

Abstract
SURP domains are exclusively found in splicing‐related proteins in all eukaryotes. SF3A1, a component of the U2 snRNP, has two tandem SURP domains, SURP1, and SURP2. SURP2 is permanently associated with a specific short region of SF3A3 within the SF3A protein complex whereas, SURP1 binds to the splicing factor SF1 for recruitment of U2 snRNP to the early spliceosomal complex, from which SF1 is dissociated during complex conversion. Here, we determined the solution structure of the complex of SURP1 and the human SF1 fragment using nuclear magnetic resonance (NMR) methods. SURP1 adopts the canonical topology of α1–α2–310–α3, in which α1 and α2 are connected by a single glycine residue in a particular backbone conformation, allowing the two α‐helices to be fixed at an acute angle. A hydrophobic patch, which is part of the characteristic surface formed by α1 and α2, specifically contacts a hydrophobic cluster on a 16‐residue α‐helix of the SF1 fragment. Furthermore, whereas only hydrophobic interactions occurred between SURP2 and the SF3A3 fragment, several salt bridges and hydrogen bonds were found between the residues of SURP1 and the SF1 fragment. This finding was confirmed through mutational studies using bio‐layer interferometry. The study also revealed that the dissociation constant between SURP1 and the SF1 fragment peptide was approximately 20 μM, indicating a weak or transient interaction. Collectively, these results indicate that the interplay between U2 snRNP and SF1 involves a transient interaction of SURP1, and this transient interaction appears to be common to most SURP domains, except for SURP2.
Keywords: bio‐layer interferometry, complex structure, NMR, SF1, SF3A1, SF3a120, splicing, SURP, SWAP, U2 snRNP
Short abstract
PDB Code(s): 7VH9;
Abbreviations
- BLI
bio‐layer interferometry
- S1BR
“SURP1‐binding site”‐containing region
- S1BRp
a peptide corresponding to a “SURP1‐binding site”‐containing region
1. INTRODUCTION
Pre‐mRNA splicing is an essential event in post‐transcriptional gene expression. This reaction is catalyzed by the spliceosome, a huge multi‐megadalton ribonucleoprotein (RNP) complex observed in eukaryotic nuclei. Spliceosome assembly occurs in a stepwise manner on pre‐mRNA and is facilitated by concerted interactions between five small nuclear RNP particles (U1, U2, U4, U5, and U6 snRNPs) and more than 100 non‐snRNP proteins. A typical process prior to the spliceosome activation stage is briefly described below. 1 Spliceosome assembly begins with the binding of (1) U1 snRNP to the 5′ splice site of pre‐mRNA, 2 (2) splicing factor 1 (SF1) to the branch point, 3 and (3) U2 auxiliary factor 65 (U2AF65 or U2AF2) and U2AF35 (U2AF1) to the polypyrimidine tract and 3′ splice site, respectively (Figure 1a). 4 This process creates the first spliceosomal complex, referred to as the early or E complex. U2 snRNPs are then recruited to the E complex in an ATP‐independent manner. 5 The initial association of U2 snRNP involves an interaction with SF1, 6 which was previously associated with the branch point sequence (BPS) at the 3′ splice site in the E complex. The E complex, to which the U2 snRNP remains bound, is converted into the pre‐splicing complex in an ATP‐dependent manner, forming what is referred to as the A complex. During the conversion process, SF1 is displaced by U2 snRNP, which consequently forms a stable association with the BPS of the pre‐mRNA. Accordingly, SF1 is completely released from the spliceosomal complex. The A complex subsequently proceeds to the spliceosome activation stage.
FIGURE 1.
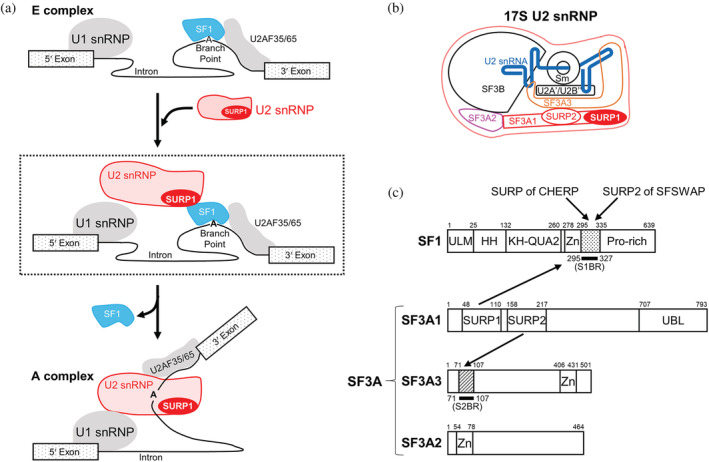
Splicing factors involved in early spliceosome assembly. (a) Schematic diagram of the process of transition from the E to A complex. U2 snRNP binds to SF1 via SURP1 for the recruitment of U2 snRNP to the E complex, shown in the dashed box. During the E to A conversion process, which occurs in an ATP‐dependent manner, SF1 is displaced by U2 snRNP and released from the spliceosome. (b) Schematic diagram of 17S U2 snRNP. U2 snRNA is represented by a blue bold line. SF3A1, SF3A2, and SF3A3 proteins, which are components of the SF3A protein complex, are depicted in red, magenta, and orange, respectively. Two tandem SURP domains, SURP1, and SURP2, in SF3A1 are represented by red oval shapes. (c) Schematic diagrams of the domain architecture of SF1, SF3A1, SF3A2, and SF3A3 from humans. SF1 has five structural domains, namely, the UHM ligand motif (ULM), helix hairpin (HH), K homology and Quaking homology 2 domain (KH‐QUA2), and zinc‐finger domain (Zn). In addition, SF1 has a proline‐rich region (Pro‐rich) in the C terminus. Regarding the SF3A complex, SF3A1 has three structural domains, SURP1, SURP2, and a Ubiquitin‐like domain (UBL) whilst SF3A2 and SF3A3 have a single structural domain, a zinc‐finger domain (Zn). No other known motifs have been found in the three components of SF3A. Bold black lines indicate a “SURP1‐binding site”‐containing region (S1BR, dotted) and the SURP2 counterpart (S2BR, hatted). A peptide corresponding to each region was used to determine the structures of the SURP1 and SURP2 complexes. Each of the binding partners of the four SURP domains is indicated by an arrow; SURP1 of SF3A1 and the SURP domains of CHERP and SFSWAP bind to S1BR in SF1, while SURP2 binds to S2BR in SF3A3.
Splicing‐related proteins generally possess various structural domains that play key roles in specific interactions with RNAs and/or proteins. The present study focuses on the structure and function of SURP domains, also known as SWAP (suppressor‐of‐white‐apricot) domains, 7 which are exclusively found in splicing‐related proteins from all eukaryotes (Table S1). In humans, there are a total of only six proteins that possess one or two tandem SURP domains (Table S1). 8 Among SURP‐containing proteins, the best‐characterized protein is currently SF3A1, which is a component of the SF3A protein complex in U2 snRNP (Figure 1b,c). U2 snRNP is a large protein‐RNA complex containing the core 12S U2 snRNP particle (comprised of U2 snRNA, Sm core proteins, and the U2A’ and U2B” complexes) and SF3A and SF3B protein complexes. 9 The core 12S U2 snRNP particle binds to SF3B to form a premature 15S U2 snRNP, which then binds to SF3A to form a functionally mature 17S U2 snRNP. 10 , 11 The SF3A protein complex is composed of three proteins: SF3A3 (SF3a60), SF3A2 (SF3a66), and SF3A1 (SF3a120), each of which contains one or three structural domains (Figure 1c). 12 SF3A3 and SF3A2 contain a zinc finger domain, whereas SF3A1 contains two tandem SURP domains and a ubiquitin‐like domain. 13 The two SURP domains of human SF3A1 are hereafter referred to as SURP1 and SURP2.
In the SF3A protein complex, binding of SURP2 to a specific region of SF3A3 is required for the stable and permanent association between SF3A1 and SF3A3. 11 In a previous study, we determined the solution structure of SURP2 (res. 48–110) in complex with the SF3A3 fragment (res. 71–107) and the solution structure of SURP1 alone (res. 48–110). 8 SURP domains are helical proteins composed of approximately 60 amino acid residues, and they adopt a unique topology of α1–α2–310–α3. SURP2 stably binds to the 17‐residue amphipathic α‐helix of the SF3A3 fragment via a surface formed mainly by α1 and α2. The study also revealed that a single residue Leu169 in α1, which is unique to the SURP2 domains of SF3A1 orthologs, is a critical determinant for SURP2 complex formation 8 (Figure 2, Figure S1a). This finding was confirmed by a particular mutation in SURP1, in which Lys was replaced with Leu at the corresponding position, allowing the SURP1 mutant to bind to the SF3A3 fragment.
