

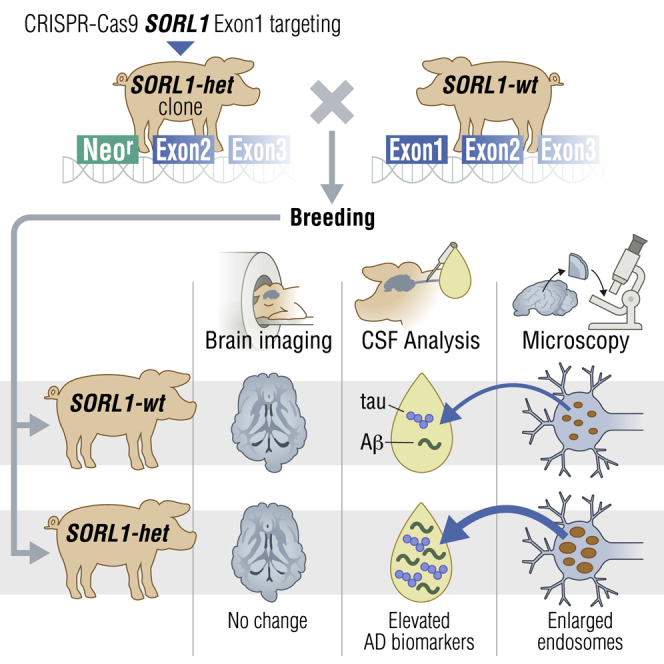
Summary
The established causal genes in Alzheimer’s disease (AD), APP, PSEN1, and PSEN2, are functionally characterized using biomarkers, capturing an in vivo profile reflecting the disease’s initial preclinical phase. Mutations in SORL1, encoding the endosome recycling receptor SORLA, are found in 2%–3% of individuals with early-onset AD, and SORL1 haploinsufficiency appears to be causal for AD. To test whether SORL1 can function as an AD causal gene, we use CRISPR-Cas9-based gene editing to develop a model of SORL1 haploinsufficiency in Göttingen minipigs, taking advantage of porcine models for biomarker investigations. SORL1 haploinsufficiency in young adult minipigs is found to phenocopy the preclinical in vivo profile of AD observed with APP, PSEN1, and PSEN2, resulting in elevated levels of β-amyloid (Aβ) and tau preceding amyloid plaque formation and neurodegeneration, as observed in humans. Our study provides functional support for the theory that SORL1 haploinsufficiency leads to endosome cytopathology with biofluid hallmarks of autosomal dominant AD.
Keywords: SORLA, retromer-dependent endosomal recycling, Alzheimer’s disease, SORL1, genome editing, CRISPR-Cas9, large animal model
Graphical abstract
Highlights
-
•
Minipig model of Alzheimer’s disease by CRISPR knockout of the causal gene SORL1
-
•
Young SORL1 het minipigs phenocopy a preclinical CSF biomarker profile of individuals with AD
-
•
SORL1 haploinsufficiency causes enlarged endosomes similar to neuronal AD pathology
-
•
A minipig model bridging the translational gap between AD mouse models and affected individuals
Andersen et al. develop a Göttingen minipig model for Alzheimer’s Disease (AD) with SORL1 haploinsufficiency and demonstrate that their young (<3 years) minipigs have enlarged endosomes and elevated Aβ peptide and tau CSF levels but are unaffected by AD brain pathologies and neurodegeneration, as assessed by PET and MRI scanning methods.
Introduction
Until recently, only APP (encoding the amyloid precursor protein), PSEN1 (presenilin 1), and PSEN2 (presenilin 2) have been identified as autosomal dominant risk genes for Alzheimer’s disease (AD).1 SORL1 (sortilin-related receptor 1) has now been proposed to be included in this exclusive list of AD causal genes.2 Individuals harboring mutations in APP or PSEN1/2 have been investigated by cerebrospinal fluid (CSF) analysis and neuroimaging, establishing an in vivo signature of the earliest preclinical stage of disease. This early disease stage is characterized by CSF elevation of APP’s β-amyloid (Aβ) peptide and tau, before onset of amyloid plaques and neurodegeneration.3,4 This presumably initiating pathogenic profile was confirmed in mouse models genetically engineered to express these mutations,5 but validation is made difficult by the complexity of tracking the onset and progression of AD’s slowly worsening pathophysiology via CSF analyses and imaging in mice.
Endosome dysfunction is strongly implicated in the underlying pathogenesis of the sporadic and familiar forms of AD.6,7 Relying on studies suggesting that the SORL1-encoded protein SORLA can act as a receptor of the endosome’s retromer complex,8,9 the first genetic link of SORL1 to AD emerged from an association analysis that investigated genes encoding components of the retromer complex, SORL1 , and other retromer-associated receptors.10 Since then, numerous studies have confirmed SORL1’s genetic link to AD through hypothesis-free genome and exome screening.11,12 Most recently, genetic epidemiology studies have identified rare truncation mutations in SORL1 almost exclusively in individuals with AD, suggesting that SORL1 functions as a causal gene.12
In parallel, numerous subsequent studies have confirmed that SORLA functions in endosomal recycling of cargo proteins,13, 14, 15, 16, 17 which, when disrupted, is a dominant upstream pathogenic pathway in AD.18, 19, 20 Recent studies have identified how impaired endosome recycling can increase cellular secretion of Aβ and tau with a subsequent detectable increase in CSF.21,22
The main component of the retromer complex is the receptor-binding core unit assembled by vacuolar protein sorting 35 (VPS35) at the center of the core, to which the VPS26 and VPS29 proteins bind.23 Two paralogs of VPS26 have been identified,24 of which VPS26B is enriched in the brain and mainly engaged in retromer-mediated recycling of cargo from endosomes to the cell surface, whereas VPS26A predominantly assists retrograde trafficking of cargo from endosomes to the Golgi/trans-Golgi network.16
Because SORL1 truncation mutations typically cause partial loss of protein function, inducing protein haploinsufficiency has turned out to be a valid and convenient model of these mutations. Previous studies using mouse models or neuronal culture studies have shown that SORL1 deficiency can phenocopy the cell biology of the other causal genes (APP, PSEN1, and PSEN2). In particular, and just like the disease-causing variants of these causal genes,25 SORL1 deficiency has been shown to accelerate Aβ production26,27 and cause abnormal swelling of endosomes in induced pluripotent stem cell (iPSC)-derived neurons,28,29 both hallmark features of sporadic and familiar forms of AD.30
Because similar studies have not yet been performed in human carriers of SORL1 loss-of-function variants prior to disease onset, it remains unknown whether SORL1 loss of function phenocopies the in vivo preclinical signature (i.e., increased CSF levels of Aβ and tau before onset of amyloid plaques and neurodegeneration) of the established causal genes. Multiple large-scale and long-term global consortia were required to establish this earliest preclinical in vivo signature of AD in individuals harboring the established causal genes. It will take an equally extensive infrastructure and a long time to ultimately test whether individuals harboring the putatively causal SORL1 genetic variants phenocopy the in vivo preclinical signature. Until then, as with APP and the PSENs, this signature can be tested for in appropriate animal models.
Here we investigated the causality of SORL1 in relation to AD in a Göttingen minipig model genetically engineered to be SORL1 haploinsufficient by heterozygous knockout (KO) of the endogenous porcine SORL1 gene, mimicking the genetic status of individuals with AD with heterozygous SORL1 truncating mutations. Besides the general public acceptance of using pigs rather than non-human primates for biomedical research, pigs have, in contrast to mice, gyrencephalic brains closely resembling the human brain31 and also share more ultra-conserved genomic regions with humans than mice.32 Of special relevance for modeling AD, human and pig APP sequences have very high similarity, sharing identical secretase cleavage sites for production of Aβ fragments found in humans.33 The APOE allele status of pigs in general (including Göttingen minipigs; C.B.S., unpublished data) has been reported as APOE-e4;34,35 thus, they may have an inherent genetic disposition for developing AD in analogy to humans, in which this isoform is associated with high risk of the disease. Importantly, the pig has a body size and longevity enabling deployment of CSF and neuroimaging markers used in the clinical setting of AD diagnosis and allowing longitudinal monitoring of these, as the lifespan can reach as high as 15 years. Göttingen minipigs grow from young (3–4 months of age) to sexually mature (4–8 months of age) and into fully developed adults within approximately 2 years.36
Results
SORL1 expression in porcine neurons
SORLA is predominantly expressed in neurons in the mouse and human brain.37 To confirm that this is also true for the pig, we examined sections of frontal cortex obtained from wild-type (wt) Göttingen minipigs by immunohistochemistry. As expected, SORLA immunoreactivity was observed predominantly in the neuronal soma (Figure 1A).
Figure 1.

Generation of SORL1-deficient Göttingen minipigs
(A) Immunohistochemical detection of endogenous SORLA predominantly in neuronal cell somata (indicated by arrowheads) in the frontal Cx from an adult wt Göttingen minipig. Scale bar, 20 μm.
(B) Schematic of porcine SORL1 transcripts from Ensembl Sscrofa 11.1. Four SORL1 isoforms are listed, of which SORL1 -202, comprising 48 exons, is considered the reference transcript.
(C) RT-PCR validation of the porcine reference SORL1 -202 transcript. cDNA obtained from RT of total RNA, isolated from the Cx, hippocampus, and Cb from a wt Göttingen minipig, was used as a template in RT-PCR using primers specific for the 5′ end of the reference SORL1 -202 transcript (exons 1–3) or for porcine GAPDH. Reactions carried out in parallel using RNA without RT (−RT) or with water (H2O) as template served as negative controls.
(D) Schematic of the endogenous SORL1 locus (top panel). The three first exons of the endogenous porcine SORL1 gene are shown as black boxes. Primers used for RT-PCR (F1+R1 and F1+R2, respectively) to validate the presence of the reference SORL1-202 transcript in the Göttingen minipig breed and in SORL1 wt, het, and ko animals, are illustrated as horizontal arrows. Also shown is a schematic of the endogenous SORL1 locus, the gene-targeting vector, and the targeted SORL1 locus (bottom panel). The targeting vector comprises a left and right homology arm (shown as blue boxes; LHA and RHA, respectively) flanked by loxP sites (green triangles). The LHA and RHA are separated by a PGK-neor/EM7-zeor expression cassette for mammalian and bacterial selection, respectively. A region of 609 bp, comprising the entire SORL1 exon 1 and its flanking regions, is replaced by the PGK-neor/EM7-zeor cassette upon successful gene editing, resulting in DNA fragments of 3,248 bp and 6,865 bp when BlpI-digested genomic DNA is hybridized with the SORL1 and neor probe, respectively. Positions of the PCR screening primer pairs F3/R3 and F4/R4 are illustrated as horizontal arrows. BlpI restriction sites are indicated as vertical arrows, and Southern blot probes (SORL1probe and neorprobe) are shown as black horizontal bars.
(E) Generation of SORL1-deficient Göttingen minipigs. Gene editing was performed by co-transfecting primary porcine fibroblasts, isolated from newborn female Göttingen minipigs, with the gene-targeting vector, the hCas9 plasmid, and the gRNA vector. Two days after transfection, the transfected cells were trypsinized, and half of the cell suspension was subjected to limiting dilution by reseeding the cells into 96-well plates. After selection for 2 weeks, G418-resistant cell clones were trypsinized and, one third of the resulting cell suspension was transferred to 96-well PCR plates for PCR screening, one third was cultured for Southern blot analysis, and one third was cultured in 96-well plates for freezing at early passages and subsequent usage as nuclear donor cells for cloning of minipigs by SCNT. Two surviving piglets with heterozygous KO of SORL1 (SORL1 het) and one surviving piglet with homozygous KO of SORL1 (SORL1 ko) were obtained after SCNT and one re-cloning. The cloned SORL1 het founders (F0 generation) were used for conventional breeding to obtain SORL1 het and SORL1 wt offspring (F1 generation).
(F) Eight-day-old cloned Göttingen minipigs. All piglets were born with no apparent gross abnormalities, but apart from one SORL1 ko piglet, all SORL1 het piglets died neonatally. Fibroblasts isolated from an ear biopsy from the SORL1 het piglet shown at the front in the photo were used for re-cloning to obtain viable SORL1 het pigs.
The human SORL1 gene gives rise to several protein-coding transcripts. In the pig, four SORL1 transcripts (SORL1 -201, -202, -203, and -204) exist, according to the Sus scrofa genome assembly 11.1 (Figure 1B). To experimentally confirm the presence of exon 1 in the reference transcript (SORL1 -202) for subsequent gene targeting, reverse-transcriptase PCR (RT-PCR) analysis was performed on cDNA obtained from cortex, hippocampus, and cerebellum tissue in wild-type Göttingen minipigs using primers specific for the 5′ and 3′ ends of this SORL1 transcript. Bands corresponding to the expected size of the SORL1 -202 reference transcript were observed in all three tested regions of the minipig brain for the 5′ RT-PCR (exons 1–3; Figure 1C) and 3′ RT-PCR (exons 46 and 47; data not shown), verifying the presence of exon 1 in pig SORL1 transcripts.
Generating SORL1 gene-edited Göttingen minipigs
We used CRISPR-Cas9-mediated gene editing and cloning to create Göttingen minipigs with compromised SORL1 expression by heterozygous deletion of 609 bp, including the entire SORL1 exon 1 (Figure 1D). Gene-edited Göttingen Minipig female fibroblasts were used for cloning by somatic cell nuclear transfer (SCNT) to generate SORL1-compromised animals (Figures 1E and 1F).
SCNT resulted in female SORL1+/− animals, as expected, but also in delivery of a single female piglet with homozygous KO of SORL1. In contrast to the remaining piglets, this cloned (F0) SORL1−/− piglet survived the post-natal period and served as a control in the study (from here on referred to as knockout [ko]). Re-cloning of fibroblasts obtained from one of the newborn cloned SORL1+/− piglets resulted in delivery of live-born founder (F0) piglets without visible gross abnormalities and harboring the expected SORL1+/− genotype (from here on referred to as heterozygous [het]), as validated by Southern blot analysis and PCR-based genotyping (Figure S1). Two sexually mature SORL1- het female founders were used for conventional breeding with wild-type Göttingen minipig boars, yielding two litters of naturally bred offspring (F1, 6 het and 4 wt; details regarding sex, genotypes, and ID numbers for all animals included in the study are provided in Table S1).
To verify that the CRISPR-Cas9 gene editing strategy employed did not introduce unintended off-target effects, we analyzed genomic DNA isolated from the re-cloned het piglets and a wt Göttingen minipig. Eight potential off-target sites residing in annotated genes (listed in Table S2) were identified when allowing for up to three mismatches between the single-guide RNA (sgRNA) and the genomic sequence. These genomic sequences, comprising the potential sgRNA binding sites, were amplified by PCR, and the resulting amplicons were sequenced. We found no evidence of Cas9-induced off-target effects in any of the genomic sites analyzed, nor did we, by PCR analyses, observe random integration of the vectors employed (encoding the sgRNA and humanized Cas9, respectively) in the genomic DNA of the cloned SORL1 het and SORL1 ko animals (data not shown).
