

Abstract
The replication of human immunodeficiency virus type 1 in mononuclear phagocytes (blood monocytes, tissue macrophages, and dendritic cells) is an important feature of HIV-1 pathogenesis. Although most primary HIV-1 isolates are able to productively infect monocytes, some reports suggest that rates of viral DNA synthesis and virus replication are reduced in HIV-1-infected monocytes as compared to infected T cells. In this study we compare kinetics of viral DNA synthesis in CD4+ T cells and monocytes following HIV-1 infection. Our results indicate that reverse transcription of viral nucleic acids following infection of monocytes occurs at rates equal to or greater than that observed following infection of T cells. These studies reveal no postentry restrictions to HIV-1 replication following infection in monocytes. Moreover, the results support the notion that both monocytes and CD4+ T cells are equally permissive for virus replication in infected individuals.
The tropism of HIV-1 for cells of macrophage lineage is an important feature of lentivirus pathogenesis (1, 2). HIV-1 infection of tissue macrophages may play a central role in virus establishment as macrophages are one of the first cells infected by HIV-1 in the human host (2, 3). In contrast to the rapid cytopathic manifestations of HIV-1 replication in T cells (4, 5) macrophages appear less susceptible to virus-induced cytopathicity (2). Thus, macrophages may serve as a stable reservoir for the dissemination and persistence of HIV-1 in brain, lymph nodes, and lung throughout subclinical infection and disease (6-8). The characteristics of macrophage infections are not well defined. Exposure of macrophages to any of the multitude of exogenous and endogenous differentiation stimuli regulates viral replication (9-11) and the control of HIV-1 persistence. A variety of reports demonstrate that cellular control mechanisms influence permissiveness (12-14) of macrophages to productive lentiviral infection and underscore differences between macrophage and T cell infections. For example, low levels of IFN-β and γ and/or other negative regulatory factors may be induced after exposure of monocytes to HIV-1 (9, 15). These factors may regulate early stages of virus replication in monocytes and may underlie the differences in susceptibility of monocytes to HIV-1 infection.
Several studies suggest that macrophages are less permissive than T cells to productive HIV-1 infection (5, 11, 16). Since macrophages are terminally differentiated nondividing host cells for HIV-1, viral nucleic acids must traverse the nuclear membrane before provirus establishment and viral replication can occur. Since the first clear report demonstrating that nondividing macrophages permitted provirus establishment (17), it has been shown that nucleophilic components associated with the preintegration complex of HIV-1 facilitate nuclear localization of viral nucleic acids in the absence of mitosis (18-20). Taken together, these reports would indicate that the restriction to virus replication in macrophages is not at the level of host cell mitosis. Several studies have demonstrated that reverse transcription of viral nucleic acids is inefficient when nucleotide precursor levels in the cell are limited as in quiescent (Go) T-lymphocytes. This has prompted the suggestion that limited levels of nucleotide precursors in macrophages may influence viral DNA synthesis and limit the kinetics of spreading infection. In support of this hypothesis, recent studies have demonstrated inefficient synthesis of viral DNA following infection of monocyte-derived macrophages (34, 11, 21). Although reverse transcription rates in infected macrophages could be increased by the addition of exogenous nucleotide precursors, they did not reach rates seen in activated T cells. This suggested that the kinetics of HIV-1 replication in blood macrophages is influenced by limitations in the cellular nucleotide pool (11, 21).
The replication parameters of monocyte tropic (HIV-1ADA; HIV-1DJV) HIV-1 in monocyte-derived macrophages and lymphoblasts are shown in Fig. 1. Both viruses elicited a spreading infection in macrophages with kinetics similar to that observed in lymphoblasts (Fig. 1). The replication rate of monocyte tropic HIV-1 variants in lymphoblasts was similar to that for the IIIB-derived T cell line-adapted HIV-1MF virus variant (not shown). Thus, under the experimental conditions utilized here, there did not appear to be any restriction in the ability of HIV-1 to establish a spreading infection in macrophages when compared to lymphoblasts.
Fig. 1.