FIGURE 2.
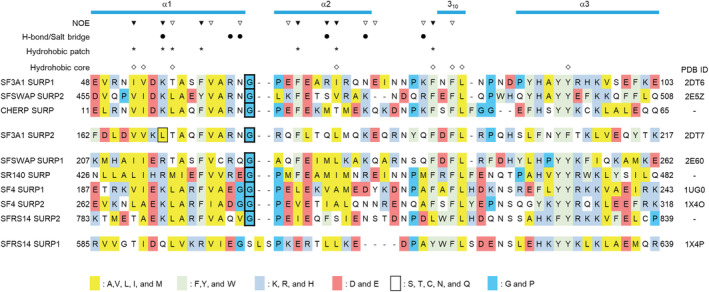
Structure‐based sequence alignment of SURP domains from humans. Amino acid sequence alignments of SURP domains from humans were performed using the ClustalW program 55 and then manually modified based on the structures of the SURP domains. The accession codes used in the sequence alignment were as follows: SF3A1 (UniProt accession No. Q15459), SFSWAP (Q12872), CHERP (Q8IWX8), SR140 (O15042), SF4 (Q8IWZ8), and SFRS14 (Q8IX01). All structures were previously published by our group, and each protein data bank (PDB) code is shown. The secondary structure elements are shown in cyan at the top. The alignments are colored according to the amino acid type shown in the alignment legend. Each box with thick black lines indicates a Gly residue connecting α1 and α2, which forms the G bend. The single box with thin black lines indicates Leu169 in α1 of SURP2, which is the determinant of the SURP2 complex formation. The inverted triangles indicate SURP1 residues for which intermolecular NOEs from S1BR were observed (open inverted triangle: less than 10 NOEs, filled inverted triangle: 10 or more NOEs). Filled round marks indicate SURP1 residues that participate in hydrogen bonds or salt bridges with S1BR. Asterisks indicate SURP1 residues that form a hydrophobic patch that interacts with S1BR. Diamonds indicate residues that form a hydrophobic core common to SURP domains.
While the binding partner of SURP2 is a constitutive component of U2 snRNP as described above, that of SURP1 is one of the components of the E complex, namely, SF1. Co‐immunoprecipitation experiments on HeLa cell nuclear extracts and yeast two‐hybrid screening assays revealed that SF1 binds to each SURP domain of three splicing‐related proteins, one of which is the SURP1 of SF3A1. 6 Moreover, GST pull‐down assays using several deletion mutants of SF1 showed that SF1 composed of 639 amino acid residues interacts with SF3A1 via a specific short region (res. 295–335) between a Zn‐finger domain and a proline‐rich region, in which neither a structural domain nor a signal sequence has been found (Figure 1c). These results indicate that SURP1 directly binds to the region of SF1, allowing U2 snRNP to interact with SF1. This interaction appears to facilitate the initial recruitment of U2 snRNPs to the spliceosome during E complex formation. 6
The other two splicing‐related proteins that bind to SF1 via SURP‐mediated interactions are calcium homeostasis endoplasmic reticulum protein (CHERP) and splicing factor, suppressor of white‐apricot homolog (SFSWAP). 6 CHERP, with a single SURP domain, is one of the U2 snRNP‐associated proteins and is involved in alternative splicing regulation. 14 , 15 SFSWAP, with two SURP domains, is an alternative splicing regulator that also autoregulates the splicing of its own pre‐mRNA 7 ; with the second domain only binding to SF1. However, limited information is currently available regarding the role of the interactions between CHERP, SFSWAP, and SF1 in each splicing process. Based on the results of the SF1 binding experiments, 6 the three SURP domains of SF3A1, CHERP, and SFSWAP were supposed to have some common points in the binding mode.
Structural studies using high‐resolution cryo‐electron microscopy revealed not only the molecular architecture, but also extensive conformational changes in U2 snRNPs during the splicing process. The SURP1 structure was confirmed in the human Bact and pre‐B complexes, followed by the A complex, 16 , 17 but not in the U2 snRNP alone. 18 Additionally, the yeast SURP1 counterpart is not found in the pre‐A complex. 19 The current structural data do not include SF1 and are therefore not applicable for examining the interaction between SURP1 and SF1.
In the present study, to clarify the structural basis of the SURP‐mediated interaction between U2 snRNP and SF1, we elucidated the solution structure of the complex of SURP1 and the SF1 fragment from humans using NMR methods. The solution structure was determined as that of a chimeric protein of the domain and the fragment connected by a linker peptide. To confirm whether the residues comprising the region associated with SURP1's interaction with the SF1 fragment were involved in ligand binding affinity, we constructed SURP1 mutants in which each of the residues was changed. We then measured the dissociation constants of the mutants with the SF1 fragment using bio‐layer interferometry (BLI) assays. We also constructed a SURP1 mutant in which Ala57 was mutated to Ser. This was chosen since the corresponding gene mutation has been detected in the SF3A1 gene of patients with myelodysplasia. 20 The present results depict the interaction mode of SURP1 with the SF1 fragment involved in the interplay between U2 snRNP and SF1 and also present a peptide interaction module for SURP domains.
2. RESULTS
2.1. Design and expression of a chimeric protein of SURP1 and the SF1 fragment
The region between residues 43 and 137 in SF3A1, which contains SURP1, interacts with residues 295 and 335 in SF1. 6 Hereafter, this region is termed a “SURP1‐binding site”‐containing region or S1BR (res. 295–335), and the peptide corresponding to S1BR is termed S1BR peptide or S1BRp (res. 295–335). We previously reported the solution structure of the free form of SURP1 composed of 63 residues (res. 48–110). 8 As the ligand peptide for binding to the 63‐residue SURP1, we used the 33‐residue S1BRp (res. 295–327) instead of the 41‐residue S1BRp (res. 295–335). These eight residues (328SVGSTSGP335) that were omitted from S1BRp (res. 295–335) are not well‐conserved among SF1 orthologs from various species (Figure S1b). BLI experiments confirmed that the dissociation constant (K D) of S1BRp (res. 295–327) for SURP1 was almost the same as that of S1BRp (res. 295–335) (Table 1), which is described in a later section. This suggests that none of the eight omitted residues are involved in the interaction with SURP1. Additionally, to check whether the 33‐residue S1BRp and 41‐residue S1BRp had any specific structures in the free form, we performed circular dichroism (CD) spectroscopy experiments. Each peptide displayed a CD spectrum with a single minimum at ~200 nm, which is indicative of the so‐called random‐coil conformation (Figure S2a).
TABLE 1.
Dissociation constants between SURP1 or mutants and S1BRp
| Analyte | K D (μM) | Ratio |
|---|---|---|
| To S1BRp (res. 295–335) | ||
| WT | 17.3 ± 1.1 | 1.1 |
| To S1BRp (res. 295–327) | ||
| WT | 15.2 ± 0.6 | 1 |
| R62E | 393 ± 90 | 25.9 |
| R62Q | 187 ± 17 | 12.3 |
| N63E | 251 ± 26 | 16.5 |
| N63Q | 139 ± 31 | 9.1 |
| K80D | 132 ± 37 | 8.7 |
| K80Q | 64.2 ± 1.2 | 4.2 |
Note: Binding of SURP1 (res. 48–110) to immobilized S1BRp (res. 295–335) and that of SURP1 or each mutant to immobilized S1BRp (res. 295–327) were measured by bio‐layer interferometry. The dissociation constants (K D) were assessed by equilibrium analysis using nonlinear regression (Figure S5). The K D values are the average of three independent experiments. Errors indicate standard deviations. The ratio means “in each case”; ratio of the K D value for wild‐type SURP1 binding to S1BRp (res. 295–327).
Abbreviations: KD: dissociation constant; S1BRp: a peptide corresponding to a “SURP1‐binding site”‐containing region.
Considering labeling with 15N and/or 13C, we initially attempted to overexpress S1BRp (res. 295–327) fused with different tags including His6, GST, and MBP in Escherichia coli. However, all the tagged peptides showed minimal expression in E. coli even though the codon usage of the gene was optimized for E. coli. Instead, we designed chimeric SURP1 protein (res. 48–110) and S1BRp (res. 295–327) connected by a 14‐residue linker region by referring to a method by Li et al. (Figure 3a). 21 The resultant chimeric protein was composed of 111 residues, comprised of an additional Gly residue (derived from the TEV protease cleavage site; see also the Methods section), 63‐residue SURP1 (res. 48–110), a 14‐residue linker (SGSSGSSGSSGSSG), and 33‐residue S1BR (res. 295–327). We obtained sufficient amounts of both unlabeled and labeled chimera samples in the E. coli expression system. Note that the residue numbering in the chimera was still in accordance with that of SURP1 of SF3A1 (res. 48–110) and S1BRp (res. 295–327) of SF1 (Figure 3a).
