

Abstract
Adoptive cell-based immunotherapy typically utilizes cytotoxic T lymphocytes (CTLs), expanding these cells ex vivo. Such expansion is traditionally accomplished through the use of autologous APCs that are capable of interactions with T cells. However, incidental inhibitory program such as CTLA-4 pathway can impair T cell proliferation. We therefore designed a nanobody which is specific for CTLA-4 (CTLA-4 Nb 16), and we then used this molecule to assess its ability to disrupt CTLA-4 signaling and thereby overcome negative costimulation of T cells. With CTLA-4 Nb16 stimulation, dendritic cell/hepatocellular carcinoma fusion cells (DC/HepG2-FCs) enhanced autologous CD8+ T cell proliferation and production of IFN-γ in vitro, thereby leading to enhanced killing of tumor cells. Using this approach in the context of adoptive CD8+ immunotherapy led to a marked suppression of tumor growth in murine NOD/SCID hepatocarcinoma or breast cancer xenograft models. We also observed significantly increased tumor cell apoptosis, and corresponding increases in murine survival. These findings thus demonstrate that in response to nanobody stimulation, DC/tumor cells-FC-induced specific CTLs exhibit superior anti-tumor efficacy, making this a potentially valuable means of achieving better adoptive immunotherapy outcomes in cancer patients.
Keywords: CTLA-4, Nanobody, Tumor-Specific CTL, Adoptive Immunotherapy
1. INTRODUCTION
Tumor is one of the diseases that threaten human health in today’s society [1, 2]. Currently, in addition to surgery, chemotherapy, and irradiation, immunotherapy has become a promising way to treat cancers [3]. Many ways for cancer immunotherapy have been established [4] including (i) application of specific killing effects of CTLs on cancer cells, (ii) functional improvement of DCs, (iii) inhibition of tumor angiogenesis and cancer cell growth by downregulating the expression of tumor-associated macrophages (TAM) with relevant drugs [5], and (iv) stimulation of DCs and helper T cells (Ths) by cytokines to improve the anti-cancer effects of immunocytes, such as the stimulation of interferon-γ (IFN-γ), interleukin-4 (IL-4), IL-10, and granulocyte-macrophage colony-stimulating factor (GM-CSF)[6–9]. Among these methods, tumor specific cytotoxic T lymphocyte adoptive therapy has received increasing attention [10–12]. The traditional adoptive immunotherapy generally uses CTLs, however, their marginal efficacy with the killing of tumor cells compromises their application making it necessary to find other ways to improve the effects of adoptive immunotherapy [13, 14]. It is known that enhancing the quality and number of adaptive T cells is a valid way to improve therapeutic effects. By transfecting total RNA, introducing specific liver cancer antigen peptides [15, 16] or cell lysates [17] from liver cancer cells to dendritic cells, or generating fusion cells between DCs and liver cancer cells [18], the specific liver cancer CTLs could be successfully induced. Although the fusion tumor/DCs can express specific tumor antigens and surface molecules of DCs effectively stimulating T cell proliferation [19, 20], the efficiency of inducing CTLs for cancer therapy [21] is low. In our previous study, we established the technology to effectively generate the fusion tumor/DCs that induced specific liver cancer T lymphocytes that were effective for liver cancer cell killing [22]. The curative effect of our strategy, however, needed further improvement.
CTLA-4 is a key negative costimulatory molecule in T cells and is expressed in activated T cells and on some cancer cells. CTLA-4 can competitively bind with B7 and inhibit further activation of T cells. It inhibits early T cell expansion and opposes the action of CD28-mediated costimulation [23]. Upon T cell activation, it is quickly upregulated and can bind B7 molecules with affinity superior to that of CD28 [24, 25]. The anti-cancer effects of CTLA-4 make it aan ideal tumor immunotherapy target by using anti-CTLA-4 antibody [26, 27]. Although the targeted therapy using anti-CTLA-4 antibody has achieved some success for some cancers, there are many limitations of this strategy. These include non-specific binding between the antibody and normal tissues, heterogeneity of tumor antigens, and poor penetration of antibody in solid tumors [28, 29]. Therefore, there is an urgent need to significantly improve this strategy by developing novel antibodies with high efficiency and low toxicity for cancer therapy exploiting the current progress in this field [30]. To this end, antibody humanization, high efficiency, and miniaturization are the major considerations for the development of novel therapeutic antibodies for cancers [31].
Nanobodies are single chain antibodies with one heavy chain variable region, which is the minimal unit to stably bind to antigen [32–35]. As small genetically engineered antibodies, nanobodies have many advantages for targeted immunotherapy of tumors including high expression, solubility, stability, strong tissue penetration and weak immunogenicity [36, 37]. In our previous study, we developed a nanobody specific for CTLA-4 (CTLA-4 Nb16) [38]. The efficacy and mechanism of CTLA-4 Nb16, however, needed further investigation. In this study, we exploredthe anti-tumor function and mechanism of tumor-specific CD8+ T lymphocytes induced by DC/HepG2-FCs or DC/MCF 7-FCs upon stimulation with CTLA-4 Nb16. The induced tumor-specific CD8+ T lymphocytes were further applied for adoptive immunotherapy in NOD/SCID mice with cancer xenografts to understand the mechanism by which CTLA-4 Nb16 boost the antitumor effects of tumor-specific T lymphocytes induced by DC/HepG2-FCs and DC/MCF 7-FCs. We presume that nanobody CTLA-4 Nb16 may represent a promising tool for inducing highly efficient CTLs and promoting the anti-tumor immune response (Scheme 1).
Scheme 1. Illustration of therapy with FC+ Nanobody.
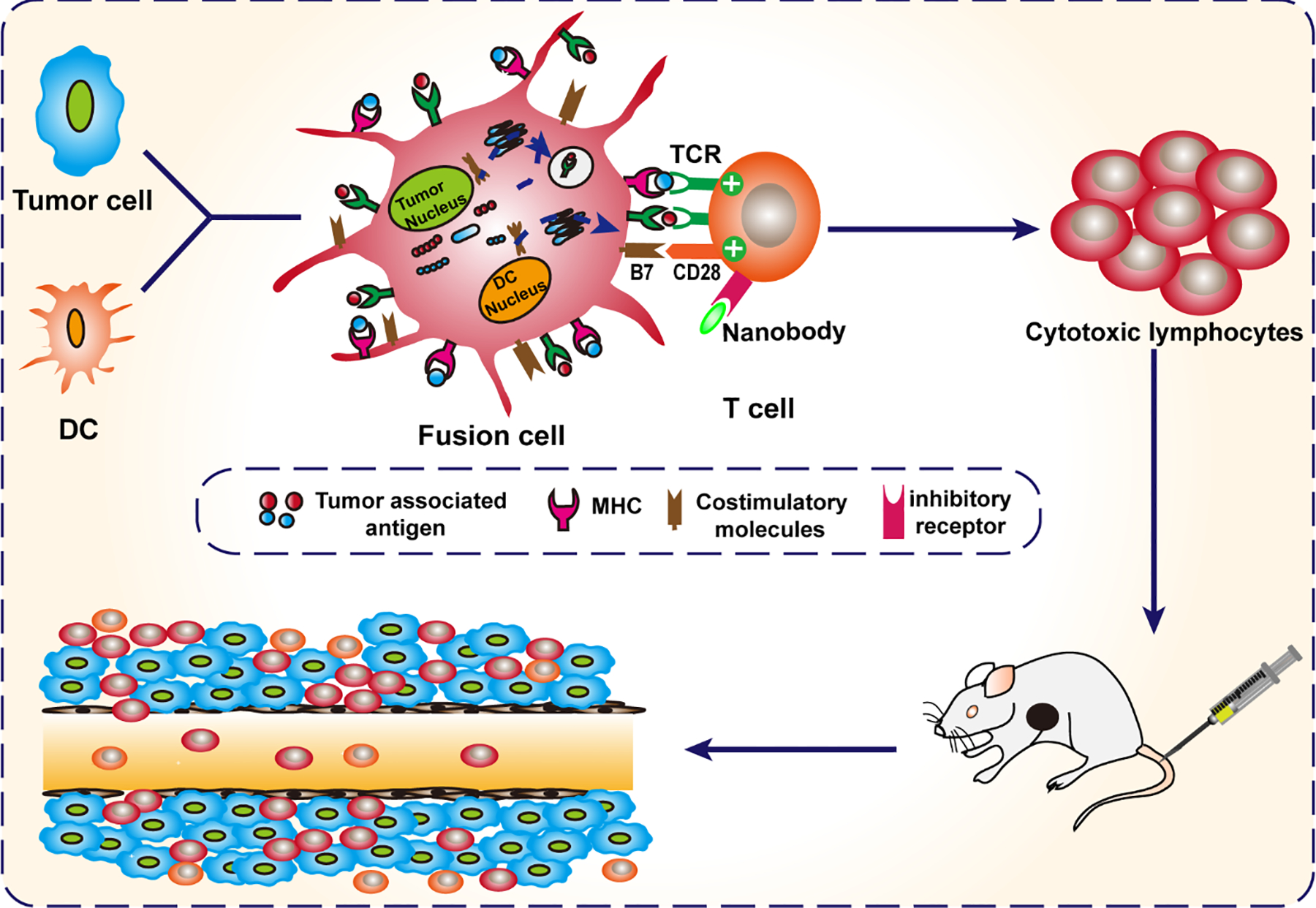
DC fusion cells (DC/tumor fusion cells, FCs) are generated. Nanobody against CTLA-4 (CTLA-4 Nb16) eliminates immunosuppression via disrupting CTLA-4-mediated negative costimulation in T cells induced by dendritic cell/tumor fusion cells. Finally, the induced-CTLs were transferred to kill tumor cells.
