

Abstract
Objectives
A subset of chronic obstructive pulmonary disease (COPD) patients have increased numbers of airway eosinophils associated with elevated markers of T2 inflammation. This analysis focussed on mast cell counts and mast cell‐related gene expression in COPD patients with higher vs lower eosinophil counts.
Methods
We investigated gene expression of tryptase (TPSAB1), carboxypeptidase A3 (CPA3), chymase (CMA1) and two mast cell specific gene signatures; a bronchial biopsy signature (MCbb) and an IgE signature (MCIgE) using sputum cells and bronchial epithelial brushings. Gene expression analysis was conducted by RNA‐sequencing. We also examined bronchial biopsy mast cell numbers by immunohistochemistry.
Results
There was increased expression of TPSAB1, CPA3 and MCbb in eosinophilhigh than in eosinophillow COPD patients in sputum cells and bronchial epithelial brushings (fold change differences 1.21 and 1.28, respectively, P < 0.01). Mast cell gene expression was associated with markers of T2 and eosinophilic inflammation (IL13, CLCA1, CST1, CCL26, eosinophil counts in sputum and bronchial mucosa; rho = 0.4–0.8; P < 0.05). There was no difference in MCIgE gene expression between groups. There was no difference in the total number of bronchial biopsy mast cells between groups.
Conclusion
These results demonstrate that eosinophilic inflammation is associated with altered mast cell characteristics in COPD patients, implicating mast cells as a component of T2 inflammation present in a subset of COPD patients.
Keywords: eosinophils, epithelial cells, sputum, type 2 inflammation
Our results demonstrate that eosinophilic inflammation is associated with altered mast cell characteristics in chronic obstructive pulmonary disease (COPD) patients, implicating mast cells as a component of T2 inflammation present in a subset of COPD patients.

Introduction
Chronic obstructive pulmonary disease (COPD) is a highly heterogeneous and complex condition characterised by chronic airway inflammation and remodelling. 1 A subset of COPD patients have increased eosinophil infiltration into the lungs. 2 , 3 Blood eosinophil counts (BEC) are a biomarker of pulmonary eosinophil numbers, as these two parameters are correlated in COPD patients. 4 , 5 , 6 Randomised controlled trials have shown that higher blood or sputum eosinophil counts are associated with greater clinical benefits of inhaled corticosteroids (ICS). 6 , 7 BEC are used in clinical practice to help guide ICS use towards COPD patients who are more likely to gain benefit.
Recent evidence has shown greater type 2 (T2) inflammation in the lungs of COPD patients with higher BEC, including increased levels of the eosinophil chemoattractant C‐C motif chemokine ligand (CCL) 24, the eosinophil activator interleukin (IL)‐5 and greater expression of the T2 genes IL13, CCL26, chloride channel accessory 1 (CLCA1) and cystatin SN (CST1). 5 , 8 , 9 Importantly, periostin (POSTN) and serpin family B member 2 (SERPINB2), which are well‐known T2 genes in asthma, were not associated with BEC in COPD, highlighting that T2 inflammation in eosinophilic COPD and asthma are not identical.
Mast cells contain several granular proteases including tryptase, carboxypeptidase A3 and chymase. Anatomical location determines mast cell protease expression with compartmental differences observed in the lungs. 10 Mucosal mast cells are tryptase+chymase− (MCT), whereas connective tissue mast cells are tryptase+chymase+ (MCTC). Mast cells are well known for their role in IgE‐mediated allergic inflammation. However, mast cells also demonstrate IgE‐independent functions including tissue repair and antimicrobial responses. 11 The role of mast cells in COPD is unclear with some, but not all, studies showing increased mast cell numbers in the lungs of COPD patients compared to controls. 12 , 13 , 14 These contrasting results may be because of patient selection, as higher sputum mast cell gene expression and tryptase protein levels have been observed in COPD patients with > 3% sputum eosinophils. 15 , 16 This suggests a role for mast cells in T2 inflammation in COPD.
Using single‐cell RNA‐sequencing of lung tissue, Vieira‐Braga et al. identified mast cell genes present in asthma patients including genes encoding the proteases tryptase (TPSAB1), carboxypeptidase A3 (CPA3) and haematopoietic prostaglandin D synthase (HPGDS), the enzyme which produces prostaglandin D2. 17 In a subsequent study using these samples, Jiang et al. proposed an 11‐gene mast cell signature with differential expression in asthma patients than in controls, which was then examined in bulk RNA‐sequencing of bronchial biopsies from asthma patients. This mast cell bronchial biopsy [MCbb] signature was related to the number of mast cells present and also appeared to be related to the activity of the cells as it was reduced in individuals using ICS. 18 Additionally, signatures which identify mast cell‐specific genes involved in IgE‐mediated inflammation have been identified following repeated activation of the mast cell IgE receptor, FcεRI (mast cell IgE [MCIgE] signature). 19 These genes do not overlap with the MCbb signature, and so may enable analysis of IgE‐mediated vs non‐IgE‐mediated mast cell activation. Both the MCbb and the MCIgE signatures have been associated with eosinophilic inflammation in the sputum of severe asthma patients 20 but may represent different mechanisms of mast cell activation.
The aim of this analysis was to further investigate mast cell numbers and characteristics in COPD, and their associations with T2 inflammation. To characterise mast cells, common genes used to differentiate mast cell populations (TPSAB1, CPA3 and CMA1) were evaluated, as well as the gene expression signatures MCbb and MCIgE (Table 1). The effects of current smoking and ICS use were evaluated, and mast cell numbers assessed using immunohistochemistry/fluorescence. These analyses were performed using data and samples from a previously published study 5 comparing bronchial and sputum samples from COPD patients with higher vs lower BEC.
Table 1.