FIGURE 3.
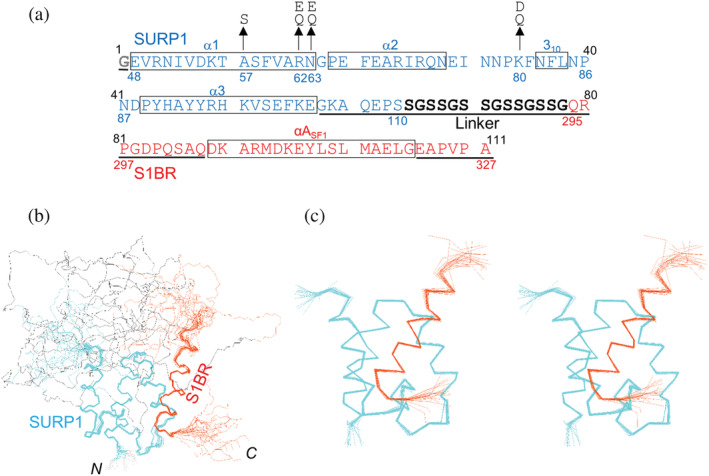
Amino acid sequence and solution structure of the chimera. (a) Amino acid sequence of the chimera used in this study. The numbers above the sequence indicate the residue numbers of the chimera. Blue and red residues/numbers correspond to SURP1 of SF3A1 and S1BR of SF1, respectively. Residues of the linker are indicated in black, and the first residue, Gly, which is derived from the TEV protease cleavage site is indicated in gray. Boxes indicate secondary structural elements according to the determined structure. Residues in the disordered region in the chimera structure are underlined. Substituted residues are indicated by arrows. (b) A trace of the backbone atoms for the 20 superimposed lowest‐energy conformers of the whole chimera composed of SURP1 (res. 48–110), the 14‐residue linker, and S1BR (res. 295–327). Backbone atoms of residues 48–103 of SURP1 and residues 305–322 of S1BR were fitted by a least‐square method. Cyan, black, and orange lines represent the Cα traces of SURP1, the linker, and S1BR, respectively. (c) Stereoview showing a trace of the backbone atoms for an ensemble of the 20 lowest energy conformers for SURP1 (res. 48–105) and S1BR (res. 302–324) in the chimera. Most of the disordered regions, including the linker, are omitted from the trace in (b) and the rest has been zoomed in.
2.2. Determination of the solution structure of the chimera that exhibits the complex structure of SURP1 and the SF1 fragment
Because the chimera is a single‐chain protein, its structure can be determined in a conventional manner using 13C/15N‐labeled protein. Using standard multidimensional heteronuclear NMR spectroscopy, we assigned the main‐chain and side‐chain resonances to 96 and 89% of the main chains and 96 and 97% of the side chains for residues 48–110 of SURP1 and residues 295–327 of S1BR, respectively. Tertiary structures were calculated using the CYANA software package based on the 1,825 1H–1H distance constraints from nuclear Overhauser effect spectroscopy (NOESY) and 160 torsion angle restraints 22 (Figure S3a). Of the 200 independently calculated structures, 40 conformers with the lowest CYANA target function values, the structural statistics of which are summarized in Table S2, were refined using restrained energy minimization. The 20 conformers that were most consistent with the experimental restraints were used for further analyses. The best‐fit superposition of the 20 conformer ensembles is shown in Figure 3b,c. The statistics of the structures, as well as the distance and torsion angle constraints used for the CYANA program, are summarized in Table 2. The root‐mean‐square deviation (RMSD) from the mean structure was 0.32 ± 0.08 Å for the backbone (N, Cα, C′) atoms and 0.97 ± 0.10 Å for all heavy (non‐proton) atoms in the well‐ordered region, composed of residues 48–103 of SURP1 and residues 305–322 of S1BR. In contrast, in addition to the N‐terminal (an additional Gly) and C‐terminal (322EAPVPA327) regions, a disordered region was observed in a consecutive sequence composed of seven residues (104GKAQEPS110) in the C‐terminal region of SURP1, all 14 residues of the linker, and 10 residues (295QRPGDPQSAQ304) in the N‐terminal region of S1BR (Figure 3a,b). Moreover, none of the linker residues had any intermolecular nuclear Overhauser effects (NOEs) with SURP1 or S1BR residues (Figure S3a). Thus, it appears that the linker does not affect the interaction between SURP1 and S1BR in the chimera structure. It follows that the well‐ordered region of the conformers of the chimera displays the complex structure of individual SURP1 and SF1 fragments (or equivalently, S1BRp).
TABLE 2.
Summary of conformational constraints and structural statistics for 20 energy‐refined conformers of the chimera of SURP1 and S1BRp (res. 295–327)
| NMR distance and dihedral angle constraints | |
|---|---|
| Distance constraints | |
| Total NOE | 1,825 |
| Intra‐residue | 407 |
| Inter‐residue | |
| Sequential (|i – j| = 1) | 456 |
| Medium‐range (1 < |i – j| < 4) | 517 |
| Long‐range (|i – j| ≥ 5) | 445 |
| NOEs between SURP1 and S1BR | 163 |
| φ/ψ dihedral angle constraint (TALOS) | 107 |
| χ 1 dihedral angle constraint, | 53 |
| Structure statistics | |
|---|---|
| AMBER energies (kcal/Mol) | |
| Mean Amber energy | −3,802.9 ± 9.5 |
| Mean restraint violation energy | 4.37 ± 0.31 |
| Mean distant violation energy | 3.59 ± 0.21 |
| Mean angle violation energy | 0.79 ± 0.19 |
| Ramachandran plot statistics (%) | |
| Residues in most favored regions | 98.6 |
| Residues in additionally allowed regions | 1.4 |
| Residues in generously allowed regions | 0.0 |
| Residues in disallowed regions | 0.0 |
| Average r.m.s.d. from mean coordinates (Å) a | |
| Backbone | 0.32 ± 0.08 |
| Heavy atoms | 0.97 ± 0.10 |
Abbreviations: NOE: nuclear Overhauser effect; S1BR: “SURP1‐binding site”‐containing region.
For residues 48–103 of SURP1 and residues 305−322 of S1BRp.
To confirm this finding, we conducted further experiments. For clarity and convenience, in this paragraph, individual SURP1 is termed SURP1i, while SURP1 contained in the chimera is termed SURP1c. First, we performed a chemical shift perturbation analysis of SURP1i upon S1BRp binding using 1H–15N HSQC to identify the binding surface of SURP1i (Figure S4a). By comparing the 1H–15N HSQC spectra of SURP1i between the free and bound forms, the absolute weighted chemical shift changes in the amide 1H and 15N of each residue between the two spectra were plotted versus the residue number (Figure 4a). The addition of S1BRp to SURP1i resulted in significant chemical shift changes in the cross peaks from the following residues: Ile52, Val53, and Arg62 in α1; Phe67 and Glu68 in α2; and Phe81 in 310. Mapping of these residues onto the ribbon drawing of SURP1i in free form demonstrates that these residues converge on a specific surface of SURP1i, which is primarily formed by α1 and α2 (Figure 4b). This result agreed well with the interaction mode between SURP1c and S1BR shown in the chimera structure, as described later. Additionally, we compared 1H–15N HSQC spectra between SURP1i in the bound form and SURP1c of the chimera to confirm the structural identity between the two SURP1 structures (Figure S4b). The absolute weighted chemical shift differences of all residues were less than 0.15 ppm, indicating no significant variation in structures between SURP1i in a bound form and SURP1c of the chimera (Figure 4c). Taken together, these findings confirmed that the chimera structure is a good representation of the structure of SURP1i complexed with S1BRp. For simplicity, we hereafter used “as the complex structure” instead of “as the chimera structure.”
FIGURE 4.
