

Summary
Microglia, the resident immune cells of the brain, play important roles during development. Although bi-directional communication between microglia and neuronal progenitors or immature neurons has been demonstrated, the main sites of interaction and the underlying mechanisms remain elusive. By using advanced methods, here we provide evidence that microglial processes form specialized contacts with the cell bodies of developing neurons throughout embryonic, early postnatal, and adult neurogenesis. These early developmental contacts are highly reminiscent of somatic purinergic junctions that are instrumental for microglia-neuron communication in the adult brain. The formation and maintenance of these junctions is regulated by functional microglial P2Y12 receptors, and deletion of P2Y12Rs disturbs proliferation of neuronal precursors and leads to aberrant cortical cytoarchitecture during development and in adulthood. We propose that early developmental formation of somatic purinergic junctions represents an important interface for microglia to monitor the status of immature neurons and control neurodevelopment.
Keywords: microglia, neuronal development, doublecortin, somatic junction, purinergic, P2Y12R
Graphical abstract
Highlights
-
•
Microglial processes contact specialized areas of developing neuronal cell bodies
-
•
Somatic junctions possess specialized molecular patterns and ultrastructure
-
•
Somatic junctions are dynamic and require P2Y12R signaling to shape neurodevelopment
-
•
Lack of microglial P2Y12Rs leads to erratic cortical cytoarchitecture in adulthood
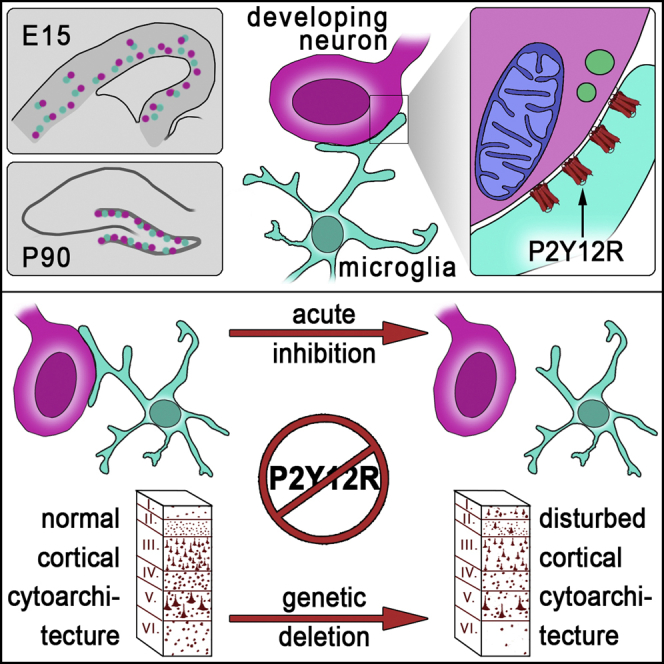
Cserép et al. show that microglial processes contact specialized areas of immature postmitotic neurons during neurogenesis. Somatic purinergic junctions possess unique nano-architecture and are highly dynamic and P2Y12R-dependent. These intercellular interactions enable microglia to monitor and shape neurodevelopment and promote neuronal integration within cortical networks.
Introduction
Microglia, the brain’s resident immune cells, have essential physiological functions in the healthy brain (Kierdorf and Prinz, 2017; Li and Barres, 2018) and contribute to brain disorders (Salter and Stevens, 2017). A role of microglia in CNS development is also emerging (Lenz and Nelson, 2018; Mosser et al., 2017; Thion and Garel, 2017; Thion et al., 2018). Microglial progenitors appear in the CNS as early as the 5th gestational week in humans and embryonic day 9 (E9) in mice (De et al., 2018; Ginhoux et al., 2010; Verney et al., 2010). The complexity of the CNS emerges from a precisely regulated developmental program, which involves neuro- and gliogenesis, migration of postmitotic neuroblasts, formation and refinement of synapses, and other forms of cell-cell connections as well as brain vasculature maturation (Martynoga et al., 2012; Silbereis et al., 2016; Urbán and Guillemot, 2014). In accordance with the complexity of brain development, multiple studies have confirmed that microglia regulate embryonic and adult neurogenesis (Cunningham et al., 2013; Diaz-Aparicio et al., 2020; De Lucia et al., 2016; Sato, 2015; Sellner et al., 2016; Sierra et al., 2010; Walton et al., 2006), direct neuronal differentiation and migration (Aarum et al., 2003), contribute to synaptogenesis and refinement (Gunner et al., 2019; Paolicelli et al., 2011; Rodríguez-Iglesias et al., 2019; Schafer et al., 2012), and drive formation of cortical layers (Ueno et al., 2013). Microglia are also responsible for removal of apoptotic neurons (Marín-Teva et al., 2004; Sierra et al., 2010), activity-dependent synapse formation (Parkhurst et al., 2013), synapse and spine remodeling (Weinhard et al., 2018), and axonal guidance (Pont-Lezica et al., 2014; Squarzoni et al., 2014). Although all of these developmental processes require spatiotemporally coordinated interactions between microglia, neuronal progenitors, and immature neurons, the key sites of intercellular communication and underlying mechanisms are unclear.
Communication between neurons and microglia takes place via multiple routes, including indirect intercellular interactions through soluble messengers and direct, anatomically defined membrane-to-membrane contacts (Cserép et al., 2021). For example, microglia have been shown to modulate developmental and adult neurogenesis through extracellular release of cytokines (interleukin-1β [IL-1β], IL-6, tumor necrosis factor alpha [TNF-α], interferon γ [IFN-γ], or transforming growth factor β [TGF-β]; Battista et al., 2006; Butovsky et al., 2006; Shigemoto-Mogami et al., 2014) and growth factors (Araki et al., 2020). In line with this, their specialized, direct interactions with neuronal synapses are also required to shape brain circuit formation. For example, by contacting the dendrites of developing neurons with preference for more distal neurites (Chugh and Ekdahl, 2016), microglia induce spine outgrowth and synaptogenesis (Miyamoto et al., 2016). During synaptic pruning, microglial processes engulf and phagocytose non-essential synapses, which is also indispensable for proper activity-dependent brain circuit maturation (Paolicelli et al., 2011; Schafer et al., 2012; Weinhard et al., 2018). Development of axon initial segments and inhibition of axonal outgrowth also takes place via direct microglial contacts on growth cones (Baalman et al., 2015; Kitayama et al., 2011). However, these interactions do not explain how microglia are able to monitor and influence the activity of developing neurons without assessing the overall status of the perisomatic neuronal compartment. Microglial interactions with the nucleus-containing cell body could be specifically important for postmitotic neuronal differentiation, cell fate decisions about neuronal survival or elimination by programmed cell death, as well as phagocytosis of damaged neuronal cell bodies, all shown to require functional microglia. Microglial regulation of the activity of immature neurons devoid of complex synaptic inputs can be essential for proper network formation.
Recently, we discovered specialized morpho-functional interaction sites in the adult brain, called somatic purinergic junctions, that are key mediators of microglia-neuron crosstalk (Cserép et al., 2020). These junctions possess a unique ultrastructural and molecular composition that is perfectly suited for bi-directional communication, enabling microglia to readily monitor neuronal status and dynamically influence neuronal functions in the adult brain. In the present study, we tested the hypothesis that somatic purinergic junctions also exist in the developing brain on postmitotic, immature neurons. We aimed to determine their precise developmental time course and characterize their nanoscale molecular architecture, subcellular anatomical organization, and functional relevance for embryonic, early postnatal, and adult neurogenesis.
Results
Microglial processes establish somatic contacts with DCX-positive postmitotic immature neurons
To assess microglia-neuron somatic junctions throughout developmental neurogenesis, we used samples from embryonic E15 (ventricular zone [VZ]/subventricular zone [SVZ]) and early postnatal (post-natal day 1 [P1], P8, and P15) mice (neocortex). To study adult neurogenesis, we analyzed the subgranular layer of the dentate gyrus (DG), a neurogenic niche that persist into adulthood (Ming and Song, 2011). Microglia were identified based on expression of the P2Y12 receptor (P2Y12R) and IBA1, which are present in yolk sac-derived microglia from early embryonic days (Hirasawa et al., 2005; Konishi et al., 2017; Mildner et al., 2017), showing almost perfect colocalization (Figure S1). To visualize neuroblasts and developing/immature postmitotic neurons, we labeled for doublecortin (DCX; Gleeson et al., 1999; Yoo et al., 2011; Figure 1). We did not discriminate between the different maturational stages within the DCX-positive pool; thus, we refer to these cells collectively as DCX-positive (DCX+) developing neurons throughout the paper. At E15, we observed that microglia were mostly confined to the VZ and SVZ and rarely present in the cortical plate (Figure 1A). In adult mice, we could also detect strong enrichment of microglia in the subgranular zone of the hippocampal dentate gyrus (Figure 1B). Microglial processes frequently contacted the cell bodies and neurites of DCX+ neurons during developmental (Figure 1C) and adult neurogenesis (Figure 1D).
Figure 1.

Microglial processes contact the cell bodies, neurites, and synapses of DCX+ developing neurons
(A) Confocal laser scanning microscopy (CLSM) image shows the distribution of IBA1+ microglia (green) in E15 pallium. Postmitotic neurons are labeled for doublecortin (DCX, red), NeuN is shown in blue, and white arrows point to the enrichment of microglia in the SVZ and VZ.
(B) IBA1+ microglia are enriched in the subgranular zone of the DG in P90 mice. Staining is as in (A), and white arrows point to some microglia. The area within the white box in the top panel is enlarged in the bottom panel.
(C and D) High-resolution CLSM images show some examples of microglia contacts in the E15 VZ (C) and the DG in P90 mice (D); white arrows point to contact sites.
(E) CLSM image shows the lack of synapses in an E15 cortical plate. Samples are labeled for IBA1 (green), DCX (red), and VGLUT1 (cyan).
(F) Staining is the same as in (E); the image shows the abundant presence of glutamatergic synapses outside of the granule cell layer.
(G) IBA1+ microglial processes (yellow) contact developing glutamatergic synapses, as identified by the appositions of VGLUT1 (magenta) and Homer1 (green) on a DCX+ dendrite (red) in the stratum moleculare of a P90 DG. White arrows point to contacts.
(H) Staining is the same as in (G); microglial processes contact synapses of mature neurons in the stratum moleculare of a P90 DG.
(I–M) CLSM images show some examples of microglia-neuron somatic contact sites during both developmental and adult neurogenesis. Nuclei are visualized by DAPI (blue), microglia are stained for IBA1 (green) and postmitotic neurons for DCX (red), and white arrows point to contact sites. (I), E15; (J), P1; (K), P8; (L), P15; (M), P90.
(N) Division of a DCX+ neuron’s cell body into “soma” and “proximal tuft” compartments.
(O) Ratio of DCX+ neurons contacted by microglial processes on their somata, proximal tufts, or both. Percentage values next to the diagrams represent the ratio of cells that were contacted either way.
(P) Somatic contact prevalence on DCX+ cell bodies (individual values represent different animals). Images and measurements are from the cortical plate in E15 mice, neocortex from P1–P15 mice, and hippocampal dentate gyrus from P90 mice; n = 3 mice in each age group.
Scale bars represent 200 μm in (A); 100 μm in (B) and (F); 5 μm in (C); 10 μm in (D), (J), and (L); 50 μm in (E), 1 μm in (G) and (H); 15 μm in (I), (K), and (M); and 2 μm in (N). See also Figure S1 and Table S1.
Because apoptosis is often associated with high proliferative activity during neurodevelopment, we investigated whether formation of somatic microglia-neuron junctions might be induced by cells committed to apoptosis. However, the vast majority of DAPI-stained nuclei of the microglia-contacted DCX+ cells possessed a healthy chromatin structure (Figures 1I–1N), and apoptotic cells were clearly visualized based on signs of chromatin condensation (Sierra et al., 2010). To strengthen this observation, we visualized Annexin V in apoptotic cells by immunofluorescence (Dénes et al., 2008). We found that DCX+ cells with somatic microglial contacts are Annexin V− (Figures S2A and S2C), whereas Annexin V+ cells show signs of chromatin condensation (Figure S2B). Thus, microglia contact viable developing neurons, and the frequent occurrence of somatic purinergic junctions at DCX+ cells is not explained by phagocytosis of apoptotic cells.
