

SUMMARY
The potential of small molecules to localize within subcellular compartments is rarely explored. To probe this question, we measured the localization of Hsp70 inhibitors using fluorescence microscopy. We found that even closely related analogues had dramatically different distributions, with some residing predominantly in the mitochondria and others in the ER. CRISPRi screens supported this idea, showing that different compounds had distinct chemogenetic interactions with Hsp70s of the ER (HSPA5/BiP) and mitochondria (HSPA9/mortalin) and their co-chaperones. Moreover, localization appeared to determine function even for molecules with conserved binding sites. Compounds with distinct partitioning have distinct anti-proliferative activity in breast cancer cells compared with antiviral activity in cellular models of Dengue virus replication, likely because different sets of Hsp70s are required in these processes. These findings highlight the contributions of subcellular partitioning and chemogenetic interactions to small molecule activity, features that are rarely explored during medicinal chemistry campaigns.
In Brief
Specific targeting of distinct Hsp70 sub-networks could be used to treat different diseases, such as cancer and virus infection while minimizing side effects. Here, Shao and Taguwa et al. address the challenge of designing diseases-specific inhibitors by taking advantage of the sub-cellular distributions of closely related Hsp70 inhibitors.
Graphical Abstract
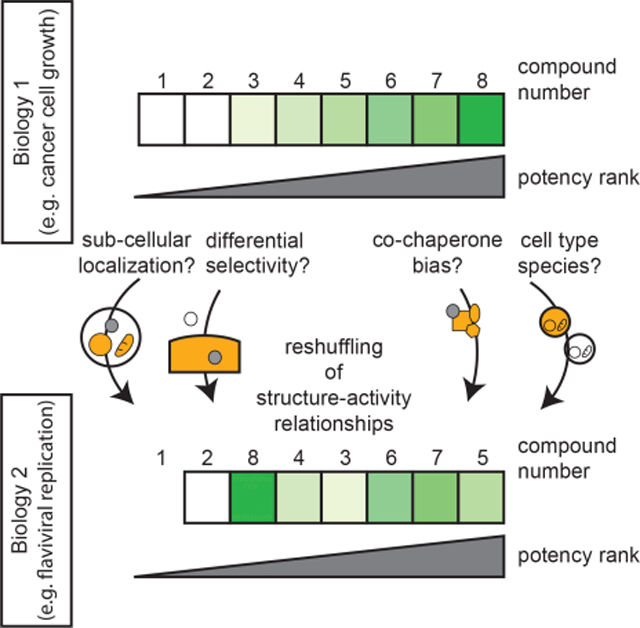
INTRODUCTION
The heat shock protein 70 (Hsp70) family of molecular chaperones plays important roles in protein homeostasis (aka proteostasis) (Rosenzweig et al., 2019). Hsp70s are ubiquitous and conserved across all eukaryotes, with homologues present in each major cellular compartment (Radons, 2016). For example, there are 17 isoforms of Hsp70 in humans, including the major examples of the cytosol and nucleus (HSPA8/Hsc70 and HSPA1A/Hsp72), endoplasmic reticulum (ER) (HSPA5/BiP) and mitochondria (HSPA9/mortalin). In these compartments, the Hsp70s aid in numerous processes related to proteostasis, including protein folding, turnover, assembly/disassembly of multiprotein complexes and regulation of protein condensate formation (Balchin et al., 2016).
Hsp70s share a conserved structural architecture and they all engage in ATP-driven cycles of substrate protein binding and release (Mayer and Gierasch, 2019; Zuiderweg et al., 2017). This cycle depends on Hsp70’s interactions with co-chaperones: J-domain proteins (JDP) promote ATP hydrolysis and substrate selection and association, while nucleotide exchange factors (NEFs) promote ADP/ATP exchange and substrate release. Each compartment of the cell (e.g. cytoplasm, nucleus, mitochondria, ER, lysosome, etc.) contains a subset of JDPs (Craig and Marszalek, 2017; Kampinga et al., 2019), NEFs (Bracher and Verghese, 2015; Craig and Marszalek, 2017; Rauch and Gestwicki, 2014) and tetratricopeptide repeat (TPR)-domain proteins (Assimon et al., 2015; Kundrat and Regan, 2010; Prodromou et al., 1999) that, together, regulate the functions of that compartment’s Hsp70s. Together, these specific sub-networks of Hsp70s and their co-chaperones are important for maintaining proteostasis in the various compartments of the cell.
Given this broad distribution and plurality of functions, it is perhaps not surprising that Hsp70s have been implicated in many diseases, including cancer, neurodegeneration, and infectious disease (Abisambra et al., 2013; Moses et al., 2018; Taguwa et al., 2015). In turn, these disease relationships have fueled a search for chemical inhibitors of Hsp70s, with a growing number being explored (Ambrose and Chapman, 2021; Evans et al., 2010; Howe et al., 2014; Rodina et al., 2013; Shrestha et al., 2016; Workman, 2020; Zaiter et al., 2019). Although these studies are ongoing, one of the early surprises of the field is that Hsp70 inhibitors are not as acutely toxic as might be expected, at least when administered to non-transformed cells (Wadhwa et al., 2000). For example, benzothiazole-based Hsp70 inhibitors, such as JG-231, are 20-fold more toxic to breast cancer cells than normal fibroblasts (Shao et al., 2018). On the surface, the relative lack of toxicity might be considered a surprising result, as Hsp70s are involved in many required housekeeping functions. One possibility is that these compounds, such as JG-231, only bind a subset of Hsp70s, such as those in a specific organelle. However, the binding site for JG-231, for example, is identical between the major members of the Hsp70 family (e.g., HSPA1, HSPA8, HSPA5 and HSPA9) (Li et al., 2013). Another possible reason for the relative safety of Hsp70 inhibitors is that cancer cells express a specific epichaperome composed of a subset of Hsp70s and co-chaperones, which might be more vulnerable to chemical inhibition (Rodina et al., 2016). The relative importance of these potential mechanisms is not well understood. Clearly, a better understanding of this question could lead to the design of improved chemical inhibitors.
Inspired by recent studies showing that some drugs selectively partition into nucleolar condensates (Klein et al., 2020), we were intrigued by the possibility that the biological activity of Hsp70 inhibitors might also be linked to selective partitioning. In the studies by Klein et al., sub-cellular localization appeared to be driven by binding to a pre-localized target protein; however, it is also known that physicochemical properties—instead of the affinity for the target per se—might also underlie the non-uniform distribution of compounds (Zheng et al., 2011). For example, cationic molecules commonly localize to the mitochondria, driven by the H+ gradient (Bernardi, 1999); such chemistry-driven partitioning has been leveraged to create chemical “addresses”, which, when appended to a molecule, favor mitochondrial accumulation (Frantz and Wipf, 2010; Wisnovsky et al., 2016). Yet, there are few examples of small molecule localization to other (i.e, non-mitochondrial) sub-cellular locations and even fewer attempts to study whether molecules in a related chemical series share the same localization patterns (Louzoun-Zada et al., 2019).
We reasoned that the members of a chemical series targeting the Hsp70 system could provide an ideal model in which to explore basic mechanisms underlying subcellular localization and specificity of pharmacological proteostasis regulators. Accordingly, we selected benchmark molecules from a 400+ compound medicinal chemistry campaign (Li et al., 2013; Shao et al., 2018). A major advantage of using compounds from this particular campaign is that they are intrinsically fluorescent (Shao and Gestwicki, 2020), which we envisioned would facilitate studies to define their subcellular distribution. Indeed, using confocal microscopy, we found that even closely related molecules have dramatically different subcellular localizations, with accumulations observed in the ER, mitochondria, secretory vesicles, and/or the nucleolus. To explore whether compounds co-located in the same compartment also target the same Hsp70 sub-networks, we performed whole genome CRISPRi screens to define their chemogenetic interactions, revealing that partially distinct cellular processes are targeted. Together, these findings suggest that both sub-cellular localization and interactions with distinct Hsp70 subnetworks (e.g., epichaperomes) may contribute to differences in compound activity. To understand the potential therapeutic implications of these findings, we tested the same series of compounds in two assays that require Hsp70s: anti-cancer and anti-viral activity (Taguwa et al., 2015; Taguwa et al., 2019). Our analyses revealed that the most potent anti-cancer compounds were not always the same as the most potent anti-viral compound, likely because these biological processes require different Hsp70 subnetworks in different cellular compartments. Thus, we speculate that variations in subcellular distribution and chemogenetic interactions may be important, but often overlooked, contributors to differences in compound activity.
RESULTS
Selection of closely related, benchmark compounds from a chemical series
To probe whether related compounds can partition into different subcellular locations, we selected benchmark Hsp70 inhibitors from a published medicinal chemistry campaign encompassing ~400 analogs. This campaign started with the benzothiazole, MKT-077 (Fig 1A), and the first reported analogs focused on improving potency, resulting in JG-98 (Fig 1A) (Li et al., 2013), while the second set of analogs focused on pharmacokinetic properties, resulting in JG-231 (Rinaldi et al., 2018; Shao et al., 2018). Importantly for the current work, these molecules are known to bind a universally conserved allosteric site that is present in all Hsp70 isoforms and this interaction disrupts co-chaperone binding (Rousaki et al., 2011; Wadhwa et al., 2000). Each analog in the reported campaign had been tested for anti-proliferative activity in both MCF7 breast cancer cells and non-transformed, mouse embryonic fibroblasts (MEFs). Thus, to guide our selection of benchmark compounds, we plotted the published potency values (EC50, MCF7) and safety index (SI; EC50, MCF7:EC50, fibroblasts) for each analog (Fig 1B). We then selected molecules at periodic points throughout the numbered chemical series (e.g., MKT-077, JG-40, JG-98, JG-194, JG-231, JG-294 and JG-345; denoted in red in Fig 1B). Here, we refer to analogs that are generally in the beginning of the series as “early” compounds (herein Type I), while molecules with generally higher numbers are termed “late” (herein Type II). The Type I compounds tend to have lower molecular weight (<500 Da) and weaker potency and safety, while later, Type II compounds have higher mass (>500 Da) and improved potency and safety. In addition to molecular mass and potency, this Type I - Type II nomenclature is also based on the subcellular distributions of the compounds, which is described below. Together, we envisioned that these benchmark compounds (together termed the “JG series”) could be used to probe the possible relationships between potency, safety, and subcellular partitioning.
Figure 1: Structure-guided design of allosteric Hsp70 inhibitor MKT-077 analogs.
