

SUMMARY
Despite therapeutic interventions for glioblastoma (GBM), cancer stem cells (CSCs) drive recurrence. The precise mechanisms underlying CSC resistance, namely inhibition of cell death, are unclear. We built on previous observations that the high cell surface expression of junctional adhesion molecule-A drives CSC maintenance and identified downstream signaling networks, including the cysteine protease inhibitor SerpinB3. Using genetic depletion approaches, we found that SerpinB3 is necessary for CSC maintenance, survival, and tumor growth, as well as CSC pathway activation. Knockdown of SerpinB3 also increased apoptosis and susceptibility to radiation therapy. SerpinB3 was essential to buffer cathepsin L-mediated cell death, which was enhanced with radiation. Finally, we found that SerpinB3 knockdown increased the efficacy of radiation in pre-clinical models. Taken together, our findings identify a GBM CSC-specific survival mechanism involving a cysteine protease inhibitor, SerpinB3, and provide a potential target to improve the efficacy of GBM therapies against therapeutically resistant CSCs.
In brief
Lauko et al. identify SerpinB3 as an inhibitor of lysosomal-mediated cell death in glioblastoma cancer stem cells. The authors demonstrate that SerpinB3 inhibition of cathepsin L released from lysosomes leads to radiation resistance. This axis may be targeted to improve the efficacy of radiotherapy in glioblastoma and other cancers.
This study did not generate new unique reagents.
Graphical Abstract
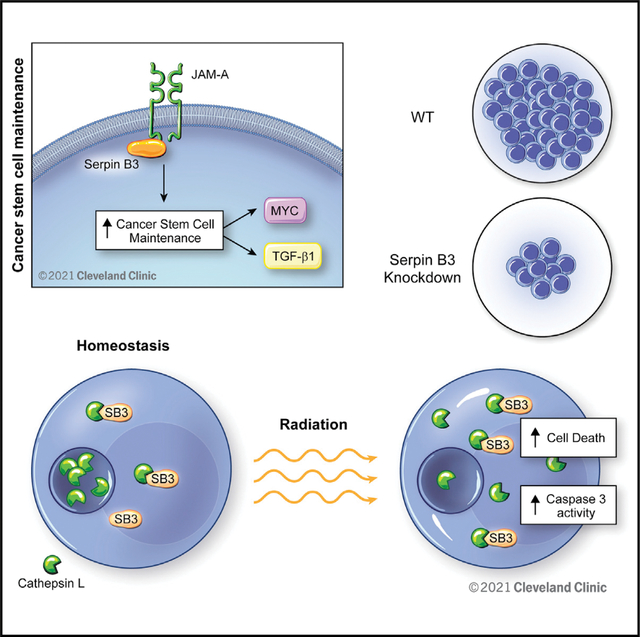
INTRODUCTION
Glioblastoma (GBM; World Health Organization [WHO] grade 4 glioma) is the most common primary malignant brain tumor and remains uniformly lethal. Despite aggressive therapies, including maximal safe surgical resection, radiation, and chemotherapy, GBM patients experience a median survival of approximately 20 months (Stupp et al., 2005, 2017). GBM therapeutic resistance has been associated with poor brain penetration of compounds due to the blood-brain barrier (Bellettato and Scarpa, 2018; Harder et al., 2018), cellular heterogeneity and plasticity (Lauko et al., 2021), and limited immune infiltration (Martinez-Lage et al., 2019; Pombo Antunes et al., 2020). The cellular heterogeneity is driven by populations of cancer stem cells (CSCs) (Gimple et al., 2019; Lathia et al., 2015), and recent studies have demonstrated that GBM contains a high degree of plasticity, with the CSC state being linked to cellular programs, including wound healing, development, and metabolic fluidity (Garnier et al., 2019; Mitchell et al., 2021; Pelaz et al., 2020) that underlie tumor growth and therapeutic resistance.
CSCs are functionally defined by their ability to self-renew and initiate a tumor upon secondary transplantation. Moreover, CSCs possess enhanced molecular mechanisms of therapeutic resistance, including DNA repair (Bao et al., 2006) and drug efflux pumps (Mason, 2015). In addition, CSCs can be subject to stressful environments, including hypoxia and necrosis, and, although some mechanisms have been proposed as to how these cells can thrive under these stressful conditions (Alvarado et al., 2017; Hsieh et al., 2011), the precise mechanisms as to how CSCs evade cell death are unclear. Cell adhesion represents a cellular mechanism that promotes pro-survival signaling and is enhanced in CSCs (Lathia et al., 2010, 2014). Specifically, GBM CSCs present elevated expression of integrins (Lathia et al., 2010), cadherins (Siebzehnrubl et al., 2013), and junctional adhesion molecule-A (JAM-A) (Alvarado et al., 2016; Lathia et al., 2014), which drive self-renewal and promote resistance to conventional therapies (Bao et al., 2006; Colak and Medema, 2014). However, the CSC-specific intracellular signaling networks that link adhesion to resistance of cell death remain poorly defined.
Cell death is a complex and tightly regulated series of cellular programs that cancer cells have evolved to evade (Castelli et al., 2021; Safa, 2016). Cell death can be triggered through a variety of mechanisms, including DNA damage, extrinsic ligands, and stressors (including hypoxic, metabolic, and endoplasmic reticulum stress). An understudied trigger of cell death is increased lysosomal permeability, which leads to the release of reactive oxygen species and cathepsins (Wang et al., 2018), a diverse family of proteases (Yadati et al., 2020) that can initiate a variety of cell death programs. Under physiologic conditions, cathepsin proteolytic activity is buffered by protease inhibitors, including a family of serine(cysteine)-protease inhibitors called serpins (Heit et al., 2013). In normal physiology, cathepsins and serpins exist in equilibrium to prevent aberrant cell death and damage to healthy tissues (Heit et al., 2013; Strnad et al., 2020). In GBM, there is limited information on lysosome-mediated cell death, which led us to hypothesize that CSCs may use serpins to counteract this mechanism of cell death. Here, we show that the CSC programs regulated by JAM-A engage SerpinB3 downstream to simultaneously maintain the CSC phenotype and inhibit lysosome-mediated cell death. Suppression of SerpinB3 increases cell death, decreases self-renewal and tumor initiation, and enhances the response of CSCs to radiation via lysosomal-mediated cell death.
RESULTS
SerpinB3 complexes with JAM-A in GBM
A series of cell surface receptors has been identified, including CD133 (Liu et al., 2006; Singh et al., 2004), CD15 (Son et al., 2009), CD49f (Lathia et al., 2010), CD44 (Beier et al., 2007), L1CAM (Bao et al., 2008), and JAM-A (Lathia et al., 2014) that regulate the expression of the CSC phenotype. With rare exceptions, the molecular cascades that connect these receptors to the downstream pluripotency machinery has not been made clear. In previous work, we established that GBM tumor cells expressed JAM-A when cultured under CSC-enriching conditions and that JAM-A expression was both necessary and sufficient for in vitro self-renewal. Previous work has suggested JAM-A signals via multiple different adaptor proteins, leading to a variety of downstream signals (Lauko et al., 2020). Subsequently, we revealed that Akt activation functions downstream of JAM-A and could be inhibited by microRNA-145 (miR-145) (Alvarado et al., 2016). While these studies have also shown that JAM-A regulates expression of the pluripotency machinery in GBM through Akt activation, our understanding of this molecular cascade is far from complete.
To expand our understanding of JAM-A signaling, we sought out additional binding partners using a histidine (His)-tagged JAM-A that we introduced into the T4121 GBM patient-derived xenograft (PDX) model. We then pulled down the His-tagged JAM-A and identified a series of binding partners using liquid chromatography coupled to mass spectrometry (MS; Figure 1A; Table S1). As expected, the protein with the greatest number of peptides identified was JAM-A itself, and we narrowed down candidate hits from the initial list, removing those with reported non-specific binding (via CRAPome; Mellacheruvu et al., 2013). Post-data filtration, our next strongest hit was a cysteine-protease inhibitor, SerpinB3 (also known as squamous cell carcinoma antigen 1 [SCCA1]), which has known roles in tumor progression in cervical, head and neck cancers, and hepatocellular carcinoma (HCC) (Cannito et al., 2015; Pontisso, 2014). SerpinB3 has been observed in the cytoplasm and nucleus of cells and is expressed primarily in the esophagus, nasopharynx, and female reproductive organs in normal human physiology (Gomes et al., 2014; Sun et al., 2017; Uhlén et al., 2015). We validated the binding of SerpinB3 to His-tagged JAM-A (Figure 1B). We observed SerpinB3 expression in JAM-A+ CSCs in a PDX model (Figure 1C), as well as in human GBM patient tissue (Figures S1A and S1B). Transcriptional profiling indicated that SerpinB3 expression was not specifically associated with any given molecular subtype (Figure S1C). When we interrogated multiple PDX models grown in CSC conditions, over 95% of cells were double positive for SerpinB3 and JAM-A (Figure S1D). In addition, we observed that SerpinB3 was more highly expressed in PDX models than in human astrocytes (Figure S1E). Furthermore, SerpinB3 and JAM-A appear to stabilize each other, as reduction of JAM-A in cultured GBM tumor cells resulted in the reduction of SerpinB3 (Figure 1D). We also observed the reverse effect (Figure S1F). We then measured SerpinB3 stability using a cycloheximide chase assay. When JAM-A was knocked down, SerpinB3 levels decreased more quickly than in control non-targeting (NT) conditions, further suggesting that JAM-A may function to stabilize SerpinB3 protein (Figure 1E). Together, these data indicate that SerpinB3 is expressed under CSC-optimized conditions in vitro and heterogeneously in CSCs and GBM patient tumor specimens and that JAM-A is involved in the stabilization of SerpinB3.
Figure 1. SerpinB3 is necessary for glioblastoma.
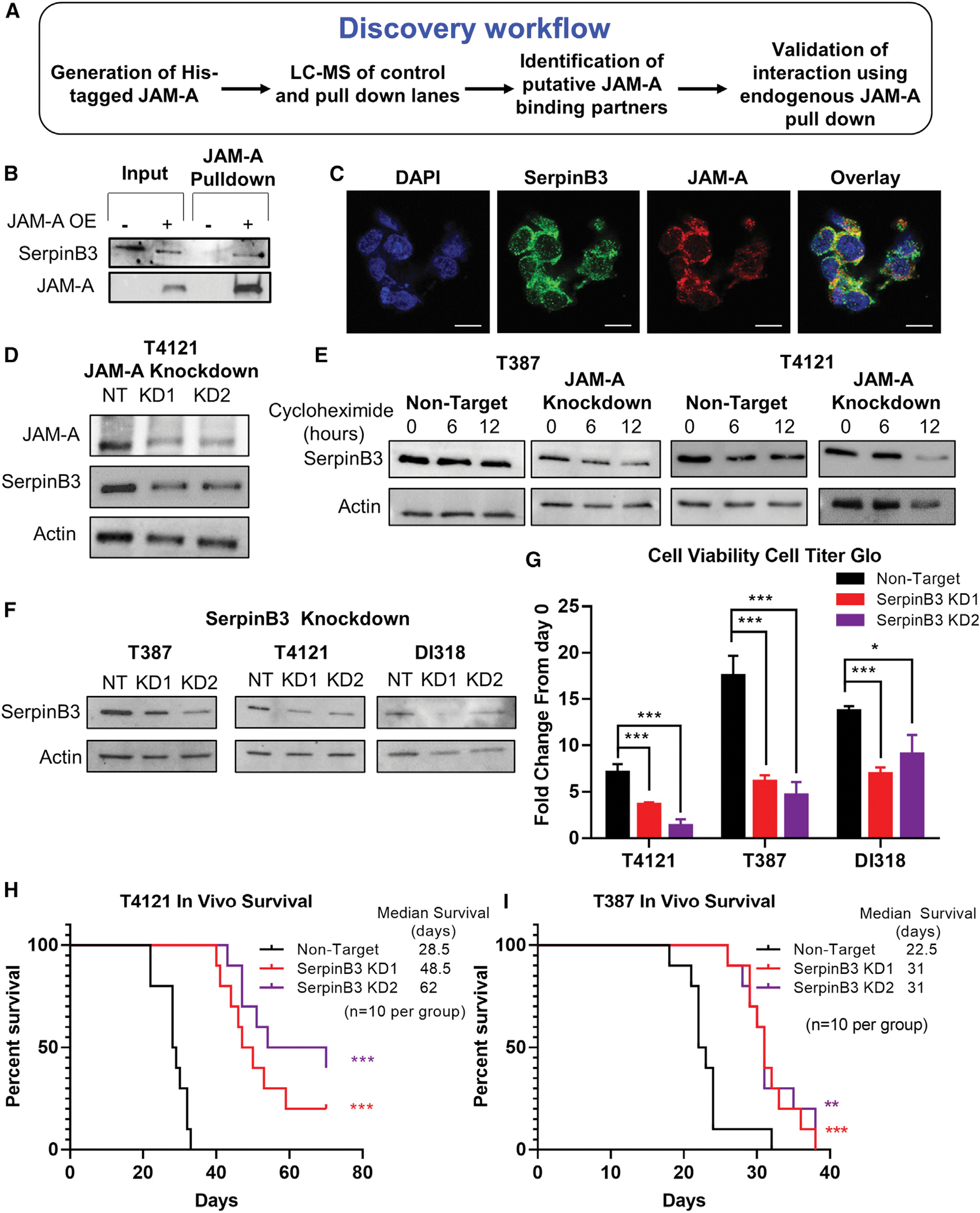
(A) Graphical abstract of His-JAM-A pulldown and liquid chromatography-mass spectrometry (LC-MS) procedure.