To test whether microglia-synapse contacts could be present in the vicinity of developing neuronal cell bodies, we labeled for IBA1, DCX, and VGLUT1 and found no glutamatergic synapses at E15 (Figure 1E), and the DG granule cell layer was also devoid of VGLUT1+ synapses (Figure 1F). Nevertheless, co-labeling for IBA1, DCX, VGLUT1, and Homer1 revealed that microglial processes contact synapses of immature (DCX+; Figure 1G) and mature neurons (Figure 1H) in the molecular layer of the adult DG. Next, we examined the prevalence of contacts between microglial processes and 3D reconstructed DCX+ cell bodies through multiple developmental stages (Figures 1I–1P). DCX+ cells possess a main, thick dendritic branch whose first short segment (maximum as long as the cell body itself; Figure 1N) we termed “proximal tuft.” Somatic microglial contacts were found already at the earliest time point examined (E15), when glutamatergic synapses are absent (Figure 1E). Somatic contact prevalence showed a progressive increase during development from more than one-third of developing neurons being contacted at a given time at E15 and P1 to about two-thirds at P8, whereas at P15, virtually all DCX+ cells received somatic microglial input. This was also the case during adult neurogenesis in the dentate gyrus of P90 mice (Figures 1Oand 1P; detailed data in Table S1.).
Direct membrane-membrane contacts between microglia and cell bodies of DCX+ developing neurons are enriched with mitochondria at all stages of development
To determine whether the large number of putative direct contacts between microglial processes and DCX+ immature neurons at the light microscopy level indeed represent somatic purinergic junctions (Cserép et al., 2020), we first exploited correlated light and electron microscopy (CLEM; Figures 2A–2K).
Figure 2.

Microglial processes form direct membrane-membrane contacts with the cell bodies of DCX+ developing neurons at sites enriched with mitochondria
(A) Schematic of correlated light and electron microscopy workflow.
(B–D) Maximum intensity projection of a 1.5-μm-thick volume from a CLSM stack from an E15 mouse shows an example of identified microglia-neuron somatic junctions (B). IBA1+ microglia are shown in green, DCX+ neurons are shown in cyan, the area in the white box is shown on a correlated transmission electron microscopy (TEM) image in (C), and red arrows point to corresponding microglia. The somatic junction within the red box in (B) is enlarged in the TEM image in (D). White arrows point to the direct membrane-membrane contact, and white arrowheads mark neuronal mitochondria close to the junction. The small CLSM inset shows the single confocal image plane closest to the TEM image. TEM images are pseudo-colored (microglia in green, developing neurons in cyan, and mitochondria in red). All 6 CLSM-identified contacts proved to be direct membrane-membrane contacts after TEM assessment.
(E–H) Same as (B)–(D) from a P90 mouse; the somatic junction within the left red box in (E) is enlarged in the TEM image in (G), and the junction within the right red box in (E) is enlarged in the TEM image in (H). All 10 CLSM-identified contacts proved to be direct membrane-membrane contacts after TEM assessment.
(I–K) Areas within red boxes in (D), (G), and (H), respectively, are enlarged from subsequent ultrathin sections.
(L) CLSM image showing an example of IBA1-labeled (green) microglia contacting the cell body of a DCX+ postmitotic neuron (blue) exactly where a large mitochondrion (TOM20 labeling, red) resides within the neuronal cell body. The area within the red dashed line is enlarged in (M).
(M) The process of a semi-automated unbiased analysis of fluorescence intensity area.
(N) The intensity values are plotted along the perimeter of the neuron.
(O) Example of a microglial process (green) contacting the proximal tuft of a DCX+ neuron in an E15 brain. Results show that TOM20 fluorescence intensity is significantly higher within the contact sites than outside of them. Each line represents results from one neuron; somata are represented by black and proximal tufts by red lines. Median values and interquartile range are marked by gray boxes (n = 164 cells from 3 mice).
(P–S) CLSM examples of mitochondrial enrichment at somatic (som.) and proximal tuft (p.t.) junctions in cortical brain samples from P1, P8, P15, and P90 mice and corresponding results. Images and measurements are from the cortical plate in E15 mice, neocortex from P1–P15 mice, and hippocampal dentate gyrus from P90 mice (3 mice/age group). Wilcoxon matched-pairs test; ∗∗∗p < 0.001.
Scale bars represent 30 μm in (B); 4 μm in (C); 1 μm in (D), (G), and (H); 500 nm in (I)–(K); 20 μm in (E), 3 μm in (L); 2 μm in (F) and (O)–(S); this also applies to insets. See also Figures S2J–S2M and Table S2.
We reconstructed 6 microglia-neuron contacts from the E15 VZ or SVZ and 10 contacts from the P90 dentate gyrus from serial sections. In all cases, we could verify the presence of direct membrane-membrane contacts between microglial processes and neuronal cell bodies.
This CLEM approach also revealed that neuronal mitochondria are frequently enriched at these intercellular contacts (Figures 2D and 2G–2K), which is also a key morphological feature of somatic purinergic junctions (Cserép et al., 2020). To quantify mitochondrial accumulation at the junctions, we applied multiple immunolabeling, correlated confocal laser scanning (CLSM), and a semi-automated unbiased method (Figures 2L–2S). We found that, at each investigated age, TOM20 (a mitochondrial protein) immunofluorescence intensity in the cell bodies and proximal tufts of DCX+ neurons was substantially higher at sites of microglial contact compared with adjacent areas (Table S2). These results confirmed that neuronal mitochondria are strongly enriched at somatic contacts. We also observed that DCX+ cells contacted by microglia possessed a healthy (non-condensed) chromatin structure and normal mitochondrial appearance (Figures 2D, 2G–2K, and S2D–S2M), confirming that developmental microglia-neuron somatic junctions are formed between microglial processes and normal (non-apoptotic) post-mitotic neurons.
Microglial P2Y12Rs define somatic purinergic junctions on DCX+ developing neurons
Next, we tested the possibility that enrichment of P2Y12Rs, characteristic microglial signaling molecules, is a defining feature of developmental somatic microglia-neuron contacts, as demonstrated earlier in the adult brain (Cserép et al., 2020). We applied CLSM and stochastic optical reconstruction microscopy (STORM) super-resolution imaging (Figures 3A–3F). P2Y12R immunofluorescence labeling was completely absent in brain sections obtained from P2Y12R knockout mice, confirming the specificity of the antibody against P2Y12R (Figure S3). Next, we performed a quantitative analysis in all five age groups, and we found that virtually all IBA1+ microglial processes that were in contact with DCX+ neuronal cell bodies also expressed P2Y12Rs. Most importantly, correlated CLSM and STORM super-resolution imaging revealed that P2Y12R expression was concentrated strongly on the contacting side of the microglial processes at all age groups examined (Figures 3A–3F and 3G; Table S3).
Figure 3.

Microglial P2Y12Rs define somatic purinergic junctions on DCX+ developing neurons; these cells do not express Kv2.1 but contain LAMP1+ lysosomes in the vicinity of microglial contacts
(A–F) Correlated STORM super-resolution and CLSM images showing the abundant expression of the microglia-specific P2Y12Rs on microglial processes contacting DCX-+ cell bodies. DCX is shown in cyan, IBA1 in green, P2Y12R in yellow, and STORM localization points (LPs) for P2Y12R in red in the insers. Counting frames are marked with red lines in main panels and white grids in insets. Images are from E15 (A), P1 (B), P8 (C), P15 (D), P90 (E), and P90 P2Y12R knockout (F) mice. Note the complete absence of CLSM and STORM signals for P2Y12R in the knockout mice in (F). All IBA1+ microglial processes in contact with DCX+ neuronal cell bodies were expressing P2Y12Rs. In the VZ and SVZ of E15 brains n = 72; in the neocortex of P1 n = 60, P8 n = 85, and P15 n = 74; and in the dentate gyrus of P90 animals n = 93 contacts were tested (n = 2 mice in each age group, altogether n = 10 mice).
(G) Spatial analysis of super-resolution data shows the enrichment of P2Y12R labeling on the contacting side of microglial processes. Black dots represent data points, a blue bracket is the interquartile range, and median is shown by a red segment; n = 35 processes from 5 mice (Table S3). Kruskal-Wallis test followed by Tukey’s comparison; n.s., no significant difference; ∗p < 0.05, ∗∗∗p < 0.001.
(H) CLSM image showing the complete lack of Kv2.1 (yellow) expression in an E15 cortical plate (142 fully reconstructed DCX+ cells tested from two mice, zero Kv2.1+). DCX (blue), IBA1 (green); the ventricle (v) is delineated by a thin red line. Rectangular areas labeled with numbers are enlarged on the right.
(I) CLSM image showing robust expression of Kv2.1 (yellow) in the dentate granule cells of P90 mice, but DCX+ (blue) cells are completely devoid of Kv2.1 labeling (136 fully reconstructed DCX+ cells tested from two mice, zero Kv2.1+). The rectangular area is enlarged on the right. n, neuron; d, DCX+ cell.
(J and K) CLSM images showing examples of IBA1-labeled (green) microglial processes contacting the cell body of DCX+ postmitotic neurons (blue) with LAMP1+ puncta (red, white arrows) in the vicinity. The images are from the SVZ/VZ of E15 (J) and the DG of P90 mice (K).
(L) 69% of all contacts in E15 and 63% in P90 mice contained LAMP1+ vesicles within the DCX+ cell bodies in the close vicinity of microglial process contact. The red and green columns together show the number of measured contacts (49 for E15, 71 for P90, 2 mice for each group), and the red columns represent the number of contacts with LAMP1 labeling (34 for E15, 45 for P90).
Scale bars represent 2 μm in (A)–(F), 30 μm in (H) (5 μm in insets), 15 μm in (I) (5 μm in insets), 2 μm in (J), and 2 μm in (K). See also Figure S3.
Our previous studies demonstrated that somatic purinergic junctions are preferentially formed at sites of neuronal exocytosis-promoting Kv2.1 clusters in the adult brain (Cserép et al., 2020). To determine whether accumulation of Kv2.1 is also a defining feature of somatic purinergic junctions during embryonic development, we performed multiple immunofluorescence labeling on samples from E15 and P90 mice (Figures 3H and 3I). We found that Kv2.1 is not expressed in VZ/SVZ at E15 (Figure 3H). Although we found robust expression of Kv2.1 in the mature granule cells in the dentate gyrus in P90 mice, but none of the DCX+ cells expressed Kv2.1 proteins (Figure 3I), suggesting that alternative routes also exist through which microglia recognize sites of intense exocytosis at the neuronal cell body.
Mitochondrion-derived vesicles (MDVs) or other mitochondrion-derived cargo often integrate into the endo-lysosomal pathway (Sugiura et al., 2014), and these vesicles are positive for the lysosomal marker LAMP1 (Soubannier et al., 2012). LAMP1+ vesicles were identified to be part of mature somatic junctions (Cserép et al., 2020), and we could also observe these organelles in DCX+ cell bodies in the vicinity (within 2 μm) of somatic contacts in the VZ/SVZ of E15 mice (Figure 3J) and in the DG of P90 mice (Figure 3K). 69% and 63% of the examined somatic contacts contained adjacent LAMP1+ puncta in E15 and P90 (Figure 3L) mice, respectively, suggesting that cellular content could also be released at developmental somatic junctions from immature neurons.
Somatic microglia-neuron communication with active, developing neurons is dynamic and P2Y12R dependent
Next, we performed ex vivo 2-photon imaging of acute brain slices prepared from postnatal CX3CR1-GFP mice, loaded with the calcium-sensitive fluorescent dye Calbryte-590 AM (Cal-590; Figures 4A and 4B). In cortical slices of P1 mice, we could observe dynamic formation of somatic junctions between microglial processes and cell bodies of developing neurons (Figure 4C; Video S1), similar to those we observed in adult mice in vivo (Cserép et al., 2020). The dynamic appearance, disappearance, and reappearance of these contacts suggests continuous, active communication between microglia and developing neurons that are not formed because of cellular injury or cell death.
Figure 4.

Developmental microglia-neuron somatic junctions are dynamic and depend on P2Y12R function
(A) Schematic depicting the experimental procedure.
(B) In vitro 2-photon imaging of mouse neocortex from P1 animals confirmed the presence of dynamic somatic junctions between microglia and developing neurons. The area in the white box is enlarged in the right panel, which is also shown in the 3D model below. The contact site marked with a white arrow in the 3D model is shown at z2, with image planes marked by z1 and z3 above and below the contact, respectively.
(C) The slice was scanned over 1 h; insets show representative time frames from a smaller area. Microglial processes (green, CX3CR1-GFP) establish somatic contacts on cell bodies of multiple developing neurons (red, Cal-590). White arrows point to contacts, some of which are re-connected multiple times by microglial processes.
(D) Schematic of the P2Y12R-inhibition experiment (E) and representative measurement of a “PSB” experiment. The calcium trace and coverage values, measured over the 30-min experiment, are superimposed, and red arrows show the respective temporal positions (t1–t6) of the insets of the measured cell. White arrows point to contacts.
