

Abstract
Background
Complex genomic profiling (CGP) has transformed cancer treatment decision making, yet there is a lack of robust and quantifiable evidence for how utilisation of CGP improves patient outcomes.
Objective
This study evaluated cohort level clinical effectiveness of CGP to improve overall survival (OS) in real-world advanced cancer patients using a registry-based matched control population.
Patients and methods
Two cohorts of advanced and refractory cancer patients were seen in consecutive series for early phase trial enrolment consideration. The first cohort (CGP group) accessed tumour profiling via a research study; while the second cohort that followed was not profiled. Overall survival between cohorts was compared using Kaplan-Meier curves and Cox proportional hazard models. Potential confounding was analysed and adjusted for using stabilised weights based on propensity scores.
Results
Within the CGP group, 25 (17.6%) patients received treatment informed by CGP results and this subgroup had significantly improved survival compared with CGP patients in whom results did not impact their treatment (unadjusted HR = 0.44, (0.22–0.88), p = 0.02). However, when comparing the entire CGP cohort with the No CGP cohort, no significant survival benefit was evident with adjusted median OS for CGP of 13.5 months (9.2–17.0) compared with 11.0 (9.2–17.4) for No CGP (adjusted HR = 0.92, (0.65–1.30), p = 0.63).
Conclusions
This study utilised real-world data to simulate a control arm and quantify the clinical effectiveness of genomic testing. The magnitude of survival benefit for patients who had CGP result-led treatments was insufficient to drive an overall survival gain for the entire tested population. Translation of CGP into clinics requires strategies to ensure higher rates of tested patients obtain clinical benefit to deliver on the value proposition of CGP in an advanced cancer population.
Supplementary Information
The online version contains supplementary material available at 10.1007/s11523-022-00910-0.
Key Points
| Complex genomic profiling can facilitate meaningful clinical benefit for a subset of tested advanced and refractory cancer patients who access clinical trials and targeted therapy. |
| The proportion of tested patients who receive personalised treatment and derive a survival benefit from complex genomic profiling must be substantial to ensure the value of testing this population in a real-world setting. |
Introduction
The transformative role of genomics for informing tailored treatment decisions in oncology is underpinned by increasingly sophisticated profiling methods using next-generation sequencing (NGS) technologies. Complex genomic characterisation of cancers, whether by large, targeted panels, whole genome, or multi-omics assays, facilitates efficient identification of established targetable variants, but also allows identification of novel predictive biomarkers driving development of therapeutics for specifically defined subpopulations. The proportion of Phase 3 trials in common cancers, including a molecularly targeted agent, shows a dramatic increasing trend from 26% in 2010 to 62% in 2020 [1]. This is confirmed by the multitude of recent international approvals for targeted agents, including against pan-cancer biomarkers, validating the significance of the molecular profile in systemic treatment decisions in oncology [2, 3].
Globally, there are many large-scale pan-cancer precision oncology studies offering complex genomic profiling (CGP), often in the advanced/refractory setting with the rationale of identifying targets for experimental treatment options. Notable studies such as the ProfiLER study in Europe or NCI-Match and I-PREDICT in the USA used molecular profiling results to recommend therapies via clinical trials or access programmes for patients with actionable biomarkers. These studies have largely demonstrated positive clinical outcomes for the subset of tested patients who received result-led therapy. The ProfiLER study reported that 44 % of matched therapy lines initiated achieved disease control or partial response and a recent I-PREDICT publication showed doubling of disease control rates and progression-free survival for those initiating a treatment regimen with higher matching scores (proportion of identified biomarkers targeted through combination therapy) [4–7].
Despite growing support for genomic-informed treatment in a research context [4, 7–9], there has not been broad adoption of CGP into routine care and research even shows rates of simpler standard profiling are suboptimal [10-–13]. Widespread access to testing still requires a significant investment and infrastructure to facilitate at scale. To justify investment in CGP, public health systems and insurance companies require evidence of comparative effectiveness of using complex testing to improve health outcomes in a tested population. Currently, our ability to gather this evidence is limited by the fact that studies are non-randomised, and largely in advanced and refractory populations where there are often low rates of initiating result directed therapy (~ 15%) as a proportion of the tested population [4, 9, 10, 12, 14–16]. A lack of robust and quantifiable evidence for how utilisation of CGP improves patient outcomes, means that there is a paucity of economic evaluations of complex testing in the literature [11, 14, 15]. Together these evidence gaps result in uncertainty and delays in funding at a health system level, which are critical to facilitate integration of CGP to clinical care.
The evidentiary gold standard of randomised controlled trials (RCTs) for pan-cancer molecular profiling directed treatment compared to standard of care are difficult to conduct due to the heterogeneity in cohorts and outcomes and the prioritisation of testing to maximise genomic discovery [4, 14, 17]. The landmark SHIVA trial performed molecular profiling on patients who were then randomised to receive a targeted therapy or standard treatment, and is the only randomised precision oncology study to date. Although this study demonstrated no survival benefit, these results were published in 2015 and there has been a substantial growth in testing capability and access to targeted therapies since that time [18]. Precision oncology studies are typically single arm with heterogeneous patient populations, and diverse treatment histories. Therefore, demonstrating the relative effectiveness of complex testing is difficult as there is no easily defined counterfactual cohort. Commonly, reported outcomes are between those receiving targeted versus untargeted therapy, which suffers from potential biological and clinical selection bias, and the use of a fixed progression-free survival ratio with patients as their own control results in outcomes that are dependent on a patients’ treatment history [4, 5]. Outcomes are also rarely described across the entire tested population, where the majority of tested patients do not benefit from CGP because of patient deterioration and the lack of availability of targeted treatments [6, 19, 20].