Confirming SORL1 haploinsufficiency in the SORL1- het minipig model
To verify that genomic deletion of SORL1 exon 1 resulted in loss of function and reduced SORL1 expression, RT-PCR was performed on cDNA obtained from total RNA isolated from the cerebellum (Cb) and frontal cortex (Cx) of brains from three sacrificed pigs (F1 wt [6478]/5 months; F1 het 6473]/5 months, and the cloned F0 ko [6304]/33 months) using a primer pair spanning the exon 1-2 boundary. The analysis confirmed that our KO strategy had resulted in complete removal of exon 1 comprising SORL1 transcripts in the cloned ko minipig. Further analysis using a primer pair spanning SORL1 exons 46 and 47 revealed that no other SORL1 transcripts were present in the ko sample, strongly indicating the lack of alternative ways to initiate transcription and that exon 1 is essential for SORL1 transcription in these brain regions of pigs (Figure 2A). Quantitative PCR (qPCR) analysis using brain specimens from eight 24- to 30-month-old minipigs (4 het and 4 wt) confirmed that the SORL1 mRNA expression levels were reduced by approximately 40% in the Cb (Figures 2B) and 55% in the Cx (Figure 2C) of het versus wt SORL1 minipigs.
Figure 2.

Decreased SORLA expression in the brain of SORL1-deficient Göttingen minipigs
(A) RT-PCR validation of SORL1-depleted Göttingen minipigs. cDNA obtained from reverse transcription of total RNA isolated from the cerebellum (Cb) or frontal cortex (Cx) from wt, het, and ko SORL1 Göttingen minipigs was used as a template in RT-PCR using primers specific for SORL1 exons 1 and 2 (see primers F1+R2 in Figure 1D), SORL1 exons 46 and 47 of the SORL1 -202 transcript, or for porcine GAPDH (reference gene). Negative controls (ctrl) correspond to reactions carried out in parallel using −RT) or with H2O as templates.
(B and C) Relative SORL1 mRNA expression in SORL1 het (n = 4) minipigs was measured by qPCR, calculated relative to the reference gene HPRT1, and normalized to SORL1 expression in SORL1 wt minipigs (n = 4) for samples from the Cb (B) and Cx (C).
(D) WB analysis confirming complete absence of SORLA expression in Cb or frontal Cx homogenates from the SORL1 ko compared with a wt and a het SORL1 Göttingen minipig (D).
(E) Homogenates of Cx from het and wt SORL1 minipigs were analyzed by WB to determine expression levels of SORLA, APP, VPS26B, and VPS26A, using the level of ACTIN as a loading ctrl.
(F–I) The expression levels of SORLA (F), APP (G), VPS26B (H), and VPS26A (I) were quantified relative to the level of ACTIN.
(J) Homogenates of Cb from het and wt SORL1 minipigs were analyzed by WB to determine expression levels of SORLA, APP, and VPS26B, using the level of ACTIN as a loading ctrl.
(K–M) The expression levels of SORLA (K), APP (L), and VPS26B (M) were quantified relative to the level of ACTIN.
(N and O) Equal amounts of CSF from het and wt SORL1 minipigs (n = 4 in both groups) were immunoprecipitated and analyzed by WB using an antibody for SORLA detection (N), and the relative levels of sSORLA were quantified (O).
Identification numbers of individual minipigs are provided below every lane for all WB analyses. Two-tailed unpaired Student’s t test was used for all statistical analyses, with p values below 0.05 considered significantly changed. Data are expressed as mean ± SEM.
Translation of the SORL1-202 transcript is predicted to produce a SORLA protein of 2,213 amino acids that shares 92.2% identity with the 2,214 residues of human SORLA (Figure S2). SORLA is a transmembrane protein but is also found in a soluble form, sSORLA, resulting from proteolysis and shedding from the neuronal cell surface.38,39
To corroborate our qPCR findings at the protein level, we next analyzed membrane-bound SORLA levels in homogenates of frontal Cx and Cb, isolated from brains of the three sacrificed minipigs (F1 wt [6478], F1 het [6473], and F0 ko [6304]) by western blot (WB). As expected, SORLA could not be detected in samples from the ko minipig (Figure 2D). A more detailed investigation by WB of homogenates from the Cx (Figure 2E) and Cb (Figure 2J) of the four het and four wt animals (24–30 months old) demonstrated a strong and significant reduction of SORLA protein of almost 70% in Cx from het compared with wt SORL1 minipigs (Figure 2F). The level of SORLA in the Cb was unaffected by the presence of only a single functional SORL1 copy (Figure 2K), suggesting that SORLA levels can be maintained in the heterozygous state in the Cb, a finding in line with it being a region free of most pathological defects and with intact SORLA expression in individuals with AD.40,41
We recently showed how deficiency of the neuronal retromer core unit VPS26B leads to a decrease in functional SORLA maturation,16,42 so here we wanted to determine whether the reverse is also true: whether SORL1 deficiency affects the expression level of VPS26 proteins. WB analysis of Cx homogenates (Figure 2E) showed strong and significantly reduced expression of both isoforms: VPS26B (Figure 2H) and VPS26A (Figure 2I). There was no consistent alteration in VPS26B expression in the Cb between wt and het SORL1 minipigs (Figures 2J and 2L), in agreement with this brain region also not showing any change in SORLA protein level. These findings support a model where SORLA and the retromer can stabilize each other, suggesting that they act as a functional unit for endosome recycling. We also determined the level of APP holoprotein in homogenates from the wt and het SORL1 minipigs, and, as expected, based on previous findings from studies of cells and mice,26 neither the Cx (Figure 2G) nor the Cb (Figure 2M) showed consistent alteration of APP expression levels.
In line with the reduced SORLA expression in the Cx of SORL1 het minipigs, WB analysis of CSF isolated from young adult (24–30 months old) animals used above showed significantly reduced sSORLA levels in het minipigs (Figures 2N and 2O). We also confirmed complete absence of sSORLA in CSF from the ko animal (Figure S1C).
As part of the validation of the model, we performed neuropathological examination and histology on brain sections from the frontal, temporal, and occipital lobes and the brain stem and Cb obtained from a het (6473) and a wt (6478) F1 SORL1 minipig at 5 months of age, respectively. Macroscopically, the ventricular system was not found to be dilated, and no apparent brain pathology was observed in the Cx in these animals. Subsequent microscopy examination of hematoxylin and eosin-stained sections also revealed normal brain morphology, including a normal ventricular ependymal layer, normal hippocampi, and no neuron loss or neuron atrophy in either of the animals (Figure S3).
From these analyses, we conclude that the SORL1 het minipigs have decreased expression of functional SORLA protein in the Cx and CSF, similar to individuals with AD harboring SORL1 truncating mutations (O.M.A., unpublished data). The reduction of SORLA in the Cx was accompanied by a reduction of VPS26A and VPS26B, suggestive of decreased functional retromer levels and compromised endosome recycling activity.
Elevated CSF Aβ and tau in SORL1 het minipigs
The level of Aβ in CSF is a biomarker of AD and known to mirror endosomal defects in neurons from individuals with AD.22 The sequence identity between human and porcine APP is 97.8% (Figure S4), and the 42 amino acids that correspond to the Aβ peptide are 100% conserved. Accordingly, we determined the level of the Aβ peptides (Aβ38, Aβ40, and Aβ42) in CSF isolated and combined from a cohort of our minipigs (wt, n = 10; het, n = 5; ko, n = 1; 5–38 months of age) using an available mesoscale discovery (MSD) assay developed for the human peptides. For analysis, samples from the het minipigs and the single ko animal were combined and compared with the group of wt minipigs. Despite the fact that samples were collected from animals across different ages (wt, mean age = 22.4 ± 3.0 months; het + ko, mean age = 23.2 ± 4.5 months), we found a robust increase in the level of Aβ peptides (170% for Aβ38, 149% for Aβ40, and 169% for Aβ42) in CSF from SORL1-deficient (combined het and the single ko) minipigs (Figures 3A–3C). Statistical analysis omitting the sample isolated from the ko minipig (data marked in red in Figure 3) showed a significant (p = 0.0016 [Aβ38], p = 0.0056 [Aβ40], and p = 0.0023 [Aβ42]) increase in CSF Aβ in the cohort of SORL1 het minipigs. The ratio between Aβ42 and Aβ40 is used as an indication of AD because the longer isoform in the brains of affected individuals is increased because of deposition into dense core plaque structures, but, as shown in Figure 3D, we did not observe any difference in this ratio when comparing het + ko and wt SORL1 minipigs, suggesting that deposition has not yet occurred in the animals.
Figure 3.

Increased Aβ and tau levels in CSF from SORL1-deficient Göttingen minipigs
(A–F) Quantification of the APP processing products Aβ38 (A), Aβ40 (B), Aβ42 (C), Aβ42/Aβ40 ratio (D), and soluble APPα (E) and APPβ (F) in CSF from SORL1-deficient (n = 6) and age-matched wt (n = 9/10) Göttingen minipigs. The average ages of the two groups of pigs were similar. The group of SORL1 het minipigs is depicted, including data obtained from the ko pig (6304) shown in red and statistical analysis shown for data excluding (black) or including (red) the data point for the SORL1 ko pig. Quantifications were performed using MSD assays for human APP fragments because of 100% conservation of the 42 amino acids comprising the Aβ sequence.
(G) WB analysis of tau in CSF (first-time isolation) from wt and het SORL1 minipigs at 5, 18, 24, or 30 months of age. Identification numbers of individual minipigs are provided below every lane. Detection was performed with the 5E2 anti-tau antibody that binds to a region of tau that is 100% conserved between the human and pig protein (Figure S5A).
(H) Quantification of CSF tau WB analysis for the 18-month-old (1 wt/het pair), 24-month-old (2 wt/het pairs), and 30-month-old (2 wt/het pairs) SORL1 minipigs. The signal for SORL1 wt pigs was set to 100% for each age, and the signal for the paired SORL1 het was expressed relative to this.
(A–F and H) Two-tailed unpaired Student’s t test was used for all statistical analyses, with p values below 0.05 considered significantly changed. Data are expressed as mean ± SEM.
(I) RT-PCR analysis of genes involved in generation of amyloid and tau. Gene expression of APP, α-secretases (ADAM10 and ADAM17), β-secretase (BACE1), subunits of the γ-secretase (PSEN1 and PSEN2), and MAPT (encoding tau) were analyzed in Cx tissue from a wt (6475), a het (6469), and the ko (6304) SORL1 minipig, validating that CRISPR-Cas9 had no detrimental effect on these genes. GAPDH served as a ctrl for successful cDNA synthesis, whereas −RT samples and water were used as a negative ctrl.
From work done in mice and cells, we know that decreased SORLA activity also leads to increases in production of sAPPα and sAPPβ,26,43 but we never assessed whether that also changes the levels of these two APP fragments within the CSF. We therefore also measured the level of sAPPα and sAPPβ in the CSF in SORL1 het animals by an MSD assay. Although both sAPP fragments showed an increased trend, only the sAPPβ fragment that is generated from endosomal β-secretase cleavage of APP showed a significant elevation in our analysis when comparing levels between wt and het + ko SORL1 minipigs (Figures 3E and 3F). This finding is in agreement with increased APP proteolysis as a result of SORL1 deficiency, as we and others have demonstrated previously, and these larger sAPP fragments are not as suitable as CSF biomarkers of AD as the smaller Aβ fragments.44,45
Because tau secretion to CSF is known to follow an unconventional pathway when endosome recycling is affected by AD,22 we next analyzed tau levels in CSF from pairs of age-matched het and wt minipigs with CSF isolated at 5, 18, 24, or 30 months of age (Figure 3G). We applied a WB analysis using an antibody (5E2) for tau, recognizing an ∼50-kDa fragment, to test pairs of CSF samples from 5- to 30-month-old het and wt SORL1 minipigs (Figure 3G) and also quantified total tau levels in CSF from 18- to 30-month-old minipigs (wt, n = 5; het, n = 5) using this approach (Figure 3H).
In all four comparisons, elevated ∼50-kDa tau levels were observed in the SORL1 het samples (Figure 3G), and the tau levels were found to be significantly increased when analyzing the samples as groups irrespective of age (p = 0.0137; Figure 3H). We also assessed the level of total tau using a commercial MSD assay developed for total tau. Because there is no assay available specifically directed to the porcine tau protein, we followed recommendations from the supplier to use an MSD assay for mouse tau for analysis of our entire set of minipig CSF samples. Using this mouse assay, we found a small but significant increase in the level of total tau in our het (n = 8) in comparison with wt (n = 11) SORL1 minipigs (109%; p = 0.0431) (Figure S5B).
We were also interested in determining whether phosphorylation of tau in the CSF was changed. However, when performing WB analysis on our CSF samples with the known AT8 antibody specific for phospho-tau, we did not obtain consistent signals (data not shown). This may reflect different phosphorylation patterns, lack of phospho-tau in the porcine CSF, or that the antibody was unsuitable for detection of the modified porcine tau protein because of the relatively low sequence homology between porcine and human tau (89.8% sequence identity; Figure S5A). Addressing this in more detail would need additional future investigations. We did, however, observe a specific tau isoform in our WB analysis using the 5E2 antibody, suggesting that some modifications of the tau protein are specific to our SORL1 het minipigs, but we were not able to address this further (Figure S5C).
Finally, to exclude potential CRISPR-Cas9 off-target effects on genes encoding proteins involved in generation of amyloid and tau, we performed RT-PCR on APP, relevant α-secretases (ADAM10 and ADAM17), β-secretase (BACE1), subunits of the γ-secretase (PSEN1 and PSEN2), and MAPT on Cx tissues from a wt (6475), a het (6469), and the ko (6304) SORL1 minipig (Figure 3I). In accordance with the findings from the initial CRISPR-Cas9 off-target analysis, the expression of these genes did not appear to be altered in the gene-edited minipigs.
These findings of elevated levels of Aβ and tau in the CSF of the SORL1 het minipigs follow observations of increased amyloid and tau pathology in cellular models of endosomal dysfunction46,47 and are in accordance with previous findings of increased levels of CSF Aβ in the very early phase of disease progression in individuals with sporadic AD,3 those with autosomal dominant AD,4 and in transgenic AD mouse models prior to amyloid plaque formation.5
SORL1 het minipigs have enlarged endosomes
The elevated secretion of Aβ and tau into CSF indicates a defect in neuronal endosome activity, as identified previously when the retromer has been inactivated.22 It is well established that the size of early endosome structures is increased in neurons of brains from individuals with AD,48,49 and similar swollen endosomes have been observed in iPSC-derived neurons with pathogenic variants in APP, PSEN1, or PSEN225 as well as in iPSC-derived neurons with SORL1 truncating variants.28,29 We therefore performed an immunohistochemistry analysis on a set of cortical brain regions from the 4 het and 4 wt SORL1 minipigs, sacrificed at 24–30 months of age, using an anti-Rab5 antibody to identify early endosomes (Figures 4A and 4B). Staining of Cx from SORL1 het minipigs showed much stronger immunoreactivity against the anti-Rab5 antibody, and quantification of the size of Rab5-positive structures (i.e., early endosomes) confirmed a significant increase in the mean area of neuronal endosomes in het compared with wt SORL1 minipigs (Figure 4C).
Figure 4.