2. MATERIALS AND METHODS
2.1. Animals and Cells
Female NOD/SCID mice of 4–6 weeks age were from Beijing Vital River Lab Animal Technology Co. Ltd (Beijing, China), under SPF grade raise. The Institutional Animal Care and Use Committee of Guangxi Medical University approved all animal studies. HepG2 and MCF7 cells were from ATCC and were grown in DMEM containing 10% FBS and penicillin/streptomycin at 37 °C in a 5% CO2 incubator.
2.2. Antibodies and Nanobodies
The human CTLA-4 Nb16 and CD105 nanobodies were developed in our laboratory. CTLA-4 recombinant protein was from Abcam (Cambridge, UK). The anti CTLA-4 monoclonal antibody was acquired from BD Biosciences (NJ, USA).
2.3. T Cells and DC Generation
Density gradient centrifugation was used to collect healthy donor peripheral blood mononuclear cells (PBMCs), which were grown in RPMI-1640 medium (Gibco) containing 10% FBS and penicillin/streptomycin. Following 2 h of culture, the suspended cells (T cells) were grown in RPMI-1640 containing 100 U/mL recombinant human IL-2 (Sigma-Aldrich). In addition, cells which adhered were used for dendritic cell differentiation via culture in RPMI supplemented with 1000 U/mL recombinant human GM-CSF (rhGM-CSF; R&D) and 500 U/mL rhIL-4 (R&D). The Institutional Review Board (Guangxi Medical University) approved all human sample research.
2.4. Fusion cell preparation
Fusion cell preparation mixed with HepG2 or MCF-7 cells labelled using PKH26 at a 5:3 ratio. After spinning at 1500 rpm for 10 min, preheated (40 °C) polyethylene glycol (PEG) (Sigma, USA) was used to treat cells for 3 minutes, then 180 μL PBS and 5 ng/mL of collagen were added following centrifugation. The cells were washed and stimulated using500 U/mL rhGM-CSF and 100 U/mL rh IL-4 in RIPM 1640 for 24 h. DAPI staining was then conducted prior to fluorescent microscopic examination. The DC/HepG2 fusion cells (DHFC) or the DC/MCF-7 fusion cells (DMFC) were prepared. Furthermore, the DC/HepG2 fusion cells at day 7 were stained using FITC-labeled antibodies specific for CD80, CD86, and MHCII (eBiosciences, CA, USA) as well as immature DC, and expression of these markers was assessed on DHFC via flow cytometry.
2.5. CTL Preparation
CD8+ T cells isolated from peripheral blood lymphocytes were plated in 24-well plates (5×106 /mL) and IL-2 (100 U/mL), IL-4 (500 U/mL) and GM-CSF (1000 U/mL) were used to treat cells. Human CD8+ T cells were first incubated with DHFC followed by stimulation with CTLA-4 Nb16, CTLA-4 antibody, or negative control CD105 Nb. According to incubate with various antibodies, cells were divided into five groups: T cells alone, DHFC, DHFC +CD105 Nb, DHFC +CTLA-4 mAb, and DHFC +CTLA-4 Nb16.
2.6. Cell Proliferation Assay
How CTLA-4 Nb16 and CTLA-4 antibody stimulation affected human lymphocyte proliferation in vitro was assessed via flow cytometry Human CD8+ T cells were grown using RPMI containing 10% FBS, 100 U/mL rhIL-2 at 37 °C with 5% CO2 in 6-well plates (cell confluence 106/mL). Human CD8+ T cells were first incubated with DHFC followed by stimulation with CTLA-4 Nb16, CTLA-4 antibody, or negative control CD105 Nb. The DHFC to CD8+ T cells marked with CFSE ratio was 1:10. CTLA-4 Nb16, CTLA-4 antibody, or CD105 Nb was added to the media (50 μg/mL). Five days later, the content of CFSE positive CD8+ T cells in cell suspension was examined viaflow cytometry analysis.
2.7. ELISPOT Assay
CD8+ T cells with IFN-γ secretion was examined by ELISPOT. Briefly, 100 μL of cell suspension was maintained in 96-well plate for 16–20 h at 37 °C with 5% CO2. Subsequently, cell suspension from each group was incubated with biotin-labeled anti-IFN-γ monoclonal antibody. By adding AEC solution, the antibody binding was examined. The spots of ELISPOT plate were counted with CTL instrument.
2.8. Cytotoxicity Assay
HepG2 and MCF-7 cells, used as target cells, were maintained in 48-well plates. Human CD8+ cells were first incubated with DHFC followed by stimulation with CTLA-4 Nb16, CTLA-4 antibody, or negative control CD105 Nb. After collection of CD8+ T lymphocytes, the E:T ratio of 5:1, 10:1 or 20:1 was set by mixing CD8+ T lymphocytes and HepG2 or MCF-7 cells. Target cells without staining, with PKH26 staining, and were treated with high temperature and stained with PI as a control for establishing flow regulation compensation. After co-culture 6 h at 37 °C with 5% CO2, target cells were stained with PI and then apoptosis was analyzed by flow cytometry. The killing rate was then calculated.
2.9. Anti-tumor Effect Experiments In Vivo
Female NOD/SCID mice aged 4–6 weeks were used. For subcutaneous tumor models, cells were collected and resuspended with PBS to implantation. Each mouse was inoculated with HepG2 cells by subcutaneous injection (2 × 106 cells/injection). For MCF-7 models, 5 × 106 cells were implanted. The length and width of tumor were measured with a caliper every 6 days. When tumors reached ~100 mm3, NOD/SCID mice were randomly divided into 6 groups (5 mice in each group). The mice treatment in each group were tail vein injected with induced CD8+ T lymphocytes every 7 days (5 × 106 cells each injection) for 3 times. Mice injected with non-stimulated CD8+ T cells or with only PBS were set as PBS and T cells group, respectively. Tumor size were measured by calipers for tumor growth and mouse survival time were recorded for survival rate analysis.
2.10. Immunohistochemistry
Immunohistochemical analysis was used to evaluate the proliferation and apoptosis of tumor. Tumor tissue was fixed in 4% Faure Marin buffer overnight. After paraffin embedding, tumor tissue was sectioned with 4 μm thickness. Tumor tissue sections were dewaxed in dimethylbenzene and ethanol solution. To examine Ki67 expression, sections were incubated with anti-Ki67 monoclonal antibody overnight at 4 °C. After washing, sections were further probedwith the HRP-labeled secondary antibody. Cell apoptosis analysis was performed by TUNEL assay. Sections were inspected with an inverted fluorescence microscope and photographed. Five cross sections were randomly selected in each tumor tissue section, and each experiment was repeated three times.
2.11. Statistical Analysis
The statistical difference was measured by t-test. One way ANOVA for more than three experiments and the value was indicated as mean ± SD. Survival was assessed via the Kaplan–Meier test. Statistical analyses were processed with Prism GraphPad 6 software. P < 0.05 indicated significance.
3. RESULTS
3.1. Preparation and Characterization of the DC/HepG2 Fusion Cells (DHFC)
We analyzed the efficiency of fusion through both fluorescent microscopic imaging and flow cytometry. Cell nucleus was stained with DAPI, then DCs were stained with CFSE (green) and HepG2 were stained using PKH26 (red) such that fused cells exhibit orange fluorescence in fluorescent images. We achieved DC/HepG2 fusion cells percentages over 75%, and DC/HepG2 fusion cells exhibited a larger volume and multiple nuclei (Figure 1A). Relative to immature DC cells, mature DHFC expressed higher CD80, CD86 and MHCII levels after 500 U/mL rhGM-CSF and 100 U/mL rhIL-4 stimulation (Figure 1B–C). This indicated that FC were still capable of responding to factors which stimulate DCs, meaning they may still be able to effectively present antigen to T cells.
