

Summary
Masking of host cell receptors following retroviral infection is the basis for the phenomenon of virus interference. Amphotropic retrovirus vectors were used to express the HIV envelope glycoprotein in a human CD4+ cell line. Envelope expression is accompanied by a reduction in the level of surface CD4 receptor molecules and correlates with the presence of intracellular envelope–CD4 receptor complexes. Cells expressing the HIV envelope acquire a cytolysis-resistant phenotype such that infection with HIV leads to a noncytopathic persistent virus infection. Furthermore, phorbol ester–mediated stimulation of viral replication in persistently infected cells results in renewed cytolytic effects which, due to the absence of CD4 in the cell population, are absolutely independent of syncytium formation. This study elucidates the mechanism by which viral persistence is initiated and maintained in the course of AIDS.
Introduction
The human acquired immune deficiency syndrome (AIDS) is characterized by a depletion of T4 positive helper lymphocytes. The resulting immune dysfunction leads to the development of life-threatening opportunistic infections which are ultimately the cause of death in AIDS patients (Gottlieb et al., 1981; Masur et al., 1981). HIV, the etiologic agent of AIDS (Barre-Sinoussi et al., 1983; Levy et al., 1984; Gallo et al., 1984), appears to play a direct role in the CD4 cell depletion process. In vitro, HIV exhibits a profound tropism for human CD4 helper lymphocytes (Dalgleish et al., 1984). Infection of these CD4 positive lymphocytes results in the appearance of multinucleated giant cells or syncytia, and, in certain cell lines, the complete destruction of the infected culture. Cell lines do, however, vary in their susceptibility to AIDS virus–mediated cytopathic effects and, in certain cases, infection of the cell line by HIV results in a noncytopathic persistent infection rather than destruction of the host cell (Dalgleish et al., 1984; Casareale et al., 1987; Kikukawa et al., 1986; McDougal et al., 1985; Hoxie et al., 1985). A common feature of persistently infected helper T lymphocyte cell lines is a loss or dramatic reduction in expression of the host CD4 receptor glycoprotein (Dalgleish et al., 1984; Folks et al., 1986; Stevenson et al., 1987; Klatzmann et al., 1984a; Hoxie et al., 1986). The reduction in CD4 receptor expression by the infected host cell appears to be directly mediated by the virus, either by interfering with transcription of the CD4 cell receptor gene itself (Hoxie et al., 1986), or more probably by posttranscriptional events that interfere with localization of the CD4 receptor on the host cell surface (Stevenson et al., 1987). One possible explanation for the ability of persistently infected cell lines to resist cytopathic effects of HIV could be the inability of these CD4 depleted cells to form syncytia, since syncytium formation is absolutely dependent upon the expression of HIV envelope on infected cells and the expression of CD4 receptors on uninfected cells (Lifson et al., 1986; Sodroski et al., 1986a). However, we propose that the reduction in CD4 receptor expression, which accompanies the persistent infection, is indicative of superinfection resistance such that these persistently infected cells are no longer susceptible to second round infection by progeny virus. In this paper, direct evidence is provided for the ability of the HIV envelope glycoprotein to modulate CD4 expression and induce interference in cytolysis sensitive CD4+ cells by restricting accumulation of unintegrated viral DNA. We also demonstrate that cytolysis can be incluced in persistently infected cells by a process that is completely independent of syncytium formation. It is proposed that envelope-mediated interference and superinfection resistance provides the basis for viral persistence and latency in the course of AIDS. Furthermore, interruption of latency by factors that induce viral replication and subsequent cytolysis may lead to the CD4 cell depletion without the involvement of syncytium formation.
Results
Amphotropic retroviral vectors were used to insert and express the HIV envelope gene in the acute lymphoblastic CD4 positive T cell line CEM. Initial attempts to express the HIV envelope gene in the absence of the tat and art/trs genes were unsuccessful due to the extremely low production of envelope glycoprotein in CEM cells. Subsequently, we used a 3.1 kb Sall–Xhol fragment excised from an almost full-length proviral HIV DNA clone (N1G-F) (Figure 1) for the efficient expression of HIV envelope glycoprotein. This fragment was inserted in forward orientation into the Sall site of the retroviral expression vector pDOL (Korman et al., 1987). Recent work has demonstrated the absolute requirement of the tat and art/trs viral gene products for the efficient translation of envelope glycoprotein from the cognate messenger RNA (Knight et al., 1987). In expectation of possible problems encountered with low expression of the HIV envelope because of low transcriptional activity of the murine leukemia virus (MLV) long terminal repeat (LTR) sequences in certain cell lines, the highly active constitutive promoter of the cytomegalovirus immediate early gene (Boshart et al., 1985) was inserted 5′ of the HIV envelope coding sequences. The presence of this CMV promoter gives an approximately 3-fold increase in CAT expression in CEM cells when compared with that produced from the retroviral long terminal repeat (M. Stevenson, unpublished data). In addition, the presence of the CMV promoter did not appear to affect greatly packaging of the retroviral vector as determined by colony forming assays on infected G418 selected NIH 3T3 cells. For comparison, the tat gene (obtained as a 2700 base EcoRI–BamHI fragment of N1G-F) was also inserted into pDOL. The retroviral vectors were packaged in the amphotropic packaging line PA317 (Miller and Buttimore, 1986), and 2 days following transfection, CEM cells were infected by cocultivation with the transfected PA317 cells. Following selection in G418, cell lines were analyzed for the presence of integrated vector sequences by Southern blot hybridization (Figure 2A). Use of a number of restriction enzymes demonstrated that the HIV envelope and promoter sequences had been transferred faithfully into the CEM cell lines and that no major deletions had occurred during retroviral packaging and insertion of the provirus (Figure 2). In addition, use of the HindIII enzyme, which cuts only once between the MLV LTRs (Figure 1), demonstrated that the G418 resistant cell population was clonal and contained a single copy of the pDOL CMV·env and pDOL·tat provirus (Figure 2). Polyadenylated RNA fractions were purified from the G418 resistant cell lines, immobilized on nylon membranes, and hybridized with an HIV-specific DNA probe (Figure 3A). The blot was also hybridized to a β-tubulin probe (Hall et al., 1983) to demonstrate that equivalent amounts of RNA were analyzed in each sample. Only CEM cells infected with retroviral vectors carrying HIV envelope and tat genes hybridized to an HIV probe spanning the complete envelope open reading frame (Figure 3A). Thus, correct integration of the provirus was accompanied by constitutive promoter activity with the generation of transcripts containing HIV sequences. The presence of the internal CMV promoter downstream of the MLV LTR sequences did not appear to influence viral packaging or the level of gene transcripts generated. Northern blot analysis (data not shown) indicated that both the MLV LTR and CMV promoter were initiating transcription in the infected cell lines. This is to be expected since initiation of transcripts from the LTR is necessary for inclusion of retroviral encapsidation sequences (ψ) on the transcript. CEM cell lines expressing the HIV envelope (pDOL CMV·env) and the HIV tat gene (pDOL·tat) retained the ability to transactivate an HIV LTR driven chloramphenicol acetyl transferase (CAT) expression plasmid (PU3RIII, Sodroski et al., 1985a) as determined by CAT measurements following transfection of the plasmid into the selected cells (data not shown). This indicated that functional tat III was being produced from both retroviral constructs. The expression of HIV envelope glycoprotein on the cell surface was confirmed by immunofluorescence staining using a high titer serum from an HIV seropositive hemophiliac (Figure 3B). CEM cells infected with the pDOL CMV·env construct reacted weakly and displayed a membrane type fluorescence (Figure 3B), whereas control CEM cells had no reactivity with the patient’s serum (Figure 3B). By radioimmunoprecipitation, we could identify the precursor gp160 glycoprotein and gp120 in metabolically labeled whole cell extracts of pDOL CMV·env CEM cells (not shown) but not in pDOL·tat infected CEM cells. We could not identify with any certainty the presence of the transmembrane gp41 in the pDOL CMV·env infected CEM cells. However, this is probably due to differential reactivity of the serum used for the analysis. Additional confirmation for the expression of functional HIV envelope was provided by the presence of syncytia in the envelope vector infected CEM cells (Lifson et al., 1986; Sodroski et al., 1986b) but not the tat infected lines. However, although most of the cells in the population were producing envelope (by IF analysis), syncytium formation was only apparent in 1%–2% of the cells (see giant cell, Figure 3B). Thus, it appears that expression of HIV envelope in our cell line did not lead to syncytium formation in every cell expressing envelope glycoprotein.
Figure 1.