Individual genes used in mast cell signatures
| Signature | Genes | Study |
|---|---|---|
| MCbb | TPSAB1, TPSD1, TESPA1, RGS13, SLC18A2, MS4A2, HPGDS, ADCYAP1, HDC, CPA3, TPSB2 | 17 |
| Repeated IgE (MCIgE) |
TMEM45B, EMR3, CH25H, LINC01272, DGAT2, MRC1, DPP4, DYSF, FPR2, MCEMP1, CCL18, TREM2, OLFM4, MX1, TGFBI, FOLR3, CYP4F3, S100A8, NUPR1, S100A12, VSTM1, HLADOA, LGMN, GLT1D1, CEBPE, S100A9, PADI4, CDA, F13A1, S100B |
19 |
| Acute IgE (2 h IgE sensitisation 2 h FcεR1 activation) |
ATP8B4, BPI, CD33, CLC, CMA1, CPA3, CTSG, FCER1A, GZMB, HDC, HOXA1, HPGDS, IL18R1, IL1RL1, IL3, IL5, LINC00597, MARCH3, MS4A2, MS4A3, MYB, NOX3, NTRK1, P2RX1, RAB27B, RGS13, SLC12A8, STXBP6, TEC, TPSAB1 |
20, 35 |
| Acute IgE (24 h IgE sensitisation 6 h FcεR1 activation) |
XIRP1, CRTAM, CNN1, AADAC, MIR3122, FASLG, CH25H, RAI2, LINC01010, CSF1, GZMB, CCL4L2, SERPINE2, TSPAN13, FHL2, CCL3L3, TGM2, LTBP4, HBEGF, EGR2, ASPHD1, CXCL8, SLC37A2, MIR221, KCNK5, ANGPTL4, SERPINE1, CCL7, DTL, MAGEB2, LINC01160, LRRC8B, CCL3, TNFSF4, CLCF1, LINC01433, SMOX, VGF, TIE1, GEM, RBFOX2, RAB7B, ZCCHC12, NRCAM, LRIG1, TRIB1, MIR103A2, MIR27A, DYRK3, PDGFA, CST6, KIAA0226L, NTN1, DGUOK‐AS1, CTXN1, CCDC147‐AS1, MAP1B, RGCC, GAL, SLC6A8, SPP1, PKIA, LRRC8C, NT5E, RRAD |
19, 20 |
| Acute IgE (overnight IgE sensitisation 24 h FcεR1 activation) |
CSF1, IL1R1, IL27RA, TNFRSF12A, CCL7, CCRL2, CXCL8, BL34, LIF, CD69, LAT, ADORA2A, CRIP1, HLA‐DQB1, HKE2, NKG7, FCGR2B, PTGER2, CLECSF5, EBI2, TREM1, FCAR, TLR2, HIVEP1, FOSB, FBJ, EGR3, PHLDA2, FGFR1, EGR2, INSIG1, PDGFA, IER3, PDGFB, BTG2, TIEG, CNK, PBEF1, TNFAIP8, MLP, CRABP2, IRF2, RASAL1, FLRT2, SMARCD3, ARHE, KAL1, FLNB, CD151, ARF6, ALCAM, MAFF, BCL6, NFATC1, ATF3, NFKBIE, MYC, ELL2, TOP2A, GTF2H2, THBD, NR4A2, GEM, SPHK1, NDUFA7, HBEGF, JAG1, LDLR, MADH7, MALT1, SPRY2, DUSP6, MAP2K3, CREM, DUSP1, MAP3K14, FUT4, JUN, PSCDBP, FYN, PGGT1B, VRK2, TTK, ENC1, SLC16A6, HIST2H2AA, CYP3A4, HIST1H1C, HEC, STK17A, PELI1, KCNAB1, B4GALT4 |
20, 36 |
| IL‐33 activation |
IL13, EBI3, CD70, IL5, CXCL2, CXCL8, TNFRSF8, TNFRSF18, CCL3, IL6, IL3, WNT2B, TNF, KITLG, GDF11, IL1B, TNFSF11, TNFRSF1B, TNFSF14, TNFSF4, CCL3L3, GDF3, IL36G, WNT11, CXCL6, IL17D, EGF, TNFRSF17, AREG, NDP, EREG, UBD, FGD6, SSTR2, KIAA1324, CEP135, NFKBIZ, GADD45B, LIMS2, KLF5, ABTB2, ITGA1, P2RX5, NFKBIA, C15orf48, LARGE2, IL2RG, RMI1, KCNH2, RIPOR3, CFLAR, HIVEP2, ZC3H12A, MIIP, RGS9, TTC39C, PMAIP1, PTX3, TLR2, RANBP9, NAPB, TNIP1, EGLN1, METAP2, ASNS, IER3, AMMECR1, MPZL1, FLVCR1, MT2A, PIM2, TMEM64, VWA8, PEAK1, ADIPOR2, TEX30, HAPLN3, KISS1R, ADAM8, TAP2, NFKB1, SLC4A7, CDC42EP2, SLC25A45, MMP9, DES |
20, 37 |
| LPS activation |
GNG4, IRF4, CSF2, IR155HG, IL6, MIR212, IL1RN, LINC01215, LOC731424, GGT5, CCL3L1, FFAR2, F3, PPBP, MIR146A, IL7R, TARP, MIR222, TM4SF1, MAOA, CCL4L1, MAMLD1, STEAP4, C3, TNFAIP6, STAT4, EBI3, IGSF6, IL2RA, NFKBIA, AQP9, ABTB2, IRAK2, EMR1, SNORA66, NFKB1, REL, CCL20, RGS9, MFSD2A, CXCL1, TNIP1, C17orf96, NFKB2, TNFRSF18, PXYLP1, LY6G6C, HILPDA, C15orf48, DUSP2, CXCL2, CYP27B1, DUSP5, PTX3, MSC, TNFSF14, GCH1, TMEM88, SLAMF1, EMP1, CLEC4A, RASGRP1, MAP3K8, FLVCR2, EDIL3, MREG, AQP2, CD40, BATF3, CD274, TNFRSF8, SLAMF7, METTL1, IL15RA, CCL24, IL1B, RNF144B, SH3RF2, SERPINB2, CISH, POU2F2, SCARF1, CCDC147, PRG2, PEAK1, TNIP3, GPR84, CXCL5, CD83, LPL, TRIP10, PPARG, SNORA61, GSAP, NLRP3, TNFAIP3, ECE1, ATP9A, PDLIM4, SDC4, RELB, APOL1, SNORA6, SERPINA1, ELOVL7, TNFAIP2, BCL2A1, GBP2, FAS, SLC2A6, ECEL1, SPATA13, STON2, CKB, PIM2, FPR2, SGPP2, THBS1, RSAD2, GPC1, FGL2, CA12, ETV3, SOCS2, IL10RA, ENTPD7, BACH2, DPP4, LUCAT1, SLCO4A1, PRDX1, RASAL2, NINJ1, ESPL1, RAB38, OLR1, TMEM163, BATF, CLEC5A, ARHGAP31, ICAM1, STAMBPL1, RASSF4 |
20, 38 |
| IFN‐γ activation |
MARKS, MT1E, MUC1, SDCCAG8, SGPL1, ITGA1, MYB, CD40, MSL3, CCL8, AP1G1, PDZD2, APOL1, P2RX5, B3GNT7, CLIP1, SLC2A6, KYNU, TIMP1, CYB5A, SLC11A2, EZH2, TNFSF10, AGT, TPBG, ARG2, CEBPD, CD48, FSTL3, PRPSAP2, FCGR2A, JAK1, IGF2R, PLSCR1, TNFAIP2, ALCAM, EHD1, ABCG4, BID, G0S2, CRYBG1, MAOA, NOP16, METAP2, NFKB2, PLA2G4A, CSF2, CFLAR, AMACR, CREB3, GADD45B, CITED2, TANK, CXCL8, MT1H, TMEM123, MT2A, IPO5, TTC39A, ANXA2, EIF4A3, ADAP1, CYLD, DLEU2, RBM7, TMEM165, TNFAIP3, ICAM1, GBP2, GCH1, BUB3, SH3BP5, STAG1, HSPA13, ACSL1, SLC7A1, CHST7, NEURL1, MAMLD1, CCL3, MAP3K8, BCL2A1, ZSCAN21, TSFM, H2AC18, EIF5B, EIF5A, DCBLD2, EMD, CRY1, HLA‐DRB4, ARL6IP1, TASP1, SINHCAF, UBIAD1, CDK17, GLS, RAB31, RBM8A, CHORDC1, PELO, SMARCA5, KLF10, DNAJB4, GSPT1, ZNF222, HMGB2, EIF5, PPP2CA, CD44, MKLN1, IL7R, RCE1, FGL2, RRAD, ATG12, ISG20, CXCL11, HLA‐DRA, STAT1, CD74, MX1, HLA‐DQA1, IFIT1, HLA‐DQB1, IL3RA, AKR1C3, RSAD2, CBR3, TAP1, ISG15, SERPING1, HBD, SECTM1, HLADPB1, DHX58, OAS1, OAS3, WARS1, CXCL10, IFIH1, HBB, HLADPA1, TRIM14, IFI44, DDX58, FOG2, LGALS3BP, PSMB9, IFI35, IFIT4, HCP5, USP18, HLA‐DOB, TRAFD1, SAMHD1, HLA‐DRB5, HLA‐DMA, OASL, DOCK9, HLA‐DRB3, FKBP11, AVIL, PSME2, POLA2, IRF7, LY6E, NOD2, CTSS, PLAAT4, BTN3A3, C4B, HLADRB1, XAF1, HLA‐DMB, OAS2, TRIM58, PSMB8, BST2, TAP2, ZC3HAV1, UBE2L6, RCBTB2, IFITM1, CD38, HLA‐G, CASP1, NKX3‐1, SQOR, NBN, FOSB, IRF1, TRIM22, TNIP2, TNFAIP6, CEP135, RTP4, TKT, IFI6, MXI1, SNN, PSME1, MT1G, KLF4, FADD, MDS019, POLR3D, DOP1A, RPS6KC1, ADGRE5, TYMP, OFD1, IFNGR2, CASP10, PSMB10, RAB33A, IL13, DNAJA1, NDP52, TIA1, DUSP1, STK39, SP100, BAZ1A, EIF1AY, TNFAIP8, SOCS1, RNF114, MAP3K7, ARHGAP25, IRLB, ATF3 |
20, 38 |
Results
Study subjects
The clinical characteristics of the study participants have been previously reported 8 and are shown in Table 2. Patients with a prior asthma diagnosis or a positive skin prick test were excluded. The groups were generally well matched for clinical characteristics, with significant differences in sputum and BAL eosinophil percentage as expected (P < 0.0001 and P = 0.02, respectively).