A potential avenue to overcome the evidentiary limitations of single-arm precision oncology trials is to apply a registry-based synthetic control arm to a group receiving CGP, which permits a health outcome assessment across the entire tested population rather than one focusing on selected patients who receive treatment [12, 21]. The use of real-world registry or health record data as a source of external or ‘synthetic’ control is expanding in clinical research and is recognised by various international regulatory bodies including the US Food and Drug Administration (FDA) and the National Institute for Health and Care Excellence (NICE) [22–24]. This study aims to estimate the overall survival following the use of a CGP programme for advanced and refractory cancer patients compared to a similar real-world patient cohort who did not receive CGP.
Methods
This was a retrospective observational study utilising two series of advanced and refractory cancer patients who were referred for consideration of early phase trials through a specialised tertiary centre clinic after progression on standard of care treatment. One of these cohorts was offered routine CGP through a research programme. We simulated a synthetic trial using these (CGP and No CGP) cohorts to compare overall survival (OS) using propensity score methods to balance the two groups based on clinical characteristics.
CGP Cohort
The cohort receiving CGP was a subset of a national single-arm prospective complex testing study recruited via the Peter MacCallum Cancer Centre Phase 1 Trials Programme in Melbourne between March 2017 and August 2018. The clinic offered profiling via the CGP study to uncover targeted treatment options to pursue via clinical trials or access pathways after progression on standard of care treatment. Informed consent for testing was obtained with oncologists from the study team. The CGP assay was an in-house 391 gene comprehensive cancer DNA panel (see Supplementary Notes). Treatment recommendations were made after curation of results and multidisciplinary review at a molecular tumour board. Participants were followed for a minimum of 12 months post-test to capture result-led treatment decisions and overall survival.
Control Cohort (No CGP)
Patients attending the same clinic in the 12 months following the recruitment of the CGP study population (recruited between September 2018 and September 2019) were identified as a potential counterfactual advanced-stage pan-cancer cohort and did not have access to complex profiling through the clinic. Any standard of care (defined as publicly reimbursed) profiling and treatment had been actioned before referral for trials, as was the case for the CGP group. Therefore, this cohort presented a unique control sample with minimal selection bias around employing complex genomics to uncover treatment options. Baseline and survival data were collected via an administrative register for these patients.
Study Eligibility
Cohorts were refined further for a cohesive population based on eligibility of (a) metastatic or incurable stage, (b) Eastern Cooperative Oncology Group (ECOG) less than or equal to two, (c) failure of a standard initial line of systemic therapy where this exists, and (d) non-synchronous cancers (Fig. 1). Colorectal cancers were excluded from analysis due to intentionally limited recruitment in the CGP cohort leading to major imbalance between cohorts and likely violation of the positivity assumption of propensity score analysis (see below) [25]. Patients in the CGP study where testing failed due to technical issues or a lack of sample available for testing were included in the No CGP cohort as there was no probability of result impacting treatment in these cases and their experience more closely reflected that of the No CGP cohort. Patients were excluded from the No CGP cohort if they presented to clinic with previous complex testing such as self-funded commercial panels.
Fig. 1.
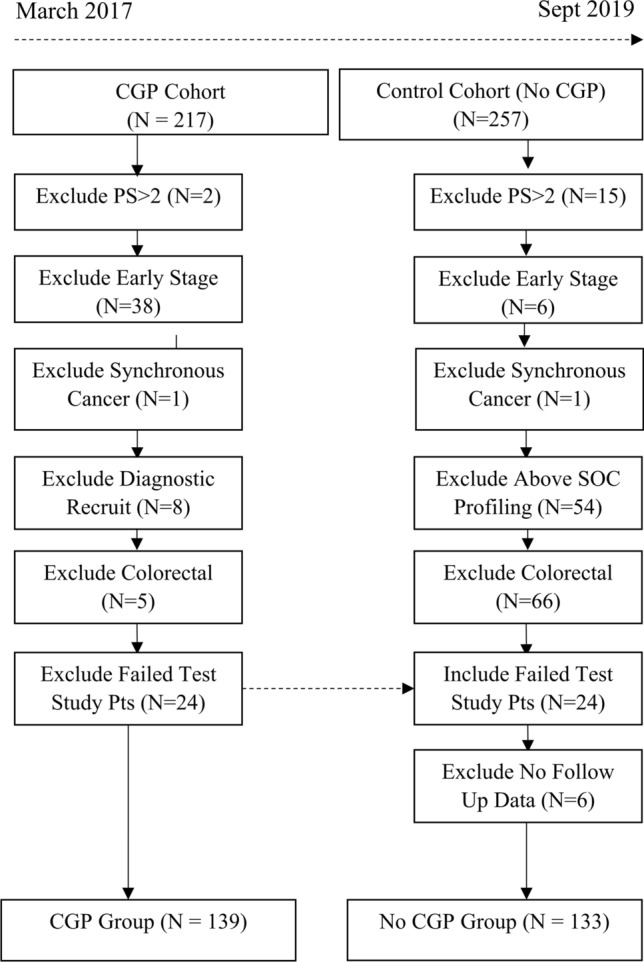
CONSORT diagram. CGP complex genomic profiling, PS performance status, Pts patients
Derivation of Propensity Score Matching and Weighting