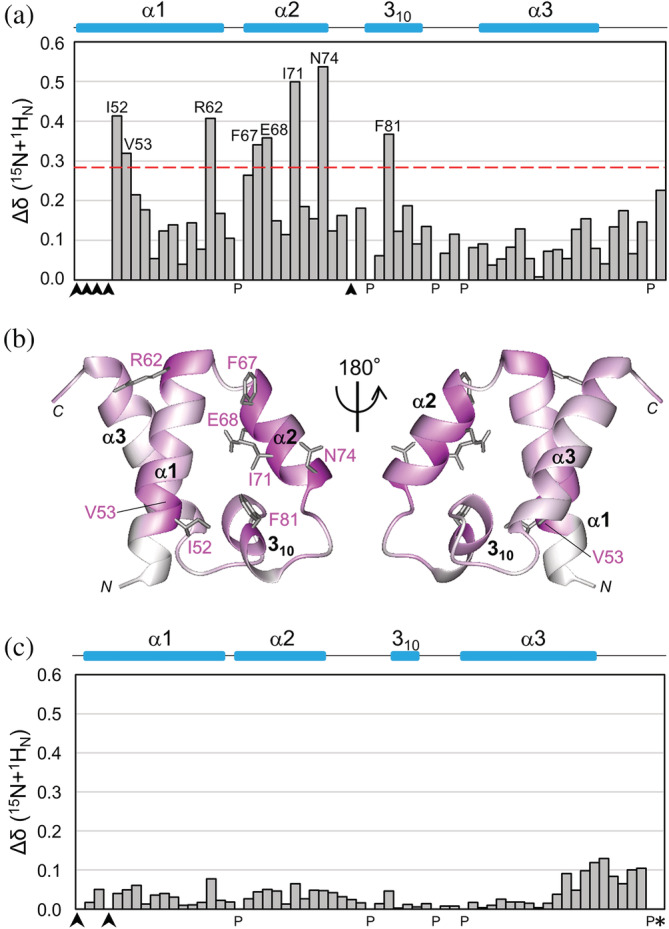
NMR chemical shift perturbations of labeled SURP1 upon binding of non‐labeled S1BRp, and comparison of chemical shift values between complex structure SURP1 and SURP1 of the chimera. (a) Quantification of the chemical shift perturbation values of labeled SURP1 upon S1BRp binding. Perturbation values were obtained from [1H,15N]‐HSQC spectra in the absence and presence of S1BRp (Figure S4a). The absolute values of the chemical shift change Δδ(15 N + 1 H N) are shown. The value for each residue was calculated as follows: Δδ(15 N + 1 H N) = [(δ 15N/6.5)2 + δ 1H 2]1/2. Perturbation values greater than the average (0.16 ppm) plus the standard deviation (0.12 ppm) were defined as significant perturbations (i.e., the significance level of 0.28 ppm is indicated by a red dotted line). Residues with resonances that were not assigned are indicated by arrowheads, and proline is indicated by P. Only residues with significant chemical shift changes are shown. (b) Mapping of residues with chemical shift changes on the ribbon representation of individual SURP1 in its free form [PDB ID 2DT7]. Residues are colored based on the magnitude of the chemical shift change upon S1BRp binding, ranging from white (not assigned or measured) to magenta (largest chemical shift change). Only the side chains of the residues with significant chemical shift changes are represented in gray. (c) Differences of the chemical shift values between the S1BRp‐bound form of SURP1 and the chimeric form of SURP1 in the chimera. The values were obtained from [1H,15N]‐HSQC spectrum of labeled SURP1 in the presence of S1BRp and from that of the labeled chimera (Figure S4b). The absolute values of the chemical shift difference between the two forms Δδ(15 N + 1 H N) are shown. The value for each residue was calculated, as described above. The average and standard deviation among residues in SURP1 are 0.04 ppm and 0.03 ppm, respectively, except for Gly110, which is the last residue of SURP1 (indicated by an asterisk). The absolute value of the chemical shift change of Gly110 is 6.19 ppm; the corresponding bar is not indicated in this graph. Because, in the chimera, Gly110 is directly connected with the linker, the chemical shift value cannot be simply compared with that of the last residue, Gly, of individual SURP1.
2.3. Complex structure of SURP1 with the SF1 fragment
The NMR results showed that the complex structure of SURP1 is comprised of three α‐helices and one 310‐helix, with an α1–α2–310–α3 topology (Figure 5a). The three α‐helices (α1, α2, and α3) are formed by residues Val49‐Asn63, Pro65‐Asn74, and Pro89‐Glu103, respectively, whereas the 310‐helix is formed by residues Asn82‐Leu84 (Figures 2 and 3a). SURP1 in complex form adopts a similar tertiary structure as in its free form, although there is a slight variation in the 310‐containing loop that connects α2 and α3, which is hereafter termed the 310 loop or simply 310 (Figure S3b). The topology of the three α‐helices is unique to SURP domains; thus, these domains are classified into a monospecific structural family/superfamily. 23 In S1BR, the region between residues 305 and 320 formed an amphipathic α‐helix composed of 16 residues (Figure 5a), which is hereafter denoted as αASF1. αASF1 mainly interacts with α1 and α2 of SURP1 in an anti‐parallel orientation relative to α1. The complex structure revealed that the interaction between SURP1 and αASF1 not only involves hydrophobic interactions, but also salt bridges and hydrogen bonds (Figure 5b,c). In the SURP1 structure, a hydrophobic patch was formed on the surface formed by α1, α2, and 310, consisting of the side chains of Ile52, Lys55 (methylene groups), Thr56 (a methyl group), Phe59 in α1; Phe67 and Ile71 in α2; and Phe81 in 310 (Figure 6). In the αASF1 structure, the following hydrophobic amino acids are aligned on one side of the α‐helix, forming a hydrophobic cluster: Tyr313p, Leu316p, Met317p, and Leu320p (“p” indicates that the residue belongs to S1BR). Therefore, the hydrophobic cluster directly contacted the hydrophobic patch of SURP1, resulting in a hydrophobic interaction network.
FIGURE 5.
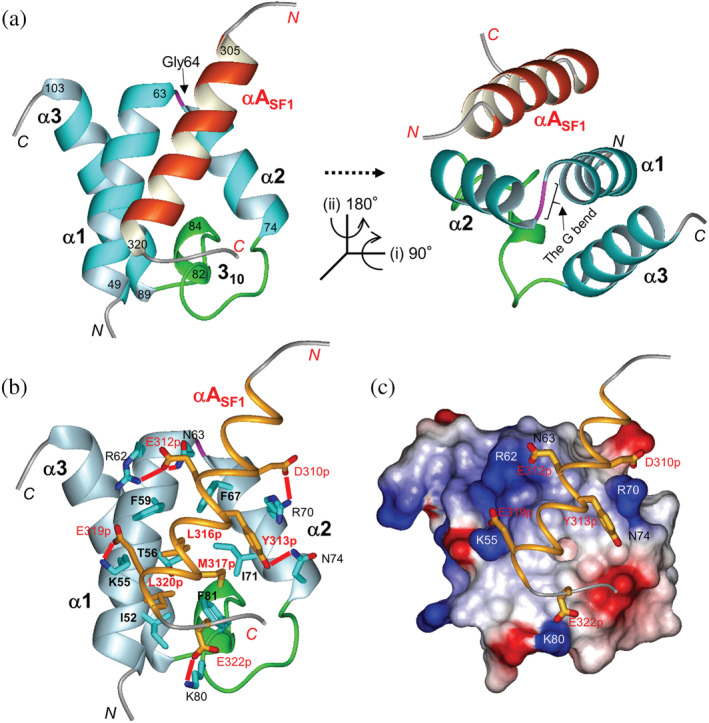
The structure of SURP1 in complex with S1BR. (a) Ribbon representation of SURP1 (res. 48–105) in complex with S1BR (res. 302–324) in different views. The structure in the left panel is in the same orientation as that of Figure 3c. The structure in the right panel is rotated as directed. The α‐helices in SURP1 and S1BR are depicted in cyan and orange, respectively. The G bend, connecting α1 and α2, and the 310 loop, connecting α2 and α3, are depicted in magenta and green, respectively. (b) Interaction between SURP1 and S1BR. Structures of SURP1 and S1BR are shown in ribbon (light blue) and thin ribbon (orange) models, respectively. Residues involved in the complex formation are shown, as described in the text. Side chains of the residues are represented as follows: carbon, cyan (SURP1), or orange (S1BR); oxygen, red; nitrogen, blue; and sulfur, yellow. Salt bridges and hydrogen bonds between SURP1 and S1BR are represented by red lines. (c) The electrostatic potential surface of SURP1 in complex with S1BR. Red and blue indicate negative and positive charges, respectively. The representation code in S1BR is the same as that in (b). The view is in the same orientation as that in (b).
FIGURE 6.
