are representative of at least three independent experiments. Data represent the mean ± SD; *p < 0.05, **p < 0.01, ***p < 0.005, and ****p < 0.0001 by one‐way ANOVA.
CBP expression was diminished in the livers of Alb/Cre‐CBP‐KO mice, whereas P300 expression was diminished in the livers of Alb/Cre‐P300‐KO mice (Figure 6A). GS expression in hepatocytes was also reduced in the livers of Alb/Cre‐CBP‐KO mice (Figure 5A,B). KO of CBP in hepatocytes did not affect S100A4 expression in immune cells or macrophage infiltration (Figure 6A,B).
FIGURE 6.
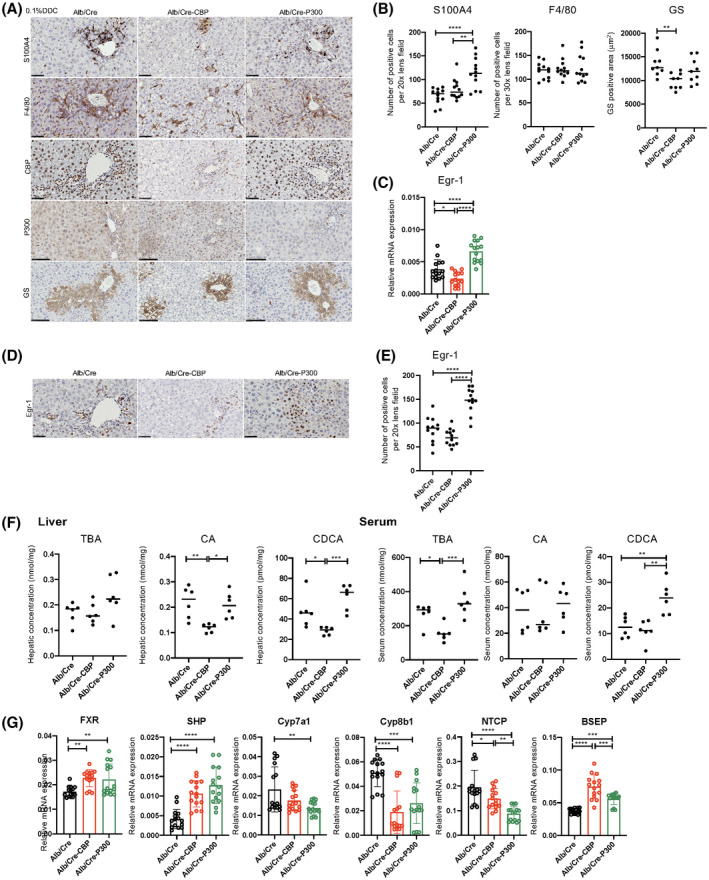
CBP KO in hepatocytes results in reduced hepatic and serum BA levels and decreased Egr‐1 expression. Alb/Cre‐CBP‐KO, Alb/Cre‐P300KO, and control Alb/Cre male mice (8 weeks old, n = 15 per group) were fed a 0.1% DDC diet for 18 days. (A) Immunohistochemical staining for S100A4, F4/80, CBP, P300, and GS (scale bars, 50 μm, except for P300 and GS [100 μm]). (B) Quantification of S100A4‐positive and F4/80‐positive cells and GS‐positive areas in the livers. (C) Egr‐1 mRNA expression in the livers as determined by real‐time quantitative PCR (n = 15 per group). (D) Immunohistochemical staining using anti‐Egr‐1 antibodies (scale bar, 50 μm). (E) Quantification of Egr‐1‐positive cells in the livers (n = 6). (F) Hepatic TBA, CA, and CDCA levels (n = 6 per group). The results shown are representative of at least three independent experiments. (G) mRNA expression of the indicated BA metabolism–related genes in the livers as determined by real‐time quantitative PCR (n = 15 per group). The results shown are representative of at least three independent experiments. Data represent the mean ± SD; *p < 0.05, **p < 0.01, ***p < 0.005, and ****p < 0.0001 by one‐way ANOVA.