Enlarged early endosomes in SORL1 het Göttingen minipigs
(A) Paraffin sections of cortical brain regions from SORL1 wt (338593 and 6475) and SORL1 het (6470 and 6469) minipigs immunolabeled with Rab5 antibody to identify early endosomes, occurring as a brown signal from 3,3′-diaminobenzidine (DAB) detection of the horseradish peroxidase (HRP)-conjugated secondary antibody.
(B) High-magnification images from bright-field microscopy of early endosome compartments (arrowheads), showing an increase in Rab5-positive structures in neurons from the het compared with the wt SORL1 minipig.
(C) The mean area of Rab5-positive (early endosome) structures was measured across neurons from wt (mean area = 0.1640 μm2 ± 0.009565, n = 27 cells) and het (mean area = 0.4887 μm2 ± 0.002815, n = 27 cells) minipig Cx areas as depicted in (B).
(D) Immunofluorescence confocal images showing SORLA expression in cultured fibroblasts from wt (6475) and het (6469) SORL1 minipigs, demonstrating lower expression levels in cells from SORL1 het minipigs, as seen by fewer positive signals.
(E) Endosome abnormalities were also present in fibroblasts from SORL1 het minipigs, as visualized by Rab5-positive structures with increased size.
(F) The mean area of Rab5-positive (early endosome) structures was measured for cultured primary fibroblasts from wt (mean area = 0.1862 ± 0.01223 μm2, n = 21 cells) and het (mean area = 0.2771 ± 0.01566 μm2, n = 21 cells) SORL1 minipigs, as depicted in (E).
Scale bars equal 50 μm (A) and 10 μm (B and D). Two-tailed unpaired Student’s t test was used for all statistical analyses, with p values below 0.05 considered significantly changed. Data are expressed as mean ± SEM.
In a parallel set of experiments, we used cultured fibroblasts derived from a wt and a het SORL1 minipig to identify early endosomes by immunocytochemistry using the anti-Rab5 antibody. We confirmed that these fibroblasts express the SORLA protein by WB (data not shown) as well as immunostaining (Figure 4D) and found a significant increase in their mean endosome areas in het compared with wt SORL1 minipigs (Figures 4E and 4F) similarly to what was observed from the neurons in the Cx of the minipig brain.
These experiments confirm that heterozygous SORL1 expression leads to endosomal defects, which is a likely explanation for the observed increases in the CSF biomarker profile.
21-month-old SORL1 het minipigs do not exhibit amyloid plaque formation
We next employed positron emission tomography (PET) imaging with the tracer [11C] N-methyl [11C] 2-(4′methylaminophenyl)-6-hydroxy-benzothiazole (PIB), which allows visual and quantitative measurement of Aβ deposition, to analyze whether amyloid plaques had started to form in the brains of the SORL1 het minipigs. We subjected four 21-month-old female minipigs (het [n = 2] and wt [n = 2]) to [11C]-PIB-PET imaging, but we did not observe any obvious increase in tracer retention in the time activity curves of the het compared with the wt animals, suggesting that fibrillar Aβ is not yet accumulating in the brains of these young adult SORL1 het animals (Figure 5A), in agreement with the measured increase rather than decrease in CSF Aβ.
Figure 5.

[11C]-PIB and [18F]-FDG PET analysis of young adult SORL1 het and SORL1 wt Göttingen minipigs
Shown is [11C]-PIB- and [18F]-FDG PET analyses of four female 21-month-old SORL1 minipigs (het, 6469 and 6470; wt, 6475 and 6477).
(A) [11C]-PIB-PET summed images from the 30- to 90-min portion of the dynamic scan, divided by averaged whole brain activity.
(B) [18F]-FDG PET standard uptake volume (SUV) images, corrected for full brain activity.
(C) Absence of amyloid plaque pathology in a wt and a het SORL1 minipig (29 and 30 months old, respectively), as evidenced by lack of signals in immunostaining for deposits using an antibody for Aβ42 that is routinely applied in clinical settings for validation of AD pathology.
(D) Absence of fibrillar tangles in the wt and het SORL1 minipigs shown in (C), visualized by lack of signals in immunostaining with anti-tau phospho-Thr231 antibody, which is routinely applied in clinical settings for validation of AD pathology.
We also investigated brain hypometabolism as a marker of neurodegeneration in the four 21-month-old female minipigs (het [n = 2] and wt [n = 2]) by PET imaging with 2-[18F]-2-deoxy-D-glucose (FDG), which measures the cerebral metabolic rates of glucose as an indicator of neuronal synaptic activity.50 We did not observe any decrease in [18F]-FDG tracer uptake in the brains of the SORL1 het minipigs (Figure 5B). Caution should be taken to interpret results on only these few animals, but it may suggest that any pathological changes occurring in the brains of these young adult SORL1 het minipigs are not sufficient to cause hypometabolism reflective of neuronal degeneration at this stage.
To validate the absence of neuropathological hallmarks of AD, immunohistochemistry analysis was performed on brain tissue sections from 30-month-old wt and het male SORL1 minipigs. In accordance with the PIB-PET analysis, amyloid plaques were not observed. Neither were fibrillary tangles detected by the AT8 antibody also employed for detection of phospho-tau in the WB analysis of CSF samples (Figures 5C and 5D).
SORL1 het minipigs display unaltered brain morphology and neuroaxonal damage up to 27 months of age
Having established that the observed CSF changes in the SORL1 het minipigs were not accompanied by amyloid deposition and brain hypometabolism, we examined whether the animals exhibited changes in brain morphology by subjecting the now 22-month-old four female minipigs (wt, n = 2; het, n = 2) as well as four 27-month-old males (wt, n = 2; het, n = 2) to anatomical magnetic resonance imaging (MRI). We observed variation between animals, mainly between males and females, but were not able to detect any sign of genotype-dependent brain atrophy in these young adult SORL1 het minipigs, including any differences in overall brain volume (Figures 6A and 6B). Likewise, individual brain structures, considered to be the first affected by atrophy in AD, did not show statistically significant differences between wt and het SORL1 minipigs, as illustrated by measurement of amygdala width, entorhinal cortical thickness, and hippocampal and ventricular volumes (Figures 6C–6F).
Figure 6.

Anatomical MRI analysis of young adult SORL1 het and SORL1 wt Göttingen minipigs
(A–F) Examples of 3D T2 FLAIR magnetic resonance images used for volume measurements (extracerebral tissue stripped for visualization) in female (6469) and male (6474) SORL1 het minipigs and female (6477) and male (338496) SORL1 wt minipigs (A). Also shown are quantification of whole brain volume (B), ventricle volume (C), amygdala width (D), entorhinal cortical thickness (E), and hippocampus volume (F) from anatomical MRI scanning. The four female minipigs (wt, 6475 and 6477; het, 6469 and 6470) and the four male minipigs (wt, 338496 and 338593; het, 6472 and 6474) were 22 and 27 months of age, respectively, when scanned. Data are expressed as mean ± SEM.
Having demonstrated normal brain anatomy with anatomical MRI, we next determined whether there were more subtle structural changes in brain microstructure using diffusion tensor imaging (DTI), a more sensitive method for detecting microscopic damage than conventional structural MRI,51 enabling detection of potential diffusivity changes because of cell loss, demyelination, and axonal injury. We subjected the eight (four female and four male) minipigs to DTI in connection with the structural MRI scan but found no changes in mean diffusivity (MD) or fractional anisotropy (FA) in the amygdala, hippocampus, corpus callosum, and deep white matter between minipigs of the two genotypes (Figures 7A–7E).
Figure 7.

Normal brain microstructure in SORL1 het and SORL1 wt Göttingen minipigs
(A) Examples of mean diffusivity (MD) and fractional anisotropy (FA) maps, representing tissue microstructure, from diffusion tensor MRI in female (6469) and male (6474) SORL1 het minipigs and female (6477) and male (338,496) SORL1 wt minipigs.
(B–E) FA and MD of the amygdala (B), hippocampus (C), corpus callosum (D), and deep white matter (E) were determined with DTI. The four female minipigs (wt, 6475 and 6477; het, 6469 and 6470) and the four male minipigs (wt, 338,496 and 338,593; het, 6472 and 6474) were 22 and 27 months of age, respectively, when scanned.
Data are expressed as mean ± SEM.
Finally, we quantified the level of neurofilament light chain (NF-L), a biomarker indicative of axonal damage. NF-L is used to monitor progression in many neurological disorders and has been suggested for tracking neuronal fitness during AD.52 In the CSF that was collected from minipigs at necropsy (wt, n = 10; het, n = 5; ko, n = 1; 5–38 months of age) and used for Aβ40/42 analyses, we did not detect any significant difference between het and wt SORL1 minipigs for this biomarker (Figure S6).
Our results suggest that the 5- to 30-month-old SORL1 het minipigs are in an early preclinical phase of AD with raised CSF Aβ and tau levels prior to progression to the more advanced AD pathology including amyloid plaque formation and neuronal loss. Our SORL1 het minipig model thus phenocopies the preclinical in vivo profile of AD observed with other established AD causal genes.4 Our findings also support endosomal dysfunction as an upstream pathway for secretion of AD CSF biomarkers.
Discussion
Here, by CRISPR-Cas9-mediated gene editing, we developed a Göttingen minipig model with SORL1 haploinsufficiency, modeling pathogenic variants from individuals with AD harboring truncating SORL1 mutations.53 Previous work employing mouse models and neuronal cultures have suggested that SORL1 deficiency phenocopies the cell biology of the established AD-causal genes encoding APP and the presenilins. Just as seen with these causal genes, SORL1 deficiency accelerates amyloid production in the early endosome of neurons,26,54 also causing endosomal traffic jams, which manifest as swollen neuronal endosomes,28 a hallmark feature of AD neurons.48,49 We observed enlarged endosome structures in neurons of SORL1 het minipigs and identified increased Rab5-positive endosomes in fibroblasts heterozygously expressing SORL1 compared with SORL1 wt cells. The fact that the increase in endosomes is not restricted to neurons is in agreement with previous findings of endo-lysosomal defects upon SORL1 silencing in cancerous cell lines15 and reports of increased endosome size in fibroblasts from individuals with AD.55
In the cases of APP and the presenilins, their mutations accelerate endosomal accumulation of the intermediary APP fragment β-CTF, the direct precursor of Aβ, and it is likely this membrane-spanning fragment, not Aβ, that causes endosomal traffic jams.6,56 In the presented SORL1-depleted minipigs, endosomal traffic jams relate to SORLA acting as a receptor for recycling cargo out of the endosome. APP and β-CTF are cargo for SORLA-dependent endosomal recycling.26,57,58 The decreased SORLA endosomal activity therefore leads to increased Aβ production, which manifests as elevated CSF Aβ levels.
We found previously that reduction in retromer activity impairs SORLA trafficking and decreases levels of mature receptor protein,13,16,42 but here we show that the opposite is also true: expression of VPS26A and VPS26B is concomitantly decreased together with SORLA in the cortical brain region vulnerable to AD, supporting a model where SORLA and the retromer act together as a functional unit.
Although these cell biology investigations are important to try to elucidate the mechanisms of the APP and presenilin genes, they have not yet been confirmed in presymptomatic carriers of SORL1 mutations. We have shown previously that complete deletion of SORL1 in mice leads to increased amyloidogenesis, but no analysis of CSF biomarkers has been performed during the course of disease progression because sampling of CSF to monitor, e.g., early changes in biomarkers is a challenging procedure in mice, hampering detection of early biochemical pathology in this animal model.26,43,54 The current study employing gene-edited Göttingen minipigs with SORL1 haploinsufficiency, mimicking the genetic status of individuals with AD with SORL1 haploinsufficiency now shows an in vivo phenocopy of SORL1 deficiency to AD’s causal genes, observed in actual affected individuals in the earliest preclinical phase of the disease (i.e., elevated Aβ and tau in CSF prior to formation of amyloid plaques and neurodegeneration). Although cell biology observations cannot be confirmed in affected individuals, their mechanistic implications can help explain the in vivo profile observed in our SORL1 minipigs. Only through cell biology do we know, for example, that amyloid is produced and first accumulates in neuronal endosomes7 when APP is cleaved by its amyloidogenic enzymes at the endosomal membranes and that intraneuronal amyloid is then secreted unconventionally by endosomal secretion.59 APP and PSEN mutations are known to accelerate amyloid production by directly affecting the amyloidogenic biochemical pathway. SORL1 deficiency, in contrast, indirectly leads to accelerated amyloid production by slowing APP recycling out of endosomes, increasing the probability that it will be cleaved at endosomal membranes harboring high β-secretase activity60 and resulting in an increase in Aβ peptides 38, 40, and 42 amino acids in length in CSF. This is also reflected in the CSF of our het + ko SORL1 minipigs, in which we observed increased levels of all three Aβ forms. In accordance with our het SORL1 pigs being young adults, we did not find intraneuronal accumulation of Aβ, but this has been reported in 10- and 18-month-old double-transgenic Göttingen minipigs overexpressing APP695sw and PSEN1M146I.61 It is therefore likely that our SORL1 minipigs would show a similar phenotype and deposit extracellular amyloid plaques upon disease progression when followed for a longer period than we were able to in the current study. In such a future study, it would also be relevant to measure whether CSF Aβ42 levels decrease in response to plaque formation.
More recent cell biology studies have clarified why tau accumulates in the CSF in the setting of mutations in APP and the presenilins. It is now known that tau is actively secreted from neurons and, just like amyloid, occurs in CSF via endosomal secretion.22,62,63 The abnormal elevation of CSF tau observed in AD is no longer thought to be driven by tangle formation or neurodegeneration per se but, rather, is an early event that reflects accelerated active secretion and one that seems to coincide with amyloid secretion.22,64,65 Our findings of increased levels of amyloid and total tau in CSF in het SORL1 minipigs are in accordance with this, but more detailed studies involving a larger number of animals would be needed to determine the exact sequence of amyloid and tau CSF alterations and whether this sequence chronology mimics the CSF changes observed in human AD progression.
Unlike the mouse, but similar to humans, the pig expresses tau isoforms comprising three (3R) and four (4R) microtubule-binding repeats.66 To detect total tau (native and phospho-tau) and phospho-tau, we employed the anti-tau 5E2 and AT8 antibodies, respectively, for WB analysis. Because only the 5E2 antibody detected tau in our porcine CSF samples, this may be due to, e.g., different phosphorylation patterns in the two species. To pursue this further requires a more thorough understanding of the phosphorylation of porcine tau as well as development of suitable assays for detection of this marker originating from pigs. The precise details of accelerated tau secretion remain unknown but likely relate to how endosomal traffic jams lead to increased translocation of tau into the endolysosomal system. Because the endosomal traffic jams induced by defects in APP, the presenilins, or SORLA have been shown to be Aβ peptide independent, amyloid and tau secretion are likely coincidental.