(B) Verification of LC-MS results by western blot. His-tagged JAM-A was overexpressed in T4121 cancer stem cells (CSCs), and protein was isolated and mixed with nickel beads. The bound fraction was subjected to immunoblotting with antibodies to SerpinB3 and JAM-A.
(C) Immunofluorescent staining demonstrating co-expression of JAM-A and SerpinB3 in T387 PDX glioblastoma tumor model (scale bar, 10 μm).
(D) JAM-A was knocked down in T4121 CSCs with 2 separate shRNA constructs, and SerpinB3 expression was measured. Actin was used as a loading control in this and all subsequent western blots.
(E) T387 and T4121 CSCs expressing JAM-A KD2 shRNA or NT control were treated with cycloheximide, and SerpinB3 expression was measured at 6 and 12 h post-treatment.
(F) Western blot demonstrating knockdown of SerpinB3 with each shRNA, KD1, and KD2.
(G) Fold change in cell viability at day 7, normalized to day 0, in 3 PDX glioblastoma models. Cell viability measured with CellTiter-Glo Luminescent Cell Viability Assay (5 technical replicates per condition, per tumor model).
(H and I) Kaplan-Meier curves depicting survival of mice with 20,000 T4121 or T387 tumor cells intracranially injected. Cells were transfected with either non-target (SHC002) or SerpinB3 (KD1 or KD2) shRNA, with n = 10 mice per group.
p < 0.05 was considered statistically significant. *p < 0.05, **p < 0.01, ***p < 0.001, as determined by 1-way ANOVA with Dunnett’s multiple comparisons test or log rank test for survival data. Error bars represent standard deviations.
Given the limited understanding of the role of SerpinB3 in GBM, we sought to assess its function using a genetic depletion approach. We used non-overlapping small hairpin RNAs (shRNAs) against SerpinB3 and were able to reduce protein levels in multiple GBM PDX-derived CSC models (T4121, T387, DI318) compared to NT controls (Figures 1F and S1G). While initial experiments were performed using four different shRNAs to demonstrate reproducibility, we subsequently focused on two constructs, knockdown 1 (KD1) and knockdown 2 (KD2). The reduction of SerpinB3 in CSCs resulted in a decreased number of viable cells in vitro (Figures 1G, S1H, and S1I) and a potent reduction in tumor initiation and growth in vivo after intracranial implantation into NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (Figures 1H, 1I, and S1J). Taken together, these data provide evidence that SerpinB3 is essential for GBM CSC growth and tumor initiation.
SerpinB3 promotes the CSC phenotype in GBM
Given the phenotypes observed upon SerpinB3 knockdown in CSCs, we also assessed changes in CSC maintenance as a result of SerpinB3 depletion. We observed that SerpinB3 knockdown reduced CSC signaling via changes in mRNA levels of core pluripotency transcription factors (OCT4, NANOG, MYC), CSC transcription factors (OLIG2), and known CSC maintenance factors (transforming growth factor-beta 1 [TGF-β1]) (Figures 2A and S2A). We then interrogated the functional consequences of SerpinB3 knockdown on self-renewal via in vitro limiting-dilution assays, a surrogate for self-renewal that can also be affected by cell proliferation and cell death, and found a potent reduction in self-renewal with SerpinB3 knockdown compared to NT control conditions across multiple CSC models (Figure 2B). To gain further insight into SerpinB3-mediated changes, we focused on c-MYC and TGF-β1 based on their reported roles in GBM CSCs (Anido et al., 2010; Bruna et al., 2007; Wang et al., 2008), essential role in cancer cell proliferation, and link to SerpinB3 in HCC (Turato et al., 2014, 2015). As predicted, knockdown of SerpinB3 reduced c-MYC expression (Figure 2C) and TGF-β1 secretion (Figure 2D). To gain additional mechanistic insight into the role of SerpinB3 in CSC-mediated cell growth and tumor initiation, we subjected SerpinB3-depleted CSCs to a cancer-focused mRNA panel using the NanoString platform. Using an unbiased clustering, we observed that NT control samples were distinct from SerpinB3 knockdown (using the KD2 construct) in two CSC models (T4121 and DI318; Figure S2B). We found a series of pathways, including cancer driver genes and a number of pathways known to regulate CSCs, including Hedgehog, Notch, and TGF-β, that were differentially expressed in NT control compared to SerpinB3-depleted cells (Figures 2E and S2B). This finding corroborated our observation of reduced TGF-β1 secretion with SerpinB3 knockdown (Figure 2D), and we additionally validated that SerpinB3 depletion reduced the expression of members of the Notch signaling network (NOTCH2 and JAGGED2; Figure S2C). We previously reported that JAM-A signals through phospho-AKT; however, when we knocked down SerpinB3, we saw no change in the phosphorylation of AKT (Figure S2D), suggesting that SerpinB3 may signal through a separate mechanism. SerpinB3 has been observed within the nucleus (Katagiri et al., 2006), where it regulates c-Jun N-terminal kinase (JNK1) kinase activity through a mechanism that has yet to be precisely elucidated. We also observed SerpinB3 expression in SOX2+ nuclei, suggesting a potential role in CSC maintenance within the nucleus (Figures 2F and S2E). These data indicate that SerpinB3 is essential for self-renewal and interacts with multiple CSCs signaling network nodes.
Figure 2. SerpinB3 regulates known CSC pathways.
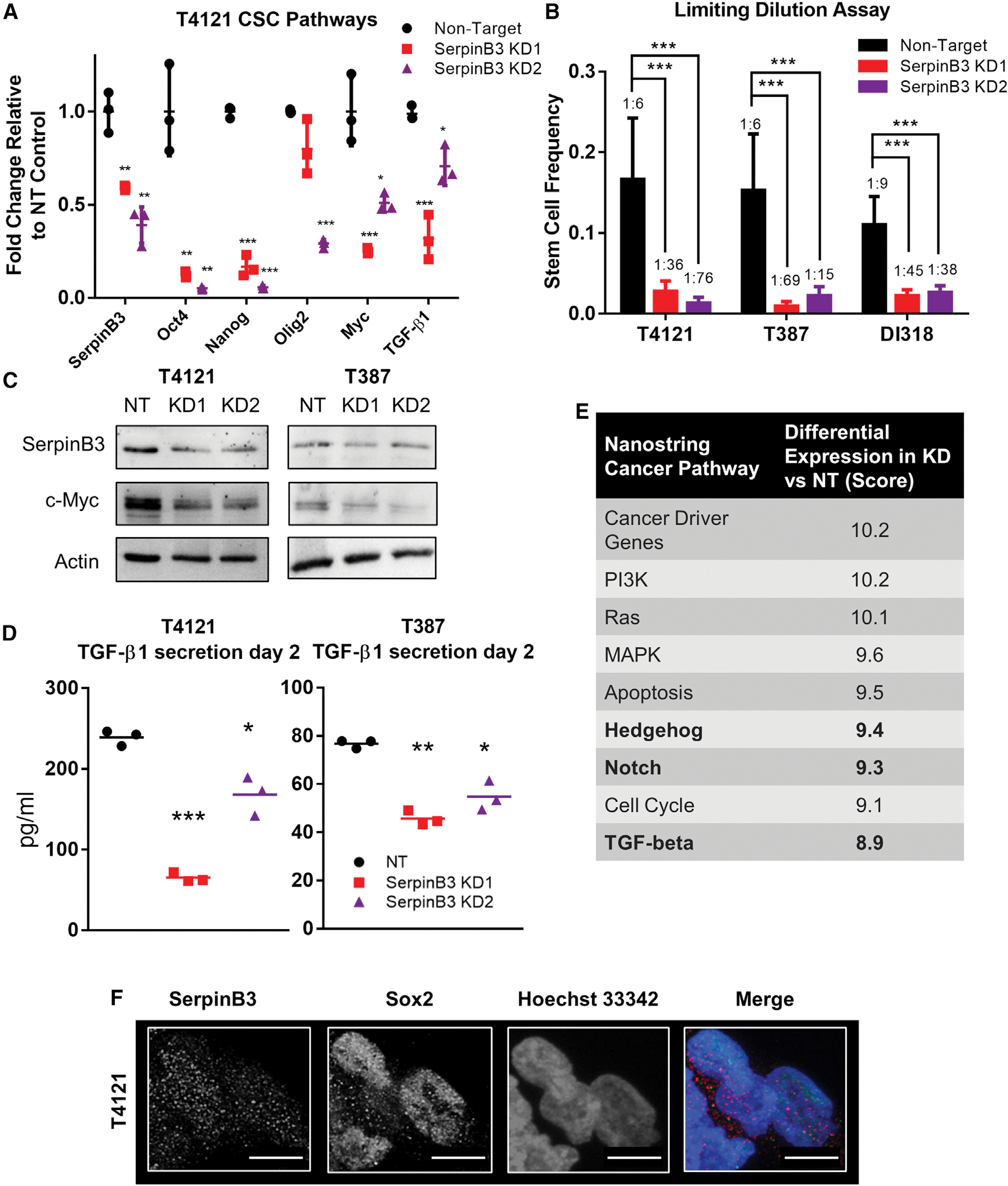
(A) RNA was isolated after SerpinB3 knockdown in T4121 cells, and qPCR was performed for SERPINB3, OCT4, NANOG, OLIG2, MYC, and TGF-β1 (3 technical replicates).
(B) Tumor cells were plated in a limiting-dilution manner, and the number of wells containing spheres was counted after 14 days and used to calculate stem cell frequencies using the online algorithm detailed in the methods. N = 12 wells per dilution.
(C) c-MYC expression after SerpinB3 knockdown was assessed via western blot, with actin as a loading control.
(D) TGF-β1 secretion was analyzed 2 days after plating and normalized to total protein (3 technical replicates per condition, per tumor model).
(E) NanoString pathway score comparing SerpinB3 knockdown to non-target control. Bolded rows represent pathways known to regulate the CSC state.
(F) Immunofluorescence staining of T4121 CSCs for SerpinB3 and SOX2. Scale bar represents 10 μM.
p < 0.05 was considered statistically significant. *p < 0.05, **p < 0.01, ***p < 0.001, as determined by 1-way ANOVA with Dunnett’s multiple comparisons, Student’s t test for qPCR data, or chi-squared p value for limiting dilution assay. Error bars represent standard deviations.
SerpinB3 protects GBM tumor cells from apoptotic death
Based on the decrease in cell viability we observed after SerpinB3 knockdown and the well-established role for SerpinB3 in inhibiting cell death, we asked whether this correlates with an increase in cell death (Villano et al., 2014). Moreover, as our NanoString analysis of cancer pathways also revealed an increase in apoptosis in cells depleted of SerpinB3 (Figure 2E), we validated this increase in apoptosis after SerpinB3 knockdown as read out by annexin V/propidium iodide (PI) double-positive cells in multiple CSC models (Figures 3A and S3A). SerpinB3 knockdown also resulted in an increase in caspase 3/7 activity compared to control conditions using the CaspaseGlo DEVD-aminoluciferin assay. DEVD is the canonical recognition site for Caspase-3 (Figures 3B and S3B). In addition, we used the IncuCyte Caspase 3/7 assay, a DEVD-tagged DNA-intercalating dye, which also demonstrated increased caspase 3/7 activity (Figure 3C). Notably, the extent to which SerpinB3 knockdown affected cellular functions appeared to correlate with the degree of knockdown, with KD2 generally exhibiting a greater effect. Together, these data demonstrate an enhanced cell death phenotype after the loss of SerpinB3.
Figure 3. SerpinB3 inhibits cell death.