The propensity score is defined as the probability of treatment assignment conditional on key covariates and is used to create a balanced sample that would normally be obtained through randomisation [26, 27]. Propensity score analysis offers advantages over more common methods for confounding adjustment, such as multiple regression, through transparent balancing of covariates across groups in a process independent from outcome analysis [26, 28, 29]. We used logistic regression to estimate the probability of receiving CGP for each patient based on factors that may impact their likelihood of being in the CGP group or that have prognostic relevance. The following patient and disease characteristics were included as covariates to account for measured confounding: age, sex, ECOG, cancer type, failed lines of systemic treatment and remaining reimbursed therapeutic options as per clinician at initial appointment. Although all these clinically relevant covariates were included, i.e., no variable selection was performed, variable transformations were considered for age and potential interactions with failed lines and cancer types to explore optimal model fit, with the final transformations selected based on Akaike Information Criterion (AIC). The resulting propensity scores were used in inverse probability of treatment weighted (IPTW) analyses. Stabilised weights were calculated and truncated at the 1st and 99th percentile to reduce excessive influence of uncommon patient profiles [25]. Inverse probability of treatment weighted methods were employed over alternatives, such as propensity score matching, due to the priority of retaining sample size. Assessment of balance achieved in baseline covariates was compared using weighted standardised differences [25].
Outcomes and Survival Analysis Methods
Date of genomic study consent for CGP was the index date for overall survival, and initial clinic visit was the index date for the No CGP group. For both cohorts, this represented the start of the process for identifying investigative treatment options. As the cohorts reflected successive time periods, potential follow-up time was systematically different. All patients had a minimum of 12 months’ follow up and survival analysis was censored at 18 months to reduce discrepancy in time at risk across cohorts. Unadjusted and IPTW adjusted Kaplan-Meier survival curves were used to generate plots and estimate survival probabilities, unadjusted and IPTW adjusted hazard ratios (HRs) were estimated using Cox proportional hazards regression. It was calculated that a HR of 0.65 could be detected with 80% power at a 5% significance level with a sample of 260 and a 65% event rate [30]. The proportional hazards assumption was assessed through significance testing of scaled Schoenfeld residuals. Robust variance estimates were used in adjusted analyses to account for the dependency induced through weighting subjects [31]. Data cleaning was performed in Stata/IC version 14.2 and analysis and graphics performed using R statistical software, version 3.6.3.
Results
Cohort Description
A total of 272 patients were included in the analysis, with 139 receiving CGP and 133 in the No CGP cohort (Table 1). Distributions of variables across groups were similar, but there was evidence of imbalance as demonstrated by standardised differences greater than 0.1 for several factors. The CGP patients were slightly younger, more frequently ECOG 0 compared to 1, had more sarcomas and non-cutaneous melanomas but fewer head and neck, genitourinary and upper gastrointestinal primaries. The CGP group had lower numbers of failed lines of systemic therapy, likely reflecting higher rates of rare subtypes of cancers without many standard systemic options. For the CGP group, 25 (17 %) patients received a result-led targeted treatment (Table S1, see Supplementary Material) largely through clinical trials or off-label access programmes. Rates of trial enrolment were similar across groups with 31 (22%) in CGP (16 result-led, 15 non-targeted) compared with 34 (26 %) in No CGP. For those who did not enrol in a clinical trial, and for whom standard treatment data were available, rates of initiating further treatment after baseline visit were also similar (CGP 32 % and No CGP 30 %). Median follow-up time in the CGP group was higher (median 13.8 [interquartile range (IQR) 6.5–18.0] months) than No CGP (median 9.4 [IQR 4.3–14.9] months) reflecting the earlier index dates in the CGP group. However, all patients had data collected between 12 and 18 months and event rates were similar with 78 (55 %) for CGP and 81 (61 %) for No CGP.
Table 1.