By showing that our gene-edited SORL1 haploinsufficient Göttingen minipigs phenocopy the biomarker profile of the earliest preclinical stage of disease observed in individuals carrying known causal mutations, we provide functional evidence that SORL1 loss-of-function mutations are causal in AD. Despite exhaustive and large-scale genetic investigations into AD, currently only four genes have been identified across our genome that can themselves cause AD: APP, PSEN1, PSEN2, and SORL1.2 Interpreting the unified in vivo profile induced by these four causal genes in the context of their cell biology consequences suggests that dysfunctional endosomal trafficking is a unified pathogenic pathway in the disease that could be pursued for drug development.19,29
Besides providing evidence that SORL1 is an AD-causing gene, the SORL1 het Göttingen minipigs allow future longitudinal studies for biochemical and neuroimaging biomarker discovery and may provide important clues regarding the sequence of events that occurs in the pharmacologically valuable treatment window between the very early preclinical stage of the disease, with raised Aβ and tau CSF levels, and the time point of amyloid brain deposition, irreversible neurodegeneration, and subsequent cognitive impairment.
Because of their similarities to humans in terms of genetics, anatomy, physiology, and biochemistry, minipigs are valuable animal models for drug testing and are being increasingly employed as non-rodent models for toxicity testing and safety pharmacology,67,68 adding to the utility of our Göttingen minipig SORL1 het model in pharmacological research targeting AD.
Limitations of the study
The microscopy method applied to assess Rab5-positive structures does not have the resolution required to determine whether the larger structures observed correspond only to enlarged endosomes, a higher number of similarly sized smaller structures clumped together, or a mixture of both. Future high-resolution microscopy could be used to address this.
Future studies should also be designed to investigate how the correlation between the decreased levels of SORLA (and accompanying retromer) activity in different brain regions translates to defects in endosome recycling capacity beyond changes in APP trafficking.
Because of the relatively low number and young age of the animals included, our study is lacking longitudinal measurements of the biomarkers examined beyond the age of 30 months. This would be relevant for determining the inter-relationship of these in further disease progression in older animals. We also did not conduct behavioral studies to examine cognitive function of our young animals, but such analyses can be performed on pigs by subjecting the animals to, e.g., spatial memory tasks using a hole board arena.69,70 Combined with longitudinal studies of the biomarkers mentioned above in a larger and older cohort of animals, such studies may add insights into the interplay between disease progression and cognitive decline and facilitate determination of optimal timing of treatment for potential cognitive rescue.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-LR11 (clone 48/LR11) | BD Biosciences | Cat#612633; RRID: AB_399670 |
| Mouse monoclonal anti-Tau (clone 5E2) | Millipore | Cat#05-348; RRID: AB_309687 |
| Mouse monoclonal anti-phospho-Tau (Ser202, Thr205, clone AT8) | Thermo Fischer Scientific | Cat#MN1020; RRID: AB_223647 |
| Rabbit monoclonal anti-Rab5 [EPR5438] | Abcam | Cat#ab109534; RRID: AB_10865740 |
| Mouse monoclonal anti-β-Actin (clone AC-15) | Sigma-Aldrich | Cat#A5441; RRID: AB_476744 |
| Rabbit polyclonal anti-SORLA (pAb_5387), in-house | DAKO | https://pabmabs.com/?p=2318 and https://doi.org/10.1074/jbc.M100857200 |
| Rabbit polyclonal anti-APP1227, in-house | Olav Andersen Lab, Aarhus University, Aarhus, Denmark | https://www.pnas.org/doi/full/10.1073/pnas.0503689102 |
| Rabbit polyclonal anti-beta Amyloid 1-42 (Aβ42) | Abcam | Cat#ab10148; RRID: AB_296881 |
| Rabbit polyclonal anti-VPS26a | Abcam | Cat #ab23892; RRID: AB_2215043 |
| Rabbit polyclonal anti-VPS26b | Abcam | Cat#ab236966; UniProt#Q4G0F5; https://www.abcam.com/vps26b-antibody-ab236966.html |
| Rabbit polyclonal anti-Tau Phospho-Thr231 | Sigma-Aldrich | Cat#AB9668; UniProt#P10636; https://www.sigmaaldrich.com/DK/en/product/mm/ab9668 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fischer Scientific | Cat#A-21206; RRID: AB_2535792 |
| Goat anti-Rabbit IgG/Biotin-Conjugated Secondary Antibody | Sigma-Aldrich | Cat#B8895; RRID: AB_258649 |
| Rabbit anti-mouse IgG/HRP-Conjugated Secondary Antibody | Agilent | Cat#P0260; RRID: AB_2636929 |
| Chemicals, peptides, and recombinant proteins | ||
| AmnioMax™ C-100 Complete Medium | Thermo Fischer Scientific | Cat#12558011 |
| Human FGF-basic Recombinant Protein (bFGF) | Thermo Fischer Scientific | Cat#PHG0266 |
| Critical commercial assays | ||
| V-Plex Aβ Peptide Panel 1 (6E10) Kit | MSD | Cat#K15200E |
| sAPPα/sAPPβ Kit | MSD | Cat#K15120E |
| Mouse Total Tau Kit | MSD | Cat#K151DSD |
| Simple Plex Human NF-L Cartridge | Protein Simple | Cat# SPCKB-PS-002448 |
| Experimental models: Cell lines | ||
| Göttingen Minipigs Primary Fibroblasts | Ellegaard Göttingen Minipigs | https://minipigs.dk/ |
| SORL1 Göttingen Minipigs Primary Fibroblasts | This study | N/A |
| Human Embryonic Kidney 293T Cell Line | ATCC | Cat#CRL-3216 |
| Experimental models: Organisms/strains | ||
| Göttingen Minipigs | Ellegaard Göttingen Minipigs | https://minipigs.dk/ |
| Oligonucleotides | ||
| Primers and probes | This paper | Table S2 |
| Recombinant DNA | ||
| pNeDaKo-Neo plasmid | Bert Vogelstein Lab, The Johns Hopkins University Medical Institutions, Baltimore, MD, USA | https://doi.org/10.1093/nar/gnh009 |
| pAAV-MCS vector | Agilent | Cat#240071 |
| Human codon-optimized Cas9 plasmid | Mali et al., 2013, https://www.science.org/doi/10.1126/science.1232033 | Addgene plasmid #41815 |
| rAAV/SORL1 KO-Neo vector | This study | N/A |
| C-check vector | Zhou et al., 2016 | https://doi.org/10.1007/s00018-015-2128-3 |
| sgRNA1 | This study | N/A |
| Software and algorithms | ||
| Zink Finger Targeter (ZiFiT) | Sander et al., 2010, https://doi.org/10.1093/nar/gkq319 | http://bindr.gdcb.iastate.edu/ZiFiT/ (discontinued) |
| CRISPR RGEN Cas-OFFinder | Bae et al., 2014, https://doi.org/10.1093/bioinformatics/btu048 | http://www.rgenome.net/cas-offinder/ |
| ImageJ2 | NIH | https://imagej.net/downloads |
| GraphPad Prism 6.0 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| BTX Microslide 0.5 mm Fusion Chamber, model 450 | BTX | https://www.btxonline.com/ |
| ELLA Automated Immunoassay System | Protein Simple | https://www.bio-techne.com/p/simple-plex/ella-automated-immunoassay-system_600-100 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Olav M. Andersen (o.andersen@biomed.au.dk).
Materials availability
The SORL1 Göttingen Minipigs are exclusively bred and can be obtained from Ellegaard Göttingen Minipigs A/S, Dalmose, Denmark.
Experimental model and subject details
Animals
Permissions for generation and breeding as well as for neuroimaging and CSF sampling of the SORL1 Göttingen Minipigs were granted by the Danish Animal inspectorate (2015-15-0202–00028 and 2019-15-0201–00264). All Göttingen Minipigs were housed under standard conditions in specific pathogen-free housing facilities at Aarhus University and maintained on a restricted minipig diet (SDS Diet, UK) throughout the study. The minipigs were all acclimatized for at least one week prior to experiments and housed, in groups after weaning until 4 months of age and thereafter as single animals, under environmental conditions of 20-22-C, 50–55% relative humidity, 12:12 h of light and darkness, and with change of air at least eight times per hour. They had free access to tap water, and their well-being was monitored twice per day. The welfare of the animals was improved by access to “easy-strø” bedding (Easy-AgriCare A/S, Denmark) and chewing toys (e.g. sisal ropes). All experiments conducted included both male and female animals of each genotype. Unrelated (n = 8) or litter-mate (n = 4) wild-type Göttingen Minipigs served as controls in the study. Details on gender, genotypes, and ID numbers for all animals included in the study are provided in Table S1.
Primary cell cultures
Primary porcine fibroblasts for gene editing were established by explant culture of ear biopsies from newborn female Göttingen Minipigs obtained from Ellegaard Göttingen Minipigs A/S. Until fibroblast outgrowth, ear biopsies were cultured in AmnioMAX-C100 complete medium (#12558011, Thermo Fischer Scientific). Upon isolation, the porcine fibroblasts were cultured at 37°C (5% CO2) in DMEM (#BE12-604F, Lonza Biowhittaker) supplemented with 15% heat-inactivated fetal bovine serum (#F7524, Sigma-Aldrich), 100 U/mL penicillin, 100 μg/mL streptomycin (#15140122, Gibco), and 2 mM glutamine (#25030024, Gibco). The cell culture medium was additionally supplemented with 5 ng/mL recombinant basic fibroblast growth factor (bFGF, #PHG0266, Thermo Fischer Scientific) for gene targeting experiments. Upon delivery of piglets, fibroblasts were isolated from ear biopsies taken on the day of birth from all cloned piglets and F1 animals following the protocol described above. The resulting cell lines from individual piglets were expanded and the first passage cryopreserved at −140°C for subsequent use in genotyping, off-target analyses and staining protocols.
For analysis of early endosomes, primary fibroblasts were isolated from ear biopsies obtained from a SORL1+/+, a SORL1+/− and a SORL1−/− minipig, from here on referred to as wt, het and ko, respectively, as described above. The fibroblasts (passage number 3–5) were seeded on poly-L-lysine coated glass coverslips in DMEM (#41965062, Gibco) supplemented with 15% fetal bovine serum (#10270106, Gibco), 100 U/mL penicillin, 100 μg/mL streptomycin (#15140122, Gibco), and 2 mM glutamine (#25030024, Gibco). Following culturing at 37°C (5% CO2) for 48 h, the cells were fixed in 4% paraformaldehyde solution, and stained with relevant antibodies (see Immunohistochemistry/Immunocytochemistry section below).
Cell lines
Human embryonic kidney 293T (HEK293T, #CRL-3216, ATCC) cells, employed for testing of the constructed sgRNAs, were cultured at 37°C (5% CO2) in DMEM (#41965062, Gibco) supplemented with 10% heat-inactivated fetal bovine serum (#F7524, Sigma-Aldrich), 100 U/mL penicillin, 100 μg/mL streptomycin (#15140122, Gibco), and 2 mM glutamine (#25030024, Gibco).
Method details
Construction of gene targeting vector and single-guide RNAs
A gene targeting vector was generated essentially as previously described.71 The left and right porcine SORL1 (pSORL1) homology arms (LHA and RHA, 880 bp and 998 bp, respectively) flanking SORL1 exon 1 were amplified by PCR using wild type Göttingen Minipigs genomic DNA as template and porcine SORL1-specific primers. These two homology arms were subsequently linked to a neomycin (neo)/zeomycin (zeo) resistance gene cassette allowing for G418 selection by a three-way fusion PCR. The neo/zeo resistance genes were comprised in a 4 kb PvuI fragment isolated from a pNeDaKO-Neo plasmid (a generous gift from Bert Vogelstein & Kenneth W. Kinzler, The Johns Hopkins University Medical Institutions, Baltimore, MD 21231, USA). The 3-way fusion PCR was performed as described71 using a Platinum Pfx polymerase (#11708–013, Thermo Fischer Scientific) and the following PCR protocol: 1 cycle of 94°C for 1 min; 25 cycles of 94°C for 30 s, 59°C for 30 s, and 68°C for 4 min; 1 cycle of 68°C for 7 min. Fusion products were digested with NotI and ligated to a NotI cleaved pAAV-MCS plasmid backbone (#240071, Agilent). The final rAAV/SORL1 KO-Neo plasmid construct was verified by NotI digestions and sequencing. Successful gene targeting using the rAAV/SORL1 KO-Neo plasmid vector results in deletion of 609 bp in the endogenous porcine SORL1 gene including the entire exon 1.
The CRISPR/Cas9 single-guide RNAs (sgRNAs) targeting the porcine SORL1 gene were designed employing the online designing tool ZiFiT.72 The human codon-optimized Cas9 (kindly made available by George Church, Addgene plasmid # 41815)73 and the sgRNA (encoded by a pFUS-U6 vector) were expressed from two individual plasmids. Two different sgRNAs, sgRNA1 and sgRNA2, were designed to target the porcine SORL1 exon 1 region. For each sgRNA construct, two pSORL1-specific complementary oligonucleotides were denatured and slowly annealed prior to ligation of the annealed oligonucleotides to a sgRNA scaffold plasmid (pFUS-U6-sgRNA) based on BsaI assembly as previously described.74 XL-2 Blue ultracompetent bacterial cells (#200150, Agilent) were subsequently transformed with the ligation mixture and the resulting bacterial cell clones were screened by PCR. Positive sgRNA clones were validated by DNA sequencing of purified plasmid DNA. PCR primers for generating the rAAV/SORL1 KO targeting vector as well as oligonucleotides and target sites used for generation of the sgRNA vectors are listed in Table S2.
Generation of pSORL1-specific C-check vector for sgRNA testing
The two pSORL1-specific sgRNAs, sgRNA1 and sgRNA2, were functionally validated employing a previously developed single strand annealing (SSA)-directed, dual fluorescent surrogate reporter system entitled C-check.74 This vector comprises two expression cassettes: an AsRED expression cassette for measuring transfection efficiency and normalization, and a truncated EGFP expression cassette for detection of double strand break (DSB)-induced SSA events. The EGFP cassette is interrupted by the two target sites for the pSORL1 sgRNAs. The C-check reporter construct will express AsRED and EGFP upon sgRNA-induced DSB and SSA repair in this target region, whereas only AsRED will be expressed if no DSB is induced, or repair occurs by non-homologous end-joining. To construct the pSORL1-specific C-check vector, two complementary oligonucleotides comprising the sgRNA target sites were annealed and cloned into the BsaI-digested C-check vector as previously described.74 XL-2 Blue Ultracompetent cells were transformed with the ligated plasmid, and resulting bacterial cell clones were screened by PCR. Positive C-check clones were validated by DNA sequencing upon purification of plasmid DNA. The oligonucleotides employed to construct the pSORL1-specific C-check vector comprising the overlapping sgRNA1 and sgRNA2 target sites are listed in Table S2.
Flow cytometric analysis of pSORL1 gRNA efficiency
The efficiencies of the two generated pSORL1-specific sgRNAs were evaluated by transfection of the sgRNA-encoding plasmid into HEK293T cells and subsequent flow cytometry of the transfected cells as previously described.74 Briefly, cells were seeded into 6-well plates (3 × 105 cells/well) and co-transfected the next day with one of the two sgRNA plasmids (75, 150, and 300 ng) together with the hCas9 plasmid (75, 150, and 300 ng), the pSORL1-specific C-check plasmid (100, 200, and 300 ng), and stuffer plasmid DNA (to adjust the amount of total DNA to 1 μg) using X-tremeGENE 9 DNA transfection reagent (#6365779001, Sigma-Aldrich). For controls, cells were transfected with only the hCas9 or C-check plasmid, or with the sgRNA construct together with the C-check plasmid. Cells were harvested 48 h post transfection by trypsinization and analyzed by flow cytometry using a BD LSRFortessa Flow Cytometer (BD Bioscience) at the FACS Core Facility, Dept. of Biomedicine, Aarhus University, to quantify the efficiency (EGFP expression) of the two sgRNAs in transfected (AsRED+) cells. Based on these results, sgRNA1 was chosen for CRISPR-based gene editing of primary Göttingen Minipigs fibroblasts.