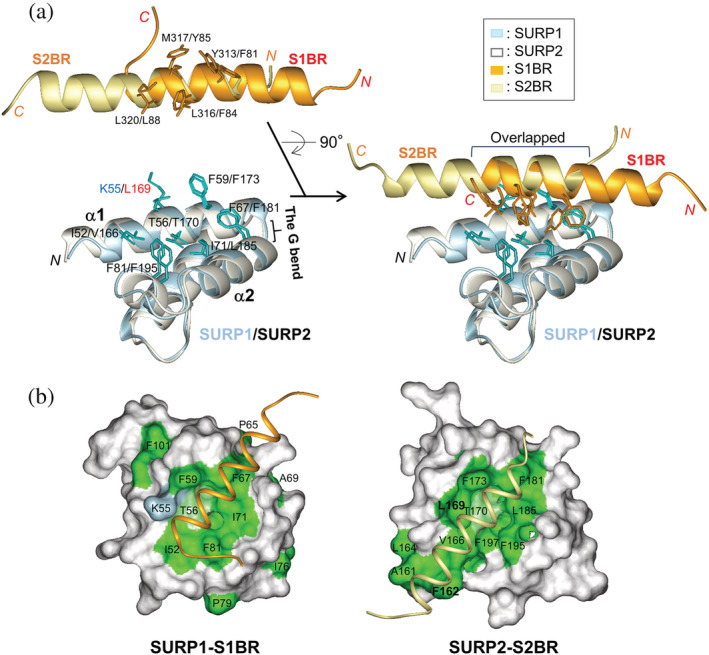
Comparison between SURP1‐S1BR and the SURP2–S2BR complexes. (a) Superposition of the structures of SURP1 (res. 48–105, light blue) in complex with S1BR (res. 302–324, orange) and of SURP2 (res. 160–217, white) in complex with S2BR (res. 77–99, light yellow) (PDB ID 2DT7). The left panels show the separation of complexes into the SURP domains and bound peptides for the clarification of residues involved in the hydrophobic interaction network. The side chains of the SURP domains and ligand peptides involved are colored cyan and orange, respectively. (b) Molecular surface representations showing hydrophobic residues of SURP1 (res. 48–105) (left panel) and SURP2 (res. 160–217) (right panel) with stick representations used for the ligand. Hydrophobic residues (Ala, Ile, Leu, Met, Phe, Pro, Trp, and Val) are colored green. In addition, a threonine residue containing a methyl group and lysine residues containing methylene groups are colored light green and light blue, respectively. The views are in the same orientation as that in Figure 5.
Salt bridges and hydrogen bonds between SURP1 and αASF1 were identified according to the strict criteria in the present study. Putative salt bridges were defined by one side‐chain carboxyl oxygen atom of Asp or Glu (i.e., one of Oδ1 or Oδ2 of Asp, or Oε1 or Oε2 of Glu) and one side‐chain nitrogen atom of Arg, Lys, or His (i.e., one of Nζ, Nη1 or Nη2 in Arg, Nζ in Lys, or Nδ1 or Nε2 of His) being within a distance of 0.4 nm. 24 Hydrogen bonds were defined by the distance between the hydrogen atom of the donor and the acceptor atom being within 0.24 nm, and the “donor atom”–“H”–“acceptor atom” angle through a bonding hydrogen (H) being within 35°. 25 Putative salt bridges were found between Lys55 in α1 and Glu319p; Arg62 in α1 and Glu312p; Arg70 in α2 and Asp310p; and Lys80 in the 310 loop and Glu322p (Figure 5b,c, Table S3). Putative hydrogen bonds were found between the side chains of Asn63 in α1 and Glu312p, and those of Asn74 in α2 and Tyr313p. All the residues were well conserved among SURP1 domains and the regions corresponding to αASF1 from various species, except for Saccharomyces cerevisiae (Figure S1), suggesting a highly conserved mode of interaction between SURP1 and S1BR throughout evolution (the exception is described in the Discussion section).
2.4. Effects of mutations in residues involved in salt bridges and hydrogen bonds on binding affinity between SURP1 and the SF1 fragment
Since most SURP domains from humans have similar amino acid residues in the positions forming the hydrophobic patch (Figure 2), hydrogen bonds and salt bridges between residues of the SURP domain and the peptide ligand, which are present in other positions, may be essential for assessing binding specificity. To confirm whether salt bridges and hydrogen bonds contribute to the specific interaction between SURP1 and S1BR, we constructed SURP1 mutants in which point mutations were introduced into each of the three residues involved (Arg62, Asn63, and Lys80) and BLI assays were performed on the BLItz platform (Figure 3a). The choice of substitution for each of the three residues depended on the type of residue at the corresponding position in the other SURP domains. For example, Arg62 was replaced with Glu, which is present in SF4 SURP1, or Gln, which is present in SFRS14 SURP2. Six SURP1 mutants were constructed as follows: R62E, R62Q, N63E, N63Q, K80D, and K80Q.
We initially evaluated the kinetics of the interaction between wild‐type SURP1 and S1BRp (res. 295–327) using BLI. Biotinylated S1BRp (ligand) was immobilized onto a streptavidin biosensor surface and the binding events of SURP1 (analyte) were monitored in real time. A concentration‐dependent box‐shaped sensorgram was obtained for the interaction between SURP1 and S1BRp (Figure S5a). This response is typical of weak or transient interactions characterized by rapid association upon sample injection, a steady equilibrium phase, and rapid dissociation, which restores the buffer baseline. 26 This type of response generally makes it difficult to perform kinetic analysis. The dissociation constant (K D) was instead assessed using an equilibrium analysis of binding responses plotted as a function of total analyte concentrations and was calculated from a non‐linear regression curve fit. K D is defined as the concentration of the analyte that results in 50% binding response at equilibrium. The results showed that the K D value for the wild‐type was 15.2 μM (Figure S5b, Table 1), which is reasonable because K D for a weak interaction is approximately greater than 10−6 M. 27 , 28
For comparison, we similarly assessed K D for each mutant (Figure S5b, Table 1). The R62E and R62Q mutations in the R62–E312p pair, which may affect the original electronic interaction, resulted in 26‐ and 12‐fold increases in K D from that of the wild‐type, respectively. Regarding the N63–E312p pair, the N63E, and N63Q mutations resulted in a 17‐ and 9.1‐fold increase in K D, respectively. The K80D and K80Q mutations in the K80‐E322p pair exerted weaker effects on K D than the other mutations, resulting in 8.7‐ and 4.2‐fold increases in K D, respectively.
These results indicate that three residues, R62, N63, and K80 (R/N/K), participate in the specific interactions between SURP1 and S1BRp via salt bridges or hydrogen bonds. Accordingly, the human SURP domains that bind to S1BRp may be explained in terms of these three key residues. The SURP domain of CHERP and the second SURP domain of SFSWAP, which reportedly binds to S1BRp, have the same residues at the corresponding positions. In contrast, the other SURP domains have more than one different residue, namely, R/Q/Q in SFSWAP SURP1, R/E/M in SR140 SURP, E/G/A in SF4 SURP1, D/G/A in SF4 SURP2, E/G/A in SFRS14 SURP1, and Q/V/D in SFRS14 SURP2 (Figure 2). These differences in the three residues appear to be one of the main reasons for the failure of the six SURP domains to bind S1BRp.
2.5. Effects of the A57S mutation on the SURP1 structure
A mutation from Ala57 to Ser was detected in the SF3A1 gene of a patient with myelodysplastic syndrome. 20 In view of the SURP1 structure, Ala57 is included in α1 and is completely buried inside the structure, contributing to hydrophobic interactions with α3 (Figure S2b). To examine the effects of the mutation on the SURP1 structure, we constructed an A57S mutant in which Ala57 was replaced with Ser (Figure 3a) and compared the CD spectra between the wild‐type and mutant. Wild‐type SURP1 showed a CD spectrum with a minimal negative peak at ∼208 nm and strong positive ellipticity at ∼195 nm, indicative of a helical conformation (Figure S2a). Deconvolution analysis using the Contin‐LL software 29 , 30 yielded values of 96.8% helix, 3.2% strand, 0% turn, and 0% unordered. The estimated secondary structure composition agreed with the helix‐rich structure of SURP1.
In contrast, the CD spectrum of the A57S mutant displayed a negative band with a single minimum at ~200 nm, which is indicative of the so‐called random‐coil conformation (Figure S2a). Deconvolution analysis yielded values of 26.1% helix, 1.2% strand, 9.1% turn, and 63.6% unordered (Figure S2a). These results indicate that the A57S mutation markedly altered the SURP1 structure, which was predominantly disordered or unstructured.