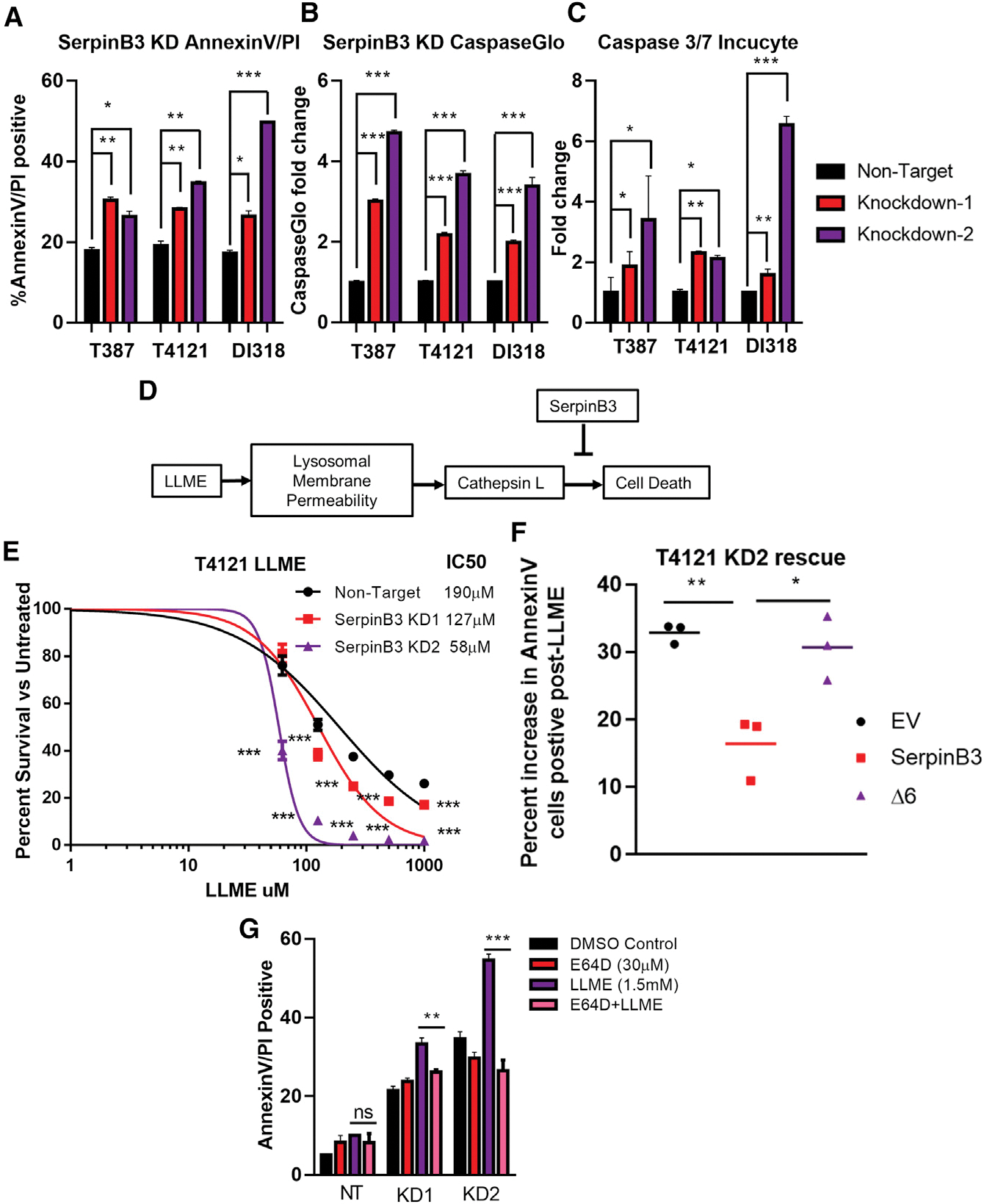
(A) The percentage of annexin V+ and propidium iodide-positive (PI+) cells after SerpinB3 knockdown was compared to the non-target control group (3 technical replicates per condition, per tumor model).
(B) Activity of caspase 3/7 was measured using Caspase-Glo after SerpinB3 knockdown, and the fold change compared to the non-target control is shown (3 technical replicates per condition, per tumor model).
(C) Quantification of caspase 3/7 IncuCyte assay normalized to cell confluence per well and compared to the NT condition (4 technical replicates per condition, per tumor model).
(D) Schematic of lysosomal-mediated cell death after treatment with l-leucyl-l-leucine methyl ester (LLME).
(E) T4121 cells were treated with LLME at varying concentrations for 7 days. On day 7, cell viability was quantified via Cell Titer Glo and compared to untreated DMSO controls for each condition. From these values, the half-maximal inhibitory concentration (IC50) was determined (5 replicates per condition).
(F) T4121 KD2 cells were transfected with DNA encoding either SerpinB3, an empty vector (EV), or a protease inhibitor-null mutant (Δ6). Cells were then treated for 6 h with either 1.5 mM LLME or DMSO control, and annexin V+ cells were quantified. The relative percentage increase compared to DMSO-treated control was quantified, and the results of 3 independent experiments are graphed.
(G) T387 cells were treated with LLME or DMSO for 6 h with or without E64D. After 6 h, annexin V+/PI+ cells were quantified, n = 3 per condition.
p < 0.05 was considered statistically significant. *p < 0.05, **p < 0.01, ***p < 0.001, as determined by 1-way ANOVA with Dunnett’s multiple comparisons or Student’s t test (F and G). Error bars represent standard deviations.
SerpinB3 inhibits lysosomal-mediated apoptosis
As apoptosis can be initiated via multiple pathways (i.e., intrinsic versus extrinsic), we sought to better understand the molecular mechanism through which SerpinB3 prevents cell death. SerpinB3 is a known inhibitor of cathepsin L (Sun et al., 2017), a cysteine protease that relocalizes to the cytoplasm after disruption of the lysosomal membrane or other acidic compartments, triggering “lysosomal-mediated cell death” (Figure 3D) (Fehrenbacher et al., 2004; Oberle et al., 2010; Piazza et al., 2007). To test whether SerpinB3 protects against lysosomal-mediated cell death, we compromised lysosomal membrane integrity using l-leucyl-l-leucine methyl ester (LLME) and observed a potent decrease in cell viability, which was further enhanced with SerpinB3 knockdown (Figures 3E, S3C, and 3D). To further determine whether the susceptibility of SerpinB3 knockdown cells to LLME was due to a loss of the cysteine cathepsin inhibitory function of SerpinB3, we used a mutant of SerpinB3 with a deletion of six amino acids (Δ6) within the hinge region of the reactive site loop (RSL). This SerpinB3 mutant protein is unable to inhibit cathepsin L activity (Sheshadri et al., 2014). SerpinB3 KD2 cells were transfected with empty vector (EV), wild-type SerpinB3, or SerpinB3 Δ6 and then treated with either DMSO or LLME for 6 h. Following treatment, the percentage of annexin V+ cells was quantified. While wild-type SerpinB3 was able to partially rescue cell death induced by LLME treatment, the Δ6 mutant did not (Figure 3F). To determine whether cathepsin L is driving in the cell death induced by LLME, PDX tumor cells were treated with a high dose of LLME (3 mM) for 6 h, and the percentage of annexin V+ and PI+ cells was calculated. Two distinct cathepsin L inhibitors, E64D and Z-FY-CHO, reduced LLME-induced cell death (Figures S3E and S3F). Finally, at a lower dose of LLME (1.5 mM), we observed a larger increase in cell death induced by LLME in cells depleted of SerpinB3, with only a minor increase in NT cells. This increase was rescued by E64D treatment, demonstrating the role of SerpinB3 in protecting cathepsin L-mediated cell death after lysosomal membrane permeability (LMP) (Figure 3G). These data demonstrate that SerpinB3 functions as a cathepsin L inhibitor in GBM CSCs and is responsible for inhibiting lysosomal-mediated apoptosis.
Radiation induces lysosomal-mediated apoptosis
We next investigated whether standard-of-care radiation treatment could affect lysosomal membrane integrity (Figure 4A). We observed an increase in LMP 6 h post-irradiation with a single dose of 5 Gy as readout by a shift in acridine orange localization (Figures 4B, 4C, and S4A). Acridine orange fluoresces red in acidic compartments and green in the remainder of the cell. In addition, we observed a relocalization of cathepsin L from the lysosome and other acidic compartments to the cytoplasm at 6 h after irradiation with a single 5-Gy dose (Figures 4D, 4E, and S4B). This can be observed as a shift in cathepsin L from a primarily punctate structure to a more diffuse localization throughout the cytoplasm. Importantly, we did not observe an increase in overall cathepsin L or SerpinB3 levels at this time point or a meaningful increase in cell death (Figures S4C and S4D). As radiation is part of the standard of care for GBM and as CSCs are resistant to radiation, we next assessed whether the depletion of SerpinB3 increases the efficacy of radiation in CSCs. SerpinB3 knockdown potently increased the sensitivity of CSCs to radiation compared to control NT conditions (Figures 4F and S4E). We also observed increased cell death 24 h post-1.5 Gy radiation in SerpinB3 knockdown cells compared to NT controls (Figures S4F and S4G). This increase in cell death was rescued by both cathepsin L inhibitors E64D and Z-FY-CHO in SerpinB3 KD1 cells (Figures 4G and S4H). Notably, temozolomide (TMZ) did not result in increased cell death, as TMZ does not induce LMP (Figure S4I) (Kanzawa et al., 2004). Taken together, these data indicate that SerpinB3 protects against LMP induced by radiation.
Figure 4. SerpinB3 buffers cells from lysosomal membrane permeability.
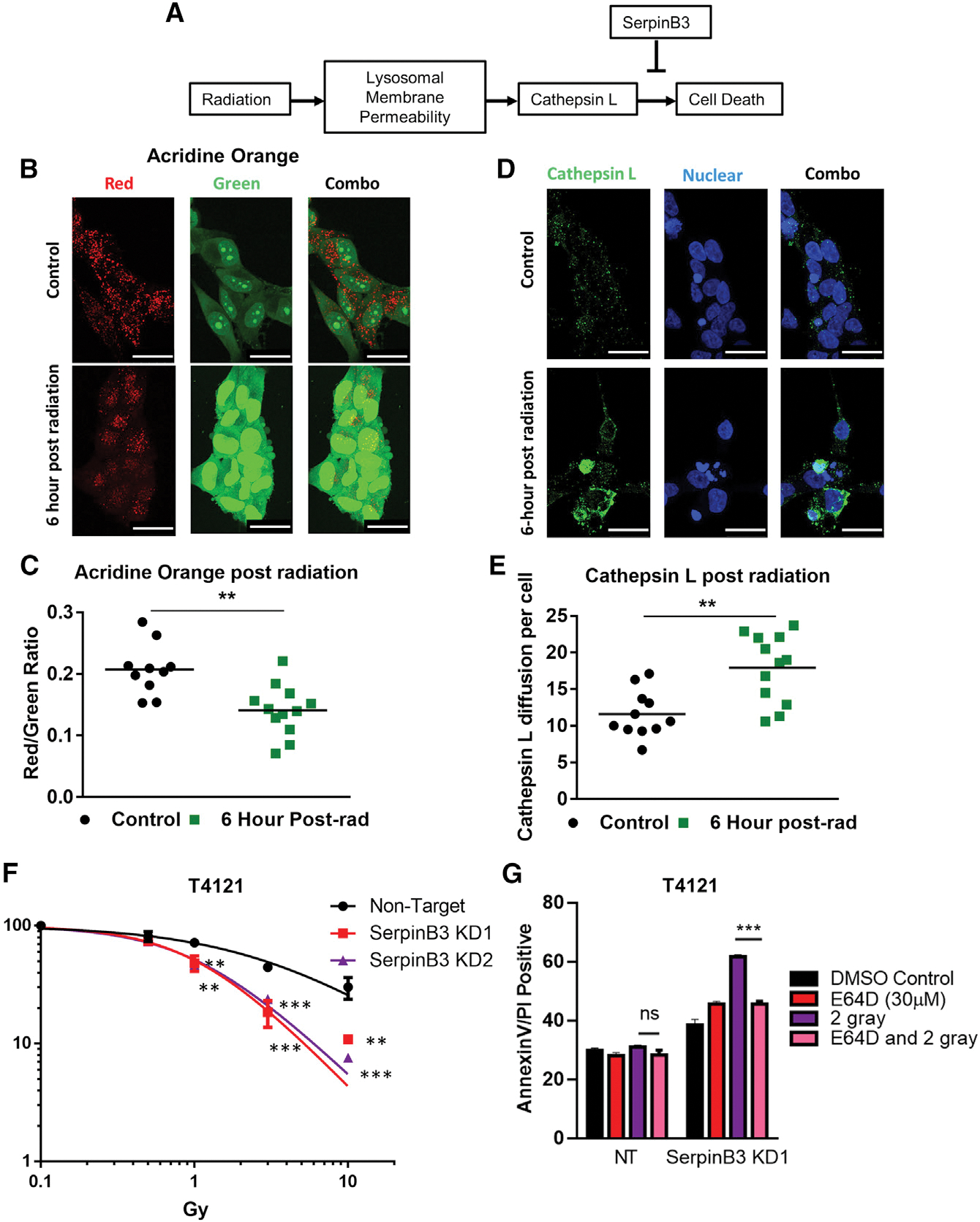
(A) Schematic of lysosomal-mediated cell death after radiation.