Real‐time PCR array analysis revealed that MMP‐2, Egr‐1, Wnt7a, and Wnt10a expression levels were decreased in Alb/Cre‐CBP‐KO mice, whereas they were increased in Alb/Cre‐P300‐KO mice compared with those in control mice (data not shown). Of note, Egr‐1 expression was again increased in expression, as observed in Mdr2‐KO and BDL mice. As expected, BA levels in the livers and sera were decreased in Alb/Cre‐CBP‐KO mice (Figure 6F), which was accompanied by a decrease in Egr‐1 expression (Figure 6C–E) and changes in BA metabolism‐regulatory genes (Figure 6G). However, Alb/Cre‐CBP‐KO did not affect BA levels in mice fed normal chow (data not shown). As mentioned previously, KO of CBP and P300 in hepatocytes resulted in different outcomes of DDC‐induced fibrosis. Microarray analysis revealed that KO of CBP and P300 in hepatocytes resulted in differential transcriptional profiles in mice on a DDC treatment (Figure S5). Thus, a specific inhibitor of CBP/β‐catenin interaction, such as PRI‐724, but not a conventional β‐catenin inhibitor, is suitable for treating cholestatic liver disease.
Roles of hepatocyte Egr‐1 in BA metabolism and liver fibrosis in the DDC diet–induced cholestatic liver disease model
As mentioned previously, PRI‐724 reduced BA levels and liver fibrosis in all three cholestatic liver disease models, accompanied by the suppression of Egr‐1 expression. Alb/Cre‐CBP‐KO mice also showed decreased fibrosis and Egr‐1 expression after DDC treatment. To determine whether Egr‐1 plays an important role in cholestatic liver fibrosis, AAV serotype 8 (AAV8)/shEgr‐1 was generated and administered to mice on a DDC diet to knock down Egr‐1 expression in hepatocytes (Figure 7A). AAV8/shEgr‐1 diminished Egr‐1 expression induced by DDC (Figure 7B,D). Egr‐1 knockdown inhibited liver injury, immune cell infiltration, and fibrosis induced by DDC, accompanied by a reduction in BA levels and changes in mRNA levels of BA metabolism‐regulatory genes (Figure 7B–H). These results suggest that Egr‐1 plays a crucial role in cholestatic liver injury by mediating BA accumulation and that the antifibrotic effects of PRI‐724 or CBP‐KO in hepatocytes are, at least in part, mediated via a reduction in Egr‐1.
FIGURE 7.
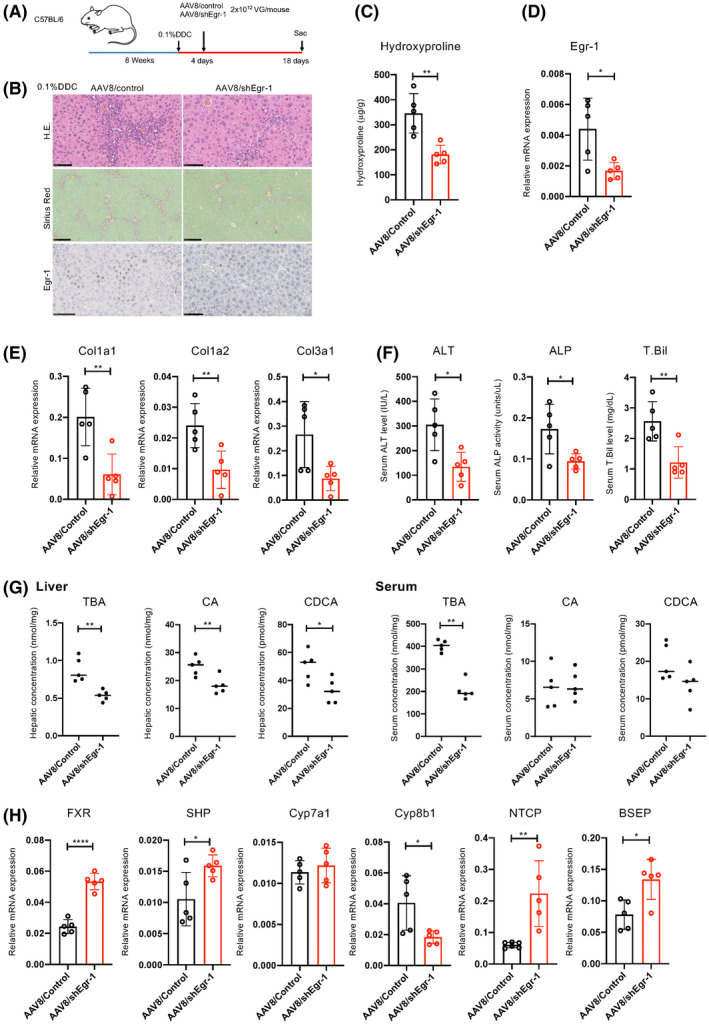