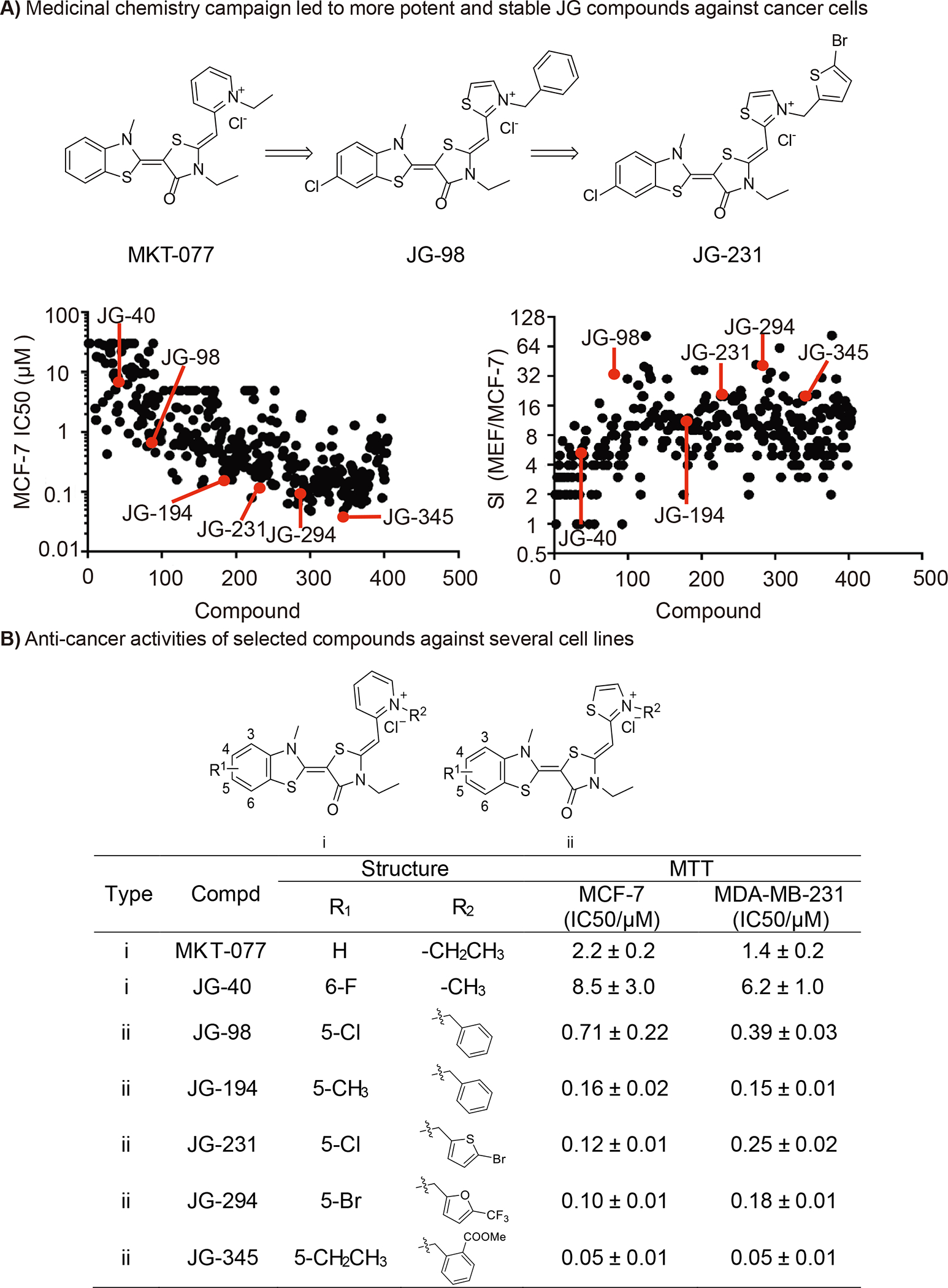
(A) Medicinal chemistry campaign led to more potent molecules against breast cancer cells; (B) Anti-cancer activities of benchmarked molecules. Anti-proliferative activity by 72h MTT assay and the data given are mean values derived from at least two independent replicates.
Differential compound activity is not linked to activation of a stress response
In each cellular compartment, Hsp70 aids in protein folding, and the accumulation of misfolded or unfolded proteins in the cytosol or ER upon Hsp70 inhibition can activate stress responses, including the heat shock response (HSR) and the unfolded-protein response (UPR) (Terrab and Wipf, 2020). The UPR is regulated through three distinct sensors: PKR-like endoplasmic reticulum kinase (PERK), inositol-requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6) (Terrab and Wipf, 2020) and the activation of each sensor has different transcriptional outputs (Walter and Ron, 2011). Therefore, we tested whether the activity of compounds in the JG series could be related to a differential activation of the distinct UPR branches or the HSR. As a control we used bortezomib, a proteasome inhibitor known to promote accumulation of misfolded proteins and to induce the HSR (Mitsiades et al., 2002). For these experiments, we first tested benchmark compounds for anti-proliferative activity against K562 leukemia cells (Fig 2B). These cells were chosen to enable CRISPR screens against K562 cells stably expressing dCas9-CRAB (see below). To provide another measure of SI, we also tested these compounds against non-tumorigenic breast epithelial MCF10A cells (Supplemental Fig. 1). We found that cellular potency against K562 and MCF10A cells roughly correlated with that reported for other cancer and normal cell lines (Shao et al., 2018).
Figure 2: JG compounds activate unfolded protein response.
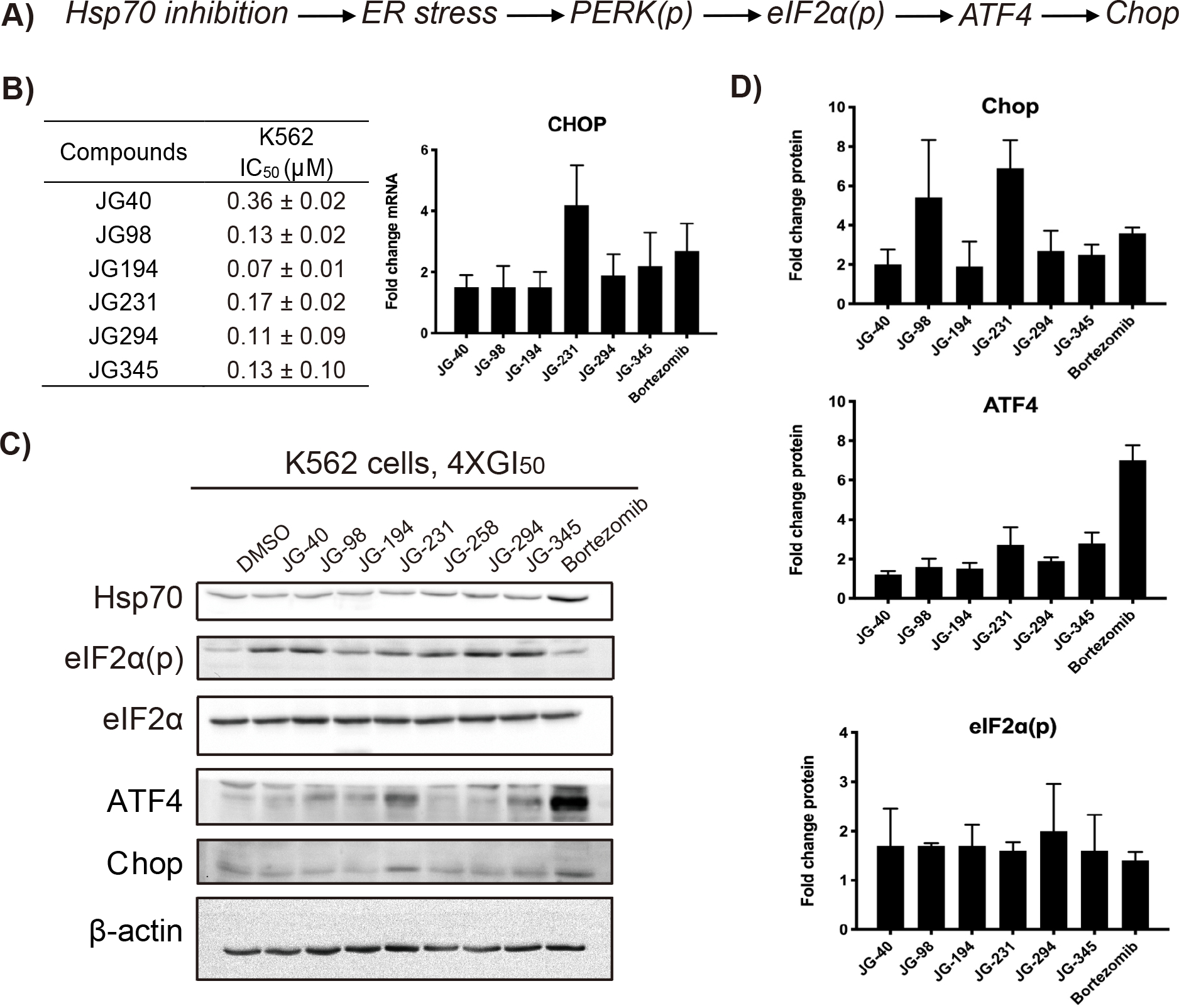
(A) Inhibition of Hsp70s led to CHOP activation; (B) Anti-proliferative activities of benchmarked molecules against K562 cells by 72h CellTiter-Glo assay and relative expression levels of CHOP between JG compounds treated and DMSO control analyzed by RT-qPCR. The data given are mean values derived from at least two independent replicates and the error bars represent S.D. (C) K562 cells were treated with the indicated compounds at 4XGI50 for 6h, lysed and blotted for proteins of interests. Results are representative of at least two independent experiments; (D) Quantification of western blot results. The error bars represent S.D.
Prior data indicated that pharmacological inhibition of BiP/HSPA5 with HA15 led to UPR-dependent cell death (Cerezo et al., 2016), and inhibition of Hsp70 with MAL3–101 triggered UPR-mediated cell death through CHOP induction (Sabnis et al., 2016). Therefore, we first checked whether the JG series induced CHOP mRNA expression by qPCR 6 h after treatment at 4XGI50 in K562 cells. As shown in Fig 2B, the examined JG compounds increased the expression of CHOP with a range of potencies. Next, we measured the levels of cytosolic Hsp70 as a proxy for induction of the HSR. In contrast to bortezomib, western blot analyses showed that none of the JG compounds increased Hsp70 levels, consistent with previous reports (Shao et al., 2018). We then examined the effect of the compounds on activation of the PERK–eukaryotic translation initiation factor α (eIF2α)–CHOP signaling axis. As above, the compounds activated this leg of the UPR with a range of effects, as shown by increased phosphorylation of the PERK substrate eIF2α and induction of ATF4 and CHOP (Fig 2C and D). Overall, this analysis indicates that the progressive increase in anticancer activity of compounds in the JG series is unrelated to the relative induction of the UPR or the HSR.
Closely related molecules exhibit distinct patterns of subcellular localization
We next examined whether the JG compounds might partition to distinct subcellular compartments. To this end, we exploited the intrinsic fluorescence of these compounds (excitation ~ 450 to 500 nm; emission ~550 to 600 nm; Suppl Fig. 2) to visualize their location. Importantly, the fluorescence of these molecules is not dependent on pH over physiological ranges (pH 5 to 8; Supplemental Fig. 2), so their intensity should not be affected by the pH ranges of various organelles. To allow for co-localization of this fluorescence with major organelles, we used Huh7 cells expressing Turquoise2 blue fluorescent protein fused to KDEL (for ER labelling) or a mitochondria targeting sequence (for mitochondrial labelling). These cells were then incubated with 5 μM of compound at 37 °C for 120 min and imaged for both compound and organelle fluorescence using confocal microscopy. Strikingly, we found that analogs in the early/Type I portion of the chemical series (Fig. 3E) co-localized with tubular mitochondria and nucleoli (Fig. 3A and 3C), while late/Type II compounds co-localized with the ER, as well as vesicle-like structures (Fig. 3B and 3C). Interestingly, while not associated with mitochondria, the Type II compounds affected mitochondrial morphology, yielding a fragmented phenotype (e.g., Fig 3D for JG-345; Supplemental Fig. 3). Together, these results show that even closely related compounds can have dramatically different sub-cellular localizations (Fig. 3E and Supplemental Table 3). Moreover, differences in partitioning tended to correlate with potency and safety (early/Type I vs. late/Type II), suggesting that it might be functionally important.