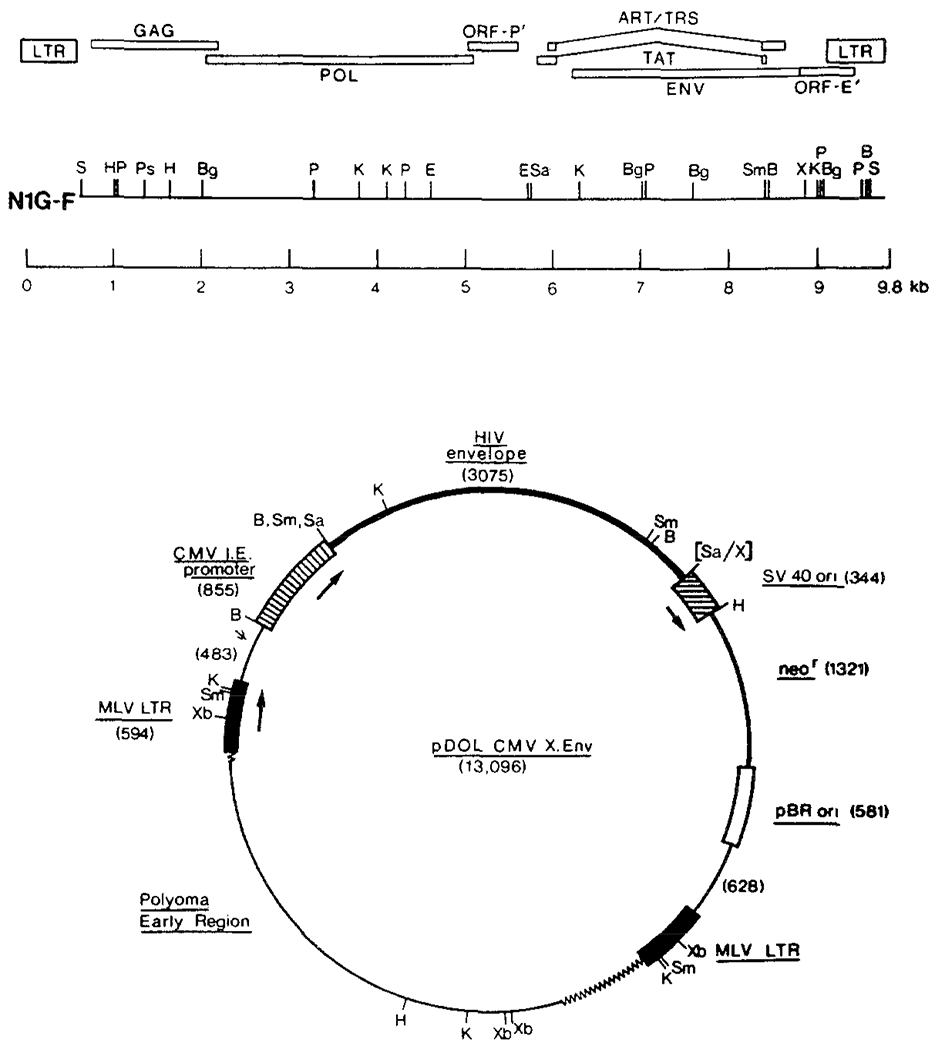
Construction of Retroviral Vector Containing HIV Envelope Coding Sequences
The complete HIV envelope gene from HIV clone N1G-F was inserted into the Sall site of the retrovirus expression vector pDOL, and the cytomegalovirus immediate early promoter was inserted at the BamHI site. Respective regions of the vector are shown to scale (sizes in base pairs in brackets). Boxed areas represent promoter/enhancer sequences and arrows indicate direction of transcription. Symbols: MLV, murine leukemia virus; CMV, cytomegalovirus; B, BamHI; Sm, Smal; Sa, Sall; K, Kpnl; X, Xhol; Xb, Xbal; H, HindIII; Ps, Pstl; ψ, retroviral packaging sequences; p, Pvull; Bg, BgIII; S, Sacl.
Figure 2.
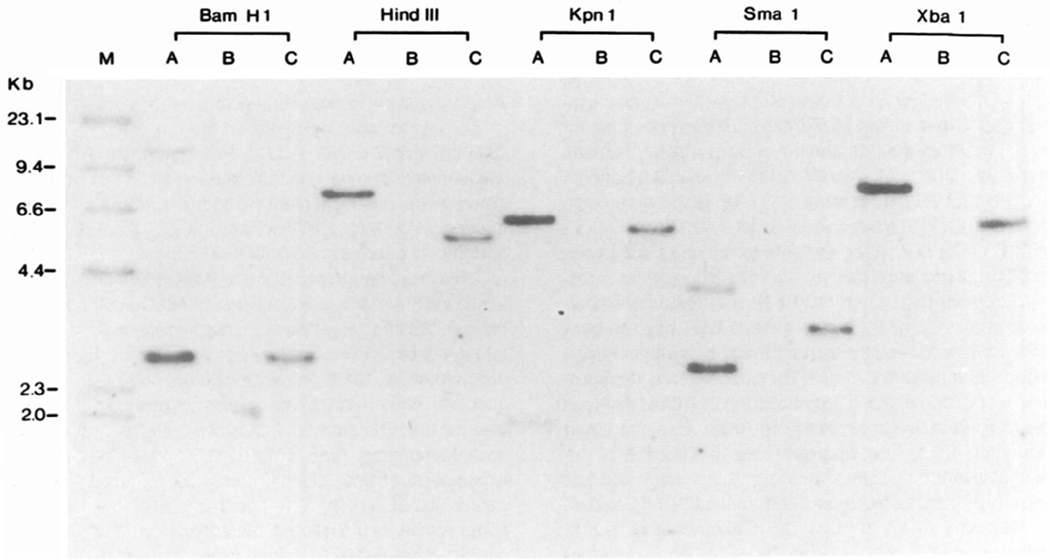
Southern Blot Analysis of Integrated HIV Envelope Sequences in CEM Cells
CEM cells were infected with retroviral env and tat expression vectors by cocultivation with DNA transfected amphotrophic packaging cell line PA317 in the presence of polybrene. Infected cells were selected in G418 and analyzed for envelope sequences. a, CEM cells infected with pDOL CMV·env; b, uninfected CEM cells; c, CEM cells infected with pDOL·tat. Genomic DNA was prepared by guanidine thiocyanate lysis, restriction endonuclease digested, electrophoresed, and transferred to nylon membranes. Blots were hybridized under high stringency conditions to a 32P-labeled probe prepared by nick translation of a Sall–Xhol fragment of λ-N1G-F. Molecular weight markers are provided by 32P end-labeled HindIII digests of λDNA.
Figure 3.
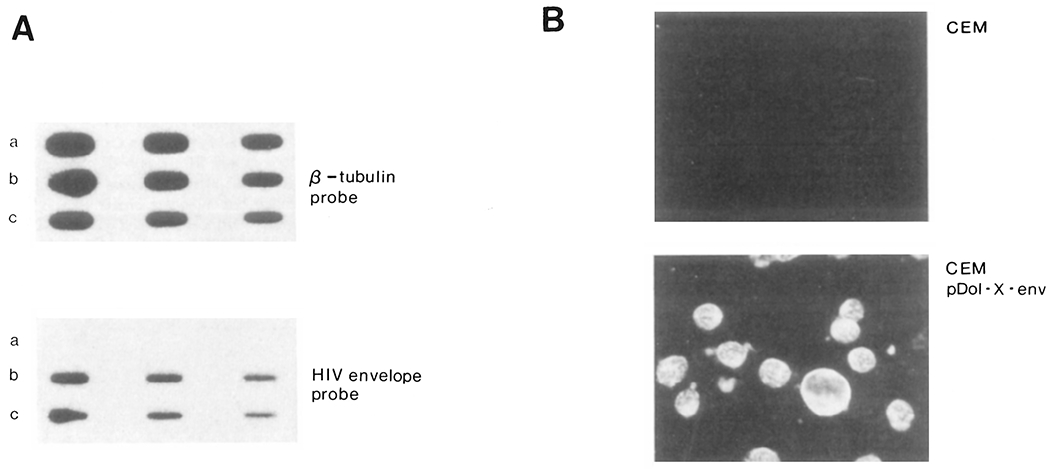
Expression of HIV Envelope–Specific RNA and Antigen in CEM Cells
(A) Poly (A)+ mRNA from guanidine thiocyanate cell lysates was selected by passage over oligo d-T cellulose columns, spotted onto nylon membranes (2, 1, and 0.5 μg) and hybridized with a β-tubulin probe (Hall et al., 1983) or HIV probe as in Figure 2: a, uninfected CEM cells; b, CEM cells infected with pDOL CMV·env; c, CEM cells infected with pDOL·tat. Blots were autoradiographed for either 3 hr (β-tubulin probe) or 18 hr (HIV probe).
(B) Indirect immunofluorescence visualization of HIV envelope in G418 selected cells. Acetone fixed cells were reacted with HIV-positive serum from an HIV seropositive hemophiliac (antibody titer, 1:5160). Bound HIV-specific IgG was detected with FITC conjugated goat anti-human IgG. Uninfected CEM cells were used as a negative control. Photomicrographs were taken on Kodak Pan film (20 sec exposure at 400 ASA). Note syncytium in CEM pDOL·env photomicrograph.
Effect of HIV Envelope on CD4 Expression
We next determined whether the expression of the HIV envelope gene in CEM cells affected the level of surface CD4 receptor expression. This was performed by fluorescence-activated cell sorter analysis (FACS analysis) using fluorescein conjugated antibodies reactive to two epitopes of the CD4 receptor (OKT4, OKT4A), and as a control we included a pan-T cell reactive monoclonal antibody (Leu 1). The OKT4 monoclonal antibody recognizes an epitope on the CD4 receptor distinct from that involved in the binding of HIV and is unable to block virus infection, while the OKT4A monoclonal recognizes the HIV binding site and blocks infection. In addition, OKT4 antibodies are able to coprecipitate the viral gp120–CD4 complex (Dalgleish et al., 1984; Klatzmann et al., 1984a; McDougal et al., 1986) and bind CD4 in presence of complexed gp120 (Mathews et al., 1987), indicating that the OKT4 epitope is not sterically blocked in the CD4-HIV gp120 complex. Fluorescence profiles showing the reactivity of pDOL CMV·env and pDOL·tat infected CEM cells and normal uninfected CEM cells with these monoclonal antibodies are shown in Figure 4, with the mean fluorescence channel number indicated on each profile. In CEM cells expressing the HIV envelope glycoprotein, reactivity with both the OKT4 and the OKT4A monoclonal antibodies was reduced by approximately 60% when compared with control CEM cells or CEM cells expressing the HIV tat gene. Since the reactivity with both OKT4 and OKT4A antibodies was reduced, simple binding of HIV envelope glycoprotein to CD4 at the cell surface could not account for the observed decrease in CD4 expression. In comparison, reactivity of the three cell lines with the fluoresceinated Leu 1 antibody was equivalent. The profiles shown are representative profiles (n = 3) and do not reflect variations in growth rates of the cells at the point of analysis, or the viability of the cells, which in all cases was greater than 98%.
Figure 4.