Table 2.
Clinical characteristics of the study population
| Eosinophillow | Eosinophilhigh | P‐value | |
|---|---|---|---|
| Age (year) | 62 ± 6 | 62 ± 4 | 0.7 |
| Gender: Male (%) | 57 | 70 | 0.5 |
| BMI (kg/m2) | 29 (18–34) | 25 (19–32) | 0.1 |
| Current smokers (%) | 43 | 60 | 0.4 |
| Pack‐years history | 42 ± 15 | 38 ± 14 | 0.4 |
| ICS use (%) | 76 | 55 | 0.2 |
| Post bronchodilator FEV1 (L) | 1.8 ± 0.4 | 1.9 ± 0.4 | 0.45 |
| Post bronchodilator FEV1 (% predicted) | 62 ± 11 | 66 ± 11 | 0.4 |
| Post bronchodilator FVC (L) | 3.7 ± 1.2 | 3.6 ± 0.9 | 0.8 |
| Post bronchodilator FEV1/FVC ratio | 50 ± 10 | 53 ± 7 | 0.3 |
| Reversibility (mL) | 190 ± 183 | 220 ± 154 | 0.6 |
| FeNO50 (ppm) | 15 (5–30) | 21 (2–61) | 0.2 |
| Atopy (% positive) | 0 | 0 | N/A |
| Total SGRQ | 42 ± 15 | 37 ± 21 | 0.4 |
| mMRC | 1 (1–4) | 1 (0–4) | 0.1 |
| CAT | 17 (5–35) | 17 (4–32) | 0.5 |
| Exacerbation rate, 12 months prior | 0 (0–3) | 0 (0–3) | 0.7 |
| Blood eosinophils (cells μL−1) | 100 (60–140) | 410 (280–890) | < 0.001 |
| Sputum eosinophils (%) | 0.5 (0.5–2.5) | 4.5 (0.25–70) | < 0.001 |
| BAL eosinophils (%) | 0 (0–3.25) | 0.75 (0–8) | 0.02 |
Data are presented as %, mean ± standard deviation, or median (range).
BMI, body mass index; CAT, COPD Assessment Test; ICS, inhaled corticosteroids; FeNO50, fractional exhaled nitric oxide at 50 mL s−1 flow rate; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; mMRC, modified Medical Research Council; PPM, parts per million; SGRQ, St George's Respiratory Questionnaire.
Bronchial brushing gene expression
In bronchial epithelial brushings, TPSAB1 and CPA3 expressions were significantly higher in eosinophilhigh than in eosinophillow patients, with fold change differences of 1.21 and 1.28, respectively (P = 0.002 and P < 0.0001, respectively; Figure 1). CMA1 expression showed a trend towards lower expression in eosinophilhigh than in eosinophillow COPD patients (P = 0.07). The MCbb signature was significantly higher in eosinophilhigh than in eosinophillow patients (P = 0.0003; Figure 1), whilst there was no difference between groups for the MCIgE signature (P = 0.3; Figure 1). To ensure TPSAB1 and CPA3 expressions were not driving the MCbb signature results, we performed a subanalysis that removed these genes; the differences between the groups remained significant (P = 0.0005; data not shown).
Figure 1.
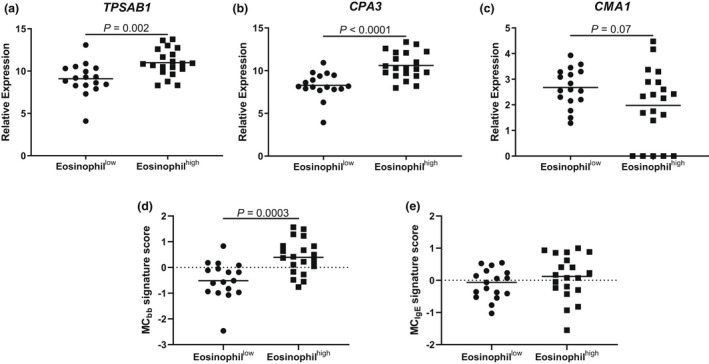
Bronchial brushing mast cell gene expression in eosinophillow and eosinophilhigh chronic obstructive pulmonary disease (COPD) patients. RNA‐sequencing was used to examine the expression of (a) TPSAB1, (b) CPA3, (c) CMA1, (d) MCbb signature and (e) MCIgE signature in n = 17 eosinophillow and n = 20 eosinophilhigh COPD patients. Data are presented as individual values where the black horizontal line represents the mean (a–c) or median (d and e).
There were no differences in the expression of other mast cell signatures between eosinophilhigh and eosinophillow COPD patients (Supplementary figure 1), apart from an acute IgE activation signature (2 h IgE sensitisation 2 h FcεR1 activation: P = 0.0003).