3. DISCUSSION
3.1. A common structural feature of SURP domains for providing an interaction surface
The complex structure shows that the surface formed by α1, α2, and the 310 loop (termed the α1/α2 surface) functions as the main interaction site for SURP1. Notably, the relative positions of α1 and α2 appear to be unique to SURP domains, in that the region connecting α1 and α2 is occupied by a single Gly residue (Figure 2, Figure S1a). The shape of α1‐[Gly]‐α2 in the ribbon model resembles a hand grip, and it appears as if one α‐helix is bent just at the Gly residue, such that a part of the α‐helix expected to be bent is wound in a left‐handed direction (Figure S6a,b). This bend is thus termed the “G bend” in the present study. This peculiar bend required the Gly residue to adopt the βPR conformation (φ = ~100°, ψ = ~170°). In the Ramachandran plot, the βPR region is a reflection of the βP region, which corresponds to the left‐handed polyproline II helix, and only Gly residues are also allowed in the βPR region (Figure S6c). 31 Consequently, the G bend connects α1 and α2 at an acute angle (approximately 45°) (Figure S6a). The side chains of the residues on α1 and α2 are arranged side by side or in parallel between the helices, resulting in a specific pattern (Figure S6d). More importantly, the α1/α2 surface has a characteristic shape that resembles a bowl‐shaped depression with a collapsed region, and its surface is used as the ligand interaction site for SURP1, as described below. The Gly residue is highly conserved among all SURP domains with only a few exceptions, indicating the conservation of not only the G bend but also the positions of the side chains of residues on the α1/α2 surface (Figure 2, Figures S1a and S7a).
An exception in humans is that the first of the two SURP domains of the SFRS14 protein, which is a putative splicing factor, 32 possesses an ordered loop composed of Ser‐Leu‐Ser connecting α1 and α2, instead of the G bend (Figure 2, Table S1). As expected, the relative positions of α1 and α2 differed from those of the SURP domains with a G bend (Figure S7b), suggesting that the peptide recognition mode of SFRS14 SURP1 or the peptide sequence differs from that of the SURP domains with a G bend. Hence, this SURP domain is classified as a different type from the canonical SURP domains, namely, Type 3, as described later.
3.2. Comparison between SURP1 and SURP2 complex structures: A common rule of the interaction mode of SURP domains
Although SURP1 has a sequence and structure similar to that of SURP2, the interaction between SURP1 and the ligand peptide is weak or transient, whereas with SURP2 it is strong or permanent. Detailed structural comparisons between the two complex structures revealed the reason for the notable difference in binding affinity and commonalities in the peptide interaction mode. Figure 6a shows an excellent superimposition of the solution complex structure of SURP1 and the ligand peptide with the SURP2 counterpart, although the position of the α‐helix of one bound peptide relative to that of the α‐helix of the other shifts by approximately two helical turns. The overlapping part of the two α‐helices is the C‐terminal half of αASF1 and the N‐terminal half of an α‐helix bound to SURP2 (termed αASF3A3), in which a similar hydrophobic interaction network, is observed between the SURP1 and SURP2 complexes. On the α1/α2 surface of each SURP domain, a hydrophobic patch was formed by the side chains of the residues (SURP1/SURP2): Ile52/Val166, Lys55 (methylene groups)/Leu169, Thr56 (a methyl group)/Thr170, and Phe59/Phe137 in α1; Phe67/Phe181 and Ile71/Leu185 in α2; and Phe81/Phe195 in 310 (Figure 5a). Leu169 is the determinant of SURP2 complex formation, as described in the Introduction. Each patch contacts a hydrophobic cluster on the α‐helix of the ligand peptide (αASF1/αASF3A3) formed by Tyr313p/Phe81p, Leu316p/Phe84p, Met317p/Tyr85p, and Leu320p/Leu88p. Because the hydrophobic moieties from amino acid residues involved in this hydrophobic patch are well preserved in many SURP domains displaying a G bend (Figure 2), the hydrophobic patch appears to have been conserved as a primary peptide interaction site throughout evolution.
This conservation in the SURP domains may reflect the sequences of the ligand peptides, consequently generating a specific sequence pattern or consensus sequence. Referring to the interacting residues of αASF1 and αASF3A3 (Figure S1b), the consensus sequence of the ligand peptides that interact with the patch may be estimated as [Y/F]‐X‐X‐[#]‐[#]‐X‐X‐[L] (#: a hydrophobic residue or Tyr, but not Trp or Pro). In addition, regarding the residues involved in salt bridges, positions 55 and 62 in all SURP domains, except for SF3A1 SURP2 and SFRS14 SURP1, are occupied by K/R and R (major residues)/E/D, respectively. Provided that the salt bridges are conserved in the SURP‐peptide interactions, an estimated consensus sequence of the ligand peptides may be extended to [E/R/K]‐[Y/F]‐X‐X‐[#]‐[#]‐X‐[E/D]‐[L].
The consensus sequence, however, is not true for SURP1 or SURP2 of the SF3A1 ortholog from S. cerevisiae because the sequences of the ligand peptides corresponding to αASF1 and αASF3A3 markedly differ from those of Schizosaccharomyces pombe and other species (Figure S1b). Additionally, the sequences of SURP1 and SURP2 from S. cerevisiae exhibited the greatest differences from those of the other species (Figure S1a). Such an exceptionality of S. cerevisiae is yet another example that indicates that proteins from S. cerevisiae are generally less similar to those from animals as compared to those from S. pombe. 33 , 34 In addition, co‐elimination of functionally related groups of spliceosome components appears to have occurred based on genome comparisons among S. cerevisiae, S. pombe, and Neurospora crassa. 35 , 36 After divergence from Saccharomyces, the interaction mode between the SURP domain and ligand peptide may have evolved in a unique manner.
3.3. Factors conferring specificity to individual SURP domains
In contrast, differences in the peptide interaction mode between the two complexes were highlighted in the non‐overlapping parts of the ligand α‐helices. Centering around the overlapped part, the interaction sites extended to the N‐ and C‐termini of αASF1 and αASF3A3, respectively (Figure 6a). In the SURP1 complex, the N‐terminal side, followed by the overlapped part, interacts with the end of α1 and the beginning of α2, which are linked by the G bend (Gly64). Arg62 and Asn63 in α1 were found next to the G bend and appeared to form a salt bridge and a hydrogen bond with Glu312p, respectively (Figure 5c). In addition, Lys80 in the 310 loop appears to form a salt bridge with Glu322p. Mutational analysis showed that Arg62, Asn63, and Lys80 were required for efficient interaction with αASF1. Because the residue combination at the three positions is unique to CHERP SURP, SFSWAP SURP2, and SF3A1 SURP1, it is conceivable that the residues confer SF1‐specificity to the three SURP domains.
In contrast, in the SURP2 complex, the C‐terminal side, found next to the overlapping part, interacted with the N‐terminal helical turn of α1 in SURP2 (Figure 6a). The length of α1 in SURP2 was longer by one helical turn compared to SURP1, and the aromatic ring of Phe162, in an additional helical turn, extended into the hydrophobic patch (Figure 6b). In addition, Leu169 in α1 expands the patch. The total hydrophobic patch of SURP2 was larger than that of SUPR1, resulting in an increase in hydrophobic interactions between SURP2 and αASF3A3 (Figure 6b). Interestingly, there were no apparent hydrogen bonds or salt bridges in the SUPR2 complex. 8 Thus, strong or permanent interactions between SURP2 and αASF3A3 appear to be achieved only by hydrophobic interactions via the expanded patch.
However, it is quite unlikely that this interaction mode is applicable to any SURP domain other than SURP2 of SF3A1 because Phe162 and Leu169 predominantly occur in the SURP2 domains of SF3A1 orthologs from various species. Considering that other SURP domains with the G bend are similar in sequence to SURP1 of SF3A1 (Figure 2), we infer that SURP domains with G bend are generally engaged in transient interactions, with the exception of SF3A1 SURP2. By changing the number and type (residue combination) of salt bridges and/or hydrogen bonds, individual SURP domains may have acquired adequate ligand affinity and specificity that involves a transient interaction, with the hydrophobic patch on the α1/α2 surface being conserved.