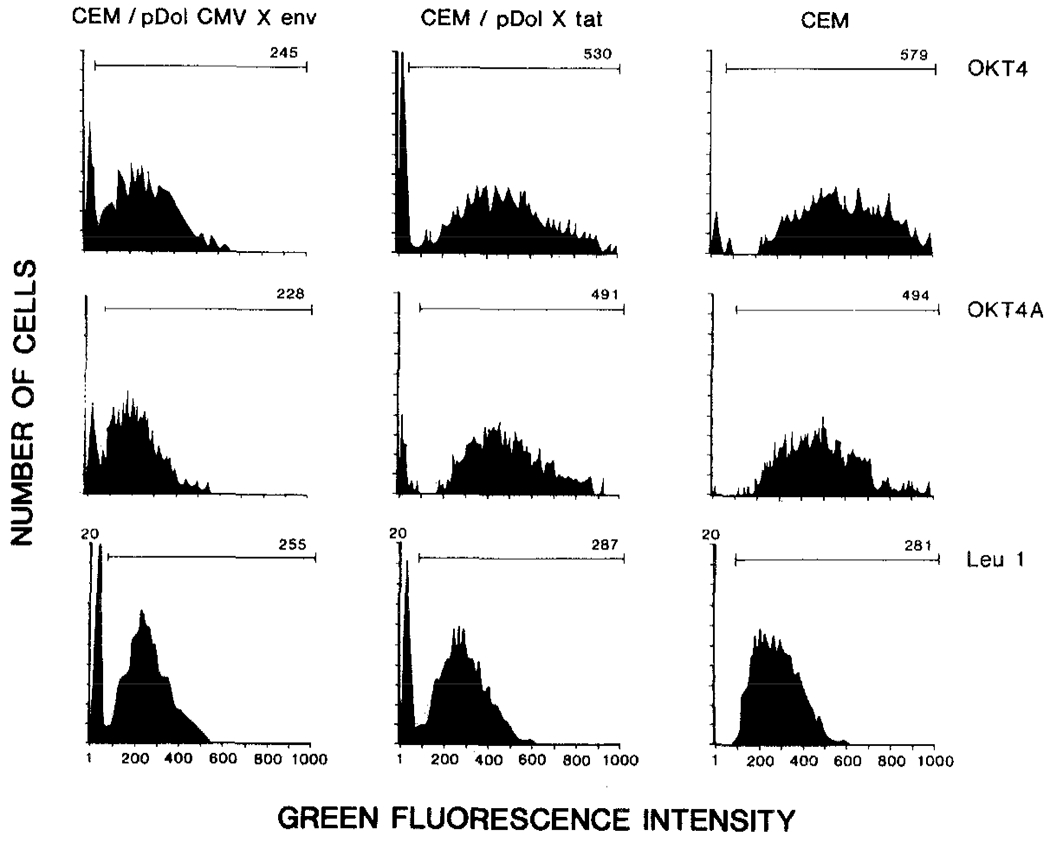
Reduced Surface CD4 Expression in Envelope-Expressing CEM Cells
Immunofluorescence flow cytometry analysis of CD4 expression with OKT4 and OKT4A antibodies. G418 selected cells were incubated with the respective monoclonal antibody (4°C, 30 min) and analyzed on an Ortho model 50H cytofluorograph. The graph displays relative fluorescence intensity versus cell number. Horizontal bar indicates channels analyzed, and mean channel number of cells in this area are indicated on each plot. Background fluorescence (using FITC conjugated second antibody alone) was less than 7%. Plots are representative results of three separate analyses.
Analysis of the configuration of the β-chain of the T cell receptor gene and of the T4 locus in cell lines expressing HIV envelope or tat, or in control CEM cultures, demonstrated identical restriction patterns between the cell lines (Figure 5A). It did not appear that expression of envelope or tat genes followed by G418 selection had resulted in an outgrowth of genetically distinct subclones, thus demonstrating that the reduced CD4 expression in HIV envelope-expressing CEM cells was not caused by the outgrowth of a CD4 receptor-depleted subpopulation that preexisted in the CEM cell line (Folks et al., 1985; Folks et al., 1986). In addition, Northern blot analysis of total cellular RNA demonstrated equivalent amounts of CD4 receptor mRNA when hybridized to a CD4 specific cDNA (Figure 5B). The results, taken together, indicate that reduced CD4 mRNA transcription could not account for the reduced surface CD4 expression in envelope-expressing CEM cell lines.
Figure 5.
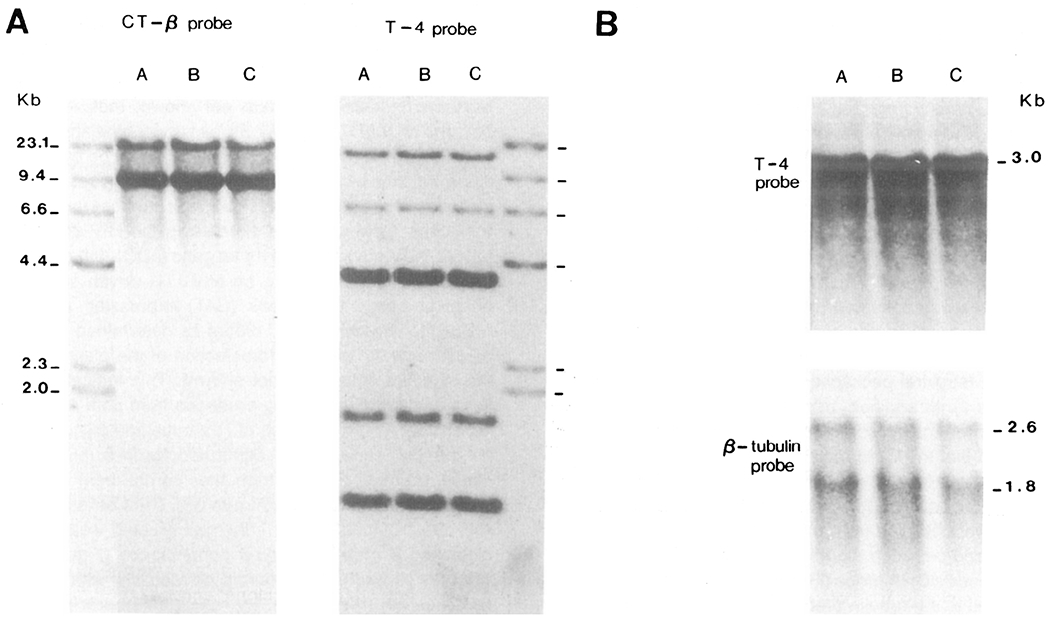
Envelope-Expressing CEM Cells Display Expected T Cell Receptor Gene Rearrangements and Have Abundant CD4 mRNA
(A) A, uninfected CEM cells; B, CEM cells infected with pDOL CMV·env; C, CEM cells infected with pDOL·tat. Cellular DNA was blotted and hybridized to a T cell receptor β-chain probe (Yanagi et al., 1984) or CD4 cDNA (Maddon et al., 1985) as described in Figure 3A.
(B) Northern blot analysis of relative T4 levels was performed by electrophoresis of total cellular RNA (10 μg/lane) on 1% agarose gels followed by transfer to nylon membranes. Blots were hybridized to 32P-labeled CD4 specific cDNA or β-tubulin. Autoradiography times were 36 hr and 4 hr for CD4 and β-tubulin probes, respectively.
Envelope/CD4 Receptor Complexing Accounts for CD4 Modulation
We next analyzed the production of CD4 receptor protein in envelope-expressing CEM cells and in control CEM cells (Figure 6). Immunoprecipitations of 35S-cysteine and 35S-methionine labeled cell lysates were performed using both the OKT4 and OKT4A antibodies. From lysates of HIV envelope–expressing CEM cells (Figure 6, lanes A and B), the OKT4A (lane A) and OKT4 (lane B) both precipitated a 58 kd protein corresponding to the size reported for the CD4 glycoprotein (Terhorst et al., 1980). In addition to the 58 kd band, the OKT4 antibody reacted with two high molecular weight proteins corresponding to an approximate molecular weight of 120 and 160 kd (Figure 6, lane B). These high molecular weight bands reactive with the OKT4 monoclonal antibody are proposed to represent HIV envelope glycoproteins (Hoxie et al., 1986). In support of this, no significant reactivity was observed with the OKT4A monoclonal antibody (Figure 6, lane A). This is to be expected since the OKT4A monoclonal antibody, unlike the OKT4 monoclonal, competes with the viral envelope for binding to the CD4 receptor (McDougal et al., 1985; McDougal et al., 1986). Thus, the two high molecular weight precipitates appear to represent a complex which is formed between the envelope gene expressed in the CEM cells and the CD4 receptor protein. We did not observe any similar complex formation in control CEM cells immunoprecipitated with either OKT4 or OKT4A monoclonal antibody (Figure 6, lanes C and D). Immunoprecipitation analysis of cell culture supernatants from control CEM cells or envelope-expressing CEM cells did not reveal the presence of HIV envelope glycoprotein in abundance nor did it reveal the presence of CD4/envelope complexes in the extracellular medium. We would interpret this as suggesting that little HIV envelope was being shed into the culture supernatant and, as such, the precipitates observed (Figure 6) most probably represent intracellular complexes between the envelope and CD4 glycoproteins rather than extracellular complexes. Thus the reduced CD4 expression in envelope-expressing CEM cells would appear to be a direct consequence of this complex formation between the envelope moiety of HIV and the CD4 receptor in CD4 positive cells. Consequently, one could propose that modulation of surface CD4 expression in this system would be a direct function of the level of HIV envelope attained in respective cell lines using the retroviral vector system described here. The level of HIV envelope expression was sufficient to reduce surface CD4 expression in CEM cells by about 60%. However, there were still available CD4 receptors on the cell surface to mediate HIV infection, allowing us to analyze the relative susceptibility of the envelope-expressing CEM cell line to direct infection with HIV and HIV-mediated cytopathicity.
Figure 6.
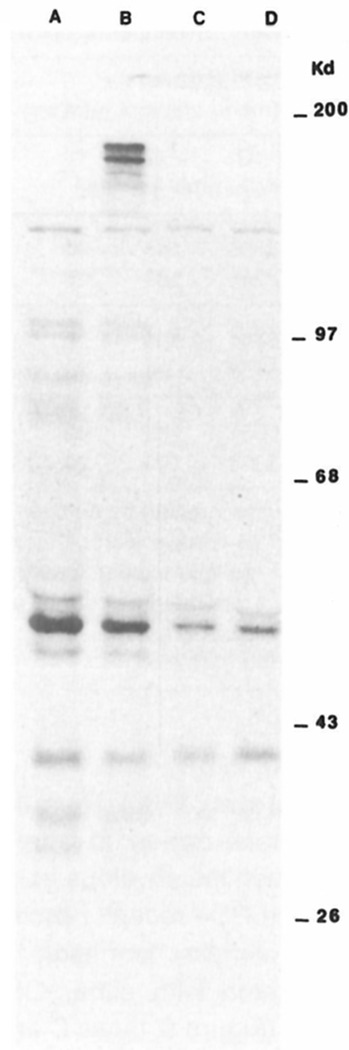
CD4/HIV Envelope Complexes in G418 Selected CEM Cells
CEM cells infected with pDOL CMV·env (A and B) or uninfected CEM cells (C and D) were metabolically labeled with 35S-methionine and 35S-cysteine for 6 hr and immunoprecipitated with OKT4A (A and C) or OKT4 (B and D) monoclonal antibodies as described in Experimental Procedures. Cell lysates were preabsorbed with protein A-Sepharose beads overnight at 4°C and then incubated with monoclonal antibody–treated beads with gentle rotation at 4°C for 12 hr. Typically, 100 μg Sepharose was adsorbed with 200 μl monoclonal antibody and conjugated with extract of 107 cells. Conjugated beads were washed, and immunoprecipitates eluted in sample buffer at 65°C for 30 min. Electrophoresis was performed on a 15% polyacrylamide gel with a 3.5% stacking gel. Molecular weights were provided by prestained protein markers (Bethesda Research Laboratories).