We analysed associations between T2 and eosinophil‐related markers and the MCbb signature. The MCbb signature was significantly associated with the COPD T2 signature (rho = 0.6; P = 0.0001; Figure 2), and the individual genes within the T2 signature (data shown in Supplementary figure 2), BEC (rho = 0.6; P = 0.0004), sputum eosinophil counts (rho = 0.4; P = 0.04) and tissue eosinophil counts (rho = 0.6; P = 0.0009; Figure 2). There were no associations with IL5 or FeNO (Supplementary figure 3; rho = −0.07; P = 0.7 and rho = 0.3 and P = 0.08, respectively). There was no association between the COPD T2 signature, eosinophil counts and the MCIgE signature (Supplementary figure 4). TPSAB1 and CPA3 expressions and the MCbb signature were significantly lower in ICS users (n = 25) than in non‐users (n = 12; Figure 3; P < 0.01 for all comparisons). In the eosinophilhigh group, the MCbb signature was lower in ICS users (n = 11) vs non‐users (n = 9), and there was a trend for reduced TPSAB1 and CPA3 expressions but this did not reach statistical significance (Supplementary figure 5; P = 0.05–0.1). The lower sample size in the eosinophillow group (ICS users and non‐users; n = 14 and n = 3, respectively) prevented comparisons. There were no differences between current smokers and ex‐smokers (Supplementary figure 6).
Figure 2.
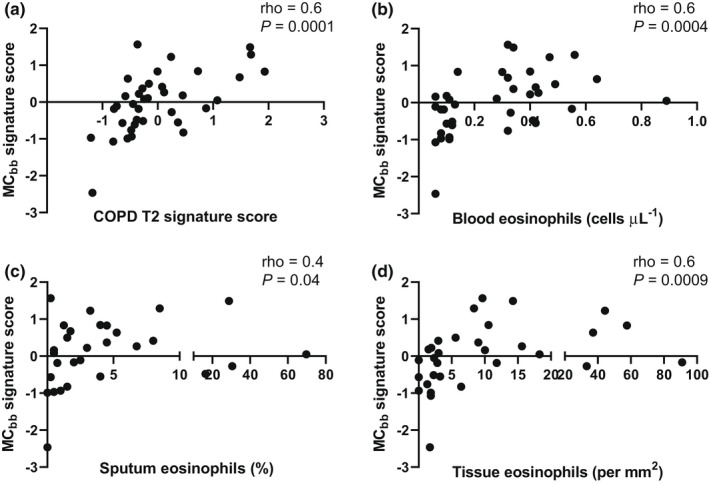
Correlations between bronchial brushing mast cell gene expression and T2 biomarkers. RNA‐sequencing was used to examine correlations between the MCbb signature and (a) chronic obstructive pulmonary disease (COPD) T2 signature, (b) blood eosinophils, (c) sputum eosinophils and (d) bronchial biopsy eosinophilsin n = 17 eosinophillow and n = 20 eosinophilhigh COPD patients. Data were analysed by Spearman's correlation.
Figure 3.
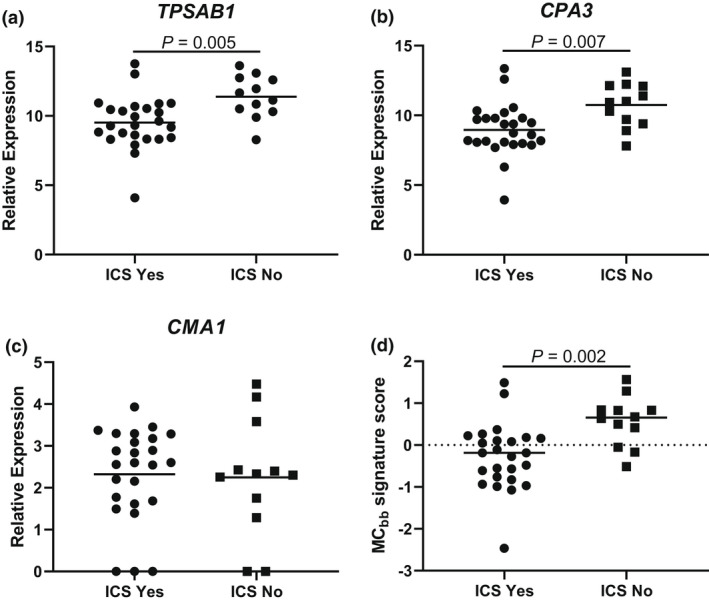
Bronchial brushing mast cell gene expression in inhaled corticosteroids (ICS) users and non‐users. RNA‐sequencing was used to examine the expression of (a) TPSAB1, (b) CPA3, (c) CMA1, (d) MCbb signature and (e) MCIgE signature in n = 25 ICS users (ICS Yes) and n = 12 non‐users (ICS No). Data are presented as individual values where the black horizontal line represents the mean (a–c) or median (d).
Mast cell counts in bronchial biopsies
Mast cell numbers in bronchial biopsies were examined by dual immunofluorescence. There was no difference in the total number of mast cells (MT + MTC), the number of MT or the number of MTC in the subepithelium of eosinophilhigh compared with eosinophillow COPD patients (Figure 4). In ICS users (n = 16), the total number of mast cells, the number of MT and the number of MTC were significantly lower than in non‐users (n = 7; Figure 4; P = 0.02, P = 0.02 and P = 0.04, respectively). In eosinophilhigh patients, there was a numerical reduction in mast cell numbers in ICS users (n = 7) than in non‐users (n = 6) but this did not reach statistical significance (Supplementary figure 7; P > 0.05). There were no differences due to current smoking (Supplementary Figure 7). The number of MT was significantly higher than MTC in eosinophilhigh and eosinophillow groups (P = 0.007 and P = 0.05, respectively).
Figure 4.
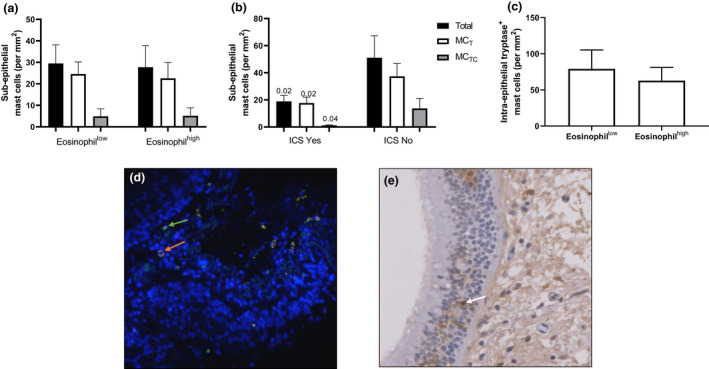
Mast cell quantification in bronchial biopsies. The number of subepithelial tryptase+ (MT), tryptase+chymase+ (MTC) and the total number of mast cells (MT + MTC) were quantified by immunofluorescence (a, b) and the number of intra‐epithelial tryptase+ mast cells were quantified by immunohistochemistry (c). Representative images of immunofluorescence where green and orange arrows indicate MT and MTC cells respectively (d) and immunohistochemistry where white arrows indicate tryptase‐positive mast cells (e). Comparisons were made between eosinophillow vs eosinophilhigh patients (a) n = 10 vs n = 13; (c) n = 14 vs n = 15; ICS users vs non‐users (b) n = 16 vs n = 7. P‐values in b signify differences between the same mast cell populations in ICS Yes vs ICS No groups.