Figure 3: Distinct subcellular distribution of JG compounds in the cells.
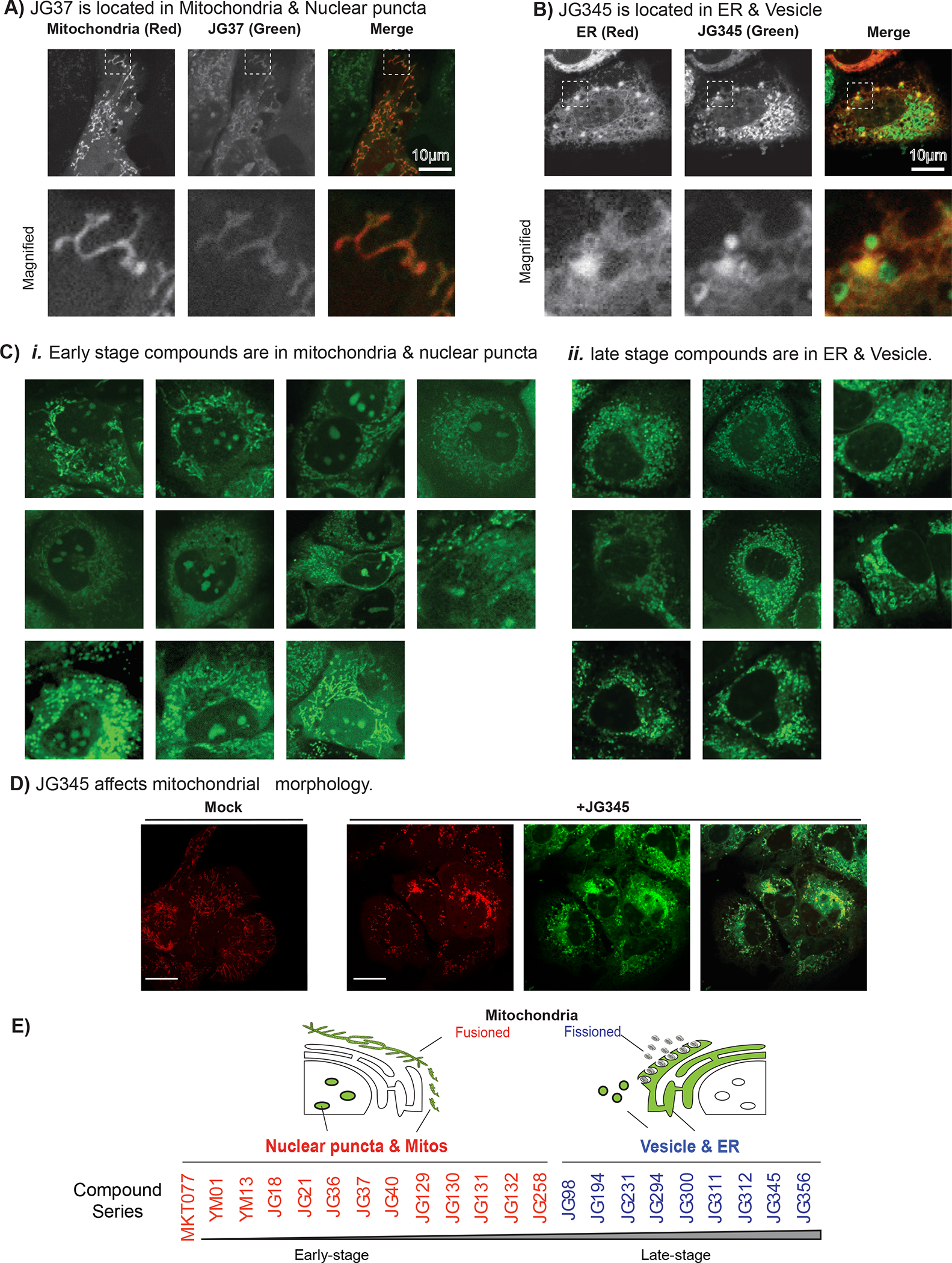
(A-B) Two representative JG compounds with distinct cellular localizations. (A) JG-37 colocalizes with fusioned mitochondria. Turquoise2-MTS expressing Huh7 cells were treated with 1μM JG37 in the culture medium at 37 °C incubator. The localizations of JG-37 (green) and mitochondria (red) were examined at 120 min post treatment. (B) JG-345 (green) is colocalized with ER (red) and forms vesicular structure. Turquoise2-KDEL expressing Huh7 cells were treated with JG-345 the same as (A). The boxed areas in the top images are magnified and displayed on the bottom. Scale bar, 10 μm. (C) The subcellular localization patterns of JG compounds. i) Early-stage JG compounds (Type I, green) were localized at fusioned mitochondria and puncta in the nucleus. ii) Late-stage JG compounds (Type II, green) were localized at ER and vesicular structure. Scale bar, 10 μm. (D) Late-stage JG compounds alters the morphology of mitochondria. Turquoise2-MTS expressing Huh7 cells were treated with JG-345 the same as (A). Scale bar, 20 μm. The shape of mitochondria and late-stage JG compounds are also shown in Supplemental Figure 3.
A CRISPRi screen reveals different chemogenetic interactions of JG compounds
The distinct subcellular partitioning of the compound series may also reflect their distinct interactions with subsets of Hsp70 subnetworks. We thus examined if the different Hsp70 inhibitors have chemogenetic interactions with distinct Hsp70 subnetworks. CRISPRi functional genomic screens are emerging as a robust way to identify the chemogenetic interactions of small molecules (Jost et al., 2017; Jost and Weissman, 2018; Kampmann, 2017). In these experiments, CRISPRi is used to systematically knockdown each gene in the presence of the compound-of-interest and changes in the cell’s growth rate are used to identify genes associated with relative resistance or sensitivity to the compound treatment (see Methods). In this case, we took advantage of the anti-proliferative activity of the benchmark Hsp70 inhibitors to perform genome-wide CRISPRi screens (Fig 4A) in K562 cells treated with JG-98, JG-194 and JG-294. These three compounds were selected because they all localize to the ER & vesicle compartments; therefore, we could minimize inter-compound differences due to broadly distinct in subcellular localization and focus on any similarities and differences in the perturbation of local Hsp70 subnetworks.
Figure 4. CRISPRi screen reveals genetic sensitivities and resistance to JG compound treatment in K562 cells.
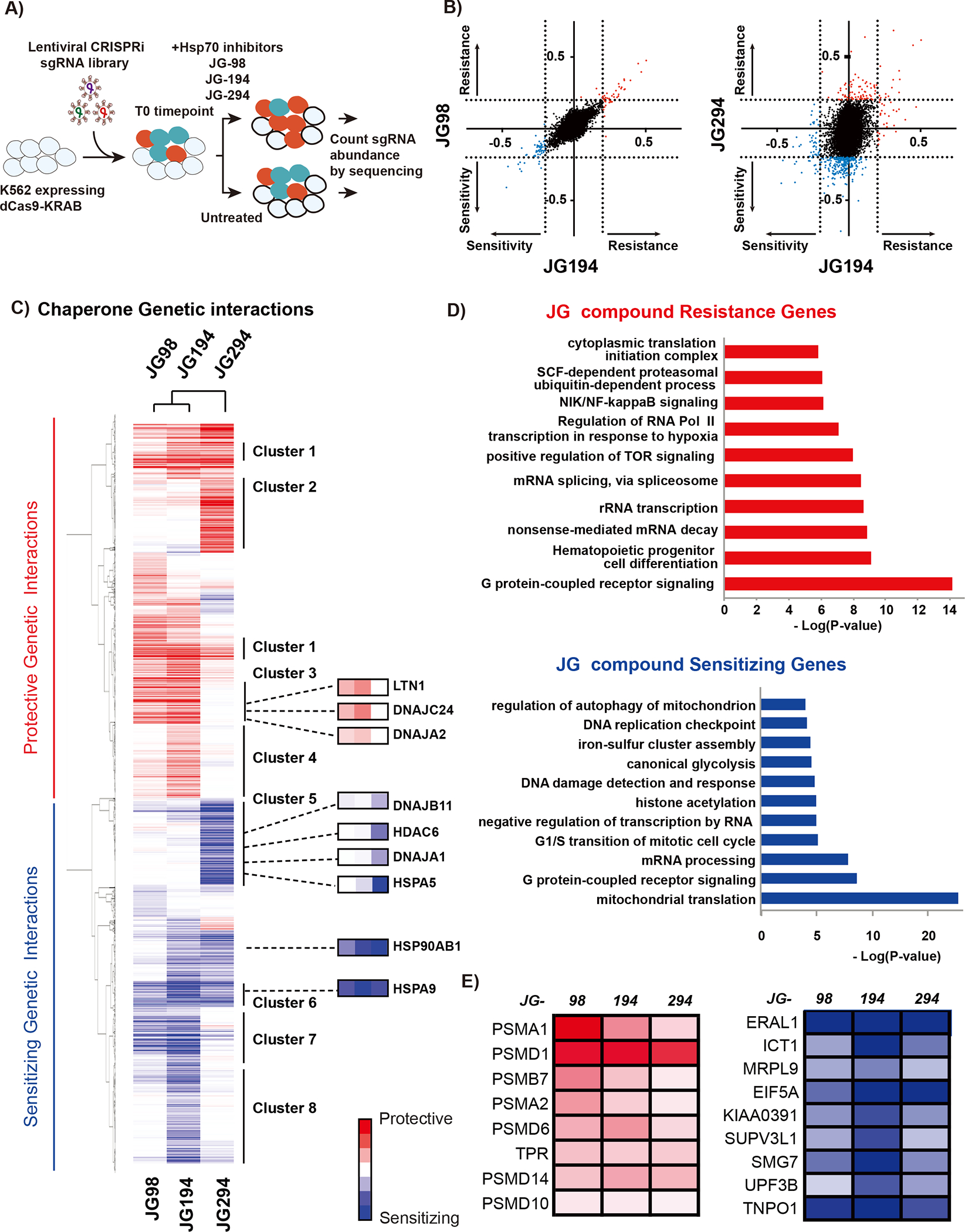
(A) Schematic of CRISPRi screen. dCas9 expressing cells were transduced with the sgRNA library, co-treated with either JG-98, JG-194, or JG-294, and deep sequenced at the initial and final time point to quantify sgRNA counts. (B) Comparison of CRISPRi screen of JG-98 with JG-194 and JG-294, respectively. JG-98 and JG-194 have very similar CRISPRi profiles, while JG-294 is very different. (C) GO analysis reveals differences in classes of genes conferring resistance or sensitivity to JG compounds. (D) Hierarchical clustering of gene score. Only significant genes (p-value > 0.01) in at least 1 condition were included in clustering. Highlighted clusters and genes show patterns of genetic vulnerabilities that drive sensitivity or resistance to JG compounds. (E) Selected protective and sensitizing genes that were shared amongst all three drug treatments. Multiple proteasome subunits were identified as protective, and mitochondrial and translation proteins were identified as sensitizing.