3.4. SURP1‐mediated interaction between U2 snRNP and SF1
BLI assays showed a transient interaction between SURP1 and S1BR peptide S1BRp, which underwent a disorder‐to‐helix transition upon binding to SURP1. In full‐length SF1, S1BR is located between a Zn‐finger domain and a proline‐rich region (Figure 1c). Based on the results that showed that S1BRp is unstructured in its free form, S1BR may be situated in an intrinsically disordered region (IDR) in SF1. Generally, an interaction between IDR and the structural binding partner accompanied by a disorder‐to‐order transition is typical of transient protein–protein interactions, which are formed and broken easily or continuously in cells. 27 , 28 The transient interaction between SURP1 and the S1BR peptide may play a significant role in the SURP1‐mediated interaction between U2 snRNP and SF1. This interaction property is presumably beneficial and necessary not only for the initial recruitment of U2 snRNP to the E complex, but also for the dissociation of SF1 from U2 snRNP during the E to A complex transition. Thus, since the A57S mutant of SURP1 is largely disordered, it seems unlikely that the U2 snRNP containing the mutant is recruited to SF1. Additionally, considering that SF1 is not involved in the splicing of all introns but influences alternative splicing decisions, 37 this putative binding failure might induce some abnormal alternative splicing that leads to myelodysplastic syndromes.
3.5. Concluding remarks
Based on the findings obtained in this study, SURP domains can be divided into three types, depending on the presence or absence of the G‐bend and levels of the peptide binding affinity: Type 1, contains G‐bend and displays low affinity; Type 2, contains G‐bend and displays high affinity (a permanent interaction); and Type 3, no G‐bend (Table 3). In humans, SF3A1 SURP2 belongs to Type 2, whereas SFRS14 SURP1 belongs to Type 3. The former is conserved in all eukaryotes, whereas the latter only occurs in mammals. In contrast, Type‐1 SURP domains increase in number from yeast to humans. Presumably, as the splicing machinery became more complex during evolution, the number of SURP‐containing proteins and number of SURP domains per protein increased. However, it still remains unclear why the number of SURP domains is limited to only 10, why SURP domains only occur in splicing‐related proteins, and why they do not diffuse to various other proteins. Assuming that the transient interaction property is an important attribute of Type‐1 SURP domains and, seemingly, Type‐3 domains, further studies on SURP domains and SURP‐containing proteins may provide key insights for understanding not only transient protein–protein interactions that often occur in various steps of the splicing process but also a special relationship between SURP domains and splicing.
TABLE 3.
Classification of SURP1 domains in humans
| Connection between α1 and α2 | Classification | SURP‐containing protein | SURP domain | Type‐specific residues | A binding protein |
|---|---|---|---|---|---|
| G‐bend | Type 1a a | SF3A1 | SURP1 | ||
| SFSWAP | SURP2 | R62, N63, K80 b | SF1 | ||
| CHERP | SURP | ||||
| Type 1b | SFSWAP | SURP1 | – c | – | |
| SR140 | SURP | – | – | ||
| SF4 | SURP1 | – | – | ||
| SF4 | SURP2 | – | – | ||
| SFRS14 | SURP2 | – | – | ||
| Type 2 | SF3A1 | SURP2 | F162, L169 | SF3A3 | |
| Loop | Type 3 | SFRS14 | SURP1 | – | – |
Currently, Type‐1 SURP domains are divided into two subtypes, depending on whether they can bind to SF1.
The residue number is according to SF3A1 SURP1.
“–” refers to “unknown.”
4. MATERIALS AND METHODS
4.1. Protein expression and purification
In the chemical shift perturbation and biochemical experiments, 15N‐ and 13C‐labeled and non‐labeled SF3A1 SURP1 were prepared as previously described. 8 Briefly, DNA encoding SURP1 (res. 48–110) of human SF3A1 was subcloned by PCR from a human full‐length cDNA clone. This DNA fragment was cloned into the expression vector pET‐15b (Novagen) as a fusion protein with an N‐terminal His affinity tag. In the protein construct, the TEV protease cleavage site (ENLYFQG) was placed between the tag and SURP1 sequence. The TEV protease was used to cleave the amide bond of the Gln‐Gly dipeptide, resulting in an additional Gly residue at the N‐terminus in the final protein product. Single amino acid mutations were introduced into SURP1 using PCR with 27‐mer primers spanning the site of the desired mutation, as described in the manufacturer's protocol for the PrimeSTAR Mutagenesis Basal Kit (Takara Bio Inc.). Each mutation was confirmed using DNA sequencing. A series of mutant proteins were prepared as previously described. 8 Note, however, that in column chromatography, RESOURCE S (GE Healthcare) was used for the wild type and mutants, except for the R62E and K80D mutants, for which RESOURCE Q was used.
For structural determination, we designed a chimeric protein comprising SURP1 (res. 48–110), a 14‐residue linker (SGSSGSSGSSGSSG), and S1BR (res. 295–27) of SF1. Using PCR, the sequence of AGCGGCAGCAGCGGCAGCAGCGGCAGCAGCAGCGGCAGCGGC encoding the linker (SGSSGSSGSSGSSG) was added to the 5′ end of the synthesized gene encoding the fragments of human SF1 (accession number: Q15637), namely S1BR (res. 295–327). The PCR product was cloned into the plasmid vector for the expression of recombinant SURP1, which was used in the above experiments, using NEBuilder HiFi DNA Assembly (NEW ENGLAND BioLabs). The chimeric protein, namely the 15N and 13C‐labeled SURP1‐Linker‐S1BRp protein, was also prepared as previously described, 8 except for the use of gel filtration in place of ion exchange.
4.2. NMR spectroscopy
The samples were concentrated to 1.4 mM in 20 mM sodium phosphate buffer (pH 7.0) containing 1 mM 1,4‐dl‐dithiothreitol‐d10 (d‐DTT) dissolved in 90% 1H2O/10% 2H2O using an Amicon Ultra‐15 filter (3,000 MWCO, Millipore). NMR experiments were performed at 25°C using a Bruker 600‐MHz spectrometers (Bruker AVANCE 600). 1H, 15N, and 13C chemical shifts were referenced relative to the frequency of the 2H lock resonance of water. The backbone and side‐chain assignments of individual SURP1 and chimeric protein were obtained using a combination of standard triple resonance experiments. 38 , 39 The spectra used for structural determination are listed in Table S4. Briefly, 2D [1H,15N]‐HSQC, 3D HNCO, HN(CA)CO, HNCA, HN(CO)CA, HNCACB, and CBCA(CO)NH spectra were used for the 1H, 15N, and 13C assignments of the protein backbone. The 1H and 13C assignments of the non‐aromatic side chains, including all the prolines, were obtained using 2D [1H,13C]‐HSQC, 3D HBHA(CO)NH, H(CCCO)NH, (H)CC(CO)NH, HCCH‐COSY, HCCH‐TOCSY, and (H)CCH‐TOCSY spectra. The assignments were checked for consistency with 3D 15N‐edited [1H,1H]‐ and 13C‐edited [1H,1H]‐NOESY spectra. The 1H and 13C spin systems of the aromatic rings of Phe, Trp, His, and Tyr were identified using 3D HCCH‐COSY and HCCH‐TOCSY experiments, and 3D 13C‐edited [1H, 1H]‐NOESY was used for the sequence‐specific resonance assignment of the aromatic side chains. NOESY spectra were recorded with a mixing time of 80 ms. The NMR data were processed using NMRPipe. 40 Analyses of the processed data were performed using the NMRView 41 and KUJIRA. 42
In the chemical shift perturbation experiments, S1BRp (res. 295–335), a 41‐residue synthesized peptide, was dissolved in 20 mM sodium phosphate buffer (pH 7.0) containing 1 mM d‐DTT to prepare a 2 mM stock solution. 2D [1H,15N]‐HSQC spectra of SURP1 at a concentration of 180 μM were recorded in the absence of S1BRp, whereas those at a concentration of 900 μM were recorded in the presence of S1BRp at the same concentration. The spectra for the sequence‐specific assignments of the resonances are listed in Table S4.
4.3. Structural calculations
The 3D structure of the chimeric protein was determined by a combination of automated NOESY cross‐peak assignments 43 and structure calculations with torsion‐angle dynamics 44 implemented in CYANA 2.1. 22 The dihedral angle constraints for φ and ψ were obtained from the main chain and13Cβ chemical shift values using the TALOS program 45 and by analyzing the NOESY spectra. Stereospecific assignments for isopropyl methyl and methylene groups were determined based on patterns of inter‐ and intra‐residual NOE intensities. 46 No hydrogen‐bond constraints were used. Structural calculations were started from 200 randomized conformers and the standard CYANA was used to simulate annealing schedule. 44 Further refinements by restrained molecular dynamics, followed by restrained energy minimization, were performed for the 40 conformers with the lowest final CYANA target function values using the Amber12 program with the Amber 2012 force field and a generalized Born model, 47 as previously described. 48 Finally, the 20 conformers with the lowest Amber energy values were selected as the final structures and deposited in the Protein Data Bank under PDB accession code 7VH9. PROCHECK‐NMR 49 and MOLMOL 50 were used to validate and visualize the final structures, respectively.