Figure 1. Identification of DC/HepG2 fusion cells and express increased CD80, CD86 and MHC II.
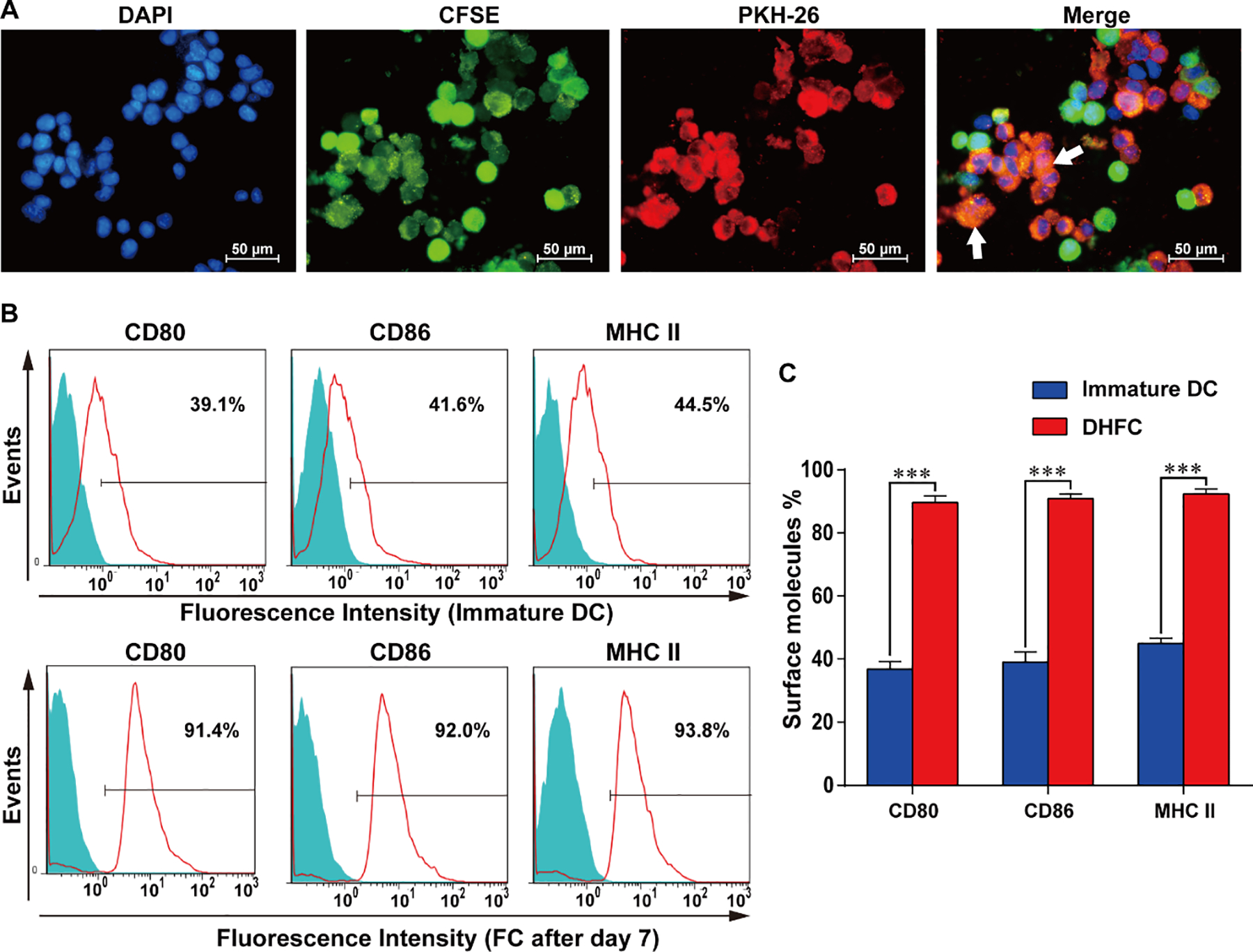
(A) The DC2.4/HepG2 fusion cell identification approach was as follows: CSFE (green) and PKH26 (red) were used for the respective labeling of DCs and HepG2 cells, after which PEG was used to mediate fusion. DAPI (blue) was then used for nuclear staining, followed by fluorescence microscopy with fused cells marked by white arrows (× 400). Images are representative of three independent experiments. (B) DCs exhibited MHC II and costimulatory molecule expression at day 7 in FCs. (C) The status of three membrane molecules, MHC II, CD80 and CD86 were high at day 7 in FCs. Data are means ± SD, n = 3. *** P < 0.001.
3.2. CTLA-4 Nb16 Promoted Proliferation of Induced CD8+ T Cells
Initially, we examined whether FC-induced CD8+ T cells proliferation was promoted by CTLA-4 Nb16. CFSE-labeled CD8+ cells were mixed with FCs, stimulated with different antibodies and then co-cultured for 5 days. Proliferation of CD8+ T cells was analyzed by flow cytometry. As shown in Figure 2A–B, the CFSE proliferation assay indicated that the cell proliferation under DHFC+CTLA-4 Nb16 induction was significantly higher than that under DHFC or DHFC+CD105 Nb induction. These data suggested that DHFC +CTLA-4 Nb16 promoted proliferation of CD8+ T lymphocytes.
Figure 2. CTLA-4 Nb16 promoted CD8+ T cell proliferation.
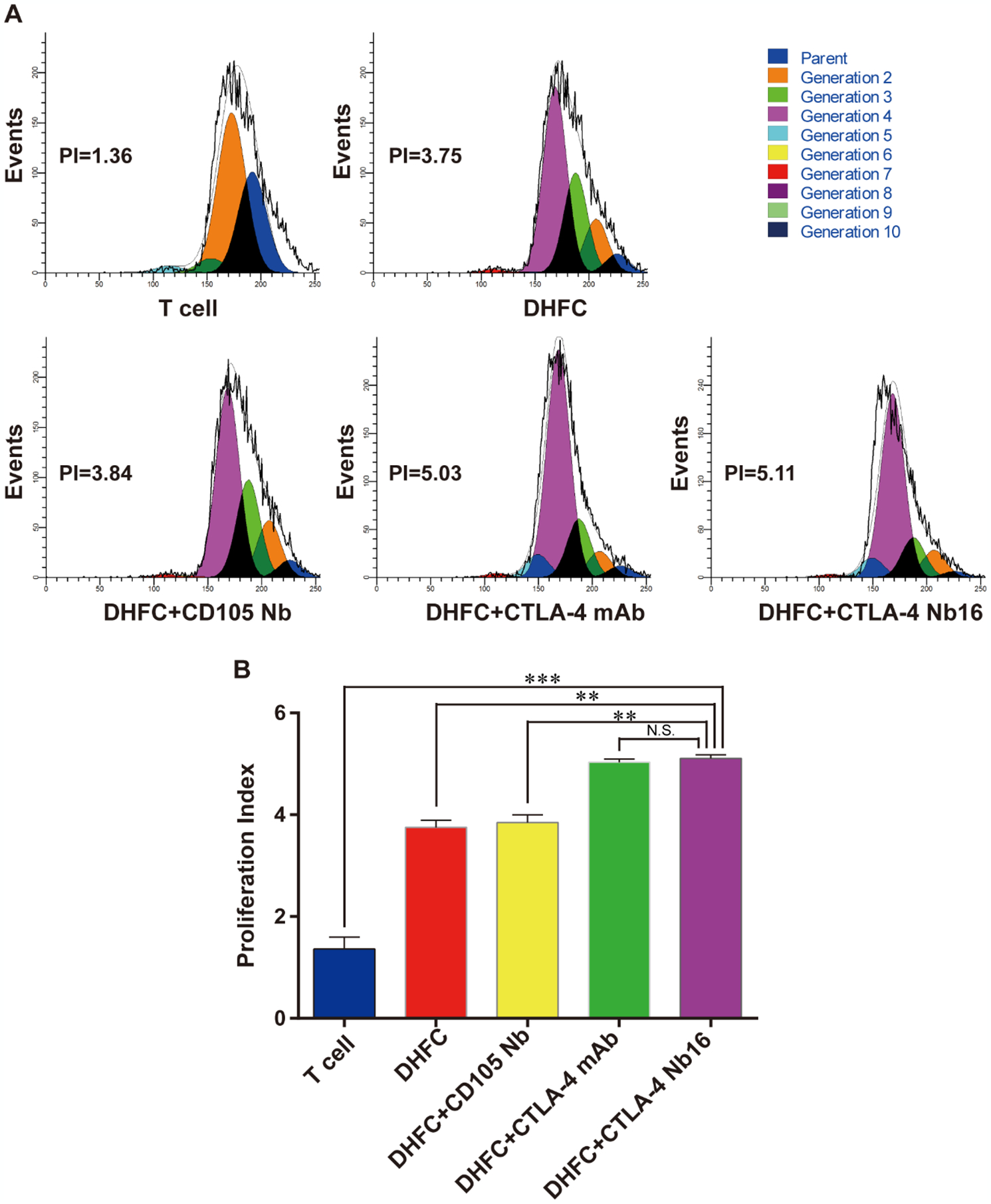
(A) CFSE-labeled CD8+ cells were mixed with different antibodies following with DHFC, and then co-cultured for 5 days. Proliferation of CD8+ T cells was assessed via flow cytometry. (B) CD8+ T Cell proliferation Index in DHFC+CTLA-4 Nb16 group was higher than that in other’s groups, except DHFC+CTLA-4 mAb group. Data are means ± SD, n = 3. NS stand for P > 0.05, ** P < 0.01, *** P < 0.001.