HIV Envelope Induces Cytolysis Resistance
CEM cells expressing either HIV envelope or tat genes and control CEM cells were infected with HIV at a low multiplicity of infection by suspending the CEM cells in virus-containing medium. After 24 hr at 37°C, infected cells were washed and resuspended in fresh medium. At designated time intervals, samples of the culture were removed to determine total cell count, cell viability, HIV antigen expression by the immunofluorescence method, expression of surface CD4 receptor, and viral production by reverse transcriptase activity in culture supernatants.
By seven days postinfection, between 60% and 75% of the cells in each cell line were HIV antigen positive (by immunofluorescence analysis; Table 1). Increase in viral antigen positivity in infected CEM cell lines was accompanied by a dramatic reduction in cell viability in control CEM cells and in CEM cells transfected with the pDOL·tat vector (Table 1). In both cases, the culture had been almost completely wiped out by viral infection at day 18 (Table 1), while reverse transcriptase analysis revealed the presence of high titers of HIV in the supernatants of these infected cultures (Table 1). In contrast, CEM cells transfected with the HIV envelope expression vector were not susceptible to HIV-mediated cytopathic effects. There was no major difference in susceptibility to direct HIV infection when compared with control CEM cultures or tat-expressing CEM cells (Table 1). However, after a slight reduction in cell viability at day 7, the cells had fully recovered by day 18, and despite being greater than 95% HIV antigen positive (Table 1), cell viability was almost 100%. In addition, the reverse transcriptase activity in envelope-expressing CEM cell supernatants was almost an order of magnitude lower than that of the CEM control cells or the tat-expressing CEM cells (Table 1). Thus it appeared that expression of the HIV envelope and concomitant reduction in CD4 receptor expression in the envelope-expressing CEM cell line had conferred a cytolysis-resistant phenotype. Upon infection with HIV, a persistent infection characterized by a sustained release of viral particles was obtained in envelope-expressing CEM cells, rather than a lytic infection as observed in the control CEM cells or the CEM cells expressing the HIV tat gene. Virus harvested from the persistently infected envelope-expressing CEM cell line was fully cytopathic when passaged onto fresh CEM cells. Thus, the presence of the HIV envelope gene in CEM cells had not attenuated the infecting virus, and the resistant phenotype of the envelope-expressing CEM cell was a direct consequence of the host cell phenotype rather than reduced cytotoxic properties of the infecting virus.
Table 1.
Susceptibility of HIV Envelope-Expressing CEM Cell Lines to HIV-Mediated Cytopathic Effects
| CD4 Expressiona (mean channel number) | HIV Antigensb (% cells positive) | Cell Viabilityc (%) | Reverse Transcriptase in Culture Supernatants (counts/ml) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell Line | 0 | 7 | 18 | 0 | 7 | 18 | 0 | 7 | 18 | 0 | 7 | 18 |
| days after infection | ||||||||||||
|
|
||||||||||||
| CEM Infected with pDOL CMV ● env | 240* | 147 | 15 | |||||||||
| 224# | 160 | 25 | 0 | 70 | >95 | >95 | 60 | >97 | 0 | 2–4 × 104 | 3–5 × 105 | |
|
|
||||||||||||
| CEM Control | 540* | 105 | 10 | |||||||||
| 515# | 80 | 20 | 0 | 75 | – | >95 | <35 | <2 | 0 | 1–3 × 105 | 2.5–3 × 106 | |
|
|
||||||||||||
| CEM Infected with pDOL ● tat | 505* | 85 | 30 | |||||||||
| 580# | 100 | 20 | 0 | 60 | – | >95 | <40 | <2 | 0 | 8–10 × 104 | 2–3 × 106 | |
G418 selected cell lines were infected by an over-night incubation in virus containing medium (2 × 105 cells/ml) from CEM producer lines. Titer of virus was standardized as relative dilution that induced HIV antigen positivity in 20%–30% of CEM cells 2 days after infection. Results are representative analysis of multiple experiments (n = 4).
Analyzed by fluorescence activated cell sorter analysis using OKT4* and OKT4A# antibodies.
Analyzed by indirect immunofluorescence using sera from an HIV seropositive hemophiliac with high gag reactivity (titer 1:2580).
Determined by trypan blue exclusion.
HIV Envelope Restricts Second Round Infection
To determine the effects of HIV envelope expression on the relative level of HIV infection, CEM cells carrying the pDOL CMV·env vector and control CEM cells were infected with HIV as outlined in Table 1. At designated time intervals, aliquots of 5 × 106 cells were withdrawn for analysis of unintegrated viral DNA (Hirt extracts) by Southern blotting and expression of viral antigens by immunofluorescence. By day 3 postinfection, unintegrated linear viral DNA of approximately 9.5 kb was evident in uncut Hirt extracts from both infected CEM and CEM·env cell lines (Figure 7). At this time, approximately 35% of the cells in each line were positive for viral antigens. By day 6, postinfection viral antigen positivity was almost 90%, and this was reflected by a dramatic increase in the level of unintegrated linear viral DNA in CEM cells (Figure 7) and to a much lesser extent in the presence of circular unintegrated viral DNA of approximately 5.5 kb (Figure 7). The level of unintegrated viral DNA in control CEM cells was increased further by day 9 postinfection, indicating that reinfection was occurring at this time since by day 6 postinfection cells were already 90% virus antigen positive. In comparison, the accumulation of unintegrated viral DNA in CEM cells expressing the HIV envelope gene was significantly lower than in infected CEM cells. The amount of unintegrated viral DNA was approximately 5-fold less than in control CEM cells by day 6 (Figure 7). In addition, the amount of unintegrated viral DNA was not significantly increased by day 9 postinfection (Figure 7). Levels of unintegrated viral DNA in CEM·env cells at 21 days compared with those at 9 days postinfection. By 44 days postinfection, the amount of unintegrated viral DNA had dropped to extremely low levels, while an abundance of integrated viral DNA was evident in Xbal digests of genomic DNA from CEM·env cells at this time (Figure 7).
Figure 7.
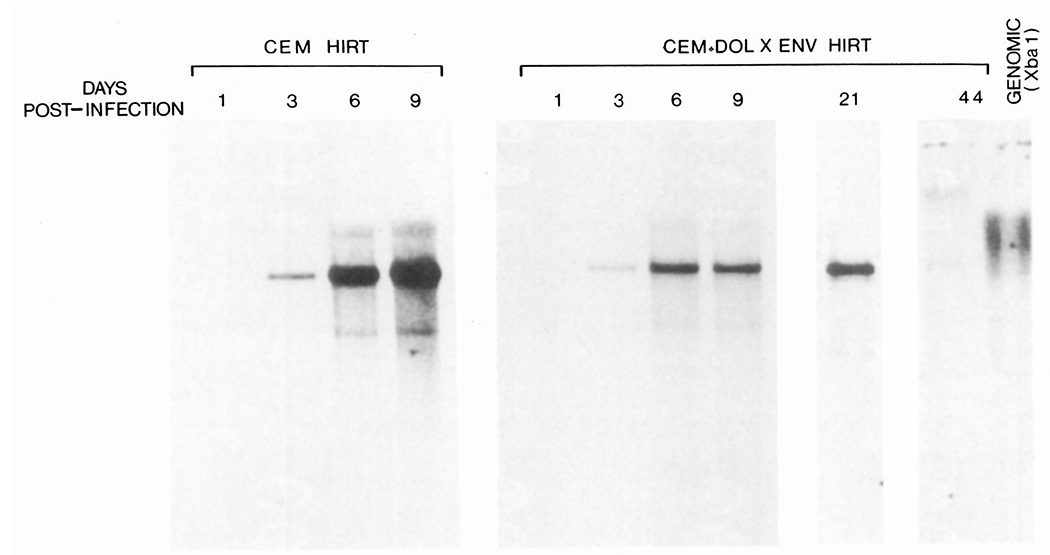
Accumulation of Unintegrated Viral DNA in Infected CEM Cells
At the indicated times following infection, unintegrated viral DNA was analyzed in Hirt extracts from aliquots of 5 × 106 cells. Samples were resolved on 0.8% agarose gels and hybridized with an HIV probe. All Hirt samples were run undigested, while Xbal endonuclease digested genomic DNA from persistently infected CEM·env cells 44 days postinfection is shown for comparison.
Phorbol Ester Induces Virus-Mediated Cytolysis in Persistently Infected CD4 Cells
We next determined whether cytolytic effects could be induced in persistently infected CEM cells following stimulation of virus replication. The tumor-promoting agent phorbol 12-myristate 13-acetate (TPA) has the ability to greatly enhance HIV replication and cytopathic effects (Harada et al., 1986), and operates at the level of transcription by acting on a 10 bp functional enhancer element within the HIV LTR (Kaufman et al., 1987). Persistently infected CEM cells, established following infection of HIV envelope-expressing cells, were washed and resuspended in medium containing 1 ng/ml TPA at 3–5 × 105 cells per ml. This concentration was used since higher doses of TPA were toxic for both infected and uninfected CEM cells. After 24 hr, cells were washed and cultured in fresh medium. Reverse transcriptase assay of culture supernatants demonstrated a 4-fold increase in virus production 24 hr after addition of TPA when compared with untreated cultures (Table 2). This was directly reflected by an almost 4-fold increase in viral RNA as determined by dot blot hybridization of total cellular RNA (Figure 8). Seventy-two hours following the addition of TPA, a dramatic cytopathic effect was observed in approximately 50% of the cells, and by 96 hr, the majority of cells in the culture had completely succumbed to cytolysis (Table 2). The dramatic cytopathic effect was independent of syncytium and no multinucleated giant cells were observable in these cultures. This was not unexpected since the persistently infected cells lacked membrane expression of the CD4 receptor: an absolute prerequisite for syncytium formation. Thus, the induced cytopathicity was a consequence of single cell killing, where death of individual cells correlated with a rapid release of progeny virus as determined by electron microscopic visualization of induced cell cultures (Figure 9). Furthermore, the induction of viral cytopathic effects appeared to be a direct consequence of stimulated virus replication and not an indirect effect of TPA. To investigate this, persistently infected cells were treated with TPA in the presence of α-amanitin. α-amanitin is a specific inhibitor of nuclear RNA polymerase II (Lindell et al., 1970) upon which the promoter elements of retroviruses are dependent. The presence of 1 or 5 βg/ml α-amanitin in the culture medium inhibited TPA-mediated stimulation of viral replication as determined by dot blot quantitation of viral RNA (Figure 8) and reverse transcriptase activity in culture supernatant (Table 2). More importantly, the presence of the α-amanitin (1 μg/ml) also completely inhibited onset of cytopathic effects (Table 2). This indicates that TPA-induced virus replication and concomitant cell lysis were a direct consequence of high level virus replication and not due to secondary effects on membrane stability by TPA.