In these experiments, K562 cells stably expressing dCas9-CRAB were infected with a CRISPRi sgRNA library such that each cell expresses on average a single sgRNA (Gilbert et al., 2014). The infected cells were then treated with puromycin to remove uninfected cells and then allowed to recover for 3 days. At time zero (T0) populations of cells were separated and frozen (T0), treated with DMSO or treated with compounds JG-98, JG-194 and JG294 and passaged for ~14 days. Genomic DNA was extracted from time zero, DMSO and JG-treated samples and then the pool of sgRNAs present in each sample was amplified by PCR and quantified by quantitative sequencing for sgRNA quantification. The normalized difference in sgRNA abundance between the DMSO control and JG compounds treated cells was calculated (Gilbert et al., 2014; Kampmann et al., 2013). The phenotype for each gene is plotted as the Rho value against the p-value of its significance in a volcano plot (Supplemental Fig. 3 and Table 4). In this experiment, genes whose knock-down enhances sensitivity to compound treatment are depleted. As partial reduction of the target protein often makes cells more sensitive to compound treatment, the molecular targets of the compounds are likely to be included in these sets of genes. By contrast, genes whose depletion confers resistance to the compound will be enriched in the compound-treated population compared to control cells.
For all three compounds, genes encoding Hsp70s were strongly sensitizing hits, consistent with these proteins being important targets (Fig. 4C). Another prominent set of hit genes consisted of ABC transporters, suggesting these compounds might use transporters to enter cells. Strikingly, we found that JG-98 and JG-194 had similar CRISPRi profiles, while JG-294’s profile was distinct (Fig. 4B). We also found varying degrees of preference for Hsp70 isoforms among the three compounds. All three compounds showed enhanced sensitivity upon mtHsp70/HSPA9 depletion, the Hsp70 isoform that resides in the mitochondria, along with BiP/HSPA5, the Hsp70 in the ER. However, JG-98 and JG-194 were most sensitive to mtHsp70/HSPA9 depletion while JG-294 showed a stronger preference for BiP/HSPA5. In addition, we observed genetic interactions with Hsp70 co-chaperones. However, the different JG compounds had differential and even opposite genetic interactions with specific co-chaperones within the same category, such as J-domain proteins. For instance, depletion of DNAJA2 and DNAJC24 offered protection from JG98 and JG-194, whereas depletion of DNAJA1 and DNAJB11 sensitized cells to JG-294 treatment. Our analysis also revealed that the different Hsp70 inhibitors differentially affect the proteostasis network, including interactions with Hsp90, VCP, distinct ubiquitination components and proteasome subunits (Fig. 4C and 4E). In addition, we discovered that depletion of HSP90AB1 exacerbated the impact of all three compounds. By contrast, depletion of proteasome components fueled resistance to all compounds. Together, these results suggest that, despite targeting a conserved site in Hsp70, the different JG compounds compromise the functions of specific chaperone complexes and processes.
To more broadly explore similarities and differences in the response of cells to JG compounds, we clustered the sensitive and resistant genes and performed GO term enrichment analysis (Fig. 5; Supplemental Fig. 5). Clusters 1 and 6 were shared by all three compounds (JG-98/194/294) and they were defined by processes of protein trafficking and quality control, as well as RNA processing. JG-98 and JG-194 shared Clusters 3 and 7, which were defined by translation/ribosome biogenesis (Cluster 3) and mitochondrial proteostasis (Cluster 7). These clusters suggest that the JG compounds do share some common chemogenetic interactions. However, individual responses were also observed. For example, JG-194 was associated with Clusters 4 and 8 (DNA/RNA replication and processing), and JG-294 with Clusters 2 and 5 (cytokinesis and oxidative phosphorylation). These signatures will require more validation to determine which ones are on- and off-target, and which ones are direct or indirect. Yet, it is striking that closely related Hsp70-targeting compounds within the same series yield partially distinct signatures in chemogenetic profiling. This property is usually invisible to chemical biologists and medicinal chemists embarking on synthetic campaigns for new chemical matter because full profiling is typically performed only at the conclusion of the campaign on the most advanced molecule. As CRISPRi screens become more routinely available, it seems feasible to expand the analysis to “earlier” compounds, perhaps providing a route to discover analogs that act upon specific sub-cellular locations and sub-networks.
Figure 5: GO analysis of gene clusters from CRISPRi screen.
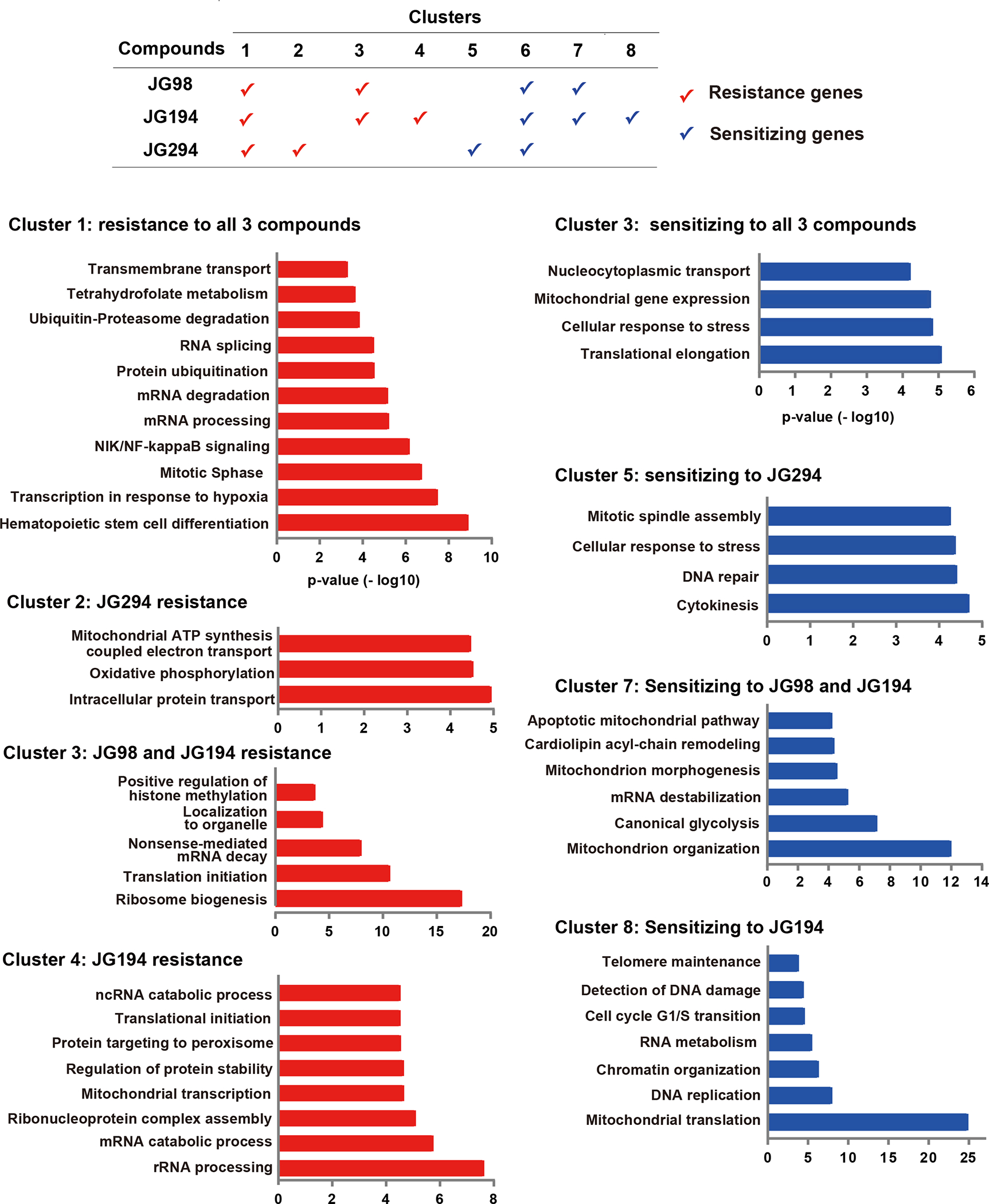
JG-98/194/294 shared gene clusters 1 and 6; JG-98/194 shared gene clusters 3 and 7. Clusters 4 and 8 are unique for JG-194, and clusters 2 and 5 are unique for JG-294. Gene clusters were analyzed and annotated by Gene Ontology Enrichment Analysis through the PANTHER v15 database.
The JG compounds inhibit dengue virus replication, but with a different rank ordering than that observed in cancer cells.
Because JG compounds have different chemogenetic signatures, we hypothesize that the rank-order of the compound’s potency, as originally determined in cancer cell growth assays, might not always be the same in other Hsp70-dependent assays. Hsp70 functions are required for Dengue virus (DENV) infection at several steps of its lifecycle, including entry, RNA replication and particle production (Taguwa et al., 2015). Thus, we tested the effect of the JG compounds on DENV replication to compare the rank-ordering to that obtained in the anti-cancer assays. To enable further comparisons, we also performed these DENV studies in cells from two distinct, relevant hosts: human and insect. Specifically, Huh7 (human) and C6/36 (mosquito) cells were infected with DENV and treated with increasing concentrations of JG compounds from 2 h post infection. Then, virus production was examined as previously described (Taguwa et al., 2015; Taguwa et al., 2019) (Fig 6A, 6B, and Supplemental Table 5). At the same time, we also determined the effects of compounds on cell viability, to exclude confounding effects from cell death (Supplemental Fig. 5D). Strikingly, the JG compounds with the strongest inhibitory effect on DENV propagation tended to be different from those showing the greatest potency in cancer cells (Fig 6D). Generally, early-stage/Type I compounds showed better potency and an improved SI in both human (Huh7) and mosquito (C6/36) cell lines, when compared to late stage/Type II compounds (Fig. 6C). Thus, the “best” Hsp70 inhibitor is not always the same in different biological models (i.e., anti-cancer vs anti-viral). These differences might be due to differences in the compound’s chemogenetic interactions within specific cell types, because we found that the relative potencies were even different in human and mosquito cells (Fig. 6A–C). For example, MKT-077, JG21, JG36, and JG132 suppressed DENV2 propagation in C6/36 by more than two logs, while JG40, JG98, JG194 and JG345 were the most active molecules in Huh7 cells. However, we also considered the possibility that these differences might also be due to subcellular localization. For example, DENV proteins are known to assemble within viral factories that promote replication in an Hsp70 dependent manner (Taguwa et al., 2015; Welsch et al., 2009). Thus, differential co-localization of compounds with these regions might underlie variation in potency. To test this idea, we repeated the confocal microscopy studies in human A549, Huh7 and mosquito C3/36 cells. Strikingly, we found that the compounds had indistinguishable subcellular localizations in all three cells types (Supplemental Fig. 5A–B) and that expression of DENV in the Huh7 cells did not alter these localizations (Supplemental Fig. 5C). Taken together, these results suggest that partitioning may be exploited to more safely inhibit Hsp70 subnetworks.
