

Abstract
Adoptive cell therapy using engineered T cell receptors (TCRs) is a promising approach for targeting cancer antigens, but tumor-reactive TCRs are often weakly responsive to their target ligands (pMHCs). Affinity-matured TCRs can enhance the efficacy of TCR-T therapy but can also cross-react with off-target antigens resulting in organ immunopathology. We developed an alternative strategy to isolate TCR mutants that exhibited high activation signals coupled with low-affinity pMHC binding, through the acquisition of catch bonds. Engineered analogs of a tumor antigen MAGE-A3-specific TCR maintained physiological affinities, whilst exhibiting enhanced target killing potency and undetectable cross-reactivity, compared to a high-affinity clinically-tested TCR that exhibited lethal cross-reactivity with a cardiac antigen. Catch bond engineering is a biophysically-based strategy to generate potent TCRs for T cell therapy with reduced potential for adverse cross-reactivity.
One Sentence Summary:
T cell receptor responses to peptide-MHC antigens are enhanced through catch bond engineering while retaining physiological affinity and specificity.
Introduction
T cells mediate many important aspects of cellular immunity, including elimination of cells expressing cancer-related self-antigens. T cells express clonotypic T cell receptors (TCRs) that interact with specific peptides bound to and presented on the cell surface by major histocompatibility complex (MHC) molecules (pMHCs). Recognition of pMHC by the TCR leads to activation of downstream signaling and effector functions in T cells, including cytokine secretion and target cell killing. The molecular and structural parameters that determine TCR sensitivity in response to pMHC have been extensively studied but remain incompletely defined (1). TCR activation potency is often correlated with pMHC binding affinity, and TCR affinity maturation can result in TCRs with enhanced responsiveness to pMHC targets. However, the 3D binding affinity generally fails to predict sensitivity, suggesting additional mechanisms modulate TCR-pMHC interactions that result in functional intracellular signaling (2–4).
Mechanical force has been recently shown to play a key role as a biophysical determinant of TCR triggering and signaling (5–7), with the TCR transforming cellular shear forces into biochemical signals when binding to agonist pMHC (5–8). Single molecule force measurements on cells have shown that there is extended bond lifetime during productive antigenic pMHC-TCR interactions, termed “catch bonds” (6, 9, 10). Indeed, there is a close correlation between the detection of catch bonds with a given TCR on a T cell and the agonist potency of a particular pMHC (6). Non-stimulatory pMHC ligands have also been identified that do not exhibit catch bonds, but bind TCRs with solution affinities characteristic of many agonist TCR/pMHC interactions (6). Mutants of these non-stimulatory pMHC ligands that show agonist activity were shown to have acquired catch bonds with the TCR, yet not substantially higher 3D affinities (11). Thus, in the environment of the T cell membrane, the presence or absence of catch bonds can act as a “switch” for TCR signaling, and is not coupled to pMHC binding affinity (11). We aimed to take advantage of this cellular TCR triggering mechanism to address the limitations of current clinical TCRs used for cancer immunotherapy.
Adoptive T cell transfer (adoptive cell therapy or ACT) with engineered T cells (TCR-T or Chimeric Antigen Receptor (CAR)-T) is currently being used for cancer treatment (12, 13). In this regimen, T cells are transduced with a tumor antigen-specific TCR or CAR, respectively, and then after in vitro expansion of cell number, administered into cancer patients (14). One advantage of TCR-T ACT over CAR-T is the natural sensitivity of TCRs to very low antigen densities on tumors. However, a drawback is that many tumor antigen-specific TCRs have low affinity for tumor-associated pMHCs and only weakly activate the TCR-T cells they bind to. To overcome this problem, a common strategy is to increase the affinity of the TCR for the tumor pMHC (15). However, in some cases affinity-matured TCRs have shown substantial off-target toxicities (14, 16, 17). Indeed, an ‘affinity-matured’ TCR to MAGE-A3, a promising tumor antigen, showed lethal off-target cross-reactivity with a cardiac peptide from the TITIN protein. High-affinity TCRs likely have a higher propensity to engage off-target pMHC ligands, so alternative approaches that bypass affinity maturation will be valuable for improving ACT with TCR-T cells. Here we report an alternative TCR engineering strategy that we call “catch bond fishing” that harnesses a biophysical parameter mediating many adhesive cell surface protein-protein interactions.
Results
Design of ‘catch bond fishing’ libraries
Our previous studies showed that TCR55 does not produce measurable T cell activation although it binds to an HIV peptide (Pol448-456) presented by the HLA-B35 MHC molecule with physiological affinities. This TCR-pMHC interaction, does not form catch bonds during the binding event (11). However, HIV peptide mutants isolated from HLA-B35 yeast pMHC libraries, such as pep20, gained the capacity to form catch bonds with TCR55 and potently activated T cells bearing this receptor, while maintaining comparable affinity to the non-stimulatory parent pMHC (Figure 1A–1B, Figure S1, Figure S2) (11). Here we asked if, in a reciprocal manner, a functional screen could isolate mutants of TCR55 that gain catch bond capacity and enable functional T cell responses evoked by the ‘non-stimulatory’ HIV peptide.
Figure 1. The design of catch bond fishing libraries and selection strategy.
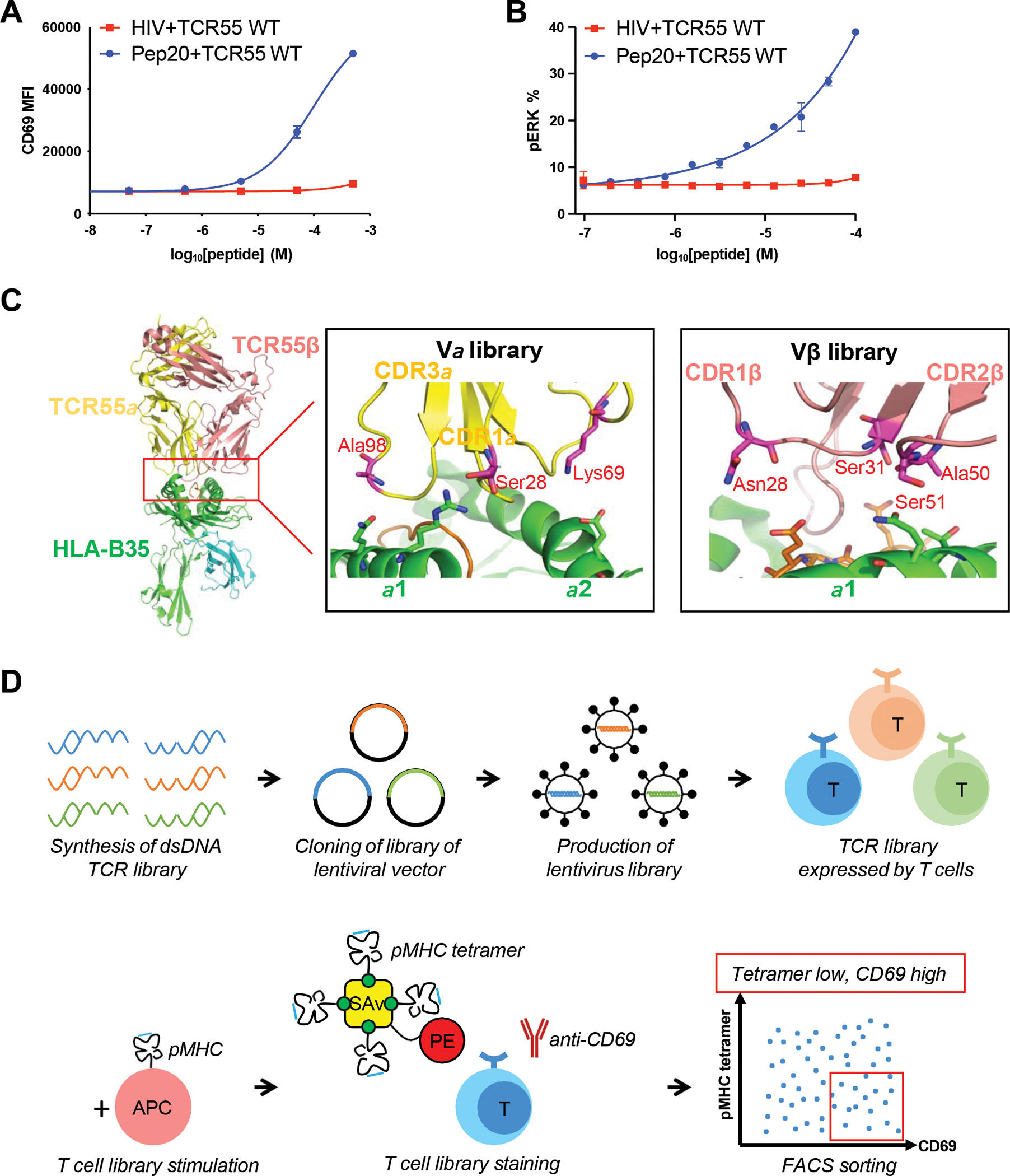
(A) TCR55-transduced SKW3 T cells were stimulated by KG-1 cells pulsed with titrated HIV or Pep20 peptides for 14 hours. Anti-CD69 staining was performed on the SKW3 T cells and analyzed by flow cytometry.
(B) TCR55-transduced SKW3 T cells were stimulated by KG-1 cells pulsed with titrated HIV or Pep20 peptides for 15 min. Anti-phospho-ERK staining was performed on the SKW3 T cells and analyzed by flow cytometry.
(C) The design of TCR55 libraries. Each library has 3 or 4 residues selected to be randomized. The side chains of the residues selected for mutation on TCR55 are shown as sticks in the figures.
(D) Workflow of catch bond engineering of TCR.
(A-B) Data are representative of 3 independent experiments. Data are shown as mean ± SD of technical triplicates.
While the source of catch bonds in force-dependent triggering has been attributed to multiple structural elements of the TCR (18), we focused our library design on the TCR-pMHC interface. Our TCR library design was guided by the biophysical characteristics of catch bonds, which are mediated by the transient formation of hydrogen bonds or salt bridges encountered during the TCR/pMHC shearing step preceding disengagement. This leads to extended bond lifetimes that manifest as a transient resistive force before unbinding (19). Thus, our strategy was to lightly mutate the complementarity determining regions (CDR) residues of TCR55 to encode polar or charged amino acids that would act as fishhooks (bait) to probe for H-bonding and/or salt bridging residues (prey) on the pMHC binding surface during disengagement. We chose TCR CDR residue positions for the libraries that were too distant from the pMHC to form direct contacts in the bound state, in order to minimize simply selecting for affinity-matured TCRs.
Based on the structure of the TCR55-HIV-B35 complex (11), three residues on the TCR55 α chain and four residues on the TCR55 β chain were selected for the library positions (Figure 1C). Our library consisted of mainly charged and polar residues including glutamine, glutamate, asparagine, aspartate, arginine, lysine, serine, and histidine to increase the chances of forming new polar interactions. The three randomized residues on the TCR55α chain were combined as one library with diversity of 1,728 (Vα library), and the four randomized residues on TCR55β chain were combined as a second library with diversity of 20,736 (Vβ library). Full-length TCR55 libraries were synthesized and cloned into a lentiviral backbone vector. Lentivirus libraries were constructed and used to infect the SKW3 T cell line at low multiplicity of infection, and TCR libraries were expressed on the surface of T cells. The Vα library was paired with the wild type TCR55β chain, and the Vβ library was paired with the wild type TCR55α chain in the transduced SKW3 cells. The libraries were stimulated with 10 μM HIV peptides and sorted for pMHC tetramer-staining low (no higher than the pMHC tetramer staining of WT TCR55) together with co-staining for activation antigen CD69-high (top 5% population based on anti-CD69 mean fluorescent intensity (MFI)) populations to enrich for “low affinity/high potency” TCR mutants (Figure 1D).
Single amino acid substitutions in TCR55 trigger activation through catch bond formation
We carried out three rounds of FACS-sorting selections on the TCR55α CDR library (diversity: 1,728) and enriched a population with a tetramer-low, CD69-high staining phenotype (Figure 2A, Figure S3A–S3B). Approximately 100 single cell clones were recovered and individually tested for activation by the HIV(Pol) peptide. The two clones (clone 8 and clone 17) that showed the most potent response to this pMHC ligand (Figure S3C), encoded identical TCR mutations on the TCR55α chain – S28G and A98H. To directly examine if the identified mutations conferred increased potency, SKW3 T cells were transduced with the TCR55α-S28G A98H and WT TCR55 β chain and stimulated by B35-associated HIV peptide (Figure S3D). To deconvolute which mutation was responsible for the activation, we tested the mutations individually (Figure S3D–S3E) and found that the single mutation of alanine to histidine in the TCR55α CDR3 was sufficient to endow the non-responsive TCR55 with the ability to be activated upon exposure to the B35-HIV pMHC (Figure 2B, Figure S4).
Figure 2. A hotspot on the TCR can tune TCR signaling strength.