Gene editing
The day before transfection, primary fibroblasts isolated from newborn female Göttingen Minipigs were seeded (1.5 × 106) into a gelatin-coated 10 cm cell culture dish. The next day, the culture medium was changed and supplemented with bFGF (5 ng/μL) and gene editing was performed by co-transfecting the cells with the gene targeting rAAV/SORL1 KO-Neo vector (5600 ng), the hCas9 plasmid (1200 ng) and the sgRNA1 vector (1200 ng) using Lipofectamin LTX Reagent (#15338500, Thermo Fischer Scientific). The transfected cells were trypsinized 48 h post transfection, and ½ of the cell suspension was subjected to limiting dilution by reseeding the cells into 5 gelatin-coated 96-well plates resulting in approx. 300 cells per well. Selection with G418 (0.8 mg/mL, #ant-gn, Invivogen) was initiated the following day and continued for two weeks. Following selection, the G418-resistant cell clones were trypsinized, 1/3 of the resulting cell suspension was transferred to 96-well PCR plates for PCR screening, and 1/3 was cultured in gelatin-coated 96-well cell culture plates for Southern blot analysis. The remaining 1/3 of the cell suspension was cultured in gelatin-coated 96-well plates, frozen at early passages and subsequently used as nuclear donor cells for SCNT.
PCR screening of gene-edited donor cells
PCR screening for successful SORL1 gene targeting was performed on lysates of individual G418-resistant cell clones with primer pairs F3+R3 and F4+R4 amplifying the 5′ KO- and 3′ KO regions, respectively (see position of primers and primer sequences in Figure 1D and Table S2, respectively). First, G418-resistant cells in the 96-well PCR plates were centrifuged and re-suspended in 25 μL lysis buffer (50 mM KCl, 1.5 mM MgCl2, 10 mM Tris-Cl, pH 8.5, 0.5% Nonidet P40, 0.5% Tween, 400 μg/mL Proteinase K) prior to lysis (65°C for 30 min, 95°C for 10 min). The lysate (3 μL) was subsequently used as template in a PCR screening using a Platinum Pfx DNA polymerase (#11708–013, Thermo Fischer Scientific) using the following PCR conditions: (1) SORL1 5′ KO screening: 1 cycle of 94°C for 2 min, 35 cycles of 94°C for 20 s, 63°C for 30 s, and 68°C for 1.5 min followed by 1 cycle of 68°C for 7 min; (2) SORL1 3′ KO screening: 1 cycle of 94°C for 2 min, 35 cycles of 94°C for 20 s, 56°C for 30 s, and 68°C for 1 min followed by 1 cycle of 68°C for 7 min. Primers used for the SORL1 gene targeting screening are listed in Table S2.
Southern blot analysis
The gene edited SORL1-het Göttingen donor cells, and cloned SORL1 Göttingen piglets, were further validated by Southern blotting using a porcine SORL1-specific probe (887 bp) located upstream of the targeted region. Also, a neor-specific probe (1162 bp), detecting the neor encoding cassette in the targeting vector, was used to examine if the gene edited cell clones and resulting cloned piglets also harbored additional unwanted random integrations of the vector (see Figure S1A). Both Southern blot probes were generated by standard PCR and subjected to random labelling using a Prime-It II Random Primer Labelling Kit (#300385, Agilent) according to the manufacturer’s instructions. Genomic DNA (15 μg) isolated from cultured gene edited fibroblasts, or from ear biopsies taken from newborn piglets, was digested with BlpI restriction enzyme overnight. The digested samples were subjected to gel electrophoresis on a 0.7% agarose gel followed by vacuum blotting onto a nitrocellulose membrane. Pre-hybridization, and hybridization with the individual probes, were carried out at 42°C and all washing procedures were performed at 53°C. Primers for generating the SORL1 and neor probes, respectively, are listed in Table S2.
Cloning and embryo transfer
Two of the validated SORL1 3′ KO/SORL1 5′ KO gene edited cell clones were used as nuclear donor cells for somatic cell nuclear transfer (SCNT) by handmade cloning as previously described.75 Cumulus-oocyte complexes harvested from slaughterhouse-derived sow ovaries were in-vitro matured and treated to remove cumulus cells and partially zonae pellucidae. The oocytes were bisected manually, and the cytoplasts without chromatin were collected. Each cytoplast was first attached to one nuclear donor cell before being fused (BTX microslide 0.5 mm fusion chamber, model 450; BTX San Diego, US). After 1 h of incubation, each cytoplast-donor cell pair was fused with an additional cytoplast creating the reconstructed embryo. All reconstructed embryos were then incubated in culture medium for 5–6 days after which the blastocysts and morulae were selected based on morphology. Two pools of cloned gene edited embryos were prepared, and 82 and 90 blastocysts/morulae, respectively, were transferred surgically into two recipient landrace surrogate sows.76 Pregnancy was diagnosed in both sows by ultrasonography after approx. 25 days. Farrowing was hormonally initiated at day 114 by intra-muscularly injected prostaglandin (Estrumate, 2 mL/sow), and the sows farrowed 8 live piglets (7 and 1, respectively) in total. All piglets, apart from one, died post-natally or over the next few weeks. The surviving piglet was genotyped as SORL1 −/− (SORL1-ko) demonstrating that the donor cell clone used for SCNT was not derived from a single gene targeted cell. Further examination of genomic DNA isolated from this SORL1-ko piglet showed, an on-target, but only partial, gene targeting obstructing one allele, whereas a large deletion was found on the other allele possibly induced by non-homologous end-joining of the free DNA ends resulting from CRISPR-mediated double-strand cleavage.
Fibroblasts were isolated from ear biopsies taken on the day of birth from all cloned piglets. SORL1+/− fibroblasts isolated from one of these piglets were used for re-cloning upon sequence validation following the same protocol as described above. Sixty-eight and 69 re-cloned embryos were transferred to two surrogate sows, respectively. Both were diagnosed pregnant, but one aborted later. The remaining pregnant sow gave birth to 9 piglets, of which 6 were alive. These cloned (F0) piglets were genotyped as SORL1-het by PCR and gene targeting was validated by Southern blotting (Figures S1A and S1B). Two of these 6 cloned founder piglets survived the post-natal period and were, upon sexual maturity, mated with wild-type Göttingen Minipigs boars (referred to as “breeding boars” in Table S1) resulting in two naturally bred F1 litters of het and wt SORL1 piglets (het, n = 6 and wt, n = 4 in total). The surviving cloned (F0) SORL1-ko minipig was included as a control in the study. The 2 breeding boars and the 4 F1 wt animals were used as controls in addition to 6 naturally bred wild-type Göttingen Minipigs obtained from Ellegaard Göttingen Minipigs A/S.
Genotyping
Genotyping of newborn cloned and naturally bred SORL1-het and SORL1-wt Göttingen Minipigs were performed on genomic DNA extracted from ear biopsies, or from fibroblasts derived from these, by standard PCR using a Platinum Superfi DNA polymerase (#12351010, Thermo Fischer Scientific) and primer sets for detecting the 5′- and -3′-SORL1 KO region, respectively, in addition to a primer set detecting the wild-type SORL1 gene. The following PCR conditions were employed: 1 cycle of 98°C for 30 s, 35 cycles of 98°C for 10 s, 60–66°C (depending on the primer set) for 10 s, and 72°C for 30 s followed by 1 cycle of 72°C for 7 min. Primers utilized for genotyping are listed in Table S2.
CRISPR off-target analysis
The online CRISPR RGEN Cas-OFFinder algorithm77 was used for identifying potential off-target sites for the employed pSORL1 sgRNA1. In addition to the targeted pSORL1 gene, eight potential off-target sites residing in annotated genes on chromosomes 2 (JUNB and ARHGAP26), 5 (XRCC6), 6 (GSE1), 8 (PCDH7), 9 (HEPACAM), 14 (TXNRD2) and 15 (TWIST2) were identified when allowing for up to 3 mismatches between the sgRNA and genomic sequence. These potential off-target regions were amplified by standard PCR using genomic DNA isolated from wt or cloned het SORL1 Göttingen Minipigs, Platinum Pfx DNA polymerase (#11708–013, Thermo Fischer Scientific), and primer pairs for the specific genomic region. The resulting amplicons, comprising the sequence region surrounding the sgRNA binding site, were purified using a Nucleospin Gel and PCR Clean-up kit (#740609, Macherey-Nagel) and subjected to DNA sequencing to verify if off-target activity had occurred.
Genomic DNA isolated from cloned het SORL1 Göttingen Minipigs was in addition analyzed for potential unwanted random integration of the plasmid constructs used for co-transfection (sgRNA, hCas9) by standard PCR using primer sets specific for the individual plasmids used for transfection. Primers used for off-target and random integration analyses are shown in Table S2.
Sampling of cerebrospinal fluid and tissues
The SORL1 Göttingen Minipigs were anesthetized with Zoletil-mix (1 mL/10 kg) prior to placing the animals in sternal recumbency with the neck flexed, and the relevant part of the neck region was surgically prepared. The anatomical landmarks were the occipital protuberance and the two lateral sides of the atlas wings. A spinal needle (BD 20 Gauge 3.50 in.) was passed perpendically down to the atlanto-occipital intervertebral space and stopped when a weak reflex from the minipig was felt. The minipig was then carefully turned to right lateral recumbency and the cerebrospinal fluid (CSF) was collected by gravity and capillary action. Apart for 4 female F1 minipigs (6469, 6470, 6475, and 6477), which were subjected to CSF sampling twice, all minipigs were euthanized immediately after CSF sampling with an intravenous injection of 30% pentobarbital (0.25 mL/kg). For all minipigs, including the 4 female F1 animals mentioned above, CSFs from the first sampling were used for analyses. CSF from one of the cloned SORL1-het animals (6402) was, however, excluded from the analyses due to contamination with blood during sampling. The brains were immediately and carefully removed from the scull and halved with a scalpel. One-half was immersed in 4% phosphate-buffered formaldehyde for 2 weeks, after which formaldehyde was replaced with PBS prior to storage at 4°C. The remaining half was used for dissection of relevant brain regions and subsequent snap-freezing of these tissues in liquid nitrogen.
RT-PCR analyses
For RT-PCR analysis of porcine SORL1 transcripts, total RNA was isolated from cortex, hippocampus and cerebellum from a wild-type Göttingen Minipig using an RNeasy Micro Kit (#74004, Qiagen). cDNA was produced using 1 μg of total RNA and a RevertAid First Strand cDNA Synthesis Kit (#K1622, Thermo Fischer Scientific) prior to performing RT-PCR analyses to validate the presence of the reference Sscrofa 11.1 SORL1 -202 transcript. RT-PCR analyses were performed using GoTaq DNA polymerase (#M7841, Promega) in a reaction volume of 25 μL comprising 1 μL of cDNA and primer pairs specific for the 5′ end (exon 1–3, see primers F1+R1 in Figure 1D) and 3′ end (exon 46–47), respectively, of the SORL1-202 transcript or with primers specific for the reference gene GAPDH. The following PCR conditions were employed: 95°C for 2 min for 1 cycle followed by 35 cycles of 95°C for 30 s; 58°C/60°C/65°C (depending on the primer set) for 30 s, 72°C for 30 s followed by 1 cycle of 72°C for 7 min. The resulting SORL1-202 amplicons were spin-column purified using a Nucleospin Gel and PCR Clean-up Kit (#740609, Macherey-Nagel) prior to validation by DNA sequencing. Sequences of primers used for the RT-PCR analyses are provided in Table S2.
For analysis of the SORL1 transcript in the generated gene-edited Göttingen Minipigs, cerebellum and cortex samples were dissected from the ko and a randomly selected wt and het SORL1 minipig, respectively, and RNA was extracted using the RNeasy Mini Kit (Qiagen, USA). 1 μg of total RNA was subsequently converted to cDNA using High Capacity RNA-to-cDNA Kit (#4387406, Applied Biosystems, USA) following manufacturer’s instructions. The resulting cDNA was used as template for amplification of porcine SORL1, using primer pairs located in the 5′ end (exon 1–2) and in the 3′ end (exon 46–47) of the transcript. GAPDH served as control for successful cDNA synthesis whereas samples without inclusion of reverse transcriptase (-RT) and water were used as negative controls. PCR was performed on a Veriti Thermal Cycler (Applied Biosystems) with Herculase II Fusion DNA Polymerase (#600675, Agilent) according to the following optimized conditions: 95°C for 1 min, 30 cycles of amplification (95°C for 20 s, 65°C for 20 s, 68°C for 1 min), and final extension at 68°C for 4 min.
For RT-PCR analysis of MAPT, APP and genes involved in APP processing, RNA was extracted from cortex samples dissected from the ko and a randomly selected wt and het SORL1 minipig, respectively, using a NucleoSpin RNA Plus kit (#740984.50, Macherey-Nagel). cDNA was synthesized using 1 μg of total RNA and a RevertAid First Strand cDNA Synthesis kit (#K1622, Thermo Scientific) according to the manufacturer's instructions. The resulting cDNA (1 μL) was used as template for amplification of porcine APP (103 bp), ADAM10 (186 bp), ADAM17 (173 bp), BACE1 (171 bp), PSEN1 (118 bp), PSEN2 (140 bp), and MAPT (162 bp) sing a GoTaq G2 DNA polymerase (#M7845, Promega) and the following optimized conditions: 95°C for 2 min, 35 cycles of amplification (95°C for 30 s, 58°C for 30 s, 72°C for 20 s), and final extension at 72°C for 7 min. Also here, GAPDH (with an amplicon of 218 bp) served as control for successful cDNA synthesis whereas samples without inclusion of reverse transcriptase (-RT) and water were used as negative controls. All primers used for RT-PCR are listed in Table S2.
Quantitative PCR analysis
Total RNA was purified from cerebellum and cortex tissue isolated from all the wt and het SORL1 Göttingen minipigs (wt, n = 4; het, n = 4) subjected to PET- and MRI imaging, respectively, using a NucleoSpin RNA Plus kit (#740984.50, Macherey-Nagel). cDNA was synthesized from 1 μg of purified total RNA using a RevertAid First Strand cDNA Synthesis kit (#K1622, Thermo Scientific) according to the manufacturer's instructions. For quantitative PCR analysis of porcine SORL1, cDNA samples (2 μL cDNA) were in duplicates subjected to qPCR amplification using an iTaq Universal SYBR Green Supermix (2×) (#1725120, BIO-RAD) on a CFX Opus 96 Real-Time PCR System (BIO-RAD). Standard curves were generated individually and in duplicate for each tissue by diluting cDNA from cortex or cerebellum successively and applying it into separate wells. The qPCR protocol consisted of an initial polymerase activation/denaturation at 95°C for 30 s, followed by 40 cycles of denaturation (5 s at 95°C), annealing and extension (30 s at 60°C). The primer set used in RT-PCR for amplification of 3′ end of the porcine SORL1 transcript was used to amplify SORL1, whereas HPRT1 served as reference gene. All primers used for qPCR are listed in Table S2. SORL1 mRNA expression levels were calculated relative to the reference gene HPRT1 and shown relative to the normalized SORL1 mRNA levels in wt pigs.
