

 This work is licensed under a
This work is licensed under a Abstract
Atherosclerosis is characterised by abnormal lipid and cell accumulation within arterial layers that leads to disturbed blood flow. Modified cholesterol forms such as oxidised low-density lipoprotein (oxLDL) enter cells altering their phenotype, triggering over-exuberant repair and arterial occlusion, myocardial infarction or stroke. We hypothesised that oxLDL enters vascular wall cells and induces interleukin-1β (IL-1β) secretion, potentially via a caspase-1/NLRP3 mechanism. Human coronary artery endothelial cells (HCAEC) and smooth muscle cells (HCASMC), isolated from different donors, were cultured and stimulated (primed) with pro-inflammatory cytokines TNFα and IL-1α (10 ng/mL each, for 48 h), followed by incubation with human oxLDL (10–50 ug/mL) for up to 6 h. Inhibitors of caspase-1 (YVAD), NLRP3 (MCC950) and gasdermin D (disulfiram) were added 1 h before oxLDL. Cell lysates and culture supernatants were collected and analysed for IL-1β using ELISA. Microscopy imaging showed oxLDL entered stimulated cells and formed particles. OxLDL at 20 and 50 ug/mL induced the maximum release of IL-1β from stimulated HCASMCs and HCAECs, respectively, compared to control. Inhibition of either NLRP3, caspase-1 or gasdermin D significantly reduced the release of IL-1β (4-fold, P < 0.0001; 14-fold, P < 0.0001, 1.5-fold, P < 0.0003, respectively) in HCAEC. In contrast, in HCASMCs, only caspase-1 inhibition reduced the release of IL-1β (2.1-fold, P < 0.0001). HCAECs and HCASMCs elicited the release of IL-1β in response to the same stimulus via different mechanisms. In HCAECs, released IL-1β potentially exits via a GSDMD-induced membrane pore. These data suggest that caspase-1 or gasdermin D inhibition is likely to be effective vessel wall cell-specific strategies for the reduction of atherosclerosis.
Keywords: HCAECs, HCASMCs, atherosclerosis, oxLDL, caspase-1, gasdermin D, IL-1β
Introduction
Atherosclerosis is a multifactorial chronic inflammatory disease characterised by excess blood lipids which become modified, entering the vessel wall and driving the disease process (1). A key modification is that of LDL to oxidised low-density lipoprotein (oxLDL) which then accumulates inside cells (2) causing interleukin-1β (IL-1β) production and release (3). Experimental studies in animal models (4) and recent trials, e.g. CANTOS (5), have indicated that the IL-1β pathway is causal in the pathogenesis of atherosclerosis. Since the MAB against IL-1β was not approved in patients with coronary artery disease due to cost–benefit issues, alternative routes to inhibit IL-1β are being sought.
An area of increasing interest is the mechanisms by which IL-1β is released on demand. Although there is considerable work on the mechanisms involved in IL-1β release in macrophages (6, 7), few studies have been done in vascular wall cells. This is surprising since it has been shown that IL-1β accumulates in vascular wall cells in human atherosclerotic lesions and that the more severe the disease, the more IL-1 is released (8). It is crucial, therefore, to perform experiments in vascular wall cells from the coronary bed to understand which mechanisms could be involved in IL-1β release and usefully targeted.
IL-1β production is a two-step process requiring a stimulus combination to promote transcription/translation of proIL-1β (6) which is cleaved to active IL-1β by caspase-1 (9). Active IL-1β can then be associated with the NLRP3 inflammasome (10). Only active IL-1β is released from cells and there is a myriad of mechanisms proposed, mainly in monocytic cells, including through microvesicles, multivesicular bodies, exosomes, channels or pores depending on the stimulus and cell type (11, 12). There are also caspase-1-independent mechanisms including roles for specific proteases (13), and we have previously shown that neutrophil elastase promotes the release of IL-1β from coronary artery endothelial cells via a caspase-1-independent vesicle-based mechanism (14).
oxLDL is a general term which covers many oxidative changes to LDL and ApoB, the principal protein carriers of the LDL particle (15). The types of alterations are many in vivo, but most laboratories use copper-oxidised LDL which is recognised as a useful moiety in cell culture-based studies (15). oxLDL may be oxidatively modified in the circulation or released from plaques (16). It is proposed that regions of the vascular wall with a greater oxLDL burden may be at higher risk of future plaque rupture (17).
Given the importance of oxLDL in the development of atherosclerosis (18), in this study, we sought to determine if oxLDL promoted ‘on demand’ release of IL-1β from vascular cells. We show that for the same stimulus, a subtly different mechanism of IL-1β release is employed by human coronary artery endothelial cells (HCAECs) and human coronary artery smooth muscle cells (HCASMCs). In HCAEC, oxLDL enters cells and, after 6 h, leads to substantial IL-1β release via a canonical NLRP3-caspase-1 pathway with final exit likely via a gasdermin pore. In HCASMC, the same stimulus appears to be dependent on caspase-1 activity alone. This difference may reflect the level of control needed by each cell type to limit ‘damage’ arising from the untoward release of IL-1β in the immediate microenvironment. This study is the first to compare ‘on demand’ IL-1β release in human vascular cells exposed to human oxLDL and provides additional insight into potential targets for inhibition of release of IL-1β from the major cell types in the vessel wall.