(F) Statistical analysis confirmed that acute inhibition of microglial P2Y12Rs induced a rapid and robust decrease of microglial process coverage on developing neurons (median, 38% decrease from baseline; interquartile range, 0.36–0.92; n = 30 cells, 3 mice). Coverage in control is 1, and PSB effect is plotted as an increase or decrease compared with the control. Wilcoxon matched-pairs test; ∗∗p < 0.001.
(G) Same as (E) but from a “vehicle” experiment.
(H) Statistical analysis confirmed that vehicle addition did not induce a significant change in microglial coverage (median, 1.06-fold increase over baseline; interquartile range, 0.75–1.66; n = 25 cells, 3 mice). Wilcoxon matched-pairs test.
Scale bars represent 10 μm.
Microglial processes (green, CX3CR1-GFP) establish somatic contacts on cell bodies of multiple developing neurons (red, Cal-590). Some of the contact sites are readily re-connected multiple times by microglial processes. Imaging duration: 1 h.
Because the microglia-specific P2Y12Rs are enriched on microglial processes involved in formation of somatic junctions and strongly determine junction formation/maintenance in the adult brain (Cserép et al., 2020), we tested the effect of acute pharmacological blockade of these receptors on the dynamics of the developing junctions on samples prepared from P8 mice. After 15-min baseline imaging, PSB-0739 (a selective and potent P2Y12R inhibitor) or vehicle was added to the perfusion solution (Figure 4D) for 15 min. We observed dynamic formation of somatic junctions between microglial processes and cell bodies of developing neurons (Figures 4E and 4G).
During baseline imaging, the lifetime of these somatic contacts (median contact lifetime was 15 min, and 53% of the tested contacts were present throughout the whole observation period) was similar to what we measured previously for mature somatic junctions in vivo (Cserép et al., 2020). Inhibition of microglial P2Y12Rs caused a significant, rapid decrease of microglial coverage on developing neuronal cell bodies (Figures 4E and 4F; Videos S2 and S3.). In contrast, addition of vehicle did not induce a significant change in microglial coverage (Figures 4G and 4H; Video S4). These results suggest that formation and maintenance of developing microglia-neuron somatic junctions depends on P2Y12R signaling.
After 15 min “baseline imaging” PSB-0739 (selective inhibitor of microglial P2Y12R-s) was added to the perfusion, and 15 min further imaging was performed. Acute inhibition of microglial P2Y12Rs induced a rapid and robust decrease of microglial process coverage.
After 15 min “baseline imaging” PSB-0739 (selective inhibitor of microglial P2Y12R-s) was added to the perfusion, and 15 min further imaging was performed. Acute inhibition of microglial P2Y12Rs induced a rapid and robust decrease of microglial process coverage.
After 15 min “baseline imaging” vehicle was added to the perfusion, and 15 min further imaging was performed. Vehicle addition did not induce any significant change in microglial coverage.
Absence of P2Y12R signaling results in aberrant cortical distribution of DCX+ cells during development and leads to erratic cytoarchitecture in adulthood
Next, we wanted to test the effect of genetic P2Y12R deletion on postnatal neurodevelopment. We compared the density of DCX+ cells in cortical layers of wild-type (WT) and P2Y12R−/− mice at P8 (Figure 5). Ctip2 and Satb2 immunofluorescence staining was used to delineate cortical layers (Figures 5B and 5C), and the density of DCX+ cells was assessed in layers 6, 4/5, and 2/3 (Figures 5C–5E). In layer 6, there was no difference in DCX+ cell density between WT and P2Y12R−/− mice. However, in layer 4/5, we observed a significant increase in DCX+ cell density in P2Y12R−/− mice compared with WT mice.
Figure 5.

Microglial P2Y12Rs are necessary for formation of proper cortical cytoarchitecture
(A) CLSM images showing triple immunofluorescence staining for Iba1, GFP, and P2Y12Rs on the different genotypes used in this experiment. Merged images also show cell nuclei labeled with DAPI.
(B) CLSM image showing triple immunofluorescence staining for Iba1, Ctip2, and Satb2 in P8 mice.
(C–E) Ctip2 and Satb2 immunofluorescence staining was used to delineate cortical layers, and the density of DCX+ cells was assessed in layers 6, 4/5, and 2/3.
(D and E) In layer 6, there was no difference in DCX+ cell density between WT, P2Y12R−/−, and CX3CR1−/− mice (n = 9 mice). However, in layers 4/5, we observed a significant (200% of WT) increase in DCX+ cell density in P2Y12R−/− mice compared with WT mice, which did not differ from CX3CR1−/− mice (n = 9 mice). On the contrary, in layers 2/3, the density of DCX+ cells was significantly lower (50% of WT) in P2Y12R−/− mice compared with WT or CX3CR1−/− mice, which did not differ from each other (n = 9 mice). Median values and interquartile range are marked by boxes and whiskers. Kruskal-Wallis test followed by Tukey’s comparison; ∗p < 0.05, ∗∗∗p < 0.001.
(F) Kv2.1 staining in adult mouse neocortex in WT and P2Y12R KO animals; insets show enlarged areas from layers 1 and 6.
(G) Lack of P2Y12Rs causes robustly elevated neuronal density in layer 1 (300% over WT) and leads to a significant decrease of neuronal density in layer 6 (75% of WT, n = 6 mice). Median values and interquartile ranges are marked by boxes and whiskers. Mann-Whitney U test; ∗∗p < 0.01.
Scale bars represent 5 μm in (A), 100 μm in (B) and (C), 20 μm in (D), 150 μm in (F), and 20 μm in insets.
On the contrary, in layer 2/3, the density of DCX+ cells was significantly lower in P2Y12R−/− mice compared with WT mice. The increased DCX+ cell density in layers 4/5 and strongly decreased density in layers 2/3 in P2Y12R−/− mice confirms an aberrant cellular distribution pattern.
According to previous studies, another microglial receptor, CX3CR1, appears to be important for developmental processes (Paolicelli et al., 2011; Ueno et al., 2013). Based on this, we wanted to test whether effects of CX3CR1 deletion may be similar to the P2Y12−/− phenotype, suggesting a general microglial effect. We found that, in layers 2/3, deletion of CX3CR1 resulted in a milder phenotype than the clear effects of P2Y12R deletion because no significant effect was observed in CX3CR1−/− mice. Similarly, in layer 4/5, only P2Y12R knockout (KO), but not CX3CR1−/− mice, showed altered DCX+ cell numbers.
To study the functional consequence of P2YR12R deficiency on the cortical cytoarchitecture in adulthood, we performed a measurement of neuronal density over the cortical layers using high-resolution stereology on CLSM image stacks. We found that the lack of P2Y12R causes robustly elevated neuronal density in layer 1, did not cause significant differences in layers 2–5, but led to a significant decrease of neuronal density in layer 6 (Figures 5F and 5G).
These results suggest that specific P2Y12R-dependent microglial assistance is important for proper cortical distribution of DCX+ cells during postnatal development as well as for the establishment of proper layer-specific neuronal density in adulthood. These microglia-related effects are likely to be mediated at least in part by developing somatic purinergic junctions, where P2Y12R are strongly enriched and are functionally important.
Microglial P2Y12R signaling regulates formation of cortical cytoarchitecture by interfering with neuronal proliferation
Finally, we wanted to test whether the observed aberrations in cortical cytoarchitecture would be due to deficits in microglial control of proliferation of neuronal precursors or apoptosis. Because these processes are more active at P1, first we tested whether the distribution of DCX+ cells would differ between WT and P2Y12R−/− mice at this age. We found that, in the deeper cortical layers close to the SVZ, the lack of microglial P2Y12Rs leads to a significantly decreased density of DCX+ cells (Figures 6A–6D). These results confirmed that microglial actions through P2Y12R signaling are involved in formation of cortical cytoarchitecture as early as P1.
Figure 6.

Microglial P2Y12R signaling is necessary for proper neuronal proliferation in the developing neocortex
(A–C) Maximum intensity projection of a 20-μm-thick volume from a CLSM stack from an P1 mouse (A). DCX+ neurons are shown in white and IBA1+ microglia in red, and nuclei are visualized by DAPI (blue). The area within the white box is enlarged in (B) and (C). The cortex is divided into 10 zones, numbered from the border of the SVZ to the pial surface.
(D) Lack of P2Y12Rs causes decreased density of DCX+ cells in the lower zones (to 51% of WT in zone 1, 72% of WT in zone 2, and to 58% of WT in zone 3; n = 6 mice). Mann-Whitney U test; ∗p < 0.05, ∗∗p < 0.01.
(E) CLSM images showing triple immunofluorescence staining for Ki67 (green), IBA1 (red), and DAPI (blue).
(F) The genotypes and the areas of measurements are the same as above.
(G) Lack of P2Y12Rs causes decreased density of Ki67+ cells to 82% of WT; n = 6 mice. Mann-Whitney U test, ∗∗p < 0.01.
(H) A population of DCX+ cells is also positive for Ki67.
(I) Lack of P2Y12Rs causes a significant decrease of the density of Ki67/DCX double-positive cells (to 0% of WT, n = 6 mice). Mann-Whitney U test, ∗p < 0.05.
(J) CLSM image showing immunofluorescence staining for Iba1 (red), DCX (white), Annexin V (green), and DAPI (blue). The images below show microglia contacting a DCX+ cell (1) and an Annexin V/DCX double-positive cell without microglial contact (2).
(K) Density of Annexin V+ cells in WT and P2Y12R−/− animals (n = 6 mice).
(L) Density of Annexin V/DCX double-positive cells in WT and P2Y12R−/− animals (n = 6 mice).
(M) Density of Annexin V+ cells contacted by microglia in WT and P2Y12R−/− animals (n = 6 mice). Mann-Whitney U test. Median values and interquartile ranges are marked by boxes and whiskers.
Scale bars represent 300 μm in (A) and (E), 5 μm in (H), and 10 μm in (J). Insets are 200 μm wide in (B) and (C) and 150 μm wide in (F). See also Figures S4 and S5.
Next, we tested the possible effect of the lack of P2Y12R on neuronal proliferation using the Ki67 marker (Miller et al., 2018; Figure 6E). We measured the density of Ki67+ cells in the cortex of P1 mice (Figure 6F) and the density of Ki67/DCX double-positive cells (Figure 6H; a smaller subpopulation of developing neurons, shortly after division; Seki et al., 2019). We found that deletion of microglial P2Y12Rs leads to a significant decrease in the density of Ki67+ cells (Figure 6G) as well as to a remarkable decrease in the density of Ki67/DCX double-positive cells (Figure 6I). Virtually none of the DCX-Ki67 double-positive cells expressed glial fibrillary acidic protein (GFAP) (Figures S4A, S4B, and S4D), and only a small percentage of the Ki67+ cells (11%, interquartile range; Figures S4C and S4E) was also positive for GFAP, suggesting that these cells are not proliferating astrocytes but developing neurons, as also suggested by earlier studies (Gleeson et al., 1999; Seki et al., 2019). These results indicate that microglia influence proliferation of neuronal progenitors in a P2Y12R-dependent manner.
Finally, we tested the possible influence of microglial P2Y12R signaling on the rate of cell death in the developing cortex. We found that the absence of P2Y12R did not cause a difference in the number of terminal deoxynucleotidyltransferase dUTP nick end labeling (TUNEL)-labeled puncta at E15 (Figures S5A–S5D) or at P1 despite the fact that a trend of an increase in KO was observed at this time point (Figures S5E–S5G). We also utilized Annexin V immunofluorescence labeling and visualized postmitotic neurons that had already exposed phosphatidylserine on their outer membrane leaflet, showing signs of commitment toward apoptosis followed by thorough, unbiased 3D analysis on high-resolution CLSM image stacks (Figures 6J–6M). We found that the cortical density of Annexin V+ cells, the density of Annexin V/DCX double-positive cells, and the density of microglial contacts onto Annexin V+ cells did not differ between WT and P2Y12R−/− mice (Figures 6K–6M). Although these data show that there are no robust differences between WT and P2Y12R−/− mice regarding cortical cell death at E15 and P1, we cannot exclude the possibility that microglia would also influence cell death and phagocytosis during cortical development via P2Y12R at other time points and/or in other brain areas.
Discussion
Direct microglia-neuron interactions at synaptic/dendritic and axonal levels have been implicated previously in various developmental processes. However, these interactions could not fully explain the entire spectrum of the diverse developmental roles of microglia because the majority of cell fate decisions are linked to cell bodies (Donato et al., 2019; Hobert et al., 2010; Terenzio et al., 2017), necessitating interactions between microglial processes and the somatic compartment. Microglial regulation of neuronal development at early stages takes place in cells that are still devoid of synapses. The ratio of contacted DCX+ cell bodies showed a substantial increase during postnatal development, with a sharp rise between P1 and P8, matching the most intensive period of synaptogenesis (Fiala et al., 1998; Khazipov et al., 2001; Tyzio et al., 1999), switching of depolarizing GABA effects to hyperpolarization, and robust changes in network activity patterns (Ben-Ari et al., 2012; Kaila et al., 2014). We suggest that, through these contacts, microglia can readily monitor the status and function of neurons, as demonstrated in the adult brain, allowing microglia to dynamically influence neuronal functions and cell fate decisions through robust bi-directional communication (Cserép et al., 2020) and assist activity-dependent neuronal integration.