Egr‐1 knockdown inhibits liver injury and fibrosis induced by DDC. Male wild‐type C57BL/6 mice (8 weeks old) were fed a 0.1% DDC diet for 18 days. The mice received a single injection of adeno‐associated virus 8 (AAV8)/shEgr‐1 and AAV8/control (n = 5 in each group) at a dose of 2 × 1012 VG/mouse 4 days after the start of the DDC diet. (A) Scheme of the treatment protocol. (B) HE, sirius red, and immunohistochemical staining for Egr‐1 (scale bars, 100, 250, and 100 μm, respectively, from the top figure) (n = 5). (C) Collagen deposition as assessed by hydroxyproline measurement (n = 5 per group). (D,E,H) mRNA expression of the indicated genes in the livers as determined by real‐time quantitative PCR (n = 5 per group). (F) Serum ALT, ALP, and T.Bil levels (n = 5 per group). (G) Hepatic TBA, CA, and CDCA levels (n = 5 per group). The results shown are representative of at least three independent experiments. Data represent the mean ± SD; *p < 0.05, **p < 0.01, ***p < 0.005, and ****p < 0.0001 by unpaired Student's t test. Abbreviations: BSEP, bile salt export pump; FXR, farnesoid X receptor; NTCP, sodium taurocholate cotransporting polypeptide.
Egr‐1 expression is enhanced in human cholestatic liver disease
As we had found that Egr‐1 plays a crucial role in the progression of cholestatic liver disease in the mouse models, finally, we comparatively examined Egr‐1 expression in liver tissues from patients with PBC and normal liver tissues (non‐tumor tissues from patients with metastatic liver tumors) as a control (Figure 8A). In patients with PBC, the numbers of Egr‐1‐positive hepatocytes were significantly increased, accompanied by an increase in CBP expression (Figure 8B). Of note, P300 expression was also increased in the livers of these patients.
FIGURE 8.
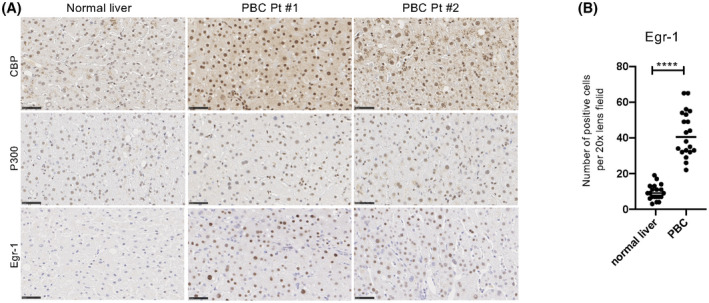
Egr‐1 expression is enhanced in human cholestatic liver disease. (A) Immunohistochemical staining for CBP, P300, and Egr‐1 in liver biopsy tissues from patients with metastatic liver tumors (non‐cancerous tissues) and patients with primary biliary cholangitis (PBC). Scale bars, 50 μm. (B) Quantification of Egr‐1‐positive hepatocytes. Data represent the mean ± SD; *p < 0.05, **p < 0.01, ***p < 0.005, and ****p < 0.0001 by an unpaired Student's t test.
DISCUSSION
In the present study, we found that PRI‐724 selectively inhibited the interaction between CBP and β‐catenin and prevented liver injury and fibrosis by suppressing BA production through a reduction in Egr‐1 expression in cholestatic liver disease. The safety of PRI‐724 has been confirmed in a phase 1 study in patients with hepatitis C virus (HCV)–related cirrhosis[ 22 ] and in a current phase 1/2a study in patients with HCV and hepatitis B virus–related cirrhosis.[ 19 ] These findings suggest a therapeutic possibility for mitigating liver fibrosis progression in cholestatic liver diseases.