Materials and methods
Ethical approval
This study does not require local ethical approval as it uses commercially obtained primary cells derived under ethical approval obtained by the manufacturer.
Experimental procedures
Human coronary artery endothelial (HCAECs) and smooth muscle cells (HCASMCs) were purchased from PromoCell (Heidelberg, Germany) and maintained according to the manufacturer’s instructions. For HCAECs, lot numbers 440Z021.1 (male, 52 years old, Caucasian), 411Z027.6 (male, 58 years old, Caucasian) and 458Z035.3 (female, 61 years old, Caucasian) were used, and for HCASMCs, lot numbers 416Z048.4 (female, 63 years old, Asian), 440Z021.2 (male, 52 years old, Caucasian) and 452Z013.3 (male, 49 years old, Caucasian) were used. Viable cells, at passage 2–5, were seeded into 12-well plates (5000 cells/cm2) and grown at 37°C/5% CO2 (v/v) until 70–80% confluent. ProIL-1β production was stimulated by cytokine incubation (TNF-α/IL-1α; 10 ng/mL each) for 48 h (14). Cells were then washed to remove all traces of stimulating cytokines, and the media was replaced with serum-free media (without additional supplements) containing oxLDL (10–50 μg/mL) (Invitrogen) for periods up to 6 h. According to the oxLDL product datasheet, native LDL was isolated from human plasma, which is sourced from a blood bank and tested for HIV, hepatitis B and C, syphilis and other infectious diseases, and subsequently oxidised via a copper sulphate-mediated process to the optimal degree of oxidation. Oil red O staining was used to determine the extent of oxLDL uptake in cells as previously described (19). It was determined previously that the maximum IL-1β release from HCAEC occurred at 6 h (14). For HCASMCs, this occurred at 2 h (data not shown). Non-cytokine stimulated and non-oxLDL treated cells were used as control. In some experiments, cells were pre-incubated with NLRP3 inflammasome inhibitor (MCC950; 10 μM), caspase-1 inhibitor I (YVAD; 50 μM) or gasdermin D inhibitor (disulfiram; 0.5–1.0 μM) for 1 h before the addition of oxLDL.
Cells were imaged using bright-field microscopy (Leica DMI4000 Inverted) prior to the collection of supernatants. Following collection, supernatants were centrifuged at 500 g for 5 min to remove all cell debris. Cells were then lysed in ice-cold 1% (v/v) Triton-X100 lysis buffer. Both supernatants and cell lysates were stored at −80°C prior to analysis.
Determination of cell viability
Cell viability was evaluated by measurement of lactate dehydrogenase (LDH) levels in supernatants. Levels of LDH were analysed using CytoTox 96 Non-Radioactive cytotoxicity kit (Promega) following the manufacturer’s instructions.
Determination of IL-1β levels
Levels of mature IL-1β (17 kDa) (pg/mL) in the supernatants and lysates were quantified using the Human IL-1 beta/IL-1F2 DuoSet ELISA from R&D Systems© (catalogue number DY201) following the manufacturer’s instructions.
Statistical analyses
Data are shown as mean ± s.e.m. Analyses were performed using Graphpad Prism version 9.0 (Graphpad). For multiple comparison tests, a one-way ANOVA followed by the Bonferroni test was performed. Statistical significance was achieved when P < 0.05.
Results
OxLDL induces the release of IL-1β in cytokine-stimulated HCAECs and HCASMCs
When activated with a dual cytokine stimulus, oxLDL appeared as light reflective particles inside both HCAECs and HCASMCs which stained positive with Oil Red O (Fig. 1A, B, C and D). Based on the extent of Oil red O staining., uptake appeared greater in HCAECs than in HCASMCs. HCAECs did not take up oxLDL at all without prior stimulation, and HCASMCs showed very minor uptake (data not shown).
Figure 1.
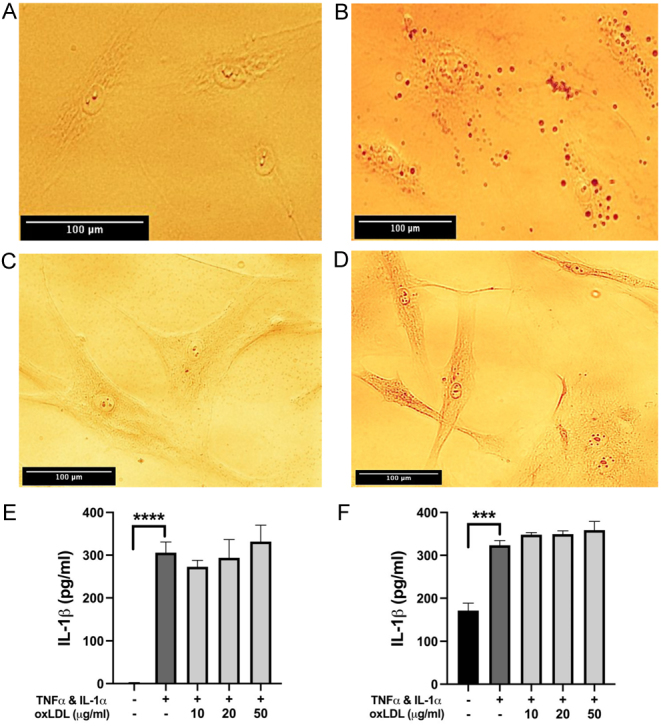
(A, B, C and D) Representative bright-field microscope images of stimulated, untreated HCAECs (A) and HCASMCs (C) compared with cytokine-stimulated (10 ng/mL of TNF-α and IL-1α for 48 h) HCAECs (B) and HCASMCs (D) treated with 50 µg/mL oxLDL for 6 h (HCAECs) or 2h (HCASMCs) and stained with Oil red O. Droplets stained with Oil red O were observed within both cell types upon stimulation and treatment. Images were taken using a Leica© bright-field microscope and are representative of 3 independent experiments, scale bar = 100 µm. (E and F) ELISA analysis of intracellular IL-1β in HCAECs (E) and HCASMCs (F) showed a significant increase following cytokine priming but no effect with oxLDL treatment when compared to the cytokine-only control. Data are presented as mean ± s.e.m. and analysed using a one-way ANOVA followed by a Bonferroni post-hoc test, ****P < 0.0001, ***P < 0.001, n = 3 independent donors.