The important role of neuronal mitochondria in developmental and adult neurogenesis has become evident over the last years (Arrázola et al., 2019; Khacho and Slack, 2018; Rangaraju et al., 2019). Mitochondrial metabolism fundamentally regulates developmental and adult neurogenesis and differentiation (Beckervordersandforth, 2017; Lorenz and Prigione, 2017; Petrelli et al., 2022), and mitochondrial structural dynamics of postmitotic neurons have been shown to drive developmental neurogenesis (Iwata et al., 2020), being instrumental for cell fate decisions at the same time (Bhola and Letai, 2016). Mitochondria are also critical for injury-related responses in the developing brain (Hagberg et al., 2014) through induction of programmed cell death (Yamaguchi and Miura, 2015) or morphogenesis of neurons (Kimura and Murakami, 2014). Thus, somatic mitochondria may provide an ultimate readout for microglia and function as possible sites to influence neuronal physiology or cell death, as demonstrated in the adult brain (Chandel, 2014; Whelan and Zuckerbraun, 2013). We found a strong association between somatic mitochondria of DCX+ cells and the contact sites formed by microglial processes during development and adult neurogenesis, similar to that seen in mature neurons (Cserép et al., 2020). Microglia are thus in an ideal position at somatic junctions to sense mitochondria-derived signals, including purine metabolites (Bajwa et al., 2019; Cserép et al., 2020; Davalos et al., 2005; Gouveia et al., 2017), and influence cellular functions. Through these junctions, microglia could modulate the activity or morphogenesis of postmitotic cells by altering their metabolism or mitochondrial function.
MDVs, other mitochondria-related substances, or signaling molecules released from neuronal cell bodies are carrying important information of the source cell’s overall status. The somato-dendritic region of developing neurons has been shown to be responsible for the majority of exocytotic events (Urbina and Gupton, 2020; Urbina et al., 2018). In the case of mature purinergic junctions, Kv2.1-channel clusters seem to be important regulators of somatic exocytosis (Mohapatra et al., 2007). Although we could not detect expression of these Kv2.1 clusters in DCX+ cells, there could be other delayed rectifier Kv-proteins present or several other molecular scaffolds providing a sufficient exocytotic surface. Our results, showing the presence of LAMP1+ lysosomes in the close vicinity of somatic junctions on DCX+ cell bodies, suggest that cellular content could be released here, acting as a local readout for microglial processes assessing cellular status. The vesicular nucleotide transporter, which is responsible for packing purine nucleotides into vesicles and has been functionally linked to somatic neuronal ATP release, has also been shown to be expressed in DCX+ developing neurons, colocalizing with LAMP1+ vesicles (Cserép et al., 2020; Menéndez-Méndez et al., 2017). This suggests that, among the wide variety of signaling molecules and mitochondrion-related substances that are presumably released at these somatic junctions, purinergic nucleotides are key candidates.
Purinergic signaling and microglial P2Y12R have been implicated in regulation of neurogenesis and brain development (Ribeiro et al., 2019; Rodrigues et al., 2019). We found robust expression of P2Y12Rs on microglial processes involved in somatic junctions throughout development and during adult neurogenesis. These microglial receptors are also expressed during human development (Mildner et al., 2017), suggesting that this feature is evolutionarily conserved. Microglial P2Y12 signaling has been shown to regulate neurogenesis and immature neuronal projections (Mo et al., 2019) and promote proliferation of adult mouse SVZ cells (Suyama et al., 2012). On the other hand, microglia couple phagocytosis with progression of apoptosis via P2Y12R signaling during development (Blume et al., 2020). All of these data suggest a role of purinergic and P2Y12R-dependent signaling processes during microglial regulation of neurodevelopment. Because these receptors are robustly expressed on microglial processes in somatic junctions, and these contact sites have been shown to release purinergic metabolites in mature neurons (Cserép et al., 2020), it is likely that similar signaling pathways within these junctions could be involved in microglial regulation of neurodevelopment. Our in vitro 2-photon microscopy results confirm that formation and maintenance of developmental somatic microglia-neuron junctions depend on P2Y12R signaling because acute receptor inhibition induced a rapid decrease in somatic microglial coverage of developing neurons. These results are in line with our previous results, confirming that the lifetime of somatic junctions showed a significant decrease upon P2Y12R inhibition in adult mice in vivo (Cserép et al., 2020). This implies that similar communication pathways can be functional during neurodevelopment. Our observation that these developmental somatic junctions are formed and re-formed in a dynamic manner suggests that microglia can readily monitor and influence neuronal activity through these specialized contact sites.
During the developmental program of neurons, basic changes in activity patterns parallel the most active period of synaptogenesis (Fiala et al., 1998). During the first postnatal week, depolarizing GABAergic effects switch to hyperpolarization, and glutamatergic synapses appear, whereas the primitive synchronous developmental oscillatory activity gradually changes to more mature activity patterns (Ben-Ari et al., 2012). Monitoring and regulation of neuronal activity by microglia through somatic junctions may provide means to exert developmental regulation during this critical period and assist formation of complex neuronal networks. This idea is also supported by the steep increase of somatic contact prevalence exactly during the first postnatal week. Several studies suggested that microglia regulate the migration of developing neurons and act as special “guideposts” for neuronal migration (Aarum et al., 2003; Squarzoni, 2015). Some of these actions require microglial CX3CR1, which appears to be important for developmental processes such as synaptic pruning, establishment of proper neuronal connectivity, and survival of layer 5 cortical neurons (Paolicelli et al., 2011; Ueno et al., 2013). Our results suggest that the effects of genetic P2Y12R deletion in the developing neocortex at least in part differ from those mediated by CX3CR1. P2Y12R deletion, but not CX3CR1 deletion, resulted in an aberrant cortical distribution of DCX+ cells at P1 as well as at P8, suggesting that somatic junctions (where P2Y12Rs are expressed abundantly) may provide an instrumental communication surface for microglia to control neuronal migration. This idea is supported by the marked, layer-specific, P2Y12R-related changes in neuronal density observed in adult animals, showing that neuronal density in layer 1 is significantly higher, whereas in layer 6, it is significantly lower in P2Y12R-deficient mice compared with their WT littermates.
Our results regarding neuronal proliferation and apoptosis suggest that, during formation of the cortical cytoarchitecture, microglia influence proliferation of neuronal progenitors and tentatively other cells in a P2Y12R-dependent manner. Virtually none of the DCX-Ki67 double-positive cells expressed GFAP, and only a small percentage of the Ki67+ cells were also positive for GFAP, confirming that these cells are not proliferating astrocytes but developing neurons, as also shown by others (Gleeson et al., 1999; Seki et al., 2019). Our results showing that microglia influence neuronal proliferation in a P2Y12R-dependent manner are in line with earlier observations showing that the absence of microglial P2Y12Rs leads to a decreased density of DCX+ cells and Ki67+ cells in the adult dentate gyrus (Diaz-Aparicio et al., 2020). Although we could not detect differences regarding apoptosis in P2Y12R-deficient mice in the present study, it is possible that microglia influence cell death and elimination of apoptotic neurons by phagocytosis in the developing neocortex at different stages of development or via pathways other than P2Y12R.
Besides microglial involvement in regulation of proliferation, migration of immature cells could also be affected by microglia. It is possible that, without proper microglia-neuron communication via somatic junctions, migrating neurons during development fail to receive proper “arrest” signals, which could lead to a migrational “overrun,” manifesting as the higher overall position of the neuronal population toward the cortical surface. The difference between the direction of density changes of DCX+ cells at P8 and mature neurons in the adult brain could be explained by a disturbed neuronal maturation course being altered in the absence of proper microglia-neuron communication. It can also be hypothesized that the temporal lack of microglia between E15 and E16 in the cortical plate described earlier (Hattori et al., 2020) could be the reason why the density change manifests only in layers 1 and 6 in the adult cortex, affecting the first and last waves of pyramidal cells. Alternatively, this could also be the result of uneven preplate splitting, which separates the marginal zone (future layer 1) and subplate (future layer 6b). The precise mechanisms through which P2Y12R-mediated effects act on the developing neuronal networks and adult cytoarchitecture should be investigated in further studies. Our observations are also supported by earlier studies that have implicated purinergic signaling in regulation of neurogenesis, although these results were mainly restricted to adult neurogenesis in the dentate gyrus and olfactory bulb (Ali et al., 2021; Stefani et al., 2018). Such mechanisms could also be important for development of complex pathological states such as seizure-induced adult neurogenesis (Mo et al., 2019). Our present results imply a key role of P2Y12R-dependent direct, cell-cell interactions that are formed between microglia and the somatic compartment of developing neurons in physiological neurodevelopment, strengthening the assumed broad roles of somatic purinergic junctions in development and adulthood (Cserép et al., 2020, 2021).
Outlook
Microglia populate the brain early during development (Ginhoux et al., 2010; Verney et al., 2010) and maintain a self-renewing, long-lived population thereafter (Füger et al., 2017; Réu et al., 2017), with marked transcriptome changes throughout life (Hammond et al., 2019; Matcovitch-Natan et al., 2016). How microglia shape neurodevelopment under different conditions via different interactions will need to be investigated in the future. Because even transient perturbations of developmental microglia-neuron interactions can lead to complex and persistent disruptions of neuronal networks (Paolicelli and Ferretti, 2017; Park et al., 2020; Prins et al., 2018; Xu et al., 2020), the clinical relevance, especially in neurodevelopmental disorders, to better understand the underlying complex neuro-immune communication and compartment-specific microglia-neuron interactions in health and disease is evident.