The number of biopsies with intact epithelium available for new experiments was low (n = 9 eosinophilhigh vs n = 4 eosinophillow), making it difficult to compare intra‐epithelial mast cell counts with these samples. We therefore counted intra‐epithelial mast cells using slides already stained with single label tryptase from the original publication, where only subepithelial mast cell counts were reported previously. 5 In 15 eosinophilhigh vs 14 eosinophillow patients, there was no difference in the number of intra‐epithelial mast cell numbers when normalised to epithelial thickness (Figure 4; P = 0.6) or basement membrane length (Supplementary figure 7; P = 0.8).
There was no difference in epithelial thickness or PAS+ cells between eosinophilhigh vs eosinophillow patients (Supplementary figure 8).
Sputum cell gene expression
In sputum, TPSAB1 and CPA3 expressions trended higher in eosinophilhigh than in eosinophillow COPD patients (Supplementary figures 9; Figure 5; P = 0.07). The expression of CMA1 was not different between groups (Supplementary figure 9; P = 0.9). The MCbb signature was significantly higher in eosinophilhigh than in eosinophillow COPD patients (Figure 5; P = 0.004) and significantly associated with the COPD T2 signature (rho = 0.6 = P = 0.001; Figure 5) and the individual genes within the T2 signature (Supplementary figure 10). The MCbb signature was also significantly associated with blood and sputum eosinophil counts (Figure 5; rho = 0.5; P = 0.009; rho = 0.8; P < 0.0001). There was a significant association with IL5 gene expression but not FeNO (Supplementary figure 11; rho = 0.8; P < 0.0001 and rho = 0.3; P = 0.2, respectively). The sputum MCbb signature was significantly associated with the bronchial brushing MCbb signature (Supplementary figure 11; rho = 0.5; P = 0.01). When TPSAB1 and CPA3 were removed from the MCbb signature, expression remained significantly higher in the eosinophilhigh patients than in the eosinophillow patients (P = 0.006; data not shown).
Figure 5.
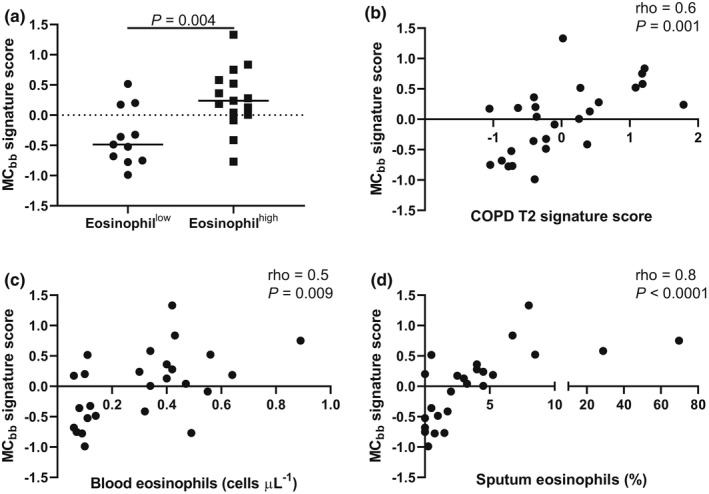
Sputum mast cell gene expression in eosinophillow and eosinophilhigh COPD patients. RNA‐sequencing was used to examine the expression of (a) MCbb signature and correlations between the MCbb signature and (b) COPD T2 signature, (c) blood eosinophils and (d) sputum eosinophils in n = 11 eosinophillow and n = 15 eosinophilhigh COPD patients. Data are presented as individual values where the black horizontal line represents the median (a). Data are analysed by Spearman's correlation (b–d).
There was no difference between groups for the MCIgE signature (P = 0.7; Supplementary figure 12), and there was no correlation between MCIgE signature and the COPD T2 signature or eosinophil counts (Supplementary figure 12). We found no differences in the expression of other mast cell signatures between eosinophilhigh and eosinophillow COPD patients (Supplementary figure 13), apart from an acute IgE activation signature (2 h IgE sensitisation 2 h FcεR1 activation: P = 0.01). There was no difference between ICS users and non‐users (Supplementary figure 14) and current and ex‐smokers (Supplementary figure 15) for individual sputum gene expression or the sputum MCbb signature.
Discussion
Mast cell gene expression, namely TPSAB1, CPA3 and a mast cell‐specific MCbb signature, was increased in eosinophilhigh compared to eosinophillow COPD patients. These findings were consistent in both bronchial epithelial brushings and sputum samples, with the MCbb signature also being correlated with T2 biomarkers. Bronchial mast cell numbers did not differ between groups. These findings suggest that the phenotypic characteristics of mast cells are different in eosinophilhigh vs eosinophillow COPD patients.
Studies in asthma patients have shown that mast cell numbers and mast cell gene expression do not consistently correlate. 18 , 20 Berry et al. reported no difference in the number of submucosal tryptase+ mast cells in eosinophilic vs non‐eosinophilic asthma patients. 21 We observed altered mast cell gene expression (TPSAB1, CPA3 and MCbb) in eosinophilhigh compared with eosinophillow COPD patients, but no differences in mast cell numbers. We interpret these COPD results as showing altered mast cell phenotype despite no changes to intra‐ and subepithelial mast cell numbers. It is possible that our results also indicate increased mast cell activity in eosinophilhigh COPD patients, although other measures of mast cell activation would be needed to properly investigate this. The association between mast cell gene expression and both the T2 signature (IL13, CLCA1, CST1 and CCL26) and eosinophilic airway inflammation (measured in sputum and bronchial mucosa) indicates that altered mast cell characteristics are part of a T2/eosinophilic profile in a subset of COPD patients.
We observed decreased mast cell gene expression and lower mast cell numbers in ICS users than in non‐users. Lower mast cell numbers associated with ICS use have also been reported in both asthma and COPD studies. 18 , 22 , 23 , 24 The reduction in mast cell numbers with ICS use provides evidence of the sensitivity of our immunohistochemistry stains to detect changes in subepithelial mast cell numbers, and therefore indirectly further supports the robustness of the finding of no difference in total mast cell numbers in eosinophilhigh vs eosinophillow COPD patients.
There is previous evidence from sputum analysis of greater mast cell activation in COPD patients with > 3% vs < 3% eosinophils, with higher levels of tryptase measured. 16 We analysed the bronchial epithelium, in addition to sputum, to enable detailed analysis of mast cell characteristics in COPD. Immunohistochemistry indicated a predominance of mucosal (MCT) over connective tissue mast cells (MCTC), compatible with previous findings in the endobronchial biospies of asthma patients. 25 There was a similar pattern for gene expression analysis where TPSAB1 (tryptase) was higher than CMA1 (chymase) expression. Furthermore, the increased gene expression of TPSAB1 and CPA3, with no increase in CMA1 expression, in eosinophilhigh vs eosinophillow COPD patients, is similar to previous observations in asthma patients with increased eosinophil counts. 25 , 26 Eosinophilic inflammation in both asthma and COPD therefore appears to be associated with a predominance of the MCT phenotype.