As expected from our previous work (14), cytokine priming induced high levels of IL-1β inside the cells. However, there was no significant change in this with the application of oxLDL, indicating that oxLDL does not directly affect the production of IL-1β in stimulated HCAECs or HCASMCs (Fig. 1E and F). Interestingly, in the absence of cytokine priming, there was an accumulation of some IL-1β inside HCASMC (Fig. 1F). After stimulation, intracellular accumulation of IL-1β was at a similar level in both cell types.
After intracellular accumulation, the release of IL-1β was achieved using oxLDL as an ‘on-demand’ release stimulus. In HCAECs, the release of IL-1β was significantly increased (4-fold) in supernatants from cells incubated with 50 µg/mL oxLDL (Fig. 2A), compared to cytokine-stimulated cell controls. HCAECs treated with 10 and 20 µg/mL oxLDL did not increase IL-1β release above levels detected in cytokine-only stimulated controls or in unstimulated cells. Thus, oxLDL induced-IL-1β release in HCAECs appears to occur after a stimulus concentration threshold has been reached.
Figure 2.
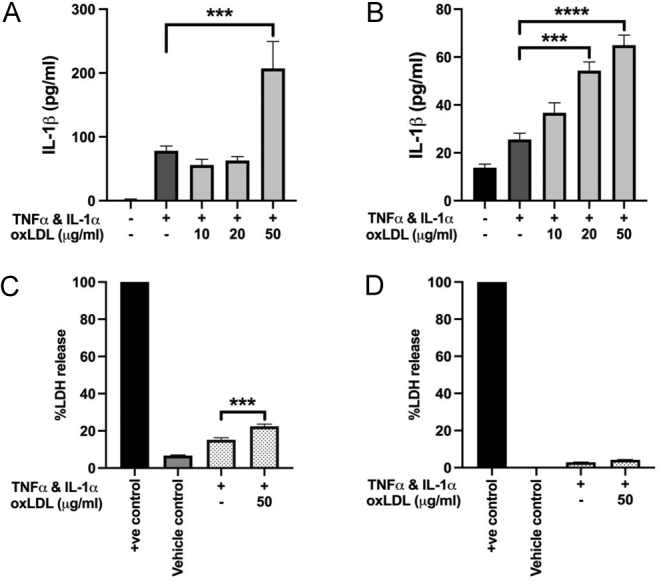
(A and B) Cytokine stimulation and treatment with higher concentrations of oxLDL for 6 h (HCAECs) or 2 h (HCASMCs) induced significant IL-1β release from both HCAECs (A) and HCASMCs (B) compared to the cytokine-only control. (C and D) Increased release of IL-1β following cytokine-priming and treatment with 50 µg/mL oxLDL is associated with elevated LDH release in HCAECs (C) but not in HCASMCs (D). Samples are compared to the cytokine-only controls. Data are presented as mean ± s.e.m. and analysed using a one-way ANOVA followed by a Bonferroni post-hoc test, ****P < 0.0001, ***P < 0.001, n = 3 independent donors.
In HCASMCs, levels of released IL-1β were lower than those observed in HCAECs. However, a clear concentration-dependent release was observed with stimulated cells treated with similar concentrations of oxLDL, with 50 µg/mL showing a 2.53-fold increase in the release of IL-1β compared to cytokine-stimulated controls, P < 0.001 (Fig. 2B).
To test whether the release seen in each cell type was not simply due to oxLDL toxicity, LDH release was determined. In stimulated HCAECs, LDH release was significantly increased in cells treated with 50 µg/mL oxLDL (Fig. 2C) compared to cytokine-only stimulated control cells and may indicate pyroptosis is occurring. This was not the case in HCASMCs where LDH release was generally low 4–6% and considered minimal (Fig. 2D).
OxLDL-induced release of IL-1β is caspase-1 dependent but in HCAEC, NLRP3 is also important
Inhibitors of caspase-1 (YVAD) and the NLRP3 inflammasome (MCC950) were used to determine whether these pathways were involved in the oxLDL-induced release of IL-1β in stimulated HCASMCs and HCAECs.
Cytokine-stimulated HCAECs, treated with oxLDL (50 µg/mL for 6 h (previously optimised for the highest level of release)), showed significantly reduced IL-1β release, back to baseline levels, when incubated with YVAD (50 μM, Fig. 3A) or MCC950 (10 μM, Fig. 3B) clearly indicating that both these pathways play a role in the release of IL-1β.