In cholestatic liver disease models, PRI‐724 reduced the synthesis of BAs, resulting in decreased liver damage and fibrosis. However, it has been reported that mice lacking β‐catenin in hepatocytes and cholangiocytes showed increased hepatic BA levels[ 26 ] and that the knockdown of β‐catenin exacerbated liver injury in Mdr2‐KO mice.[ 10 ] These discrepant findings may be explained by the differential roles of CBP and P300. In the DDC‐induced cholestatic liver disease model, Alb/Cre‐CBP‐KO mice showed reduced liver damage and fibrosis accompanied by decreased BA synthesis, whereas these features were exacerbated in Alb/Cre‐P300‐KO. These results suggest that CBP and P300 have opposite roles in BA synthesis and that PRI‐724 shifts the balance from CBP/β‐catenin to P300/β‐catenin‐mediated transcription, resulting in reduced BA synthesis and liver fibrosis. FXR reportedly plays a key role in synthesizing BAs by reducing CYP7A1 and CYP8B1 expression via SHP and regulating FXR via β‐catenin.[ 27 ] Knockdown of β‐catenin prevented BDL‐induced liver fibrosis due to loss of the FXR/β‐catenin complex, which resulted in FXR activation and reduced BA levels.[ 11 ]
BAs are synthesized from cholesterol through a series of reactions, with Cyp7A1, which conjugates with glycine and taurine, being the rate‐determining enzyme in hepatocytes. BAs are excreted in the bile canaliculus by the bile salt export pump (BSEP).[ 28 ] FXR suppresses sodium taurocholate cotransporting polypeptide (NTCP) expression and induces the expression of BSEP, which excretes BA to the bile duct side.[ 28 ]
We made interesting observations regarding the relationship between CBP/P300‐β‐catenin hepatocyte signaling and BA metabolism–related enzymes during cholestasis (Figure 6G). First, because Alb/Cre‐P300‐KO mice developed cholestasis after DDC diet administration, results indicating the activation of FXR‐SHP, a decrease in Cyp7A1 NTCP expression, and an increase in BSEP expression were to be expected, but the actual results were similar to those obtained from Alb/Cre mice. This indicates that BA metabolism–related enzymes function as expected if CBP/β‐catenin signaling occurs in hepatocytes during cholestasis. On the other hand, although cholestasis was suppressed in Alb/Cre‐CBP‐KO mice after DDC diet administration, changes in BA metabolism–related enzymes similar to those in Alb/Cre‐P300‐KO mice were observed. This indicates that BA metabolism functions in the presence of hepatocyte P300/β‐catenin signaling when DDC diet induces bile duct injury. Briefly, this means that the expression of genes involved in BA metabolism changes regardless of the absence of hepatocyte CBP/P300‐β‐catenin signaling but that individual gene expression is enhanced compared with that in Alb/Cre mice. It is also interesting to note that Alb/Cre‐CBP‐KO mice showed increased expression of NTCP and BSEP compared with Alb/Cre‐P300‐KO mice.
Using PCR arrays, we found that BA synthesis is regulated by Egr‐1 expression via β‐catenin‐mediated transcription. Egr‐1 has important roles in cholestatic liver injury.[ 12 ] In Egr‐1‐KO mice, liver injury induced by BDL was reduced.[ 29 ] CBP and P300, which are transcriptional coactivators of Egr‐1,[ 30 ] showed reduced expression in Alb/Cre‐CBP‐KO mice but an increased expression in Alb/Cre‐P300 mice. Thus, it is evident that Egr‐1 expression is regulated by a CBP‐mediated signal. SHP represses Cyp expression and BA synthesis by inhibiting the activity of liver receptor homolog‐1, whose expression is increased by Egr‐1.[ 15 ] In accordance herewith, knockdown of Egr‐1 using AAV8/shEgr‐1 reduced BA synthesis and enhanced SHP expression. Thus, we hypothesize that PRI‐724 reduces BA synthesis via these mechanisms.
As MMPs have fibrolytic effects, the induction of MMPs may be one of the mechanisms underlying the antifibrotic effects of PRI‐724. Macrophages are known to secrete MMPs.[ 31 ]
DDC‐treated Alb/Cre‐CBP‐KO mice showed suppressed liver fibrosis. Moreover, an increase in MMP‐9 expression and a decrease in TIMP‐1 expression were expected, but TIMP‐1 expression was increased contrary to expectation.