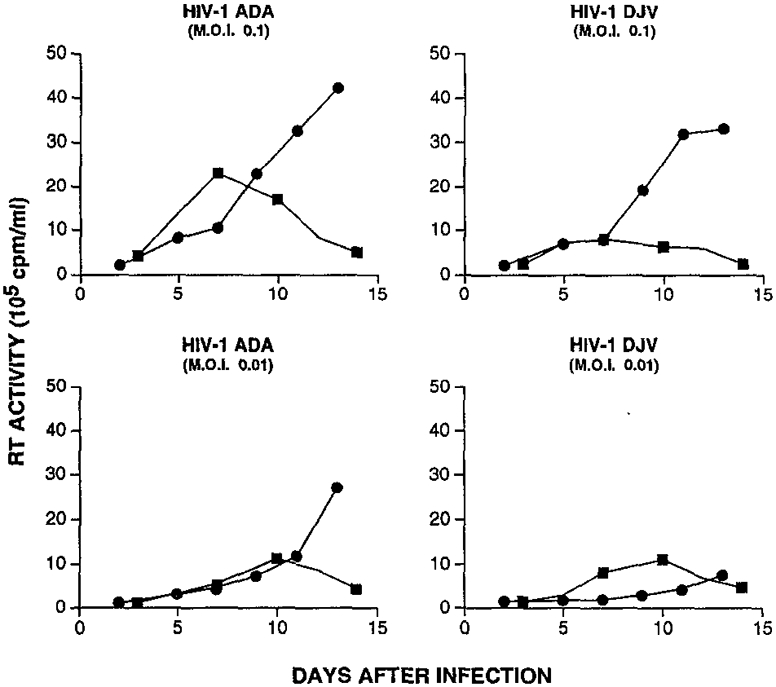
Replication of monocyte tropic HIV-1 variants in primary monocyte-derived macrophages (circles) and phytohemagglutinin (PHA)-activated peripheral blood lymphocytes (squares). Lymphocytes and monocytes were isolated by counter-current centrifugal elutriation of mononuclear leukocyte-rich cell preparations obtained from normal HIV-1 and hepatitis B seronegative donors by leukapheresis. Lymphocytes were activated by PHA (2 μg/ml) overnight and then cultured in the presence of interleukin 2 (IL-2). Elutriated monocytes were cultured in 24-well microtiter plates as adherent monolayers in medium containing MCSF (1000 U/ml, a generous gift from Genetic Institute, Cambridge, MA) for 7 to 10 days prior to HIV-1 infection (10). Cells were infected with the indicated HIV-1 isolate at two multiplicities of infection (0.1 (top) and 0.01 per cell (bottom), which is equivalent to 1 × 105 and 1 × 104 cpm per 7.5 × 105 cells). HIVADA has been described previously (42); HIV-1DJV is a clinical monocyte tropic isolate which was obtained at autopsy from the brain of an individual who had died of AIDS. Two hours after infection, culture supernatants containing virions were removed and replaced with fresh culture medium. At 2- to 3-day intervals, culture supernatants were harvested in triplicate for measurement of reverse transcriptase activity.