Figure 3.
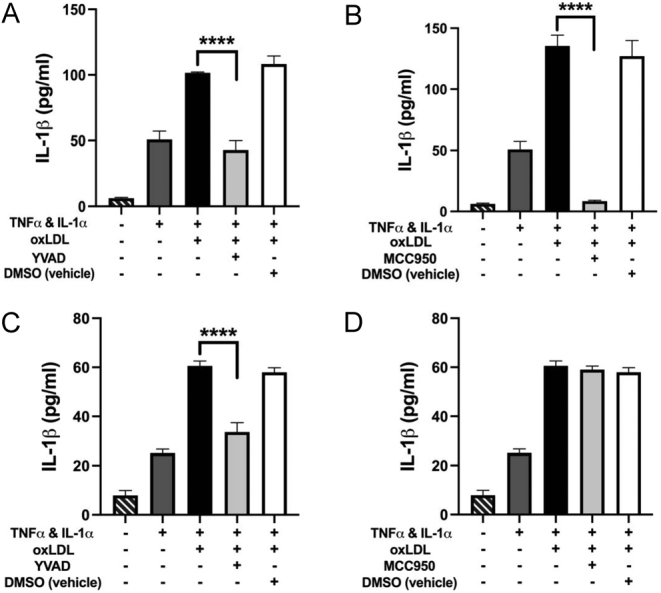
(A and B) ELISA analysis showed a significant reduction in oxLDL-induced IL-1β release from HCAECs following inhibition of both caspase-1 (YVAD) (A) and the NLRP3 inflammasome (MCC950) (B). (C and D) The release of IL-1β from oxLDL-treated HCASMCs was only significantly reduced upon inhibition of caspase-1 (C) and not the NLRP3 inflammasome (D). Data are expressed as mean ± s.e.m. and analysed using a one-way ANOVA and a Bonferroni post-hoc test, ****P < 0.0001, n = 3 independent donors.
Similarly, cytokine-stimulated HCASMCs treated with oxLDL (20 µg/mL for 2 h, optimised for maximal IL-1β release) showed a similar significant reduction in IL-1β release following YVAD treatment (Fig. 3C). However, surprisingly, the NLRP3 inflammasome inhibitor MCC950 (10 μM) had no effect on IL-1β release after oxLDL treatment. This suggests that on-demand release of IL-1β from HCASMC is dependent on caspase-1 activity alone (Fig. 3D).
OxLDL-induced release of IL-1β is gasdermin D dependent and linked to pyroptosis, but only in endothelial cells
Gasdermins are becoming more widely known as a contributor to sterile inflammation (20) and gasdermin D exhibits pore-forming properties in cell membranes (21, 22). Disulfiram, an inhibitor of gasdermin D which blocks pyroptosis in inflammatory cells (23), was used to determine whether a gasdermin-D-specific pyroptosis pathway was involved in oxLDL-induced release of IL-1β in stimulated HCASMCs and HCAECs.
Cytokine-stimulated HCAECs, treated with oxLDL (50 µg/mL for 6 h (previously optimised for the highest level of release)), showed significantly reduced IL-1β release after treatment with disulfiram at 1 µM, clearly indicating the release mechanism involves activation of the gasdermin D pathway (Fig. 4A). This corresponded with a reduction in pyroptosis in these cells exposed to 1 µM disulfiram. (Fig. 4C). In contrast, disulfiram had no effect on IL-1β release from HCASMCs (Fig. 4B) and there was no LDH release under these conditions (Fig. 4D), showing the cell-specific effect of this inhibitor on IL-1β release under the conditions studied.
Figure 4.
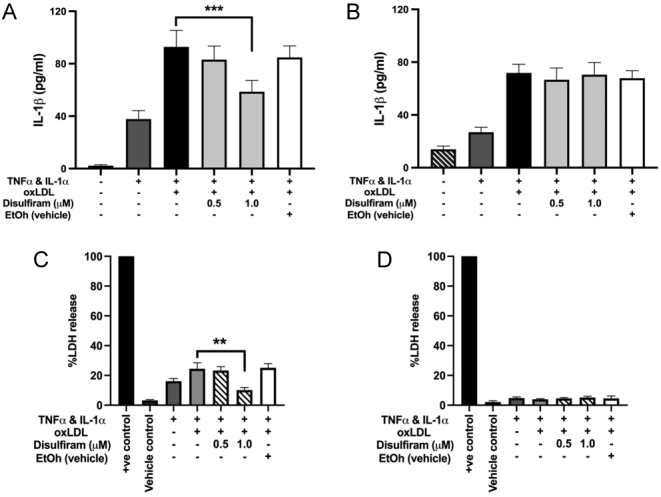
(A and B) ELISA analysis showed a significant reduction in oxLDL-induced IL-1β release from HCAECs following inhibition of gasdermin D (disulfiram) (A) but disulfiram was without effect on the release of IL-1β from HCASMCs (B). (C and D) The release of LDH was measured in HCAEC after cytokine treatment which was significantly reduced by 1 µM disulfiram treatment (C). There was no LDH release under any condition from HCASMC. Data are expressed as mean ± s.e.m. and analysed using a one-way ANOVA and a Bonferroni post-hoc test, ***P < 0.001, ** P < 0.01, n = 3 independent donors.