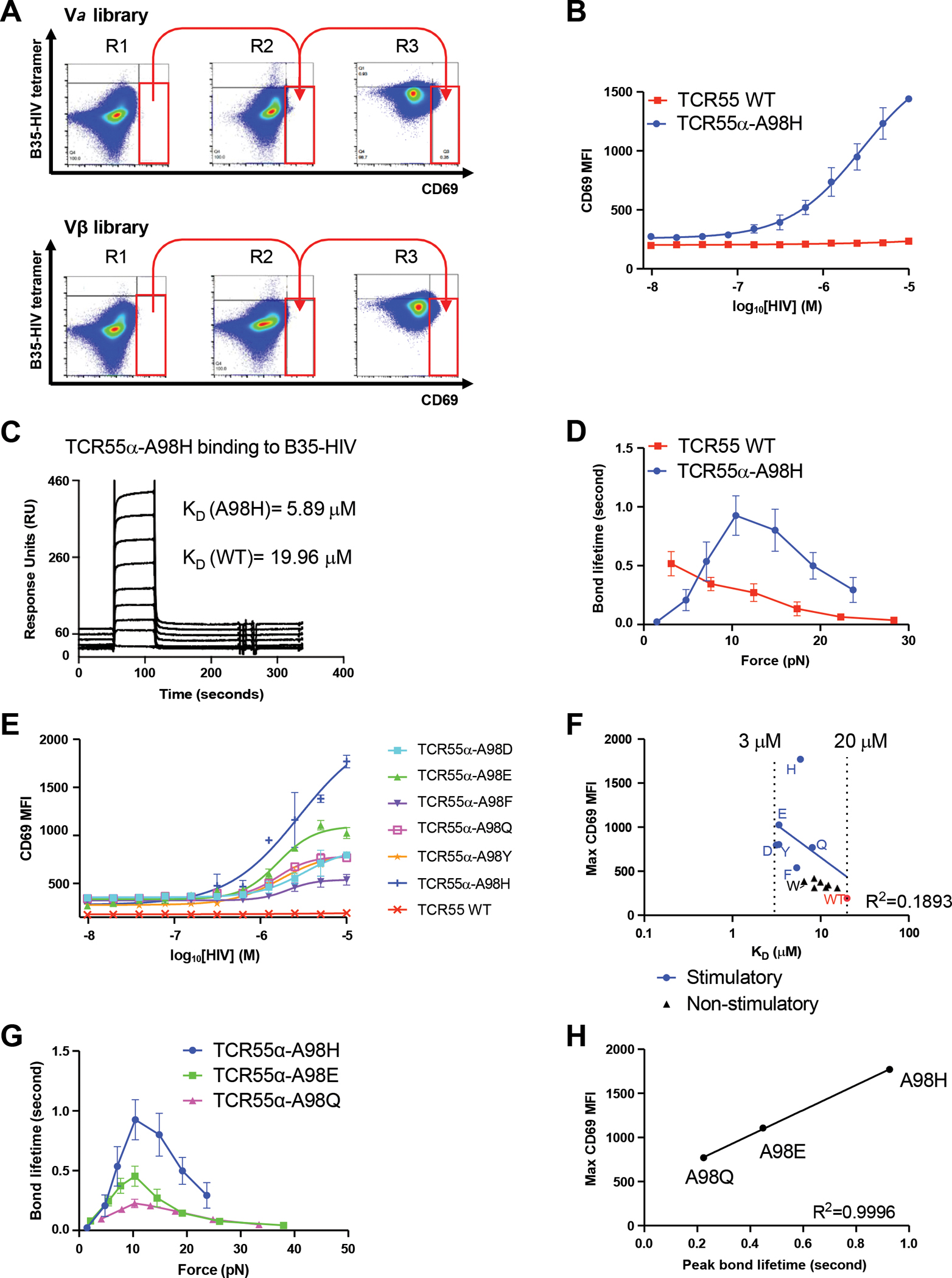
(A) B35-HIV tetramer staining and anti-CD69 staining of cells transduced with library clones in each round of selection. The gate is based on the staining of WT TCR55.
(B) A stimulatory clone, TCR55α-A98H, was selected from the library and was stimulated by KG-1 cells pulsed with titrated HIV peptides for 14 hours. Anti-CD69 staining was performed on the transduced SKW3 T cells and analyzed by flow cytometry.
(C) SPR experiments of TCR55α-A98H protein binding to B35-HIV. Biotinylated B35-HIV monomer was immobilized on the streptavidin chip and the TCR55α-A98H protein was flowed through the chip.
(D) Biomembrane force probe experiments to measure bond lifetime force curves for TCR55α-A98H or TCR55 WT binding to B35-HIV.
(E) TCR55α-A98 was mutated to D, E, F, Q, Y, H and used to transduce SKW3 T cells with WT TCR55β. The transfectants were stimulated by KG-1 cells pulsed with titrated HIV peptides for 14 hours. Anti-CD69 staining was performed on the transduced SKW3 T cells and analyzed by flow cytometry.
(F) Mean value of maximal anti-CD69 MFI versus 3D binding affinity KD of TCR55α-A98 mutants transfectants. The linear correlation analysis was performed for stimulatory mutants and TCR55 WT.
(G) Biomembrane force probe experiments to measure bond lifetime force curves for TCR55α-A98H, TCR55α-A98E or TCR55α-A98Q T cell transfectants binding to B35-HIV.
(H) Mean value of maximal anti-CD69 MFI versus peak bond lifetime of TCR55α-A98 mutants transfectants.
(B, E) Data are representative of 3 independent experiments. Data are shown as mean ± SD of technical triplicates.
(D, G) Data are shown as mean ± SEM of 500+ individual bond lifetimes per force curve.
The 3D affinity of TCR55α-A98H binding to the B35-HIV pMHC was measured by surface plasmon resonance (SPR) as KD = 5.9 μM, which is approximately three-fold lower than the WT TCR55 binding to B35-HIV (KD = 17 μM) but still in the physiological affinity range for TCR/pMHC interactions, and higher than measured for binding of TCR589 to B35-HIV (KD = 4 μM), a receptor-ligand pair with agonist qualities (Figure 2C, Figure S3F) (11). Biomolecular force probe (BFP) experiments were conducted to determine if TCR55α-A98H forms catch bonds with B35-HIV. The non-responsive WT TCR55 showed progressively shorter bond lifetime with increasing force, consistent with slip bond formation. In contrast, application of force increased bond lifetime between TCR55α-A98H and B35-HIV, indicating catch bond formation (Figure 2D). Analysis of the previously published structure of TCR55 bound to B35-HIV (11) suggests that the residues Q65 and T69 on the B35 MHC heavy chain molecule might form new bonds with H98 on TCR55α (Figure S3G). Q65 or T69 was mutated to alanine, and only the Q65A mutation significantly abrogated the activation of TCR55α-A98H, suggesting the triggering catch bond may involve an interaction between B35-Q65 and TCR55α-A98H (Figure S3H). BFP showed that B35-Q65A-HIV formed catch bonds with TCR55α-A98H but exhibited shorter peak bond lifetimes than the B35-HIV/ TCR55α-A98H interaction (Figure S3I).
Calibrating TCR55 signaling strength by bond lifetime
The acquisition of T cell activation by B35-HIV(Pol) coincident with catch bond formation by a single point mutant of TCR55 provided an opportunity to investigate structure-function relationships between amino acid substitutions and activation strength. We mutated the TCR55α-A98 to 12 different amino acids to investigate how residue identity at this position affected the strength of TCR signaling. In addition to histidine, mutations to aspartate, glutamate, phenylalanine, glutamine, and tyrosine also enabled TCR55 signaling through B35-HIV(Pol) engagement for lymphocyte activation, albeit to different extents (Figure 2E). In contrast, mutations to cysteine, lysine, asparagine, arginine, serine, threonine, and tryptophan did not activate TCR55 (Figure S5A). Therefore, only select polar, aromatic, and charged amino acids replacing residue TCR55α-A98 enabled effective signaling in response to B35-HIV. To investigate if there was a correlation between signaling capacity and binding strength, we measured the 3D affinity by SPR for each of the different TCR55α-A98 mutants binding to B35-HIV pMHC. Most mutants have an affinity in a narrow range between KD = 3 μM and KD = 20 μM (Figure S6, Table S1). Neither the maximum CD69 MFI (R2 = 0.1893) or EC50 (R2 = 0.02855) of stimulatory mutants were correlated to the SPR affinity of stimulatory mutants, suggesting that 3D affinity could not explain the gain-of-function exhibited by the stimulatory mutants (Figure 2F; Figure S5B). Indeed, TCR55α-A98W (KD = 6.5 μM), a variant that exhibited higher affinity than WT-TCR55 (KD = 19 μM), did not enable TCR-dependent activation in response to B35-HIV(Pol). Furthermore, the most ligand-sensitive of the TCR mutants, TCR55α-A98H (KD = 5.9 μM), did not have the highest affinity (Figure 2F, Table S1). Based on BFP measurements for two B35-HIV responsive mutants: TCR55α-A98E and TCR55α-A98Q (Figure 2G), we found that Emax was correlated with the peak bond lifetime (R2 = 0.996), rather than affinity (Figure 2H). Thus, the strength of the catch bonds is a key parameter for the discrimination between agonist and non-agonist TCR-pMHC interactions.
We carried out a parallel screen on the TCR55β CDR library (diversity: 20,736), and identified a TCR55 variant, clone 36, that exhibited a high level of T cell activation by B35-HIV(Pol) (Figure S7A–S7B). Clone 36 contained two mutations: a CDR1 mutation TCR55β-N28Q, and a CDR2 mutation TCR55β-A50D. We identified the isolated TCR55β-A50D mutation as necessary and sufficient to enable T cell activation by B35-HIV (Figure S7C). Replacing the TCR55β-A50 position with alternative amino acids showed that aspartate, glutamate, phenylalanine, histidine, asparagine, glutamine, serine, threonine, and tyrosine supported TCR55 mutant responses to B35-HIV to different degrees, while cysteine, lysine, arginine, and tryptophan did not support effective signaling (Figure S7D–S7E). The SPR affinities of TCR55β-A50 mutants exhibited a range of KD = 2–20 μM, similar to the TCR55α mutants, and falling within the natural physiological range of TCR affinities (Figure S7F–S7G, Figure S8, Table S2). There was a better correlation between maximal CD69 MFI versus 3D binding affinity KD (R2 = 0.7558) among the TCR55β-A50 mutants than the TCR55α-A98 mutants (Figure S9A). However, the EC50 was not correlated with the 3D affinity (R2 = 0.3543) (Figure S9B), again suggesting that affinity alone was not sufficient to explain the gain of function with these mutant TCRs. BFP experiments with the TCR55β-A50E, TCR55β-A50D, TCR55β-A50H and TCR55β-A50T mutants (Figure S9C) again showed that peak bond lifetime correlated with Emax for TCR55β-A50 mutants stimulated by the B35-HIV pMHC ligand (R2 = 0.8644) (Figure S9D). Analysis of the crystal structure of TCR55-HIV-B35 complex (11) shows that residues T69 and Q72 on the B35-HIV pMHC potentially mediate the formation of new hydrogen bonds with TCR55β-A50E (Figure S9E). K562 cells transduced with B35-T69A prevented activation of T cells bearing TCR55β-A50E, whereas the B35-Q72A mutation had no effect (Figure S9F). Consistent with these results, BFP measurement showed that B35-T69A-HIV only formed slip bonds with TCR55β-A50E (Figure S9G).
Signaling landscape of catch bond engineered TCR
To assess how the catch bond engineered TCR55 mutants affect intracellular signaling in T cells in response to B35-HIV pMHC ligand, we employed a live cell imaging reporter system to measure the activation dynamics of the ERK, p38 and NFAT2 signaling pathways (Figure S10A–S10E). In these cells, translocation of fluorescent versions of these intracellular signaling molecules can be visualized in real time and quantified on a single cell basis. Upon engagement with HIV peptide-pulsed B35-expressing antigen-presenting cells, reporter Jurkat T cells expressing the catch bond engineered TCR variants displayed enhanced pathway activation when compared to the non-responding parent TCR55, using the stimulatory TCR589 as a positive control (Figure 3A–3C). While both TCR55α-A98H and TCR55β-A50E mutants were able to activate the ERK and p38 signaling pathways for a similar duration at the population level, substantial differences in NFAT2 activation dynamics were observed (Figure 3C). These results were quantified by single-cell AUC (area under the curve) analysis (Figure 3D–3F, Table S3–S4), which demonstrated significant differences in both ERK and NFAT2 signaling responses for all the tested TCR variants. Due to the substantially lower signal-to-noise ratio of the p38-KTR reporter, we observed more subtle p38 signaling differences that follow the same hierarchy of mean AUC distribution compared to ERK or NFAT2 activation (Figure 3D–3G). We find a strong correlation between mean ERK (R2 = 0.9370) or NFAT2 (R2 = 0.9415) AUC distribution and peak bond lifetime, again suggesting that catch bond strength plays a significant role in TCR-ligand engagements that result in functional intracellular signaling. (Figure 3H).
Figure 3. Signaling landscape of catch bond engineered TCRs.