3.3. CTLA-4 Nb16 Increased the Number of FC-induced IFN-γ-secreting CD8+ T Cells
We investigated the effects of CTLA-4 Nb16 on stimulating IFN-γ production from CD8+ T cells via ELISPOT. CD8+ cells were mixed with FC cells, stimulated with different antibodies, and then co-cultured for 7 days. The results clearly indicated that the positive cell numbers in DHFC+CTLA-4 Nb16 group were markedly more than that in other groups (Figure 3A), a conclusion further verified by the statistical analysis (Figure 3B, P < 0.001 or P < 0.0001). These results demonstrated that DHFC+CTLA-Nb16 might induce T lymphocyte activation and increase the cell numbers with IFN-γ secretion.
Figure 3. CTLA-4 Nb16 increased the abundance of IFN-γ secreting CD8+ T lymphocytes.
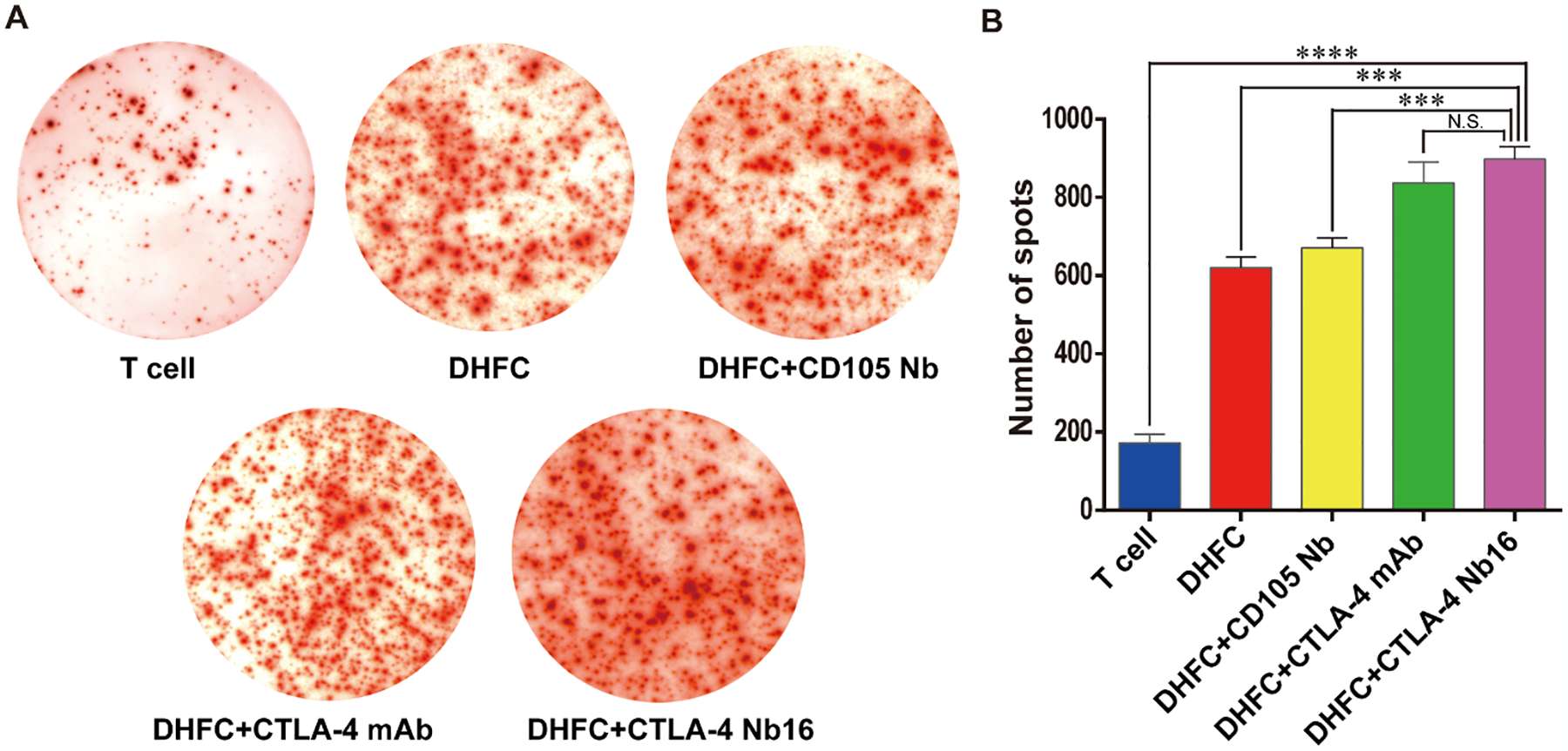
(A) CD8+ cells were mixed with different antibodies following with DHFC, and then co-cultured for 7 days. ELISPOT assay was applied to examine the number of CD8+ T lymphocytes secreting IFN-γ. (B) The number of spots in DHFC+CTLA-4 Nb16 group increased significantly than other’s groups, except DHFC+CTLA-4 mAb groups. Data are means ± SD, n = 3. *** P < 0.001, **** P < 0.0001.
3.4. CTLA-4 Nb16 Facilitated the Tumor Cell Killing by FC-induced CD8+ T Cells
A cytotoxicity assay was used to examine the impact of DHFC+CTLA-4 Nb16-induced CD8+ T lymphocytes on tumor cell killing. Figure 4A determinated, by the effector:target ratios of (E:T) 5:1, 10:1 or 20:1, that DHFC+CTLA-4 Nb16-induced CD8+ T lymphocytes exhibited much higher killing effects on HepG2 cells than the CD8+ T lymphocytes induced by other fusion cells with control nanobodies. As shown in Figure 4B, the DHFC+CTLA-4 Nb16-induced CD8+ T lymphocytes effector cells had a certain killing effect on MCF-7, but the killing ability was lower than that of HepG2.
Figure 4. CTLA-4 Nb16 promoted the tumor cell killing effects of CD8+ lymphocytes.