(B and C) Acridine orange was added to cells 6 h after irradiation with 5 Gy, and images from 12 random visual fields were taken. The red:green ratio per image was calculated comparing control to irradiated conditions. Scale bar represents 25 μM.
(D and E) Six hours post-irradiation, cells were fixed with paraformaldehyde and stained for cathepsin L. The integrated density of cathepsin L per cell was determined and compared between the radiation and control conditions (12 images per condition). Scale bar, 25 μM.
(F) Cell viability was measured after 2 days of varying doses of radiation, and the percentage of viable cells is shown compared to each group’s untreated control at each dose of radiation (3 technical replicates per condition).
(G) T4121 tumor cells were treated with 2 Gy radiation with or without 30 μM E64D. Twenty-four hours post-radiation, annexin V+/PI+ cells were quantified, n = 3 per condition.
p < 0.05 was considered statistically significant. *p < 0.05, **p < 0.01, ***p < 0.001, as determined by 1-way ANOVA with Dunnett’s multiple comparisons (F) or Student’s t test (C, E, and G). Error bars, standard deviations.
SerpinB3 loss enhances the effects of radiation in vivo
To determine whether SerpinB3 is important for radiation resistance in vivo, we transplanted NT and SerpinB3-knockdown T4121 CSCs and subjected the mice to a pre-clinical radiation paradigm (Figure 5A). We found that SerpinB3 knockdown increased tumor latency, and this was further extended by irradiation with 10 Gy over 5 days, increasing the hazard ratio in SerpinB3 knockdown compared to control conditions (Figures 5B–5E). This response was also observed with a lower dose of radiation (Figures S5A–S5E). Taken together, these data suggest that SerpinB3 prevents cell death and contributes to radiation resistance.
Figure 5. SerpinB3 contributes to radiation resistance.
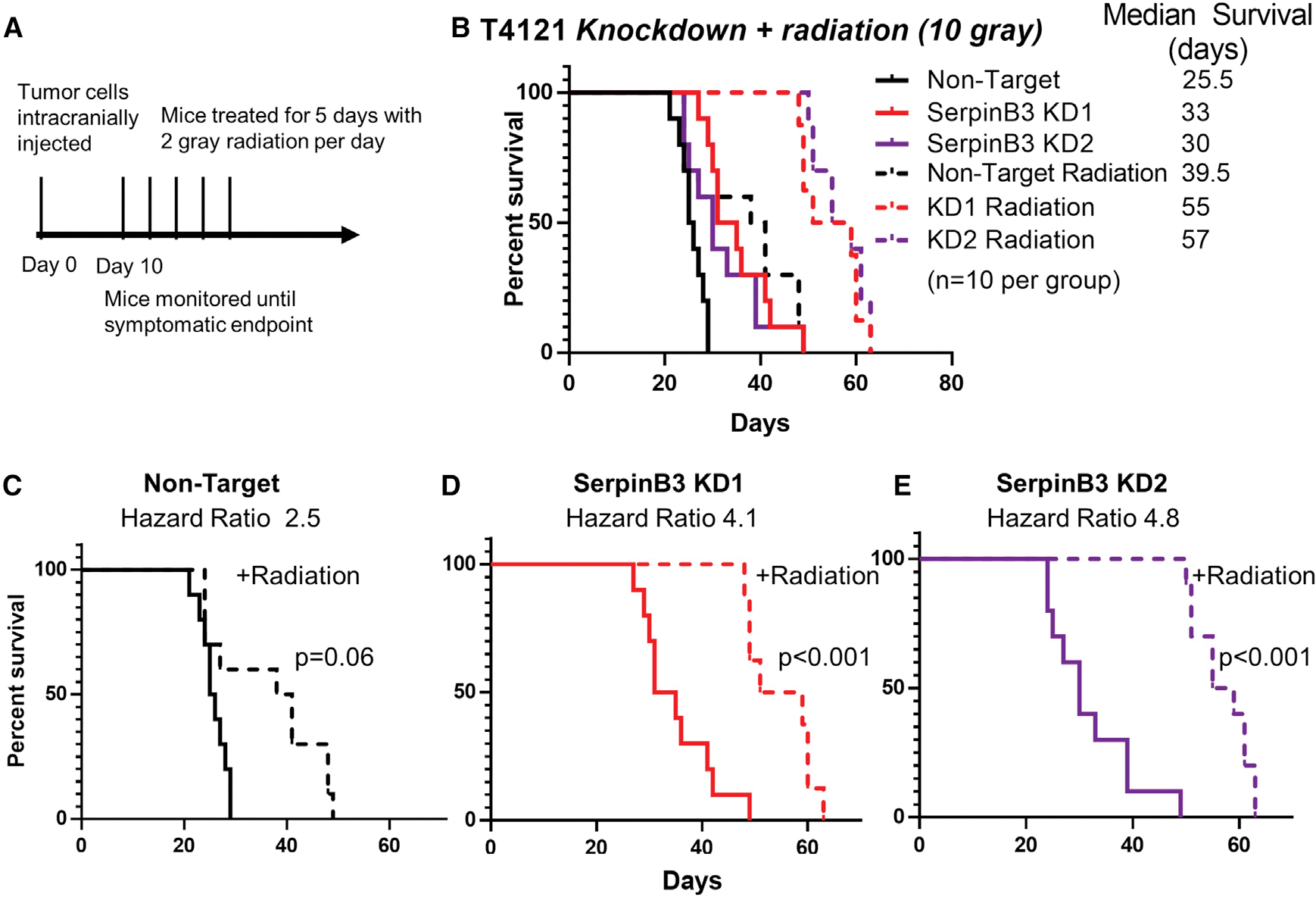
(A) Schematic of in vivo radiation experiment with 10 Gy total radiation treatment.
(B–E) A total of 20,000 tumor cells per condition were intracranially injected into 10 mice per group. Ten days after irradiation, mice received 2 Gy of radiation per day for 5 days (total of 10 Gy) to the head. (B) All of the treatment groups are shown together with median survival values given. The groups were subsequently divided into (C) non-target with or without radiation, (D) SerpinB3 KD 1 with or without radiation, and (E) SerpinB3 KD 2 with or without radiation.
*p < 0.05 was considered statistically significant. *p < 0.05, **p < 0.01, ***p < 0.001, as determined by log rank test.
DISCUSSION
Resistance to apoptosis is a well-recognized hallmark of cancer, but specific resistance mechanisms underlying cell death have not been thoroughly investigated in CSCs. This is surprising given that the predominant phenotype of CSCs is enhanced therapeutic resistance, specifically to radiation and temozolomide in the case of GBM. SerpinB3 represents a mechanism for GBM CSC survival that may also be functionally important in other cancers. The role of SerpinB3 in cancer is not well developed, despite being originally identified as overexpressed in SCC (Kato and Torigoe, 1977). Studies in cervical cancer, non-small cell lung cancer, breast cancer, esophageal SCC, and HCC have correlated elevated SerpinB3 expression with clinical stage and decreased response to therapy (Collie-Duguid et al., 2012; Liu et al., 2015; Ngan et al., 1990; Petty et al., 2006; Shimada et al., 2003; Wang et al., 2022). The roleof SerpinB3 in GBM had not been studied until recently, when a long non-coding RNA (lncRNA), TMEM44-AS1 (Bian et al., 2021), was found to bind to SerpinB3, forming a positive feedback loop with MYC. While this suggests the importance of SerpinB3 in GBM, the detailed molecular mechanism(s) by which SerpinB3 drives oncogenesis in its role in therapy resistance have yet to be fully elucidated.
There are several hypothesized mechanisms by which SerpinB3 could affect cancer-relevant phenotypes (e.g., impact on the stem cell state, resistance to apoptosis, invasion). A recent study in cholangiocarcinoma found that SerpinB3 was expressed in a stem-like subset of cells and that knockdown of SerpinB3 resulted in decreased invasion and proliferation (Correnti et al., 2021). In addition to its roles in cancer, SerpinB3 is expressed in hepatic stem cells, where SerpinB3 expression correlates with decreased activated caspase 3 (Villano et al., 2014). SerpinB3 has been linked to the inhibition of apoptosis in the settings of endoplasmic reticulum stress (Verfaillie et al., 2013), tumor necrosis factor-α (TNF-α) release (Suminami et al., 2001), radiation (Murakami et al., 2001), and ultraviolet radiation (Katagiri et al., 2006), but the exact mechanism is unknown. In this study, we have highlighted the role of lysosomal membrane permeability and cathepsin L release as a mechanism of resistance to apoptosis. SerpinB3 inhibits cathepsin L (Sun et al., 2017), and cathepsin L released from the lysosomes and other acidic compartments has been shown to cause caspase-mediated cell death (Fehrenbacher et al., 2004; Oberle et al., 2010). Finally, while multiple mechanisms of radiation-induced cell death have been documented, our study builds on earlier work highlighting the role of lysosomal membrane permeability in sensitizing tumor cells to radiation in GBM (Zhou et al., 2020). A recent paper found a similar role for SerpinB3 in inhibiting cathepsin L in cervical cancer post-radiation (Wang et al., 2022). Taken together, these data outline a mechanism whereby SerpinB3 expressed in GBM tumor cells leads to radiation resistance by buffering lysosomal membrane permeability.
Targeting the CSC state remains a clinically interesting possibility, and our observations suggest that SerpinB3 inhibition may be a mechanism by which cells with a CSC phenotype can be sensitized to radiation. Our observations, the upregulated expression of SerpinB3 in other cancers, and the dependence of other tumor cells on SerpinB3 (Figures S5F and S5G) set the foundation for additional studies of stem-like cells in other tumor types. In this manuscript, we have demonstrated a role for SerpinB3 in both the maintenance of the CSC state and in resistance to apoptosis. We have shown that JAM-A binds to SerpinB3 and is involved in its stabilization. In addition, the loss of SerpinB3 has led to decreased expression of multiple well-established CSC transcription factors and pathways.
Limitations of the study
There are several limitations to this study. From a technical perspective, our tumor models were grown in CSC-enriching sphere conditions, which does not recapitulate the complex tumor microenvironment in terms of tumor cell state or consider other cell types such as neurons and astrocytes. Moreover, our radiation methods fail to recapitulate the hypoxic microenvironment of the tumor within the human brain. From a conceptual perspective, we used three different GBM tumor models, which does not recapitulate the entire spectrum of heterogeneity observed in GBM, and the function of SerpinB3 may vary across different tumors.
Conclusions and future directions
Future priorities include the development of brain-penetrant SerpinB3 inhibitors. The flexibility of the protease inhibitor domain represents one developmental challenge, along with the high degree of homology between SerpinB3 and SerpinB4 outside the protease inhibitor region. These challenges necessitate a further understanding of this signaling network, including JAM-A-dependent versus -independent functions of SerpinB3 and the importance of JAM-A SerpinB3 stability, localization, and function. Another consideration for therapeutic development is the impact on cell death, which is a fundamental process in organ development and homeostasis in healthy tissue. The extent to which the inhibition of SerpinB3 may serve as a transforming event for cancer initiation should be an additional consideration. Finally, the exact mechanism by which SerpinB3 maintains the CSC phenotype remains incompletely understood. This mechanism may involve the regulation of c-MYC, with the changes in CSC state downstream of MYC, or SerpinB3 may interact with other transcription factors within the nucleus that regulate the CSC state. Whether this maintenance is dependent on protease inhibition has also yet to be explored.
SerpinB3 has emerged as a molecule of interest across numerous tumor types and further research into mechanisms for targeting SerpinB3 is required moving forward. In summary, our findings suggest that SerpinB3 may be a targetable mechanism leveraged by CSCs to resist cell death.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Justin D. Lathia (lathiaj@ccf.org).