Discussion
We report that oxLDL induces the release of IL-1β in human vascular endothelial and smooth muscle cells via different caspase-1-mediated mechanisms. In endothelial cells, the release mechanism involves NLRP3, caspase-1 and potential inflammatory pyroptosis. In smooth muscle cells, which appear to have an intracellular pool of IL-1β under basal conditions, only caspase-1 is involved in IL-1β release under our experimental conditions. This suggests precise and cell-specific control of IL-1β release in response to oxLDL which may have implications for the use of inflammasome inhibitors in atherosclerosis.
Our data show that HCASMCs appear to produce a significant amount of intracellular IL-1β in response to the background culture conditions. This is similar to other work which has shown cytosolic accumulation of IL-1R1 receptors in vascular smooth muscle cells (VSMC) (24) and the production and release of IL-1β in the absence of stimulation, in growing aortic VSMC (25). In contrast, using human endothelial cells from the coronary bed, IL-1β was only produced after the cells were primed as previously described (26). In ECs, there was no IL-1β release at baseline although, when stimulated, EC and VSMC release similar amounts of IL-1β.
It is known that lipids accumulate within VSMC (27) and EC (19). We show that significant oxLDL is seen inside cells after priming, suggesting that only ‘activated’ cells (those that express increased scavenger receptors (28)), and not quiescent cells, have the potential to take up oxLDL and release IL-1β. It is already known that when human umbilical vein endothelial cells (HUVEC) take up oxLDL this leads to F-actin reorganisation, an increase in cell rigidity and lysosomal exocytosis (29), all of which may contribute to the IL-1β release process. A small amount of blebbing of the cell membrane was seen on both cell types towards the end of the period of stimulation with oxLDL as has been shown previously in epithelial cells in response to apoptotic stimuli (30) and in VSMC in response to calcium phosphate particles (31). Unlike these situations, we did not observe any cell death occurring. Our in vitro experimental conditions are particularly relevant to the known mechanisms of development of atherosclerosis where the levels of inflammatory cytokines such as IL-1 increase inside the plaque (32) and where arterial stiffness associated with increased oxLDL in plasma is increased in acute coronary syndrome leading to a 4-fold higher risk of future events (33).
After cytokine stimulation, IL-1β levels inside coronary VSMC and EC are similar in magnitude but much lower than in macrophages and in agreement with earlier studies in umbilical vein endothelial cells (14, 26). Vascular cells from the coronary bed release IL-1β in response to oxLDL in low serum media, and this amounts to 30–40% of the IL-1β contained inside the cells over short periods (hours). The magnitude and time course of release are similar to other studies in EC (14) and VSMC (34). The finding that not all the IL-1β inside the cells is released may be due to it being sequestered within the cell by intracellular binding proteins/soluble receptors (24) preventing all of the IL-1β pool from being released on demand.
We studied and compared the mechanisms of release over short periods (up to 6 h) using recognised inhibitors of key mediators of IL-1β processing such as caspase-1 and NLRP3. Previous studies (very few) using these inhibitors to study IL-1β release in vascular cells identified mechanisms including microvesicles and exosomes (14, 34) and pyroptosis (35) or necrosis.
In HCAEC, our data suggest that IL-1β secretion across a pyroptotic plasma membrane may have occurred since low, but significantly increased, LDH release compared with baseline was measured. In addition, the HCAEC remained alive for at least 24 h (end of the study period) after exposure to the stimulus. Our data also showed significant inhibition of IL-1β release in endothelial cells pre-treated with the cysteine reactive drug, disulfiram, suggesting activation of a gasdermin pore to elicit cytokine release – this process has been termed hyperactivation (36). Disulfiram is known to block pyroptosis by binding to cysteine 191, blocking oligomerisation and hence pore formation (37). Disulfiram has also been shown to alter the substrate cleavage activity of caspases through the oxidation of key thiols (38).
Thus, we conclude that in response to the specific stimulus (oxLDL), HCAECs in culture become hyperactivated (gasdermin activation, IL-1β release but remain alive) (36) and display pyroptotic features such as LDH release. It is interesting that in dendritic cells a mixture of oxidised phosphorylcholines also induces non-canonical inflammasome-dependent release of IL-1β in the absence of cell death (39). A potential mechanism involved could be the initial damage of plasma membranes by oxidised lipids leading to subsequent membrane repair moderated by factors in the immediate cellular environment, but this requires further fundamental research for a full explanation (40).
Disulfiram was without effect on IL-1β release from VSMC, and in response to oxLDL, no LDH release was measured in our experimental setting.
As summarised in Fig. 5, in HCAEC, in relation to oxLDL as an on-demand release stimulus, NLRP3, caspase-1 and gasdermin D activation were required. This contrasts with HCASMCs where the mechanism of on-demand release of IL-1β by oxLDL required caspase-1 activation only.
Figure 5.