Immunoprecipitation (IP)
CSF samples from the SORL1 Göttingen minipigs, subjected to PET-and MRI-imaging (het, n = 4; wt, n = 4) were analyzed by immunoprecipitation and Western blot. Samples (150 μg total protein) were incubated with a primary in-house polyclonal rabbit anti-SORLA antibody (pAb_5387)78 overnight at 4°C, followed by incubation with Gammabind G Sepharose beads (GE Healthcare, #17-0885-01) for 6 hs at 4°C. Beads were then washed with cold PBS, before the final elution of IP products from the beads using Nupage LDS Sample buffer 4X (Invitrogen, #NP0007) added with β-mercaptoethanol. Samples were subsequently analyzed by Western blot.
Equal volumes of precipitated proteins from CSF were loaded on Nupage 4–12% Bis-Tris gel (Invitrogen) and separated by SDS-PAGE, prior to transfer of proteins onto a nitrocellulose membrane (Biorad). Immunoblotting using a mouse anti-LR11 primary antibody (BD Transduction Laboratories, #612633) for detection of SORLA was performed overnight at 4°C followed by incubation with anti-mouse HRP secondary antibody for 1 h at RT. Detection of proteins and image acquisition were performed using the iBright Western Blot Imaging system (Invitrogen). Quantification of sSORLA signals in het and wt samples was performed using ImageJ.
Western blotting
Following dissection of cerebellum and cortex samples from the wt and het SORL1 Göttingen minipigs subjected to the PET-and MRI-imaging, or from randomly selected wt, het and the ko SORL1 minipigs, tissues were homogenized and lysed. SDS-PAGE separation of an equal amount of proteins from either tissue or CSF samples was performed using 4–12% NuPAGE Tris-Acetate gels (Invitrogen). Subsequently, proteins were transferred to nitrocellulose membranes (Amersham) and blocking of unspecific binding was performed for 1 h at room temperature in blocking buffer (TST buffer 20% (Tris-base 0.25 M, NaCl 2.5 M, 0.5% Tween 20, pH 9.0), 2% skimmed milk powder, and 2% Tween 20). The membranes were incubated overnight at 4°C with the following primary antibodies: mouse anti-LR11 (1:500) for SORLA detection, mouse-anti-tau 5E2 for tau detection (1:1000, Millipore, #05-348), rabbit anti-VPS26b (1:1000, Abcam, #ab236966), rabbit anti-VPS26a (1:1000, Abcam rabbit, #ab23892), rabbit anti-APP1227 (1:1000, polyclonal in-house antibody),26 and with mouse anti-actin (1:1000, #A5441, Sigma) as loading control. Next, the membrane was washed three times for 5 min in washing buffer (CaCl2 0.2 mM, MgCl2 0.1 mM, HEPES 1 mM, NaCl 14 mM, 0.2% skimmed milk powder, 0.05% Tween 20) and incubated with HRP-conjugated secondary antibody (1:1500, #P0260, Agilent) for 1 h at RT. Proteins were finally detected with ECL detection reagent (GE Healthcare Amersham) and visualized with iBright Western Blot Imaging system.
MSD immunoassays
Aβ peptide panel 1 V-Plex assay
The Aβ standards (Aβ1-38, Aβ1-40 and Aβ1-42) were mixed and diluted in Diluent 35 in 4-fold serial dilutions as instructed from the manufacturer (MSD, #K15200E). The precoated plate was blocked in Diluent 35 for 1 h at RT followed by four times washing. Anti-Aβ 4G8 detection antibody and standards or CSF samples (10 μL from CSF samples with equal protein amounts) were added simultaneously to the wells, and incubated at RT for 2 hs. Lastly, the wells were washed four times and 2X Read Buffer T was added for immediate analysis. Electrochemiluminescent signals were obtained on a SECTOR® Imager (MSD) and the Aβ concentrations were calculated in Microsoft Excel by fitting the samples to a standard curve. All washing steps were performed in 1X PBS +0.05% Tween 20.
sAPPα/sAPPβ duplex assay
A serial dilution of the combined sAPPα and sAPPβ standards was prepared by diluting them in a 1% MSD Blocker A solution following instructions from the manufacturer (MSD, #K15120E). The precoated sAPPα/sAPPβ plate was blocked in 3% Blocker A solution for 1 h at RT followed by four times washing. CSF samples and standards were added to the plate and incubated for 1 h at RT. Subsequently the plate was washed four times and APP detection antibody provided with the kit was added to the wells. Finally, the plate was washed four times and incubated in Read Buffer T for 10 min at RT. The plate was read with a SECTOR® Imager (MSD) and the sAPPα and sAPPβ concentrations in the CSF were calculated by fitting the sample values to a standard curve using Microsoft Excel. All washing steps were performed in 1X Tris Wash Buffer.
Mouse total tau assay
The Tau441 standard was diluted in 10% MSD Blocker A in 4-fold serial dilutions following the manufacturer’s protocol (MSD, #K151DSD). The precoated mouse total Tau plate was incubated in Blocker A solution for 1 h at RT and subsequently washed four times. Standards and CSF samples were added to the plate and incubated at RT for 1 h followed by four washes. Next, anti-Tau detection antibody provided with the kit was added to the wells and incubated for 1 h at RT. Finally, the plate was washed four times and Read Buffer T was added for immediate scanning of the plate using a SECTOR ® Imager (MSD). Tau CSF concentrations were calculated by fitting the samples to a standard curve using Microsoft Excel. All washing steps were performed in 1X Tris Wash Buffer.
Immunohistochemistry/immunocytochemistry
For the SORLA expression analysis in porcine neurons, paraffin embedded brain tissue was sectioned on a microtome (Leica RM2155) with 5 μm thickness followed by heating at 60°C for 1 h. Sections of tissue were then deparaffinated in a xylene substitute (#109843, Neo-Clear, Merck) followed by dehydration in a series of ethanol (absolute to 70%) and rinsed in TBS (Tris buffered saline, pH 7.8) for 5 min. at room temperature. The DAKO Real™ EnVision™ System (#K4010, Dako) was used for the immunostaining. The sections were treated with an antigen retrieval solution (Target Retrieval pH 9, #S2367, DAKO) for 20 min. in a microwave and rinsed in TBS for 5 min at room temperature. The antigen retrieval was followed by incubation with peroxidase block for 5 min at room temperature and rinsed in TBS for 5 min. at room temperature. The sections were then incubated with a primary in-house polyclonal rabbit anti-SORLA antibody (pAb_5387) at 4°C over night and rinsed in TBS for 5 min at room temperature. The sections were incubated with secondary antibody (Peroxidase Labelled Polymer Anti-rabbit; #P0217, Agilent) for 30 min. at room temperature and rinsed in TBS for 5 min. at room temperature. For detection, a solution of Liquid DAB+ Chromogen in Substrate Buffer was used. The sections were incubated for 10 min. at room temperature, rinsed in TBS for 5 min at room temperature followed by staining with hematoxylin (#MHS16, Sigma Aldrich) for 3 min. at room temperature. The sections were then washed in deionized water (ddH2O), dehydrated in a series of ethanol (50% to absolute) and treated with a xylene substitute (#109843, Neo-Clear, Merck). Cover glass were mounted with Eukitt mounting medium (#03989, Sigma Aldrich) and images were acquired using a standard bright-field microscope (Zeiss Apotome) (Figure 1A).
The analysis of endosome size in porcine neurons was carried out on 5 μm paraffin embedded brain sections using a standard DAB (3,3′-diaminobenzidine) immunostaining protocol. Paraffin sections were melted at 60°C for 30 min followed by deparaffination in xylene (Neoclear, Merck, #109843) and rehydration in ethanol (100%–70%) in a series of steps. Antigen retrieval was performed in prewarmed citrate buffer pH 6 (Sigma, #C9999) for 10 min at RT. The slides were treated with PBS supplemented with hydrogen peroxide (H2O2) for 10 min at RT to block endogeneous peroxidases. After washing the sections were blocked in 5% normal goat serum for 1 h at RT and incubated in rabbit anti-Rab5 (1:200, Abcam, #ab109534) primary antibody O.N at 4°C. The slides were washed and incubated in biotin-conjugated secondary antibody (1:100, anti-rabbit, Sigma, #B8895) for 2 hs at RT followed by washing in PBS +0.3% Triton X-100 (PBST). Treatment in Extra-Avidin peroxidase (1:200, Sigma, #E2886) was performed for 1 h at RT followed by sequential washes in PBS and PBST. The sections were then incubated in Tris-Base pH 7.2 for 10 min at RT and detected with a solution of 0.05% DAB supplemented with 0.03% H2O2 pH 7.2 for 5 min. Subsequently, they were counterstained with hematoxylin (Vector Labs, #H-3401-500) for 10 min and washed extensively in running tap water followed by dehydration in ethanol and xylene. Finally, the slides were mounted using Eukitt mounting medium (Sigma, #25608-33-7) and imaged using a digital slide scanner microscope (Olympus VS120). Magnified views (60×) were extracted using OlyVia software (Olympus) and image processing was performed using ImageJ software. The analysis of endosome size was performed by subtracting background using the rolling-ball algorithm followed by thresholding and watershed algorithms to identify individual puncta. The pixel area of each puncta was calculated and the mean pixel area of each neuron (n = 27 for each genotype) was presented.
For further analysis of early endosomes, primary fibroblasts were isolated from ear biopsies obtained from a wt (6475) and a het (6469) SORL1 minipig, respectively, as described above, and seeded onto coverslips in a 24-well plate. Following culturing for 48 h, cells were fixed with 4% PFA and washed with PBS, before proceeding with staining. Briefly, fibroblasts were washed twice with PBS+0.1% Triton X-100 (PBST), followed by a blocking step in PBS added with 10% fetal bovine serum for 30 min at RT. Fibroblasts were then incubated with primary antibody (rabbit anti-Rab5, 1:100, Abcam, # ab109534; or rabbit anti-SORLA, 1:200, in-house polyclonal pAb_5387 antibody) overnight at 4°C. The day after, cells were washed with PBST and incubated with anti-rabbit Alexa Fluor 488 (Life Technologies, #A21206) for 1 h at RT. Nuclei were counterstained using Hoechst solutionI (Invitrogen, #H3570) diluted at 1:50.000 in water, and coverslips were mounted on glass slides using Dako Fluorescent Mounting Medium (Dako, #S3023).
Images were acquired at Zeiss LSM 710 confocal at 63× magnification, and image analysis and quantification of endosome size was performed using ImageJ (n = 21 per genotype). Individual puncta were identified by applying simple thresholding and watershed algorithm. For each puncta, pixel area was calculated and the mean area of all puncta within a cell was reported.
Neurofilament light polypeptide assay
From the CSF that was collected from anesthetized minipigs at necropsy (10 SORL 1 -wt and 6 SORL1-compromised minipigs (SORL1-het, n = 5 and SORL1-ko, n = 1)), aliquots of 15 μL each were prepared and immediately frozen on dry ice prior to storage at −80°C until analysis for neurofilament light polypeptide (NF-L) levels. NF-L levels were quantified at a 1:50 dilution using the Protein Simple Platform ELLA according to the manufacturer’s instructions (LLOQ: 2.7 pg/mL, ULOQ: 10,290 pg/mL). Parallelism was observed for CSF dilutions between 1:10 and 1:100. For analysis, animals were grouped by genotype (wt versus het + ko), independent of age.
Pathology
Neuropathological examination was performed blinded to the investigator, initially macroscopically on slices of the formalin-fixed brains, and subsequently microscopically on formalin-fixed, paraffin-embedded and hematoxylin-stained brain sections from the frontal, temporal and occipital lobes, from the brain stem (mesencephalon, pons and myelencephalon) and cerebellum obtained from 2 randomly selected F1 female minipigs (1 SORL1-het and 1 SORL1-wt) at 5 months of age, respectively.
To evaluate presence of neuropathological changes such as neurofibrillary tangles and amyloid plaques, 4 μm thick sections containing frontal lobe and hippocampus, obtained from 29–30 mo old female (n = 4) and male (n = 4) SORL1 minipigs subjected to MRI-scanning (wt, n = 4; het, n = 4), were stained with antibodies for tau phosphorylated at Thr-231 (AB9668, 1:2400, Sigma-Aldrich, Germany) and Aβ42 (ab10148, 1:100, Abcam, UK) on the automated staining platform Ventana Discovery Ultra (Ventana Medical Systems, Tucson, AZ, USA). Antigen demasking was achieved by heat induced epitope retrieval in Cell Conditioning Buffer 1 (CC1, Ventana Medical Systems, USA) at 95°C for 8 min for anti-tau and for 48 min for anti-Aβ42. In addition, for Aβ42 antigen retrieval, the sections were also incubated for 10 min in 80% formic acid. A positive immunoreactivity was detected with DISCOVERY Amplification anti-HQ Horseradish Peroxidase Multimer (Ventana Medical Systems) and 3,3′-Diaminobenzidin (Ventana Medical Systems). Subsequently, the slides were counterstained in hematoxylin and coverslipped with mounting medium (Pertex). To ensure validity of the IHC stainings, one section from an AD patient containing hippocampus was included in every run as an external control.
[18F]-FDG and [11C]-PIB-PET imaging
Four 21-month old female minipigs (wt, n = 2; het, n = 2) were randomly selected and subjected to PET neuroimaging with [18F]-FDG and [11C]-PIB at the Department of Nuclear Medicine and PET at Aarhus University Hospital. The procedure was performed blinded to the investigators conducting the preparation and PET imaging of the animals. The 16 h fasted minipigs were premedicated with an intramuscular injection of 1.3 mg/kg midazolam and 6.3 mg/kg s-ketamine. Anaesthesia was then induced through an ear vein catheter with 1.3 mg/kg midazolam and 3.1 mg/kg s-ketamine. After intubation, the anaesthesia was maintained with 2.0–2.1% isoflurane. The pigs were mechanically ventilated 15 times per minute with approximately 8 mL/kg of a 1: 2.2 oxygen-medical air mixture. The minipigs were placed prone with their heads in the center of the field of view of a Siemens Biograph Truepoint PET/CT system. A CT scan for attenuation correction of PET data was acquired. For [18F]-FDG, a dose of 5MBq/kg of was then injected via the ear vein and 45 min later, a 30-min PET scan was acquired. The injected dose in the 4 pigs ranged from 170–216MBq depending on bodyweight. Similarly, on a separate day, 138–204MBq of [11C]-PIB was injected into the ear vein and 90 min dynamic imaging began at tracer injection. Both tracers were administered in 20 mls of saline and then flushed with 10 mls of saline. After [11C]-PIB injection, the catheter was removed to avoid scanner artifacts. PET images were reconstructed using the iterative TrueX algorithm (Siemens), and CT and PET data were fused by the system. PET scans were then co-registered to an average MRI of the minipig brain using PMOD version 4.0 and regional standard uptake values were obtained and normalized to whole brain activity.