Limitations of the study
Our findings raise several questions that are beyond the scope of the present paper. First, we focused our experiments on postmitotic immature neurons, confirmed by their DCX expression. However, microglial regulation of neurogenesis, proliferation, differentiation, and survival of neural precursors or migration are active processes even at embryonal stages (Aarum et al., 2003; Bilimoria and Stevens, 2015; Cunningham et al., 2013; Erblich et al., 2011; Ueno et al., 2013), often affecting more immature progenitors. Somatic interactions have already been observed between microglial processes and the cell bodies of dividing neuronal progenitors (Cunningham et al., 2013; Noctor et al., 2019; Penna et al., 2020); thus, it needs to be addressed whether these are also functioning as somatic junctions. Second, longitudinal in vivo imaging studies will also be necessary to decipher whether developmental phagocytosis (Diaz-Aparicio et al., 2020; Sierra et al., 2010; VanRyzin et al., 2019) would require somatic junctions as specific checkpoints. Mitochondrial changes are involved in developmental apoptosis of neuronal progenitors (László et al., 2020), and these changes could also be sensed or even regulated by microglia, which, in turn, can eliminate unfit progenitors by inducing programmed cell death followed by phagocytosis. The NAD+:NADH ratio plays an important role in regulating stem cell fate (Khacho and Slack, 2018), and we have shown previously that microglia are able to interfere with mitochondrial NADH production at somatic junctions in a P2Y12R-dependent manner (Cserép et al., 2020). This also raises the possibility that microglia could, at least in part, exert developmental regulation by modifying the mitochondrial function of neuronal progenitors or immature neurons. It could also be investigated whether the changing morpho-functional properties of microglia at different developmental stages (Matcovitch-Natan et al., 2016) could play a role in the increasing contact prevalence throughout development or, on the contrary, whether the increased demand for microglial contacts by developing neurons could be a driver of microglial morphology changes. Further functional studies could also reveal what types of neuronal content could be released at these somatic junctions, which members of the exocytotic release machinery are potentially involved, and whether changes in neuronal somatic exocytosis, through which microglial quality control is achieved at somatic junctions (Cserép et al., 2020), would be altered at different stages of neurodevelopment.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Annexin V | Novusbio | Cat# NB100-1930; RRID: AB_10001784 |
| Rat anti-Ctip2 | Abcam | Cat# AB18465; RRID: AB_10973033 |
| Chicken anti-DCX | Synaptic Systems | Cat# 326-006; RRID: AB_2737040 |
| Mouse anti-DCX | Santa Cruz | Cat# sc-271390; RRID: AB_10610966 |
| Rabbit anti-GFP | Invitrogen | Cat# A11122; RRID: AB_221569 |
| Chicken anti-GFP | Invitrogen | Cat# A10262; RRID: AB_2534023 |
| Mouse anti-GFAP | Sigma | Cat# G3893; RRID: AB_477010 |
| Rabbit anti-Homer1 | Synaptic Systems | Cat# 160003; RRID: AB_887730 |
| Guinea pig anti-IBA1 | Synaptic Systems | Cat# 234004; RRID: AB_2493179 |
| Goat anti-IBA1 | Novusbio | Cat# NB100-1028; RRID: AB_521594 |
| Rabbit anti-IBA1 | Wako Chemicals | Cat# 019-19741; RRID: AB_839504 |
| Rabbit anti-Ki67 | Abcam | Cat# ab15580; RRID: AB_443209 |
| Mouse anti-Kv2.1 | NeuroMab | Cat# 75-014; RRID: AB_10673392 |
| Rabbit anti-LAMP1 | Abcam | Cat# ab24170; RRID: AB_775978 |
| Rabbit anti-P2Y12R | Anaspec | Cat# AS-55043A; RRID: AB_2298886 |
| Mouse anti-Satb2 | Abcam | Cat# AB51502; RRID: AB_882455 |
| Rabbit anti-TOM20 | Santa Cruz | Cat# sc-11415; RRID: AB_2207533 |
| Guinea pig anti-vGluT1 | Synaptic Systems | Cat# 135304; RRID: AB_887878 |
| DyLight 405 donkey anti-rabbit | Jackson | Cat# 711-476-152; RRID: AB_2632566 |
| biotinylated donkey anti-rabbit | BioRad | Cat# 644008; RRID: AB_619842 |
| Alexa 488 donkey anti-goat | Jackson | Cat# 705-546-147; RRID: AB_2340430 |
| Alexa 488 donkey anti-guinea-pig | Jackson | Cat# 706-546-148; RRID: AB_2340473 |
| Alexa 488 donkey anti-chicken | Jackson | Cat# 703-546-155; RRID: AB_2340376 |
| Alexa 488 donkey anti-rabbit | Jackson | Cat# 711-546-152; RRID: AB_2340619 |
| Alexa 594 goat anti-guinea-pig | LifeTech | Cat# A11076; RRID: AB_141930 |
| Alexa 594 donkey anti-rabbit | LifeTech | Cat# A21207; RRID: AB_141637 |
| Alexa 594 donkey anti-chicken | Jackson | Cat# 703-586-155; RRID: AB_2340378 |
| Alexa 594 donkey anti-mouse | Invitrogen | Cat# A-21203; RRID: AB_141633 |
| Alexa 594 donkey anti-rat | Jackson | Cat# 712-586-153; RRID: AB_2340691 |
| Alexa 647 donkey anti-guinea-pig | Jackson | Cat# 706-606-148; RRID: AB_2340477 |
| Alexa 647 donkey anti-chicken | Jackson | Cat# 703-606-155; RRID: AB_2340380 |
| Alexa 647 donkey anti-rabbit | Jackson | Cat# 711-605-152; RRID: AB_2492288 |
| Alexa 647 donkey anti-mouse | Jackson | Cat# 715-605-150; RRID: AB_2340866 |
| Chemicals, peptides, and recombinant proteins | ||
| Triton X-100 | Sigma-Aldrich | Cat# 9036-19-5 |
| Paraformaldehyde | Molar Chemicals Kft. | Cat# 30525-89-4 |
| Albumin from human serum | Sigma-Aldrich | Cat# 70024-90-7 |
| Aqua-Poly/Mount | Polysciences | Cat# 18606-20 |
| Ketamine | Medicus Partner Kft. | N/A |
| Xylazine- hydrochloride | Medicus Partner Kft. | N/A |
| Digitonin | Sigma-Aldrich | Cat# 11024-24-1 |
| Dulbecco’s PBS | Sigma-Aldrich | N/A |
| Glucose Oxidase from Aspergillus niger | Sigma-Aldrich | Cat# 9001-37-0 |
| Catalase from bovine liver | Sigma-Aldrich | Cat# 9001-05-2 |
| Mercaptoethylamine | Sigma-Aldrich | N/A |
| Vector® ImmPACT DAB, HRPSubstrate | Fisher Scientific | Cat# 12963644 |
| Vector® ImmPACT NovaRED, HRP-Substrate | Fisher Scientific | Cat# 15788269 |
| Durcupan™ ACM single component A, M epoxy resin |
Sigma-Aldrich | Cat# 44611-100ML |
| Durcupan™ ACM single component B, hardener 964 |
Sigma-Aldrich | Cat# 44612-100ML |
| Durcupan™ ACM single component C, accelerator 960 |
Sigma-Aldrich | Cat# 44613-100ML |
| Durcupan™ ACM single component D |
Sigma-Aldrich | Cat# 44614-100ML |
| Osmium Tetroxide, 4% Aqueous Solution | Science Services GmbH | Cat# E19190 |
| Uranyl Acetate | Electron Microscopy Sciences | Cat# 22400 |
| PSB-0739 | Tocris | Cat# 6083 |
| DAPI | Sigma-Aldrich | Cat# 28718-90-3 |
| Critical commercial assays | ||
| TUNEL-assay | Millipore | Cat# S7100 |
| ABC-HRP Kit | Vector Laboratories | Cat# PK-6100 |
| Calbryte 590 AM | AATBio | Cat# 20701/AAT |
| Experimental models: Organisms/strains | ||
| C57BL/6 mouse | The Jackson Laboratory | RRID: IMSR_JAX:000664 |
| CX3CR1GFP mouse | The Jackson Laboratory | RRID: IMSR_JAX:005582 |
| P2ry12−/− mouse | Haynes et al. (2006) | N/A |
| Software and algorithms | ||
| NIS-Elements AR 4.3 | Nikon | Cat# MQS31510 |
| Fiji software | NIH | RRID: SCR_003070 |
| N-STORM 3.4 software | N/A | N/A |
| Adobe Photoshop CS6 | N/A | N/A |
| Statistica 13.4.0.14 | TIBCO | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ádám Dénes (denes.adam@koki.hu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Animals
Experiments were carried out on E15, P1, P8, P15 and P90 old C57BL/6 (RRID:IMSR_JAX:000664), P1 and P8 old CX3CR1+/GFP (IMSR_JAX:005582), P8 old P2Y12−/− (Haynes et al., 2006) and CX3CR1GFP/GFP male mice. Animals were bred at the SPF unit of the Animal Care Unit of the Institute of Experimental Medicine (IEM, Budapest, Hungary). Mice had free access to food and water and were housed under light-, humidity- and temperature-controlled conditions. All experiments were performed in accordance with the Institutional Ethical Codex and the Hungarian Act of Animal Care and Experimentation guidelines (40/2013, II.14), which are in concert with the European Communities Council Directive of September 22, 2010 (2010/63/EU). The Animal Care and Experimentation Committee of the Institute of Experimental Medicine and the Animal Health and Food Control Station, Budapest, have also approved the experiments under the number PE/EA/1021–7/2019, PE/EA/673–7/2019. All experiments were performed in accordance with ARRIVE guidelines.
Method details
Perfusion and tissue processing for histology
Adult mice were anesthetized by intraperitoneal injection of 0.15–0.25 mL of an anesthetic mixture (containing 20 mg/mL ketamine, 4 mg/mL xylazine-hydrochloride). Animals were perfused transcardially with 0.9% NaCl solution for 1 min, followed by 4% freshly depolymerized paraformaldehyde (PFA) in 0.1 M phosphate buffer (PB) pH 7.4 for 40 min, and finally with 0.1 M PB for 10 min to wash the fixative out. Blocks containing the primary somatosensory cortex and dorsal hippocampi were dissected and coronal sections were prepared on a vibratome (VT1200S, Leica, Germany) at 50 μm thickness for immunofluorescent experiments and electron microscopy. For some experiments, 25 μm thick sections from E15 mouse brains were prepared on a cryostat, and dried onto glass slides.
Immunofluorescent labeling and CLSM
Before the immunofluorescent staining, the 50 μm thick sections were washed in PB and Tris-buffered saline (TBS). For Ctip2 and Satb stainings, this was followed by citrate-buffer treatment (10 mM Sodium-citrate, pH:6.00 at 90°C for 45 min). Thorough washing was followed by blocking for 1 h in 1% human serum albumin (HSA; Sigma-Aldrich) and 0.03–0.1% Triton X-100 dissolved in TBS and 100 ug/mL Digitonin (D141-100MG, Sigma). No Triton X-100 or digitonin were added for the Ctip2 and Satb2 samples. After this, sections were incubated in mixtures of primary antibodies overnight at room temperature. After incubation, sections were washed in TBS and were incubated overnight at 4°C in the mixture of secondary antibodies, all diluted in TBS. Secondary antibody incubation was followed by washes in TBS, PB, then sections were mounted on glass slides, and coverslipped with Aqua-Poly/Mount (Polysciences). Immunofluorescence was analyzed using a Nikon Eclipse Ti-E inverted microscope (Nikon Instruments Europe B.V., Amsterdam, The Netherlands), with a CFI Plan Apochromat VC 60× oil immersion objective (numerical aperture: 1.4) and an A1R laser confocal system. We used 405, 488, 561 and 647 nm lasers (CVI Melles Griot), and scanning was done in line serial mode, pixel size was 50 × 50 nm. Image stacks were taken with NIS-Elements AR. For primary and secondary antibodies used in this study, please see Key resources table.
TUNEL-assay
Cell death was detected by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay by using Apoptag Red In Situ Apoptosis Detection Kit according to the manufacturer’s protocol (Millipore, Cat. nr. S7100). After CLSM imaging analysis was performed with analyse particles plugin of Fiji software.
STORM super-resolution imaging
Free-floating brain sections were blocked with 1% human serum albumin followed by immunostaining with rabbit anti-P2Y12R, guinea-pig anti-IBA1 and chicken anti-DCX antibodies, followed by anti-rabbit Alexa 647, anti-guinea-pig Alexa 488 and anti-chicken Alexa 594 secondary antibodies. Sections were mounted onto #1.5 borosilicate coverslips and covered with imaging medium containing 5% glucose, 0.1 M mercaptoethylamine, 1 mg/mL glucose oxidase, and catalase (Sigma, 1500 U/mL) in Dulbecco’s PBS (Sigma), immediately before imaging. STORM imaging was performed for P2Y12R (stimulated by a 647 nm laser) by using a Nikon N-STORM C2+ super-resolution system that combines ‘Stochastic Optical Reconstruction Microscopy’ technology and Nikon’s Eclipse Ti research inverted microscope to reach a lateral resolution of 20 nm and axial resolution of 50 nm. Imaging was performed using the NIS-Elements AR 4.3 with N-STORM 3.4 software. Molecule lists were exported from NIS in txt format, and the three image planes of the ics-ids file pairs from the deconvolved confocal stacks matching the STORM volume were converted to the ome-tiff format using FIJI software. Confocal and corresponding STORM images were fitted in Adobe Photoshop CS6. Localization points exceeding a photon count of 2000 were counted as specific super-resolution localization points. Local density filter (10 neighbours within 150 nm for P2Y12R) and Z-filter (±300 nm from focal plane) was applied to the localization points.
Correlated CLSM and immune-electron microscopy
After extensive washes in PB and 0.05 M TBS sections were blocked in 1% HSA in TBS and 0,03% Triton X.100 and 50 ug/mL digitonin (D141-100MG, Sigma). Then, they were incubated in primary antibodies (chicken anti-DCX, guinea-pig anti-IBA1, together with either rabbit anti-IBA1 or rabbit anti-P2Y12R) diluted in TBS containing 0.05% sodium azide for 2–3 days. After repeated washes in TBS, the sections were incubated in fluorophore-conjugated and biotinylated secondary antibodies diluted in TBS. Secondary antibody incubation was followed by washes in TBS and PB. Sections were mounted in PB, coverslipped, sealed with nail-polish. Immunofluorescence for DCX and IBA1-Gp was analyzed using a Nikon Eclipse Ti-E inverted microscope (Nikon Instruments Europe B.V., Amsterdam, The Netherlands), with a CFI Plan Apochromat VC 60× water immersion objective (NA: 1.2) and an A1R laser confocal system. We used 488, 561 and 647 nm lasers, and scanning was done in line serial mode. Image stacks were obtained with NIS-Elements AR software with a pixel size of 50 × 50 nm in X-Y, and 150 nm Z-steps. After imaging, sections were washed thoroughly, and incubated in avidin–biotinylated horseradish peroxidase complex (Elite ABC; 1:300; Vector Laboratories) diluted in TBS for 3 h at room temperature or overnight at 4°C. The immunoperoxidase reaction against IBA1-Rb or P2Y12R was developed using 3,3-diaminobenzidine (DAB; Sigma-Aldrich) as chromogen. The sections were then treated with 1% OsO4 in 0.1 M PB, at room temperature, dehydrated in ascending alcohol series and in acetonitrile and embedded in Durcupan (ACM; Fluka). During dehydration, the sections were treated with 1% uranyl acetate in 70% ethanol for 20 min. For electron microscopic analysis, correlated tissue samples from the VZ of E15 and from the gyrus dentatus of P90 mice were glued onto Durcupan blocks. Consecutive 70 nm thick sections were cut using an ultramicrotome (Leica EM UC7) and picked up on Formvar-coated single-slot grids. Ultrathin sections were examined in a Hitachi 7100 electron microscope equipped with a Veleta CCD camera (Olympus Soft Imaging Solutions, Germany). During the correlation a large number of wildfield images were taken from the samples and several maps were constructed to ensure that the very same tissue volumes are processed for electron microscopy that have been imaged using CLSM. Somatic junctions were randomly selected in the confocal stacks, and these contacts were found on the electron microscopic images to test the prevalence of direct membrane-membrane contacts. For correlation and montage stitching Adobe Photshop CS6 and Fiji were used.