Baseline characteristics by CGP group
| Unadjusted | Adjusted using IPTW | ||||||
|---|---|---|---|---|---|---|---|
| CGP (N = 139) | No CGP (N = 133) | p-value | SD | CGP (N = 141.4) | No CGP (N = 125.4) | SD | |
| Age | 57.4 [21.8–86.1] | 61.3 [30.8–82.5] | 0.014* | 0.342 | 60.7 [21.8–86.1] | 59.7 [30.8–82.5] | 0.005 |
| Sex | |||||||
| Male | 73 (52.5) | 74 (55.6) | 0.693 | 0.063 | 77.1 (54.5) | 69.3 (55.3) | 0.015 |
| ECOG | |||||||
| 0 | 58 (41.7) | 36 (27.1) | 0.032* | 0.332 | 49.2 (34.8) | 42.4 (33.8) | 0.031 |
| 1 | 72 (51.8) | 89 (66.9) | 83.5 (59.1) | 75.8 (60.5) | |||
| 2 | 9 (6.5) | 8 (6.0) | 8.7 (6.2) | 7.1 (5.7) | |||
| Failed lines of systemic treatment | |||||||
| 0 [0–6] | 1 [0–7] | < 0.001** | 0.539 | 1 [0–6] | 1 [0–7] | 0.085 | |
| Therapeutic options remainingϯ | |||||||
| 0 | 87 (62.6) | 92 (69.2) | 0.158 | 0.235 | 93.1 (65.8) | 81.5 (65.0) | 0.040 |
| 1 | 39 (28.1) | 36 (27.1) | 39.3 (27.8) | 36.7 (29.3) | |||
| 2 or more | 13 (9.4) | 5 (3.8) | 9.0 (6.4) | 7.1 (5.7) | |||
| Cancer primary | |||||||
| CUP | 7 (5.0) | 10 (7.5) | 0.021* | 0.534 | 9.7 (6.9) | 8.5 (6.7) | 0.103 |
| GU | 4 (2.9) | 9 (6.8) | 4.5 (3.2) | 5.9 (4.7) | |||
| Gynae | 15 (10.8) | 11 (8.3) | 13.5 (9.5) | 11.6 (9.2) | |||
| H&N/NM Skin | 19 (13.7) | 24 (18.0) | 25.6 (18.1) | 22.1 (17.6) | |||
| Lung | 17 (12.2) | 16 (12.0) | 16.7 (11.8) | 14.4 (11.5) | |||
| Melanoma | 12 (8.6) | 3 (2.3) | 7.4 (5.2) | 5.0 (4.0) | |||
| Other rare | 17 (12.2) | 9 (6.8) | 12.8 (9.1) | 11.5 (9.2) | |||
| Sarcoma | 22 (15.8) | 11 (8.3) | 16.7 (11.8) | 14.4 (11.5) | |||
| UGI | 26 (18.7) | 40 (30.1) | 34.4 (24.4) | 32.5 (25.9) | |||
| Rare | |||||||
| Yes | 106 (76.3) | 85 (63.9) | 0.036* | 0.272 | 102.4 (72.4) | 87.1 (69.5) | 0.066 |
Data presented as n (%) or median [range], p-value determined by Chi squared statistic or Mann Whitney U test as appropriate
Rare classification defined by specific histology as incidence < 6 per 100,000
CGP complex genomic profiling, CUP cancer of unknown primary, ECOG Eastern Cooperative Oncology Group, GU genitourinary cancer, H&N/NM head and neck non-melanoma, IPTW inverse probability of treatment weighted, SD standardised difference, UGI upper gastrointestinal cancer
ϮAs per referring clinician or study clinician review, *p < 0.05, **p < 0.01
Inverse Probability of Treatment Weights and Balance Diagnostics
The propensity score model is presented in Table S2 (see Supplementary Material). The IPTWs ranged from 0.54 to 2.59, with a mean of 0.98, and the distribution of weights was similar across treatment groups (Figure S1, Supplementary Material), indicating there were no signs of individuals with a disproportionately large weight in the analysis. Applying IPTWs to our sample resulted in well-balanced cohorts, this weighting resulted in slightly adjusted denominators (Table 1).
Survival Analysis
Results for all survival analyses are presented in Table 2. Kaplan-Meier curves for the CGP and No CGP cohorts (Fig. 2a) and further stratified by whether result-led treatment (RLT) was initiated for the CGP cohort (Fig. 2b), display the survival benefit for CGP. The unadjusted assessment of OS within the CGP group indicated potential benefits of receiving a RLT, with median OS not reached for those with RLT and 13.6 months for those where CGP results did not influence subsequent treatment (HR [95 % CI] = 0.44 [0.22–0.88], p = 0.02). In the unadjusted comparison, the CGP group demonstrated significantly improved median OS (14.4 vs 9.9 months, HR [95 % CI] = 0.71 [0.53–0.96], p = 0.03) compared to no CGP. Figure2b displays the benefit from CGP in the RLT subgroup, but also shows improved OS for the CGP (No RLT) subgroup compared to No CGP.
Table 2.
Survival analyses by CGP groups
| Within CGP group (N = 139) | CGP result-led impact (n = 25) | No result-led impact (n = 114) | p value |
|---|---|---|---|
| Median survival (months) | NR | 13.6 (10.4–17.8) | |
| 12-month survival probability | 80.0% (65.8–97.3) | 52.5% (44.0–62.7) | |
| Log rank test | 9 events | 68 events | 0.02* |
| Univariate hazard ratio | 0.44 (0.22–0.88) | 0.02* |
| Unadjusted CGP vs no CGP (N = 272) | CGP (n = 139) | No CGP (n = 133) | p value |
|---|---|---|---|
| Median survival (months) | 14.4 (12.0–NR) | 9.9 (8.6–13.4) | |
| 12-month survival probability | 57.5% (49.8–66.5) | 41.6% (33.7–51.2) | |
| Log rank test | 77 events | 81 events | 0.03* |
| Univariate hazard ratio | 0.71 (0.52–0.97) | 0.03* |
| IPTW (adjusted) CGP vs no CGP (N = 276) | CGP (n = 141) | No CGP (n = 125) | p value |
|---|---|---|---|
| Median survival (months) | 13.5 (9.2–17.0) | 11.0 (9.2–17.4) | |
| 12-month survival probability | 52.5% (43.9–62.8) | 45.8% (37.2–56.4) | |
| Adjusted univariate hazard ratio | 0.92 (0.65–1.30) | 0.63 |
CGP complex genomic profiling, IPTW inverse probability of treatment weighted, NR not reached
Hazard ratio determined by Cox proportional hazards regression
Survival probabilities determined by Kaplan-Meier methods
95 % confidence intervals presented in brackets, *p < 0.05
Fig. 2.