Magnetic resonance imaging
The four female 22-month old minipigs (wt, n = 2; het, n = 2), already subjected to PET scanning, and four randomly selected 27-month old male minipigs (wt, n = 2; het, n = 2) were subsequently subjected to Magnetic Resonance Imaging (MRI). The procedure was performed blinded to the investigators conducting the preparation and MRI imaging of the animals. The pigs were prepared in the same ways as prior to PET imaging, but instead of isoflurane, the anaesthesia was maintained with intravenous propofol (approximately 8 mg/kg/h) infusion. MRI was carried out on a 3T scanner (MR750, GE Healthcare) in one session. We obtained structural proton images and DTI for assessment of volumes and diffusivity using a flexible 16-channel coil (GE Healthcare). Anatomy and volumes were evaluated using T1-weighted (TR/TE = 14/500 ms, 0.8 × 0.8 × 1.4 mm3) and T2-weighted (TR/TE/TI = 6300/103/1750 ms, 0.8 × 0.8 × 1.4 mm3) fluid attenuation inversion recovery (FLAIR) 3D imaging. Further, we performed DTI (TR/TE = 8000/65 ms, 30 directions, b = 1000 s/mm2, 2.1 × 1.9 × 4 mm3), which was processed and analyzed using FSL (Analysis Group, FMRIB, Oxford).79
Quantification and statistical analysis
Statistical analyses were performed using the GraphPad Prism version 6.0 for windows (GraphPad software). For comparison between two different datasets a two-tailed unpaired Student's t-test was used. p values below 0.05 were considered significant. The statistical details of each experiment can be found in the figure legends, including the statistical test used and the exact number of animals or cells analyzed. In all figures, data are expressed as means with error bars representing standard errors of the mean.
Acknowledgments
We thank Kjeld Dahl Winther (SEGES, Denmark) for excellent veterinary assistance during CSF sampling. We are also grateful to Bert Vogelstein and Kenneth W. Kinzler (Johns Hopkins University, Baltimore, MD) for kindly providing the pNeDaKO plasmid vector. We also thank Trine S. Petersen, Lisa Maria Røge, Dorte Qualmann, Anette M. Pedersen, Janne Adamsen, Klaus Villemoes, Adrian Zeltner, Michelle Sørensen, Sandra Bonnesen, and Benedicte Vestergaard for skilled technical assistance and Anne Mette V. Toft, Mette Bak, and Martin A. Fredsted for skillful assistance with animal handling and transport to scanning facilities. We are grateful to Asad Jan and Ken P. Kragsfeldt for help with microscopy analysis and illustrations, respectively. This study was carried out in accordance to the ARRIVE guidelines and was supported by grants to C.B.S. from the Lundbeck Foundation (R100-A9209), The Danish Heart Association (16-R107-A6813-22997), the Ellegaard Göttingen Minipigs Research Foundation, and Ellegaard Göttingen Minipigs A/S.
Author contributions
O.M.A. performed initial cloning of targeting constructs, conducted analysis on CSF, collected animal samples, and performed WB experiments, preparation of illustrations, and interpretation of data. N.B., A.K.O.A., E.S.S.H., and C.L. performed MRI analyses. A.M.L., A.K.O.A., and D.J.B. conducted PET analyses. G.G.P. genotyped animals, performed CRISPR off-target analysis, and established primary fibroblast cultures from Göttingen minipig ear biopsies. A.M.G.J. performed MSD assays for CSF samples and IHC for brain tissue. G.M. performed RT-PCR, WB analysis for brain homogenates, and ICC analysis on cultured fibroblasts. B.P.U. performed neuropathological analysis of the 5-month-old animals. J.R.N. consulted on the neuropathological analysis. K.R.J. supervised selection of control Göttingen minipigs and control tissue to include in the study. M.M.J. and I.E.H. dissected minipig brains and performed neuropathological analysis for amyloid plaques and fibrillary tangles. M.L.K. performed IHC on brain tissue. C.E.T. consulted on CSF AD biomarkers. L.B. and M.D. conducted the neurofilament light analyses on CSF. Y.L. performed manual cloning and culturing of the reconstructed embryos. H.S.P. and H.C. performed embryo transfer and supervised delivery and monitoring of the cloned piglets. Y.L. provided vectors for construction of sgRNA and C-check plasmids. L.B. consulted on the initial study design. S.A.S. consulted on data interpretation. L.F.M. provided Göttingen minipig fibroblasts for genetic modification and control animals for the study. C.B.S. constructed sgRNA vectors; generated the genetically modified donor cells for cloning; conducted southern blot analyses, animal sampling, genotyping, RT-PCR, and qPCR analyses on brain tissues; prepared illustrations; and interpreted data. O.M.A. and C.B.S. conceptualized and supervised the entire study and wrote the manuscript together with S.A.S.
Declaration of interests
L.B. and M.D. are employees of AbbVie and own AbbVie stock. AbbVie participated in the design and study conduct for this research as well as in the interpretation of data, review, and approval of the publication. Ellegaard Göttingen Minipigs A/S has the commercialization rights to the genetically altered Göttingen minipig SORL1 model. S.A.S. is a co-founder of Retromer Therapeutics, has equity in the company, and is a paid consultant to the company. O.M.A. also has commercial interests in Retromer Therapeutics, but the company was not involved in any aspects of the study.
Published: September 12, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2022.100740.
Contributor Information
Olav M. Andersen, Email: o.andersen@biomed.au.dk.
Charlotte B. Sørensen, Email: cbs@clin.au.dk.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Cacace R., Sleegers K., Van Broeckhoven C. Molecular genetics of early-onset Alzheimer's disease revisited. Alzheimers Dement. 2016;12:733–748. doi: 10.1016/j.jalz.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 2.Scheltens P., De Strooper B., Kivipelto M., Holstege H., Chételat G., Teunissen C.E., Cummings J., van der Flier W.M. Alzheimer's disease. Lancet. 2021;397:1577–1590. doi: 10.1016/S0140-6736(20)32205-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jensen M., Schröder J., Blomberg M., Engvall B., Pantel J., Ida N., Basun H., Wahlund L.O., Werle E., Jauss M., et al. Cerebrospinal fluid A beta42 is increased early in sporadic Alzheimer's disease and declines with disease progression. Ann. Neurol. 1999;45:504–511. doi: 10.1002/1531-8249(199904)45:4<504::aid-ana12>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 4.Bateman R.J., Xiong C., Benzinger T.L.S., Fagan A.M., Goate A., Fox N.C., Marcus D.S., Cairns N.J., Xie X., Blazey T.M., et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N. Engl. J. Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maia L.F., Kaeser S.A., Reichwald J., Lambert M., Obermüller U., Schelle J., Odenthal J., Martus P., Staufenbiel M., Jucker M. Increased CSF Abeta during the very early phase of cerebral Abeta deposition in mouse models. EMBO Mol. Med. 2015;7:895–903. doi: 10.15252/emmm.201505026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nixon R.A. Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer's disease: inseparable partners in a multifactorial disease. FASEB. J. 2017;31:2729–2743. doi: 10.1096/fj.201700359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Small S.A., Gandy S. Sorting through the cell biology of Alzheimer's disease: intracellular pathways to pathogenesis. Neuron. 2006;52:15–31. doi: 10.1016/j.neuron.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Small S.A., Kent K., Pierce A., Leung C., Kang M.S., Okada H., Honig L., Vonsattel J.P., Kim T.W. Model-guided microarray implicates the retromer complex in Alzheimer's disease. Ann. Neurol. 2005;58:909–919. doi: 10.1002/ana.20667. [DOI] [PubMed] [Google Scholar]
- 9.Muhammad A., Flores I., Zhang H., Yu R., Staniszewski A., Planel E., Herman M., Ho L., Kreber R., Honig L.S., et al. Retromer deficiency observed in Alzheimer's disease causes hippocampal dysfunction, neurodegeneration, and Abeta accumulation. Proc. Natl. Acad. Sci. USA. 2008;105:7327–7332. doi: 10.1073/pnas.0802545105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rogaeva E., Meng Y., Lee J.H., Gu Y., Kawarai T., Zou F., Katayama T., Baldwin C.T., Cheng R., Hasegawa H., et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat. Genet. 2007;39:168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raghavan N.S., Brickman A.M., Andrews H., Manly J.J., Schupf N., Lantigua R., Wolock C.J., Kamalakaran S., Petrovski S., Tosto G., et al. Whole-exome sequencing in 20, 197 persons for rare variants in Alzheimer's disease. Ann. Clin. Transl. Neurol. 2018;5:832–842. doi: 10.1002/acn3.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holstege H., Hulsman M., Charbonnier C., Grenier-Boley B., Quenez O., Drozeva D., et al. Exome sequencing identifies novel AD-associated genes. Preprint at medRxiv. 2020 doi: 10.1101/2020.07.22.20159251. [DOI] [Google Scholar]
- 13.Fjorback A.W., Seaman M., Gustafsen C., Mehmedbasic A., Gokool S., Wu C., Militz D., Schmidt V., Madsen P., Nyengaard J.R., et al. Retromer binds the FANSHY sorting motif in sorLA to regulate amyloid precursor protein sorting and processing. J. Neurosci. 2012;32:1467–1480. doi: 10.1523/JNEUROSCI.2272-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rohe M., Hartl D., Fjorback A.N., Klose J., Willnow T.E. SORLA-mediated trafficking of TrkB enhances the response of neurons to BDNF. PLoS One. 2013;8:e72164. doi: 10.1371/journal.pone.0072164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pietilä M., Sahgal P., Peuhu E., Jäntti N.Z., Paatero I., Närvä E., Al-Akhrass H., Lilja J., Georgiadou M., Andersen O.M., et al. SORLA regulates endosomal trafficking and oncogenic fitness of HER2. Nat. Commun. 2019;10:2340. doi: 10.1038/s41467-019-10275-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simoes S., Guo J., Buitrago L., Qureshi Y.H., Feng X., Kothiya M., Cortes E., Patel V., Kannan S., Kim Y.H., et al. Alzheimer's vulnerable brain region relies on a distinct retromer core dedicated to endosomal recycling. Cell Rep. 2021;37:110182. doi: 10.1016/j.celrep.2021.110182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mishra S., Knupp A., Szabo M.P., Williams C.A., Kinoshita C., Hailey D.W., Wang Y., Andersen O.M., Young J.E. The Alzheimer's gene SORL1 is a regulator of endosomal traffic and recycling in human neurons. Cell. Mol. Life Sci. 2022;79:162. doi: 10.1007/s00018-022-04182-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pensalfini A., Kim S., Subbanna S., Bleiwas C., Goulbourne C.N., Stavrides P.H., Jiang Y., Lee J.H., Darji S., Pawlik M., et al. Endosomal dysfunction induced by directly overactivating Rab5 recapitulates prodromal and neurodegenerative features of Alzheimer's disease. Cell Rep. 2020;33:108420. doi: 10.1016/j.celrep.2020.108420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Small S.A., Petsko G.A. Endosomal recycling reconciles the Alzheimer's disease paradox. Sci. Transl. Med. 2020;12:eabb1717. doi: 10.1126/scitranslmed.abb1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burrinha T., Martinsson I., Gomes R., Terrasso A.P., Gouras G.K., Almeida C.G. Upregulation of APP endocytosis by neuronal aging drives amyloid-dependent synapse loss. J. Cell Sci. 2021;134:jcs255752. doi: 10.1242/jcs.255752. [DOI] [PubMed] [Google Scholar]
- 21.Small S.A., Petsko G.A. Retromer in Alzheimer disease, Parkinson disease and other neurological disorders. Nat. Rev. Neurosci. 2015;16:126–132. doi: 10.1038/nrn3896. [DOI] [PubMed] [Google Scholar]
- 22.Simoes S., Neufeld J.L., Triana-Baltzer G., Moughadam S., Chen E.I., Kothiya M., Qureshi Y.H., Patel V., Honig L.S., Kolb H., Small S.A. Tau and other proteins found in Alzheimer's disease spinal fluid are linked to retromer-mediated endosomal traffic in mice and humans. Sci. Transl. Med. 2020;12:eaba6334. doi: 10.1126/scitranslmed.aba6334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kovtun O., Leneva N., Bykov Y.S., Ariotti N., Teasdale R.D., Schaffer M., Engel B.D., Owen D.J., Briggs J.A.G., Collins B.M. Structure of the membrane-assembled retromer coat determined by cryo-electron tomography. Nature. 2018;561:561–564. doi: 10.1038/s41586-018-0526-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bugarcic A., Zhe Y., Kerr M.C., Griffin J., Collins B.M., Teasdale R.D. Vps26A and Vps26B subunits define distinct retromer complexes. Traffic. 2011;12:1759–1773. doi: 10.1111/j.1600-0854.2011.01284.x. [DOI] [PubMed] [Google Scholar]
- 25.Kwart D., Gregg A., Scheckel C., Murphy E.A., Paquet D., Duffield M., Fak J., Olsen O., Darnell R.B., Tessier-Lavigne M. A large panel of isogenic APP and PSEN1 mutant human iPSC neurons reveals shared endosomal abnormalities mediated by APP beta-CTFs, not Abeta. Neuron. 2019;104:256–270.e5. doi: 10.1016/j.neuron.2019.07.010. [DOI] [PubMed] [Google Scholar]
- 26.Andersen O.M., Reiche J., Schmidt V., Gotthardt M., Spoelgen R., Behlke J., von Arnim C.A.F., Breiderhoff T., Jansen P., Wu X., et al. SorLA/LR11, a neuronal sorting receptor that regulates processing of the amyloid precursor protein. Proc. Natl. Acad. Sci. USA. 2005;102:13461–13466. doi: 10.1073/pnas.0503689102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andersen O.M., Rudolph I.M., Willnow T.E. Risk factor SORL1: from genetic association to functional validation in Alzheimer's disease. Acta Neuropathol. 2016;132:653–665. doi: 10.1007/s00401-016-1615-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knupp A., Mishra S., Martinez R., Braggin J.E., Szabo M., Kinoshita C., Hailey D.W., Small S.A., Jayadev S., Young J.E. Depletion of the AD risk gene SORL1 selectively impairs neuronal endosomal traffic independent of amyloidogenic APP processing. Cell Rep. 2020;31:107719. doi: 10.1016/j.celrep.2020.107719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hung C., Tuck E., Stubbs V., van der Lee S.J., Aalfs C., van Spaendonk R., Scheltens P., Hardy J., Holstege H., Livesey F.J. SORL1 deficiency in human excitatory neurons causes APP-dependent defects in the endolysosome-autophagy network. Cell Rep. 2021;35:109259. doi: 10.1016/j.celrep.2021.109259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nixon R.A. Endosome function and dysfunction in Alzheimer's disease and other neurodegenerative diseases. Neurobiol. Aging. 2005;26:373–382. doi: 10.1016/j.neurobiolaging.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 31.Villadsen J., Hansen H.D., Jørgensen L.M., Keller S.H., Andersen F.L., Petersen I.N., Knudsen G.M., Svarer C. Automatic delineation of brain regions on MRI and PET images from the pig. J. Neurosci. Methods. 2018;294:51–58. doi: 10.1016/j.jneumeth.2017.11.008. [DOI] [PubMed] [Google Scholar]