Ex vivo 2-photon imaging in acute brain slices
Acute brain slices were prepared from 1 and 8 days old (P1 and P8) CX3CR1+/GFP mice. Animals were placed on ice for 5–6 min, decapitated and the brains were transferred rapidly to ice-cold artificial cerebrospinal fluid (ACSF; containing in mM: 126 NaCl, 2.5 KCl, 10 glucose, 1.25 NaH2PO4, 2 MgCl2, 2 CaCl2 and 26 NaHCO3) saturated with carbogen (95% O2-5% CO2). Coronal (300 μm thick) brain slices were cut with a Leica VT1200S Vibratome in ice-cold ACSF. The slices were loaded with 40 μM Cal-590-AM dissolved in ACSF, for 60–90 min, at RT. The Calbryte 590-AM, this robust fluorescence-based assay tool for detecting intracellular calcium mobilization was chosen for its brightness, high signal to noise ratio, improved intracellular retention and loading efficiency as well as homogenous cytoplasmic distribution. To ensure adequate oxygenation of the submerged slices during dye incubation, the loading chamber (25 mm diameter; 1 mL volume) was continuously oxygenated with carbogen.
Multiphoton imaging was accomplished by a Nikon Eclipse FN1 upright microscope, equipped with a Nikon A1R MP + multiphoton system using a Chameleon Vision II Ti:Sapphire tunable (680–1080 nm) laser and a 25× water dipping objective lens [CFI75 Apo LWD NA = 1.1 WD = 2 mm]. The excitation wavelength was set to 980 nm to measure GFP (peak at 920 nm) and Calbryte-590 (Cal-590, peak at 1050 nm) signals simultaneously. The emission light was split at 593 nm and recorded with two separate detectors. For GFP 550/88 and for Cal-590 AM 641/75 emission filters were used. The Z-stack (2 μm step size, 10 μm range) images were taken at a 0.25 frame per second rate, for 15 min in ACSF (3 mL/min perfusion rate) followed by a 15 min perfusion with vehicle or 4 μM PSB-0739, a selective P2Y12R antagonist. Maximum intensity projections were used for calcium data analysis using Fiji software within the boundaries of Cal-590-AM loaded cell bodies. The microglial process coverage of individual cells was determined in a 1.2 μm region around the cell body. Here, the area fraction of thresholded pixels (corresponding to GFP positive microglial processes) were determined in time, for each individual neuron. Coverage in control is 1, and PSB or vehicle effect is plotted as increase or decrease compared to control (Figures 4E and 4G, respectively).
Quantification and statistical analysis
Quantitative analysis of CLSM data
Quantitative analysis of each dataset was performed by at least two observers, who were blinded to the origin of the samples, the experiments and did not know of each other’s results. For the colocalization measurement of microgial markers during development in the mouse brain, confocal stacks with triple immunofluorescent labeling (IBA1-Gp, IBA1-Gt and P2Y12R) were used, acquired from the VZ/SVZ region of E15, neocortex of P1-P15 and dentate gyrus of P90 mice. During the analysis cells with a DAPI-labeled nucleus in the IBA1-Gp channel were randomly selected. After that we measured the colocalization with the other two microglia labelings (IBA1-Gt, P2Y12R) in each age group.
For the analysis of somatic junction prevalence, confocal stacks with double immunofluorescent labeling (DCX and IBA1) were acquired from the VZ/SVZ region of E15, neocortex of P1-P15 and dentate gyrus of P90 mice. All labeled and identified cells or proximal tufts (the proximal segment of the thickest dendrite of the cell, not longer than the cell body itself) were counted, when the whole cell body or tuft was located within the Z-stack. Given somata or proximal tufts were considered to be contacted by microglia, when a microglial process clearly touched it (i.e. there was no space between neuronal soma and microglial process) on an at least 0.5 μm long segment.
TOM20 fluorescent intensity profiles were analyzed using a semi-automatic method. Confocal stacks with triple immunofluorescent labeling (microglia, DCX and TOM20) were collected. The section containing the largest cross-section of a neuron was used to trace the cell membrane according to DCX-labeling. This contour was then expanded and narrowed by 0.5 μm to get an extracellular and an intracellular line, respectively (Figure 2M). The intensity of fluorescent labeling was analyzed along these lines. After normalizing and scaling, microglial contact was identified where microglial fluorescent intensity was over 20% of total, for at least 500 nm. Then the contact area was extended 500-500 nm on both sides, and TOM20 fluorescent intensity within these areas was measured for “contact” value.
For the measurement of the density of DCX-positive cell bodies in the developing cortex of different genotypes at P8, Ctip2 and Satb2 immunofluorescent staining was used to delineate cortical layers, and the density of DCX + developing neurons was assessed in layers 6, 4/5 and 2/3. DCX + -cells were counted with the optical dissector method within the 50 μm thick CLSM stacks, using the NIS-Elements software and 100 × 100 μm counting frames.
For the measurement of the density of Kv2.1-positive neuronal cell bodies in the adult cortex of different genotypes, Kv2.1 and Kv2.2 immunofluorescent stainings, together with DAPI-labeling were used to delineate cortical layers (as described in (Bishop et al., 2015)), and the density of neurons was assessed in layers 6, 5, 4, 2/3 and 1. Neuronal cell bodies were counted with the optical dissector method within the 50 μm thick CLSM stacks, using the NIS-Elements software and 50 × 50 μm counting frames.
For the measurement of DCX-positive cell distribution in P1 cortex, the measurement of Ki67-positive cell density, Ki67/DCX colocalization, density of contacts between microglial processes and Ki67-positive cells, the measurement of Annexin-V-positive cell density, Annexin-V/DCX colocalization, density of contacts between microglial processes and Annexin-V-positive cells, 20 μm thick cryostate sections from brains of WT and P2Y12R −/− mice were imaged, and the width of the cortex divided into 10 equal parts (Figures 6B and 6C). The objects within these ROI-s were counted with the optical dissector method. In all experiments it was ensured, that measurements are performed on sections with identical rostro-caudal position.
Quantitative analysis of STORM data
For the spatial analysis of super-resolution data, contacting processes were divided into 4 equal quarters based on their distance from the neuronal membrane, the 1st being in direct contact with the neuron, the 4th being the opposite side, (Figures 3A–3F, schematic representation and data: G). STORM channels with the outline of the quarters were exported, the STORM channel was binarized in Fiji, and the integrated density was measured for each quarter, and percentages calculated.
Statistical analysis
All quantitative assessment was performed in a blinded manner wherever it was possible. We applied the nonparametric Wilcoxon signed rank test to compare two dependent data groups, Mann-Whitney U-test to compare two independent groups, and Kruskal-Wallis test to compare multiple independent groups. Statistical analysis was performed with the Statistica 13.4.0.14 package (TIBCO), differences with p < 0.05 were considered significant throughout this study. (Significance was labeled on the figures the following way: not significant: n.s., p < 0.05: ∗, p < 0.01: ∗∗, p < 0.001: ∗∗∗).
Acknowledgments
We thank Drs. László Barna and Pál Vági for kindly providing microscopy support and Dóra Gali-Györkei, Balázs Pintér, and Erika Tischler for excellent technical assistance. The study was supported by “Momentum” research grants from the Hungarian Academy of Sciences (LP2016-4/2016 and LP2022-5/2022 to Á.D.), ERC-CoG 724994 (to Á.D.), the János Bolyai Research Scholarship of the Hungarian Academy of Sciences (to C.C.), ÚNKP-20-3-II and ÚNKP-21-4 (to B.P.) and ÚNKP-20-5 and ÚNKP-21-5 (to C.C.), the New National Excellence Program of the Ministry for Innovation and Technology, the Gedeon Richter's Talentum Foundation (to A.D.S.), and by 2020-1.2.4-TÉT-IPARI-2021-00005 (to Á.D.). I.K. was supported by the National Brain Research Program (2017-1.2.1-NKP-2017-00002) and by the National Research, Development and Innovation Office, Hungary (Frontier Program 129961). The funding institutions had no role in the conceptualization, design, data collection, analysis, decision to publish, or preparation of the manuscript.
Author contributions
C.C., B.P., Z.I.L., Z.L., and Á.D. conceived the project. Tissue samples were prepared by Z.I.L. and Z.L. Immunohistochemistry and CLSM were performed by C.C., B.P., and A.D.S. STORM microscopy was performed by B.P. Correlated light and electron microscopy were performed by C.C. and A.D.S. In vitro imaging was performed by Z.K. and M.K. Analysis was performed by C.C., B.P., A.D.S., A.K., Z.K., and M.N. I.K. provided resources and essential intellectual contributions and revised the manuscript. Á.D. obtained funding and supervised the project. C.C. and Á.D. wrote the paper with input from all authors.
Declaration of interests
The authors declare no competing interests.
Published: September 20, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111369.
Contributor Information
Csaba Cserép, Email: cserep.csaba@koki.hu.