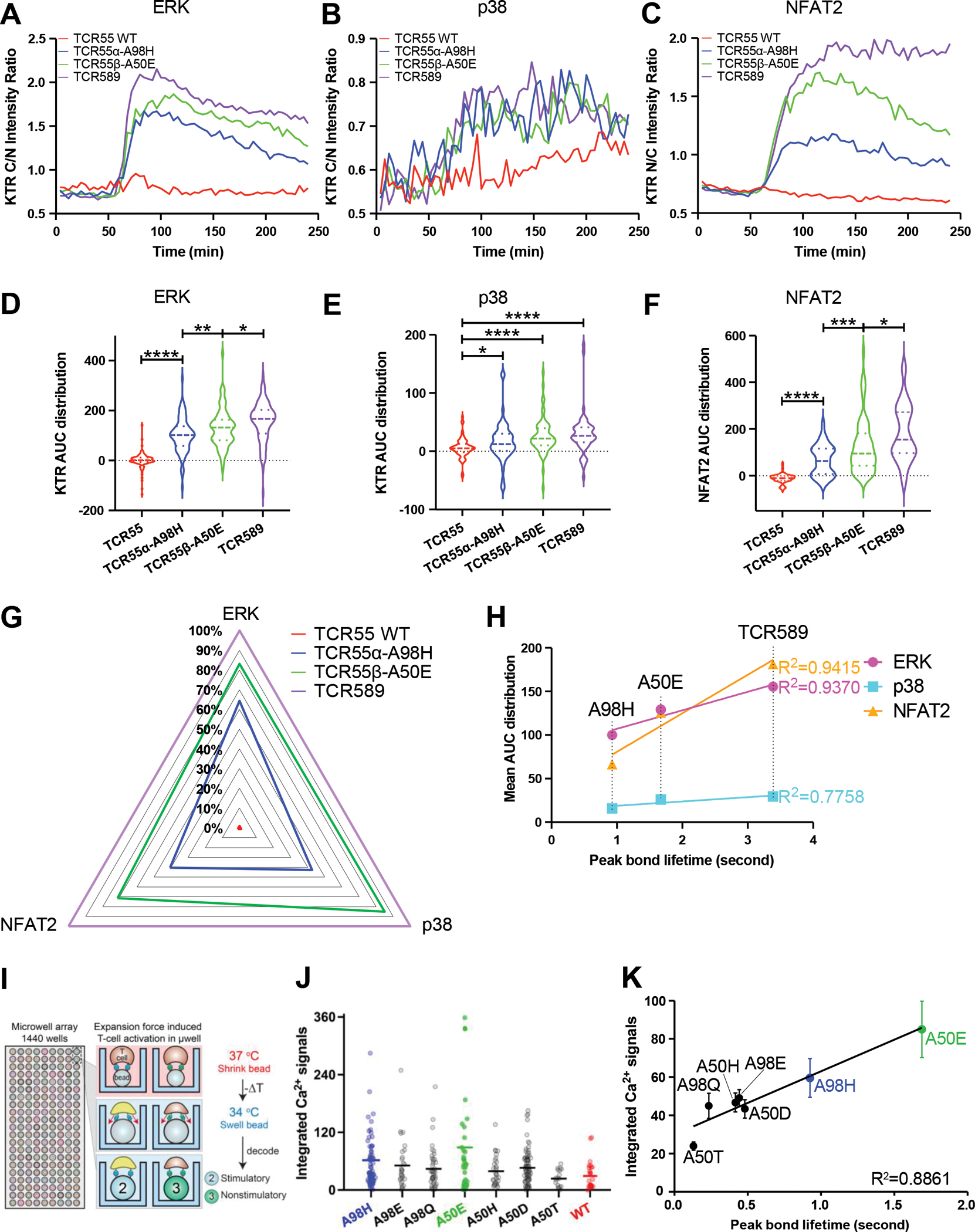
(A) ERK activation dynamics induced by B35-HIV engagement with the indicated TCR55 variant or TCR589, measured by ERK-KTR-mScarlet cytoplasmic/nuclear intensity ratio over imaging time.
(B) p38 activation dynamics measured by p38-KTR-mScarlet cytoplasmic/nuclear intensity ratio over imaging time.
(C) NFAT2 activation dynamics measured by GFP1-11-NFAT2 nuclear/cytoplasmic intensity ratio over imaging time.
(D) AUC (area under the curve) distribution of single-cell ERK activation dynamics.
(E) AUC distribution of single-cell p38 activation dynamics.
(F) AUC distribution of single-cell NFAT2 activation dynamics.
(G) Radar summary plot with normalized mean AUC values to illustrate the signaling landscape of TCR55 variant or TCR589 in response to B35-HIV engagement.
(H) Mean ERK/p38/NFAT2 AUC distribution versus peak bond lifetime measurement.
(I) Schematic illustration of bead-T cell interaction in BATTLES.
(J) Calcium flux signaling strength of different TCR55 mutants transfectants. Individual cell signals are shown as circular markers; lines represent the mean values.
(K) The correlation between calcium flux signaling strength and peak bond lifetime of different TCR55 mutants transfectants. Errors represent standard error of the mean.
(A-F, I) Data are representative of 2 independent experiments.
Applied force activation of TCR at physiological pMHC density
To investigate the triggering of catch bond engineered TCR55 at extremely low but physiologically relevant levels of pMHC (HIV/HLA-B35), we used the BATTLES technique (Biomechanically-Assisted T-cell Triggering for Large-scale Exogenous-pMHC Screening) (20). The BATTLES technique uses temperature-sensitive polymer beads coated with pMHC proteins displayed at physiological densities (3–4.5 pMHCs/cell) to apply ramping forces (estimated maximum magnitude = 20–27.5 pN/s) to T cells interacting with bead surfaces (Figure 3I) (21). Upon activation of force, we monitored Ca2+ signaling (which is correlated with initial T cell triggering) for >1,000 SKW3 T cells transduced with engineered TCR55s containing either TCR55α-A98H, TCR55α-A98E, TCR55α-A98Q, TCR55β-A50E, TCR55β-A50H, TCR55β-A50D or TCR55β-A50T substitutions interacting with HIV peptides (Figure 3J). While some T cells exhibited sustained increases in cellular Ca2+ flux (Figure S10F, top and middle rows), most cells showed decreasing fluorescence intensities and resulted in negative accumulated signals, indicating no triggering (Figure S10F, bottom row). This is consistent with prior literature showing that only a small fraction of T cells are activated at low pMHC densities, even with optimal force (21). All tested substitutions except TCR55β-A50T yielded higher integrated per-cell Ca2+ signals as compared with WT, with the magnitude of the integrated signal showing a strong correlation with measured peak bond lifetimes (Figure 3K). These results, using force-induced activation of single T cells, provide evidence that engineered TCRs can drive efficient activation under the low-density pMHC conditions encountered in vivo.
Application of TCR catch bond engineering to TCR-T cell therapy
Catch bond engineering has implications for ACT with TCR-T cells because many wild-type tumor-reactive TCRs have low affinity binding to tumor pMHC and low sensitivity to signaling in response to relevant tumor-associated antigens, resulting in inefficient tumor killing (22–24). The melanoma antigen MAGE-A3-specific TCR (WT) was chosen for catch bond engineering. The antigen is HLA-A1-restricted with a reported 3D SPR binding affinity of KD = 500 μM to the WT TCR (16, 25). This TCR shows extremely poor T cell activation in response to the tumor antigen MAGE-A3, while an affinity-matured mutant of the WT MAGE-A3 TCR, A3A TCR, mediates greatly enhanced T cell activation by the same ligand (Figure 4A). However, in clinical trials for melanoma the A3A TCR was found to cross-react with presented TITIN peptide, which is expressed mainly in cardiovascular tissue, leading to a high level of cardiotoxicity (Figure 4A) (16, 17). We asked whether we could use catch bond engineering to improve the sensitivity of the poorly-responsive parental wild-type TCR to MAGE-A3 ligand while maintaining low affinity to avoid cross-reactivity with TITIN.
Figure 4. Catch bond engineering of MAGE-A3-specific TCR.
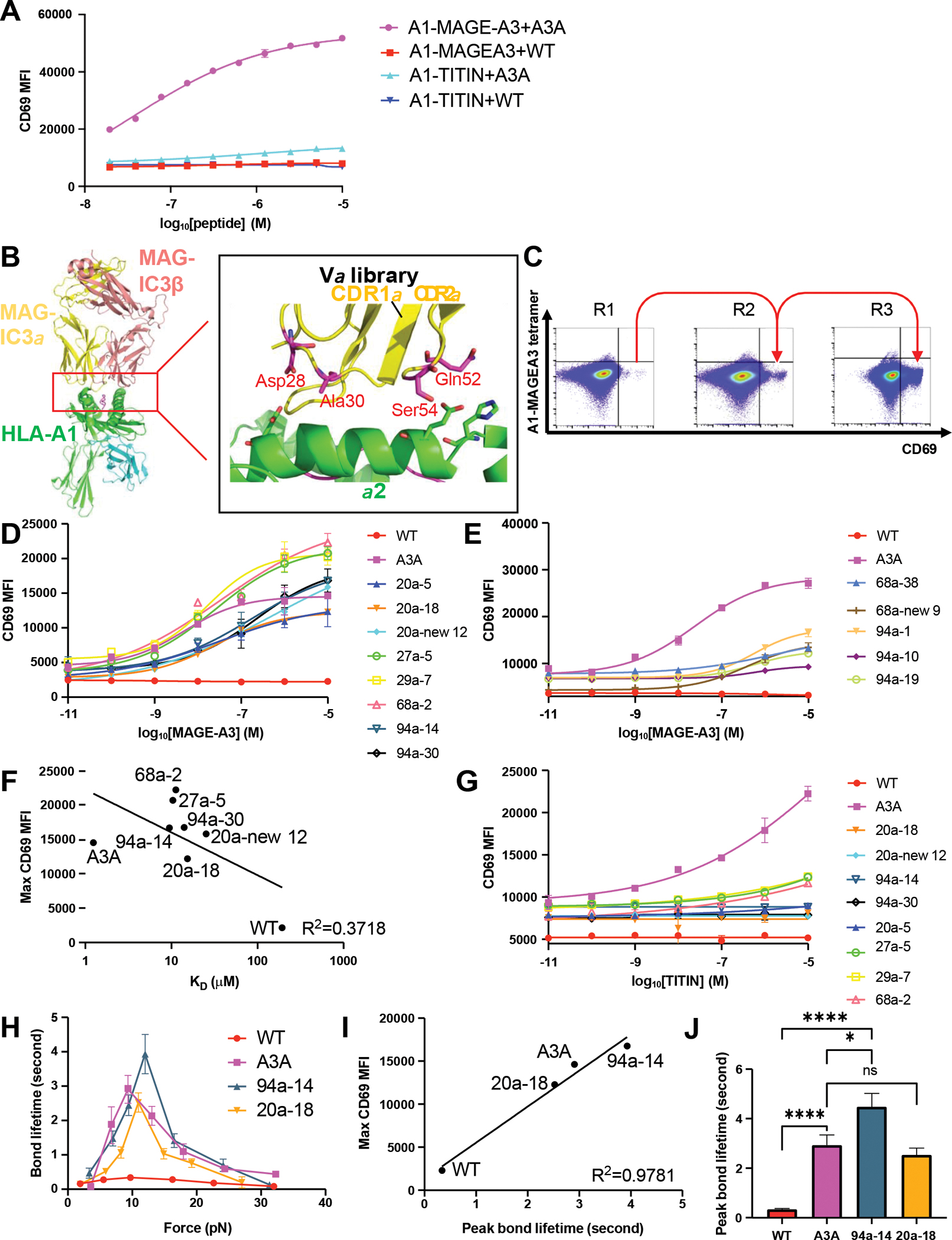
(A) The WT TCR or A3A TCR chains were transduced in SKW3 T cells. The transfectants were stimulated by HLA-A1+ 293T cells pulsed with titrated MAGE-A3 peptide or TITIN peptide. Anti-CD69 staining was performed on the T cells and analyzed by flow cytometry.
(B) The design of MAGE-A3 TCR Vα library. The library has 4 residues picked to be randomized. The side chains of the selected resides on the TCR were shown as sticks in the figures.
(C) 3 rounds of selection of the MAGE-A3 TCR Vα library on tetramer staining-low and anti-CD69 staining-high gate. The gate is based on the staining of MAGE-A3 WT TCR.
(D) The 8 high-potency MAGE-A3 TCR mutants were transduced into SKW3 T cells. The transfectants were stimulated by HLA-A1+ 293T cells pulsed with titrated MAGE-A3 peptide. Anti-CD69 staining was performed on the T cells and analyzed by flow cytometry.
(E) The 5 intermediate-potency MAGE-A3 TCR mutants were transduced into SKW3 T cells. The transfectants were stimulated by HLA-A1+ 293T cells pulsed with titrated MAGE-A3 peptide. Anti-CD69 staining was performed on the T cells and analyzed by flow cytometry.
(F) The correlation between mean value of maximal anti-CD69 MFI and 3D affinity of selected MAGE-A3 TCR mutants binding to HLA-A1-MAGE-A3.
(G) The 8 high-potency MAGE-A3 TCR mutants were transduced in SKW3 T cells. The transfectants were stimulated by HLA-A1+ 293T cells pulsed with titrated TITIN peptide. Anti-CD69 staining was performed on the T cells and analyzed by flow cytometry.