We next compared the kinetics of reverse transcription of viral nucleic acids following infection of lymphoblasts and monocytes. Synthesis of early, intermediate, and late products of reverse transcription were identified by polymerase chain reaction (PCR) and primers to LTR U3/R, pol, and LTR R/gag regions of the viral genome, respectively (22, 23). The examination of de novo DNA synthesis following acute HIV infection is limited by the presence of preexisting viral DNA (proviral (23) and virion-associated DNA (24, 25)) in the inoculum. Treatment of the inoculum with DNase (23) does not completely eliminate input proviral DNA forms and, in particular, has little effect on virus-associated DNA (M. Stevenson, unpublished observations). Therefore, the contribution of virion-associated DNA was excluded by comparing viral DNA synthesis in drug free with that in AZT containing lymphoblast cultures. To provide more definitive evidence for the presence of full-length plus and minus strand viral DNA we compared the synthesis of 1 and 2 LTR circle forms of episomal viral DNA in infected cells. Although these episomal forms of viral DNA do not appear to represent provirus precursors (26-28), they are formed only after synthesis of full-length viral DNA and its transport to the host cell nucleus (22, 26). In addition, such episomal forms are not found within the virus inoculum but are formed only within acutely infected cells (M. Stevenson, unpublished observations). The accumulation of early, intermediate, and late products of reverse transcription including nuclear episomal forms of viral DNA in HIV-1-infected lymphoblasts is illustrated in Fig. 2. De novo synthesis of early, intermediate, and late reverse transcription intermediates (when compared to levels observed in AZT-treated cultures) was evident by 8 hr after infection with the HIV-1ADA isolate. In particular, when compared with AZT-treated cultures, there was a clear increase in the PCR signal generated by LTR U3/R and poll/J primers at 8 hr postinfection and at 24 hr postinfection using LTR U3/gag (late) primers (Fig. 2). Similar rates of accumulation of reverse transcription intermediates were observed following infection of lymphoblasts by HIV-1DJV (compare LTR U3/gag products at 8 and 24 hr postinfection with AZT controls). Episomal HIV-1 DNA forms containing 1 LTR (1 LTR circle) were evident by 24 and 36 hr postinfection with HIV-1ADA and HIV-1DJV isolates, respectively. Episomal forms of viral DNA containing 2 LTRs (2 LTR circle) which are typically observed in lower abundance in retrovirus-infected cells (29) were detectible by 36 and 48 hr after infection with HIV-1ADA and HIV-1DJV isolates, respectively. This suggests that under culture conditions and multiplicities of infection utilized here, viral DNA synthesis in HIV-1ADA- and HIV-1DJV-infected lymphoblasts was complete by at least 24 and 36 hr postinfection, respectively.
Fig. 2.
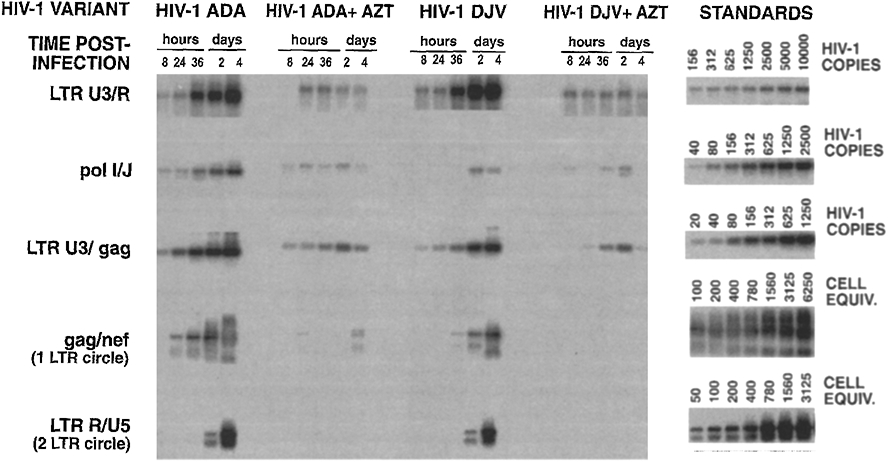
Synthesis of viral DNA in lymphoblasts following HIV-1 infection. At the indicated times postinfection, lymphoblasts (5 × 105 cells) were taken for isolation of total cellular DNA and analysis by PCR (19). In AZT-treated cultures, drug (1 μM) was added to lymphoblast and monocyte cultures for 18 hr prior to infection and was maintained in cultures throughout the period of the experiment. DNA from 5 × 104 cells was subjected to 30 cycles of PCR with primers to LTR U3 and R (coordinates 9194–9214 and 9591–9610 of HIV HXB2; (43) pol/l and J (coordinates 2131–2149 and 2592–2610); LTR U3 and gag (coordinates 9194–9214 and 794–815) for amplification of early, intermediate and late reverse transcription products, respectively. Amplification of episomal forms of viral DNA containing 1 and 2 LTRs was performed using gag and nef (coordinates 912–931 and 9008–9028) and LTR R and U5 (coordinates 9591–9610 and 9650–9679) primers, respectively. For LTR primers, only 3′ LTR coordinates are given. PCR amplification products were visualized after Southern blot transfer and hybridization with 32P-labeled oligonucleotide probes. Hybridized blots were visualized on a molecular phosphorimager SF (Molecular Dynamics). Standards for LTR U3/R, poll/J, and LTR R/gag products were generated by PCR on doubling dilutions of DNA from 8E5 cells which contain one defective viral genome per cell (44). Standards for 1 and 2 LTR circle forms of viral DNA were generated from doubling dilutions of HIV-1-infected CD4+ MT-4 cells.