Kaplan-Meier survival curves displaying overall survival stratified by complex genomic profiling (CGP) group and result-led treatment (RLT)
The survival benefit for the CGP cohort in the unadjusted analysis was no longer demonstrated in the IPTW-adjusted survival analyses that accounted for imbalance in prognostic variables between the cohorts (median OS 13.5 vs 11.0, HR [95 % CI] = 0.9 [0.65–1.31], p = 0.63) (Fig. 2c). When the adjusted CGP group is stratified into RLT and no RLT (Fig. 2d) the balancing to represent a synthetic trial, achieved through weighting cohorts is evident as the ‘CGP (No RLT) and No CGP curves overlap. Although Fig. 2d shows survival gains for those patients where actionable targets were identified and actioned in the CGP cohort, this was of benefit to only 17 % of patients and is insufficient to drive a survival gain for the entire CGP cohort. There was no evidence of violation of the proportional hazards assumption across analyses.
Discussion
This study demonstrates the potential value of real-world data to facilitate comparative effectiveness analyses that are critical to the evaluation and translation of CGP for advanced cancer populations. Despite our results supporting earlier findings that patients with RLT initiated from CGP have improved OS, this proportion was not sufficient to establish a significant OS benefit across the entire tested cohort.
The novelty of this study was the identification of a suitable control cohort and use of endorsed methodology to adjust for confounding in a real-world setting [22–24]. This analysis builds on literature supporting the potential of complex profiling to enable viable treatment options for a proportion of advanced cancer patients, as it extends on the common ‘within group’ survival comparisons that are often reported for single-arm cohorts. Through achieving a balanced control cohort, we were able to evaluate the effectiveness of testing versus no testing, rather than a more biased and restricted ‘targeted versus untargeted’ testing subgroup evaluation. This comparison differs from conventional ‘targeted versus untargeted’ comparisons, as patients who do not initiate treatment after testing are not excluded from analysis. We retained these patients in our CGP (No RLT) and No CGP subgroups, as the primary aim was assessing the value of CGP in the entire tested population, irrespective of the treatment outcome. This minimises selection bias from restricting evaluation to treated patients, as all tested patients are deemed suitable for treatment at the time of initiating testing.
The use of real-world control arms is emerging in oncology as an approach to evaluate rare and challenging cohorts such as clinical outcomes for rare biomarker-positive populations with the Flatiron Health database [32, 33]. To our knowledge the only other study using real-world data to estimate the comparative effectiveness of CGP to inform treatment in advanced pan-cancer is a recent article by Weymann et al [21]. They compared survival outcomes for patients undergoing whole genome and transcriptome sequencing with propensity-score matched controls from a population-based cancer registry in Canada. Findings were consistent with our study, where a minority (15 %) of tested patients received result-led treatment, and no OS gain for tested patients was seen compared to matched controls. The use of a larger dataset permitted a comprehensive matching approach for control selection but required estimation of temporal data such as the index date for the control group. Our study utilised propensity score weighting to retain maximum data. As observational data become more commonly employed for complex evaluations, it will be critical to ensure clinical assumptions informing analyses hold to leverage the potential of observational data for reducing rather than amplifying uncertainty in estimating intervention effects. Further research exploring study designs such as simulated controls will support much-needed evaluation of the relative benefits of complex genomic testing strategies.
With a rate of RLT for CGP patients of 17 % and a dramatic survival benefit demonstrated in this RLT subgroup, a lack of survival gain in the overall CGP cohort in our adjusted comparative analysis supports commonly cited concerns around the current limited clinical effectiveness, and by extension the cost effectiveness, of complex genomics from a payer or public health perspective [4, 11, 14]. Given that the proportion and impact of RLT change in this CGP cohort is comparable to international studies in the literature, such as ProfiLER and NCI-Match who reported treatment initiation in tested patient rates of 6 % and 12 %, respectively, the likelihood of similar limitations on effectiveness of using CGP to improve survival in these larger advanced and refractory cancer cohorts is real [4, 6, 21, 35].
These results highlight the importance of increasing the rate and clinical impact of actionable findings to achieve real-time targeted treatment survival benefits that support genomic testing. Commonly reported reasons for low rates of RLT initiation in precision oncology studies include lack of biomarkers identified, patient deterioration, continuation of standard therapy and lack of trial and drug access [4, 15, 34]. Efficiency gains and cost reductions of scaling up complex testing in conjunction with more comprehensive and sensitive testing technology and growing targeted therapy options will support increasing use of CGP. However, implementation challenges such as inequity and barriers to trial access, prohibitive drug costs and lack of decision support for clinicians mean CGP impact beyond informing standard of care treatments may be even lower in a community setting and must be considered [11, 12, 16, 35, 36]. Studies exploring the use of CGP to inform initial systemic treatment options rather than the more common refractory populations may better capture the relative survival benefits of CGP informed access to both standard and investigative targeted therapies, particularly for patients with lung cancer or sarcoma where molecular profiles can define the entire treatment pathway [11]. Notably improved evidence for the comparative effectiveness of CGP in terms of improved survival is only an initial step towards translation as subsequent economic evaluations will be largely influenced by therapeutic access and costs. Further research utilising linked clinical and genomic real-world data will be key to identifying cancer populations and timepoints when using CGP to inform treatment will be particularly valuable and encourage large-scale testing access for those most likely to yield real-time survival benefits [12, 36–39].