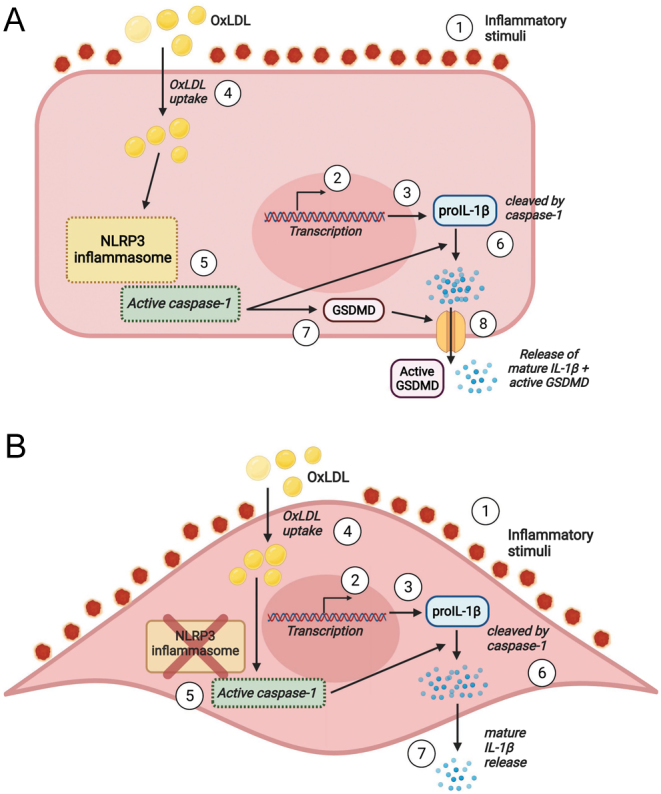
Graphical summary of the release mechanism of IL-1β in vascular cells in response to oxLDL as an on-demand stimulus. In (A), primed EC (1), gene transcription (2) leads to the synthesis of proIL-1β (3) and receptor expression for uptake of oxLDL (4). oxLDL activates the NLRP3 inflammasome including caspase 1 (5) which both cleaves proIL-1β (6) and activates gasdermin D (7) leading to the release of IL-1β (8). In (B), primed VSMC through inflammatory stimuli (2) causes gene transcription (2) and leads to the synthesis of proIL-1β (3) and receptor expression for uptake of oxLDL (4). OxLDL activates caspase 1 (5) leading to activation of IL-1β (6), and mature IL-1β is released (7).
These data suggest a greater level of control of IL-1β release in the endothelial cells of coronary arteries than in vascular smooth muscle cells, in relation to the same on-demand release stimulus. A limitation of our work is that it was not possible to produce high-quality and reproducible Western blots despite concentration of our protein samples. Had this been possible it would have been ideal to investigate the effect of disulfiram on caspase-1 processing in the two cell types. A further limitation is that we were unable to study intact monolayers and therefore barrier function due to the experimental workflow. Our in vitro model is therefore more akin to an eroded plaque surface rather than a stable plaque where the endothelium is intact. Despite these, this is the first comparison of IL-1β release from human coronary vascular cells using an identical stimulation and release regime, and extrapolation of these data suggests that NLRP3 inhibition may not curtail IL-1β release in the media or plaque of the vessel wall where the endothelium is intact, or where there are few macrophages, e.g. in early atherosclerosis. In support, the study by Sharma and coworkers in a mouse model with MCC950 only showed inhibitory effects when atherosclerosis was combined with diabetes, and there was no effect on lesion size at the aortic root, or on IL-1β or IL-6 production in lesions in atherosclerotic controls with mild disease (41). We note that in the first paper reporting MCC950 inhibition in experimental atherosclerosis (42), studies were conducted on lesions in carotid arteries and not the aortic bed or in human vascular cells. We have shown in our work presented here that, in relation to ‘on demand’ release of IL-1β and the potential mechanisms involved, the specific vascular bed studied and, therefore, the treatment chosen matters.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work was supported by a PhD scholarship to Dr Majid Almansouri from the Embassy of Saudi Arabia KAU1571-00016603. This work was also partly supported by British Heart Foundation grant PG/13/55/30365.
Acknowledgements
M A and P P undertook experiments and generated and interpreted data. S E F and J C conceived the original idea and generated funding, supervised the experiments and analysed data. All authors were involved in writing and editing of the manuscript.