From the results of KO mice, MMP‐9 is increased when the CBP signal acts on macrophages and stellate cells, and decreases when the CBP signal does not act on macrophage. This result suggests that macrophages are involved in the production of MMP‐9. Similarly, TIMP‐1 increases when macrophage and stellate cells are affected by the CBP signal, and decreases when the macrophage CBP signal does not act. This observation therefore suggests that macrophage CBP signals are necessary for the production of TIMP‐1. The target cells of PRI‐724 are presumed to be only macrophage and stellate cells, but the data on hepatocytes are still insufficient. We would like to confirm the effect of PRI‐724 on hepatocytes in a future investigation.
In a previous study, knockdown of P300 led to decreased MMP‐9 and MMP‐2 mRNA levels,[ 32 ] suggesting that P300/β‐catenin is involved in MMP expression.
This study had some limitations. First, Egr‐1 may have various roles in organ fibrosis, but we did not investigate the direct fibrotic effects of Erg‐1 other than the regulation of BA synthesis. Egr‐1‐KO mice are protected from bleomycin‐induced skin and lung fibrosis.[ 33 ] However, Egr‐1 expression is reportedly increased in the livers of CCI4‐treated mice,[ 34 ] suggesting that Egr‐1 plays a different role in liver fibrosis. Second, our data did not provide information on the sources of MMPs that are inhibited by CBP/β‐catenin. It remains unclear whether the MMPs produced are involved in fibrosis resolution in our model. Finally, the effects of PRI‐724 on hepatic stellate cells in murine cholestatic liver diseases were not investigated. Further studies are needed to resolve these uncertainties.
In summary, this study showed that the CBP/β‐catenin inhibitor PRI‐724 alleviates cholestasis and suppresses liver injury and fibrosis in murine models of cholestatic liver disease. In addition, we confirmed that fibrosis was suppressed in mice lacking CBP in hepatocytes and identified Egr‐1 as an important transcription factor for BA synthesis.
Finally, P300 inhibition in hepatocytes induced a hepatic pathophysiology that was completely different from that induced by CBP inhibition. Thus, PRI‐724 has an advantage over classic β‐catenin inhibitors in treating cholestatic liver diseases.
AUTHOR CONTRIBUTIONS
Research funding, research design, and experimental plan: Kiminori Kimura. Experiments and data analysis: Masamichi Kimura, Koji Nishikawa, Yosuke Osawa, Jun Imamura, Kenzaburo Yamaji, Hiroshi Yatsuhashi, Kazumoto Murata, and Kouichi Miura. Data collection and histological interpretation: Kenichi Harada. Study supervision: Atsushi Tanaka, Tatsuya Kanto, Michinori Kohara, and Terumi Kamisawa. Mice experiments: Michinori Kohara, Yosuke Osawa, and Kiminori Kimura. Data interpretation and manuscript draft: Yosuke Osawa and Kiminori Kimura. All authors approved the final version of the manuscript.
FUNDING INFORMATION
Supported by the Japan Agency for Medical Research and Development (JP18pc0101024h0001, JP20ek0109457h0001, and JP20lm0203057h0003).
CONFLICT OF INTEREST
Nothing to report.
ETHICS STATEMENT
All experiments were conducted in accordance with the institutional guidelines of the National Academy of Sciences (Guide for the Care and Use of Laboratory Animals). The study protocol was approved by the Research Committee of Tokyo Metropolitan Komagome Hospital. All analyses using human samples were approved by the ethics committee of Tokyo Metropolitan Komagome Hospital (No. 2831). This study adhered to the principles of the Declaration of Helsinki.
Supporting information
Figure S1.
Figure S2.
Figure S3.
Figure S4.
Figure S5.
Figure S6.
Appendix S1
ACKNOWLEDGMENT
The authors thank K. Kozuka, Y. Hayashi, E. Kojika, N. Okamoto, and H. Miyatake for the technical support, and their lab members for their continuous support and constructive criticism. They also thank Editage (www.editage.jp) for the English language editing.