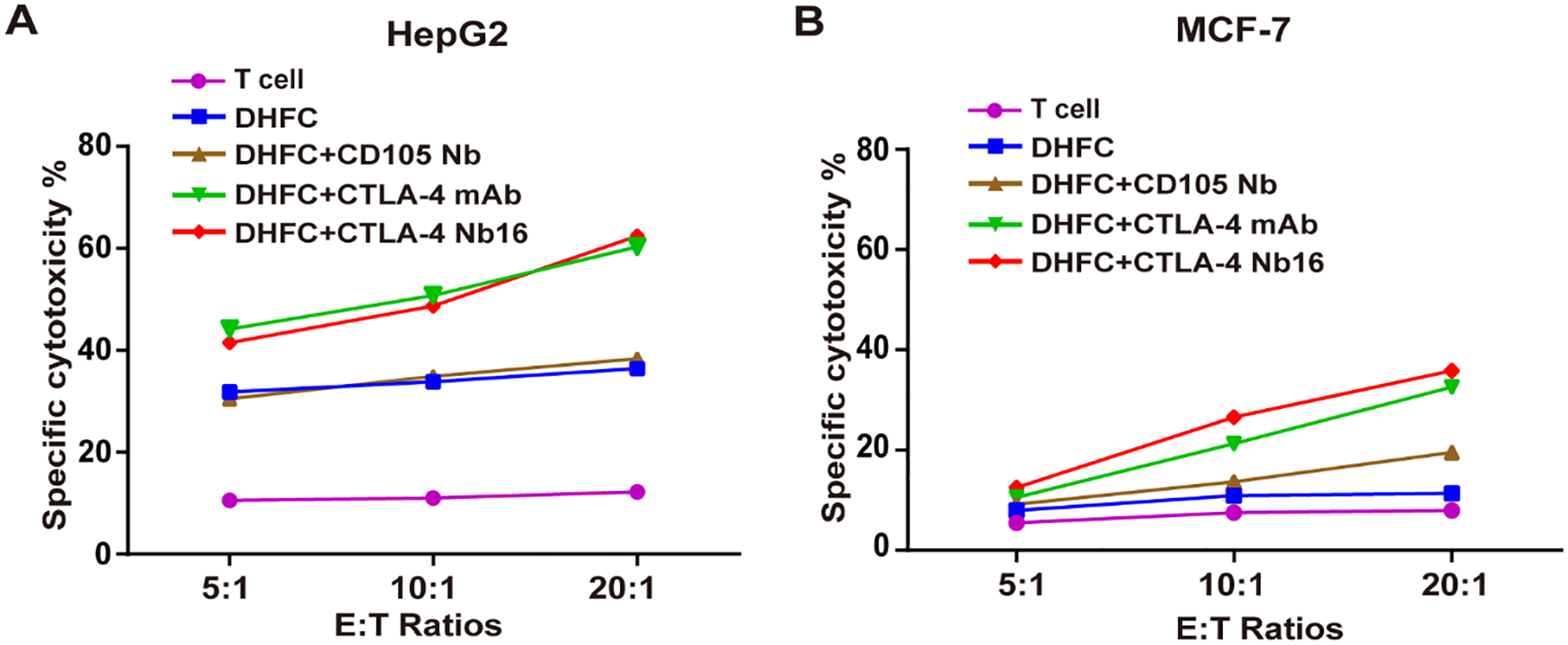
(A) Comparison of cell killing effects of CD8+ T lymphocytes induced by indicated treatments on HepG2 in different E:T raitos of 5:1, 10:1 and 20:1. DHFC+CTLA-4 Nb16 enhanced cytotoxic T cell killing function on target HepG2 cells. (B) Comparison of cell killing effects of CD8+ T lymphocytes induced by indicated treatments on MCF-7. It had weaker killing effect on MCF-7 than HepG2. Data are means ± SD, n = 3.
3.5. CTLA-4 Nb16 Increased the Anti-tumor Efficacy of DHFC-induced CD8+ T Cells In Vivo
Model therapy experiments were conducted as a means of assessing the anti-tumor effects in animal model. We examined the effects of DHFC+CTLA-4 Nb16-induced CD8+ T lymphocytes in vivo. As shown in Figure 5A & B, adoptive therapy using DHFC+CTLA-4 Nb16-induced CD8+ T lymphocytes resulted in the smallest average tumor size and the longest survival time in all groups. These results indicated that the adoptive therapy using DHFC+CTLA-4 Nb16-induced CD8+ T lymphocytes significantly suppressed tumor growth and enhanced murine survival in animals with human liver cancer. Similar results were observed in mice with human breast cancer xenografts treated with DMFC+CTLA-4 Nb16-induced CD8+ T lymphocytes. DMFC+CTLA-4 Nb16 treatment group could inhibit breast cancer growth and prolong the survival of mice, the results showed a certain therapeutic effect (Figure 5 C - D).
Figure 5. CTLA-4 Nb16 increased the anti-tumor effects of CD8+ T lymphocyte by inhibiting HepG2 and MCF-7 tumor growth and enhancing survival.
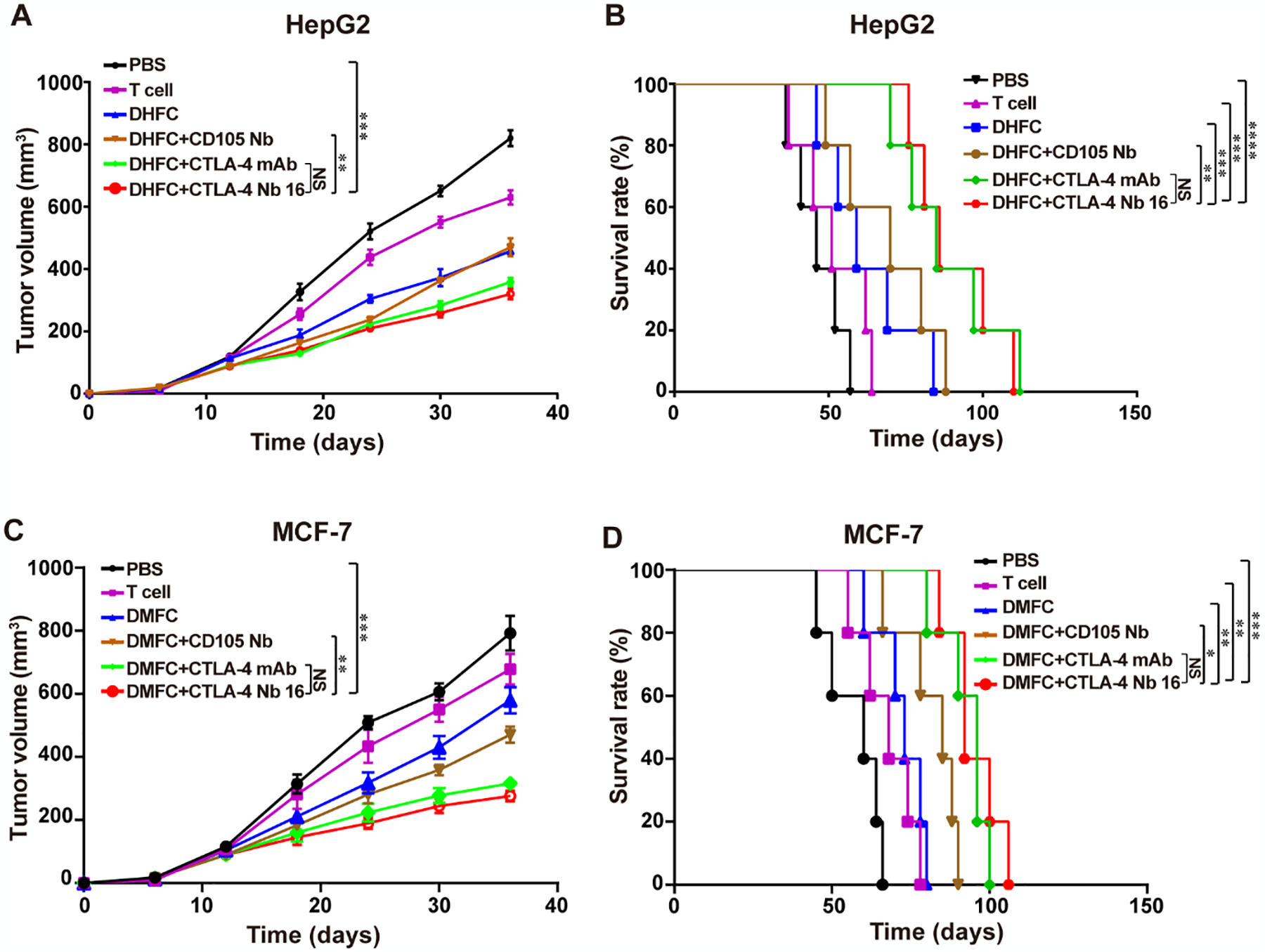
(A) The Therapy with DHFC+CTLA-4 Nb16 significantly delayed the tumor growth in mice bearing liver cancer xenograft. (B) Kaplan-Meier survival curves of mice reported in DHFC+CTLA-4 Nb16 group therapy significantly increased survival. (C) The Therapy with DMFC+CTLA-4 Nb16 significantly delayed the tumor growth in mice bearing breast cancer xenograft. (D) Kaplan-Meier survival curves of mice reported in DMFC+CTLA-4 Nb16 group therapy significantly increased survival. Data are means ± SD or percentage survival (n = 5 mice/group). **P < 0.01, *** P < 0.001.
3.6. CTLA-4 Nb16 Promoted Tumor Cell Apoptosis by FC-induced CD8+ T Cells
Immunohistochemical analysis showed fewer Ki67 positive cells in tumor tissues of mice with adoptive therapy using DHFC+CTLA-4 Nb16 induced CD8+ T lymphocytes than those in other groups (Figure 6A–B). While TUNEL assays indicated that the number of TUNEL positive cells was higher compared to those in other groups (Figure 6C). Statistical analysis of an average number of apoptotic cells and standard derivation further confirmed that the difference was significant (Figure 6D). These results demonstrated that treatment with DHFC +CTLA-4 Nb16 efficiently impaired tumor cell proliferation, mediating their apoptotic death in mice by induced CD8+ T lymphocytes.
Figure 6. CTLA-4 Nb16 stimulation suppressed proliferation of tumor cells and promoted tumor cell apoptosis in mice.