The MCIgE signature did not differ between groups. This signature is derived from a repeated IgE exposure mast cell activation model, and we therefore conclude that chronic IgE‐mediated mast cell activation is not involved in eosinophilic inflammation in COPD. Of note, the groups in this study had similar serum IgE levels, and only non‐atopic individuals were included. We also examined other in vitro mast cell signatures described by Tiotiu et al. 20 which examined acute IgE stimulation (three signatures), IL‐33 stimulation, LPS stimulation and IFN‐γ stimulation of mast cells. There were no differences in the expression of these signatures between eosinophilhigh and eosinophillow patients, apart from an acute IgE signature (2 h IgE sensitisation 2 h FcεR1 activation) which contains several genes that overlap with the MCbb signature (CPA3, HDC, HPGDS, RGS13 and TPSAB1) and also includes CLC which is also strongly expressed by eosinophils. However, the general lack of positive results from the Tiotiu et al. 20 signatures, which were primarily used to investigate asthma, likely reflect different pathophysiological processes in COPD.
It has been reported that chymase‐only positive mast cells are present in the bronchi, 27 although none were observed in the current study. Our findings are in agreement with another study in COPD patients. 14 It should also be noted that the type of tissue fixative used can influence mast cell identification in the lungs; Kleinjan et al. 28 observed that the percentage of chymase‐only mast cells in the lung parenchyma of tissue fixed with acetone was 1%, whereas in tissue fixed with Carnoy's fluid, chymase‐only mast cells were undetectable. Overall, it appears that the relative numbers of these cells are small and that methodology influences successful detection.
It has been reported that mucosal mast cell numbers in the small airways of COPD GOLD stage 1–3 patients are similar to controls, but there are reduced numbers in GOLD 4 patients. 14 We included GOLD 1–3 patients and found differences in mast cell gene expression associated with eosinophil counts, highlighting mast cell heterogeneity within COPD patients.
Our study has limitations. We used bulk RNA‐sequencing data to investigate mast cell signatures. However, the mast cell signatures used were developed from single‐cell analysis and further validated in mixed‐cell samples. 18 It will be important to confirm our findings in COPD using single‐cell RNA‐sequencing. Whilst our sample size is modest, the well‐phenotyped nature of the subjects enables evaluation of eosinophilhigh vs eosinophillow individuals. Due to sample availability, we were unable to measure extracellular markers of mast cell activation, for example prostaglandin D2 and tryptase. We conducted multiple testing (correlations) which can increase the number of type 1 errors. However, these correlations were exploratory in nature to supplement the main analyses. A healthy control group analysis may aid interpretation of the level of mast cell activation, although it has been reported that mast cell numbers in healthy subjects are very low. 18 Finally, the aim of our study was to investigate mast cells in eosinophilic COPD; other cell types may also be relevant (e.g. Th2 cells), which could be the focus of future investigations.
In conclusion, the data reported here show altered mast cell phenotype and characteristics in COPD patients with eosinophilic inflammation. There is a growing understanding of the nature of T2 and eosinophilic inflammation in COPD, which represents a potential target for pharmacological treatment. 29 , 30 Our findings help understand the complexity of T2 and eosinophilic inflammation in COPD, which appears to encompass changes in mast cell characteristics.
Methods
Study subjects
Chronic obstructive pulmonary disease patients aged > 40 with a smoking history of > 10 pack‐years, a postbronchodilator forced expiratory volume in 1 s (FEV1) and forced vital capacity (FVC) ratio < 0.7, and with no history of asthma were recruited. Patients receiving oral corticosteroids or antibiotics within 6 weeks of the study were excluded. Atopy was determined by a positive skin prick test against house dust mite extract, cat dander or grass pollen. Here, we include samples from n = 20 eosinophilhigh (> 250 cells μL−1) and n = 17 eosinophillow (< 150 cells μL−1) COPD patients because of sample availability. This study was conducted in accordance with the Declaration of Helsinki 1975. Sample collection was approved by the local research ethics committees (REC): South Manchester REC 06/Q1403/156 (brushings), Tameside and Glossop local REC 05/Q1402/41 (sputum) and NRES Committee North West–Preston 10/H1016/25 (blood).
Sputum and bronchoscopy samples
Sputum was induced using 3%, 4% and 5% saline, inhaled in sequence for 5 min, up to 15 min via an ultrasonic nebuliser (EASYneb II, Flaemnouva, Italy) and processed as previously reported. 31 To minimise contamination of saliva, all subjects were instructed to thoroughly rinse their mouth with distilled water and perform coughing prior to sputum expectoration. Sputum plugs were isolated from the saliva component, combined proportionately with phosphate buffered saline (PBS) and vortexed for 10 s, rocked for 15 min and centrifuged (790 g for 10 min at 4°C). PBS supernatants were removed and 0.2% dithiothreitol (DTT) was added and the suspension was vortexed for 10 s, rocked for 15 min and filtered using a 48 μm filter (Sefar Ltd, Manchester, UK). The suspension was centrifuged (790 g for 10 min at 4°C), and DTT supernatants were removed. The cell pellet was lysed in RLT buffer (Qiagen, Crawley, UK) plus β‐mercaptoethanol added according to the manufacturer's instructions prior to RNA extraction. Before lysing, a small number of cells were removed to prepare cytospins. Slides were air dried for 30 min and then fixed in methanol for 10 min before staining with RapiDiff (Triangle, Skelmersdale, UK) for differential cell counting.
Some patients included in this study have been used in previous publications. 5 , 8
Bronchoscopy was performed after the subjects had been sedated as previously described. 31 BAL was collected from the right and/or left upper lobe. The bronchoscope was wedged in the bronchus and a maximum of 4 × 60 mL aliquots of prewarmed sterile 0.9% NaCl solution were instilled per lobe. The aspirated fluid was stored on ice before filtration (100 μm filter, Becton Dickenson, Oxford, UK). The filtrate was centrifuged (400 g for 10 min at 4°C), and the BAL fluid removed. The cell pellet was lysed in RLT buffer plus β‐mercaptoethanol prior to RNA extraction. Bronchial brushings were collected in bronchial epithelial basal medium (Lonza, Basel, Switzerland) and stored on ice before centrifugation (400 g for 10 min at 4°C). The cell pellet was lysed in RLT buffer plus β‐mercaptoethanol prior to RNA extraction. Endobronchial biopsies were collected from airway generations 2–5 using 2‐mm radial jaw biopsy forceps (Boston Scientific, Hemel Hempstead, UK) and immediately fixed in 4% neutral buffered formalin, before being processed and embedded in paraffin.
RNA extraction and cDNA analysis
Total RNA was extracted from sputum cells and bronchial epithelial brushings using ZR RNA MicroPrep kit (Zymo Research, Orange, CA, USA) and RNA‐seq libraries were prepared using TruSeq Stranded mRNA Prep kit (Illumina, San Diego, CA, USA) per manufacturers' protocols. To confirm RNA quality, RIN scores ≥ 7 were evaluated using the 2100 Bioanalyzer system (Agilent, Santa Clara, CA, USA). Paired‐end sequencing (75 base pairs per end) with sequencing depth at 80 million reads was performed on the HiSeq2000 platform (Illumina) to generate FASTQ files. These were aligned to human genome (version HG19) using HiSAT2 (John Hopkin's University, Baltimore, USA) and SAMtools (Genome Research Limited, Cambridge, UK). Normalised read counts were generated per transcript using DESeq2. 32 Read counts were then transformed to log2 scale (after adding 1 to account for zero read counts).