Figure 6: Antiviral activity of JG compounds in distinct host cells.
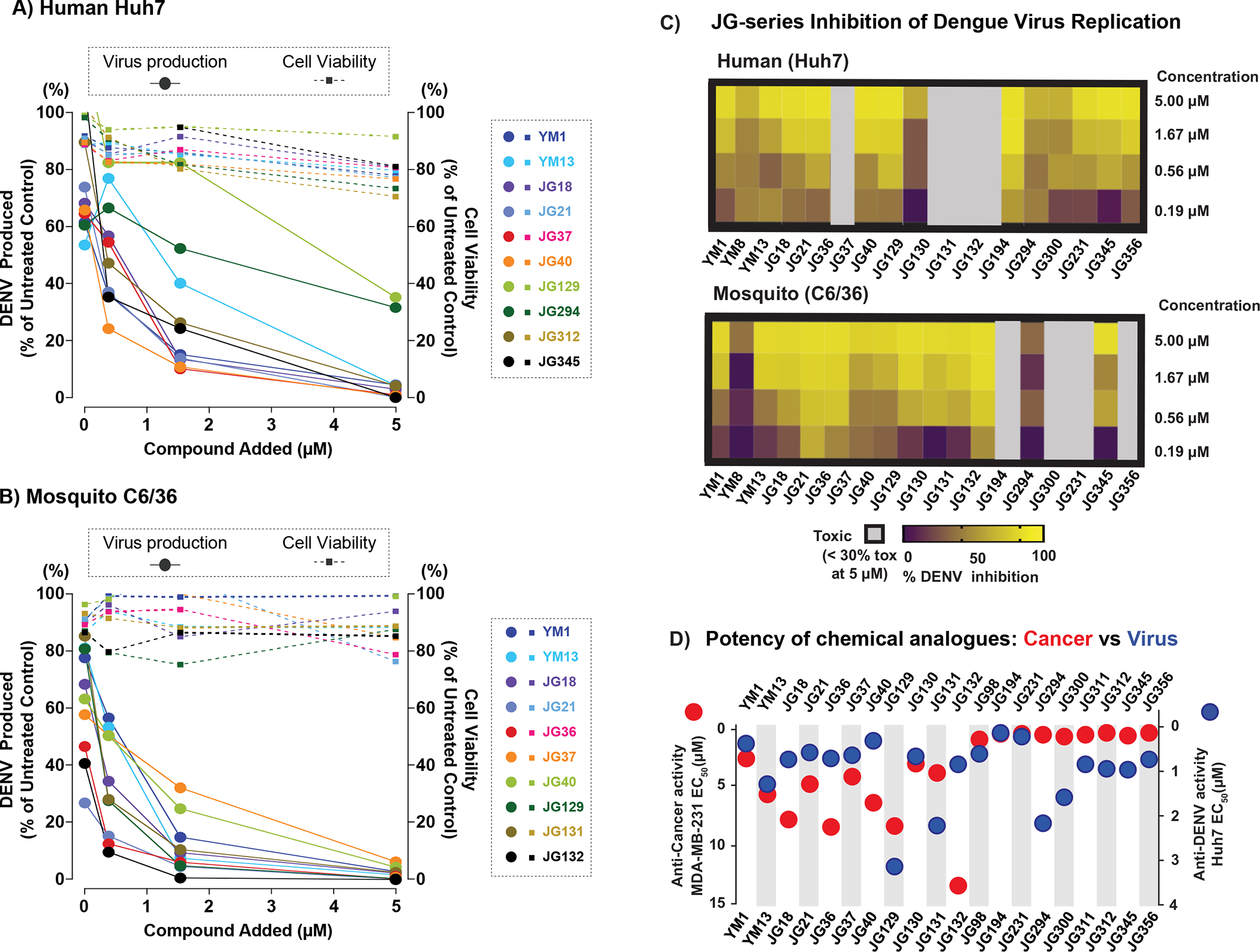
(A-C) Effect of JG compounds on DENV productivity and cell viability in mammalian Huh7 (A) and mosquito C6/36 (B) cells. Each virus productivity was shown as square and dashed line (left Y axis) and cell viability as circle and solid line (right Y-axis). X-axis: drug concentration (0.56, 1,66 and 5 μM). (C) Heat map illustrating anti DENV effect in Huh7(i) and C6/36(ii). Highly toxic JG compounds showed as grey color. (D) The comparison of two distinct activities of JG compounds. Anti-cancer activity in MDA-MB-231 cells (red circles, left axis) and anti-DENV activity in Huh7(blue circles, right axis) were shown with EC50.The data shown are average of three independent experiments.
DISCUSSION
Dysfunction in proteostasis is emerging as a major driver of human diseases, ranging from cancer, to viral infection, neurodegeneration, and cystic fibrosis (Balch et al., 2008). Accordingly, compounds that target proteostasis factors hold promise as therapeutics for a broad range of diseases. However, this strategy also presents a number of challenges. For example, core proteostasis factors, such as Hsp70 and Hsp90, are located in multiple cellular compartments and collaborate with a range of co-chaperones to perform complex tasks. Some of these tasks are likely to be related to the disease, while others are housekeeping functions. Thus, one concern is that toxicity might arise when therapeutically targeting these factors (Sanchez et al., 2020). One possible avenue to circumvent these challenges is to identify inhibitors that are specific for disease-relevant proteostasis pathways, meaning that they target only a subset of the chaperone pool – as defined by both (i) sub-cellular location and (ii) the specific subset of co-chaperones bound to the chaperone.
Here, we explored whether subcellular localization provide an approach to focus compound activity. Indeed, drug discovery campaigns against the mitochondrial Hsp90 chaperone (Joshi et al., 2018) have used chemical addresses, appended to the inhibitor, to achieve a similar goal. We focused our efforts on a set of closely related Hsp70 inhibitors because the binding site of these compounds is conserved across Hsp70 family members found in all major compartments (Li et al., 2013; Rousaki et al., 2011; Shao et al., 2018). Moreover, it was already known that these compounds are relatively well-tolerated in non-transformed cells and even in animal models (Moses et al., 2018; Taguwa et al., 2019), indicating their activity may be higher against the cancer Hsp70 epichaperome. Using microscopy, we found that partitioning to distinct compartments might contribute to their relative safety. Specifically, Type II compounds tended to have superior potency and SI values and were localized to ER/vesicles. These findings resonate with recent reports that some small molecules partition to phase-separated membrane-less compartments (Klein et al., 2020). Indeed, we see some of the JG compounds partitioning to nucleoli, a nuclear membrane-less compartment that exhibits properties consistent with phase separation. Multi-protein complexes can form such condensates via liquid–liquid phase separation, which creates a unique biochemical environment and offers a means to regulate cellular processes (Alberti et al., 2019). Thus, small molecules that partition into specific subcellular organelles and/or into condensates can likely act on their targets more effectively and specifically by achieving higher local concentrations than those available in the rest of the cell. This conceptual paradigm might be extended to protein assemblies as well as virus-induced compartments (Etibor et al., 2021).
What is the mechanism of compound partitioning? We found that early-stage/Type I JG compounds were generally found in mitochondria and nucleoli, whereas late-stage/Type II JG compounds colocalized with ER and vesicles. These subcellular distributions could theoretically be affected by many factors, including (but not limited to) physicochemical properties (e.g., charge, lipophilicity), uptake/efflux transporters, protein binding and differential metabolism. Indeed, our CRISPRi screens demonstrated that knockdown of ABC transporters impacts the potency of these molecules, suggesting that drug uptake/efflux might be one factor driving differential localization. In addition, we found a correlation between molecular weight and the cellular distribution of JG compounds, with small (<500 Da) examples (early/Type I) localized to the mitochondria and nucleus. It is not clear why this relationship exists, but it might relate to the ability of ABC transporters to accommodate specific compounds. Despite these speculations, the molecular mechanisms that govern partitioning are not known and further work will be required to define their specificity. Rather, we propose that the strength of the current work is to suggest that such mechanistic studies are worthwhile because partitioning appears to be correlated with function.
CRISPRi screens are being increasingly used to identify chemogenetic interactions of compounds with cellular pathways (Jost and Weissman, 2018; Kampmann, 2017). Many groups are now routinely performing such studies on the “final” chemical probe or clinical candidate, to confirm target engagement, reveal biomarkers and identify potential off-targets. Our work suggests that periodic screens performed on intermediate compounds within an ongoing medicinal chemistry campaign, not just the final candidate, could inform the evolution of these properties. For example, we observed a reliance on Hsp70s, as expected (Shao et al., 2018), but also identified compound-specific patterns of sensitivity and resistance related to specific cellular complexes and processes, including mitochondria, glycolysis, and RNA metabolism, among others. If such CRISPRi studies had been performed during the medicinal chemistry campaign, we would have been attuned to these changing chemogenetic interactions. This concept seems especially important for the Hsp70 system, because CRISPRi approaches provide insight into the Hsp70 subnetworks (epichaperomes) that are important for a compound’s activity (Fig 7A). For example, we found that knockdown of genes linked to the proteasome provided resistance to all JG compounds tested, potentially because inhibiting proteasomal degradation gives non-native polypeptides more time to fold. While that result might be expected, other findings were more interesting. For example, while knockdown of translation initiation components and mRNA biogenesis provided resistance to JG-98 and JG-194, perhaps by reducing the nascent chain load on available Hsp70 capacity; knockdown of translation elongation components enhanced sensitivity to JG-294, perhaps by increasing release of truncated incomplete nascent chains. All three of these compounds (JG-98, JG-194 and JG-194) localize to the ER/vesicles, so their differential chemogenomic interactions are not explained by distribution alone. It seems likely that these compounds could affect specific Hsp70 subcomplexes (Fig 7A–B), through mechanisms that are not clear. One possibility in that differences in post-translational modifications (PTMs) of Hsp70s and their co-chaperones, which are known to be widespread (Nitika et al., 2020), could alter the relative binding of compounds to these complexes.
Figure 7: A model for Hsp70 functions and selective functional inhibition of Hsp70s by JG compounds in different diseases models.