(H) Biomembrane force probe experiments to measure bond lifetime force curves for WT, A3A, 94a-14 or 20a-18 TCR binding to HLA-A1-MAGE-A3. Data are shown as mean ± SEM of 500+ individual bond lifetimes per force curve.
(I) Mean value of maximal anti-CD69 MFI versus peak bond lifetime of MAGE-A3 TCR mutants transfectants.
(J) Multiple measurements of bond lifetime at 10 pN for WT, A3A, 94a-14 and 20a-18 TCR. ns: not significant; ✱: P<0.05; ✱✱: P<0.01; ✱✱✱: P<0.001; ✱✱✱✱: P<0.0001
(A, D-E, G) Data are representative of 3 independent experiments. Data are shown as mean ± SD of technical triplicates.
We did not have a crystal structure of the low affinity WT TCR complex with HLA-A1-MAGE-A3, but a structure of the affinity-matured version of the TCR with the HLA-A1-MAGE-A3 complex was available (25). We thus modeled the WT TCR binding to HLA-A1-MAGE-A3 and designed a library on the TCR α chain (Figure 4B). Following the design strategy for TCR55, the residues chosen (CDR1α positions 28, 30; CDR2α positions 52, 54) for the library fall within the CDR loops and are relatively close to the pMHC but do not directly contact the pMHC (Figure 4B). The SKW3 T cell line was transduced with the library at low MOI and CD69-hi/tetramer-lo clones were selected as described earlier (Figure 4C, Figure S11A). After three rounds of selection, 96 single cell clones were selected from the enriched population and tested for TCR-dependent activation. We isolated 13 distinct mutant-transduced SKW3 clones that showed enhanced responsiveness to the MAGE-A3 peptide at a concentration unable to trigger T cells expressing the parental WT TCR (Figure 4D–4E, Table S5). By comparing the Emax of the TCR mutants, we defined 8 clones as “high-potency” mutants compared to the A3A TCR (Figure 4D, Figure S11B), and 5 clones as “intermediate-potency” mutants (Figure 4E). We measured SPR 3D KD for six high-potency mutants and two intermediate-potency mutants binding to HLA-A1-MAGE-A3. The affinities ranged from KD = 10 to 50 μM, significantly lower affinities than that of A3A (KD = 1.24 μM) (Figure S12, Table S6). We did not observe a correlation between Emax vs 3D affinity (R2 = 0.3718) (Figure 4F) but observed a weak correlation between EC50 and affinity (R2 = 0.5998) (Figure S11C). We tested if the 8 high-potency mutants showed cross-reactive functional responses to the TITIN peptide. The A3A-transduced SKW3 cells were strongly activated by the TITIN pMHC ligand (Figure 4G). Four mutants (20a-18, 20a-new 12, 94a-14, and 94a-30) exhibited no cross-reactivity with the TITIN peptide, whereas the remaining four displayed very weak activation by TITIN only at high peptide concentrations (Figure 4G).
We also measured the binding affinity of all catch bond engineered TCR mutants to HLA-A1-TITIN and they had very low or unmeasurable 3D binding affinity (KD > 100 μM), whereas A3A affinity for TITIN was KD = 7.7 μM (Table S7, Figure S13). BFP experiments were performed for WT TCR, A3A TCR, TCR mutants 94a-14 and 20a-18, and all formed catch bonds with HLA-A1-MAGE-A3, with the mutant 94a-14 having a higher peak bond lifetime than A3A and WT TCR (Figure 4H). The peak bond lifetimes of WT, A3A, 94a-14, and 20a-18 TCR were well correlated to the maximal CD69 MFI measured in Figure 4D (R2 = 0.9781) (Figure 4I). A force of ~10 pN for a CD8:TCR:agonist has been demonstrated to promote optimal effector signaling (6, 8–10). At ~10 pN of force 94a-14 TCR has a significantly higher peak bond lifetime than both WT and A3A TCRs (Figure 4J). BFP experiments for 94a-14 or 20a-18 TCR with HLA-A1-TITIN indicate that only slip bond formation was observed for both TCRs (Figure S14A), consistent with the loss of TITIN cross-reactivity by 94a-14 and 20a-18 TCRs.
To test if the MAGE-A3 TCR mutants could efficiently kill HLA-A1-MAGE-A3+ tumor cells, human primary T cells were transduced with the WT, A3A, and TCR mutants, and cocultured with the HLA-A1-MAGE-A3+ melanoma cell line A375 (Figure 5A–5E) or the HLA-A1-MAGE-A3+ colon cancer cell line HCT-116 (Figure 5F–5J, Figure S15). In response to A375 cells, the engineered TCRs 94a-14 and 20a-18 were superior in target killing to the WT TCR and at least comparable, and in some cases superior to A3A in target stimulated effector activity depending on the metric analyzed (IFN-γ, TNF, degranulation) (Figure 5A–5E). Similar trends were seen in response to HCT-116 cells, which express lower levels of the MAGE-A3 antigen, (Figure 5F–5J). The mutants 20a-5 and 27a-5 were also tested in human primary T cells and showed a high level of cytotoxicity against A375 melanoma cells (Figure S11D–11H) and HCT-116 colon cancer cells (Figure S11I–S11M).
Figure 5. Cytotoxicity and specificity of engineered MAGE-A3-specific TCR.
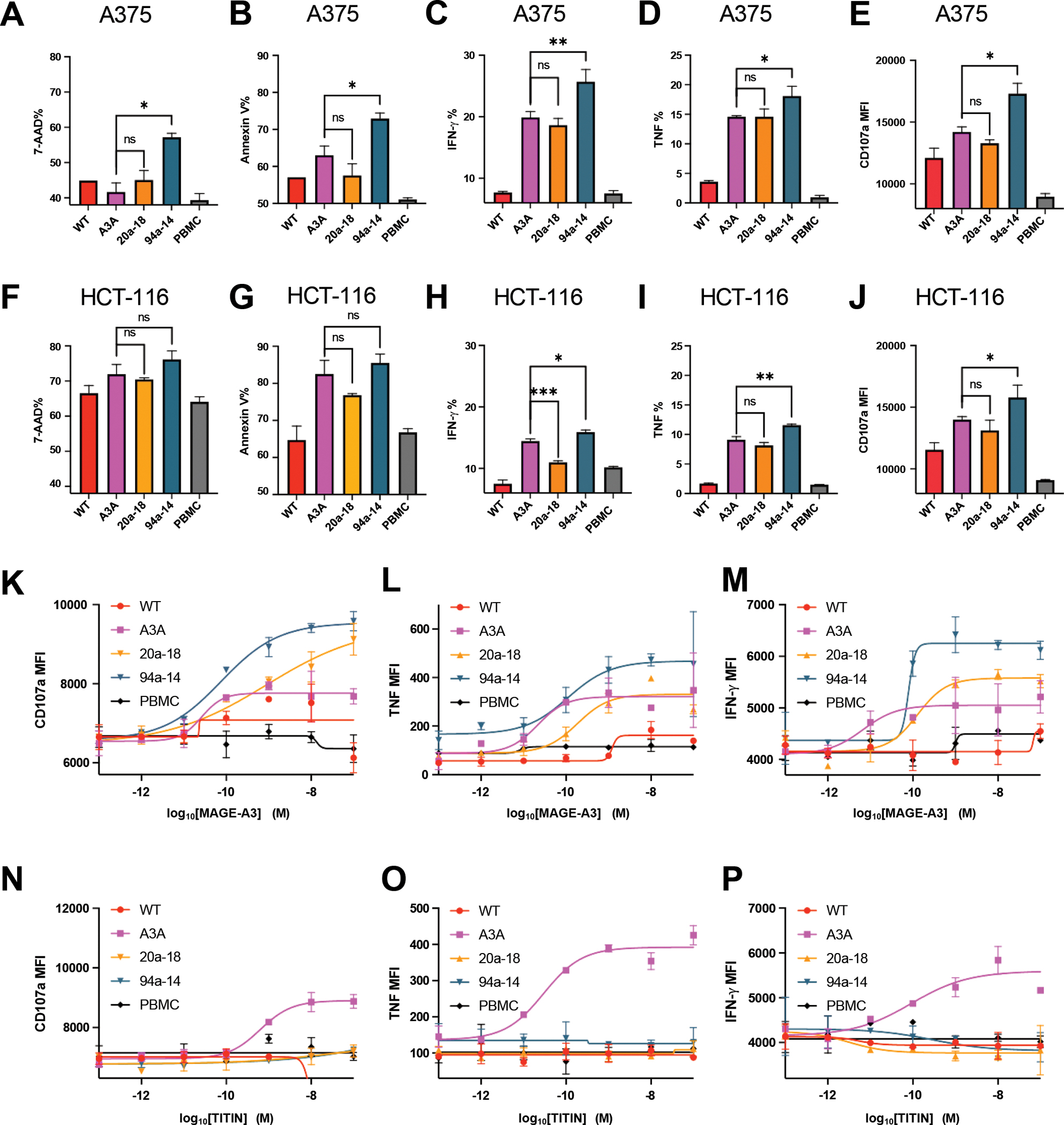
(A-B) Killing of A375 melanoma cell line by different MAGE-A3-specific TCR transduced human primary T cells.
(C-E) IFN-γ, TNF, and cytotoxic granule release (CD107a staining) by different MAGE-A3-specific TCR transduced human primary T cells, induced by the A375 melanoma cell line.
(F-G) Killing of HCT-116 colon cancer cell line by different MAGE-A3-specific TCR transduced human primary T cells.
(H-J) IFN-γ, TNF, and cytotoxic granule release (CD107a staining) by different MAGE-A3-specific TCR transduced human primary T cells, induced by the HCT-116 colon cancer cell line.
(K-M) Cytotoxic granule release (CD107a staining), TNF, and IFN-γ by different MAGE-A3-specific TCR transduced human primary T cells, induced by HLA-A1+ 293T cells pulsed with a titration of MAGE-3 peptide.
(N-P) Cytotoxic granule release (CD107a staining), TNF, and IFN-γ by different MAGE-A3-specific TCR transduced human primary T cells, induced by HLA-A1+ 293T cells pulsed with a titration of TITIN peptide.
(A-P) Data are representative of 3 independent experiments. Data are shown as mean ± SD of technical duplicates. ns: not significant; ✱: P<0.05; ✱✱: P<0.01; ✱✱✱: P<0.001; ✱✱✱✱: P<0.0001
To examine if TCR clones 94a-14 and 20a-18 exhibited cross reactivity to TITIN, primary human T cells transduced with the respective TCRs were co-cultured with MAGE-A3 or TITIN peptide-pulsed antigen-presenting cells. While 20a-18 or 94a-14 showed enhanced cytotoxicity, degranulation, and cytokine secretion (Figure 5K–5M) after coculturing with MAGE-A3 pulsed cells, none of these TCR clones responded to the presented TITIN peptide (Figure 5N–5P). Similarly, the 20a-5 and 27a-5 clones mediated potent cytotoxic responses to MAGE-A3 (Figure S11N–S11P) but only minimal cross-reactivity to TITIN at high concentrations of peptide (Figure S11Q–S11S).
Profiling the cross-reactivity of engineered MAGE-A3 TCRs
Although the engineered TCRs lacked substantial reactivity with the TITIN peptide, we asked if the engineered TCRs had acquired new, off target peptide reactivities. We turned to a yeast-display pMHC library system originally used to characterize the cross-reactivity of TCRs (26, 27), and to uncover the specificities of TCRs derived from tumor-resident T cells (28). We first generated an HLA-A*01 9-mer peptide library to survey the cross-reactive landscape of the wild-type, affinity matured A3A, and 3 catch bond engineered MAGE-A3 TCR variants (Figure 6A). The library was designed based on peptide sequences known to bind HLA-A*01, fixing anchor residues in positions P3 to aspartate and glutamate and P9 to tyrosine to ensure proper presentation of the peptides in the HLA groove. All remaining positions allowed flexibility to all 20 amino acids for a library diversity of 1.8 × 108.
Figure 6. Cross-reactivity screening of MAGE-A3 TCR variants with pMHC libraries.