We next examined the accumulation of viral reverse transcription products following infection of monocyte-derived macrophages (Fig. 3). Although some virion-associated strong stop DNA (identified using LTR U3/R primers) was evident in AZT-treated cultures, there appeared to be very little, if any, association of proviral DNA forms in the inoculum (23) (identified using poll/J and LTR U3/gag primers) with monocytes. The reason for the differences in uptake of preexisting proviral DNA forms by lymphocytes and monocytes is unclear but may relate to the different biophysical properties of these cells. Early, intermediate, and late products of reverse transcription were evident by 8 hr postinfection with the HIV-1ADA isolate, while accumulation of late, products of reverse transcription was somewhat delayed in HIV-1DJV-infected cultures (Fig. 3). One LTR circle forms of viral DNA were evident as early as 8 hr postinfection with HIV-1ADA although their appearance was greatly delayed in monocytes infected with HIV-1DJV. We do not attach much significance to differences in 1 LTR circle accumulation between the two HIV-1 variants since accumulation of 2 LTR circle forms of viral DNA occurred at relatively similar late rates in HIV-1ADA- and HIV-1DJV-infected monocytes (Fig. 3). Taken together, the data indicates that viral DNA synthesis proceeds rapidly to completion in monocytes and at rates equal to or greater than that observed in lymphoblasts. The comparable rates of reverse transcription in both cell systems are supported by comparable rates with which monocyte tropic HIV-1 variants elicited a spreading infection in lymphoblasts and monocytes as shown in Fig. 1.
Fig. 3.
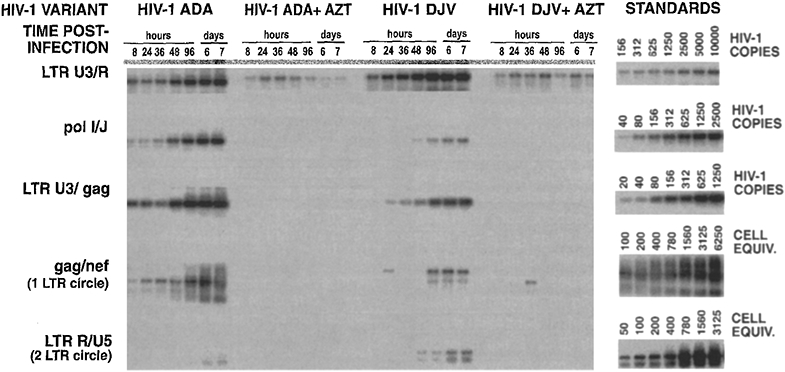
Synthesis of viral DNA in HIV-1-infected monocytes. Monocytes were established in 24-well plates. Cells were harvested by scraping and DNA was isolated as described elsewhere (19). Early, intermediate, and late products of reverse transcription and episomal forms of viral DNA were amplified using primer pairs identified in Fig. 2.
To address whether contaminating lymphoblasts in the macrophage cultures contributed to the observed patterns of virus replication and DNA synthesis, we examined viral DNA synthesis in monocytes infected with a T cell line-adapted HIV-1 variant, HIV-1MF (30). T cell line-adapted HIV-1 isolates are unable to replicate in primary monocytes, a restriction which is manifest at the level of virus entry (fusion, uncoating) and which precludes synthesis of viral cDNA (31). In monocytes infected with HIV-1MF there was no evidence of any de novo viral DNA synthesis despite efficient reverse transcription in lymphoblasts infected with this virus (Fig. 4). Since macrophage cultures were refractory to infection by the T cell line-adapted HIV-1MF isolate, it is unlikely that the presence of contaminating lymphocytes in the monocytes could account for the efficient viral DNA synthesis and virus replication observed after infection with monocyte tropic HIV-1 isolates.
Fig. 4.