Table 2.
Induction of Cytolysis in Persistently Infected CD4 Negative CEM Cells
| CD4 Expressiona (hr after TPA addition) | Reverse Transcriptase (in culture supernatant) counts 1 ml:106 | Cell Viability (%) | HIV RNAb (relative induction index) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Treatment | 12 | 72 | 24 | 48 | 24 | 72 | 96 | 12 | 24 |
|
|
|||||||||
| None | 0 | 0 | 0.6–1.0 | 0.5–1.2 | >98 | >95 | >95 | 1 | 1 |
|
|
|||||||||
| TPA 1 ng/ml | 0 | 0 | 2.0–3.5 | 2.5–4.0 | 90–95 | 40–60 | 0–25 | 0.8–1.5 | 2.0–4.7 |
|
|
|||||||||
| TPA 1 ng/ml + α-amanitin 1 μg/ml | 0 | 0 | 0.7–1.0 | 0.7–1.1 | >95 | >90 | >90 | 1 | 1.2–1.5 |
|
|
|||||||||
| α-amanitin 1 μg/ml | 0 | 0 | 0.5–1.0 | 0.2–0.4 | ND | >90 | >90 | 0.9 | 0.4–0.9 |
CEM cells (expressing pDOL CMV ● env) persistently infected with HIV were treated with TPA and/or α-amanitin for 24 hr as described in the legend to Figure 7.
Determined by FACS analysis. Values are 0 since mean fluorescence channels of cells analyzed with OKT4 and OKT4A antibodies were not significantly higher than cells incubated with FITC conjugated IgG alone.
Analyzed by ribonuclease protection. Values are expressed as levels of induction relative to nontreated culture. Ranges reflect values obtained from four separate experiments.
Figure 8.

Effect of TPA on HIV Transcription in Persistently Infected CD4 Cells
Persistently infected CEM cells (expressing pDOL · env CEM) were seeded in 25 cm3 flasks at 5–10 × 105 cells per ml in a total volume of 5 ml. Treatment was initiated by addition of TPA (in DMSO) to a final concentration of 1 ng/ml and/or α-amanitin (in methanol) to a final concentration of 1 μg/ml. After 24 hr at 37°C, total cellular RNA was extracted from the cells by guanidine thiocyanate lysis, and blotted in doubling dilutions onto nitrocellulose. Blots were hybridized with an HIV probe as indicated in Figure 2, or a β-actin probe (Gunning et al., 1983) and autoradiographed for 3 hr at −80°C. Hybridization intensity was determined by scanning densitometry; the induction index is the hybridization intensity relative to the signal obtained with untreated control cells.
Figure 9.
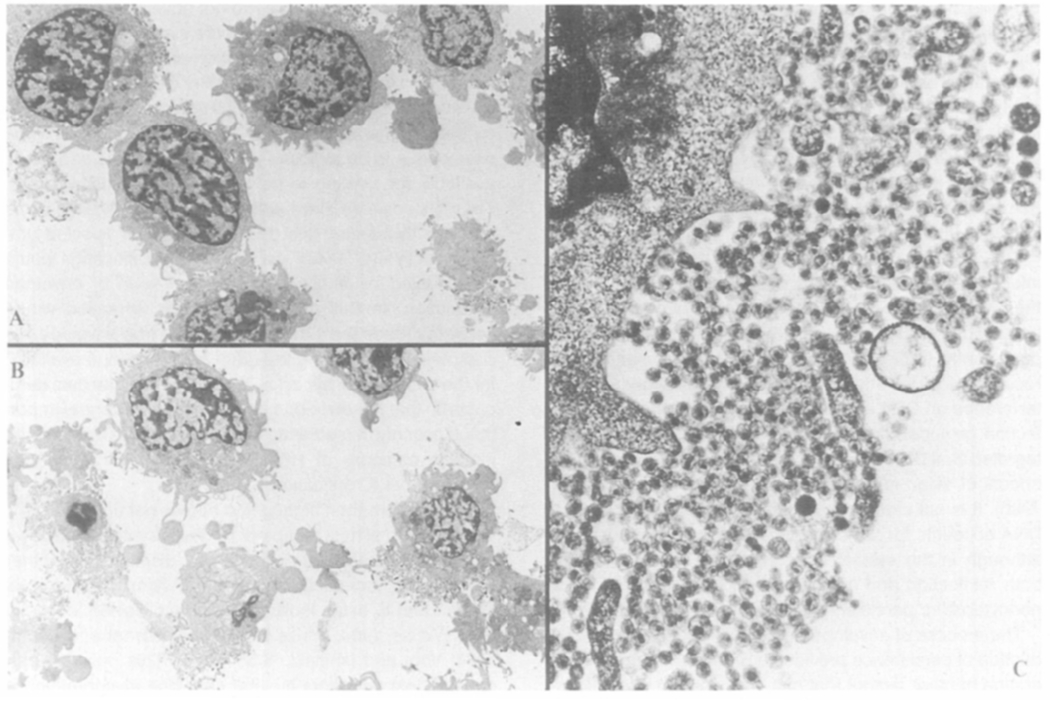
Transmission Electron Micrographs of TPA-Induced Cells Persistently Infected with HIV
pDOL CMV·env (Figure 1) expressing CEM cells persistently infected with HIV before (A) and after (B and C) a 24 hr incubation with TPA as detailed in Figure 8. Cell pellets (approximately 5 × 105 cells) were fixed in 400 μl of picric acid-paraformaldehyde-glutaraldehyde (PPG) postfixed with osmium tetroxide, dehydrated, and embedded in Araldite 6005. Thin sections were stained with uranyl acetate and lead citrate, and examined with a Philips EM 300 electron microscope. Magnifications: 1.5× K (A and B); 15× K (C).
Discussion
The results described here demonstrate that expression of the HIV envelope gene in cytolysis-sensitive CD4 positive cell lines confers a cytolysis-resistant phenotype to the host cell, such that a persistent virus infection, as opposed to rapid cytolysis, ensues following infection with HIV. The results also show that the reduced CD4 receptor expression characteristic of AIDS virus–infected CD4+ cell lines is directly attributable to an alteration by the HIV envelope in the processing and localization of the CD4 glycoprotein at the cell surface. Thus, the observed loss of CD4 receptor expression is an indirect effect of viral infection and replication. In the results described here, we were able to observe a 60% reduction in surface T4 receptor expression without any significant reduction in the amounts of CD4 receptor mRNA. The work by Hoxie et al. (1985) indicated an effect of HIV infection on steady-state levels of CD4 receptor mRNA and postulated that reduced CD4 receptor gene expression was a major mechanism for the loss in CD4 receptor expression following HIV infection. However, we have demonstrated that the reduction in surface CD4 expression is a rapid phenomenon and is not accompanied by a similar rapid reduction in CD4 RNA expression (Stevenson et al., 1987) and, as such, posttranscriptional interference of CD4 receptor localization appears to be the major mechanism of CD4 modulation in HIV infected cell lines.
Expression of the HIV envelope in CD4 positive cell lines, in addition to modulating surface CD4 receptor expression, greatly modifies the host cell’s response to viral cytopathic effects following HIV infection. It is most probable that the acquisition of a cytolysis-resistant phenotype is a direct consequence of the reduced CD4 receptor expression in the envelope-expressing host cell. A correlation does exist between the absolute level of CD4 receptor expression and the host cell’s sensitivity to viral cytopathic effects, such that cell lines expressing high levels of this receptor (for example, HTLV I and HTLV II transformed T cell lines, and Sup T-1 cells) are exquisitely sensitive to the cytolytic effects of HIV, while cells expressing relatively low CD4 receptor levels (CR-10, H9, Jurkat) are more resistant to cytolysis. Our results provide direct evidence for this hypothesis in that an envelope-mediated CD4 receptor reduction is followed by a resistance to HIV-induced cytopathicity. The level of reduction in CD4 expression was modest in our CEM/envelope cell line (60%); this reduction was, however, sufficient to modify greatly the host cell phenotype.
A direct consequence of this envelope-mediated CD4 depletion appears to be a restriction on the kinetics of HIV infection and reinfection. Following infection of susceptible cells by HIV, high levels of unintegrated viral DNA accumulate within the cell. In control CEM cells that were already fully virus antigen positive, additional increases in the relative amounts of unintegrated viral DNA were evident, thus demonstrating that reinfection was taking place in this cell system. In comparison, the accumulation of unintegrated viral DNA was greatly reduced in CEM cells expressing the envelope glycoprotein, and since viral antigen positivity was not affected, this indicates a restriction imposed upon viral reinfection.
The absence of unintegrated viral DNA in persistently infected CEM cells (44 days postinfection, Figure 7) further demonstrates that reinfection does not occur in a full persistent infection. This is not unexpected since these persistently infected cells do not have available CD4 receptors for HIV infection due to envelope-mediated interference on CD4 expression. The susceptibility to reinfection or superinfection and the accumulation of unintegrated viral DNA has been correlated with the cytopathic effects of avian leukosis virus subgroups (Weller et al., 1980). It is not clear if accumulation of unintegrated viral DNA accounts for the cytopathic effects of HIV infection, although in the system described here a restriction on both reinfection and accumulation of viral DNA led to a noncytopathic persistent infection.