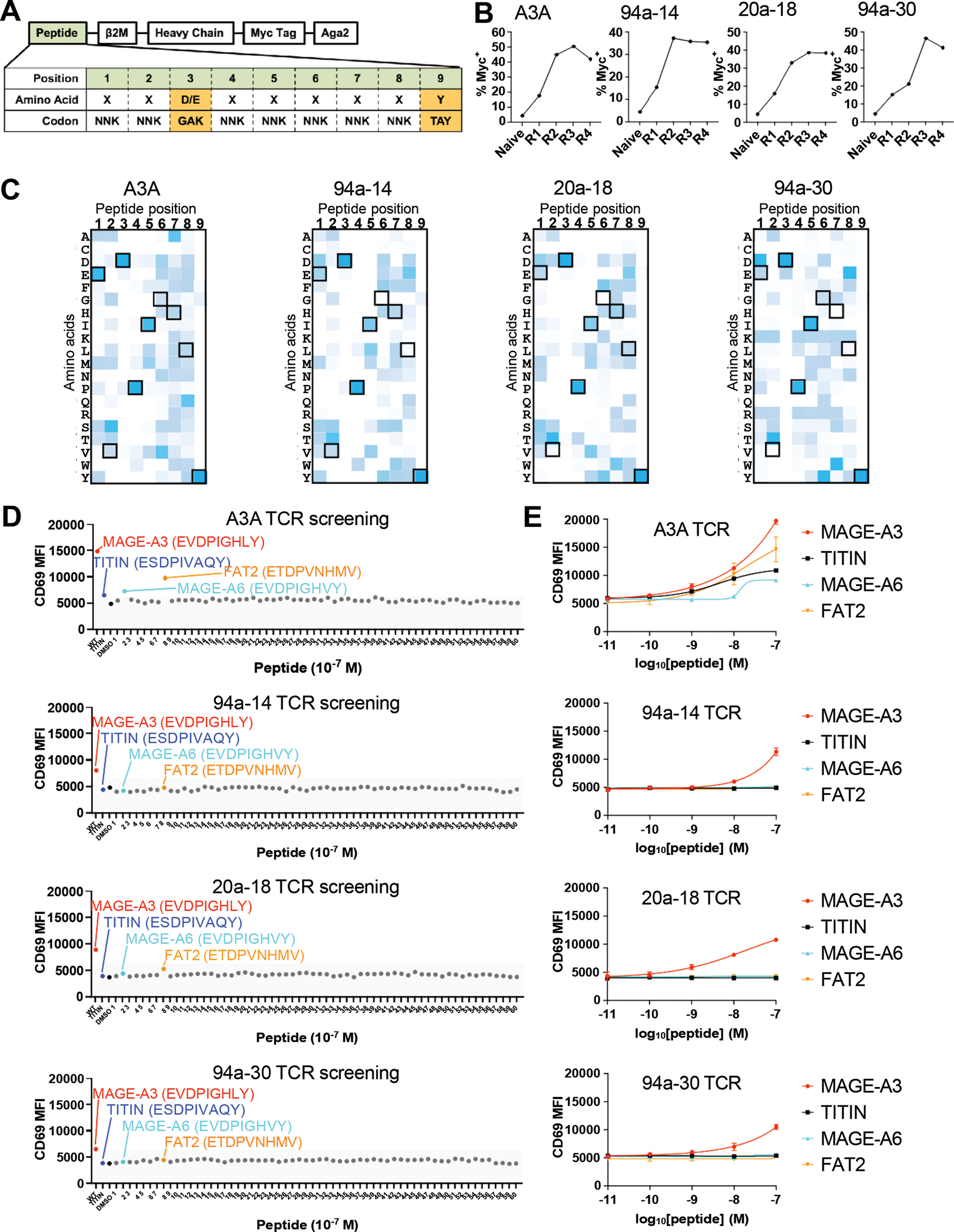
(A) Design of the single-chain HLA-A*01 yeast-display peptide library. The DNA peptide library design shows an NNK codon library for all positions except anchor positions P3 (GAK) and P9 (TAY) to maximize peptides displayed by HLA-A*01. The single-chain trimer construct is N-terminal to the Myc tag fused to Aga2 for expression on yeast.
(B) Increasing myc tag expression on yeast over rounds of selection represents enrichment of peptide HLA-A*01 and positive selection of the library.
(C) Heat map of round 4 selected peptides showing peptide position by amino acid accounting for the number of reads detected per peptide. Boxed amino acids represent the MAGE-A3 peptide EVDPIGHLY. Dark blue represents a more enriched amino acid in that position.
(D) MAGE-A3 (red dot), TITIN (blue dot), DMSO (black dot) and 60 predicted peptides (MAGE-A6: cyan dot; FAT2: orange dot) were used to pulse 293T-HLA-A1 cells to stimulate SKW3 T cells expressing different TCRs for 14 hours. Anti-CD69-APC staining was performed and analyzed on flow cytometry.
(E) 293-HLA-A1 cells were pulsed with titrated MAGE-A3, TITIN, MAGE-A6 or FAT2 peptides to stimulate SKW3 T cells expressing MAGE-A3 TCR variants for 14 hours. Anti-CD69-APC staining was performed and analyzed on flow cytometry.
(D) Data are representative of 2 independent experiments.
(E) Data are representative of 2 independent experiments. Data are shown as mean ± SD of technical duplicates.
We performed selections following established methods with soluble, recombinant forms of the wild-type MAGE-A3 (WT) TCR, A3A, 94a-14, 20a-18, or 94a-30. While the WT TCR failed to enrich any yeast clones, presumably due to its very low 3D binding affinity (KD > 500 μM) for MAGE-A3 (16), the high affinity A3A and the engineered mutants strongly enriched populations of yeast clones (Figure 6B). The selected library pools were sequenced to isolate individual sequences. The selected peptides showed strong convergence at the N-terminal end for all the TCR variants, with a lack of C-terminal specificity, as previously described for A3A (29) (Figure 6C). Aside from the fixed anchor residues, P1 GLU, P4 PRO, and P5 ISO showed strong conservation, and notably exist in both MAGE-A3 and TITIN peptides. The three catch-bond engineered TCR variants showed very similar sequence preferences, indicating that the specificities of the TCRs were minimally changed by catch bond engineering. The deep sequencing data was used to make off-target predictions using previously developed statistical methods (Table S8–S11). For the A3A TCR, both TITIN and MAGE-A3 were top ranked predictions, ranking as 1 and 7 respectively (Table S8). However, for the 3 catch bond engineered TCRs, TITIN was not predicted in the top 35 peptides, while the MAGE-A3 peptide was predicted to bind to all 3 catch bond engineered TCRs: ranking 1st for TCR 94a-14 (Table S9), ranking as 2nd for TCR 20a-18 (Table S10), and ranking as 34th for TCR 94a-30 (Table S11).
We tested the top 20 putative off-target predictions for the A3A TCR and catch-bond engineered TCRs with T cell activation assays. The top 20 predicted peptides for each TCR were synthesized and used for screening each TCR (60 peptides in total after removing repetitive peptides, listed in Table S12). For the A3A TCR, we found that, in addition to MAGE-A3 and TITIN, it was also activated by two previously discovered epitopes MAGE-A6 and FAT2 (30) (Figure 6D–6E). For the 3 catch bond engineered TCRs (94a-14, 20a-18, and 94a-30), only the MAGE-A3 peptide significantly activated the T cells over baseline (Figure 6D–6E). For the WT TCR, none of the peptides significantly stimulated the T cells compared to the DMSO control (Figure S16A). The collective results of these cross-reactivity profiling experiments shows that the screen could identify both known on- and off-target specificities for the high-affinity A3A TCR, and that catch bond engineering did not introduce off-target specificities corresponding to known sequences in the human proteome. Although we cannot formally rule out the possibility that different types of cross-reactivity screens could identify off-target specificities that we did not find, the yeast-display pMHC screen represents a stringent test that shows the absence of unanticipated human antigen cross-reactivity while clearly identifying the source of cardiac toxicity seen with the A3A TCR.
Discussion
In environments where cell-cell interactions are subject to shear stresses, mechanical force plays an important role in signal transduction by a variety of receptor-ligand systems. Indeed, catch bonds have been observed as a natural signal potentiating mechanism in various low affinity cell surface adhesion systems such as those involving cadherins, selectins, and Notch, and more recently the TCR (31–33). Effective TCR signaling upon T cell engagement with an agonist pMHC ligand on an antigen presenting cell involves the formation of catch bonds that extend receptor-ligand interaction lifetime upon application of a pulling force (5, 6, 8–10, 27, 28). Indeed, the presence or absence of catch bonding residues in peptide antigens can decouple TCR triggering from conventional measurements of pMHC binding strength (11). Here, using by screening for mutant TCRs with a combination of modest solution affinity but high sensitivity to ligand-induced signaling, we show that TCRs with increased catch bond formation, as measured by BFP on T cells, dominate among the effective mutant TCRs isolated. These newly acquired catch bonds have not predisposed the TCRs to increased human antigen cross-reactivity evident from screening pMHC libraries. This suggests that while slow off rate per se can enable effective TCR signaling upon pMHC binding (34, 35), catch bonds can play a deterministic role for antigen responsive TCRs expressed on T cells. The degree to which catch bonds are contributed to by cellular factors such as membrane fluidity remains unknown (36).
The ease of identifying such TCRs in the screen, suggests that catch bonds may play a substantial role in the overall operational TCR repertoire and help explain the existing discrepancies in the literature between measured solution binding affinities for specific pMHC and the capacity of those pMHC to show agonist properties in terms of T cell activation (2). The motility of T lymphocytes when scanning for ligand on antigen-presenting or target cells, along with the activity of cellular filipodia (37), provide tugging or shear forces that would favor prolongation of TCR-ligand interaction by catch bond formation to enable effective phosphatase exclusion as compared to intrinsic slow-off rate binding that could be disrupted by such forces. This finding has direct implications for the emerging field of TCR-T therapy (12, 13, 38, 39), where the inherently weak “self” tumor reactivity of TCRs presents limitations to clinical activity.
Our selection strategy was critical to successful isolation of ligand-sensitive yet low-affinity clones for several reasons. First, we focused our libraries on polar and charged residues that can maximize the likelihood of mutant substitutions engaging in adventitious polar interactions during TCR/pMHC disengagement. Second, we designed the libraries to focus on residues that were not in direct contact with the pMHC so that the selection did not simply isolate high-affinity (especially slow off-rate) TCRs. We chose residues that were in the “second shell” of TCR CDR residues, in close proximity to the pMHC surface but too distant to form direct interactions in the ground state complex. These residues would be ideally positioned to act as “hooks” during shearing of the TCR/pMHC interface. Third, our functional selection strategy directly isolated signaling active (CD69-hi), but low affinity (tetramer low) clones. Although the 3D binding affinity KD of the isolated clones does trend to slightly higher affinity than the WT TCRs, the affinities remain firmly in the physiological regime and the 3D binding affinity KD does not correlate with activity, validating the screening principles.
For our proof-of-concept studies we used the TCR55-B35-HIV system because of the physiological binding affinity (KD = 17 μM) of this TCR with the B35-HIV pMHC, but undetectable TCR activation after ligand binding (11, 40). All the stimulatory single-site TCR mutants had affinities within the physiological regime (3D KD ~ 2 μM to 20 μM), comparable to the WT TCR55, and showed different degrees of bond lifetime extension that correlated with activation strength. These results show that catch bond engineered TCRs can be “tuned” for sensitivity through scanning different amino acid substitutions at hotspot positions. Such tunability allows for careful curation of clones with the desired balance of activation versus affinity. We emphasize that TCR signaling can be affected by both TCR affinity maturation or catch bond engineering. Indeed, there was a weak positive correlation between the TCR mutants’ sensitivity and affinity. However, catch bond engineering enables potency enhancement whilst maintaining physiologic affinity, reducing predisposition towards off-target cross-reactivity compared to affinity-matured TCRs.
While the safety of engineered T cell therapy will ultimately depend on the degree of preferential expression of the target tumor antigen versus healthy tissue, the strategy of catch bond engineering to maintain physiological affinity yet strong agonist signaling responses may help to reduce the chance of unwanted cross-reactivity with other pMHC for clinically-directed TCRs. Enhancing the efficacy of clinical TCRs has generally involved affinity maturation (15, 17, 41, 42). However, some affinity-matured TCRs have displayed off-target toxicity (17, 43). The extreme peptide selectivity of catch-bond engineered TCRs may even be helpful in mitigating on-target/off-tumor reactivities. For example, by enhancing therapeutic indices based on relative expression levels of unmutated self tumor antigens, or neoantigens with very close sequence similarity to a self antigen, expressed in healthy versus cancerous tissues (43). Given the relative ease that we isolated such mutants, and the simplicity of the screen, this lends itself well to a general approach in the TCR-T clinical development pipeline without requiring specialized structural insights.
Materials and Methods
Cell lines
SKW3 T cells (DSMZ) were cultured in RPMI-1640+GluMax (Thermo Fisher Scientific) complemented with 10% fetal bovine serum (FBS, Sigma-Aldrich), 10 mM HEPES and 50 U/mL Pen-Strep (Thermo Fisher Scientific) at 37 °C and 5% CO2.
LentiX cells and 293T cells were cultured in DMEM (Thermo Fisher Scientific) supplemented with 10% FBS, 2 mM L-Glutamine, 10 mM HEPES and 50 U/mL Pen-Strep (Thermo Fisher Scientific) at 37 °C and 5% CO2.
KG-1 cells (ATCC) were cultured in IMDM (Thermo Fisher Scientific) supplemented with 10% FBS and 50 U/mL Pen-Strep (Thermo Fisher Scientific) at 37 °C and 5% CO2.
SF9 cells were cultured in SF900-III media (Thermo Fisher) supplemented with 10% FBS and 10 mg/mL gentamicin sulfate (Thermo Fisher) at 27 °C and atmospheric CO2.
Hi5 cells were grown in insect cell culture medium (Expression Systems) supplemented with 10 mg/mL gentamicin sulfate (Thermo Fisher) at 27 °C and atmospheric CO2.
Jurkat cell lines were cultured in RPMI 1640 supplemented with 10% FBS, 2 mM L-Glutamine, 50 U/mL Penicillin, 50 μg/mL Streptomycin, and 50 μM β-mercaptoethanol at 37 °C and 5% CO2.