Gene expression analysis
All the gene expression analysis was conducted using bulk RNA‐sequencing data generated from bronchial brushings or sputum cells. We compared the individual expression of common mast cell genes (TPSAB1, CPA3 and CMA1) along with mast cell signatures generated from asthma bronchial biopsies (MCbb signature) and cord blood derived mast cells sensitised with IgE for 24 h followed by repeated activation of FcεRI with anti‐IgE for 2 weeks (MCIgE signature). 17 , 20 We also included other mast cell signatures including acute IgE activation (three signatures), IL‐33 activation, lipopolysaccharide (LPS) activation and interferon (IFN)‐γ activation as per Tiotiu et al. Full details of the genes included are shown in Table 1.
We correlated the expression of mast cell signatures (MCbb signature and MCIgE signature) with our previously validated COPD T2 signature (IL13, CLCA1, CST1 and CCL26) which was elevated in the sputum and bronchial brushings of COPD patients with higher BEC. 8 We also correlated mast cell signatures with IL5 gene expression and FeNO. The gene expression scores for each signature were calculated as per Bhakta et al. and Southworth et al. 33 , 34
Immunohistochemistry and immunofluorescence
All antibodies were validated to determine the optimal conditions prior to sample staining using tonsil as a positive control tissue. The optimal antigen retrieval method was selected from either; citrate buffer pH 6, EDTA pH 8, tris EDTA buffer pH 9, no retrieval or pepsin enzyme digestion. This was followed by optimal antibody concentration and finally testing the specificity of the antibody using an isotype control and omission of the primary antibody.
Immunohistochemistry for tryptase+ mast cells was previously reported. 5 Briefly, biopsies were cut into 3‐μm sections, dewaxed and rehydrated before heat‐induced antigen retrieval in a citrate pH 6 buffer. Mast cells were identified using mouse monoclonal anti‐tryptase (clone AA1, Dako, Stockport, UK). The immune reaction was detected using the Imm‐PRESS Excel Anti‐mouse kit (Vector, Peterborough, UK). Bronchial biopsies were imaged and analysed using ImagePro Plus‐6.0 at a magnification of x200. The number of tryptase‐positive cells were quantified in the bronchial epithelium by a single blinded observer and expressed per mm2.
Double‐immunofluorescence staining for mast cell tryptase and mast cell chymase was conducted on 3 μm bronchial biopsy sections. Deparaffinisation and antigen retrieval were carried out for 20 min at 97C in a PT‐Module (Thermo Fisher, Runcorn, UK) using the Thermo Dewax and HIER Buffer M (Thermo Fisher). Following incubation with protein blocking solution (Abcam, Cambridge, UK), the slides were incubated in mouse monoclonal antitryptase (clone AA1, Dako 1:200) and mouse monoclonal antichymase (Clone CC1, Abcam 1:100) antibodies, stained sequentially. To increase the sensitivity of the staining, secondary antibodies were employed in the form of goat anti‐mouse IgG followed by the detection of the primary–secondary antibody reaction using donkey anti‐goat IgG H&L (Alexa Fluor® 488) and donkey anti‐goat IgG H&L (Alexa Fluor® 568) The omission of primary antibodies was used as negative controls. Finally, Hoescht 33258 (Abcam) was added to each sample for nuclear staining. Digital micrographs were obtained using a Nikon Eclipse 80i microscope (Nikon UK Ltd, Surrey, UK) equipped with a QI imaging digital camera and analysed using ImagePro Plus‐6.0 at a magnification of x200. The number of tryptase and/or chymase‐positive cells was quantified in the bronchial epithelium and subepithelial layer by a single blinded observer and expressed mm2. Counts were quality control checked with an interuser agreement of < 10%.
Goblet cells were identified as periodic acid Schiff‐positive cells, and staining was conducted according to the manufacturer's instructions. Briefly, sections were rinsed with distilled water for, oxidised in 0.5% periodic acid solution, followed by washing with distilled water. Slides were then placed in Schiff's reagent, washed in warm running tap water, then counterstained in Gill 3 Haematoxylin.
Statistical analysis
Statistical analyses were performed using GraphPad InStat software (GraphPad Software Inc, La Jolla, CA, USA). Data distributions were determined by the D'Agostino and Pearson normality test. Comparisons between groups were made by an unpaired t‐test, the Mann–Whitney U‐test or the Chi‐squared test where indicated. Pearson or Spearman's correlations were performed to determine associations between gene expression and other markers of T2 and eosinophilic inflammation.
Conflicts of interest
DS has received sponsorship to attend and speak at international meetings, honoraria for lecturing or attending advisory boards from the following companies: Aerogen, AstraZeneca, Boehringer Ingelheim, Chiesi, Cipla, CSL Behring, Epiendo, Genentech, GlaxoSmithKline, Glenmark, Gossamerbio, Kinaset, Menarini, Novartis, Pulmatrix, Sanofi, Teva, Theravance and Verona. AH and JD have no conflicts of interest. T‐HP and CM are employees of AstraZeneca.
Author Contributions
Andrew Higham: Conceptualization; data curation; formal analysis; methodology; writing – original draft; writing – review and editing. Josiah Dungwa: Methodology; writing – review and editing. Christopher McCrae: Resources; writing – review and editing. Tuyet‐Hang Pham: Resources; writing – review and editing. Dave Singh: Conceptualization; data curation; formal analysis; funding acquisition; investigation; methodology; supervision; writing – original draft; writing – review and editing.
Supporting information
Supplementary figures 1‐15
Acknowledgments
DS is supported by the National Institute for Health Research (NIHR) Manchester Bio‐medical Research Centre (BRC). The authors acknowledge the North West Lung Centre Charity for supporting this project. In addition, we thank the study participants for their contribution. This report is independent research and the views expressed in this publication are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health. Data included in this study were obtained from samples collected from a previous bronchoscopy study funded by Medimmune.