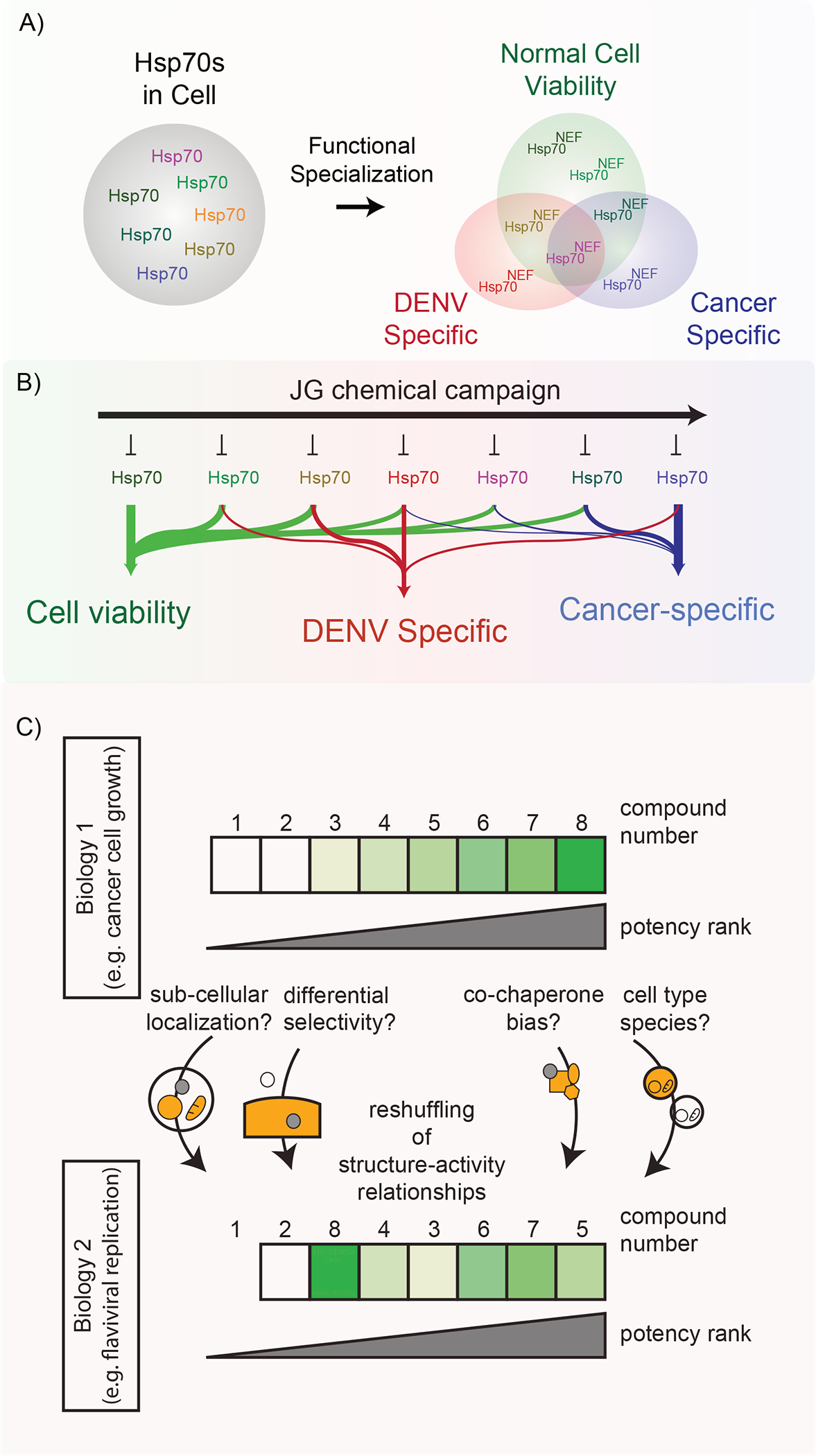
(A) Multiple Hsp70 isoforms exists in cells, and different diseases might rely on distinct Hsp70 and its co-chaperones. (B) The optimal JG compounds for anti-cancer and anti-viral activities are different. (C) A chemical campaign targeting a conserved proteostasis factor may achieve specificity towards distinct biological activities through subcellular localization and/or partitioning to distinct subcomplexes.
An important, practical conclusion of this study is that the top ranking Hsp70 inhibitor for one application (e.g., cancer cell growth) might not be best compound for another assay (e.g., DENV replication; Fig. 7C). Why might this be? Our results suggest that the anti-cancer potency of an Hsp70 inhibitor is likely a product of multiple factors, including the reliance of that cell’s viability on specific Hsp70 complexes within specific subcellular locations. For anti-viral therapy, however, it seems that different Hsp70 sub-networks, in different locations, could be involved. Thus, for Hsp70 systems, it seems unlikely that a single “optimal” chemical probe is possible or even desired. Rather, a better framework might be to consider a chemical probe as only optimal for a specific complex/epichaperome (Gestwicki and Shao, 2019).
More broadly, these results add to our understanding of how to safely target major nodes of the proteostasis network. One classic way to achieve this goal has been to take advantage of small structural differences between closely related isoforms of the same target, expressed in different subcellular regions, to design compounds that only bind one isoform. Indeed, this strategy has been done with great effect for Hsp90 inhibitors, improving their safety (Khandelwal et al., 2018; Mishra et al., 2021; Que et al., 2018; Zhang et al., 2019). However, we propose that specificity could also arise from physical properties of the compounds themselves, such as size, charge, and the ability to partition into specific structures, such as membraneless condensates, cellular assemblies, and/or viral replication/assembly compartments. While these latter properties cannot currently be achieved rationally, a better understanding of this process may open new avenues in drug design. Given that the mechanism and rules driving the subcellular distribution of small molecules are unclear, the differences in potency of general inhibitors for various applications needs, for the moment, to be empirically determined. If chemical biologists could learn the “rules” of compound localization, we could likely build superior probes and drugs.
Limitations of the study:
Our findings raise the exciting possibility of exploiting specific properties, such as size or subcellular localization, to identify disease- and function specific inhibitors while targeting a broadly acting cellular factor, such as a chaperone. One limitation of our findings that should be allayed by further investigation is that the molecular mechanisms that govern molecule partitioning and diseases specificity are not yet clear. Thus, at this point identification of the right compounds for a given indication will require broad screening of a family of compounds.
SIGNIFICANCE:
It has been difficult to identify inhibitors of proteostasis targets that are both effective and safe, likely because these targets play multiple roles. One potential way to circumvent this issue is to localize inhibitors to specific subcellular compartments and/or direct them to specific sub-networks. Here we show that despite the conservation of Hsp70s, chemical campaigns can yield inhibitors that act on only a subset of Hsp70-requiring processes, as revealed by CRISPR-based chemogenomics. This selectivity seems to originate, in part, from differential sub-cellular localization of the compounds. We suggest that subcellular partitioning provides a way to impart selectivity to inhibitors that target otherwise ubiquitous proteins.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Judith Frydman (jfrydman@leland.stanford.edu) and Jason Gestwicki (jason.gestwicki@ucsf.edu).
Materials availability
Correspondence and requests for materials should be addressed to the Lead Contact.
Data and code availability
All data are available in the main text or as Supplemental information. The full list of Rho- and p-values for the CRISPRi screens are in Table S4. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL and SUBJECT DETAILS
Cell lines
293T, K562 and Huh7 cells were cultivated in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% nonessential amino acids, 100 U/mL penicillin and 100 mg/mL streptomycin. C6/36 (larva whole, Aedes albopictus mosquito) cells were maintained in minimum essential medium (MEM) with 10% FBS, nonessential amino acids, 100 U/ml penicillin and 100 mg/ml streptomycin. 293T, K562 and Huh7 cells were maintained at 37 °C and C6/36 cells at 32°C in a humidified 5% CO2 atmosphere.
Viruses
The infectious cDNA clone pD2/IC-30P-A of DENV2 (strain 16681) was used to transcribe viral RNA in vitro that was electroporated into Huh7 cells to produce infectious DENV2. The supernatant containing infectious virus were harvested and and stored in −80°C.
METHOD DETAILS
Chemical synthesis
Compounds used in this manuscript were synthesized as reported previously (Li et al., 2013; Shao et al., 2018) and characterized by 1H NMR and LC-MS/MS for identity. All compounds were >95% pure by HPLC.
qPCR analysis
For qPCR analysis, cell pellets were thawed and processed using the RNeasy Mini Kit (Qiagen) with the optional on column RNase free DNAase (Qiagen) treatment. cDNA was then generated using the RETROscript Reverse Transcription Kit (Ambion, ThermoScientific). A 1:80 dilution of cDNA was used for Real-Time PCR using a StepOnePlus System (Applied Biosystems, ThermoScientific) using the primers in Supplemental Table 2, and the data were analyzed using an efficiency normalized relative expression ratio (Pfaffl, M. Nucleic Acid Research 2001;29(9):e45 PMCID: PMC55695) to determine fold change in mRNA compared to a DMSO-treated control.
Western blotting
K562 cells were seeded onto 6-well plates at a density of 200K cells/ml and grown for 24 hrs in DMEM + 10% FBS. The cells were then treated with 4 times the GI50 (calculated by CellTiter Glo Assay (Promega)) for an additional 6 hrs. Following treatment, cells were harvested by centrifugation for 1 min @ 18,000 g. After aspirating the culture media, cell pellets were stored at −80 °C. For Western blotting, cell pellets were thawed on ice and lysed in radioimmunoprecipitation assay (RIPA) buffer (25 mM Tris·HCl, pH 7.6, 150 mM NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS supplemented with complete Mini, EDTA-free protease inhibitor cocktail (Roche)) by 10 passes through a 28 gauge needle. Unbroken cells and debris were pelleted for 15 min at 18,000 g at 4°C. Protein concentration was determined using the Pierce BCA Protein Assay Kit (ThermoScientific) using bovine serum albumin for the standard curve. Equal protein was loaded and resolved using 10% SDS-polyacrylamide gels followed by semi-dry transfer onto nitrocellulose membranes for protein detection using the following primary antibodies: anti-CHOP, anti-ATF4, anti-phospho-eIF2α (Cell Signaling Technology at 1:1000), anti-eIF2α (Cell Signaling Technology at 1:2000), and anti-Hsp70 (StressMarq Biosciences at 1:1000). Anti-β-actin (Cell Signaling Technology, at 1:2000) was used as loading control. HRP-conjugated secondary antibodies (anti-rabbit and anti-mouse IgG, Cell Signaling Technology at 1:4000) were then applied for 2 hrs at room temperature prior to imaging. Bound antibodies were detected with SuperSignal West Pico or West Femto Chemiluminescent Substrate (Pierce, ThermoScientific), and images were acquired using a ChemiDoc XRS+ (BioRad). Data were analyzed using ImageJ software (National Institutes of Health, NIH).
Fluorescence microscopy
mTurquoise2 blue fluorescent protein fused with KDEL or Mitochondria targeting sequences were designated as cellular organelle markers of ER or mitochondria, respectively. The cells expressing each turquoise2 cultured on glass bottom plate were incubated with media containing JG compounds at 37°C for 120 min and imaged using a LSM-700 scanning confocal microscope (Zeiss). The images were merged with image J software.