The process of envelope-mediated interference and induction of persistence provides a precedent for the cooperation of other factors that can contribute to viral persistence in AIDS. The ability of the product of the F (3′ orf) gene to reduce CD4 expression has been demonstrated (Guy et al., 1987). It is plausible that the combined effects of the envelope and F gene products serve to augment the down modulation in CD4. CD4 modulation is not a property restricted to HIV gene products, but is displayed by gangliosides (a ubiquitous component of vertebrate cell membranes; Offner et al., 1987) and lymphocyte chemoattractant factor, LCF (W. Cruickshank, personal communication). The contribution of these agents to viral interference and the initiation of persistence remains to be established, although such a contribution would be of central importance in understanding the pathophysiology of latent HIV infections.
The underlying mechanism for the ability of cells expressing reduced CD4 receptors to resist viral cytopathic effects is a matter for discussion. However, we propose that HIV-mediated cytopathic effects are a direct consequence of virus replication and are not dependent upon syncytium formation (Lifson et al., 1986; Sodroski et al., 1986b). An abundance of CD4 receptors during the initial stages of viral infection provide the conditions for reinfection of the host cell by newly released progeny virus, thus initiating superinfection. The kinetics of the initial infection appear to be the important factor in determining whether a lytic or persistent type infection will be initiated. Thus, following initial infection, if CD4 receptor expression were sufficiently reduced due to envelope/CD4 complex formation, then the paucity of CD4 receptors on the cell surface would restrict second round infection with progeny virus. Noteworthy is that in our cell line, which was stably expressing HIV envelope, we did not observe a widespread formation of syncytia. One possible explanation is that the complex formation between envelope and CD4 receptor not only limits the localization of the CD4 receptor on the cell surface, but limits the localization of the HIV envelope on the cell membrane, and this must be achieved if the envelope is to be exposed on the cell surface and made available for binding to neighboring CD4 positive cells. Thus it is possible that a similar situation is initiated in the persistently infected host cell. In persistently infected lymphocytes in vivo, a very low level of HIV replication would be reflected by a concomitantly low level of envelope production. In this instance, complex formation would serve to reduce the intracellular pool of free envelope glycoprotein, thus limiting the amount of envelope available for the formation of syncytia. Taken together, the data demonstrate that the envelope gene of HIV is of central importance not only in mediating viral infection, but in determining the outcome of HIV infection, i.e., a lytic versus persistent viral replication.
The phenomenon highlighted here is not unique to HIV. The masking of host receptors by newly synthesized envelope glycoproteins appears to be displayed by other retroviruses, including the human T lymphotropic virus Types I and II, avian leukosis virus, and bovine leukemia virus (Weiss, 1984; Weiss et al., 1985; Dorner and Coffin, 1986; Vogt and Ishizaki, 1965, 1966). This masking process of host receptors by viral envelope glycoproteins is collectively termed interference and leads to the establishment of superinfection resistance (Weller et al., 1980). We demonstrate here that the induction of superinfection resistance by the HIV envelope gene is accompanied by a reduced sensitivity to HIV-mediated cytolytic effects. Thus this phenomenon has important implications for both the pathogenesis of and treatment strategies for AIDS. Superinfection resistance and the initiation of a persistent infection may be the underlying basis for viral persistence in vivo following initial exposure to HIV. The AIDS complex is characterized by a long incubation period, whereby HIV is harbored in a minimally replicative state in CD4 lymphocytes. The mechanism by which high level virus replication is initiated in persistently infected cells during the course of AIDS remains to be established. However, such a stimulus of the T cell may be provided by some change in the immunological status of the infected individual (Nabel and Baltimore, 1987; Zagury et al., 1986) or by the removal of a block on viral transactivation, thus leading to renewed high level viral replication and subsequent cell death. Our results provide evidence for this hypothesis. A cytopathic response ensues following initiation of a high level virus replication in persistently infected cells, despite the absence of CD4 and syncytium formation. This suggests that cytopathicity may be due to a cytotoxic viral gene product that manifests its effects only when expressed at very high levels. An ideal candidate for this toxic product would be the envelope glycoprotein. A high concentration of envelope molecules on the cell membrane may destabilize membrane structure in a syncytium independent fashion. Experiments are in progress to evaluate this possibility directly.
The CD4 receptor itself has received considerable attention since strategies for approaches to AIDS therapy may involve the design of agents that interfere with viral binding to the CD4 epitope (Lasky et al., 1987; Sattentau et al., 1986; Kowalski et al., 1987) or modulation of the expression of the CD4 receptor itself. In view of the data presented here, the phenomenon of interference must be taken into consideration. Interference with viral infection provides the conditions for the initiation of a persistent infection such that persistently infected host cells provide a slow release reservoir for the continued generation of HIV in the infected individual.
Experimental Procedures
Construction of Retrovirus Envelope Expression Vector
Standard recombinant techniques were used to construct the vector described. The retrovirus vector used (pDOL or pLJ; Korman et al., 1987) is an MLV-based packaging defective retrovirus which has had 5′ and 3′ splice donor/acceptor sequences removed. To ensure efficient expression of HIV envelope in human lymphoid cells, the CMV immediate early promoter (Boshart et al., 1985) was used as the promoter for expression of HIV sequences. The CMV promoter was excised as an 855 bp Stul–HindIII fragment from plasmid pBC12 CMV IL2 (Cullen, 1986). After conversion of Stul and HindIII sites to BamHI using synthetic linkers (New England Biolabs), the CMV promoter was inserted into the BamHI site of pDOL lying immediately 3′ of the viral packaging sequences (ψ). HIV envelope coding sequences were derived from an almost full-length proviral clone (λNIG-F; Volsky et al., 1986) as a 3.1 kb Sall–Xhol fragment, and inserted into the Sall site of pDOL immediately 3′ of the CMV promoter. The 3.1 kb Sall–Xhol fragment of N1G-F also contains the viral regulatory genes tat (Sodroski et al., 1985b), and art/trs (Sodroski et al., 1986a; Feinberg et al., 1986). Initial attempts to express envelope in the absence of tat and art/trs genes resulted in very low envelope production, thus necessitating their inclusion in the envelope expression vector. The pDOL·tat vector (data not shown) was derived by subcloning a 2700 bp EcoRI–BamHI fragment of N1G-F into the BamHI site of pDOL after conversion of the EcoRI site to BamHI. In this vector, the viral 5′ LTR is utilized as the promoter element for expression of tat sequences.
Cell Culture and Retrovirus Packaging and Infection
CEM cells (human leukemic CD4+ cell line) and PA317 cells (Amphotropic retrovirus packaging cell line; Miller and Buttimore, 1986) were maintained at 37°C in an atmosphere of 5% CO2 in RPMI 1640 and DMEM (GIBCO, Grand Island, NY), respectively, supplemented with 10% fetal bovine serum, 7 mM L-glutamine, 2 mM HEPES (N-2-hydroyethyl piperazine-N’-2-ethanesulfonic acid), penicillin (100 μg/ml), and streptomycin (100 μg/ml). For packaging of retrovirus vectors, 1–2 × 106 PA317 cells were plated onto a 60 mm culture flask 1 day prior to transfection with 10 μg vector DNA using calcium phosphate coprecipitation. Two days after transfection, medium in the flasks was removed and replaced with 5 ml RPMI 1640 containing 2 × 106 CEM cells and 8 μg/ml polybrene. After a 12 hr cocultivation, CEM cells were washed off the PA317 monolayer and cultured in RPMI 1640 containing 1 mg/ml G418 (GIBCO; approximately 50% active). Residual PA317 cells harvested with the CEM cells were allowed to adhere to the flask, allowing the removal of CEM cells to fresh flasks. G418 selection continued for 4 weeks before analysis of selected cell phenotype.
RNA/DNA Isolation and Analysis
Preparation of Cellular DNA and RNA
Total cellular RNA was prepared by the CsCI-guanidine thiocyanate procedure (Chirgwin et al., 1979). Cellular RNA, which pelleted through the CsCI cushion, was washed twice with 200 μl of absolute alcohol at room temperature, suspended in 400 μl of diethyl pyrocarbonate-treated H2O, and precipitated with 2 volumes of absolute ethanol at −20°C in the presence of 0.3 M sodium acetate (pH 5.2). Cellular DNA remaining at the CsCl/guanidium thiocyanate interface was diluted with 2 volumes of Tris-EDTA, extracted once with an equal volume of phenol-chloroform-isoamyl alcohol (25:24:1, vol/vol/vol) and once with an equal volume of chloroform-isoamyl alcohol (24:1, vol/vol), and precipitated with 5 volumes of 80% alcohol at −20°C.
Preparation of Unintegrated Viral DNA
Unintegrated HIV DNA was isolated by the method of Hirt (1967) with modifications of Chinsky and Soeiro (1981). Briefly, aliquots of 5 × 106 cells were lysed at 65°C in 0.6% SDS, and Hirt supernatants were extracted twice with phenol, once with chloroform, precipitated with ethanol, and analyzed by Southern blotting.
Northern and Southern Blot Analysis
RNA (10 μg per tube) was denatured in 50% formamide, 6% formaldehyde, 20 mM MOPS (morpholine-propanesulfonic acid, pH 7.0), 5 mM sodium acetate, 1 mM EDTA for 15 min at 55°C, chilled on ice, and fractionated by electrophoresis in 1% agarose gels containing 6% formaldehyde, 20 mM MOPS (pH 7.0), 5 mM sodium acetate, and 1 mM EDTA for 16 hr at 45 V. The gels were treated in 0.05 N NaOH–0.6 M NaCl for 20 min, followed by soaking in 0.5 M Tris-HCl (pH 7.5), 0.6 M NaCl for 45 min. RNA was transferred to Nytran nylon membranes (Schleicher and Schuell, Keene, NH) in 20× SSC (1× SSC is 0.015 M sodium citrate plus 0.15 M NaCl). For Northern (RNA) blot hybridizations, RNA blots were prehybridized for 4 hr at 42°C in a solution containing 5× SSC, 50% formamide, 3× Denhardt’s reagent, 0.25% SDS, and 100 μg of denatured salmon sperm DNA per ml. Hybridization continued for 24 hr in hybridization cocktail (as above) supplemented with 1 × 106 to 2 × 106 cpm of 32P-labeled DNA probe per ml (specific activity, 2 × 108 to 5 × 108 cpm/μg). Hybridized filters were washed with brisk agitation in three changes of 0.2× SSC, 0.1% SDS at 55°C to 60°C. The washed filters were blotted dry, wrapped in Saran Wrap cling film, and subjected to autoradiography with Kodak X-Omat intensifying screens at −80°C.