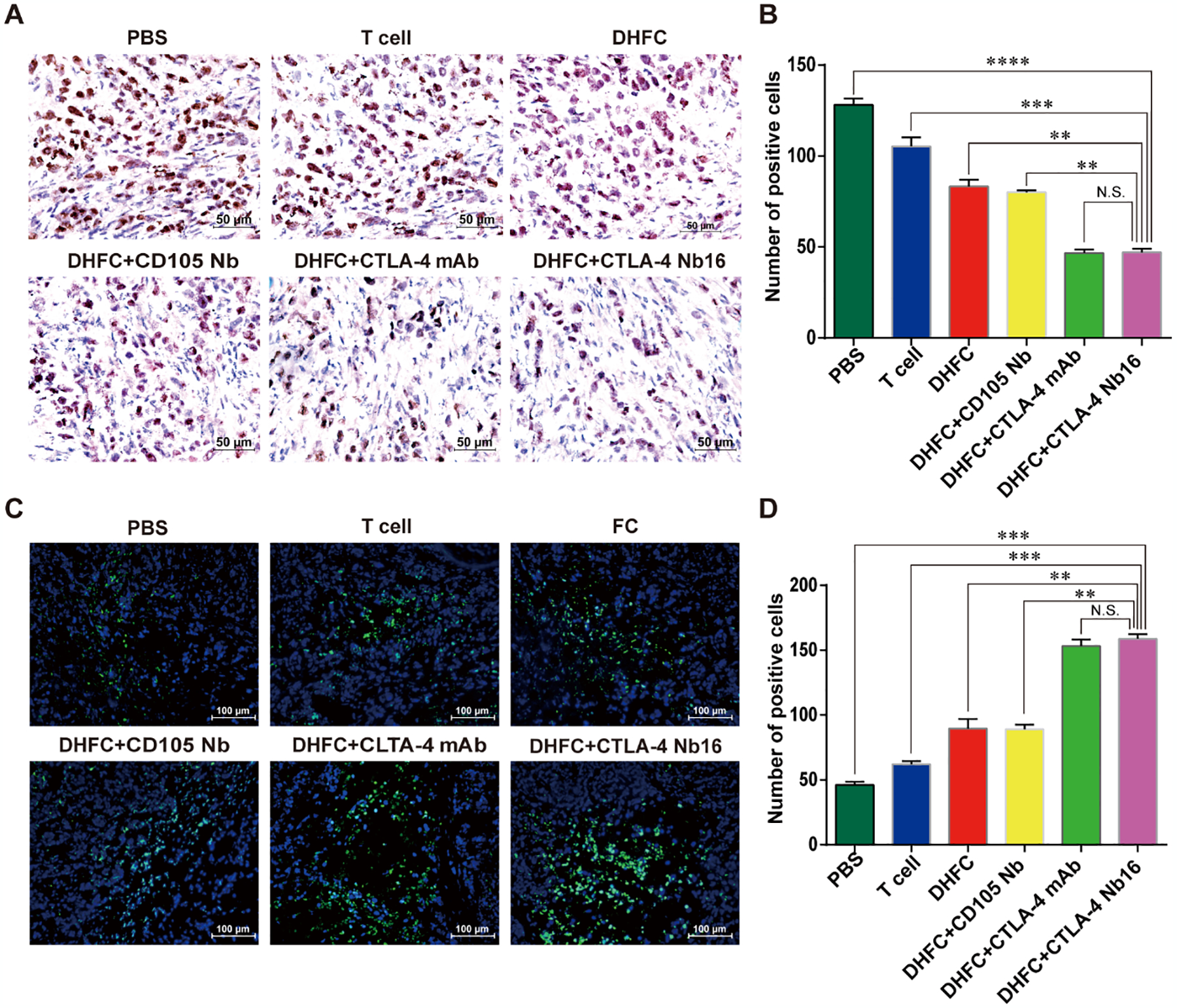
(A) Immunohistochemical assay of Ki67 staining in different groups (x 40). Brown in nuclear indicates the positive staining. (B) The Ki67 expression of tumor cells in DHFC+CTLA4-Nb16 group was significant lower than in other groups, except in DHFC+CTLA4-mAb group. (C) TUNEL assay was used to monitor cell apoptosis in different groups. Blue indicates nuclear and green indicates the positive signal (×20). (D) The apoptosis of tumor cells in DHFC+CTLA4-Nb16 group was significant higher than that in other’s groups, except in DHFC+CTLA4-mAb group. Data are measn ± SD. **P < 0.01, *** P < 0.001, **** P < 0.0001.
4. DISCUSSION
Due to the intrinsic disadvantages of traditional cancer therapy, tumor immunotherapy has currently become the promising strategy to treat malignant cancers. For cancer immunotherapy, it has been demonstrated that the prognosis and the comprehensive therapy effects could be improved if the activated anti-tumor effector cells were adoptively introduced into patients [39, 40]. Among the different strategies of tumor immunotherapy, the adoptive therapy using tumor-specific CTLs has received increasing attention [41]. For instance, the in vitro cultured and amplified tumor-specific CTLs were used for tumor treatment [42]. In this strategy, the preparation of fusion cells from DCs and tumor cells is one of the effective ways to induce CTLs [43, 44]. DCs are specialized functional antigen presenting cells, which can be used to induce liver cancer-specific CTLs by transfecting with total RNA from liver cancer cells [15], introducing liver cancer-specific antigen peptides [16, 45] or tumor lysates [17], and generating fusion cells with liver cancer cells [46]. In clinical trials, these methods were safe, but the improvement of clinical results was limited [47, 48]. Despite the advantage of fusion DC/liver cancer cells in presenting tumor antigens and initiating efficient Th1 reaction [46, 49], few reports indicate that this method was clinically effective. Therefore, it is necessary to establish new strategies to improve the efficiency of CTL adoptive therapy induced by fusion cells [21, 44, 50].
CTLA-4 negatively regulates T cells whose activation can be inhibited. As a means of disrupting CTLA-4 inhibition, an anti-CTLA-4 antibody was applied for tumor immunotherapy. However, the inherent disadvantages of antibodies include large size/molecular weight, poor penetrability in tumor tissues and vascular barrier, and low effective concentration in solid tumors significantly compromising the effectiveness of antibody drugs in tumor treatment [51]. To overcome many of these disadvantages of traditional antibodies, genetically engineered nanobodies have been developed. Nanobodies exhibit higher affinity and antigen specificity, superior tissue penetration, and weak immunogenicity and are suited for tumor immunotherapy. Nanobodies and other recombinant antibodies offer a number of advantages. One can take advantage of normal in vivo immune maturation (in this case in a camelid), but also utilize the high throughput screening with phage in order to optimize their properties and efficacy. The small size of nanobodies allows them to reach restricted binding pockets and may facilitate cell entry. Their known structures and stability to heat and to solvents simplify chemical derivatization using tools such as PEGalation and click chemistry reagents. Nanobody production in bacteria leads to a dramatic cost reduction of the reagent but also simplifies genetic modification of the nanobody to improve pharmacokinetics and efficacy. As a reagent, the small and defined sequence of nanobodies helps us to avoid many problems of irreproducibility associated with classical antibodies [52, 53].
CTLA-4, an important negative co-stimulatory molecule on the surface of T cells, represents a good target for tumor immunotherapy. In our previous study, we developed CTLA-4 Nb16 and showed its good binding affinity with CTLA-4. We further investigated whether CTLA-4 Nb16 promoted the anti-tumor effects of T cells by blocking CTLA-4 suppression and promoting the specificity of CTLs. By binding to CTLA-4 on the surface of CD8+ T lymphocytes, CTLA-4 Nb16 increased CD8+ T lymphocyte activation, with stronger cytotoxicity and anti-tumor effects against HepG2 cells. CTLA-4 Nb16 could block CTLA-4 suppression and increase the immune response of CD8+ T lymphocytes. Data in this study clearly demonstrated that when DHFC or DMFC were stimulated with CTLA-4 Nb16, CD8+ T lymphocyte-mediated tumor killing was facilitated which subsequently inhibited tumor growth and prolonged murine survival in animals with human liver cancer or breast cancer. In the previous study, we have done lots of research on nanomaterials, fusion vaccines and combination to enhance cancer adoptive therapy of DCs and T cells [54], this novel nanobody combination nanomaterials to induce tumor-specific CTLs for adoptive immunotherapy may offer a new approach for in the clinic.
5. CONCLUSIONS
In summary, we applied nanobody against CTLA-4 to eliminate immunosuppression for adoptive cellular immunotherapy. By blocking T cell induced by DHFC with CTLA-4 Nb16, the suppression of CTLA-4-mediated T cell activation could be inhibited during the process of CTL-activated killing. When DC/HepG2 fusion cells or DC/MCF-7 fusion cells were treated with CTLA-4 Nb16, the induced CD8+ T cells markedly suppressed tumor growth and prolonged survival time of mice with human cancer by inhibiting tumor cell proliferation and driving apoptosis. This suggests that the nanobody CTLA-4 Nb16 increases tumor-specific CD8+ T Cell activity and offers unique advantages for adoptive cancer immunotherapy. This novel nanobody-based strategy to induce tumor-specific CTLs for adoptive immunotherapy may offer a new approach for cancer adoptive therapy.