As a retrospective study, this work has certain limitations. As with any observational study, there is potential for unmeasured confounding that has not been adjusted for [23]. As a single-site study, the sample size was limited and with the adjusted HR of 0.92, the study was not sufficiently powered to detect if this is a minor yet statistically significant clinical benefit demonstrated by the CGP group. The analysis relies on the assumption that refractory cancer patients recruited through the clinic for CGP represent a similar distribution of patients referred for clinical trial assessment to those in the No CGP group. To negate the impact of this limitation, thorough eligibility criteria were applied to ensure the final cohorts represented similar patient groups. The two series of patients that immediately followed each other across a total recruitment period of 32 months, had a large overlap in trials available, which reduces but cannot remove the potential for favourable bias in survival for the No CGP group through improved treatments or supportive care. In this study, completion of available data was high; however, additional prognostic clinical data such as liver function or sites of metastatic disease and a larger overall sample size could have added greater validity to the specification of the propensity score model and prognostic comparability of the two groups, particularly with such a heterogenous population [40]. To enable a comparison of cohorts with CGP results versus no CGP, we included CGP patients with failed testing in the No CGP cohort, and while this may minimise potential bias due to the differing recruitment time periods, other sources of bias such as biological factors with low volume disease cannot be ruled out. Finally, the study involved patients seen at an academic centre who had failed standard of care treatment, and while this was consistent across the CGP and No CGP cohorts, the results may therefore not be generalisable to a community setting.
Conclusions
In summary, this study utilised a synthetic control cohort to assess the relative survival benefit of using CGP in an advanced real-world pan-cancer population to overcome the bias in precision oncology utility estimates and determine the value of testing at a population level. Although currently the use of CGP may result in limited gains to an unselected, late-stage population, further real-world evidence exploring how CGP can impact certain cancer subpopulations at higher rates and through earlier initiation in the treatment pathway, will allow identification of more effective testing strategies and support translation of genomics into oncology care where it is of most clinical value.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors acknowledge all contributing members of the Melbourne Genomics Health Alliance Solid Tumour Flagship team and the Molecular Pathology department of the Peter MacCallum Cancer Centre. We thank all patients and their families for their involvement in the study.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Declarations
Funding
The CGP study was funded the Melbourne Genomics Health Alliance. No funding was received for performing this analysis. Sophie O’Haire was supported by an Australian Government Research Training Program (RTP) Fee-Offset Scholarship through The University of Melbourne.
Conflicts of interest
SOH, KD, FF, CG, SJL, SF and JD declare no potential conflicts of interest. MIJ receives unrestricted research funding to the University of Melbourne from Illumina. BT reports grants and personal fees from Amgen, grants and personal fees from Astra Zeneca, grants from Astellas, grants and personal fees from BMS, grants and personal fees from Janssen, grants and personal fees from Pfizer, grants and personal fees from MSD, grants and personal fees from Ipsen, personal fees from IQVIA, personal fees from Sanofi, personal fees from Tolmar, personal fees from Novartis, grants and personal fees from Bayer, personal fees from Roche, outside the submitted work.
Ethics approval
The CGP study was approved by Melbourne Health HREC (approval no. HREC/13/MH/326) and approval with waiver of consent granted for de-identified clinical data collection of the No CGP cohort.
Consent to participate
Informed consent was obtained for all participants in the CGP cohort. The requirement for consent was waived for the collection and use of de-identified health outcomes data for the No CGP cohort.
Consent for publication
Not applicable.
Data and code availability
The dataset analysed during the current study is available from the corresponding author on reasonable request.
Author’s contribution
Conceptualisation (SOH, KD, FF), Methodology (SOH, KD, FF), Formal Analysis (SOH, KD, FF), Investigation (SOH, BT, SL, KS, SF, JD), Writing—Original Draft (SOH, KD, FF), Writing—Review & Editing (SOH, KD, FF, BT, SL, CG, KS, SF, JD, MIJ), Visualisation (SOH, FF), Supervision (CG, SF, JD, MIJ), Funding Acquisition (CG, SF, JD, MIJ).
References
- 1.Del Paggio JC, Berry JS, Hopman WM, Eisenhauer EA, Prasad V, Gyawali B, et al. Evolution of the randomized clinical trial in the era of precision Oncology. JAMA Oncol. 2021;7(5):728–734. doi: 10.1001/jamaoncol.2021.0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.U.S. Food & Drug Administration. Hematology/Oncology (Cancer) Approvals & Safety Notifications. 2021. https://www.fda.gov/drugs/resources-information-approved-drugs/hematologyoncology-cancer-approvals-safety-notifications. Accessed 6 Jun 2021.
- 3.European Medicines Agency. Medicines. 2021. https://www.ema.europa.eu/en/medicines. Accessed 6 Jun 2021.