Ádám Dénes, Email: denes.adam@koki.hu.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Aarum J., Sandberg K., Haeberlein S.L.B., Persson M.A.A. Migration and differentiation of neural precursor cells can be directed by microglia. Proc. Natl. Acad. Sci. USA. 2003;100:15983–15988. doi: 10.1073/pnas.2237050100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali A.A.H., Abdel-Hafiz L., Tundo-Lavalle F., Hassan S.A., von Gall C. P2Y2 deficiency impacts adult neurogenesis and related forebrain functions. FASEB J. 2021;35:e21546. doi: 10.1096/fj.202002419RR. [DOI] [PubMed] [Google Scholar]
- Araki T., Ikegaya Y., Koyama R. The effects of microglia- and astrocyte-derived factors on neurogenesis in health and disease. Eur. J. Neurosci. 2020;54:5880–5901. doi: 10.1111/ejn.14969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrázola M.S., Andraini T., Szelechowski M., Mouledous L., Arnauné-Pelloquin L., Davezac N., Belenguer P., Rampon C., Miquel M.C. Mitochondria in developmental and adult neurogenesis. Neurotox. Res. 2019;36:257–267. doi: 10.1007/s12640-018-9942-y. [DOI] [PubMed] [Google Scholar]
- Baalman K., Marin M.A., Ho T.S.Y., Godoy M., Cherian L., Robertson C., Rasband M.N. Axon initial segment–associated microglia. J. Neurosci. 2015;35:2283–2292. doi: 10.1523/JNEUROSCI.3751-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajwa E., Pointer C.B., Klegeris A. The role of mitochondrial damage-associated molecular patterns in chronic neuroinflammation. Mediators Inflamm. 2019;2019:1–11. doi: 10.1155/2019/4050796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battista D., Ferrari C.C., Gage F.H., Pitossi F.J. Neurogenic niche modulation by activated microglia: transforming growth factor β increases neurogenesis in the adult dentate gyrus. Eur. J. Neurosci. 2006;23:83–93. doi: 10.1111/j.1460-9568.2005.04539.x. [DOI] [PubMed] [Google Scholar]
- Beckervordersandforth R. Mitochondrial metabolism-mediated regulation of adult neurogenesis. Brain Plast. 2017;3:73–87. doi: 10.3233/BPL-170044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y., Woodin M.A., Sernagor E., Cancedda L., Vinay L., Rivera C., Legendre P., Luhmann H.J., Bordey A., Wenner P., et al. Refuting the challenges of the developmental shift of polarity of GABA actions: GABA more exciting than ever. Front. Cell. Neurosci. 2012;6:35. doi: 10.3389/fncel.2012.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhola P.D., Letai A. Mitochondria—judges and executioners of cell death sentences. Mol. Cell. 2016;61:695–704. doi: 10.1016/j.molcel.2016.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilimoria P.M., Stevens B. Microglia function during brain development: new insights from animal models. Brain Res. 2015;1617:7–17. doi: 10.1016/j.brainres.2014.11.032. [DOI] [PubMed] [Google Scholar]
- Bishop H.I., Guan D., Bocksteins E., Parajuli L.K., Murray K.D., Cobb M.M., Misonou H., Zito K., Foehring R.C., Trimmer J.S. Distinct cell-and layer-specific expression patterns and independent regulation of kv2 channel subtypes in cortical pyramidal neurons. J. Neurosci. 2015;35:14922–14942. doi: 10.1523/JNEUROSCI.1897-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume Z.I., Lambert J.M., Lovel A.G., Mitchell D.M. Microglia in the developing retina couple phagocytosis with the progression of apoptosis via P2RY12 signaling. Dev. Dyn. 2020;249:723–740. doi: 10.1002/dvdy.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O., Ziv Y., Schwartz A., Landa G., Talpalar A.E., Pluchino S., Martino G., Schwartz M. Microglia activated by IL-4 or IFN-γ differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol. Cell. Neurosci. 2006;31:149–160. doi: 10.1016/j.mcn.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Chandel N.S. Mitochondria as signaling organelles. BMC Biol. 2014;12:34. doi: 10.1186/1741-7007-12-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chugh D., Ekdahl C.T. Interactions between microglia and newly formed hippocampal neurons in physiological and seizure-induced inflammatory environment. Brain Plast. 2016;1:215–221. doi: 10.3233/BPL-150014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cserép C., Pósfai B., Dénes Á. Shaping neuronal fate: functional heterogeneity of direct microglia-neuron interactions. Neuron. 2021;109:222–240. doi: 10.1016/j.neuron.2020.11.007. [DOI] [PubMed] [Google Scholar]
- Cserép C., Pósfai B., Lénárt N., Fekete R., László Z.I., Lele Z., Orsolits B., Molnár G., Heindl S., Schwarcz A.D., et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science. 2020;367:528–537. doi: 10.1126/science.aax6752. [DOI] [PubMed] [Google Scholar]
- Cunningham C.L., Martínez-Cerdeño V., Noctor S.C. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J. Neurosci. 2013;33:4216–4233. doi: 10.1523/JNEUROSCI.3441-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D., Grutzendler J., Yang G., Kim J.V., Zuo Y., Jung S., Littman D.R., Dustin M.L., Gan W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- De Lucia C., Rinchon A., Olmos-Alonso A., Riecken K., Fehse B., Boche D., Perry V.H., Gomez-Nicola D. Microglia regulate hippocampal neurogenesis during chronic neurodegeneration. Brain Behav. Immun. 2016;55:179–190. doi: 10.1016/j.bbi.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S., Van Deren D., Peden E., Hockin M., Boulet A., Titen S., Capecchi M.R. Two distinct ontogenies confer heterogeneity to mouse brain microglia. Development. 2018;145:dev152306. doi: 10.1242/dev.152306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dénes Á., Ferenczi S., Halász J., Környei Z., Kovács K.J. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J. Cereb. Blood Flow Metab. 2008 doi: 10.1038/jcbfm.2008.64. [DOI] [PubMed] [Google Scholar]
- Diaz-Aparicio I., Paris I., Sierra-Torre V., Plaza-Zabala A., Rodríguez-Iglesias N., Márquez-Ropero M., Beccari S., Huguet P., Abiega O., Alberdi E., et al. Microglia actively remodel adult hippocampal neurogenesis through the phagocytosis secretome. J. Neurosci. 2020;40:1453–1482. doi: 10.1523/JNEUROSCI.0993-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato A., Kagias K., Zhang Y., Hilliard M.A. Neuronal sub-compartmentalization: a strategy to optimize neuronal function. Biol. Rev. Camb. Philos. Soc. 2019;94:1023–1037. doi: 10.1111/brv.12487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erblich B., Zhu L., Etgen A.M., Dobrenis K., Pollard J.W. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS One. 2011;6:e26317. doi: 10.1371/journal.pone.0026317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala J.C., Feinberg M., Popov V., Harris K.M. Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J. Neurosci. 1998;18:8900–8911. doi: 10.1523/JNEUROSCI.18-21-08900.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Füger P., Hefendehl J.K., Veeraraghavalu K., Wendeln A.-C., Schlosser C., Obermüller U., Wegenast-Braun B.M., Neher J.J., Martus P., Kohsaka S., et al. Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat. Neurosci. 2017;20:1371–1376. doi: 10.1038/nn.4631. [DOI] [PubMed] [Google Scholar]
- Ginhoux F., Greter M., Leboeuf M., Nandi S., See P., Gokhan S., Mehler M.F., Conway S.J., Ng L.G., Stanley E.R., et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson J.G., Lin P.T., Flanagan L.A., Walsh C.A. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 1999;23:257–271. doi: 10.1016/s0896-6273(00)80778-3. [DOI] [PubMed] [Google Scholar]
- Gouveia A., Bajwa E., Klegeris A. Extracellular cytochrome c as an intercellular signaling molecule regulating microglial functions. Biochim. Biophys. Acta. Gen. Subj. 2017;1861:2274–2281. doi: 10.1016/j.bbagen.2017.06.017. [DOI] [PubMed] [Google Scholar]
- Gunner G., Cheadle L., Johnson K.M., Ayata P., Badimon A., Mondo E., Nagy M.A., Liu L., Bemiller S.M., Kim K.-W., et al. Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nat. Neurosci. 2019;22:1075–1088. doi: 10.1038/s41593-019-0419-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg H., Mallard C., Rousset C.I., Thornton C. Mitochondria: hub of injury responses in the developing brain. Lancet Neurol. 2014;13:217–232. doi: 10.1016/S1474-4422(13)70261-8. [DOI] [PubMed] [Google Scholar]
- Hammond T.R., Dufort C., Dissing-Olesen L., Giera S., Young A., Wysoker A., Walker A.J., Gergits F., Segel M., Nemesh J., et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity. 2019;50:253–271.e6. doi: 10.1016/j.immuni.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori Y., Naito Y., Tsugawa Y., Nonaka S., Wake H., Nagasawa T., Kawaguchi A., Miyata T. Transient microglial absence assists postmigratory cortical neurons in proper differentiation. Nat. Commun. 2020;11:1631. doi: 10.1038/s41467-020-15409-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes S.E., Hollopeter G., Yang G., Kurpius D., Dailey M.E., Gan W.-B., Julius D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006;9:1512–1519. doi: 10.1038/nn1805. [DOI] [PubMed] [Google Scholar]
- Hirasawa T., Ohsawa K., Imai Y., Ondo Y., Akazawa C., Uchino S., Kohsaka S. Visualization of microglia in living tissues using Iba1-EGFP transgenic mice. J. Neurosci. Res. 2005;81:357–362. doi: 10.1002/jnr.20480. [DOI] [PubMed] [Google Scholar]
- Hobert O., Carrera I., Stefanakis N. The molecular and gene regulatory signature of a neuron. Trends Neurosci. 2010;33:435–445. doi: 10.1016/j.tins.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata R., Casimir P., Vanderhaeghen P. Mitochondrial dynamics in postmitotic cells regulate neurogenesis. Science. 2020;369:858–862. doi: 10.1126/science.aba9760. [DOI] [PubMed] [Google Scholar]
- Kaila K., Price T.J., Payne J.A., Puskarjov M., Voipio J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 2014;15:637–654. doi: 10.1038/nrn3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khacho M., Slack R.S. Mitochondrial dynamics in the regulation of neurogenesis: from development to the adult brain. Dev. Dyn. 2018;247:47–53. doi: 10.1002/dvdy.24538. [DOI] [PubMed] [Google Scholar]
- Khazipov R., Esclapez M., Caillard O., Bernard C., Khalilov I., Tyzio R., Hirsch J., Dzhala V., Berger B., Ben-Ari Y. Early development of neuronal activity in the primate hippocampus in utero. J. Neurosci. 2001;21:9770–9781. doi: 10.1523/JNEUROSCI.21-24-09770.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierdorf K., Prinz M. Microglia in steady state. J. Clin. Invest. 2017;127:3201–3209. doi: 10.1172/JCI90602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T., Murakami F. Evidence that dendritic mitochondria negatively regulate dendritic branching in pyramidal neurons in the neocortex. J. Neurosci. 2014;34:6938–6951. doi: 10.1523/JNEUROSCI.5095-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitayama M., Ueno M., Itakura T., Yamashita T. Activated microglia inhibit axonal growth through RGMa. PLoS One. 2011;6 doi: 10.1371/journal.pone.0025234. e25234-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi H., Kobayashi M., Kunisawa T., Imai K., Sayo A., Malissen B., Crocker P.R., Sato K., Kiyama H. Siglec-H is a microglia-specific marker that discriminates microglia from CNS-associated macrophages and CNS-infiltrating monocytes. Glia. 2017;65:1927–1943. doi: 10.1002/glia.23204. [DOI] [PubMed] [Google Scholar]
- László Z.I., Lele Z., Zöldi M., Miczán V., Mógor F., Simon G.M., Mackie K., Kacskovics I., Cravatt B.F., Katona I. ABHD4-dependent developmental anoikis safeguards the embryonic brain. Nat. Commun. 2020;11:4363. doi: 10.1038/s41467-020-18175-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz K.M., Nelson L.H. Microglia and beyond: innate immune cells as regulators of brain development and behavioral function. Front. Immunol. 2018;9:698. doi: 10.3389/fimmu.2018.00698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Barres B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018;18:225–242. doi: 10.1038/nri.2017.125. [DOI] [PubMed] [Google Scholar]
- Lorenz C., Prigione A. Mitochondrial metabolism in early neural fate and its relevance for neuronal disease modeling. Curr. Opin. Cell Biol. 2017;49:71–76. doi: 10.1016/j.ceb.2017.12.004. [DOI] [PubMed] [Google Scholar]
- Marín-Teva J.L., Dusart I., Colin C., Gervais A., Van Rooijen N., Mallat M. Microglia promote the death of developing purkinje cells. Neuron. 2004;41:535–547. doi: 10.1016/s0896-6273(04)00069-8. [DOI] [PubMed] [Google Scholar]
- Martynoga B., Drechsel D., Guillemot F. Molecular control of neurogenesis: a view from the mammalian cerebral cortex. Cold Spring Harb. Perspect. Biol. 2012;4:a008359. doi: 10.1101/cshperspect.a008359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matcovitch-Natan O., Winter D.R., Giladi A., Vargas Aguilar S., Spinrad A., Sarrazin S., Ben-Yehuda H., David E., Zelada González F., Perrin P., et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science. 2016;353:aad8670. doi: 10.1126/science.aad8670. [DOI] [PubMed] [Google Scholar]
- Menéndez-Méndez A., Díaz-Hernández J.I., Ortega F., Gualix J., Gómez-Villafuertes R., Miras-Portugal M.T. Specific temporal distribution and subcellular localization of a functional vesicular nucleotide transporter (VNUT) in cerebellar granule neurons. Front. Pharmacol. 2017;8:951. doi: 10.3389/fphar.2017.00951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A., Huang H., Radke J., Stenzel W., Priller J. P2Y12 receptor is expressed on human microglia under physiological conditions throughout development and is sensitive to neuroinflammatory diseases. Glia. 2017;65:375–387. doi: 10.1002/glia.23097. [DOI] [PubMed] [Google Scholar]