HEK293T cell line was cultured in DMEM supplemented with 10% FBS, 2 mM L-Glutamine, and 18 mM HEPES at 37 °C and 5% CO2.
Packaging of lentivirus
HEK293T-derived LentiX cells were seed in 6-well plate at a density of 3×105 cells/mL (2 mL in total). On the next day, for each well of cells, 750 ng plasmid of interest, 500 ng psPAX, 260 ng pMD2.G were mixed with 4.5 μL Fugene transfection reagent (Promega) in 100 μL Opti-MEM and rested for 20 min. Fresh cRPMI media were added to each well. Then, the DNA/Fugene mixture was added to each well. Optionally, 12 hours after the transfection, the supernatant of each well was replaced with 2 mL fresh cRPMI. 48 hours after the transfection, the supernatant was ready to infect 106 cells.
Cloning of TCR library
The dsDNA of the TCR library was synthesized commercially by GeneArt technology (Thermo Fisher Scientific) and was cloned into pHR lentiviral vector by HiFi assembly (New England Biolabs). Specifically, 20 ng dsDNA of TCR library, 100 ng linearized pHR vector and 10 μL HiFi assembly mastermix were mixed and incubated at 50 °C for 1 hour (do 8 replicates). 10 μL assembly product was analyzed on agarose gel to check the success of assembly. The remaining assembly product was purified by PCR product clean up kit (Qiagen) and eluted in 30 μL water. The electrocompetent cells MegaX DH10B™ T1R Electrocomp™ Cells (Thermo Fisher Scientific) was defrosted on ice for 30 min. Then, 50 μL MegaX cells were mixed with 5 μL (>100 ng) HiFi assembly product. The tube was tapped for three times and incubated on ice for 30 min. The bacteria/DNA mix was then transferred to chilled electroporation cuvette. The electroporation was conducted at 2.0 kV, 200 Ω, 25 μF. The cells were immediately recovered in 1000 μL SOC media. The competent cells culture was then recovered at 37 °C, 225 rpm for 1 hour. After the recovery, 10 μL and 1000 μL cell culture was plated on the square bioassay dish (Corning) and cultured at 37 °C overnight. The square bioassay dish plated with 10 μL culture was used for calculating the colony forming unit (cfu). All the colonies were scraped from the square bioassay dish and the plasmids were extracted by maxiprep (Qiagen).
TCR library display by T cells
Lentivirus of the TCR library was packaged by the method above. Lentivirus of TCR55 Vα library was titrated and coinfected SKW3 T cells with wild-type TCR55β lentivirus. Lentivirus of TCR55 Vβ library was titrated and coinfected SKW3 T cells with wild-type TCR55α lentivirus. Lentivirus of MAGE library was titrated and coinfected SKW3 T cells with wild-type MAGE-A3 TCRβ lentivirus. 48 hours after the infection, the percentage of TCR-positive population was determined by anti-CD3 (clone OKT3, BioLegend) staining and analyzed by flow cytometry. The titration of lentivirus that led to 20% infection efficiency was used to infect 100–200 million SKW3 T cells to have a low MOI. TCR-positive cells were sorted (Sony SH800S) and used for further sorting selection.
TCR library selection
10 million KG-1 cells were labelled with CFSE according to manufacturer’s protocol (Thermo Fisher Scientific). The KG-1 cells were then pulsed with 10 μM HIV peptide for 3 hours at 37 °C, 5% CO2. The KG-1 cells were resuspended at 5×105 cells/mL and aliquoted into 96-well plate at 200 μL per well. The KG-1 cells were washed once to remove excess peptides. The library of 10 million T cells were resuspended at 5×105 cells/mL and aliquoted into the 96-well plate with KG-1 cells at 200 μL per well. After 14-hour activation, the cells were stained with anti-CD69-APC (clone FN50, BioLegend) and B35-HIV-PE tetramer (the method of making pMHC tetramer is described below) on ice for 30 min. Cells were sorted to select tetramer-staining-low (comparable to TCR55 WT T cell’s tetramer staining), anti-CD69-staining-high (top 5% in terms of anti-CD69 MFI) population. Cells were sorted into FBS to maintain cell health. Sorted cells were cultured in cRPMI. It took 2 weeks to grow enough cells to continue the next round of selection. After 3–5 rounds of selection, single cell clones were obtained by diluting cells to 2.5 cells/mL and aliquoting 200 μL cell dilution to each well of 96-well U-bottom plate (Corning). It took 2–4 weeks to grow enough number of cells from single cell clone. Each single cell clone was tested by TCR55 signaling assay described below.
Sequencing of TCR mutants
Single cell clones of SKW3 T cells with expected phenotype were used to extract genomic DNA according to the manufacturer’s protocol. The TCR mutant DNA fragment was cloned by PCR and ligated into the pHR vector. The product of ligation was used to transform competent E. coli cells and 30 single colonies was picked for sequencing the TCR mutants. More than one TCR sequence might be found in each single cell clone (each T cell might still be transduced with more than one lentiviral particle at the beginning) and each TCR sequence should be tested individually by transducing SKW3 T cells for further TCR activation signaling assay.
TCR55 signaling assay
Peptide was dissolved and titrated in DMSO. KG-1 cells were labelled with CFSE and then resuspended at 5×105 cells/mL. 200 μL KG-1 cells were aliquoted to each well of 96-well U-bottom plate. KG-1 cells were pulsed with titrated peptides for 3 hours at 37 °C, 5% CO2. After that, KG-1 cells were washed once to remove excess peptides. SKW3 T cell transfectants were resuspended at 5×105 cells/mL and 200 μL T cells were added to each well with peptide-pulsed KG-1 cells. The stimulation was performed at 37 °C, 5% CO2 for 14 hours. After the stimulation, the cells were stained with anti-CD69-APC and anti-αβTCR-BV421 (clone IP26, BioLegend) on ice for 30 min and analyzed by CytoFLEX flow cytometer (Beckman). For phosphor-ERK staining, the stimulation was performed for only 15 min at 37°C, 5% CO2. After the stimulation, the cells were immediately fixed with 4% PFA and shake for 15 min. The cells were then washed with PBS (0.5% BSA) and permeabilized in ice cold methanol for 30 min on ice. The cells were then washed with PBS (0.5% BSA) for 2 times and stained with 1:50 dilution of anti-pERK1/2 (clone 197G2, Cell Signaling Technology) for 1 hour at room temperature with shaking. The cells were washed once and analyzed by CytoFLEX.
MAGE-A3-specific TCR signaling assay
MAGE-A3 (EVDPIGHLY) or TITIN (ESDPIVAQY) peptide (80% purity, Elim peptide) was dissolved and titrated in DMSO. HLA-A1-P2A-EGFP lentiviral vector was used to transfect HEK293T cells and GFP-positive cells were sorted and used as antigen-presenting cells (293T-A1). The 293T-A1 cells were resuspended at 5×105 cells/mL and pulsed with titrated peptide for 3 hours at 37 °C, 5% CO2. 200 μL KG-1 cells were aliquoted to each well of 96-well U-bottom plate. After the pulsing, the 293T-A1 cells were washed once to remove excess peptides. MAGE-A3 specific TCR mutants-transduced SKW3 cells were resuspended at 5×105 cells/mL and 200 μL T cells were added to each well with peptide-pulsed 293T-A1 cells. The stimulation was performed at 37 °C, 5% CO2 for 14 hours. After the stimulation, the cells were stained with anti-CD69-APC and anti-Vβ5.1-BV421 (clone LC4, ThermoFisher Scientific) on ice for 30 min and analyzed by CytoFLEX flow cytometer (Beckman).
Transduction of human primary T cells with TCR
Human whole blood from healthy anonymous volunteer donors was purchased from Stanford Blook Bank under the approved protocol of APB-2749-KG1018. 6-well plate was coated with 2 mL of 2.5 μg/mL anti-CD3 (OKT3 clone) overnight. The next day, human PBMC were added to the plate with 5 μg/mL anti-CD28 and cultured at 37 °C, 5% CO2 for 3 days. 4 million LentiX cells were seed in 10-cm dish and transfected with lentiviral vector of MAGE-A3-specific TCR α chain or β chain. The lentivirus was made as described above. In total 40 mL of TCR virus were concentrated to 500 μL using 100 kDa-cutoff filter. 5 million preactivated human PBMC were resuspended in 500 μL media and mixed with 500 μL concentrated TCR virus and 5 μg/mL Polybrene and 100 U/mL human IL-2. The virus/cells mixture was processed with spin infection under 2800 rpm, 32 °C for 2 hours.
Killing assay of tumor cells
20,000 A375 or HCT-116 cells were seed in each well of 96-well plate. 60,000 MAGE-A3-specific TCR-transduced human primary cells were added to each well with tumor cells and cocultured for 24 hours. The plate was washed in EDTA-free buffer and stained with 7-AAD (ThermoFisher Scientific) and Annexin V-APC (BioLegend) for 10 min. The plate was analyzed by CytoFLEX.
Cytotoxicity, cytokine, and granule release assays
200,000 tumor cells or peptide-pulsed 293T-A1 cells were seeded in each well of 96-well plate overnight. Next day, 200,000 MAGE-A3-specific TCR-transduced human primary cells were mixed with 1:100 anti-CD107a-PE (clone H4A3, BioLegend) and 1:1000 brefeldin A, and then added to each well. Coculture was done for 6 hours at 37 °C, 5% CO2. After 6 hours, the plate was stained with anti-CD8-BV421 (clone RPA-T8, BD Biosciences), anti-Vβ5.1-APC. Then the plate was fixed with IC fixation and permeabilized by permeabilization buffer. The plate was further stained with anti-IFN-γ-BV605 (clone B27, BioLegend) and anti-TNF-PE-Cy7 clone MAb11, BioLegend) on ice for 30 min. The plate was then washed and analyzed by CytoFLEX.
Production of MHC and β-2-microglobulin inclusion body
The protein of B35 MHC heavy chain and human β-2-microglobulin were made in E. coli as inclusion body. Specifically, B35 MHC heavy chain or human β-2-microglobulin was cloned into pET28a vector and transformed into BL21 (DE3) E. coli strain. Single colony was picked and resuspended in 10 mL LB media containing 50 μg/mL kanamycin and shake at 250 rpm, 37 °C for 12–16 hours. Then the 10 mL culture was added into 1 L LB media containing 50 μg/mL kanamycin and shake at 250 rpm, 37 °C for around 3 hours until the OD = 0.5 – 0.6. IPTG was added into the culture at final concentration of 1 mM and continued to shake for another 3 hours. The bacteria culture was spin down at 6000 rpm for 20 min. The bacteria pellet was resuspended in 50 mL buffer 1 (50 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM DTT, 5% Triton X-100, 1 mM EDTA, 0.2 mM PMSF). Then the bacteria were sonicated under the program of 2 min sonication plus 2 min rest. The sonication program was repeat 4 times continuously. After that, bacteria were spin 7500 rpm for 15 min. It was repeated for two more times to resuspend the bacteria pellet in buffer 1 and do the sonication. The bacteria pellet was then resuspended in 50 mL buffer 2 (50 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA). Then the bacteria were sonicated under the program of 2 min sonication plus 2 min rest. The sonication program was repeat 4 times continuously. After that, bacteria were spin 7500 rpm for 15 min. It was repeated for one more time to resuspend the bacteria pellet in buffer 2 and do the sonication. The inclusion body was pelleted and solubilized in 25 mL buffer (8 M Urea, 50 mM Tris-HCl pH 8.0, 10 mM EDTA, 10 mM DTT).
Refolding of pMHC
Refolding buffer was prepared as 100 mM Tris-HCl pH 8, 400 mM Arginine, 5 M Urea, 0.5 mM oxidized glutathione, 5 mM reduced glutathione, 2 mM EDTA. 30 mg peptide was dissolved in DMSO and added to each liter of refolding buffer. For each liter of refolding buffer, 30 mg MHC heavy chain inclusion body and 30 mg human β−2-microglobulin inclusion body were mixed in a syringe and added into each liter of refolding buffer drop by drop. Then, the refold buffer/protein were poured into dialysis tubing (Spectrum Labs) and dialyzed into 10 L 10 mM Tris pH 8.0. The 10 L 10 mM Tris pH 8.0 buffer was changed every 12 hours and repeated for 4 times in total. Then, the protein was purified by using weak anion exchange resin (DEAE Cellulose, Santa Cruz Biotechnologies). Specifically, DEAE-Cellulose was equilibrated with 10 mM Tris-HCl, pH 8.0 in a column. Then the dialyzed refolded protein solution flowed through the cellulose column drop by drop and repeated the flowing one more time. The refolded protein was eluted in 30 mL 10 mM Tris-HCl, pH 8.0 plus 0.5 M NaCl. The protein was buffer exchanged into 10 mM Tris-HCl, pH 8.0 and concentrated to 500 μL and biotinylated overnight. Biotinylated refolded protein was analyzed by size exclusion chromatography (Superdex 200, GE Healthcare) and ion exchange (MonoQ, GE Healthcare) on AKTAPurifier (GE Healthcare).
pMHC tetramer
For staining each 10 million cells, 20 μg biotinylated pMHC protein and 30 μg streptavidin-PE (Thermo Fisher Scientific) were aliquoted. 20% of total amount of streptavidin-PE were added into biotinylated pMHC each time at an interval time of one hour and repeated for 5 times. During the interval time, the tetramer was incubated on ice. The pMHC tetramer was stored at 4 °C overnight before using.