Acknowledgements
This work was supported, in part, by grants from Project of National Natural Scientific Foundation of China (No. 81773254, No. 81560494, No. 81272778); Programs for Changjiang Scholars and Innovative Research Team in University (No.IRT_15R13); International Cooperation Project of the Misnistry of Science and Technology of China (No. 2015DFA31320); Project for Innovative Research Team in Guangxi Natural Science Foundation (2015GXNSFFA139001); Project for International Nanobody Research Center of Guangxi (No.GuiKe-AD17195001); Postdoctoral Science Foundation funded project of Guangxi Medical University under Grant (No. 02306214010D); Partial supports were provided by NIH-NIEHS GRANTS ES002710 and Superfund Grant P42 ES004699.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
REFERENCES
- 1.Liedtke C, and Kolberg HC, 2015. Current Medical Treatment of Patients with Non-Colorectal Liver Metastases: Primary Tumor Breast Cancer. Visceral Medicine, 31(6), pp.424–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nam SJ, Yeo HY, Chang HJ, Kim BH, and Park JW, 2016. A new cell block method for multiple immunohistochemical analysis of circulating tumor cells in patients with liver cancer. Cancer research and treatment: official journal of Korean Cancer Association, 48(4), pp.1229–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hersey P, and Gallagher S, 2014. Intralesional immunotherapy for melanoma. Journal of surgical oncology, 109(4), pp.320–326. [DOI] [PubMed] [Google Scholar]
- 4.Mona A, Sabry S, Eman K, Ahmed E, Mohamed S, & Motawa E, 2018. A novel potential effective strategy for enhancing the antitumor immune response in breast cancer patients using a viable cancer cell-dendritic cell-based vaccine. Oncology letters, 16(1), pp.529–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang W, Zhu XD, Sun HC, Xiong YQ, Zhuang PY, Xu HX, and Tang ZY, 2010. Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clinical Cancer Research, 16(13), pp.3420–3430. [DOI] [PubMed] [Google Scholar]
- 6.Cohen JE, Merims S, Frank S, Engelstein R, Peretz T, and Lotem M, 2017. Adoptive cell therapy: past, present and future. Immunotherapy, 9(2), pp.183–196. [DOI] [PubMed] [Google Scholar]
- 7.Nishi K, and Okazaki A, 2015. Combined Use of Interferon-Gamma Release Assay and Low-Dose Computed Tomography for Tuberculosis Screening Program of Health Care Workers. Kekkaku:[Tuberculosis], 90(11–12), pp.683–687. [PubMed] [Google Scholar]
- 8.Grasse M, Meryk A, Miggitsch C, and Grubeck-Loebenstein B, 2018. GM-CSF improves the immune response to the diphtheria-component in a multivalent vaccine. Vaccine, 36(31), pp.4672–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pelak MJ, Śnietura M, Lange D, Nikiel B, and Pecka KM, 2015. The prognostic significance of indoleamine-2,3-dioxygenase and the receptors for transforming growth factor β and interferon γ in metastatic lymph nodes in malignant melanoma. Polish Journal of Pathology, 66(4), pp.376–382. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Zhou Y, Brauer F, and Heffernan J, 2012. Viral dynamics model with CTL immune response incorporating antiretroviral therapy. Journal of Mathematical Biology, 67(4), pp.901–934. [DOI] [PubMed] [Google Scholar]
- 11.Tsang DN, Lai CK, Yam WC, Chan JW, Mok YW, Seto WH, and Ng TK, 2015. Use of interferon gamma release assay to assess latent tuberculosis infection among healthcare workers in Hong Kong. Hong Kong Med J, 21(Suppl 7), pp.22–25. [PubMed] [Google Scholar]
- 12.Ruan QZ, Fu JQ, Wu XX, Huang LP, and Ruan RS, 2018. Rational combinations of in vivo cancer antigen priming and adoptive T-cell therapy mobilize immune and clinical responses in terminal cancers. Cancer Immunology, Immunotherapy, 67(6), pp.907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quezada SA, Peggs KS, Simpson TR, and Allison JP, 2011. Shifting the equilibrium in cancer immunoediting: from tumor tolerance to eradication. Immunological Reviews, 241(1), pp.104–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schreiber RD, Old LJ, and Smyth MJ, 2011. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science, 331(6024), pp.1565–1570. [DOI] [PubMed] [Google Scholar]
- 15.Zhang L, Zhang H, Liu W, Wang H, Jia J, Si X, and Ren J, 2005. Specific antihepatocellular carcinoma T cells generated by dendritic cells pulsed with hepatocellular carcinoma cell line HepG2 total RNA. Cellular Immunology, 238(1), pp.61–66. [DOI] [PubMed] [Google Scholar]
- 16.Bray SM, Vujanovic L, and Butterfield LH, 2011. Dendritic Cell-Based Vaccines Positively Impact Natural Killer and Regulatory T Cells in Hepatocellular Carcinoma Patients. Clinical and Developmental Immunology, 2011, pp.1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palmer DH, Midgley RS, Mirza N, Torr EE, Ahmed F, Steele JC, and Adams DH, 2009. A phase II study of adoptive immunotherapy using dendritic cells pulsed with tumor lysate in patients with hepatocellular carcinoma. Hepatology, 49(1), pp.124–132. [DOI] [PubMed] [Google Scholar]
- 18.Cao DY, Yang JY, Yue SQ, Tao KS, Song ZS, Wang DS, and Dou KF, 2009. Comparative analysis of DC fused with allogeneic hepatocellular carcinoma cell line HepG2 and autologous tumor cells as potential cancer vaccines against hepatocellular carcinoma. Cellular Immunology, 259(1), pp.13–20. [DOI] [PubMed] [Google Scholar]
- 19.Luo Y, Li L, Zou P, Wang J, Shao L, Zhou D, and Liu L, 2014. Rapamycin Enhances Long-Term Hematopoietic Reconstitution of Ex Vivo Expanded Mouse Hematopoietic Stem Cells by Inhibiting Senescence. Transplantation Journal, 97(1), pp.20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie S, Wu X, Zhang G, Xu K, Bian X, Zhang S, and Ye Y, 2014. Remarkable regression of a lung recurrence from an undifferentiated embryonal sarcoma of the liver treated with a DC vaccine combined with immune cells: A case report. Cellular Immunology, 290(2), pp.185–189. [DOI] [PubMed] [Google Scholar]
- 21.Kim WJ, Newman WC, and Amankulor NM, 2017. Phase I/II Trial of Combination of Temozolomide Chemotherapy and Immunotherapy With Fusions of Dendritic and Glioma Cells in Patients With Glioblastoma. Neurosurgery, 81(1), pp.11. [DOI] [PubMed] [Google Scholar]
- 22.Pang YB, Cui BY, He J, Huang XP, Liang W, Li LQ, and Luo XL, 2017. Experimental study on the immune response of fusion tumor vaccine of HepG2 and dendritic cells in vitro. Zhonghua yi xue za zhi, 97(7), pp.535–539. [DOI] [PubMed] [Google Scholar]
- 23.Taggart D, Andreou T, Scott KJ, Williams J, Rippaus N, Brownlie RJ, and Lorger M, 2018. Anti–PD-1/anti–CTLA-4 efficacy in melanoma brain metastases depends on extracranial disease and augmentation of CD8+ T cell trafficking. Proceedings of the National Academy of Sciences, 115(7), pp.E1540–E1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, and Allison JP, 2014. Localized Oncolytic Virotherapy Overcomes Systemic Tumor Resistance to Immune Checkpoint Blockade Immunotherapy. Science Translational Medicine, 6(226), pp.226ra32–226ra32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu D, Badell IR, and Ford ML, 2018. Selective CD28 blockade attenuates CTLA-4–dependent CD8+ memory T cell effector function and prolongs graft survival. JCI Insight, 3(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paterson AM, and Sharpe AH, 2010. Taming tissue-specific T cells: CTLA-4 reins in self-reactive T cells. Nature Immunology, 11(2), pp.109–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wing K, Yamaguchi T, and Sakaguchi S, 2011. Cell-autonomous and -non-autonomous roles of CTLA-4 in immune regulation. Trends in Immunology, 32(9), pp.428–433. [DOI] [PubMed] [Google Scholar]