Kimura M, Nishikawa K, Osawa Y, Imamura J, Yamaji K, Harada K, et al. Inhibition of CBP/β‐catenin signaling ameliorated fibrosis in cholestatic liver disease. Hepatol Commun. 2022;6:2732–2747. 10.1002/hep4.2043
Masamichi Kimura and Koji Nishikawa contributed equally to this work.
REFERENCES
- 1. Lleo A, Wang GQ, Gershwin ME, Hirschfield GM. Primary biliary cholangitis. Lancet. 2020;396:1915–26. [DOI] [PubMed] [Google Scholar]
- 2. Poupon RE, Poupon R, Balkau B. Ursodiol for the long‐term treatment of primary biliary cirrhosis. The UDCA‐PBC Study Group. N Engl J Med. 1994;330:1342–7. [DOI] [PubMed] [Google Scholar]
- 3. Tanaka A, Hirohara J, Nakano T, Matsumoto K, Chazouilleres O, Takikawa H, et al. Association of bezafibrate with transplant‐free survival in patients with primary biliary cholangitis. J Hepatol. 2021;75:565–71. [DOI] [PubMed] [Google Scholar]
- 4. Corpechot C, Chazouilleres O, Rousseau A, Le Gruyer A, Habersetzer F, Mathurin P, et al. A placebo‐controlled trial of bezafibrate in primary biliary cholangitis. N Engl J Med. 2018;378:2171–81. [DOI] [PubMed] [Google Scholar]
- 5. Trauner M, Fuchs CD. Novel therapeutic targets for cholestatic and fatty liver disease. Gut. 2022;71:194–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Trauner M, Fuchs CD, Halilbasic E, Paumgartner G. New therapeutic concepts in bile acid transport and signaling for management of cholestasis. Hepatology. 2017;65:1393–404. [DOI] [PubMed] [Google Scholar]
- 7. Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. [DOI] [PubMed] [Google Scholar]
- 8. Nusse R. Wnt signaling in disease and in development. Cell Res. 2005;15:28–32. [DOI] [PubMed] [Google Scholar]
- 9. Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pradhan‐Sundd T, Kosar K, Saggi H, Zhang R, Vats R, Cornuet P, et al. Wnt/beta‐catenin signaling plays a protective role in the Mdr2 knockout murine model of cholestatic liver disease. Hepatology. 2020;71:1732–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang R, Nakao T, Luo J, Xue Y, Cornuet P, Oertel M, et al. Activation of WNT/Beta‐catenin signaling and regulation of the farnesoid X receptor/beta‐catenin complex after murine bile duct ligation. Hepatol Commun. 2019;3:1642–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bhattacharyya S, Wu M, Fang F, Tourtellotte W, Feghali‐Bostwick C, Varga J. Early growth response transcription factors: key mediators of fibrosis and novel targets for anti‐fibrotic therapy. Matrix Biol. 2011;30:235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen SJ, Ning H, Ishida W, Sodin‐Semrl S, Takagawa S, Mori Y, et al. The early‐immediate gene EGR‐1 is induced by transforming growth factor‐beta and mediates stimulation of collagen gene expression. J Biol Chem. 2006;281:21183–97. [DOI] [PubMed] [Google Scholar]
- 14. Zhang Y, Xu N, Xu J, Kong B, Copple B, Guo GL, et al. E2F1 is a novel fibrogenic gene that regulates cholestatic liver fibrosis through the Egr‐1/SHP/EID1 network. Hepatology. 2014;60:919–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li J, Zhu X, Zhang M, Zhang Y, Ye S, Leng Y, et al. Limb expression 1‐like (LIX1L) protein promotes cholestatic liver injury by regulating bile acid metabolism. J Hepatol. 2021;75:400–13. [DOI] [PubMed] [Google Scholar]
- 16. Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov. 2014;13:513–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Osawa Y, Oboki K, Imamura J, Kojika E, Hayashi Y, Hishima T, et al. Inhibition of cyclic adenosine monophosphate (cAMP)‐response element‐binding protein (CREB)‐binding protein (CBP)/beta‐catenin reduces liver fibrosis in mice. EBioMedicine. 2015;2:1751–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kahn M. Symmetric division versus asymmetric division: a tale of two coactivators. Future Med Chem. 2011;3:1745–63. [DOI] [PubMed] [Google Scholar]