Materials availability
Data and code availability
Accession numbers are listed in the key resources table. DOI link to original western blot images is available in key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
|
| ||
| Antibodies | ||
|
| ||
| SerpinB3 | Invitrogen | PA5-30164; RRID:AB_2547638 |
| JAM-A | B&D Biosciences | 612120; RRID:AB_399491 |
| cathepsin L | ThermoFisher | BMS1032; RRID:AB_10596643 |
| c-Myc | Cell Signaling Technology | 5605; RRID:AB_1903938 |
| β-Actin | Bio-Rad | 12004163; RRID:AB_2861334 |
| Rabbit StarBright 700 | Bio-Rad | 12004161; RRID:AB_2721073 |
| JAM-A | Santa Cruz | sc-53623; RRID:AB_784134 |
| SOX2 | R&D | MAB2018; RRID:AB_358009 |
| AKT | Cell Signaling | 9272; RRID:AB_329827 |
| pAKT(Ser473) | Cell Signaling | 4075S; RRID:AB_916029 |
| Donkey-anti mouse Alexa Fluor 555 | ThermoFisher | A-31570; RRID:AB_2536180 |
| Donkey-anti rabbit Alexa Fluor 488 | ThermoFisher | A-21206; RRID:AB_2535792 |
| JAM-A AF647 | Santa Cruz | sc-53623 AF647; RRID:AB_784134 |
|
| ||
| Biological samples | ||
|
| ||
| GBM Fresh Tumor Samples | Cleveland Clinic Foundation and Northwestern University | This manuscript |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| N-terminal His-tagged full-length JAM-A | Sinobiologicals | HG10198-NH |
| Hoechst 33342 | Invitrogen | H3570 |
| Vectashield | Vector Labs | H-1000-10 |
| acridine orange | Sigma | A6014 |
| pH6 retrieval buffer (Reveal) | Biocare | V1000 |
| serum-free casein background block (Background Sniper) | Biocare | BS966 |
| MACH 4 Universal HRP Polymer | Biocare | M4U534 |
| Geltrex | Life Technologies | A1413301 |
| puromycin | ThermoFisher | 54-022-2100 |
| L-leucyl-L-leucine methyl ester | Cayman | #16008 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Betazoid DAB Chromogen Kit | Biocare | BDB2004 |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | G7570 |
| Caspase-Glo 3/7 | Promega | G8090 |
| RNeasy mini kit | Qiagen | 74004 |
| nCounter® PanCancer Pathways Panel | Nanostring | N/A |
| TGF-β1 DuoSet ELISA | R&D systems | DY240 |
| FITC-labeled annexin V and propidium iodide | BioLegend | 640914 |
| Caspase 3/7 | Sartorius | 4704 |
| qScript synthesis reagent | Quanta Biosciences | 95048 |
| SYBR-Green Mastermix | SA Biosciences | 4385610 |
| E64D | Enzo | BML-PI107-0001 |
| Z-FY-CHO | Med Chem Express | HY-128140 |
| Cycloheximide | Alfa Aesar | J66004 |
| LIVE/DEAD™ Fixable Blue Dead Cell Stain Kit | Invitrogen | L23105 |
| MojoSort Buffer (5X) | Biolegend | 480017 |
| eBioscience™ Fixation/Permeabilization | Invitrogen | 00-5523 |
| MycoAlert detection kit | Lonza | LT07-118 |
| CD133 selection beads | Miltenyi | 130-097-049 |
|
| ||
| Deposited data | ||
|
| ||
| Unaltered Blots | Mendeley data | https://doi.org/10.17632/6dwf49mg73.1 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| 293T | DuBridge et al., 1987 | Obtained internally, available https://www.atcc.org/products/crl-3216 |
| T387 | Bao et al., 2008 | N/A |
| T4121 | Bao et al., 2008 | N/A |
| DI318 | This manuscript | N/A |
| Human astrocytes | ScienCell Research Laboratories | #1800 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) | Jackson Labs | 005557 |
|
| ||
| Oligonucleotides | ||
|
| ||
| SERPINB3 F- CGCGGTCTCGTGCTATCTGG, R- AGAAGAGGATGCTGTTGGTC |
This manuscript | N/A |
| OCT4 F- TGAGTCAGTGAACAGGGAATG R- AATCTCCCCTTTCCATTCGG |
This manuscript | N/A |
| NANOG F- GAAATACCTCAGCCTCCAGC R- GCGTCACACCATTGCTATTC |
This manuscript | N/A |
| OLIG 2 F- AGCTCCTCAAATCGCATCC R-AAAAGGTCATCGGGCTCTG |
This manuscript | N/A |
| c-MYC F- TTCGGGTAGTGGAAAACCAG R- AGTAGAAATACGGCTGCACC |
This manuscript | N/A |
| TGF-β1 F- AAGTGGACATCAAGGGTTC R- GTCCTTGCGGAAGTCAATGT |
This manuscript | N/A |
| GAPDH F-ACATCGCTCAGACACCATG R- TGTAGTTGAGGTCAATGAAGGG |
This manuscript | N/A |
| NOTCH2 F- GTGCCTATGTCCATCTGGATGG R- AGACACCTGAGTGCTGGCACAA |
This manuscript | N/A |
| JAGGED2 F- GCTGCTACGACCTGGTCAATGA R- AGGTGTAGGCATCGCACTGGAA |
This manuscript | N/A |
| Primers also listed in Table S2 | This manuscript | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| shRNA Control Plasmid (SHC002) | Sigma | SHC002 |
| SerpinB3 KD1 | Sigma | TRCN0000373440 |
| SerpinB3 KD2 | Sigma | TRCN0000373501 |
| SerpinB3 KD3 | Sigma | TRCN0000052398 |
| SerpinB3 KD4 | Sigma | TRCN0000373500 |
| JAM-A KD1 | Sigma | TRCN0000061650 |
| JAM-A KD2 | Sigma | TRCN0000061649 |
| psPAX2 | Addgene | #12260; RRID:Addgene_12260 |
| pMD2G | Addgene | #12259; RRID:Addgene_12259 |
| pLPC-N FLAG vector | Addgene | #12521; RRID:Addgene_12521 |
| Fugene HD | Promega | E2311 |
|
| ||
| Software and algorithms | ||
|
| ||
| LASX software | Leica | N/A |
| IncuCyte SX5 Live-Cell Analysis Instrument | Sartorius | https://www.sartorius.com/en/products/live-cell-imaging-analysis/live-cell-analysis-instruments/sx5-live-cell-analysis-instrument |
| nSolver version 4.0 | Nanostring | N/A |
| ELDA | Hu and Smyth, 2009 | http://bioinf.wehi.edu.au/software/elda/index.html |
| Applied Biosystems QuantStudio 3 | Thermo Fisher | N/A |
| Fiji | Schindelin et al., 2012 | https://imagej.net/downloads |
|
| ||
| Other | ||
|
| ||
| Depmap | Tsherniak et al., 2017 | https://depmap.org/R2-D2/ |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
GBM tumor cell derivation and culture
GBM tumor models were generated by passaging primary tumor cells through immunocompromised mice as previously described (Bao et al., 2006; Lathia et al., 2010). Briefly, primary tumor cells were intracranially implanted into NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (male or female), and upon tumor formation, tumors were isolated and digested with papain (Worthington). Dissociated cells were plated overnight in Neurobasal Medium minus phenol red (ThermoFisher) with 1× B27 supplement (ThermoFisher, #17504001), 1 mM sodium pyruvate, 2 mM L-glutamine, 50 U/mL penicillin/streptomycin, 20 ng/mL human (h)EGF and 20 ng/mL hFGF2 (R&D systems). Subsequently, CD133+ cells were isolated by magnetic bead sorting (Miltenyi, #130–097-049) and cultured in the media described above. Some cell models were previously established at Duke University and obtained through approved material transfer agreements. CD133+ cells were seeded in suspension culture at 5 × 104 cells/mL and passaged no more than 10 times. After 10 passages, cells were re-implanted subcutaneously into the flank of NSG mice and enriched for CD133+ cells. Cells were routinely checked for mycoplasma using MycoAlert detection kit (Lonza, LT07–118) and all cells were prophylactically treated with low dose mycoplasma removal agent (MP bio 093050044) after initial CSC isolation. De-identified GBM specimens were collected from the Cleveland Clinic Brain Tumor and Neuro-Oncology Center in accordance with an Institutional Review Board-approved protocol, and informed consent was obtained from all GBM patients contributing tumor specimens. The gender information of the tumors is not known. Human astrocytes from cerebral cortex were obtained from ScienCell Research Laboratories (#1800).
Intracranial implantation
Intracranial tumor transplants were performed as described previously (Bayik et al., 2020). Six to 8 week old NSG mice were anesthetized with inhaled isoflurane for the duration of the procedure. Initial survival studies were done with both male and female mice. After no sex difference was observed, the remainder of the experiments were performed on male mice. A total of 20,000 T4121 or T387 CSCs infected with control or SerpinB3 shRNAs were suspended in 10 μL Neurobasal null medium and stereotactically implanted into the left hemisphere ~2.5 mm deep into the brain. In relevant experiments, on day 10 after implantation, mice were anesthetized with xylazine (0.13 mg/mouse) and ketamine (1.3 mg/mouse) and exposed to 2 Gy radiation for either 3 or 5 days (PANTAK) starting 10 days post-tumor implantation and shielding the body with lead. Mice were monitored for neurologic signs and weight loss and deemed at endpoint when exhibiting any of these symptoms. Endpoint mice were transcardially perfused using 4% paraformaldehyde, and the brains were dissected for histological analysis after at least 48 h in 4% paraformaldehyde. All experiments were performed in compliance with institutional guidelines and were approved by the Institutional Animal Care and Use Committee of the Cleveland Clinic (protocol 2019–2195 and 2019–2299).
In a separate experiment, 5 mice per group were sacrificed at a predetermined endpoint of 21 days after injection of T4121 cells (2 NT mice died before the endpoint and were not included). At 21 days, mice were perfused, and brains were collected. Sections were cut at three different levels for each brain and subjected to hematoxylin and eosin staining. The largest tumor cross-section for each brain was identified and ImageJ was utilized to quantify the area of the entire brain and area of the tumor. The tumor proportion of the cross-section was calculated and graphed.
METHOD DETAILS
Immunoblotting
Protein was isolated from cells using a lysis buffer composed of 10 mM Tris HCl, 1 mM EDTA, 150 mM NaCl, 0.5% NP-40, 1 mM PMSF, 1× protease inhibitor (Sigma, #p8340), and 1× phosphatase inhibitor cocktail (Sigma, #p5726). Cells were intermittently incubated with the lysis buffer on ice and vortexed three times before being spun down for 10 min at 14,000 rpm. Protein concentrations were measured using bovine serum albumin for the protein standard and protein assay dye (Bio-Rad). A total of 40 μg of protein per condition was denatured with SDS-PAGE sample buffer and then loaded into polyacrylamide SDS-PAGE gels. The gels were run at 120 volts for 80 min and then transferred onto PVDF membranes (Millipore). The membranes were then blocked with 5% nonfat milk and probed with the appropriate primary antibody: SerpinB3 (Invitrogen PA5–30164, 1:5000), JAM-A (B&D Biosciences 612120, 1:1000), cathepsin L (ThermoFisher BMS1032, 1:5000), AKT (Cell Signaling, 9272), phosphoAKT (Ser473)(Cell Signaling, 4075A) and c-MYC (Cell Signaling Technology 5605, 1:5000). β-Actin (Bio-Rad 12004163, 1:10,000) was used as a loading control. Secondary antibodies specific to the species of the primary antibody conjugated to horseradish peroxidase were added to the membranes: anti-rabbit (Invitrogen) and anti-mouse (EMD Millipore). Membranes were developed with Pierce ECL 2 Western Blotting Substrate (Thermo Scientific) onto film. For some experiments, secondary antibody was conjugated to StarBright 700 (Bio-Rad 12004161). For these experiments, a Bio-Rad Chemidoc MP was used to image the blots.
Affinity purification of His-tagged JAM-A
T4121 CSCs were transiently transfected with N-terminal His-tagged full-length JAM-A (Sinobiologicals, HG10198-NH). The His-tagged JAM-A was pulled down and isolated with nickel beads. Mass spectrometric analysis was used to identify binding partners that were pulled down along with JAM-A. For protein digestion, the bands were cut from the gel, washed/destained in 50% ethanol/5% acetic acid and then dehydrated in acetonitrile. The bands were then reduced with DTT and alkylated with iodoacetamide prior to in-gel digestion. Bands were digested overnight in-gel using trypsin. The peptides that were formed were extracted from the polyacrylamide in 50% acetonitrile with 5% formic acid. These extracts were combined and evaporated to <10 μL in a Speedvac and then resuspended in 1% acetic acid. The LC-MS system was a Finnigan LTQ-Obitrap Elite hybrid mass spectrometer system. The HPLC column was a Dionex 15 cm × 75 μm id Acclaim Pepmap C18, 2 μm, 100 Å reverse phase capillary chromatography column. The digest was analyzed using the data-dependent multitask capability of the instrument acquiring full-scan mass spectra to determine peptide molecular weights and product ion spectra to determine amino acid sequence in successive instrument scans. The data were analyzed by using all CID spectra collected in the experiment to search the human UniProtKB database with the search program Mascot. These partners were cross-referenced with the contaminant repository for affinity purification to remove negative controls.
Immunostaining
Cells were plated onto coverslips in 6 well plates, fixed with 4% paraformaldehyde, blocked in donkey serum with 0.1% Triton X-100, and then incubated with the appropriate primary antibody (SerpinB3; PA5–30164 Invitrogen, 1:500, JAM-A; Santa Cruz sc-53623, 1:500, SOX2; R&D, MAB2018) followed by a species-specific secondary antibody. Secondary antibodies were as follows, donkey-anti mouse Alexa Fluor 555 (ThermoFisher) and donkey-anti rabbit Alexa Fluor 488 (ThermoFisher). The cells were then stained with Hoechst 33,342 (Invitrogen H3570, 1:3000) before being mounted with Vectashield (Vector Labs) onto glass cover slides and imaged using a confocal microscope.