CRISPRi screening
A chronic myeloid leukemia (K562) cell line stably expressing dCas9-KRAB was infected with a CRISPRi sgRNA library. These cells were used because they have been shown to be readily infected and high efficiency and they have stable expression of dCas9-KRAB. The genomic DNA from a population of these cells was harvested at the onset of the experiment (t0) and used as a comparison with the subsequent time point. Half the remaining cells were treated with JG compound at the EC40–70 value and the two populations grown for 13 days. At each passage, control and drug treated populations of cells were maintained such that there was >1000 cells per sgRNA. At the end of the 7 days, genomic DNA was harvested. Then, the cassette encoding the sgRNA was amplified by PCR from the t0, t13 (untreated), and t13 (compound treated) samples. The relative sgRNA abundance was determined by next generation sequencing as previously described (Jost et al., 2017; Kampmann et al., 2013). Hit genes were ranked based on average phenotype of the three most extreme sgRNAs targeting them or by a Mann−Whitney test. A Python script for processing the CRISPRi library sequencing data is available at https://github.com/mhorlbeck/ScreenProcessing.
Go term analysis, statistics and clustering
A score was generated for each hit gene by multiplying the determined phenotype by −log(p-value). Gene scores were clustered hierarchically in Cluster, as described in (Eisen et al., 1998), and displayed by Java TreeView (Saldanha, 2004). Gene clusters were further analyzed and annotated by Gene Ontology Enrichment Analysis through the PANTHER v15 database (Ashburner et al., 2000; Gene Ontology, 2021; Mi et al., 2019).
Virus assays
DENV2 16681 strain infected 1 × 105 (Huh7) and 5 × 105 (C6/36) cells at MOI 0.5 for 1 hour. The media was replaced with media containing compounds or DMSO and the extracellular virus was harvested at 48 hours post infection by collecting culture supernatant and pelleting debris at 1800 rpm for 5 min. For virus titration by focus forming assay, confluent Huh7 or C6/36 cultured in 48-well plates were inoculated with a limiting 10-fold dilution series of virus in culture medium with 2% FBS for 1 h. The inoculum was removed and cells overlaid with media supplemented with 0.8% methylcellulose and 2% FBS. After 3 days, cells were fixed in 4% paraformaldehyde in PBS and permeabilized with 0.5% Triton X-100. Infectious foci were stained with anti-E antibody and visualized with a VECTASTAIN Elite ABC anti-mouse IgG kit with a VIP substrate (Vector Laboratories, Burlingame, CA USA).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data plotting and statistical analyses were performed using Prism 7 (GraphPad Software). Details on the number of technical and biological (independent) replicates of each experiment can be found in the figure legends.
Supplementary Material
Supplemental Table 3: Subcellular distributions and physicochemical properties of JG compounds. Related to Figure 3.
Supplemental Table 4: The genes list from CRISPRi screen with Rho and p-value. Related to Figure 4 and 5.
Supplemental Table 5 Antiviral and cell growth activity of JG compounds in Huh7 and C6/36 cells. Related to Figure 6.
Supplemental Table 1: Chemical structures and anti-proliferative activities of JG compounds in this study. Related to Figure 1.
Supplemental Table 2: The list of primers for RT-qPCR. Related to Figure 2.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HSP70 | StressMarq Biosciences | Cat # SMC-113A |
| ATF4 | Cell Signaling Technologies | Cat # 11815 |
| CHOP | Cell Signaling Technologies | Cat # 2895 |
| peIF2alpha | Cell Signaling Technologies | Cat # 9721 |
| eIF2alpha | Cell Signaling Technologies | Cat # 5324 |
| anti-mouse-HRP | Cell Signaling Technologies | Cat # 7076 |
| anti-rabbit-HRP | Cell Signaling Technologies | Cat # 7074 |
| β-actin | Cell Signaling Technologies | #4867L |
| Bacterial and Virus Strains | ||
| DENV2 16681 Strain | Gift from CDC | N/A |
| Chemicals | ||
| JG40 | (Li et al., 2013) | N/A |
| JG98 | (Li et al., 2013) | N/A |
| JG194 | (Shao et al., 2018) | N/A |
| JG258 | (Shao et al., 2018) | N/A |
| JG294 | (Shao et al., 2018) | N/A |
| JG345 | (Shao et al., 2018) | N/A |
| Rneasy mini kit | Qiagen | Cat # 74106 |
| High Capacity cDNA Reverse Transcription Kit | Thermo Fisher scientific | Cat # 4368813 |
| iTaqTM Universal SYBR Green Supermix | Bio-Rad | Cat # 1725125 |
| DMEM, high glucose, GlutaMAX™ Supplement | Thermo Fisher scientific | Cat # 10566016 |
| MEM, GlutaMAX™ Supplement | Thermo Fisher scientific | Cat # 41090036 |
| Experimental Models: Cell Lines | ||
| Huh7 | Gift from Prof. Yoshi Matsuura | N/A |
| 293T | ATCC | Cat # CRL-3216 |
| C6/36 | Gift from Prof. Yoshi Matsuura | N/A |
| K562 | Prof. Jonathan Weissman lab | N/A |
| Oligonucleotides | ||
| See Table S2 for the list of primers for qRT-PCR | ||
| Recombinant DNA | ||
| pLEF-MTS-mTurquoise2 | This study | N/A |
| pLEF-ER-mTurquoise2 | This study | N/A |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Excel | Microsoft | N/A |
| PRISM | Graphpad | https://www.graphp/ad.com |
| ImageLab | Biorad | https://www.bio-rad.com |
| Cluster | (Eisen et al., 1998) | http://bonsai.hgc.jp/mdehoon/software/cluster/software.htm |
| Java TreeView | (Saldanha, 2004) | http://jtreeview.sourceforge.net |
| Python | Python | https://www.python.org |
Highlights.
Distinct Hsp70 sub-networks are targeted by chemically related inhibitors.
Closely related Hsp70 inhibitors display distinct subcellular localizations.
Differently localized Hsp70 inhibitors have anti-cancer versus anti-viral activities.
Subcellular localization may contribute to low toxicity of certain Hsp70 inhibitors.
ACKNOWLEDGEMENTS.
This work was supported by grants from the NIH (NS059690 to J.E.G., AI127447 and AG054407 to JF, DK101584 to C.J.G., and GM131732 and DK079307 to J.L.B.), and the National Natural Science Foundation of China to H.S. (grant no. 82104001), and by a grant from the long-term European Molecular Biology Organization via a post-doctoral fellowship (ALTF 823–2016) to S.S. and Uehara Memorial Foundation and Naito foundation for S.T.
Footnotes
DECLARATION OF INTERESTS. none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abisambra J, Jinwal UK, Miyata Y, Rogers J, Blair L, Li X, Seguin SP, Wang L, Jin Y, Bacon J, et al. (2013). Allosteric heat shock protein 70 inhibitors rapidly rescue synaptic plasticity deficits by reducing aberrant tau. Biol Psychiatry 74, 367–374. 10.1016/j.biopsych.2013.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti S, Gladfelter A, and Mittag T (2019). Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 176, 419–434. 10.1016/j.cell.2018.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrose AJ, and Chapman E (2021). Function, Therapeutic Potential, and Inhibition of Hsp70 Chaperones. J Med Chem 64, 7060–7082. 10.1021/acs.jmedchem.0c02091. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29. 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assimon VA, Southworth DR, and Gestwicki JE (2015). Specific Binding of Tetratricopeptide Repeat Proteins to Heat Shock Protein 70 (Hsp70) and Heat Shock Protein 90 (Hsp90) Is Regulated by Affinity and Phosphorylation. Biochemistry 54, 7120–7131. 10.1021/acs.biochem.5b00801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch WE, Morimoto RI, Dillin A, and Kelly JW (2008). Adapting proteostasis for disease intervention. Science 319, 916–919. 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- Balchin D, Hayer-Hartl M, and Hartl FU (2016). In vivo aspects of protein folding and quality control. Science 353, aac4354. 10.1126/science.aac4354. [DOI] [PubMed] [Google Scholar]
- Bernardi P (1999). Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiological reviews 79, 1127–1155. 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- Bracher A, and Verghese J (2015). The nucleotide exchange factors of Hsp70 molecular chaperones. Front Mol Biosci 2, 10. 10.3389/fmolb.2015.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerezo M, Lehraiki A, Millet A, Rouaud F, Plaisant M, Jaune E, Botton T, Ronco C, Abbe P, Amdouni H, et al. (2016). Compounds Triggering ER Stress Exert Anti-Melanoma Effects and Overcome BRAF Inhibitor Resistance (vol 29, pg 805, 2016). Cancer Cell 30, 183–183. 10.1016/j.ccell.2016.06.007. [DOI] [PubMed] [Google Scholar]
- Craig EA, and Marszalek J (2017). How Do J-Proteins Get Hsp70 to Do So Many Different Things? Trends in Biochemical Sciences 42, 355–368. 10.1016/j.tibs.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, and Botstein D (1998). Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A 95, 14863–14868. 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etibor TA, Yamauchi Y, and Amorim MJ (2021). Liquid Biomolecular Condensates and Viral Lifecycles: Review and Perspectives. Viruses 13. 10.3390/v13030366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans CG, Chang L, and Gestwicki JE (2010). Heat shock protein 70 (hsp70) as an emerging drug target. Journal of medicinal chemistry 53, 4585–4602. 10.1021/jm100054f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantz M-C, and Wipf P (2010). Mitochondria as a target in treatment. Environmental and Molecular Mutagenesis 51, 462–475. 10.1002/em.20554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Ontology C (2021). The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res 49, D325–D334. 10.1093/nar/gkaa1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gestwicki JE, and Shao H (2019). Inhibitors and chemical probes for molecular chaperone networks. J Biol Chem 294, 2151–2161. 10.1074/jbc.TM118.002813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. (2014). Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661. 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe MK, Bodoor K, Carlson DA, Hughes PF, Alwarawrah Y, Loiselle DR, Jaeger AM, Darr DB, Jordan JL, Hunter LM, et al. (2014). Identification of an allosteric small-molecule inhibitor selective for the inducible form of heat shock protein 70. Chem Biol 21, 1648–1659. 10.1016/j.chembiol.2014.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S, Wang T, Araujo TLS, Sharma S, Brodsky JL, and Chiosis G (2018). Adapting to stress - chaperome networks in cancer. Nat Rev Cancer 18, 562–575. 10.1038/s41568-018-0020-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost M, Chen Y, Gilbert LA, Horlbeck MA, Krenning L, Menchon G, Rai A, Cho MY, Stern JJ, Prota AE, et al. (2017). Combined CRISPRi/a-Based Chemical Genetic Screens Reveal that Rigosertib Is a Microtubule-Destabilizing Agent. Mol Cell 68, 210–223 e216. 10.1016/j.molcel.2017.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost M, and Weissman JS (2018). CRISPR Approaches to Small Molecule Target Identification. ACS Chem Biol 13, 366–375. 10.1021/acschembio.7b00965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampinga HH, Andreasson C, Barducci A, Cheetham ME, Cyr D, Emanuelsson C, Genevaux P, Gestwicki JE, Goloubinoff P, Huerta-Cepas J, et al. (2019). Function, evolution, and structure of J-domain proteins. Cell Stress Chaperones 24, 7–15. 