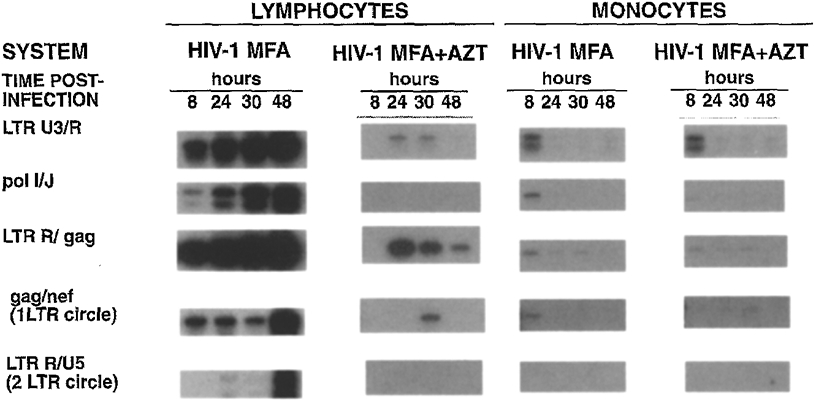
Synthesis of viral DNA in lymphoblasts and monocyte-derived macrophages after infection with a T cell line-adapted HIV-1 variant. Lymphoblasts and monocytes were infected in parallel with HIV-1MF (30) as outlined in Fig. 1. At the indicated times postinfection, cells were harvested for isolation and amplification of viral DNA from 5 × 104 cells as described in the legend to Fig. 2.
The results described in this report indicate that under the culture conditions used in this study, monocytes are as permissive to HIV-1 infection and replication as lymphoblasts and demonstrate that conditions for reverse transcription of viral nucleic acids are not rate limiting. These studies also support previous observations indicating that the restriction to macrophage infection by T cell line-adapted HIV-1 isolates is manifest at the level of virus entry, prior to the initiation of reverse transcription (31) rather than at a step which follows synthesis of viral DNA (32-34). The differences in results between this and previous studies (11, 21) likely reflects the viral isolate or the conditions for isolation and maintenance of monocytes. HIV-1ADA and HIV-1DJV are highly adapted monocyte tropic strains which replicate to high titers and are cytopathic for macrophages. Thus, the rates of viral DNA synthesis and virus replication observed in this study may be a reflection of the highly permissive infection that ensues following infection of monocytes by these variants. However, differences in rates of viral DNA synthesis are more likely to be governed by differences in the host cell environment than the virus isolate. In this regard, recombinant human macrophage colony stimulating factor (MCSF) is used in our studies for initiation of monocyte cultures. MCSF enhances long-term monocyte survival in the absence of cell proliferation (35, 36) and HIV-1 replication is greatly enhanced in monocytes containing MCSF (10). Although MCSF is used only during initiation of cultures and not during the period of spreading virus infection this may be sufficient to prime macrophages and facilitate their permissiveness to virus replication. Moreover, during the course of in vitro propagation, monocyte-derived macrophages can synthesize their own MCSF. Human sera used to cultivate monocytes contain up to 500 U/ml of MCSF (37, 38). Thus, although the addition of MCSF at the initiation of monocyte culture is likely a biologically relevant system, this alone cannot explain differences in permissiveness of cells to productive HIV-1 infection. A series of reports suggests that cytokines or other immune factors released as a consequence of monocyte/macrophage activation after HIV-1 infection regulate the initial events of viral replication in monocytes (9, 15). IFN-α, -β, or -γ, TNFα, or IL-1β produced as a consequence of HIV-1 replication could control initial events of virus/macrophage interactions (39). Although addition of many of these factors can regulate virus replication in monocytes, like MCSF, the endogenous production of cytokines as a consequence of HIV-1 infection collectively are low and ultimately fail to control virus replication (12, 40). Indeed, a number of reports show no constitutive cytokine production following HIV-1 monocyte infection (12, 40, 41). Thus, at present, no clear explanations can be forwarded for the differences in rates of virus replication in monocytes in this compared with previous studies. These issues aside, our studies indicate that under certain conditions and with certain virus isolates, the macrophage is a highly permissive cell for productive HIV-1 infection. This underscores the importance of the tissue macrophage as a reservoir for HIV-1 dissemination and persistence.