Lysosomal membrane permeability post irradiation
T4121 tumor cells were treated with 5 Gy radiation. After 6 h, 2 μg/mL acridine orange (Sigma A6014) was added for 30 min, and media was replaced before live cells were imaged.
For cathepsin L staining, tumor cells were treated with 5 Gy radiation and fixed with 4% formaldehyde after 6 h. Cathepsin L antibody (ThermoFisher BMS1032, 1:1000) was added overnight, secondary antibody was then added (donkey-anti mouse Alexa Fluor 488, ThermoFisher), and Hoechst 33342 (Invitrogen H3570, 1:3000) was utilized as a nuclear counterstain.
Confocal microscopy
All images were taken with an inverted Leica SP8 confocal microscope at 40× magnification at room temperature. The LASX software was utilized for image acquisition. For image analysis, Fiji software was utilized. For quantification, images were split into individual channels, and the “integrated density” tool was utilized to quantify intensity of each channel per image. When quantifying the total number of cells, nuclei were counted manually.
Immunohistochemistry on human glioblastoma
Standard immunohistochemistry analysis was performed on two patient specimens with a diagnosis of primary IDH-wild-type GBM using SerpinB3 antibody (Invitrogen, PA5–30164) diluted at 1:1000. Four-micrometer thick sections of FFPE tissue on charged slides were baked in the oven at 60C for 60 min before being deparrafinized and re-hydrated. Antigen retrieval was achieved using a pH6 retrieval buffer (Biocare Reveal). Slides were cooled to room temperature and washed in TBS before neutralizing endogenous peroxidase (Biocare Peroxidase 1). Slides were then treated with a serum-free casein background block (Biocare Background Sniper) before pre-incubation in a 10% goat serum block for 60 min. Primary antibody was then added to the slides for overnight incubation at 4C. After incubation, slides were washed well with TBS-T before incubating in HRP polymer (Biocare MACH 4 Universal HRP Polymer). Finally, reaction products were visualized with DAB (Biocare Betazoid DAB Chromogen Kit). Slides were then counterstained with hematoxylin, dehydrated and mounted with xylene-based mounting media.
Cycloheximide protein stability assay
T387 and T4121 CSCs were stably transduced with JAM-A KD-2 or SHC002 shRNA constructs. Cells were plated into a 12 well dish with 100,000 cells per well. Cycloheximide (Alfa Aesar, #J66004) was added to all wells except the control. Cells were collected 6 and 12 h after addition of cycloheximide and protein was isolated and run on Western blot that was blotted for SerpinB3 (Invitrogen PA5–30164, 1:5000) with actin as loading control.
SerpinB3 overexpression mutants
SerpinB3 overexpression vectors were obtained from the Zong lab and were developed as previously described (Sheshadri et al., 2014). These included a wild-type SerpinB3 in the pLPC-N Flag vector (Addgene, #12521) as well as an empty vector and a mutant SerpinB3 with amino acids 340–345 deleted (Δ6). Cells were transfected with Fugene HD (Promega, E2311) at an optimized ratio with Opti-mem. After 12 h, media was replaced, and cells recovered for 2 days before treatment with LLME (1.5 mM) and assessment of apoptosis as described below.
Stable transduction with lentiviral shRNA and overexpression construct
MISSION® pLKO.1-puro Non-Mammalian shRNA Control Plasmid (SHC002) and SerpinB3 shRNA plasmids TRCN0000373440 (KD1), TRCN0000373501 (KD2), TRCN0000052398 (KD3) and TRCN0000373500 (KD4) were purchased from Sigma. These correspond to four non-overlapping single shRNAs. Lentivirus was packaged in 293 T cells using psPAX2 and pMD2G using calcium phosphate transfection, and media containing lentiviral particles were collected. This supernatant containing lentiviral particles was concentrated using PEGit virus precipitation solution according to the manufacturer’s protocol (System Biosciences). JAM-A knockdown constructs were as follows: TRCN0000061650 (KD1) and TRCN0000061649 (KD2).
Prior to transfection, CSCs were grown adherently on 6 well plates pretreated with Geltrex (Life Technologies, A1413301). Lentivirus was added to and incubated with the cells for 24 h. Then cells were grown in their appropriate media for 24 h, after which selection with puromycin (ThermoFisher, 54–022-2100) was initiated. Transfected cells were incubated in media with puromycin (1 mg/mL stock) at 1:333 for 48 h. Stably transfected cells were maintained in their regular media plus puromycin at 1:1000.
Cellular viability
Cellular viability was measured by plating each line of interest in triplicate in a 96-well plate at a density of 1000 cells/100 μL media per well. ATP levels at day 0 and day 7 were measured using CellTiter-Glo Luminescent Cell Viability Assay (Promega). For analysis, day 7 was normalized to the day 0 measurement.
To measure cell count over time, cells were plated in triplicate in a Geltrex-coated 96-well plate at a density of 1000 cells/100 μL media per well. The 96-well plate was then placed in the IncuCyte SX5 Live-Cell Analysis Instrument, and images were taken every 8 h for 7 days. The cell-by-cell software was then utilized to determine cell count per well, and these values were normalized to the time 0 cell count.
NanoString
RNA was isolated using an RNeasy mini kit (Qiagen), and then the nCounter® PanCancer Pathways Panel was used to analyze RNA expression. Two tumor models (T4121 and DI318) were analyzed in triplicate in each condition (non-target and SerpinB3 knockdown (KD2)). nSolver version 4.0 was utilized to determine pathway alterations.
TGF-β ELISA
R&D systems human TGF-β1 DuoSet ELISA catalog# DY240 was used to quantify TGF-β1 in vitro from conditioned media isolated at day 2 after plating 200,000 cells per well in a 12 well plate with 1.5 mL of complete Neurobasal media. Output was normalized to total protein concentration in the pellet to control for changes in cell viability.
Radiation treatment
A total of 50,000 cells per well were plated in triplicate in a 12 well plate. Cells were then irradiated with varying doses of radiation. On day 2, cell viability was measured using CellTiter-Glo Luminescent Cell Viability Assay (Promega). Viability was normalized to the untreated control for each condition and graphed as a percentage of the total.
For post-radiation apoptosis assays, cell were irradiated and when indicated, cathepsin L inhibitors E64D (Enzo, #PI107–0001) and Z-FY-CHO (Med Chem Express, #HY-128140) were added immediately after radiation. Cells were incubated for 24 h, then treated with Accutase (Biolegend, #42320) to ensure a single-cell suspension and stained with annexin V and propidium iodide as described below. Double positive cells were quantified. A similar experiment was performed with Temozolomide (Santa Cruz, #CAS 85622–93-1) as a replacement for irradiation.
L-leucyl-L-leucine methyl ester IC50
A total of 4,000 cells per well was plated in 96 well plate in quintuplets. L-leucyl-L-leucine methyl ester (LLME, Cayman #16008) was added over a range of concentrations. After 7 days of treatment, cell viability was measured, and half-maximal inhibitory concentrations (IC50) for each condition were calculated.
L-leucyl-L-leucine methyl ester apoptosis assay
Cells were plated in triplicate in a 12 well dish with 50,000 cells per well. LLME or a DMSO control was added into appropriate wells. Cathepsin L inhibitors E64D (Enzo, #PI107–0001) and Z-FY-CHO (Med Chem Express, #HY-128140) were added at the same time. After 6 h, cells were stained with annexin V and propidium iodide as described below and double-positive cells were quantified.
Limiting-dilution analysis
Cells were plated at 100 cells per well in 12 wells of a 96 well plate, and two-fold serial dilutions were performed. Twelve wells of each cell dose were plated. Limiting dilution plots and stem-cell frequencies were calculated using ELDA analysis (http://bioinf.wehi.edu.au/software/elda/index.html (Hu and Smyth, 2009)).
Cell death
For caspase activity assays, cells were plated in quintuplicate at 10,000 cells/well in 96 well plates for 48 h. Caspase 3/7 activity was determined with the Caspase-Glo 3/7 assay (Promega, G8090) and caspase activity was normalized to cell number by performing the CellTiter-Glo Luminescent Cell Viability Assay on the duplicate plate.
Additionally, cells were plated for imaging in an IncuCyte as described above, and 5 μM Caspase 3/7 dye was included in the media (Sartorius, #4704). The number of red nuclei (indicating active caspase 3/7) was divided by the area confluence per well. These values were then normalized to the non-target control.
For annexin V and propidium iodide assay, 25,000 cells/well were plated in 1.5 mL of Neurobasal media. After 48 h, a single-cell suspension was obtained, and FITC-labeled annexin V and propidium iodide were added in accordance with the protocol (BioLegend, #640914). Samples were run on an LSR Fortessa flow cytometer (BD Biosciences) with a minimum of 10,000 events collected. Single cells were gated, and the percentage of annexin V- and PI-positive cells was determined.
JAM-A and SerpinB3 coexpression
The percentage of PDX GBM CSCs that co-expressed SerpinB3 and JAM-A was determined with flow cytometry. GBM CSCs were grown as spheres treated with Accutase to form a single-cell suspension. Cells were treated with LIVE/DEAD™ Fixable Blue Dead Cell Stain Kit (Invitrogen, L23105) at a 1:1,000 dilution in Mojosort buffer (Biolegend, #480017). After washing, JAM-A AF647 (Invitrogen, #00–5523) was added and incubated for 10 min. Cells were then fixed overnight. The next day, cells were washed in permeabilization buffer. Post-wash, either SerpinB3 or CD4 intracellular control was added to the cells. Cells were again washed, and donkey-anti rabbit Alexa Fluor 488 was added. Cells were then run through an LSR Fortessa flow cytometer (BD Biosciences) with a minimum of 10,000 events collected.
Real-time reverse transcription polymerase chain reaction
RNA was collected from cells using an RNeasy kit (Qiagen, 74004). RNA concentrations were measured using a NanoDrop spectrophotometer, and cDNA was synthesized with qScript synthesis reagent (Quanta Biosciences, 95048). qPCR was run with the primers shown in Table S2 using SYBR-Green Mastermix (SA Biosciences, 4385610) and an Applied Biosystems QuantStudio 3. During analysis, threshold cycle numbers were normalized to GAPDH or Actin levels.
Depmap RNAi
The RNAi DEMETER2 analysis framework was utilized to determine the gene dependency of SerpinB3 (McFarland et al., 2018). Data was accessed from https://depmap.org/R2-D2/ on 12/12/2021 and relied on three large RNAi datasets (Marcotte et al., 2016; McDonald et al., 2017; Tsherniak et al., 2017).
Bioinformatics
On May 12th, 2022, the TCGA glioblastoma Agilent-4502A dataset was accessed via http://gliovis.bioinfo.cnio.es/. SerpinB3 expression was compared across GBM subtypes.
QUANTIFICATION AND STATISTICAL ANALYSIS
For two-group comparisons, p values were calculated using Student’s t Test. For multiple group comparisons, one-way ANOVA with Dunnett’s multiple comparisons test was used as indicated in the figure legends. Log rank tests were used for survival analysis. GraphPad Prism 6 was used for statistical tests. All in vitro experiments were done in at least technical triplicates for each experimental group, and multiple independent experiments were performed. The Grubb’s test was performed to determine whether any outliers were statistically different. All figures graph mean and standard deviation. Statistical details can be found in figure legends. p < 0.05 was considered statistically significant. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Supplementary Material
Highlights.
Junctional adhesion molecule-A stabilizes SerpinB3 in glioblastoma cancer stem cells
Knockdown of SerpinB3 decreased stem cell phenotypes and increased apoptosis
Radiation induces lysosomal membrane permeability and cathepsin L release in GBM
SerpinB3 inhibition of cathepsin L-mediated cell death drives radiation resistance
ACKNOWLEDGMENTS
We thank Drs. William Schiemann, Mark Jackson, and Alex Huang (Case Western Reserve University) for critical feedback and Gary Silverman, Clifford Luke, and Stephanie Markovina (Washington University in St. Louis School of Medicine) for discussions and advice about lysosomal assessments. We thank Ms. Karen Kasler and Dr. Robert Fairchild for their assistance with the Nanostring analysis. We thank Belinda Willard for assistance with the MS and proteomics analysis. We thank Dr. Wei-Xing Zo (Rutgers University) for the SerpinB3 overexpression constructs. We also thank members of the Lathia laboratory, including Katie Troike, Samuel Sprowls, Salma Ben Salem, Kristen Kay, Juyeun Lee, and Sabrina Wang, for insightful discussions, and Sadie Johnson for assistance with mouse work. We thank Ms. Amanda Mendelsohn for illustration assistance. This work is supported by National Institutes of Health (NIH) grants F30CA250254 (to A.L.), T32GM007250 (to A.L.), K99CA248611 (to D.B.), 5T32AI007024 (to D.C.W.), R01 NS117104 (to J.D.L. and C.H.), and R35 NS127083 (to J.D.L.). Work in the Lathia laboratory is also supported by the American Brain Tumor Association, the Case Comprehensive Cancer Center, the Lerner Research Institute, and NIH grants P01 CA245705 and R01 NS109742.