- 4.Tsimberidou AM, Fountzilas E, Nikanjam M, Kurzrock R. Review of precision cancer medicine: evolution of the treatment paradigm. Cancer Treat Rev. 2020;86:102019. doi: 10.1016/j.ctrv.2020.102019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zimmer K, Kocher F, Spizzo G, Salem M, Gastl G, Seeber A. Treatment according to molecular profiling in relapsed/refractory cancer patients: a review focusing on latest profiling studies. Comput Struct Biotechnol J. 2019;17:447–453. doi: 10.1016/j.csbj.2019.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trédan O, Wang Q, Pissaloux D, Cassier P, de la Fouchardière A, Fayette J, et al. Molecular screening program to select molecular-based recommended therapies for metastatic cancer patients: analysis from the ProfiLER trial. Ann Oncol. 2019;30(5):757–765. doi: 10.1093/annonc/mdz080. [DOI] [PubMed] [Google Scholar]
- 7.Sicklick JK, Kato S, Okamura R, Schwaederle M, Hahn ME, Williams CB, et al. Molecular profiling of cancer patients enables personalized combination therapy: the I-PREDICT study. Nat Med. 2019;25(5):744–750. doi: 10.1038/s41591-019-0407-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Velden DL, Hoes LR, van der Wijngaart H, van Berge Henegouwen JM, van Werkhoven E, Roepman P, et al. The Drug Rediscovery protocol facilitates the expanded use of existing anticancer drugs. Nature. 2019;574(7776):127–131. doi: 10.1038/s41586-019-1600-x. [DOI] [PubMed] [Google Scholar]
- 9.Cobain EF, Wu YM, Vats P, Chugh R, Worden F, Smith DC, et al. Assessment of clinical benefit of integrative genomic profiling in advanced solid tumors. JAMA Oncol. 2021;7(4):525–533. doi: 10.1001/jamaoncol.2020.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsiao SJ, Sireci AN, Pendrick D, Freeman C, Fernandes H, Schwartz GK, et al. Clinical utilization, utility, and reimbursement for expanded genomic panel testing in adult Oncology. JCO Precis Oncol. 2020;4:1038–1048. doi: 10.1200/PO.20.00048. [DOI] [PubMed] [Google Scholar]
- 11.Mosele F, Remon J, Mateo J, Westphalen CB, Barlesi F, Lolkema MP, et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: a report from the ESMO Precision Medicine Working Group. Ann Oncol. 2020;31(11):1491–1505. doi: 10.1016/j.annonc.2020.07.014. [DOI] [PubMed] [Google Scholar]
- 12.Presley CJ, Tang D, Soulos PR, Chiang AC, Longtine JA, Adelson KB, et al. Association of broad-based genomic sequencing with survival among patients with advanced non-small cell lung cancer in the community Oncology setting. JAMA. 2018;320(5):469–477. doi: 10.1001/jama.2018.9824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gutierrez ME, Price KS, Lanman RB, Nagy RJ, Shah I, Mathura S, et al. Genomic profiling for KRAS, NRAS, BRAF, microsatellite instability, and mismatch repair deficiency among patients with metastatic colon cancer. JCO Precis Oncol. 2019;3:PO.19.00274. doi: 10.1200/PO.19.00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Remon J, Dienstmann R. Precision oncology: separating the wheat from the chaff. ESMO Open. 2018;3(6):e000446. doi: 10.1136/esmoopen-2018-000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weymann D, Pataky R, Regier DA. Economic evaluations of next-generation precision oncology: a critical review. JCO Precis Oncol. 2018;2:1–23. doi: 10.1200/PO.17.00311. [DOI] [PubMed] [Google Scholar]
- 16.Levit LA, Kim ES, McAneny BL, Nadauld LD, Levit K, Schenkel C, et al. Implementing precision medicine in community-based oncology programs: three models. J Oncol Pract. 2019;15(6):325–329. doi: 10.1200/JOP.18.00661. [DOI] [PubMed] [Google Scholar]
- 17.Donoghue MTA, Schram AM, Hyman DM, Taylor BS. Discovery through clinical sequencing in oncology. Nat Cancer. 2020;1(8):774–783. doi: 10.1038/s43018-020-0100-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le Tourneau C, Delord JP, Gonçalves A, Gavoille C, Dubot C, Isambert N, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015;16(13):1324–1334. doi: 10.1016/S1470-2045(15)00188-6. [DOI] [PubMed] [Google Scholar]
- 19.Le Tourneau C, Borcoman E, Kamal M. Molecular profiling in precision medicine oncology. Nat Med. 2019;25(5):711–712. doi: 10.1038/s41591-019-0442-2. [DOI] [PubMed] [Google Scholar]
- 20.Schwaederle M, Zhao M, Lee JJ, Lazar V, Leyland-Jones B, Schilsky RL, et al. Association of biomarker-based treatment strategies with response rates and progression-free survival in refractory malignant neoplasms: a meta-analysis. JAMA Oncol. 2016;2(11):1452–1459. doi: 10.1001/jamaoncol.2016.2129. [DOI] [PubMed] [Google Scholar]