10.1007/s12192-018-0948-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampmann M (2017). Elucidating drug targets and mechanisms of action by genetic screens in mammalian cells. Chem Commun (Camb) 53, 7162–7167. 10.1039/c7cc02349a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampmann M, Bassik MC, and Weissman JS (2013). Integrated platform for genome-wide screening and construction of high-density genetic interaction maps in mammalian cells. Proc Natl Acad Sci U S A 110, E2317–2326. 10.1073/pnas.1307002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelwal A, Kent CN, Balch M, Peng S, Mishra SJ, Deng J, Day VW, Liu W, Subramanian C, Cohen M, et al. (2018). Structure-guided design of an Hsp90beta N-terminal isoform-selective inhibitor. Nat Commun 9, 425. 10.1038/s41467-017-02013-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein IA, Boija A, Afeyan LK, Hawken SW, Fan M, Dall’Agnese A, Oksuz O, Henninger JE, Shrinivas K, Sabari BR, et al. (2020). Partitioning of cancer therapeutics in nuclear condensates. Science 368, 1386–1392. 10.1126/science.aaz4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundrat L, and Regan L (2010). Balance between Folding and Degradation for Hsp90-Dependent Client Proteins: A Key Role for CHIP. Biochemistry 49, 7428–7438. 10.1021/bi100386w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Srinivasan SR, Connarn J, Ahmad A, Young ZT, Kabza AM, Zuiderweg ER, Sun D, and Gestwicki JE (2013). Analogs of the Allosteric Heat Shock Protein 70 (Hsp70) Inhibitor, MKT-077, as Anti-Cancer Agents. ACS Med Chem Lett 4. 10.1021/ml400204n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louzoun-Zada S, Jaber QZ, and Fridman M (2019). Guiding Drugs to Target-Harboring Organelles: Stretching Drug-Delivery to a Higher Level of Resolution. Angewandte Chemie 58, 15584–15594. 10.1002/anie.201906284. [DOI] [PubMed] [Google Scholar]
- Mayer MP, and Gierasch LM (2019). Recent advances in the structural and mechanistic aspects of Hsp70 molecular chaperones. J Biol Chem 294, 2085–2097. 10.1074/jbc.REV118.002810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Ebert D, Huang X, and Thomas PD (2019). PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res 47, D419–D426. 10.1093/nar/gky1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra SJ, Khandelwal A, Banerjee M, Balch M, Peng S, Davis RE, Merfeld T, Munthali V, Deng J, Matts RL, and Blagg BSJ (2021). Selective Inhibition of the Hsp90alpha Isoform. Angew Chem Int Ed Engl 60, 10547–10551. 10.1002/anie.202015422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Fanourakis G, Gu X, Bailey C, Joseph M, Libermann TA, Treon SP, et al. (2002). Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc Natl Acad Sci U S A 99, 14374–14379. 10.1073/pnas.202445099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses MA, Kim YS, Rivera-Marquez GM, Oshima N, Watson MJ, Beebe KE, Wells C, Lee S, Zuehlke AD, Shao H, et al. (2018). Targeting the Hsp40/Hsp70 Chaperone Axis as a Novel Strategy to Treat Castration-Resistant Prostate Cancer. Cancer Res 78, 4022–4035. 10.1158/0008-5472.CAN-17-3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitika Porter, C.M., Truman AW, and Truttmann MC (2020). Post-translational modifications of Hsp70 family proteins: Expanding the chaperone code. J Biol Chem 295, 10689–10708. 10.1074/jbc.REV120.011666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodromou C, Siligardi G, O’Brien R, Woolfson DN, Regan L, Panaretou B, Ladbury JE, Piper PW, and Pearl LH (1999). Regulation of Hsp90 ATPase activity by tetratricopeptide repeat (TPR)-domain co-chaperones. EMBO J 18, 754–762. 10.1093/emboj/18.3.754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que NLS, Crowley VM, Duerfeldt AS, Zhao J, Kent CN, Blagg BSJ, and Gewirth DT (2018). Structure Based Design of a Grp94-Selective Inhibitor: Exploiting a Key Residue in Grp94 To Optimize Paralog-Selective Binding. J Med Chem 61, 2793–2805. 10.1021/acs.jmedchem.7b01608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radons J (2016). The human HSP70 family of chaperones: where do we stand? Cell Stress Chaperon 21, 379–404. 10.1007/s12192-016-0676-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch JN, and Gestwicki JE (2014). Binding of human nucleotide exchange factors to heat shock protein 70 (Hsp70) generates functionally distinct complexes in vitro. J Biol Chem 289, 1402–1414. 10.1074/jbc.M113.521997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi S, Assimon VA, Young ZT, Morra G, Shao H, Taylor IR, Gestwicki JE, and Colombo G (2018). A Local Allosteric Network in Heat Shock Protein 70 (Hsp70) Links Inhibitor Binding to Enzyme Activity and Distal Protein-Protein Interactions. ACS chemical biology 13, 3142–3152. 10.1021/acschembio.8b00712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodina A, Patel PD, Kang Y, Patel Y, Baaklini I, Wong MJ, Taldone T, Yan P, Yang C, Maharaj R, et al. (2013). Identification of an allosteric pocket on human hsp70 reveals a mode of inhibition of this therapeutically important protein. Chem Biol 20, 1469–1480. 10.1016/j.chembiol.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodina A, Wang T, Yan P, Gomes ED, Dunphy MP, Pillarsetty N, Koren J, Gerecitano JF, Taldone T, Zong H, et al. (2016). The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 538, 397–401. 10.1038/nature19807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig R, Nillegoda NB, Mayer MP, and Bukau B (2019). The Hsp70 chaperone network. Nat Rev Mol Cell Biol 20, 665–680. 10.1038/s41580-019-0133-3. [DOI] [PubMed] [Google Scholar]
- Rousaki A, Miyata Y, Jinwal UK, Dickey CA, Gestwicki JE, and Zuiderweg ER (2011). Allosteric drugs: the interaction of antitumor compound MKT-077 with human Hsp70 chaperones. J Mol Biol 411, 614–632. 10.1016/j.jmb.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabnis AJ, Guerriero CJ, Olivas V, Sayana A, Shue J, Flanagan J, Asthana S, Paton AW, Paton JC, Gestwicki JE, et al. (2016). Combined chemical-genetic approach identifies cytosolic HSP70 dependence in rhabdomyosarcoma. Proceedings of the National Academy of Sciences of the United States of America 113, 9015–9020. 10.1073/pnas.1603883113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha AJ (2004). Java Treeview--extensible visualization of microarray data. Bioinformatics 20, 3246–3248. 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- Sanchez J, Carter TR, Cohen MS, and Blagg BSJ (2020). Old and New Approaches to Target the Hsp90 Chaperone. Curr Cancer Drug Targets 20, 253–270. 10.2174/1568009619666191202101330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H, and Gestwicki JE (2020). Neutral analogs of the heat shock protein 70 (Hsp70) inhibitor, JG-98. Bioorganic & medicinal chemistry letters 30, 126954. 10.1016/j.bmcl.2020.126954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H, Li X, Moses MA, Gilbert LA, Kalyanaraman C, Young ZT, Chernova M, Journey SN, Weissman JS, Hann B, et al. (2018). Exploration of Benzothiazole Rhodacyanines as Allosteric Inhibitors of Protein-Protein Interactions with Heat Shock Protein 70 (Hsp70). J Med Chem 61, 6163–6177. 10.1021/acs.jmedchem.8b00583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha L, Patel HJ, and Chiosis G (2016). Chemical Tools to Investigate Mechanisms Associated with HSP90 and HSP70 in Disease. Cell chemical biology 23, 158–172. 10.1016/j.chembiol.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguwa S, Maringer K, Li X, Bernal-Rubio D, Rauch JN, Gestwicki JE, Andino R, Fernandez-Sesma A, and Frydman J (2015). Defining Hsp70 Subnetworks in Dengue Virus Replication Reveals Key Vulnerability in Flavivirus Infection. Cell 163, 1108–1123. 10.1016/j.cell.2015.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguwa S, Yeh MT, Rainbolt TK, Nayak A, Shao H, Gestwicki JE, Andino R, and Frydman J (2019). Zika Virus Dependence on Host Hsp70 Provides a Protective Strategy against Infection and Disease. Cell Rep 26, 906–920 e903. 10.1016/j.celrep.2018.12.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrab L, and Wipf P (2020). Hsp70 and the Unfolded Protein Response as a Challenging Drug Target and an Inspiration for Probe Molecule Development. ACS Med Chem Lett 11, 232–236. 10.1021/acsmedchemlett.9b00583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadhwa R, Sugihara T, Yoshida A, Nomura H, Reddel RR, Simpson R, Maruta H, and Kaul SC (2000). Selective toxicity of MKT-077 to cancer cells is mediated by its binding to the hsp70 family protein mot-2 and reactivation of p53 function. Cancer Res 60, 6818–6821. [PubMed] [Google Scholar]
- Walter P, and Ron D (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CK, Walther P, Fuller SD, Antony C, Krijnse-Locker J, and Bartenschlager R (2009). Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 5, 365–375. 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisnovsky S, Lei EK, Jean SR, and Kelley SO (2016). Mitochondrial Chemical Biology: New Probes Elucidate the Secrets of the Powerhouse of the Cell. Cell chemical biology 23, 917–927. 10.1016/j.chembiol.2016.06.012. [DOI] [PubMed] [Google Scholar]
- Workman P (2020). Reflections and Outlook on Targeting HSP90, HSP70 and HSF1 in Cancer: A Personal Perspective. Adv Exp Med Biol 1243, 163–179. 10.1007/978-3-030-40204-4_11. [DOI] [PubMed] [Google Scholar]
- Zaiter SS, Huo Y, Tiew FY, Gestwicki JE, and McAlpine SR (2019). Designing de Novo Small Molecules That Control Heat Shock Protein 70 (Hsp70) and Heat Shock Organizing Protein (HOP) within the Chaperone Protein-Folding Machinery. Journal of Medicinal Chemistry 62, 742–761. 10.1021/acs.jmedchem.8b01436. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Banerjee M, Davis RE, and Blagg BSJ (2019). Mitochondrial-targeted Hsp90 C-terminal inhibitors manifest anti-proliferative activity. Bioorg Med Chem Lett 29, 126676. 10.1016/j.bmcl.2019.126676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N, Tsai HN, Zhang X, Shedden K, and Rosania GR (2011). The subcellular distribution of small molecules: a meta-analysis. Molecular pharmaceutics 8, 1611–1618. 10.1021/mp200093z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuiderweg ER, Hightower LE, and Gestwicki JE (2017). The remarkable multivalency of the Hsp70 chaperones. Cell Stress Chaperones 22, 173–189. 10.1007/s12192-017-0776-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 3: Subcellular distributions and physicochemical properties of JG compounds. Related to Figure 3.
Supplemental Table 4: The genes list from CRISPRi screen with Rho and p-value. Related to Figure 4 and 5.
Supplemental Table 5 Antiviral and cell growth activity of JG compounds in Huh7 and C6/36 cells. Related to Figure 6.
Supplemental Table 1: Chemical structures and anti-proliferative activities of JG compounds in this study. Related to Figure 1.
Supplemental Table 2: The list of primers for RT-qPCR. Related to Figure 2.
Data Availability Statement
All data are available in the main text or as Supplemental information. The full list of Rho- and p-values for the CRISPRi screens are in Table S4. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.