References
- 1.Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, Tokgozoglu L, Lewis EF. Atherosclerosis. Nature Reviews: Disease Primers 20195 56. ( 10.1038/s41572-019-0106-z) [DOI] [PubMed] [Google Scholar]
- 2.Itabe H, Obama T, Kato R. The dynamics of oxidized LDL during atherogenesis. Journal of Lipids 20112011 418313. ( 10.1155/2011/418313) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang K, Zhang XJ, Cao LJ, Liu XH, Liu ZH, Wang XQ, Chen QJ, Lu L, Shen WF, Liu Y. Toll-like receptor 4 mediates inflammatory cytokine secretion in smooth muscle cells induced by oxidized low-density lipoprotein. PLoS ONE 20149 e95935. ( 10.1371/journal.pone.0095935) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chamberlain J, Francis S, Brookes Z, Shaw G, Graham D, Alp NJ, Dower S, Crossman DC. Interleukin-1 regulates multiple atherogenic mechanisms in response to fat feeding. PLoS ONE 20094 e5073. ( 10.1371/journal.pone.0005073) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SDet al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. New England Journal of Medicine 20173771119–1131. ( 10.1056/NEJMoa1707914) [DOI] [PubMed] [Google Scholar]
- 6.Lopez-Castejon G, Brough D. Understanding the mechanism of IL-1beta secretion. Cytokine and Growth Factor Reviews 201122189–195. ( 10.1016/j.cytogfr.2011.10.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Starling S.Innate immunity: revealing the secrets of IL-1 secretion. Nature Reviews: Immunology 2017182–3. ( 10.1038/nri.2017.155) [DOI] [PubMed] [Google Scholar]
- 8.Galea J, Armstrong J, Gadsdon P, Holden H, Francis SE, Holt CM. Interleukin-1 beta in coronary arteries of patients with ischemic heart disease. Arteriosclerosis, Thrombosis, and Vascular Biology 1996161000–1006. ( 10.1161/01.atv.16.8.1000) [DOI] [PubMed] [Google Scholar]
- 9.Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 1992356768–774. ( 10.1038/356768a0) [DOI] [PubMed] [Google Scholar]
- 10.Schroder K, Tschopp J. The inflammasomes. Cell 2010140821–832. ( 10.1016/j.cell.2010.01.040) [DOI] [PubMed] [Google Scholar]
- 11.Martin-Sanchez F, Diamond C, Zeitler M, Gomez AI, Baroja-Mazo A, Bagnall J, Spiller D, White M, Daniels MJ, Mortellaro Aet al. Inflammasome-dependent IL-1beta release depends upon membrane permeabilisation. Cell Death and Differentiation 2016231219–1231. ( 10.1038/cdd.2015.176) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monteleone M, Stow JL, Schroder K. Mechanisms of unconventional secretion of IL-1 family cytokines. Cytokine 201574213–218. ( 10.1016/j.cyto.2015.03.022) [DOI] [PubMed] [Google Scholar]
- 13.Afonina IS, Muller C, Martin SJ, Beyaert R. Proteolytic processing of interleukin-1 family cytokines: variations on a common theme. Immunity 201542991–1004. ( 10.1016/j.immuni.2015.06.003) [DOI] [PubMed] [Google Scholar]
- 14.Alfaidi M, Wilson H, Daigneault M, Burnett A, Ridger V, Chamberlain J, Francis S. Neutrophil elastase promotes interleukin-1beta secretion from human coronary endothelium. Journal of Biological Chemistry 201529024067–24078. ( 10.1074/jbc.M115.659029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parthasarathy S, Raghavamenon A, Garelnabi MO, Santanam N. Oxidized low-density lipoprotein. Methods in Molecular Biology 2010610403–417. ( 10.1007/978-1-60327-029-8_24) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hartley A, Haskard D, Khamis R. Oxidized LDL and anti-oxidized LDL antibodies in atherosclerosis – novel insights and future directions in diagnosis and therapy. Trends in Cardiovascular Medicine 20192922–26. ( 10.1016/j.tcm.2018.05.010) [DOI] [PubMed] [Google Scholar]
- 17.van Dijk RA, Kolodgie F, Ravandi A, Leibundgut G, Hu PP, Prasad A, Mahmud E, Dennis E, Curtiss LK, Witztum JLet al. Differential expression of oxidation-specific epitopes and apolipoprotein(a) in progressing and ruptured human coronary and carotid atherosclerotic lesions. Journal of Lipid Research 2012532773–2790. ( 10.1194/jlr.P030890) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Di Pietro N, Formoso G, Pandolfi A. Physiology and pathophysiology of oxLDL uptake by vascular wall cells in atherosclerosis. Vascular Pharmacology 2016841–7. ( 10.1016/j.vph.2016.05.013) [DOI] [PubMed] [Google Scholar]
- 19.Zhang CP, Ding XX, Tian T, Li BJ, Wang CY, Jiang SS, Shao JQ, Yuan YL, Tian Y, Zhang Met al. Impaired lipophagy in endothelial cells with prolonged exposure to oxidized lowdensity lipoprotein. Molecular Medicine Reports 2020222665–2672. ( 10.3892/mmr.2020.11345) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer FA, Chen KW, Bezbradica JS. Posttranslational and therapeutic control of gasdermin-mediated pyroptosis and inflammation. Frontiers in Immunology 202112 661162. ( 10.3389/fimmu.2021.661162) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016535153–158. ( 10.1038/nature18629) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen X, He WT, Hu L, Li J, Fang Y, Wang X, Xu X, Wang Z, Huang K, Han J. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Research 2016261007–1020. ( 10.1038/cr.2016.100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, Ruan J, Luo X, Lou X, Bai Yet al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nature Immunology 202021736–745. ( 10.1038/s41590-020-0669-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng Y, Humphry M, Maguire JJ, Bennett MR, Clarke MC. Intracellular interleukin-1 receptor 2 binding prevents cleavage and activity of interleukin-1alpha, controlling necrosis-induced sterile inflammation. Immunity 201338285–295. ( 10.1016/j.immuni.2013.01.