- 21.Weymann D, Laskin J, Jones SJM, Lim H, Renouf DJ, Roscoe R, et al. Matching methods in precision oncology: an introduction and illustrative example. Mol Genet Genomic Med. 2021;9(1):e1554. doi: 10.1002/mgg3.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thorlund K, Dron L, Park JJH, Mills EJ. Synthetic and external controls in clinical trials—a primer for researchers. Clin Epidemiol. 2020;12:457–467. doi: 10.2147/CLEP.S242097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Armstrong K. Methods in comparative effectiveness research. J Clin Oncol. 2012;30(34):4208–4214. doi: 10.1200/JCO.2012.42.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feinberg BA, Gajra A, Zettler ME, Phillips TD, Phillips EG, Jr, Kish JK. Use of real-world evidence to support FDA approval of oncology drugs. Value Health. 2020;23(10):1358–1365. doi: 10.1016/j.jval.2020.06.006. [DOI] [PubMed] [Google Scholar]
- 25.Austin PC, Stuart EA. Moving towards best practice when using inverse probability of treatment weighting (IPTW) using the propensity score to estimate causal treatment effects in observational studies. Stat Med. 2015;34(28):3661–3679. doi: 10.1002/sim.6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Austin PC. An introduction to propensity score methods for reducing the effects of confounding in observational studies. Multivariate Behav Res. 2011;46(3):399–424. doi: 10.1080/00273171.2011.568786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenbaum PR, Rubin DB. The central role of the propensity score in observational studies for causal effects. Biometrika. 1983;70(1):41–55. doi: 10.1093/biomet/70.1.41. [DOI] [Google Scholar]
- 28.Desai RJ, Franklin JM. Alternative approaches for confounding adjustment in observational studies using weighting based on the propensity score: a primer for practitioners. BMJ. 2019;367:l5657. doi: 10.1136/bmj.l5657. [DOI] [PubMed] [Google Scholar]
- 29.Merkow RP, Schwartz TA, Nathens AB. Practical guide to comparative effectiveness research using observational data. JAMA Surg. 2020;155(4):349–350. doi: 10.1001/jamasurg.2019.4395. [DOI] [PubMed] [Google Scholar]
- 30.Chow S, Shao J, Wang H. Sample size calculations in clinical research. 2. London: Chapman & Hall/CRC Biostatistics Series; 2008. p. 177. [Google Scholar]
- 31.Austin PC. Variance estimation when using inverse probability of treatment weighting (IPTW) with survival analysis. Stat Med. 2016;35(30):5642–5655. doi: 10.1002/sim.7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bazhenova L, Lokker A, Snider J, Castellanos E, Fisher V, Fellous M, et al. TRK fusion cancer: patient characteristics and survival analysis in the real-world setting. Target Oncol. 2021;16(3):389–399. doi: 10.1007/s11523-021-00815-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doebele RC, Perez L, Trinh H, Martinec M, Martina R, Riehl T, et al. Comparative effectiveness analysis between entrectinib clinical trial and crizotinib real-world data in ROS1+ NSCLC. J Comp Eff Res. 2021;10(17):1271–1282. doi: 10.2217/cer-2021-0131. [DOI] [PubMed] [Google Scholar]
- 34.Tan O, Shrestha R, Cunich M, Schofield DJ. Application of next-generation sequencing to improve cancer management: a review of the clinical effectiveness and cost-effectiveness. Clin Genet. 2018;93(3):533–544. doi: 10.1111/cge.13199. [DOI] [PubMed] [Google Scholar]
- 35.Flaherty KT, Gray R, Chen A, Li S, Patton D, Hamilton SR, NCI-MATCH Team et al. The molecular analysis for therapy choice (NCI-MATCH) trial: lessons for genomic trial design. J Natl Cancer Inst. 2020;112(10):1021–1029. 10.1093/jnci/djz245. [DOI] [PMC free article] [PubMed]
- 36.Malone ER, Oliva M, Sabatini PJB, Stockley TL, Siu LL. Molecular profiling for precision cancer therapies. Genome Med. 2020;12(1):8. doi: 10.1186/s13073-019-0703-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ersek JL, Black LJ, Thompson MA, Kim ES. Implementing precision medicine programs and clinical trials in the community-based oncology practice: barriers and best practices. Am Soc Clin Oncol Educ Book. 2018;38:188–196. doi: 10.1200/EDBK_200633. [DOI] [PubMed] [Google Scholar]
- 38.Hlevnjak M, Schulze M, Elgaafary S, Fremd C, Michel L, Beck K, et al. CATCH: a prospective precision oncology trial in metastatic breast cancer. JCO Precis Oncol. 2021;5:PO.20.00248. doi: 10.1200/PO.20.00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kato S, Kim KH, Lim HJ, Boichard A, Nikanjam M, Weihe E, et al. Real-world data from a molecular tumor board demonstrates improved outcomes with a precision N-of-One strategy. Nat Commun. 2020;11(1):4965. doi: 10.1038/s41467-020-18613-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garrido-Laguna I, Janku F, Vaklavas C, Falchook GS, Fu S, Hong DS, et al. Validation of the royal marsden hospital prognostic score in patients treated in the phase I Clinical Trials Program at the MD Anderson Cancer Center. Cancer. 2012;118(5):1422–1428. doi: 10.1002/cncr.26413. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The dataset analysed during the current study is available from the corresponding author on reasonable request.