Restriction endonuclease-digested DNA (10 μg) was electrophoresed in 0.8% Tris-borate (pH 8.0) agarose gels and transferred to Nytran nylon filters by the procedure of Southern. Filters were prehybridized and hybridized at 42°C in a solution containing 5× SSC, 50% formamide, 1% SDS, 3× Denhardt’s solution, 100 μg of denatured salmon sperm DNA per ml, and 5 mM EDTA (pH 8.0). Blots were washed as described for Northern blots. Bacteriophage λDNA digested with HindIII (New England Biolabs, Beverly, MA) and end labeled with DNA polymerase I large fragment was used as size markers.
RNA Slot Blots
Cesium chloride fractionated RNA samples were denatured as detailed for Northern analysis, and a dilution series prepared in 10× SSC. Samples were blotted on nitrocellulose membranes using a slot blot template (Schleicher and Schuell, Keene, NH). The filters were baked and hybridized as outlined for Northern blotting.
Immunofluorescence Staining
Infected cells were washed in PBS, spotted on glass slides, dried, and fixed in acetone at −20°C for 15 min. The fixed cells were reacted with HIV-positive serum from an asymptomatic HIV seropositive hemophiliac (antibody titer 1:5160). After a 30 min incubation at 37°C, cells were reacted with fluorescein isothiocyanate-conjugated goat anti-human immunoglobulin G, and virus antigen-positive cells were counted under an epifluorescence microscope (American Optical, Buffalo, NY).
Cell Surface Marker Analysis
Cell samples (5 × 105 total) were washed with PBS and incubated at 4°C for 30 min in the presence of unlabeled monoclonal T cell markers OKT4A, OKT4 (Ortho), or Leu 1 (Becton-Dickinson) followed by fluorescein-conjugated second antibody. Cell fluorescence was examined with an Ortho model 50H cytofluorograph.
Immunoprecipitation
Cell lines were washed twice with PBS and labeled for 6 hr in RPMI medium (1–2 × 106 cells per ml) containing 5% dialyzed calf serum and 50 to 100 μCi each of L-[35S]methionine and cysteine per ml (1000 μCi/mmol; New England Nuclear, Boston, MA). At the end of the labeling period, cells were washed with PBS containing 0.2 mM phenylmethylsulfonyl fluoride at 4°C. Cell pellets (107 cells) were incubated in 1 ml of immunoprecipitation buffer for 30 min at 4°C and centrifuged at 3,000 μ g for 20 min to remove nuclei. Immunoprecipitation buffer contained 0.02 M Tris, 0.12 M NaCl (pH 8.0), with 0.2 mM phenylmethylsulfonyl fluoride, 5 μg of aprotinin per ml, 0.2 mM EGTA, 0.2 mM sodium fluoride, 0.2% sodium deoxycholate, and 0.5% (vol/vol) Nonidet P-40. Cell lysates were preabsorbed with protein A-Sepharose beads (Pharmacia, Piscataway, NJ) overnight at 4°C and then incubated with monoclonal antibody OKT4A-treated beads with gentle rotation at 4°C for 12 hr. Typically, 100 μg of Sepharose beads were absorbed with 100 to 200 μl of monoclonal antibodies and conjugated with extract of 107 cells. Conjugated beads were washed once with immunoprecipitation buffer, once with buffer containing 0.5 M NaCl, and once in buffer containing 0.1% SDS. Washed beads were suspended in 50 μl of electrophoresis sample buffer (0.01 M Tris, pH 8.0), 2% SDS, 5% 2-mercaptoethanol, 25 μg of bromophenol blue per ml, 10% glycerol. Adsorbed material was eluted by heating beads at 65°C for 30 min. Electrophoresis was performed in a 15% polyacrylamide gel with a 3.5% stacking gel. Gels were fixed, dried, and autoradiographed with Kodak X-Omat intensifying screens. Protein molecular weight markers were myosin (200,000), phosphorylase b (97,400), bovine serum albumin (68,000), ovalbumin (43,000), α-chymotrypsin (25,700), β-lactoglobulin (18,400), and lysozyme (14,300) (Bethesda Research Laboratories, Gaithersburg, MD).
Virus Propagation and Virus Infections
The LAV-N1T strain of HIV (Casareale et al., 1987) was maintained and purified as detailed previously (Stevenson et al., 1987). Cells harvested at the exponential growth phase were infected by addition of virus containing medium (1 × 106 RT U/ml) at a 1:10 dilution (106 cells/per ml). After 24 hr at 37°C, cells were washed and resuspended in fresh medium at approximately 2 × 105 cells per ml. At designated time intervals, culture samples were removed to determine cell count, viability (trypan blue exclusion), HIV antigen expression (immunofluorescence assay), and reverse transcriptase activity in culture supernatants. The moi used typically results in HIV antigen expression in 10%–20% of CEM cells 2 days postinfection.
Solution RNA Hybridizations
Quantitation of HIV RNA in infected cells was performed by a modified ribonuclease protection assay essentially as described elsewhere (Pellegrino et al., 1987). High specific activity single-stranded antisense HIV RNA probes were generated using T7 polymerase from a HindIII linearized transcription (pGEM; Promega) plasmid containing the HIV pol gene. Probe was purified by passage over a 1 ml Sephadex G50–150 column and hybridized to infected cell lysates in 5 M guanidine thiocyanate under conditions of probe excess.
Acknowledgments
We thank Richard Mulligan for providing the pLJ vector, Bryan Cullen for the pBC 12 CMV IL2 vector, Jonathan Goldsmith for the hemophiliac patient sera, W. Cruickshank for providing information regarding activity of LCF on CD4 expression, Solon Rhode for critical reading of this manuscript and helpful discussion, Charles Kuszynski for fluorescence cell sorter analysis, Richard Wilson for electron micrographs, David Crouse for TPA, and Thais C. Chaudry for preparation of the manuscript. This work was supported by a grant from the American Foundation for AIDS Research (AmFAR) to M. S. and a grant from the Nebraska Eagles of Cancer.
References
- Barre-Sinoussi F, Chermann JC, Rey R, Nugeyre MT, Chamaret S, Gruest J, Dauget C, Axler-Blin C, Brun-Vezinet F, Rouzioux C, Rosenbaum W, and Montagnier L (1983). Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome. Science 220, 868–871. [DOI] [PubMed] [Google Scholar]
- Boshart M, Weber F, Jahn G, Dorsch-Häsler K, Fleckenstein B, and Schaffner W (1985). A very strong enhancer is located upstream of an immediate early gene of human cytomegalovirus. Cell 41, 521–530. [DOI] [PubMed] [Google Scholar]
- Casareale D, Stevenson M, Sakai K, and Volsky DJ (1987). A human T-cell line resistant to cytopathic effects of the human immunodeficiency virus (HIV). Virology 156, 40–49. [DOI] [PubMed] [Google Scholar]
- Chinsky J, and Soeiro R (1981). Fv-1 host restriction of friend leukaemia virus: analysis of unintegrated proviral DNA. J. Virol 40, 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirgwin JM, Pryzbyla AE, MacDonald RJ, and Rutter WJ (1979). Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry 18, 5294–5299. [DOI] [PubMed] [Google Scholar]
- Cullen BR (1986). Trans-activation of human immunodeficiency virus occurs via a bimodal mechanism. Cell 46, 973–982. [DOI] [PubMed] [Google Scholar]
- Dalgleish A, Beverley P, Clapham P, Crawford D, Greaves M, and Weiss R (1984). The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 312, 763–766. [DOI] [PubMed] [Google Scholar]
- Dorner AJ, and Coffin JM (1986). Determinants for receptor interaction and cell killing on the avian retrovirus glycoprotein gp85. Cell 45, 365–374. [DOI] [PubMed] [Google Scholar]
- Feinberg MB, Jarrett RF, Aldovini A, Gallo RC, and Wong-Staal F (1986). HTLV III expression and production involve complex regulation at the levels of splicing and translation of viral RNA. Cell 46, 807–817. [DOI] [PubMed] [Google Scholar]
- Folks T, Benn S, Rabson A, Theodore T, Hoggan MO, Martin M, Lightfoote M, and Sell K (1985). Characterization of acontinuous T-cell line susceptible to the cytopathic effects of the acquired immunodeficiency syndrome (AIDS)-associated retrovirus. Proc. Natl. Acad. Sci. USA 82, 4539–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folks T, Powell DM, Lightfoote M, Bena S, Martin MA, and Fauci AS (1986). Induction of HTLV-III/LAV from a nonvirus-producing T-cell line: implications for latency. Science 231, 600–602. [DOI] [PubMed] [Google Scholar]
- Gallo R, Salahuddin S, Popovic M, Shearer Q, Kaplan M, Haynes B, Palker T, Redfield R, Oleske J, Safai B, White G, Foster P, and Markham P (1984). Frequent detection and isolation of cytoplasmic retroviruses (HTLV III) from patients with AIDS and at risk for AIDS. Science 224, 200–203. [DOI] [PubMed] [Google Scholar]
- Gottleib MS, Schroff R, Schanker HM, Weisman J, Fan PT, Wolf RA, and Saxon A (1981). Pneumocystis carinnii pneumonia and mucosal candidiasis in previously healthy homosexual men: evidence of a new acquired cellular immunodeficiency. N. Engl. J. Med 305, 1426–1431. [DOI] [PubMed] [Google Scholar]
- Gunning P, Ponte P, Okayama H, Engel J, Blau H, and Kedes L (1983). Isolation and characterization of full-length cDNA clones for human α-, β-, γ-Actin mRNAs: skeletal but not cytoplasmic actins have an amino-terminal cysteine that is subsequently removed. Mol. Cell. Biol 3, 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy B, Kieny MP, Riviere Y, LePeuch C, Dott K, Girard M, Montagnier L, and Lecocq JP (1987). HIV F/3′ orf encodes a phosphorylated GTP-binding protein resembling an oncogene product. Nature 330, 266–269. [DOI] [PubMed] [Google Scholar]