ACKNOWLEDGMENTS
We thank Kelly Bowers and Karen Spiegel for outstanding administrative support. These works were funded in part by AmFAR Grant 02065-15-RGR (H.E.G.), the University of Nebraska Research Initiative start up funds (H.E.G.), NIH Grants PO1 NS31492-01 (H.E.G.), PO1 HL43628-05 (H.E.G.), AI 32890 (M.S.), and AI30386 (M.S.), and the Dana Foundation (H.E.G.). Dr. Howard Gendelman is a Carter Wallace Fellow of the Department of Pathology and Microbiology at the University of Nebraska Medical Center.
REFERENCES
- 1.Fauci A, Science 262, 1011–1018 (1993). [DOI] [PubMed] [Google Scholar]
- 2.Gendelman HE, and Morahan PS, In “The Macrophage” (Lewis CE and McGee JOD, Eds.), pp. 157–195. IRL Press, New York, 1992. [Google Scholar]
- 3.Zhu T, Hongmei M, Wang N, Nam DS, Cao Y, Koup RA, and Ho DD, Science 261, 1179–1181 (1993). [DOI] [PubMed] [Google Scholar]
- 4.Cheng-Mayer C, and Levy JA, Curr. Trends Microbiol. Immunol 160, 145–156 (1990). [DOI] [PubMed] [Google Scholar]
- 5.Levy JA, Microbiol. Rev 57, 183–289 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koenig S, Gendelman HE, Orenstein JM, DalCanto MC, Pezeshkpour GH, Yungbluth M, Janotta F, Aksamit A, Martin MA, and Fauci AS, Science 233, 1089–1093 (1986). [DOI] [PubMed] [Google Scholar]
- 7.Shaw GM, Hahn BH, Arya SK, Groopman JE, Gallo RC, and Wong-Staal F, Science 226, 1165–1170 (1984). [DOI] [PubMed] [Google Scholar]
- 8.Wiley CA, Schrier RD, Nelson JA, Lampert PW, and Oldstone MBA, Proc. Natl. Acad. Sci. USA 83, 7089–7093 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gendelman HE, Baca LM, Kubrak CA, Genis P, Burrous S, Friedman RM, Jacobs D, and Meltzer MS, J. Immunol 148, 422–429 (1992). [PubMed] [Google Scholar]
- 10.Kalter CD, Nakamura M, Turpin JA, Baca LM, Hoover DL, Dieffenbach C, Ralph P, Gendelman HE, and Meltzer MS, J. Immunol 146, 298–306 (1991). [PubMed] [Google Scholar]
- 11.O’Brien WA, Namazi A, Kalhor H, Mae S-H, Zack JA, and Chen IS, J. Virol 68, 1258–1263 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gendelman HE, Friedman RM, Joe S, Baca LM, Turpin JM, Dveksler G, Meltzer MS, and Dieffenbach C, J. Exp. Med 172, 1433–1442 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gendelman HE, Narayan O, Kennedy-Stoskopf S, Kennedy P, Ghotbi Z, Clements JE, Stanley J, and Pezeshkpour JG, J. Virol, 58, 67–74 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gendelman HE, Narayan O, Molineaux F, Clements JE, and Ghotbi Z, Proc. Natl. Acad. Sci. USA 82, 7086–7092 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gessani S, Puddu P, Varano B, Borghi P, Conti L, Fantuzzi L, and Belardelli F, J. Virol 68, 1983–1986 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valentin A, Von Gegerfelt A, Matsuda S, Nilsson K, and Asjo B, J. Acquir. Immune. Defic. Syndr 4, 751–759 (1991). [PubMed] [Google Scholar]
- 17.Weinberg JB, Matthews TJ, Cullen BR, and Malim MH J. Exp. Med 174, 1477–1482 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bukrinsky M, Haggerty S, Dempsey MP, Sharova N, Adzhubei A, Spitz L, Lewis P, Goldfarb D, Emerman M, and Stevenson M, Nature 365, 666–669 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bukrinsky MI, Sharova N, McDonald TL, Pushkarskaya T, Tarpley WG, and Stevenson M, Proc. Natl. Acad. Sci. USA 90,6125–6129 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heinzinger NK, Bukrinsky MI, Haggerty SA, Ragland AM, Lee M-A, Kewalramani V, Gendelman HE, Ratner L, Stevenson M, and Emerman M, Proc. Natl. Acad. Sci. USA 91, 7311–7315 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collin M, and Gordon S, Virology 200, 114–120 (1994). [DOI] [PubMed] [Google Scholar]