Footnotes
DECLARATION OF INTERESTS
M.S.A. is the recipient of grants/research support from AstraZeneca, Abbvie, Bristol Myers Squibb, Bayer, Incyte, Pharmacyclics, Novocure, and Merck; is a shareholder in Doctible and Mimivax; and has received honoraria or consultation fees from Wiley, Abvvie, Bayer, Elsevier, Forma Therapeutics, Karyopharm Therapeutics, Tocagen, and VBI Vaccines. The remaining authors declare no competing interests.
INCLUSION AND DIVERSITY
We worked to ensure gender balance in the recruitment of human subjects. We worked to ensure sex balance in the selection of non-human subjects. We worked to ensure diversity in experimental samples through the selection of the cell lines.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111348.
REFERENCES
- Alvarado AG, Turaga SM, Sathyan P, Mulkearns-Hubert EE, Otvos B, Silver DJ, Hale JS, Flavahan WA, Zinn PO, Sinyuk M, et al. (2016). Coordination of self-renewal in glioblastoma by integration of adhesion and microRNA signaling. Neuro Oncol. 18, 656–666. 10.1093/neuonc/nov196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarado AG, Thiagarajan PS, Mulkearns-Hubert EE, Silver DJ, Hale JS, Alban TJ, Turaga SM, Jarrar A, Reizes O, Longworth MS, et al. (2017). Glioblastoma cancer stem cells evade innate immune suppression of self-renewal through reduced TLR4 expression. Cell Stem Cell 20, 450–461.e4. 10.1016/j.stem.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anido J, Sáez-Borderías A, Gonzàlez-Juncà A, Rodón L, Folch G, Carmona MA, Prieto-Sánchez RM, Barba I, Martínez-Sáez E, Prudkin L, et al. (2010). TGF-B receptor inhibitors target the CD44(high)/id1(high) glioma-initiating cell population in human glioblastoma. Cancer Cell 18, 655–668. 10.1016/j.ccr.2010.10.023. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, and Rich JN (2006). Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760. 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, Li Z, Sathornsumetee S, Wang H, McLendon RE, Hjelmeland AB, and Rich JN (2008). Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 68, 6043–6048. 10.1158/0008-5472.CAN-08-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayik D, Lauko AJ, Roversi GA, Serbinowski E, Acevedo-Moreno L-A, Lanigan C, Orujov M, Lo A, Alban TJ, Kim A, et al. (2020). Hepatobiliary malignancies have distinct peripheral myeloid-derived suppressor cell signatures and tumor myeloid cell profiles. Sci. Rep. 10, 18848. 10.1038/s41598-020-75881-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ, Aigner L, Brawanski A, Bogdahn U, and Beier CP (2007). CD133+ and CD133− glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 67, 4010–4015. 10.1158/0008-5472.CAN-06-4180. [DOI] [PubMed] [Google Scholar]
- Bellettato CM, and Scarpa M (2018). Possible strategies to cross the blood–brain barrier. Ital. J. Pediatr. 44, 131. 10.1186/s13052-018-0563-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian E, Chen X, Cheng L, Cheng M, Chen Z, Yue X, Zhang Z, Chen J, Sun L, Huang K, et al. (2021). Super-enhancer-associated TMEM44-AS1 aggravated glioma progression by forming a positive feedback loop with Myc. J. Exp. Clin. Cancer Res. 40, 337. 10.1186/s13046-021-02129-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruna A, Darken RS, Rojo F, Ocaña A, Peñuelas S, Arias A, Paris R, Tortosa A, Mora J, Baselga J, and Seoane J (2007). High TGFβ-smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell 11, 147–160. 10.1016/j.ccr.2006.11.023. [DOI] [PubMed] [Google Scholar]
- Cannito S, Turato C, Paternostro C, Biasiolo A, Colombatto S, Cambieri I, Quarta S, Novo E, Morello E, Villano G, et al. (2015). Hypoxia up-regulates SERPINB3 through HIF-2α in human liver cancer cells. Oncotarget 6, 2206–2221. 10.18632/oncotarget.2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelli V, Giordano A, Benedetti E, Giansanti F, Quintiliani M, Cimini A, and d’Angelo M (2021). The great escape: the power of cancer stem cells to evade programmed cell death. Cancers 13, 328. 10.3390/cancers13020328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colak S, and Medema JP (2014). Cancer stem cells–important players in tumor therapy resistance. FEBS J. 281, 4779–4791. 10.1111/febs.13023. [DOI] [PubMed] [Google Scholar]
- Collie-Duguid ESR, Sweeney K, Stewart KN, Miller ID, Smyth E, and Heys SD (2012). SerpinB3, a new prognostic tool in breast cancer patients treated with neoadjuvant chemotherapy. Breast Cancer Res. Treat. 132, 807–818. 10.1007/s10549-011-1625-9. [DOI] [PubMed] [Google Scholar]
- Correnti M, Cappon A, Pastore M, Piombanti B, Lori G, Oliveira DVPN, Munoz-Garrido P, Lewinska M, Andersen JB, Coulouarn C, et al. (2021). The protease-inhibitor SerpinB3 as a critical modulator of the stem-like subset in human cholangiocarcinoma. Liver Int. 42, 233–248. 10.1111/liv.15049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBridge RB, Tang P, Hsia HC, Leong PM, Miller JH, and Calos MP (1987). Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol Cell Biol 7, 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehrenbacher N, Gyrd-Hansen M, Poulsen B, Felbor U, Kallunki T, Boes M, Weber E, Leist M, and Jäättelä M (2004). Sensitization to the lysosomal cell death pathway upon immortalization and transformation. Cancer Res. 64, 5301–5310. 10.1158/0008-5472.CAN-04-1427. [DOI] [PubMed] [Google Scholar]
- Garnier D, Renoult O, Alves-Guerra M-C, Paris F, and Pecqueur C (2019). Glioblastoma stem-like cells, metabolic strategy to kill a challenging target. Front. Oncol. 9, 118. 10.3389/fonc.2019.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimple RC, Bhargava S, Dixit D, and Rich JN (2019). Glioblastoma stem cells: lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 33, 591–609. 10.1101/gad.324301.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes S, Marques PI, Matthiesen R, and Seixas S (2014). Adaptive evolution and divergence of SERPINB3: a young duplicate in great Apes. PLoS One 9, e104935. 10.1371/journal.pone.0104935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder BG, Blomquist MR, Wang J, Kim AJ, Woodworth GF, Winkles JA, Loftus JC, and Tran NL (2018). Developments in blood-brain barrier penetrance and drug repurposing for improved treatment of glioblastoma. Front. Oncol. 8, 462. 10.3389/fonc.2018.00462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heit C, Jackson BC, McAndrews M, Wright MW, Thompson DC, Silverman GA, Nebert DW, and Vasiliou V (2013). Update of the human and mouse SERPIN gene superfamily. Hum. Genom. 7, 22. 10.1186/1479-7364-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh A, Ellsworth R, and Hsieh D (2011). Hedgehog/GLI1 regulates IGF dependent malignant behaviors in glioma stem cells. J. Cell. Physiol. 226, 1118–1127. 10.1002/jcp.22433. [DOI] [PubMed] [Google Scholar]
- Hu Y, and Smyth GK (2009). ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 347, 70–78. 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, and Kondo S (2004). Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 11, 448–457. 10.1038/sj.cdd.4401359. [DOI] [PubMed] [Google Scholar]
- Katagiri C, Nakanishi J, Kadoya K, and Hibino T (2006). Serpin squamous cell carcinoma antigen inhibits UV-induced apoptosis via suppression of c-JUN NH2-terminal kinase. J. Cell Biol. 172, 983–990. 10.1083/jcb.200508064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, and Torigoe T (1977). Radioimmunoassay for tumor antigen of human cervical squamous cell carcinoma. Cancer 40, 1621–1628. . [DOI] [PubMed] [Google Scholar]
- Lathia JD, Gallagher J, Heddleston JM, Wang J, Eyler CE, Mac-Swords J, Wu Q, Vasanji A, McLendon RE, Hjelmeland AB, and Rich JN (2010). Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell 6, 421–432. 10.1016/j.stem.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathia JD, Li M, Sinyuk M, Alvarado AG, Flavahan WA, Stoltz K, Rosager AM, Hale J, Hitomi M, Gallagher J, et al. (2014). High-throughput flow cytometry screening reveals a role for junctional adhesion molecule a as a cancer stem cell maintenance factor. Cell Rep. 6, 117–129. 10.1016/j.celrep.2013.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CLL, and Rich JN (2015). Cancer stem cells in glioblastoma. Genes Dev. 29, 1203–1217. 10.1101/gad.261982.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauko A, Mu Z, Gutmann DH, Naik UP, and Lathia JD (2020). Junctional adhesion molecules in cancer: a paradigm for the diverse functions of cell-cell interactions in tumor progression. Cancer Res. 80, 4878–4885. 10.1158/0008-5472.CAN-20-1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauko A, Lo A, Ahluwalia MS, and Lathia JD (2021). Cancer cell heterogeneity & plasticity in glioblastoma and brain tumors. Semin. Cancer Biol. 82, 162–175. 10.1016/j.semcancer.2021.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, Lu L, Irvin D, Black KL, and Yu JS (2006). Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 5, 67. 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Gao Y, Yang B, Jia X, Zhai D, Li S, Zhang Q, Jing L, Wang Y, Du Z, and Wang Y (2015). Overexpression of squamous cell carcinoma antigen 1 is associated with the onset and progression of human hepatocellular carcinoma. Arch. Med. Res. 46, 133–141. 10.1016/j.arcmed.2015.03.003. [DOI] [PubMed] [Google Scholar]
- Marcotte R, Sayad A, Brown KR, Sanchez-Garcia F, Reimand J, Haider M, Virtanen C, Bradner JE, Bader GD, Mills GB, et al. (2016). Functional genomic landscape of human breast cancer drivers, vulnerabilities, and resistance. Cell 164, 293–309. 10.1016/j.cell.2015.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Lage M, Lynch TM, Bi Y, Cocito C, Way GP, Pal S, Haller J, Yan RE, Ziober A, Nguyen A, et al. (2019). Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathol. Commun. 7, 203. 10.1186/s40478-019-0803-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason WP (2015). Blood-brain barrier-associated efflux transporters: a significant but underappreciated obstacle to drug development in glioblastoma. Neuro Oncol. 17, 1181–1182. 10.1093/neuonc/nov122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald ER, de Weck A, Schlabach MR, Billy E, Mavrakis KJ, Hoffman GR, Belur D, Castelletti D, Frias E, Gampa K, et al. (2017). Project DRIVE: a compendium of cancer dependencies and synthetic lethal relationships uncovered by large-scale, deep RNAi screening. Cell 170, 577–592.e10. 10.1016/j.cell.2017.07.005. [DOI] [PubMed] [Google Scholar]
- McFarland JM, Ho ZV, Kugener G, Dempster JM, Montgomery PG, Bryan JG, Krill-Burger JM, Green TM, Vazquez F, Boehm JS, et al. (2018). Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat. Commun. 9, 4610. 10.1038/s41467-018-06916-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellacheruvu D, Wright Z, Couzens AL, Lambert J-P, St-Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY, et al. (2013). The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 10, 730–736. 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell K, Troike K, Silver DJ, and Lathia JD (2021). The evolution of the cancer stem cell state in glioblastoma: emerging insights into the next generation of functional interactions. Neuro Oncol. 23, 199–213. 10.1093/neuonc/noaa259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami A, Suminami Y, Hirakawa H, Nawata S, Numa F, and Kato H (2001). Squamous cell carcinoma antigen suppresses radiation-induced cell death. Br. J. Cancer 84, 851–858. 10.1054/bjoc.2000.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngan HY, Chan SY, Wong LC, Choy DT, and Ma HK (1990). Serum squamous cell carcinoma antigen in the monitoring of radiotherapy treatment response in carcinoma of the cervix. Gynecol. Oncol. 37, 260–263. 10.1016/0090-8258(90)90344-k. [DOI] [PubMed] [Google Scholar]