- 19. Kimura K, Kanto T, Shimoda S, Harada K, Kimura M, Nishikawa K, et al. Safety, tolerability, and anti‐fibrotic efficacy of the CBP/beta‐catenin inhibitor PRI‐724 in patients with hepatitis C and B virus‐induced liver cirrhosis: an investigator‐initiated, open‐label, non‐randomised, multicentre, phase 1/2a study. EBioMedicine. 2022;80:104069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gijbels E, Pieters A, De Muynck K, Vinken M, Devisscher L. Rodent models of cholestatic liver disease: a practical guide for translational research. Liver Int. 2021;41:656–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Osawa Y, Kojika E, Hayashi Y, Kimura M, Nishikawa K, Yoshio S, et al. Tumor necrosis factor‐alpha‐mediated hepatocyte apoptosis stimulates fibrosis in the steatotic liver in mice. Hepatol Commun. 2018;2:407–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kimura K, Ikoma A, Shibakawa M, Shimoda S, Harada K, Saio M, et al. Safety, tolerability, and preliminary efficacy of the anti‐fibrotic small molecule PRI‐724, a CBP/beta‐catenin inhibitor, in patients with hepatitis C virus‐related cirrhosis: a single‐center, open‐label, dose escalation phase 1 trial. EBioMedicine. 2017;23:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barikbin R, Neureiter D, Wirth J, Erhardt A, Schwinge D, Kluwe J, et al. Induction of heme oxygenase 1 prevents progression of liver fibrosis in Mdr2 knockout mice. Hepatology. 2012;55:553–62. [DOI] [PubMed] [Google Scholar]
- 24. Sullivan BP, Cui W, Copple BL, Luyendyk JP. Early growth response factor‐1 limits biliary fibrosis in a model of xenobiotic‐induced cholestasis in mice. Toxicol Sci. 2012;126:267–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nakamoto N, Sasaki N, Aoki R, Miyamoto K, Suda W, Teratani T, et al. Gut pathobionts underlie intestinal barrier dysfunction and liver T helper 17 cell immune response in primary sclerosing cholangitis. Nat Microbiol. 2019;4:492–503. [DOI] [PubMed] [Google Scholar]
- 26. Yeh TH, Krauland L, Singh V, Zou B, Devaraj P, Stolz DB, et al. Liver‐specific beta‐catenin knockout mice have bile canalicular abnormalities, bile secretory defect, and intrahepatic cholestasis. Hepatology. 2010;52:1410–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thompson MD, Moghe A, Cornuet P, Marino R, Tian J, Wang P, et al. beta‐Catenin regulation of farnesoid X receptor signaling and bile acid metabolism during murine cholestasis. Hepatology. 2018;67:955–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li T, Chiang JY. Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev. 2014;66:948–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim ND, Moon JO, Slitt AL, Copple BL. Early growth response factor‐1 is critical for cholestatic liver injury. Toxicol Sci. 2006;90:586–95. [DOI] [PubMed] [Google Scholar]
- 30. Silverman ES, Du J, Williams AJ, Wadgaonkar R, Drazen JM, Collins T. cAMP‐response‐element‐binding‐protein‐binding protein (CBP) and p300 are transcriptional co‐activators of early growth response factor‐1 (Egr‐1). Biochem J. 1998;336(Pt 1):183–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fingleton B. Matrix metalloproteinases as regulators of inflammatory processes. Biochim Biophys Acta Mol Cell Res. 2017;1864:2036–42. [DOI] [PubMed] [Google Scholar]
- 32. Santer FR, Hoschele PP, Oh SJ, Erb HH, Bouchal J, Cavarretta IT, et al. Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Mol Cancer Ther. 2011;10:1644–55. [DOI] [PubMed] [Google Scholar]
- 33. Wu M, Melichian DS, de la Garza M, Gruner K, Bhattacharyya S, Barr L, et al. Essential roles for early growth response transcription factor Egr‐1 in tissue fibrosis and wound healing. Am J Pathol. 2009;175:1041–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pritchard MT, Nagy LE. Hepatic fibrosis is enhanced and accompanied by robust oval cell activation after chronic carbon tetrachloride administration to Egr‐1‐deficient mice. Am J Pathol. 2010;176:2743–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Figure S2.
Figure S3.
Figure S4.
Figure S5.
Figure S6.
Appendix S1