- 22.Bukrinsky MI, Sharova N, Dempsey MP, Stanwick TL, Bukrinskaya AG, Haggerty S, and Stevenson M, Proc. Natl. Acad. Sci. USA 89, 6580–6584 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zack JA, Arrigo S. j., Weitsman SR, Go AS, Haislip A, and Chen IS,Y Cell 61, 213–222 (1990). [DOI] [PubMed] [Google Scholar]
- 24.Lori F, Veronese FD, DeVico AL, Lusso P, Reitz MSJ, and Gallo RC. J. Virol 66, 5067–5074 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trono D, J. Virol 66, 4893–4900 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown PO, Bowerman B, Varmus HE, and Bishop JM, Cell 49, 347–356 (1987). [DOI] [PubMed] [Google Scholar]
- 27.Ellis J, and Bernstein A, J. Virol 63, 2844–2846 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lobel LI, Murphy JE, and Goff SP, J. Virol, 63, 2629–2637 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Varmus HE, and Swanstrom R, In “RNA Tumor Viruses” (Weiss R, Teich N, Varmus HE, and Coffin J, Eds.), pp. 369–512. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 1984. [Google Scholar]
- 30.Stevenson M, Haggerty S, Lamonica C, Mann AM, Meier C, and Wasiak A, J. Virol 64, 3792–3803 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cann AJ, Zack JA, Go AS, Arrigo SJ, Koyanagi Y, Green PL, Koyanagi Y, Pang S, and Chen ISY, J. Virol 64, 4735–4742 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang Z-B, Potash MJ, Simm M, Shahabuddin M, Chac W, Gendelman HE, Eden E, and Volsky DJ, J. Virol 67, 6893–6896 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mori K, Ringler D, and Desrosiers R, J. Virol 67, 2807–2814 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmidtmayerova H, Bolmont C, Baghdiguian S, Hirsch I, and Chermann J-C, Virology 190, 124–133 (1992). [DOI] [PubMed] [Google Scholar]
- 35.Becker S, Warren MK, and Haskill S, J. Immunol 139, 3703–3708 (1987). [PubMed] [Google Scholar]
- 36.Stanley ER, Ciba Found. Symp 118, 29–41 (1986). [DOI] [PubMed] [Google Scholar]
- 37.Das SK, Stanley ER, Guilbert LJ, and Forman LW, Blood 58, 630–641 (1981). [PubMed] [Google Scholar]
- 38.Hanamura T, Motoyoshi K, Yoshida K, Saito M, Miura Y, Kawashima T, Nishida M, and Takaku F, Blood 72, 886–892 (1988). [PubMed] [Google Scholar]
- 39.Rosenberg ZF, and Fauci AS, FASEB J. 5, 2382–2390 (1991). [DOI] [PubMed] [Google Scholar]
- 40.Molina J-M, Schindler R, Ferriani R, Sakaguchi M, Vannier E, Dinarello CA, and Groopman JE, J. Infect. Dis 161, 888–893 (1990). [DOI] [PubMed] [Google Scholar]
- 41.Munis JR, Richman DD, and Kornbluth RS, J. Clin. Invest 85, 591–596 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Westervelt P, Trowbridge DB, Epstein LG, Blumberg BM, Li Y, Hahn BH, Shaw GM, Price RW, and Ratner L, J. Virol 66, 2577–2582 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Myers G, MacInnes K, and Korber B, AIDS Res. Hum. Retroviruses 8, 373–386 (1992). [DOI] [PubMed] [Google Scholar]
- 44.Folks TM, Powell D, Lightfoote M, Koenig S, Fauci AS, Benn S, Rabson A, Gendelman HE, Hoggan MD, Venkatesan S, and Martin MA,J. Exp. Med 164, 280–290 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]