Production of TCR protein by Expi293
The TCR protein used for SPR was produced in Expi293 cells (Thermo Fisher Scientific). Specifically, TCR α chain was cloned into pD649 vector with basic zipper, and TCR β chain was cloned into pD649 vector with acid zipper. 15 μg TCR α chain constructs and 15 μg TCR β chain constructs were transfected into 75 million Expi293 cells according to the manufacturer’s protocol. 4 days after the transfection, the cell culture was spin down at 400 g for 5 min and the supernatant was saved. 1-fold volume of PBS was added to the supernatant and final concentration of 20 mM Tris-HCl pH 8.0 buffer was added. 2 mL Nickel-NTA was added to the supernatant and the solution was rotated overnight at 4 °C. Then, the solution was flowed through a column to collect the Ni-NTA and bounded protein. 1× HBS pH 7.2 containing 10 mM Imidazole was used to wash the Ni-NTA and protein once, and the protein was eluted by 1× HBS pH 7.2 containing 300 mM Imidazole. The protein was concentrated in a 30 kDa filter (Millipore) and buffer exchanged in 1× HBS pH 7.2. The protein was purified by size-exclusion chromatography using Superdex200 column on AKTAPurifier (GE Healthcare). The purified protein was collected from the according fraction based on the size and run on SDS-PAGE to check the size and 1:1 stoichiometry.
Production of TCR protein by insect cells
The TCR α chain was cloned into pAcGP67a vector with basic zipper, and TCR β chain was cloned into pAcGP67a vector with acid zipper. 2 μL baculovirus linear DNA and 2 μg TCR constructs were mixed with 100 μL Opti-MEM (Thermo Fisher Scientific) and 6.6 μL Fugene (Promega), and rest for 15 min. The mixture was added into 2 million SF9 cells and wait for 6–7 days. The cell culture was spin down at 2000 rpm for 8 min. The supernatant was saved as P0 virus. The P1 virus was made by adding 25 μL P0 virus to 25 mL SF9 cells at 2 million cells/mL. 25 mL media was added to the culture after 24 hours. 6–7 days later, the P1 virus was collected by spinning down the cell culture at 2000 rpm for 8 min and saving the supernatant. The P1 virus of TCR α chain and TCR β chain was used and titrated to coinfect 2 million Hi5 cells to determine the optimal amount of P1 virus used to get the highest amount of 1:1 expression. Usually, 1–4 mL P1 virus for each chain was used for 1 L Hi5 cells (2 million cells/mL). Optimal amount of P1 virus of TCR α chain and TCR β chain was added to Hi5 cells. 72 hours after the coinfection, the cell culture was spin down at 1500 rpm for 15 min. The supernatant was collected, and for each liter of supernatant, 100 mL 1 M Tris pH 8.0, 1 mL 1 M NiCl2, and 1 mL 5 M CaCl2 was added and stirred for 30 min. After that, the solution was spin down at 6000 rpm for 15 min. The supernatant was collected and 3 mL Ni-NTA was added to each liter of the solution. The solution was stirred for 5 hours or overnight. Then, the solution was filtered through Buchner funnel and the Ni-NTA was transferred to a filter column. The protein-bound Ni-NTA was washed with 500 mL 1× HBS pH 7.2 containing 20 mM Imidazole. Then, the protein was eluted with 15 mL 1× HBS pH 7.2 containing 300 mM Imidazole. The protein was concentrated in a 30 kDa filter and washed once with 1xHBS pH 7.2. The protein was purified by size-exclusion chromatography using Superdex200 column on AKTAPurifier (GE Healthcare). The purified protein was collected from the according fraction based on the size and run on SDS-PAGE to check the size and 1:1 stoichiometry.
Surface plasmon resonance
The affinity of TCR binding to the specific pMHC was measured by surface plasmon resonance on Biacore T100 (GE Healthcare). The refolded pMHC protein was biotinylated and immobilized on streptavidin chip (GE Healthcare). The TCR protein was treated with 3C protease to remove the basic/acid zipper. The pMHC protein was immobilized until a 100–200 RU increase, and the titrated TCR protein was flowed through the flow cell at 25 °C. The affinity of steady-state was determined by the Biacore software. No surface regeneration was required because the sample completely returned to the baseline after the dissociation.
BFP assay
The BFP force clamp assay has previously been described in detail (6, 44, 45). In brief, a T cell of interest were aspirated onto a piezo driven micropipette controlled by Labview (National Instrument) programs. An opposing micropipette as an aspirated RBC biotinylated with EZ-link NHS-PEG-Biotin (Thermo Fisher Scientific). At the apex of this RBC was a streptavidin-maleimide (Sigma-Aldrich) bound glass bead coated with the pMHCs of interest (HLA B35-HIV(Pol448–456), B35-Pep20, A1-MAGE-A3 or A1-TITIN). This RBC:bead complex served as a force probe sensor. Each T cell was repetitively brought into contact, held and then retracted to the distance controlled by the piezo actuator. The retraction and hold phase generated a force on the TCR:MHC bond, which could be altered, based on the distance the T cell was retracted. The position of the edge of the bead was tracked by the high-resolution camera (1,600 frames/sec) with < 3 nm displacement precision. The camera then recorded the time it took for the T cell to disengage the glass bead, which can visually be seen by the RBC retracting and the bead returning to its starting position. Multiple repeated cycles (known as force-clamp cycles) could be carried at a single force in order to generate an average bond lifetime between the TCR and peptide:MHC complex. Varying the level of force and recording lifetimes allowed for the determination of the average bond lifetime and the type of bond formation.
Molecular cloning of TCR signaling reporter plasmids
LCAG-HBG and LEG11-NFAT2 lentiviral expression plasmids were created by Gibson Assembly cloning based on a split-GFP system described previously (46, 47). EF1α-ERK-KTR-mScarlet or EF1α-p38-KTR-mScarlet lentiviral expression vector was generated by Gibson Assembly cloning based on an ERK-KTR-Clover or a p38-KTR-mCerulean3 plasmid from Markus Covert lab (Addgene #59150 or #59155) (48).
Jurkat ERK and p38-NFAT2 reporter cell lines
To create a live cell nuclear marker with GFP1-10 expression, Jurkat cell line was transduced with the LCAG-HBG lentiviral expression vector. Stable H2B-tBFP+ Jurkat cells were isolated by FACS sorting and transduced with the LE-EKS lentiviral expression vector. Stable ERK-KTR-mScarlet+ Jurkat cells were then isolated by FACS sorting to create the ERK reporter cell line. To create the p38-NFAT2 reporter cell line, H2B-tBFP+ Jurkat cells were transduced with the LE-38KS and the LEG11-NFAT2 lentiviral expression vectors. Stable p38-KTR-mScarlet+ and GFP1-11-NFAT2+ Jurkat cells were isolated by FACS sorting.
Live cell confocal microscopy
Live cell fluorescence time-lapse imaging data were collected using a Leica SP8 microscope with a 63x NA 1.4 oil objective (Biological Imaging Section, Research Technologies Branch, NIAID). Glass-bottom 8-well imaging chambers were coated with poly-D-lysine overnight at 4°C and washed twice with PBS. Cells were imaged in a heated 37°C environment with 5% CO2. Imaging data were processed by Imaris Cell module, customized Batch analysis, and TranslocQ pipelines.
BATTLES
To produce thermo-responsive ‘smart beads’ (~47 μm in diameter), we generated a mixture of N-Isopropylacrylamide (NIAPM, 9.2% w/v), poly(ethylene glycol) diacrylate (PEGDA, MW=700, 2.8437% v/v), lanthanide nanophosphors, sodium acrylate (1M, 5.5% v/v) and lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP, 39.2 mg/mL, 2.5% v/v). We then injected this mixture and a fluorinated HFE7500 oil suspension with 2% ionic Krytox 157 FSH surfactant and 0.05% v/v acrylic acid into a microfluidic droplet generator to produce water-in-oil droplets that were subsequently polymerized into solid beads under flood UV light (IntelliRay, UV0338) at 100% amplitude (7” away from the lamp, power= ~50–60 mW/cm2) for 2 minutes (49). After polymerization, carboxylated ‘smart beads’ were washed with 2 mL dimethylformamide for 20 s; 2 mL dichloromethane for 10 s; and 2 mL methanol for 20 s prior to being resuspended in 1 mL PBST buffer. To coat ‘smart beads’ with streptavidin, we preactivated ~200,000 beads with 1%w/v the N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) in 400 μL 0.1 M MES buffer (pH = 4.5) supplemented with 0.01% (v/v) Tween-20 for 3.5 hours at RT on an end-over-end rotator (10 rpm). The beads were spined down, washed with 1 mL 0.1 M borate buffer (pH = 8.5) supplemented with 0.01% (v/v) Tween-20 and subsequently resuspended in 400 μL of the same buffer. We then added 16 μL of streptavidin solution (dissolved in 1X PBS at 1 mg/mL) into the mixture and rotated the mixture overnight at 4°C. Next day, we quenched the conjugation reaction by adding 10 μL of 0.25 M ethanolamine in 0.1 M borate buffer (pH = 8.5) to the mixture and rotating for 30 minutes at 4°C. The final product was washed 3 times with PBST buffer, resuspended in 200 μL of the same buffer and stored at 4°C for further use. pMHC functionalized ‘smart beads’ were generated by mixing 0.5 μL of 10 nM biotin-pMHCs with ~20,000 streptavidin ‘smart’ beads in 50 μL PBST buffer. A PDMS microwell array (1440 wells) was then used to colocalized the pMHC coated beads and the calcium dye (Cal-250, 2μM) stained T cells. To exert mechanical load on bead-associated T cells, the chip was heated to and maintained at 37°C for 1 min and then cooled to and kept at 34°C for 2 min. Immediately after cooling, we acquired a total of 150 Ca2+ fluorescence images at 4 s intervals. Integrated Ca2+ signals for single T cells were analyzed by ImageJ and a custom-written MATLAB code.
Yeast-display HLA-A1-peptide library
The yeast-display HLA-A1-peptide library was generated similarly to previously described protocol (11, 27, 28). To express the HLA-A1-peptide, a single-chain format of peptide library, β−2-microglobulin (β2M) and A1 heavy chain connected by linkers was fused N-terminal to Aga2. The A1 heavy chain contains a Y84A mutation to allow an opening at the terminal of MHC groove and a linker can connect the peptide with β2M. For the peptide library, P3 and P9 were set as anchoring residues with limited diversity: P3 as asparate or glutamate, P9 as tyrosine only. For other positions of peptide library, NNK codon was used to allow all 20 amino acids. The peptide library was synthesized as short nucleotide primers which were amplified via PCR to generate the single chain of pMHC-Aga2 inserts. To generate yeast-display HLA-A1-peptide library, competent EBY-100 yeast cells were electroporated with pMHC-Aga2 library inserts and linear pYAL vector. The pMHC-Aga2 library inserts were ligated to pYAL vector inside yeast cells via homologous recombination. By plating the initial yeast library at 1:10,000, 1:1,000, 1:100, and 1:10, the library size was calculated to have 1.8×108 functional diversity. The yeast library was grown in SDCAA pH 4.5 media. The yeast library was then induced to express the pMHC library protein by growing in SGCAA pH 4.5 media.
Selection of yeast-displayed HLA-A1-peptide library
Yeast-display HLA-A1-peptide library was selected with streptavidin-coated magnetic beads coated with biotinylated TCR proteins. The number of yeast cells used for each round of selection should be 10 times higher than the diversity of the last selection step (Round 1 should use yeast cells number of 10 times of naïve library diversity). The yeast library was first incubated with 250 μL streptavidin magnetic beads in 10 mL PBE buffer (PBS+0.5% FBS+1 mM EDTA) and rotated at 4°C for 1 hour to do negative selection and remove unspecific binding to streptavidin magnetic beads. After incubation, the yeast-beads mixture was passed through an LS column (Miltenyi) and washed with PBE buffer for 3 times, and all the flow-through was collected. Streptavidin magnetic beads coated with TCR protein was prepared by mixing 400 nM biotinylated TCR monomer with 250 μL streptavidin beads in 4.7 mL PBE buffer for 15 min at 4 °C. The flow-through was incubated with TCR-beads for 3 hours at 4 °C on a rotator. The yeast cells were washed and pelleted down at 5000 g for 1 minute. The yeast cells were resuspended in 5 mL PBE buffer and passed through an LS column and washed with PBE buffer for 3 times. The flow-through was discarded. The cells in the column were eluted by 5 mL PBE buffer and pelleted down. The pellet was washed one time with SDCAA media and resuspend again in 3 mL SDCAA media to grow overnight. When the OD is over 2, yeast cells were induced in SGCAA for 2–3 days before the next round of selection. The yeast library was stained with specific TCR tetramer and anti-Myc antibody after each round of selection. The TCR tetramer was prepared at the final concentration of 400 nM by mixing TCR monomer and streptavidin-A647 at the ratio of 5:1. 100,000 yeast cells were stained with TCR tetramer and 2 μL anti-c-Myc-488 antibody (9402S, Cell Signaling) in 200 μL buffer. FACS plots were gated based on the yeast cells induced by SGCAA and stained with streptavidin-A647. Further rounds of selection were repeated with 10×108 yeast with only a modification done to the negative and positive selection using only 50 μL of streptavidin-coated beads with or without TCR in 500 μL of PBE.