References
- 1. Higham A, Quinn AM, Cancado JED, Singh D. The pathology of small airways disease in COPD: historical aspects and future directions. Respir Res 2019; 20: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rutgers SR, Postma DS, ten Hacken NH et al. Ongoing airway inflammation in patients with COPD who do not currently smoke. Thorax 2000; 55: 12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rutgers SR, Timens W, Kaufmann HF, van der Mark TW, Koeter GH, Postma DS. Comparison of induced sputum with bronchial wash, bronchoalveolar lavage and bronchial biopsies in COPD. Eur Respir J 2000; 15: 109–115. [DOI] [PubMed] [Google Scholar]
- 4. Higham A, Leow‐Dyke S, Jackson N, Singh D. Stability of eosinophilic inflammation in COPD bronchial biopsies. Eur Respir J 2020; 56: 2004167. [DOI] [PubMed] [Google Scholar]
- 5. Kolsum U, Damera G, Pham TH et al. Pulmonary inflammation in patients with chronic obstructive pulmonary disease with higher blood eosinophil counts. J Allergy Clin Immunol 2017; 140: 1181–1184.e7. [DOI] [PubMed] [Google Scholar]
- 6. Singh D, Bafadhel M, Brightling CE et al. Blood eosinophil counts in clinical trials for chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2020; 202: 660–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Singh D, Agusti A, Anzueto A et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease: the GOLD science committee report 2019. Eur Respir J 2019; 53: 1900164. [DOI] [PubMed] [Google Scholar]
- 8. Higham A, Beech A, Wolosianka S et al. Type 2 inflammation in eosinophilic chronic obstructive pulmonary disease. Allergy 2021; 76: 1861–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. George L, Taylor AR, Esteve‐Codina A et al. Blood eosinophil count and airway epithelial transcriptome relationships in COPD versus asthma. Allergy 2020; 75: 370–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Andersson CK, Mori M, Bjermer L, Lofdahl CG, Erjefalt JS. Novel site‐specific mast cell subpopulations in the human lung. Thorax 2009; 64: 297–305. [DOI] [PubMed] [Google Scholar]
- 11. Bradding P, Arthur G. Mast cells in asthma ‐ state of the art. Clin Exp Allergy 2016; 46: 194–263. [DOI] [PubMed] [Google Scholar]
- 12. Soltani A, Ewe YP, Lim ZS et al. Mast cells in COPD airways: relationship to bronchodilator responsiveness and angiogenesis. Eur Respir J 2012; 39: 1361–1367. [DOI] [PubMed] [Google Scholar]
- 13. Gosman MM, Postma DS, Vonk JM et al. Association of mast cells with lung function in chronic obstructive pulmonary disease. Respir Res 2008; 9: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Andersson CK, Mori M, Bjermer L, Lofdahl CG, Erjefalt JS. Alterations in lung mast cell populations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2010; 181: 206–217. [DOI] [PubMed] [Google Scholar]
- 15. Winter NA, Qin L, Gibson PG et al. Sputum mast cell/basophil gene expression relates to inflammatory and clinical features of severe asthma. J Allergy Clin Immunol 2021; 148: 428–438. [DOI] [PubMed] [Google Scholar]
- 16. Louis RE, Cataldo D, Buckley MG et al. Evidence of mast‐cell activation in a subset of patients with eosinophilic chronic obstructive pulmonary disease. Eur Respir J 2002; 20: 325–331. [DOI] [PubMed] [Google Scholar]
- 17. Vieira Braga FA, Kar G, Berg M et al. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat Med 2019; 25: 1153–1163. [DOI] [PubMed] [Google Scholar]
- 18. Jiang J, Faiz A, Berg M et al. Gene signatures from scRNA‐seq accurately quantify mast cells in biopsies in asthma. Clin Exp Allergy 2020; 50: 1428–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Suurmond J, Habets KLL, Tatum Z et al. Repeated FcepsilonRI triggering reveals modified mast cell function related to chronic allergic responses in tissue. J Allergy Clin Immunol 2016; 138: 869–880. [DOI] [PubMed] [Google Scholar]
- 20. Tiotiu A, Badi Y, Kermani NZ et al. Association of differential mast cell activation to granulocytic inflammation in severe asthma. Am J Respir Crit Care Med 2022; 205: 397–411. [DOI] [PubMed] [Google Scholar]
- 21. Berry M, Morgan A, Shaw DE et al. Pathological features and inhaled corticosteroid response of eosinophilic and non‐eosinophilic asthma. Thorax 2007; 62: 1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gizycki MJ, Hattotuwa KL, Barnes N, Jeffery PK. Effects of fluticasone propionate on inflammatory cells in COPD: an ultrastructural examination of endobronchial biopsy tissue. Thorax 2002; 57: 799–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hattotuwa KL, Gizycki MJ, Ansari TW, Jeffery PK, Barnes NC. The effects of inhaled fluticasone on airway inflammation in chronic obstructive pulmonary disease: a double‐blind, placebo‐controlled biopsy study. Am J Respir Crit Care Med 2002; 165: 1592–1596. [DOI] [PubMed] [Google Scholar]
- 24. James A, Gyllfors P, Henriksson E et al. Corticosteroid treatment selectively decreases mast cells in the smooth muscle and epithelium of asthmatic bronchi. Allergy 2012; 67: 958–961. [DOI] [PubMed] [Google Scholar]
- 25. Dougherty RH, Sidhu SS, Raman K et al. Accumulation of intraepithelial mast cells with a unique protease phenotype in TH2‐high asthma. J Allergy Clin Immunol 2010; 125: 1046–1053.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang G, Baines KJ, Fu JJ et al. Sputum mast cell subtypes relate to eosinophilia and corticosteroid response in asthma. Eur Respir J 2016; 47: 1123–1133. [DOI] [PubMed] [Google Scholar]
- 27. Weidner N, Austen KF. Heterogeneity of mast cells at multiple body sites. Fluorescent determination of avidin binding and immunofluorescent determination of chymase, tryptase, and carboxypeptidase content. Pathol Res Pract 1993; 189: 156–162. [DOI] [PubMed] [Google Scholar]
- 28. KleinJan A, Godthelp T, Blom HM, Fokkens WJ. Fixation with Carnoy's fluid reduces the number of chymase‐positive mast cells: not all chymase‐positive mast cells are also positive for tryptase. Allergy 1996; 51: 614–620. [DOI] [PubMed] [Google Scholar]
- 29. Yousuf A, Ibrahim W, Greening NJ, Brightling CE. T2 Biologics for Chronic Obstructive Pulmonary Disease. J Allergy Clin Immunol Pract 2019; 7: 1405–1416. [DOI] [PubMed] [Google Scholar]
- 30. Singh D, Agusti A, Martinez FJ et al. Blood eosinophils and chronic obstructive pulmonary disease: a global initiative for Chronic Obstructive Lung Disease Science Committee 2022 review. Am J Respir Crit Care Med 2022; 206: 17–24. [DOI] [PubMed] [Google Scholar]
- 31. Higham A, Karur P, Jackson N, Cunoosamy DM, Jansson P, Singh D. Differential anti‐inflammatory effects of budesonide and a p38 MAPK inhibitor AZD7624 on COPD pulmonary cells. Int J Chron Obstruct Pulmon Dis 2018; 13: 1279–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 2014; 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bhakta NR, Solberg OD, Nguyen CP et al. A qPCR‐based metric of Th2 airway inflammation in asthma. Clin Transl Allergy 2013; 3: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Southworth T, Van Geest M, Singh D. Type‐2 airway inflammation in mild asthma patients with high blood eosinophils and high fractional exhaled nitric oxide. Clin Transl Sci 2021; 14: 1259–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chtanova T, Newton R, Liu SM et al. Identification of T cell‐restricted genes, and signatures for different T cell responses, using a comprehensive collection of microarray datasets. J Immunol 2005; 175: 7837–7847. [DOI] [PubMed] [Google Scholar]
- 36. Jayapal M, Tay HK, Reghunathan R et al. Genome‐wide gene expression profiling of human mast cells stimulated by IgE or FcεRI‐aggregation reveals a complex network of genes involved in inflammatory responses. BMC Genomics 2006; 7: 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nagarkar DR, Ramirez‐Carrozzi V, Choy DF et al. IL‐13 mediates IL‐33‐dependent mast cell and type 2 innate lymphoid cell effects on bronchial epithelial cells. J Allergy Clin Immunol 2015; 136: 202–205. [DOI] [PubMed] [Google Scholar]
- 38. Okumura S, Kashiwakura J, Tomita H et al. Identification of specific gene expression profiles in human mast cells mediated by Toll‐like receptor 4 and FcepsilonRI. Blood 2003; 102: 2547–2554. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures 1‐15