- 28.Field CS, Hunn MK, Ferguson PM, Ruedl C, Ancelet LR, and Hermans IF, 2018. Blocking CTLA-4 while priming with a whole cell vaccine reshapes the oligoclonal T cell infiltrate and eradicates tumors in an orthotopic glioma model. OncoImmunology, 7(1), pp.e1376154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dias JD, Hemminki O, Diaconu I, Hirvinen M, Bonetti A, Guse K, and Cerullo V, 2012. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Therapy, 19(10), pp.988–998. [DOI] [PubMed] [Google Scholar]
- 30.Iwama S, De Remigis A, Callahan MK, Slovin SF, Wolchok JD, and Caturegli P, 2014. Pituitary Expression of CTLA-4 Mediates Hypophysitis Secondary to Administration of CTLA-4 Blocking Antibody. Science Translational Medicine, 6(230), pp.230ra45–230ra45. [DOI] [PubMed] [Google Scholar]
- 31.Luiz M, Pereira S, Prado N, Gonçalves N, Kayano A, Moreira-Dill L, and Zuliani J, 2018. Camelid Single-Domain Antibodies (VHHs) against Crotoxin: A Basis for Developing Modular Building Blocks for the Enhancement of Treatment or Diagnosis of Crotalic Envenoming. Toxins, 10(4), pp.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muyldermans S, 2013. Nanobodies: Natural Single-Domain Antibodies. Annual Review of Biochemistry, 82(1), pp.775–797. [DOI] [PubMed] [Google Scholar]
- 33.Faraji F, Tajik N, Behdani M, Shokrgozar MA, Zarnani AH, Shahhosseini F, and Habibi-Anbouhi M, 2018. Development and characterization of a camelid single-domain antibody directed to human CD22 biomarker. Biotechnology and Applied Biochemistry, 65(5), pp.718–725. [DOI] [PubMed] [Google Scholar]
- 34.Farasat A, Rahbarizadeh F, Hosseinzadeh G, Sajjadi S, Kamali M, and Keihan AH, 2017. Affinity enhancement of nanobody binding to EGFR: in silico site-directed mutagenesis and molecular dynamics simulation approaches. Journal of Biomolecular Structure and Dynamics, 35(8), pp.1710–1728. [DOI] [PubMed] [Google Scholar]
- 35.Arezumand R, Alibakhshi A, Ranjbari J, Ramazani A, and Muyldermans S, 2017. Nanobodies As Novel Agents for Targeting Angiogenesis in Solid Cancers. Frontiers in Immunology, 8, pp.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shinozaki N, Hashimoto R, Fukui K, and Uchiyama S, 2017. Efficient generation of single domain antibodies with high affinities and enhanced thermal stabilities. Scientific Reports, 7(1), pp.5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pruszynski M, D’Huyvetter M, Bruchertseifer F, Morgenstern A, and Lahoutte T, 2018. Evaluation of an Anti-HER2 Nanobody Labeled with 225Ac for Targeted α-Particle Therapy of Cancer. Molecular Pharmaceutics, 15(4), pp.1457–1466. [DOI] [PubMed] [Google Scholar]
- 38.Wan R, Liu A, Hou X, Lai Z, Li J, Yang N, and Zhao Y, 2017. Screening and antitumor effect of an anti-CTLA-4 nanobody. Oncology Reports, 39(2), pp.511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lissoni P, Brivio F, Fumagalli L, Messina G, Meregalli S, Porro G, and D’AMICO G, 2009. Effects of the conventional antitumor therapies surgery, chemotherapy, radiotherapy and immunotherapy on regulatory T lymphocytes in cancer patients. Anticancer research, 29(5), pp.1847–1852. [PubMed] [Google Scholar]
- 40.Singh C, Qian JM, James BY, and Chiang VL, 2019. Local tumor response and survival outcomes after combined stereotactic radiosurgery and immunotherapy in non–small cell lung cancer with brain metastases. Journal of Neurosurgery, pp.1–6. [DOI] [PubMed] [Google Scholar]
- 41.Reissfelder C, Stamova S, Gossmann C, Braun M, Bonertz A, Walliczek U, and Benner A, 2014. Tumor-specific cytotoxic T lymphocyte activity determines colorectal cancer patient prognosis. Journal of Clinical Investigation, 125(2), pp.739–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sui CG, Wu D, Meng FD, Yang MH, and Jiang YH, 2015. Anti-prostate cancer effects of CTL cell induction in vitro by recombinant adenovirus mediated PSMA/4–1BBL dendritic cells: an immunotherapy study. Genetics and Molecular Research, 14(2), pp.7208–7217. [DOI] [PubMed] [Google Scholar]
- 43.Wang X, Zhang FC, Zhao HY, Lu XL, Sun Y, Xiong ZY, and Jiang XB, 2014. Human IP10-scFv and DC-induced CTL synergistically inhibit the growth of glioma in a xenograft model. Tumor Biology, 35(8), pp.7781–7791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qian L, Tang Z, Yin S, Mo F, Yang X, Hou X, and Lu X, 2018. Fusion of Dendritic Cells and Cancer-Associated Fibroblasts for Activation of Anti-Tumor Cytotoxic T Lymphocytes. Journal of Biomedical Nanotechnology, 14(10), pp.1826–1835. [DOI] [PubMed] [Google Scholar]
- 45.Butterfield LH, Ribas A, Dissette VB, Lee Y, Yang JQ, De la Rocha P, and McBride WH, 2006. A Phase I/II Trial Testing Immunization of Hepatocellular Carcinoma Patients with Dendritic Cells Pulsed with Four -Fetoprotein Peptides. Clinical Cancer Research, 12(9), pp.2817–2825. [DOI] [PubMed] [Google Scholar]
- 46.Zhang HM, Zhang HM, Zhang LW, Liu WC, Cheng J, Si XM, and Ren J, 2006. Comparative analysis of DC fused with tumor cells or transfected with tumor total RNA as potential cancer vaccines against hepatocellular carcinoma. Cytotherapy, 8(6), pp.580–588. [DOI] [PubMed] [Google Scholar]
- 47.El Ansary M, Mogawer S, Elhamid SA, Alwakil S, Aboelkasem F, El Sabaawy H, & Abdelhalim O, 2012. Immunotherapy by autologous dendritic cell vaccine in patients with advanced HCC. Journal of Cancer Research and Clinical Oncology, 139(1), pp.39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Greten TF, Forner A, Korangy F, N’Kontchou G, Barget N, Ayuso C, Ormandy LA, Manns MP, Beaugrand M, and Bruix J, 2010. A phase II open label trial evaluating safety and efficacy of a telomerase peptide vaccination in patients with advanced hepatocellular carcinoma. BMC Cancer, 10(1), pp.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sakoda Y, Nagai T, Murata S, Mizuno Y, Kurosawa H, Shoda H, Morishige N, Yanai R, Sonoda KH, and Tamada K, 2016. Pathogenic Function of Herpesvirus Entry Mediator in Experimental Autoimmune Uveitis by Induction of Th1- and Th17-Type T Cell Responses. The Journal of Immunology, 196(7), pp.2947–2954. [DOI] [PubMed] [Google Scholar]
- 50.Atomura R, Sanui T, Fukuda T, Tanaka U, Toyoda K, Taketomi T, Yamamichi K, Akiyama H, and Nishimura F, 2016. Inhibition of Sprouty2 polarizes macrophages toward an M2 phenotype by stimulation with interferon γ and Porphyromonas gingivalislipopolysaccharide. Immunity, Inflammation and Disease, 4(1), pp.98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oyen D, Srinivasan V, Steyaert J, & Barlow JN, 2011. Constraining Enzyme Conformational Change by an Antibody Leads to Hyperbolic Inhibition. Journal of Molecular Biology, 407(1), pp.138–148. [DOI] [PubMed] [Google Scholar]
- 52.Bever CS, Dong JX, Vasylieva N, Barnych B, Cui Y, Xu ZL, Hammock BD, and Gee SJ, 2016. VHH antibodies: emerging reagents for the analysis of environmental chemicals. Analytical and Bioanalytical Chemistry, 408(22), pp.5985–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li C, Tang Z, Hu Z, Wang Y, Yang X, Mo F, & Lu X, 2018. Natural Single-Domain Antibody-Nanobody: A Novel Concept in the Antibody Field. Journal of Biomedical Nanotechnology, 14(1), pp.1–19. [DOI] [PubMed] [Google Scholar]
- 54.Hu Z, Chen J, Zhou S, Yang N, Duan S, Zhang Z, Lu X, and Zhao Y, 2017. Mouse IP-10 Gene Delivered by Folate-modified Chitosan Nanoparticles and Dendritic/tumor Cells Fusion Vaccine Effectively Inhibit the Growth of Hepatocellular Carcinoma in Mice. Theranostics, 7(7), pp.1942–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]