- 32.Wernersson R., Schierup M.H., Jørgensen F.G., Gorodkin J., Panitz F., Staerfeldt H.H., Christensen O.F., Mailund T., Hornshøj H., Klein A., et al. Pigs in sequence space: a 0.66X coverage pig genome survey based on shotgun sequencing. BMC Genom. 2005;6:70. doi: 10.1186/1471-2164-6-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen X.H., Siman R., Iwata A., Meaney D.F., Trojanowski J.Q., Smith D.H. Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am. J. Pathol. 2004;165:357–371. doi: 10.1016/s0002-9440(10)63303-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brzozowska A., Grimholt U., Kulseth M.A., Wold I., Rogne S. The sequence of porcine apolipoprotein E (APOE) cDNA. DNA Sequence. 1993;4:207–210. doi: 10.3109/10425179309015633. [DOI] [PubMed] [Google Scholar]
- 35.Ramsoondar J.J., Rucker E.B., Vasquez J.C., Gallagher D.S., Grimm D.R., Lunney J.K., Schook L.B., Piedrahita J.A. Isolation and genetic characterization of the porcine apolipoprotein E gene. Anim. Genet. 1998;29:43–47. doi: 10.1046/j.1365-2052.1998.00273.x. [DOI] [PubMed] [Google Scholar]
- 36.Ayuso M., Buyssens L., Stroe M., Valenzuela A., Allegaert K., Smits A., Annaert P., Mulder A., Carpentier S., Van Ginneken C., Van Cruchten S. The Neonatal and Juvenile pig in pediatric drug discovery and development. Pharmaceutics. 2021;13:44. doi: 10.3390/pharmaceutics13010044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Motoi Y., Aizawa T., Haga S., Nakamura S., Namba Y., Ikeda K. Neuronal localization of a novel mosaic apolipoprotein E receptor, LR11, in rat and human brain. Brain Res. 1999;833:209–215. doi: 10.1016/s0006-8993(99)01542-5. [DOI] [PubMed] [Google Scholar]
- 38.Hermey G., Sjøgaard S.S., Petersen C.M., Nykjær A., Gliemann J. Tumour necrosis factor a-converting enzyme mediates ectodomain shedding of Vps10p-domain receptor family members. Biochem. J. 2006;395:285–293. doi: 10.1042/BJ20051364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tüshaus J., Müller S.A., Kataka E.S., Zaucha J., Sebastian Monasor L., Su M., Güner G., Jocher G., Tahirovic S., Frishman D., et al. An optimized quantitative proteomics method establishes the cell type-resolved mouse brain secretome. EMBO J. 2020;39:e105693. doi: 10.15252/embj.2020105693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Offe K., Dodson S.E., Shoemaker J.T., Fritz J.J., Gearing M., Levey A.I., Lah J.J. The lipoprotein receptor LR11 regulates amyloid b production and amyloid precursor protein traffic in endosomal compartments. J. Neurosci. 2006;26:1596–1603. doi: 10.1523/JNEUROSCI.4946-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blechingberg J., Poulsen A.S.A., Kjølby M., Monti G., Allen M., Ivarsen A.K., Lincoln S.J., Thotakura G., Vægter C.B., Ertekin-Taner N., et al. An alternative transcript of the Alzheimer's disease risk gene SORL1 encodes a truncated receptor. Neurobiol. Aging. 2018;71:266.e11–266.e24. doi: 10.1016/j.neurobiolaging.2018.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Christensen S.K., Narimatsu Y., Simoes S., Goth C.K., Vægter C.B., Small S.A., Clausen H., Andersen O.M. Endosomal trafficking is required for glycosylation and normal maturationof the Alzheimer’s-associated protein sorLA. bioRxiv. 2020 doi: 10.1101/2020.07.12.199885. Preprint at. [DOI] [Google Scholar]
- 43.Dodson S.E., Andersen O.M., Karmali V., Fritz J.J., Cheng D., Peng J., Levey A.I., Willnow T.E., Lah J.J. Loss of LR11/SORLA enhances early pathology in a mouse model of amyloidosis: evidence for a proximal role in Alzheimer's disease. J. Neurosci. 2008;28:12877–12886. doi: 10.1523/JNEUROSCI.4582-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lewczuk P., Kamrowski-Kruck H., Peters O., Heuser I., Jessen F., Popp J., Bürger K., Hampel H., Frölich L., Wolf S., et al. Soluble amyloid precursor proteins in the cerebrospinal fluid as novel potential biomarkers of Alzheimer's disease: a multicenter study. Mol. Psychiatr. 2010;15:138–145. doi: 10.1038/mp.2008.84. [DOI] [PubMed] [Google Scholar]
- 45.Araki W., Hattori K., Kanemaru K., Yokoi Y., Omachi Y., Takano H., Sakata M., Yoshida S., Tsukamoto T., Murata M., et al. Re-evaluation of soluble APP-alpha and APP-beta in cerebrospinal fluid as potential biomarkers for early diagnosis of dementia disorders. Biomark. Res. 2017;5:28. doi: 10.1186/s40364-017-0108-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Young J.E., Fong L.K., Frankowski H., Petsko G.A., Small S.A., Goldstein L.S.B. Stabilizing the retromer complex in a human stem cell model of Alzheimer's disease reduces TAU phosphorylation independently of amyloid precursor protein. Stem Cell Rep. 2018;10:1046–1058. doi: 10.1016/j.stemcr.2018.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vagnozzi A.N., Li J.G., Chiu J., Razmpour R., Warfield R., Ramirez S.H., Praticò D. VPS35 regulates tau phosphorylation and neuropathology in tauopathy. Mol. Psychiatr. 2021;26:6992–7005. doi: 10.1038/s41380-019-0453-x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Cataldo A.M., Barnett J.L., Pieroni C., Nixon R.A. Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer's disease: neuropathologic evidence for a mechanism of increased beta-amyloidogenesis. J. Neurosci. 1997;17:6142–6151. doi: 10.1523/JNEUROSCI.17-16-06142.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cataldo A.M., Peterhoff C.M., Troncoso J.C., Gomez-Isla T., Hyman B.T., Nixon R.A. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 2000;157:277–286. doi: 10.1016/s0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bonvento G., Valette J., Flament J., Mochel F., Brouillet E. Imaging and spectroscopic approaches to probe brain energy metabolism dysregulation in neurodegenerative diseases. J. Cerebr. Blood Flow Metabol. 2017;37:1927–1943. doi: 10.1177/0271678X17697989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aung W.Y., Mar S., Benzinger T.L. Diffusion tensor MRI as a biomarker in axonal and myelin damage. Imag. Med. 2013;5:427–440. doi: 10.2217/iim.13.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mattsson N., Andreasson U., Zetterberg H., Blennow K., Alzheimer’s Disease Neuroimaging Initiative Association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 2017;74:557–566. doi: 10.1001/jamaneurol.2016.6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holstege H., van der Lee S.J., Hulsman M., Wong T.H., van Rooij J.G., Weiss M., Louwersheimer E., Wolters F.J., Amin N., Uitterlinden A.G., et al. Characterization of pathogenic SORL1 genetic variants for association with Alzheimer's disease: a clinical interpretation strategy. Eur. J. Hum. Genet. 2017;25:973–981. doi: 10.1038/ejhg.2017.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rohe M., Carlo A.S., Breyhan H., Sporbert A., Militz D., Schmidt V., Wozny C., Harmeier A., Erdmann B., Bales K.R., et al. Sortilin-related receptor with A-type repeats (SORLA) affects the amyloid precursor protein-dependent stimulation of ERK signaling and adult neurogenesis. J. Biol. Chem. 2008;283:14826–14834. doi: 10.1074/jbc.M710574200. [DOI] [PubMed] [Google Scholar]
- 55.Corlier F., Rivals I., Lagarde J., Hamelin L., Corne H., Dauphinot L., Ando K., Cossec J.C., Fontaine G., Dorothée G., et al. Modifications of the endosomal compartment in peripheral blood mononuclear cells and fibroblasts from Alzheimer's disease patients. Transl. Psychiatry. 2015;5:e595. doi: 10.1038/tp.2015.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Small S.A., Simoes-Spassov S., Mayeux R., Petsko G.A. Endosomal traffic jams represent a pathogenic hub and therapeutic target in Alzheimer's disease. Trends Neurosci. 2017;40:592–602. doi: 10.1016/j.tins.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mecozzi V.J., Berman D.E., Simoes S., Vetanovetz C., Awal M.R., Patel V.M., Schneider R.T., Petsko G.A., Ringe D., Small S.A. Pharmacological chaperones stabilize retromer to limit APP processing. Nat. Chem. Biol. 2014;10:443–449. doi: 10.1038/nchembio.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lauritzen I., Bécot A., Bourgeois A., Pardossi-Piquard R., Biferi M.G., Barkats M., Checler F. Targeting gamma-secretase triggers the selective enrichment of oligomeric APP-CTFs in brain extracellular vesicles from Alzheimer cell and mouse models. Transl. Neurodegener. 2019;8:35. doi: 10.1186/s40035-019-0176-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Arbo B.D., Cechinel L.R., Palazzo R.P., Siqueira I.R. Endosomal dysfunction impacts extracellular vesicle release: central role in Abeta pathology. Ageing Res. Rev. 2020:101006. doi: 10.1016/j.arr.2019.101006. [DOI] [PubMed] [Google Scholar]
- 60.Vassar R., Citron M. Abeta-generating enzymes: recent advances in beta- and gamma-secretase research. Neuron. 2000;27:419–422. doi: 10.1016/s0896-6273(00)00051-9. [DOI] [PubMed] [Google Scholar]
- 61.Jakobsen J.E., Johansen M.G., Schmidt M., Liu Y., Li R., Callesen H., Melnikova M., Habekost M., Matrone C., Bouter Y., et al. Expression of the Alzheimer's disease mutations AbetaPP695sw and PSEN1M146I in double-transgenic gottingen minipigs. J. Alzheim. Dis. 2016;53:1617–1630. doi: 10.3233/JAD-160408. [DOI] [PubMed] [Google Scholar]
- 62.Carosi J.M., Denton D., Kumar S., Sargeant T.J. Retromer dysfunction at the nexus of tauopathies. Cell Death Differ. 2021;28:884–899. doi: 10.1038/s41418-020-00727-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van der Kant R., Goldstein L.S.B., Ossenkoppele R. Amyloid-beta-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci. 2020;21:21–35. doi: 10.1038/s41583-019-0240-3. [DOI] [PubMed] [Google Scholar]
- 64.Vos S.J., Xiong C., Visser P.J., Jasielec M.S., Hassenstab J., Grant E.A., Cairns N.J., Morris J.C., Holtzman D.M., Fagan A.M. Preclinical Alzheimer's disease and its outcome: a longitudinal cohort study. Lancet Neurol. 2013;12:957–965. doi: 10.1016/S1474-4422(13)70194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Konijnenberg E., Tomassen J., den Braber A., Ten Kate M., Yaqub M., Mulder S.D., Nivard M.G., Vanderstichele H., Lammertsma A.A., Teunissen C.E., et al. Onset of preclinical Alzheimer disease in monozygotic twins. Ann. Neurol. 2021;89:987–1000. doi: 10.1002/ana.26048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Habekost M., Qvist P., Denham M., Holm I.E., Jørgensen A.L. Directly reprogrammed neurons express MAPT and APP splice variants pertinent to ageing and neurodegeneration. Mol. Neurobiol. 2021;58:2075–2087. doi: 10.1007/s12035-020-02258-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bode G., Clausing P., Gervais F., Loegsted J., Luft J., Nogues V., Sims J., Steering Group of the RETHINK Project The utility of the minipig as an animal model in regulatory toxicology. J. Pharmacol. Toxicol. Methods. 2010;62:196–220. doi: 10.1016/j.vascn.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 68.Pedersen H.D., Mikkelsen L.F. Biomarkers in Toxicolgy. Second Edition. 2019. Gôttingen minipigs as large animal model in toxicology; pp. 75–89. [Google Scholar]
- 69.Gieling E., Wehkamp W., Willigenburg R., Nordquist R.E., Ganderup N.C., van der Staay F.J. Performance of conventional pigs and Gottingen miniature pigs in a spatial holeboard task: effects of the putative muscarinic cognition impairer Biperiden. Behav. Brain Funct. 2013;9:4. doi: 10.1186/1744-9081-9-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roelofs S., Murphy E., Ni H., Gieling E., Nordquist R.E., van der Staay F.J. Judgement bias in pigs is independent of performance in a spatial holeboard task and conditional discrimination learning. Anim. Cognit. 2017;20:739–753. doi: 10.1007/s10071-017-1095-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kohli M., Rago C., Lengauer C., Kinzler K.W., Vogelstein B. Facile methods for generating human somatic cell gene knockouts using recombinant adeno-associated viruses. Nucleic Acids Res. 2004;32:e3. doi: 10.1093/nar/gnh009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sander J.D., Maeder M.L., Reyon D., Voytas D.F., Joung J.K., Dobbs D. ZiFiT (Zinc Finger Targeter): an updated zinc finger engineering tool. Nucleic Acids Res. 2010;38:W462–W468. doi: 10.1093/nar/gkq319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhou Y., Liu Y., Hussmann D., Brøgger P., Al-Saaidi R.A., Tan S., Lin L., Petersen T.S., Zhou G.Q., Bross P., et al. Enhanced genome editing in mammalian cells with a modified dual-fluorescent surrogate system. Cell. Mol. Life Sci. 2016;73:2543–2563. doi: 10.1007/s00018-015-2128-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li J., Pedersen H.S., Li R., Adamsen J., Liu Y., Schmidt M., Purup S., Callesen H. Developmental potential of pig embryos reconstructed by use of sow versus pre-pubertal gilt oocytes after somatic cell nuclear transfer. Zygote. 2014;22:356–365. doi: 10.1017/S0967199412000676. [DOI] [PubMed] [Google Scholar]
- 76.Schmidt M., Kragh P.M., Li J., Du Y., Lin L., Liu Y., Bøgh I.B., Winther K.D., Vajta G., Callesen H. Pregnancies and piglets from large white sow recipients after two transfer methods of cloned and transgenic embryos of different pig breeds. Theriogenology. 2010;74:1233–1240. doi: 10.1016/j.theriogenology.2010.05.026. [DOI] [PubMed] [Google Scholar]
- 77.Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jacobsen L., Madsen P., Jacobsen C., Nielsen M.S., Gliemann J., Petersen C.M. Activation and functional characterization of the mosaic receptor SorLA/LR11. J. Biol. Chem. 2001;276:22788–22796. doi: 10.1074/jbc.M100857200. [DOI] [PubMed] [Google Scholar]
- 79.Jenkinson M., Beckmann C.F., Behrens T.E.J., Woolrich M.W., Smith S.M. Neuroimage. 2012;62:782–790. doi: 10.1016/j.neuroimage.2011.09.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.