- Miller I., Min M., Yang C., Tian C., Gookin S., Carter D., Spencer S.L. Ki67 is a graded rather than a binary marker of proliferation versus quiescence. Cell Rep. 2018;24:1105–1112.e5. doi: 10.1016/j.celrep.2018.06.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming G.L., Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70:687–702. doi: 10.1016/j.neuron.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto A., Wake H., Ishikawa A.W., Eto K., Shibata K., Murakoshi H., Koizumi S., Moorhouse A.J., Yoshimura Y., Nabekura J. Microglia contact induces synapse formation in developing somatosensory cortex. Nat. Commun. 2016;7:12540. doi: 10.1038/ncomms12540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo M., Eyo U.B., Xie M., Peng J., Bosco D.B., Umpierre A.D., Zhu X., Tian D.-S., Xu P., Wu L.-J. Microglial P2Y12 receptor regulates seizure-induced neurogenesis and immature neuronal projections. J. Neurosci. 2019;39:9453–9464. doi: 10.1523/JNEUROSCI.0487-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra D.P., Vacher H., Trimmer J.S. The surprising catch of a voltage-gated potassium channel in a neuronal SNARE. Sci. STKE. 2007:pe37. doi: 10.1126/stke.3932007pe37. [DOI] [PubMed] [Google Scholar]
- Mosser C.-A., Baptista S., Arnoux I., Audinat E. Microglia in CNS development: shaping the brain for the future. Prog. Neurobiol. 2017;149–150:1–20. doi: 10.1016/j.pneurobio.2017.01.002. [DOI] [PubMed] [Google Scholar]
- Noctor S.C., Penna E., Shepherd H., Chelson C., Barger N., Martínez-Cerdeño V., Tarantal A.F. Periventricular microglial cells interact with dividing precursor cells in the nonhuman primate and rodent prenatal cerebral cortex. J. Comp. Neurol. 2019;527:1598–1609. doi: 10.1002/cne.24604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli R.C., Ferretti M.T. Function and dysfunction of microglia during brain development: consequences for synapses and neural circuits. Front. Synaptic Neurosci. 2017;9:9–17. doi: 10.3389/fnsyn.2017.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli R.C., Bolasco G., Pagani F., Maggi L., Scianni M., Panzanelli P., Giustetto M., Ferreira T.A., Guiducci E., Dumas L., et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- Park G.H., Noh H., Shao Z., Ni P., Qin Y., Liu D., Beaudreault C.P., Park J.S., Abani C.P., Park J.M., et al. Activated microglia cause metabolic disruptions in developmental cortical interneurons that persist in interneurons from individuals with schizophrenia. Nat. Neurosci. 2020;23:1352–1364. doi: 10.1038/s41593-020-00724-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst C.N., Yang G., Ninan I., Savas J.N., Yates J.R., Lafaille J.J., Hempstead B.L., Littman D.R., Gan W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155:1596–1609. doi: 10.1016/j.cell.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penna E., Mangum J.M., Shepherd H., Martínez-Cerdeño V., Noctor S.C. Development of the neuro-immune-vascular plexus in the ventricular zone of the prenatal rat neocortex. Cereb. Cortex. 2021;31:2139–2155. doi: 10.1093/cercor/bhaa351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrelli F., Scandella V., Montessuit S., Zamboni N., Martinou J.-C., Knobloch M. Mitochondrial pyruvate metabolism regulates the activation of quiescent adult neural stem cells. bioRxiv. 2022 doi: 10.1126/sciadv.add5220. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pont-Lezica L., Beumer W., Colasse S., Drexhage H., Versnel M., Bessis A. Microglia shape corpus callosum axon tract fasciculation: functional impact of prenatal inflammation. Eur. J. Neurosci. 2014;39:1551–1557. doi: 10.1111/ejn.12508. [DOI] [PubMed] [Google Scholar]
- Prins J.R., Eskandar S., Eggen B.J.L., Scherjon S.A. Microglia, the missing link in maternal immune activation and fetal neurodevelopment; and a possible link in preeclampsia and disturbed neurodevelopment? J. Reprod. Immunol. 2018;126:18–22. doi: 10.1016/j.jri.2018.01.004. [DOI] [PubMed] [Google Scholar]
- Rangaraju V., Lewis T.L., Hirabayashi Y., Bergami M., Motori E., Cartoni R., Kwon S.K., Courchet J. Pleiotropic mitochondria: the influence of mitochondria on neuronal development and disease. J. Neurosci. 2019;39:8200–8208. doi: 10.1523/JNEUROSCI.1157-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Réu P., Khosravi A., Bernard S., Mold J.E., Salehpour M., Alkass K., Perl S., Tisdale J., Possnert G., Druid H., Frisén J. The lifespan and turnover of microglia in the human brain. Cell Rep. 2017;20:779–784. doi: 10.1016/j.celrep.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro D.E., Glaser T., Oliveira-Giacomelli Á., Ulrich H. Purinergic receptors in neurogenic processes. Brain Res. Bull. 2019;151:3–11. doi: 10.1016/j.brainresbull.2018.12.013. [DOI] [PubMed] [Google Scholar]
- Rodrigues R.J., Marques J.M., Cunha R.A. Purinergic signalling and brain development. Semin. Cell Dev. Biol. 2019;95:34–41. doi: 10.1016/j.semcdb.2018.12.001. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Iglesias N., Sierra A., Valero J. Rewiring of memory circuits: connecting adult newborn neurons with the help of microglia. Front. Cell Dev. Biol. 2019;7:1–20. doi: 10.3389/fcell.2019.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter M.W., Stevens B. Microglia emerge as central players in brain disease. Nat. Med. 2017;23:1018–1027. doi: 10.1038/nm.4397. [DOI] [PubMed] [Google Scholar]
- Sato K. Effects of microglia on neurogenesis. Glia. 2015;63:1394–1405. doi: 10.1002/glia.22858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer D.P., Lehrman E.K., Kautzman A.G., Koyama R., Mardinly A.R., Yamasaki R., Ransohoff R.M., Greenberg M.E., Barres B.A., Stevens B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki T., Hori T., Miyata H., Maehara M., Namba T. Analysis of proliferating neuronal progenitors and immature neurons in the human hippocampus surgically removed from control and epileptic patients. Sci. Rep. 2019;9:18194. doi: 10.1038/s41598-019-54684-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellner S., Paricio-Montesinos R., Spieß A., Masuch A., Erny D., Harsan L.A., Elverfeldt D.V., Schwabenland M., Biber K., Staszewski O., et al. Microglial CX3CR1 promotes adult neurogenesis by inhibiting Sirt 1/p65 signaling independent of CX3CL1. Acta Neuropathol. Commun. 2016;4:102. doi: 10.1186/s40478-016-0374-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemoto-Mogami Y., Hoshikawa K., Goldman J.E., Sekino Y., Sato K. Microglia enhance neurogenesis and oligodendrogenesis in the early postnatal subventricular zone. J. Neurosci. 2014;34:2231–2243. doi: 10.1523/JNEUROSCI.1619-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A., Encinas J.M., Deudero J.J.P., Chancey J.H., Enikolopov G., Overstreet-Wadiche L.S., Tsirka S.E., Maletic-Savatic M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7:483–495. doi: 10.1016/j.stem.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silbereis J.C., Pochareddy S., Zhu Y., Li M., Sestan N. The cellular and molecular landscapes of the developing human central nervous system. Neuron. 2016;89:248–268. doi: 10.1016/j.neuron.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubannier V., McLelland G.L., Zunino R., Braschi E., Rippstein P., Fon E.A., McBride H.M. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 2012;22:135–141. doi: 10.1016/j.cub.2011.11.057. [DOI] [PubMed] [Google Scholar]
- Squarzoni P., Thion M.S., Garel S. Neuronal and microglial regulators of cortical wiring: usual and novel guideposts. Front. Neurosci. 2015;9:248. doi: 10.3389/fnins.2015.00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squarzoni P., Oller G., Hoeffel G., Pont-Lezica L., Rostaing P., Low D., Bessis A., Ginhoux F., Garel S. Microglia modulate wiring of the embryonic forebrain. Cell Rep. 2014;8:1271–1279. doi: 10.1016/j.celrep.2014.07.042. [DOI] [PubMed] [Google Scholar]
- Stefani J., Tschesnokowa O., Parrilla M., Robaye B., Boeynaems J.M., Acker-Palmer A., Zimmermann H., Gampe K. Disruption of the microglial ADP receptor P2Y13 enhances adult hippocampal neurogenesis. Front. Cell. Neurosci. 2018;12:134. doi: 10.3389/fncel.2018.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura A., McLelland G.-L., Fon E.A., McBride H.M. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. 2014;33:2142–2156. doi: 10.15252/embj.201488104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suyama S., Sunabori T., Kanki H., Sawamoto K., Gachet C., Koizumi S., Okano H. Purinergic signaling promotes proliferation of adult mouse subventricular zone cells. J. Neurosci. 2012;32:9238–9247. doi: 10.1523/JNEUROSCI.4001-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terenzio M., Schiavo G., Fainzilber M. Compartmentalized signaling in neurons: from cell biology to neuroscience. Neuron. 2017;96:667–679. doi: 10.1016/j.neuron.2017.10.015. [DOI] [PubMed] [Google Scholar]
- Thion M.S., Garel S. On place and time: microglia in embryonic and perinatal brain development. Curr. Opin. Neurobiol. 2017;47:121–130. doi: 10.1016/j.conb.2017.10.004. [DOI] [PubMed] [Google Scholar]
- Thion M.S., Ginhoux F., Garel S. Microglia and early brain development: an intimate journey. Science. 2018;362:185–189. doi: 10.1126/science.aat0474. [DOI] [PubMed] [Google Scholar]
- Tyzio R., Represa A., Jorquera I., Ben-Ari Y., Gozlan H., Aniksztejn L. The establishment of GABAergic and glutamatergic synapses on CA1 pyramidal neurons is sequential and correlates with the development of the apical dendrite. J. Neurosci. 1999;19:10372–10382. doi: 10.1523/JNEUROSCI.19-23-10372.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno M., Fujita Y., Tanaka T., Nakamura Y., Kikuta J., Ishii M., Yamashita T. Layer V cortical neurons require microglial support for survival during postnatal development. Nat. Neurosci. 2013;16:543–551. doi: 10.1038/nn.3358. [DOI] [PubMed] [Google Scholar]
- Urbán N., Guillemot F. Neurogenesis in the embryonic and adult brain: same regulators, different roles. Front. Cell. Neurosci. 2014;8:396. doi: 10.3389/fncel.2014.00396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbina F.L., Gupton S.L. SNARE-mediated exocytosis in neuronal development. Front. Mol. Neurosci. 2020;13:1–17. doi: 10.3389/fnmol.2020.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbina F.L., Gomez S.M., Gupton S.L. Spatiotemporal organization of exocytosis emerges during neuronal shape change. J. Cell Biol. 2018;217:1113–1128. doi: 10.1083/jcb.201709064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanRyzin J.W., Marquardt A.E., Argue K.J., Vecchiarelli H.A., Ashton S.E., Arambula S.E., Hill M.N., McCarthy M.M. Microglial phagocytosis of newborn cells is induced by endocannabinoids and sculpts sex differences in juvenile rat social play. Neuron. 2019;102:435–449.e6. doi: 10.1016/j.neuron.2019.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verney C., Monier A., Fallet-Bianco C., Gressens P. Early microglial colonization of the human forebrain and possible involvement in periventricular white-matter injury of preterm infants. J. Anat. 2010;217:436–448. doi: 10.1111/j.1469-7580.2010.01245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton N.M., Sutter B.M., Laywell E.D., Levkoff L.H., Kearns S.M., Marshall G.P., Scheffler B., Steindler D.A. Microglia instruct subventricular zone neurogenesis. Glia. 2006;54:815–825. doi: 10.1002/glia.20419. [DOI] [PubMed] [Google Scholar]
- Weinhard L., di Bartolomei G., Bolasco G., Machado P., Schieber N.L., Neniskyte U., Exiga M., Vadisiute A., Raggioli A., Schertel A., et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 2018;9:1228. doi: 10.1038/s41467-018-03566-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan S.P., Zuckerbraun B.S. Mitochondrial signaling: forwards, backwards, and in between. Oxid. Med. Cell. Longev. 2013:351613. doi: 10.1155/2013/351613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z.-X., Kim G.H., Tan J.-W., Riso A.E., Sun Y., Xu E.Y., Liao G.-Y., Xu H., Lee S.-H., Do N.-Y., et al. Elevated protein synthesis in microglia causes autism-like synaptic and behavioral aberrations. Nat. Commun. 2020;11:1797. doi: 10.1038/s41467-020-15530-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y., Miura M. Programmed cell death in neurodevelopment. Dev. Cell. 2015;32:478–490. doi: 10.1016/j.devcel.2015.01.019. [DOI] [PubMed] [Google Scholar]
- Yoo D.Y., Yoo K.Y., Choi J.W., Kim W., Lee C.H., Choi J.H., Park J.H., Won M.H., Hwang I.K. Time course of postnatal distribution of doublecortin immunoreactive developing/maturing neurons in the somatosensory cortex and hippocampal CA1 region of C57BL/6 mice. Cell. Mol. Neurobiol. 2011;31:729–736. doi: 10.1007/s10571-011-9670-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Microglial processes (green, CX3CR1-GFP) establish somatic contacts on cell bodies of multiple developing neurons (red, Cal-590). Some of the contact sites are readily re-connected multiple times by microglial processes. Imaging duration: 1 h.
After 15 min “baseline imaging” PSB-0739 (selective inhibitor of microglial P2Y12R-s) was added to the perfusion, and 15 min further imaging was performed. Acute inhibition of microglial P2Y12Rs induced a rapid and robust decrease of microglial process coverage.
After 15 min “baseline imaging” PSB-0739 (selective inhibitor of microglial P2Y12R-s) was added to the perfusion, and 15 min further imaging was performed. Acute inhibition of microglial P2Y12Rs induced a rapid and robust decrease of microglial process coverage.
After 15 min “baseline imaging” vehicle was added to the perfusion, and 15 min further imaging was performed. Vehicle addition did not induce any significant change in microglial coverage.
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.