- Hall JL, Dudley L, Dobner PR, Lewis SA, and Cowan NJ (1983). Identification of two human β-tubulin isotypes. Mol. Cell Biol 3, 854–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada S, Koyanagi Y, Nakashima H, Kobayashi N, and Yamamoto N (1986). Tumor promoter, TPA, enhances replication of HTLV III/ LAV. Virology 154, 249–258. [DOI] [PubMed] [Google Scholar]
- Hirt B (1967). Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol 26, 365–369. [DOI] [PubMed] [Google Scholar]
- Hoxie JA, Haggarty BS, Rackowski JL, Pillsbury N, and Levy JA (1985). Persistent noncytopathic infection of normal human T lymphocytes with AIDS-associated retrovirus. Science 229, 1400–1402. [DOI] [PubMed] [Google Scholar]
- Hoxie JA, Alpers JD, Rackowski JL, Huebner K, Haggarty BS, Cedarbaum AJ, and Reed JC (1986). Alterations in T4 (CD4) protein and mRNA synthesis in cells infected with HIV. Science 234, 1123–1127. [DOI] [PubMed] [Google Scholar]
- Kaufman JD, Valandra G, Roderiquez G, Bushar G, Giri C, and Norcross MA (1987). Phorbol ester enhances human immunodeficiency virus-promoted gene expression and acts on a repeated 10-base pair functional enhancer element. Mol. Cell. Biol 7, 3759–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikukawa R, Koyanagi Y, Harada S, Kobayashi N, Hatanaka M, and Yamamoto N (1986). Differential susceptibility to the acquired immunodeficiency syndrome retrovirus in cloned cells of human leukemic T-cell line Molt-4. J. Virol 57, 1159–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klatzmann D, Champagne E, Chamaret S, Gruest J, Gustard D, Hercend T, Gluckman JC, and Montagnier L (1984a). T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature 312, 767–768. [DOI] [PubMed] [Google Scholar]
- Klatzmann D, Barre-Sinoussi F, Nugeyre MT, Dauget C, Vilmer E, Griscelli C, Brun-Vezinet F, Rouzoux C, Gluckman JL, Chermann JC, and Montagnier L (1984b). Selective tropism of lymphadenopathy associated virus (LAV) for helper-inducer T lymphocytes. Science 225, 59–63. [DOI] [PubMed] [Google Scholar]
- Knight DM, Flomerfelt FA, and Ghrayeb J (1987). Expression of the art/trs protein of HIV and study of its role in viral envelope synthesis. Science 236, 837–840. [DOI] [PubMed] [Google Scholar]
- Korman AJ, Frantz JO, Strominger JL, and Mulligan RC (1987). Expression of human class II major histocompatibility complex antigens using retrovirus vectors. Proc. Natl. Acad. Sci. USA 84, 2150–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski M, Potz J, Basiripour L, Dorfman T, Goh WC, Terwilliger E, Dayton A, Rosen C, Haseltine W, and Sodroski J (1987). Functional regions of the envelope glycoprotein of human immunodeficiency virus type 1. Science 237, 1351–1355. [DOI] [PubMed] [Google Scholar]
- Lasky LA, Nakamura G, Smith DH, Fennie C, Shimasaki C, Patzer E, Berman P, Gregory T, and Capon DJ (1987). Delineation of a region of the human immunodeficiency virus type 1 gp120 glycoprotein critical for interaction with the CD4 receptor. Cell 50, 975–985. [DOI] [PubMed] [Google Scholar]
- Levy J, Hoffman AD, Kramer SM, Landis JA, Shimabukuro JM, and Oskiro LS (1984). Isolation of lymphocytopathic retroviruses from San Francisco patients with AIDS. Science 225, 840–842. [DOI] [PubMed] [Google Scholar]
- Lifson JD, Feinberg MB, Reyes GR, Rabin L, Banapour B, Chakrabarti S, Moss B, Wong-Staal F, Steimer KS, and Engleman EG (1986). Induction of CD4-dependent cell fusion by the HTLV-III/LAV envelope glycoprotein. Nature 323, 725–728. [DOI] [PubMed] [Google Scholar]
- Lindell TJ, Weinberg F, Morris PW, Roeder RG, and Rutter WJ (1970). Specific inhibition of nuclear RNA polymerase II by α-amanitin. Science 170, 447–449. [DOI] [PubMed] [Google Scholar]
- Maddon PJ, Littman DR, Godfrey M, Maddon DE, Chess L, and Axel R (1985). The isolation and nucleotide sequence of a cDNA encoding the T cell surface protein T4: a new member of the immunoglobulin gene family. Cell 42, 93–104. [DOI] [PubMed] [Google Scholar]
- Masur H, Michelis MA, Green JB, Onorato I, Vande Stouwe RA, Holzman RS, Wormser G, Brettman L, Lange M, Murray HW, and Cunningham-Rundles S (1981). An outbreak of community-acquired pneumocystis carinii pneumonia: initial manifestation of cellular immune dysfunction. N. Engl. J. Med 305, 1431–1438. [DOI] [PubMed] [Google Scholar]
- Mathews TJ, Weinhold KJ, Lyerly HK, Langlois AJ, Wigzell H, and Bolognesi DP (1987). Interaction between the human T-cell lymphotropic virus type IIIB envelope glycoprotein gp120 and the cell surface antigen CD4: role of carbohydrate in binding and cell fusion. Proc. Natl. Acad. Sci. USA 84, 5424–5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDougal J, Mawle A, Cort S, Nicholson J, Cross G, Shepple-Campbell J, Hicks D, and Sligh J (1985). Cellular tropism of the human retrovirus HTLV III/LAV. 1. Role of T cell activation and expression of the T4 antigen. J. Immunol 135, 3151–3162. [PubMed] [Google Scholar]
- McDougal JS, Kennedy MS, Sligh JM, Cort SP, Mawle A, and Nicholson JKA (1986). Binding of HTLV-III/LAV to T4+ T cells by a complex of the 110K viral protein and the T4 molecule. Science 231, 382–385. [DOI] [PubMed] [Google Scholar]
- Miller AD, and Buttimore C (1986). Redesign of retrovirus packaging cell lines to avoid recombination leading to helper virus production. Mol. Cell. Biol 6, 2895–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabel G, and Baltimore D (1987). An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 326, 711–713. [DOI] [PubMed] [Google Scholar]
- Offner H, Thieme T, and Vandenbark AA (1987). Gangliosides induce selective modulation of CD4 from helper T lymphocytes. J. Immunol 139, 3295–3305. [PubMed] [Google Scholar]
- Pellegrino MG, Lewin M, Meyer WA, Lanciotti RS, Bhaduri-Hauck L, Volsky DJ, Sakai K, Folks TM, and Gillespie D (1987). A sensitive solution hybridization technique for detecting RNA in cells: application to HIV in blood cells. Biotechniques 5, 452–459. [Google Scholar]
- Sattentau QJ, Dalgleish AG, Weiss RA and Beveriy PCL (1986). Epitopes of the CD4 antigen and HIV infection. Science 234, 1120–1123. [DOI] [PubMed] [Google Scholar]
- Sodroski JG, Rosen CR, Wong-Staal F, Popovic M, Arya S, Gallo RC, and Haseltine WA (1985a). Trans-acting transcriptional activation of the long terminal repeat of human T cell leukemia virus type III (HTLV-III). Science 227, 171–173. [DOI] [PubMed] [Google Scholar]
- Sodroski J, Patarca R, Rosen C, Wong-Staal F, and Haseltine W (1985b). Location of the trans-activating region on the genome of human T-cell lymphotropic virus type III. Science 229, 74–77. [DOI] [PubMed] [Google Scholar]
- Sodroski J, Goh WC, Rosen C, Dayton A, Terwilliger E, and Haseltine W (1986a). A second posttranscriptional trans-activator gene required for HTLV III replication. Nature 321, 412–417. [DOI] [PubMed] [Google Scholar]
- Sodroski J, Goh WC, Rosen C, Campbell K, and Haseltine WA (1986b). Role of the HTLV-III/LAV envelope in syncytium formation and cytopathicity. Nature 322, 470–474. [DOI] [PubMed] [Google Scholar]
- Somasundaran M, and Robinson HL (1987). A major mechanism of human immunodeficiency virus-induced cell killing does not involve cell fusion. J. Virol 61, 3114–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson M, Zhang X, and Volsky DJ (1987). Downregulation of cell surface molecules during noncytopathic infection of T cells with human immunodeficiency virus. J. Virol 61, 3741–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terhorst C, van Agthoven A, Reinherz E, and Schlossman S (1980). Biochemical analysis of human T lymphocyte differentiation antigens T4 and T5. Science 209, 520–521. [DOI] [PubMed] [Google Scholar]
- Vogt PK, and Ishizaki R (1965). Reciprocal patterns of genetic resistance to avian tumor viruses in two lines of chickens. Virology 26, 664–678. [DOI] [PubMed] [Google Scholar]
- Vogt PK, and Ishizaki R (1966). Patterns of viral interference in the avian leukosis and sarcoma complex. Virology 30, 368–374. [DOI] [PubMed] [Google Scholar]
- Volsky DJ, Sakai K, Stevenson M, and Dewhurst S (1986). Retroviral etiology of the acquired immune deficiency syndrome. AIDS Res. 2, S35–S48. [PubMed] [Google Scholar]
- Weiss RA (1984). Experimental biology and assay of RNA tumor viruses. In RNA Tumor Viruses, Volume 1, Second Edition, Weiss R, Teich N, Varmus H, and Coffin J, eds. (Cold Spring Harbor, New York: Cold Spring Harbor Laboratory; ), pp. 209–260. [Google Scholar]
- Weiss RA, Clapham P, Nagy K, and Hosino H (1985). Envelope properties of human T-cell leukemia viruses. Curr. Top. Microbiol. Immunol 115, 235–246. [DOI] [PubMed] [Google Scholar]
- Weller SK, Joy AE, and Temin HM (1980). Correlation between cell killing and massive second round superinfection by members of subgroups of avian leukosis virus. J. Virol 33, 494–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagi Y, Yoshikai Y, Legget K, Clark SP, Aleksander I, and Mak TW (1984). A human T-cell specific cDNA clone encodes a protein having extensive homology to immunoglobulin chains. Nature 308, 145–149. [DOI] [PubMed] [Google Scholar]
- Zagury D, Bernard J, Chenier R, Feldman M, Sarin P, and Gallo RC (1986). Immune induction of T cell death in long term HTLV-III infected cultures. A cytopathogenic model for AIDS T cell depletion. Science 231, 850–853. [DOI] [PubMed] [Google Scholar]