- Oberle C, Huai J, Reinheckel T, Tacke M, Rassner M, Ekert PG, Buellesbach J, and Borner C (2010). Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ. 17, 1167–1178. 10.1038/cdd.2009.214. [DOI] [PubMed] [Google Scholar]
- Pelaz SG, Jaraíz-Rodríguez M,Álvarez-Vázquez A, Talaverón R, García-Vicente L, Flores-Hernández R, Cedrón M.G. de, Tabernero M, Molina AR, et al. (2020). Targeting metabolic plasticity in glioma stem cells in vitro and in vivo through specific inhibition of c-Src by TAT-Cx43266–283. EBioMedicine 62. 10.1016/j.ebiom.2020.103134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petty RD, Kerr KM, Murray GI, Nicolson MC, Rooney PH, Bissett D, and Collie-Duguid ESR (2006). Tumor transcriptome reveals the predictive and prognostic impact of lysosomal protease inhibitors in non-small-cell lung cancer. J. Clin. Oncol. 24, 1729–1744. 10.1200/JCO.2005.03.3399. [DOI] [PubMed] [Google Scholar]
- Piazza MD, Mader C, Geletneky K, Herrero Y Calle M, Weber E, Schlehofer J, Deleu L, and Rommelaere J (2007). Cytosolic activation of cathepsins mediates parvovirus H-1-Induced killing of cisplatin and TRAIL-resistant glioma cells. J. Virol. 81, 4186–4198. 10.1128/JVI.02601-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pombo Antunes AR, Scheyltjens I, Duerinck J, Neyns B, Movahedi K, and Van Ginderachter JA (2020). Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. Elife 9, e52176. 10.7554/eLife.52176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontisso P (2014). Role of SERPINB3 in hepatocellular carcinoma. Ann. Hepatol. 13, 722–727. [PubMed] [Google Scholar]
- Safa AR (2016). Resistance to cell death and its modulation in cancer stem cells. Crit. Rev. Oncog. 21, 203–219. 10.1615/CritRevOncog.2016016976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheshadri N, Catanzaro JM, Bott AJ, Sun Y, Ullman E, Chen EI, Pan J-A, Wu S, Crawford HC, Zhang J, and Zong WX (2014). SCCA1/SERPINB3 promotes oncogenesis and epithelial-mesenchymal transition via the unfolded protein response and IL6 signaling. Cancer Res. 74, 6318–6329. 10.1158/0008-5472.CAN-14-0798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada H, Nabeya Y, Okazumi S.i., Matsubara H, Shiratori T, Gunji Y, Kobayashi S, Hayashi H, and Ochiai T (2003). Prediction of survival with squamous cell carcinoma antigen in patients with resectable esophageal squamous cell carcinoma. Surgery 133, 486–494. 10.1067/msy.2003.139. [DOI] [PubMed] [Google Scholar]
- Siebzehnrubl FA, Silver DJ, Tugertimur B, Deleyrolle LP, Siebzehnrubl D, Sarkisian MR, Devers KG, Yachnis AT, Kupper MD, Neal D, et al. (2013). The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol. Med 5, 1196–1212. 10.1002/emmm.201302827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, and Dirks PB (2004). Identification of human brain tumour initiating cells. Nature 432, 396–401. 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Son MJ, Woolard K, Nam D-H, Lee J, and Fine HA (2009). SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell 4, 440–452. 10.1016/j.stem.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strnad P, McElvaney NG, and Lomas DA (2020). Alpha1-Antitrypsin deficiency. N. Engl. J. Med. 382, 1443–1455. 10.1056/NEJMra1910234. [DOI] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. (2005). Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996. 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, Toms S, Idbaih A, Ahluwalia MS, Fink K, et al. (2017). Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA 318, 2306–2316. 10.1001/jama.2017.18718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suminami Y, Nagashima S, Murakami A, Nawata S, Gondo T, Hirakawa H, Numa F, Silverman GA, and Kato H (2001). Suppression of a squamous cell carcinoma (SCC)-related serpin, SCC antigen, inhibits tumor growth with increased intratumor infiltration of natural killer cells. Cancer Res. 61, 1776–1780. [PubMed] [Google Scholar]
- Sun Y, Sheshadri N, and Zong W-X (2017). SERPINB3 and B4: from biochemistry to biology. Semin. Cell Dev. Biol. 62, 170–177. 10.1016/j.semcdb.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a cancer dependency map. Cell 170, 564–576.e16. 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turato C, Vitale A, Fasolato S, Ruvoletto M, Terrin L, Quarta S, Ramirez Morales R, Biasiolo A, Zanus G, Zali N, et al. (2014). SERPINB3 is associated with TGF-β1 and cytoplasmic β-catenin expression in hepatocellular carcinomas with poor prognosis. Br. J. Cancer 110, 2708–2715. 10.1038/bjc.2014.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turato C, Cannito S, Simonato D, Villano G, Morello E, Terrin L, Quarta S, Biasiolo A, Ruvoletto M, Martini A, et al. (2015). SerpinB3 and yap interplay increases myc oncogenic activity. Sci. Rep. 5, 17701. 10.1038/srep17701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. (2015). Tissue-based map of the human proteome. Science 347, 1260419. 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Verfaillie T, Garg AD, and Agostinis P (2013). Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett. 332, 249–264. 10.1016/j.canlet.2010.07.016. [DOI] [PubMed] [Google Scholar]
- Villano G, Turato C, Quarta S, Ruvoletto M, Ciscato F, Terrin L, Semeraro R, Paternostro C, Parola M, Alvaro D, et al. (2014). Hepatic progenitor cells express SerpinB3. BMC Cell Biol. 15, 5. 10.1186/1471-2121-15-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Gómez-Sintes R, and Boya P (2018). Lysosomal membrane permeabilization and cell death. Traffic 19, 918–931. 10.1111/tra.12613. [DOI] [PubMed] [Google Scholar]
- Wang J, Wang H, Li Z, Wu Q, Lathia JD, McLendon RE, Hjelmeland AB, and Rich JN (2008). c-Myc is required for maintenance of glioma cancer stem cells. PLoS One 3, e3769. 10.1371/journal.pone.0003769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Luke CJ, Pak SC, Shi V, Chen L, Moore J, Andress AP, Jayachandran K, Zhang J, Huang Y, et al. (2022). SERPINB3 (SCCA1) inhibits cathepsin L and lysoptosis, protecting cervical cancer cells from chemoradiation. Commun. Biol. 5, 1–16. 10.1038/s42003-021-02893-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadati T, Houben T, Bitorina A, and Shiri-Sverdlov R (2020). The ins and outs of cathepsins: physiological function and role in disease management. Cells 9, E1679. 10.3390/cells9071679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Guo Y, Zhang X, and Jiang Z (2020). Lys05 induces lysosomal membrane permeabilization and increases radiosensitivity in glioblastoma. J. Cell. Biochem. 121, 2027–2037. 10.1002/jcb.29437. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Accession numbers are listed in the key resources table. DOI link to original western blot images is available in key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
|
| ||
| Antibodies | ||
|
| ||
| SerpinB3 | Invitrogen | PA5-30164; RRID:AB_2547638 |
| JAM-A | B&D Biosciences | 612120; RRID:AB_399491 |
| cathepsin L | ThermoFisher | BMS1032; RRID:AB_10596643 |
| c-Myc | Cell Signaling Technology | 5605; RRID:AB_1903938 |
| β-Actin | Bio-Rad | 12004163; RRID:AB_2861334 |
| Rabbit StarBright 700 | Bio-Rad | 12004161; RRID:AB_2721073 |
| JAM-A | Santa Cruz | sc-53623; RRID:AB_784134 |
| SOX2 | R&D | MAB2018; RRID:AB_358009 |
| AKT | Cell Signaling | 9272; RRID:AB_329827 |
| pAKT(Ser473) | Cell Signaling | 4075S; RRID:AB_916029 |
| Donkey-anti mouse Alexa Fluor 555 | ThermoFisher | A-31570; RRID:AB_2536180 |
| Donkey-anti rabbit Alexa Fluor 488 | ThermoFisher | A-21206; RRID:AB_2535792 |
| JAM-A AF647 | Santa Cruz | sc-53623 AF647; RRID:AB_784134 |
|
| ||
| Biological samples | ||
|
| ||
| GBM Fresh Tumor Samples | Cleveland Clinic Foundation and Northwestern University | This manuscript |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| N-terminal His-tagged full-length JAM-A | Sinobiologicals | HG10198-NH |
| Hoechst 33342 | Invitrogen | H3570 |
| Vectashield | Vector Labs | H-1000-10 |
| acridine orange | Sigma | A6014 |
| pH6 retrieval buffer (Reveal) | Biocare | V1000 |
| serum-free casein background block (Background Sniper) | Biocare | BS966 |
| MACH 4 Universal HRP Polymer | Biocare | M4U534 |
| Geltrex | Life Technologies | A1413301 |
| puromycin | ThermoFisher | 54-022-2100 |
| L-leucyl-L-leucine methyl ester | Cayman | #16008 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Betazoid DAB Chromogen Kit | Biocare | BDB2004 |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | G7570 |
| Caspase-Glo 3/7 | Promega | G8090 |
| RNeasy mini kit | Qiagen | 74004 |
| nCounter® PanCancer Pathways Panel | Nanostring | N/A |
| TGF-β1 DuoSet ELISA | R&D systems | DY240 |
| FITC-labeled annexin V and propidium iodide | BioLegend | 640914 |
| Caspase 3/7 | Sartorius | 4704 |
| qScript synthesis reagent | Quanta Biosciences | 95048 |
| SYBR-Green Mastermix | SA Biosciences | 4385610 |
| E64D | Enzo | BML-PI107-0001 |
| Z-FY-CHO | Med Chem Express | HY-128140 |
| Cycloheximide | Alfa Aesar | J66004 |
| LIVE/DEAD™ Fixable Blue Dead Cell Stain Kit | Invitrogen | L23105 |
| MojoSort Buffer (5X) | Biolegend | 480017 |
| eBioscience™ Fixation/Permeabilization | Invitrogen | 00-5523 |
| MycoAlert detection kit | Lonza | LT07-118 |
| CD133 selection beads | Miltenyi | 130-097-049 |
|
| ||
| Deposited data | ||
|
| ||
| Unaltered Blots | Mendeley data | https://doi.org/10.17632/6dwf49mg73.1 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| 293T | DuBridge et al., 1987 | Obtained internally, available https://www.atcc.org/products/crl-3216 |
| T387 | Bao et al., 2008 | N/A |
| T4121 | Bao et al., 2008 | N/A |
| DI318 | This manuscript | N/A |
| Human astrocytes | ScienCell Research Laboratories | #1800 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) | Jackson Labs | 005557 |
|
| ||
| Oligonucleotides | ||
|
| ||
| SERPINB3 F- CGCGGTCTCGTGCTATCTGG, R- AGAAGAGGATGCTGTTGGTC |
This manuscript | N/A |
| OCT4 F- TGAGTCAGTGAACAGGGAATG R- AATCTCCCCTTTCCATTCGG |
This manuscript | N/A |
| NANOG F- GAAATACCTCAGCCTCCAGC R- GCGTCACACCATTGCTATTC |
This manuscript | N/A |
| OLIG 2 F- AGCTCCTCAAATCGCATCC R-AAAAGGTCATCGGGCTCTG |
This manuscript | N/A |
| c-MYC F- TTCGGGTAGTGGAAAACCAG R- AGTAGAAATACGGCTGCACC |
This manuscript | N/A |
| TGF-β1 F- AAGTGGACATCAAGGGTTC R- GTCCTTGCGGAAGTCAATGT |
This manuscript | N/A |
| GAPDH F-ACATCGCTCAGACACCATG R- TGTAGTTGAGGTCAATGAAGGG |
This manuscript | N/A |
| NOTCH2 F- GTGCCTATGTCCATCTGGATGG R- AGACACCTGAGTGCTGGCACAA |
This manuscript | N/A |
| JAGGED2 F- GCTGCTACGACCTGGTCAATGA R- AGGTGTAGGCATCGCACTGGAA |
This manuscript | N/A |
| Primers also listed in Table S2 | This manuscript | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| shRNA Control Plasmid (SHC002) | Sigma | SHC002 |
| SerpinB3 KD1 | Sigma | TRCN0000373440 |
| SerpinB3 KD2 | Sigma | TRCN0000373501 |
| SerpinB3 KD3 | Sigma | TRCN0000052398 |
| SerpinB3 KD4 | Sigma | TRCN0000373500 |
| JAM-A KD1 | Sigma | TRCN0000061650 |
| JAM-A KD2 | Sigma | TRCN0000061649 |
| psPAX2 | Addgene | #12260; RRID:Addgene_12260 |
| pMD2G | Addgene | #12259; RRID:Addgene_12259 |
| pLPC-N FLAG vector | Addgene | #12521; RRID:Addgene_12521 |
| Fugene HD | Promega | E2311 |
|
| ||
| Software and algorithms | ||
|
| ||
| LASX software | Leica | N/A |
| IncuCyte SX5 Live-Cell Analysis Instrument | Sartorius | https://www.sartorius.com/en/products/live-cell-imaging-analysis/live-cell-analysis-instruments/sx5-live-cell-analysis-instrument |
| nSolver version 4.0 | Nanostring | N/A |
| ELDA | Hu and Smyth, 2009 | http://bioinf.wehi.edu.au/software/elda/index.html |
| Applied Biosystems QuantStudio 3 | Thermo Fisher | N/A |
| Fiji | Schindelin et al., 2012 | https://imagej.net/downloads |
|
| ||
| Other | ||
|
| ||
| Depmap | Tsherniak et al., 2017 | https://depmap.org/R2-D2/ |