Deep sequencing
Yeast DNA was extracted by Zymoprep II Kit (Zymo Research) for each round of selection from 50 million yeast cells. Barcoding PCR was firstly done for each DNA sample. The barcoding primes were designed as: Forward barcoding primer 5’ CTACACGACGCTCTTCCGATCTNNNNNNNN6 nucleotide barcode of your choice beginning of your sequence Tm (annealing)= 60 3’; Reverse barcoding primer 5’ end of your sequence Tm annealing=60NNNNNNNNAGATCGGAAGAGCGGTTCAGCAGGAAT 3’. The barcoding PCR product was run on agarose gel and gel purified. Illumina PCR was then done by using the barcoding PCR product as template and specific Illumina PCR primers: Illumina F 5’AATGATACGGCGACCACACGAGTCTACACTCTTTCCCTACACGACGCTCTTCCGA 3’; Illumina R (order the reverse complement)-5’GAAGAGCGGTTCAGCAGGAATGCCGAGACCGATCTCGTATGCCGTCTTCTGCTT G 3’. The PCR product was purified by gel extraction. The Illumina PCR product was quantified by nanodrop. The amount of each Illumina PCR product and water needed to obtain 40 μL 8 nM solution was calculated, aliquoted and mixed together. We used the Illumina V2 2×300 cycle kit following the manufacturer’s protocol for a low diversity library.
Analysis of deep sequencing data and prediction of wild type peptides from yeast selection
The sequencing results were firstly paired by PANDASEQ. The paired sequences were then imported into Geneious software to parse barcodes for each round of selection. Unique peptides were trimmed from the sequences and frequencies of amino acids were counted by custom Perl scripts used prior (27, 28, 50). To predict wild type peptides for each TCR, a positional frequency matrix was determined based on peptides from round 3 selection. To score 9-mer peptides in the human proteome data, unique peptides counted more than 10 were used to generate position weight matrices (PWM). Each PWM from individual TCR selections were then used to predicted wildtype peptides from human proteome. The Homo sapiens proteome used was from UniProtKB (Proteome ID UP000005640; June 2020 update). Python was used for algorithm for weighted positional frequency matrix and ranking a reference proteome (28).
Screening of predicted wild type peptides
The top 20 predicted wild type peptides for TCR A3A, 94a-14, 20a-18, 94a-30 were synthesized and there were 59 different peptides all together after removing repetitive peptides. Due to MAGE-A12 was shown to be cross-reactive in previous study (43), the HLA-A1 restricted MAGE-A12 peptide was also synthesized and tested. In total 60 different wild type peptides were used to screen activity of different TCRs. Briefly, 100,000 293-A1 cells were pulsed with different wild type peptides in each well of 96-well plate for 3 hours at 37 °C, 5% CO2. The 293-A1 cells were then washed with completed RPMI to remove excess peptides. 100,000 SKW3 cells expressing different TCRs were added to each well and cocultured for 14 hours at 37 °C, 5% CO2. Anti-CD69-APC and anti-TCR-BV421 staining of cells were done on ice and analyzed on flow cytometer. To do dose response of MAGE-A3, TITIN, MAGE-A6, and FAT2 peptides, 100,000 HLA-A1 cells were pulsed with titrated peptides in each well of 96-well plate for 3 hours at 37 °C, 5% CO2. The 293-A1 cells were then washed one time with completed RPMI to remove excess peptides. 100,000 SKW3 cells expressing different TCRs were added to each well and cocultured for 14 hours at 37 °C, 5% CO2. Anti-CD69-APC and anti-TCR-BV421 staining of cells were done on ice and analyzed on flow cytometer.
Supplementary Material
Acknowledgments
We thank Michelle Yen, Ricardo Fernandes, Caleb Glassman, Leon Su, João Rodrigues and Fan Liu for reading the manuscript, helpful discussions or reagents.
Funding:
K.C.G. is supported by NIH grant 5R01AI103867, Howard Hughes Medical Institute, Parker Foundation for Cancer Immunotherapy, and a Bio-X seed grant. W. C. and R.N.G. are supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health. B.D.E. is supported by NIH grant R01 AI147641 and R01 NS071518. P.M.F. was partially supported by NIH grants 1DP2GM123641, R01GM107132 and a Stanford Bio-X Interdisciplinary Initiatives seed grant. P.M.F. is a Chan Zuckerberg Biohub Investigator and acknowledges the support of a Sloan Research Foundation Fellowship. Y.F. is a Cancer Research Institute Postdoctoral Fellow. Part of this work was performed at the Stanford Nano Shared Facilities (SNSF), supported by the National Science Foundation under award ECCS-1542152.
Footnotes
Data and materials availability:
All data are available in the main text or the supplementary materials. Requests for resources and reagents should be directed to the corresponding author.
References
- 1.Stone JD, Chervin AS, Kranz DM, T-cell receptor binding affinities and kinetics: impact on T-cell activity and specificity. Immunology. 126, 165–176 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Degano M et al. , A functional hot spot for antigen recognition in a superagonist TCR/MHC complex. Immunity. 12, 251–261 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Kalergis AM et al. , Efficient T cell activation requires an optimal dwell-time of interaction between the TCR and the pMHC complex. Nat. Immunol. 2, 229–234 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Kolawole EM, Lamb TJ, Evavold BD, Relationship of 2D Affinity to T Cell Functional Outcomes. Int J Mol Sci. 21, 7969 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim ST et al. , The alphabeta T cell receptor is an anisotropic mechanosensor. J. Biol. Chem. 284, 31028–31037 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu B, Chen W, Evavold BD, Zhu C, Accumulation of dynamic catch bonds between TCR and agonist peptide-MHC triggers T cell signaling. Cell. 157, 357–368 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Y et al. , DNA-based nanoparticle tension sensors reveal that T-cell receptors transmit defined pN forces to their antigens for enhanced fidelity. Proc. Natl. Acad. Sci. U.S.A. 113, 5610–5615 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feng Y et al. , Mechanosensing drives acuity of αβ T-cell recognition. Proc. Natl. Acad. Sci. U.S.A. 114, E8204–E8213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Das DK et al. , Force-dependent transition in the T-cell receptor β-subunit allosterically regulates peptide discrimination and pMHC bond lifetime. Proc. Natl. Acad. Sci. U.S.A. 112, 1517–1522 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu B, Kolawole EM, Evavold BD, Mechanobiology of T Cell Activation: To Catch a Bond. Annu. Rev. Cell Dev. Biol. (2021), doi: 10.1146/annurev-cellbio-120219-055100. [DOI] [PubMed] [Google Scholar]
- 11.Sibener LV et al. , Isolation of a Structural Mechanism for Uncoupling T Cell Receptor Signaling from Peptide-MHC Binding. Cell. 174, 672–687.e27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fesnak AD, June CH, Levine BL, Engineered T cells: the promise and challenges of cancer immunotherapy. Nat. Rev. Cancer. 16, 566–581 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manfredi F et al. , TCR Redirected T Cells for Cancer Treatment: Achievements, Hurdles, and Goals. Front Immunol. 11, 1689 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morgan RA et al. , Cancer Regression in Patients After Transfer of Genetically Engineered Lymphocytes. Science. 314, 126–129 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rapoport AP et al. , NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 21, 914–921 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cameron BJ et al. , Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med. 5, 197ra103–197ra103 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Linette GP et al. , Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 122, 863–871 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hwang W, Mallis RJ, Lang MJ, Reinherz EL, The αβTCR mechanosensor exploits dynamic ectodomain allostery to optimize its ligand recognition site. Proc. Natl. Acad. Sci. U.S.A. 117, 21336–21345 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu P et al. , Mechano-regulation of Peptide-MHC Class I Conformations Determines TCR Antigen Recognition. Mol. Cell (2019), doi: 10.1016/j.molcel.2018.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feng Y, Zhao X, White AK, Garcia KC, Fordyce PM, Structure-activity mapping of the peptide- and force-dependent landscape of T-cell activation. bioRxiv, 2021.04.24.441194 (2021). [Google Scholar]
- 21.Hui KL, Balagopalan L, Samelson LE, Upadhyaya A, Cytoskeletal forces during signaling activation in Jurkat T-cells. Mol Biol Cell. 26, 685–695 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhong S et al. , T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proc. Natl. Acad. Sci. U.S.A. 110, 6973–6978 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thaxton JE, Li Z, To affinity and beyond: harnessing the T cell receptor for cancer immunotherapy. Hum Vaccin Immunother. 10, 3313–3321 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoffmann MM, Slansky JE, T-cell receptor affinity in the age of cancer immunotherapy. Mol Carcinog. 59, 862–870 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raman MCC et al. , Direct molecular mimicry enables off-target cardiovascular toxicity by an enhanced affinity TCR designed for cancer immunotherapy. Sci Rep. 6, 18851 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adams JJ et al. , T cell receptor signaling is limited by docking geometry to peptide-major histocompatibility complex. Immunity. 35, 681–693 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Birnbaum ME et al. , Deconstructing the Peptide-MHC Specificity of T Cell Recognition. Cell. 157, 1073–1087 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gee MH et al. , Antigen Identification for Orphan T Cell Receptors Expressed on Tumor-Infiltrating Lymphocytes. Cell. 172, 549–563.e16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gee MH, Yang X, Garcia KC, Facile method for screening clinical T cell receptors for off-target peptide-HLA reactivity. bioRxiv, 472480 (2018). [Google Scholar]
- 30.Kula T et al. , T-Scan: A Genome-wide Method for the Systematic Discovery of T Cell Epitopes. Cell. 178, 1016–1028.e13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buckley CD et al. , Cell adhesion. The minimal cadherin-catenin complex binds to actin filaments under force. Science. 346, 1254211 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marshall BT et al. , Direct observation of catch bonds involving cell-adhesion molecules. Nature. 423, 190–193 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Notch-Jagged complex structure implicates a catch bond in tuning ligand sensitivity. Science. 355, 1320–1324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pielak RM et al. , Early T cell receptor signals globally modulate ligand:receptor affinities during antigen discrimination. Proc. Natl. Acad. Sci. U.S.A. 114, 12190–12195 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van der Merwe PA, Dushek O, Mechanisms for T cell receptor triggering. Nat. Rev. Immunol. 11, 47–55 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Limozin L et al. , TCR-pMHC kinetics under force in a cell-free system show no intrinsic catch bond, but a minimal encounter duration before binding. Proc. Natl. Acad. Sci. U.S.A. 116, 16943–16948 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cai E et al. , Visualizing dynamic microvillar search and stabilization during ligand detection by T cells. Science. 356, eaal3118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME, Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat. Rev. Cancer. 8, 299–308 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao L, Cao YJ, Engineered T Cell Therapy for Cancer in the Clinic. Front Immunol. 10, 2250 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ueno T, Tomiyama H, Fujiwara M, Oka S, Takiguchi M, Functionally impaired HIV-specific CD8 T cells show high affinity TCR-ligand interactions. J. Immunol. 173, 5451–5457 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Poncette L, Chen X, Lorenz FK, Blankenstein T, Effective NY-ESO-1-specific MHC II-restricted T cell receptors from antigen-negative hosts enhance tumor regression. J. Clin. Invest. 5 (2018), doi: 10.1172/JCI120391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Isser A, Schneck JP, High-affinity T cell receptors for adoptive cell transfer. J. Clin. Invest. 129, 692 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morgan RA et al. , Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 36, 133–151 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen Y et al. , Fluorescence Biomembrane Force Probe: Concurrent Quantitation of Receptor-ligand Kinetics and Binding-induced Intracellular Signaling on a Single Cell. J Vis Exp, e52975 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen W, Zarnitsyna VI, Sarangapani KK, Huang J, Zhu C, Measuring Receptor-Ligand Binding Kinetics on Cell Surfaces: From Adhesion Frequency to Thermal Fluctuation Methods. Cell Mol Bioeng. 1, 276–288 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cabantous S, Terwilliger TC, Waldo GS, Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 23, 102–107 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Cabantous S et al. , A new protein-protein interaction sensor based on tripartite split-GFP association. Sci Rep. 3, 2854–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Regot S, Hughey JJ, Bajar BT, Carrasco S, Covert MW, High-sensitivity measurements of multiple kinase activities in live single cells. Cell. 157, 1724–1734 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feng Y, White AK, Hein JB, Appel EA, Fordyce PM, MRBLES 2.0: High-throughput generation of chemically functionalized spectrally and magnetically encoded hydrogel beads using a simple single-layer microfluidic device. Microsyst Nanoeng. 6, 109–13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gee MH et al. , Stress-testing the relationship between T cell receptor/peptide-MHC affinity and cross-reactivity using peptide velcro. Proc. Natl. Acad. Sci. U.S.A. 115, E7369–E7378 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supplementary materials. Requests for resources and reagents should be directed to the corresponding author.