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim EJ, Park SY, Baek SE, Jang MA, Lee WS, Bae SS, Kim K. & Kim CD.HMGB1 increases IL-1beta production in vascular smooth muscle cells via NLRP3 inflammasome. Frontiers in Physiology 20189 313. ( 10.3389/fphys.2018.00313) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilson HL, Varcoe RW, Stokes L, Holland KL, Francis SE, Dower SK, Surprenant A, Crossman DC. P2X receptor characterization and IL-1/IL-1RA release from human endothelial cells. British Journal of Pharmacology 2007151115–127. ( 10.1038/sj.bjp.0707213) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Campbell JH, Popadynec L, Nestel PJ, Campbell GR. Lipid accumulation in arterial smooth muscle cells. Influence of phenotype. Atherosclerosis 198347279–295. ( 10.1016/0021-9150(8390059-x) [DOI] [PubMed] [Google Scholar]
- 28.Cominacini L, Pasini AF, Garbin U, Davoli A, Tosetti ML, Campagnola M, Rigoni A, Pastorino AM, Lo Cascio V, Sawamura T. Oxidized low density lipoprotein (ox-LDL) binding to ox-LDL receptor-1 in endothelial cells induces the activation of NF-kappaB through an increased production of intracellular reactive oxygen species. Journal of Biological Chemistry 200027512633–12638. ( 10.1074/jbc.275.17.12633) [DOI] [PubMed] [Google Scholar]
- 29.Couto NF, Rezende L, Fernandes-Braga W, Alves AP, Agero U, Alvarez-Leite J, Damasceno NRT, Castro-Gomes T, Andrade LO. OxLDL alterations in endothelial cell membrane dynamics leads to changes in vesicle trafficking and increases cell susceptibility to injury. Biochimica et Biophysica Acta: Biomembranes 20201862 183139. ( 10.1016/j.bbamem.2019.183139) [DOI] [PubMed] [Google Scholar]
- 30.Barros LF, Kanaseki T, Sabirov R, Morishima S, Castro J, Bittner CX, Maeno E, Ando-Akatsuka Y, Okada Y. Apoptotic and necrotic blebs in epithelial cells display similar neck diameters but different kinase dependency. Cell Death and Differentiation 200310687–697. ( 10.1038/sj.cdd.4401236) [DOI] [PubMed] [Google Scholar]
- 31.Dautova Y, Kozlova D, Skepper JN, Epple M, Bootman MD, Proudfoot D. Fetuin-A and albumin alter cytotoxic effects of calcium phosphate nanoparticles on human vascular smooth muscle cells. PLoS ONE 20149 e97565. ( 10.1371/journal.pone.0097565) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Tassell BW, Toldo S, Mezzaroma E, Abbate A. Targeting interleukin-1 in heart disease. Circulation 20131281910–1923. ( 10.1161/CIRCULATIONAHA.113.003199) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meisinger C, Baumert J, Khuseyinova N, Loewel H, Koenig W. Plasma oxidized low-density lipoprotein, a strong predictor for acute coronary heart disease events in apparently healthy, middle-aged men from the general population. Circulation 2005112651–657. ( 10.1161/CIRCULATIONAHA.104.529297) [DOI] [PubMed] [Google Scholar]
- 34.Dautova Y, Kapustin AN, Pappert K, Epple M, Okkenhaug H, Cook SJ, Shanahan CM, Bootman MD, Proudfoot D. Calcium phosphate particles stimulate interleukin-1beta release from human vascular smooth muscle cells: a role for spleen tyrosine kinase and exosome release. Journal of Molecular and Cellular Cardiology 201811582–93. ( 10.1016/j.yjmcc.2017.12.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Correa R, Silva LFF, Ribeiro DJS, Almeida RDN, Santos IO, Correa LH, de Sant’Ana LP, Assuncao LS, Bozza PT, Magalhaes KGet al. Lysophosphatidylcholine induces NLRP3 inflammasome-mediated foam cell formation and pyroptosis in human monocytes and endothelial cells. Frontiers in Immunology 201910 2927. ( 10.3389/fimmu.2019.02927) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity 20184835.e6–44.e6. ( 10.1016/j.immuni.2017.11.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, Benson BL, Chirieleison SM, Huang AY, Dubyak GRet al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Science Immunology 20183 eaat2738. ( 10.1126/sciimmunol.aat2738) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nobel CS, Kimland M, Nicholson DW, Orrenius S, Slater AF. Disulfiram is a potent inhibitor of proteases of the caspase family. Chemical Research in Toxicology 1997101319–1324. ( 10.1021/tx970131m) [DOI] [PubMed] [Google Scholar]
- 39.Zhivaki D, Borriello F, Chow OA, Doran B, Fleming I, Theisen DJ, Pallis P, Shalek AK, Sokol CL, Zanoni Iet al. Inflammasomes within hyperactive murine dendritic cells stimulate long-lived T cell-mediated anti-tumor immunity. Cell Reports 202033 108381. ( 10.1016/j.celrep.2020.108381) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McNeil PL, Kirchhausen T. An emergency response team for membrane repair. Nature Reviews: Molecular Cell Biology 20056499–505. ( 10.1038/nrm1665) [DOI] [PubMed] [Google Scholar]
- 41.Sharma A, Choi JSY, Stefanovic N, Al-Sharea A, Simpson DS, Mukhamedova N, Jandeleit-Dahm K, Murphy AJ, Sviridov D, Vince JEet al. Specific NLRP3 inhibition protects against diabetes-associated atherosclerosis. Diabetes 202170772–787. ( 10.2337/db20-0357) [DOI] [PubMed] [Google Scholar]
- 42.van der Heijden T, Kritikou E, Venema W, van Duijn J, van Santbrink PJ, Slutter B, Foks AC, Bot I, Kuiper J. NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E-deficient mice-brief report. Arteriosclerosis, Thrombosis, and Vascular Biology 2017371457–1461. ( 10.1161/ATVBAHA.117.309575) [DOI] [PubMed] [Google Scholar]