4.4. BLI measurements
BLI measurements were performed at 25°C using a BLItz instrument (Fortebio Inc.). The samples were buffered with 1× kinetics buffer (1× PBS, pH 7.4, 0.01% BSA, and 0.002% Tween 20). Streptavidin biosensor tips (Fortebio Inc.) were pre‐wetted with 1× kinetic buffer for 10 min. Four microliters of biotin‐tagged SF1 S1BRp (TORAY Research Center) (10 μg/ml) was immobilized onto the biosensor tip. Measurements were performed according to the manufacturer's instructions. Raw data were exported into GraphPad Prism 9.3.1 for Windows and plateau binding values were fitted to the saturation binding equation:
| (1) |
Where B max is the maximum specific binding from one site binding model using non‐linear regression to obtain K D. The measurements were conducted in triplicate.
4.5. CD measurements
The samples used for CD measurements contained 0.1 mg/ml protein in 1× PBS buffer. The experiments were performed on a Jasco J‐820 spectrometer at 20°C using a cuvette path length of 1 mm. Spectral scans ranging between 195 and 250 nm were acquired and accumulated. The results are expressed as the mean residue ellipticity. The secondary structure content was estimated from each spectrum using the CONTIN/LL program 29 , 30 via the DICHROWEB server. 51 , 52 , 53 , 54 A reference dataset, Set 7, was used for analysis.
AUTHOR CONTRIBUTIONS
Nobukazu Nameki: Conceptualization (equal); investigation (equal); visualization (equal); writing – original draft (lead). Masayuki Takizawa: Data curation (lead); investigation (equal). Takayuki Suzuki: Investigation (supporting). Shoko Tani: Investigation (supporting). Naohiro Kobayashi: Software (lead). Taiichi Sakamoto: Investigation (supporting); resources (lead); writing – review and editing (supporting). Yutaka Muto: Investigation (equal); supervision (equal); writing – review and editing (equal). Kanako Kuwasako: Conceptualization (equal); funding acquisition (lead); investigation (equal); project administration (lead); supervision (equal); visualization (equal); writing – original draft (supporting); writing – review and editing (equal).
CONFLICT OF INTEREST
The authors declare no conflict of interests.
Supporting information
Table S1 SURP‐containing proteins and SURP domains in eukaryotic species.
Table S2 CYANA structural statistics for 40 conformers of the chimera of SURP1 and S1BRp (res. 295–327) with the lowest CYANA target function values.
Table S3 Summary of the putative salt bridges and hydrogen bonds between SURP1 and S1BR.
Table S4 Summary of NMR measurements for structural analysis of SURP1 in three forms.
Figure S1 Structure‐based multiple sequence alignment of SURP1 and SURP2 in SF3A1 orthologs, and that of S1BR in SF1 orthologs and S2BR in SF3A3 orthologs.
Figure S2 CD spectroscopy of wild‐type SURP1, the A57S mutant, and the SF1 fragments.
Figure S3 Intermolecular NOEs between SURP1 and S1BR, and comparison of SURP1 structures between free and complex forms.
Figure S4 Comparisons of [1H,15N]‐HSQC spectra between individual SURP1 in free and complex forms, and those between SURP1 of the chimera and individual SURP1 in a complex form.
Figure S5 Equilibrium analysis of binding responses for interactions of SURP1 or mutants to S1BRp using BLI.
Figure S6 A unique topology of α1 and α2 characterized by the G bend.
Figure S7 Comparison of structures among human SURP domains in terms of the G bend.
ACKNOWLEDGEMENTS
We thank Megumi Ishizuka and Yuka Yoshimura for their assistance with this series of experiments. We are also grateful to Messrs. Takeshi Seguchi and Yasuhiro Horie (PrimeTech Corp.) for their technical assistance and helpful suggestions for the use of the BLItz instrument and kinetic analyses. This work was supported by the Japan Society for the Promotion of Science KAKENHI [grant number JP15K06982] (to K.K.), a grant from the Sumitomo Foundation [No. 151227] (to K.K.), and a grant from the Takeda Science Foundation (to K.K.).
Nameki N, Takizawa M, Suzuki T, Tani S, Kobayashi N, Sakamoto T, et al. Structural basis for the interaction between the first SURP domain of the SF3A1 subunit in U2 snRNP and the human splicing factor SF1 . Protein Science. 2022;31(10):e4437. 10.1002/pro.4437
Review Editor: Carol Post
Funding information JSPS KAKENHI, Grant/Award Number: JP15K06982; Sumitomo Foundation, Grant/Award Number: 151227; Takeda Science Foundation
Contributor Information
Yutaka Muto, Email: ymuto@musashino-u.ac.jp.
Kanako Kuwasako, Email: kanameki@musashino-u.ac.jp.
DATA AVAILABILITY STATEMENT
The atomic coordinates for the ensemble of 20 energy‐refined NMR conformers, representing the solution structure of the SURP1‐Linker‐S1BRp protein, were deposited in the Protein Data Bank under accession code 7VH9. Chemical shift assignments for the SURP1‐Linker‐S1BRp protein were deposited in the BMRB database under accession number 36445.
REFERENCES
- 1. Wahl MC, Will CL, Lührmann R. The spliceosome: Design principles of a dynamic RNP machine. Cell. 2009;136:701–718. [DOI] [PubMed] [Google Scholar]
- 2. Plaschka C, Lin PC, Charenton C, Nagai K. Prespliceosome structure provides insights into spliceosome assembly and regulation. Nature. 2018;559:419–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu Z, Luyten I, Bottomley MJ, et al. Structural basis for recognition of the intron branch site RNA by splicing factor 1. Science. 2001;294:1098–1102. [DOI] [PubMed] [Google Scholar]
- 4. Yoshida H, Park SY, Sakashita G, et al. Elucidation of the aberrant 3′ splice site selection by cancer‐associated mutations on the U2AF1. Nat Commun. 2020;11:4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Das R, Zhou Z, Reed R. Functional association of U2 snRNP with the ATP‐independent spliceosomal complex E. Mol Cell. 2000;5:779–787. [DOI] [PubMed] [Google Scholar]
- 6. Crisci A, Raleff F, Bagdiul I, et al. Mammalian splicing factor SF1 interacts with SURP domains of U2 snRNP‐associated proteins. Nucleic Acids Res. 2015;43:10456–10473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Denhez F, Lafyatis R. Conservation of regulated alternative splicing and identification of functional domains in vertebrate homologs to the drosophila splicing regulator, suppressor‐of‐white‐apricot. J Biol Chem. 1994;269:16170–16179. [PubMed] [Google Scholar]
- 8. Kuwasako K, He F, Inoue M, et al. Solution structures of the SURP domains and the subunit‐assembly mechanism within the splicing factor SF3a complex in 17S U2 snRNP. Structure. 2006;14:1677–1689. [DOI] [PubMed] [Google Scholar]
- 9. Hodges PE, Beggs JD. RNA splicing. U2 fulfils a commitment. Curr Biol. 1994;4:264–267. [DOI] [PubMed] [Google Scholar]
- 10. Krämer A, Grüter P, Gröning K, Kastner B. Combined biochemical and electron microscopic analyses reveal the architecture of the mammalian U2 snRNP. J Cell Biol. 1999;145:1355–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nesic D, Krämer A. Domains in human splicing factors SF3a60 and SF3a66 required for binding to SF3a120, assembly of the 17S U2 snRNP, and prespliceosome formation. Mol Cell Biol. 2001;21:6406–6417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brosi R, Hauri HP, Krämer A. Separation of splicing factor SF3 into two components and purification of SF3a activity. J Biol Chem. 1993;268:17640–17646. [PubMed] [Google Scholar]
- 13. Krämer A, Mulhauser F, Wersig C, Gröning K, Bilbe G. Mammalian splicing factor SF3a120 represents a new member of the SURP family of proteins and is homologous to the essential splicing factor PRP21p of Saccharomyces cerevisiae . RNA. 1995;1:260–272. [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Q, Wang Y, Liu Y, et al. U2‐related proteins CHERP and SR140 contribute to colorectal tumorigenesis via alternative splicing regulation. Int J Cancer. 2019;145:2728–2739. [DOI] [PubMed] [Google Scholar]
- 15. De Maio A, Yalamanchili HK, Adamski CJ, et al. RBM17 interacts with U2SURP and CHERP to regulate expression and splicing of RNA‐processing proteins. Cell Rep. 2018;25:726–736e727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Haselbach D, Komarov I, Agafonov DE, et al. Structure and conformational dynamics of the human Spliceosomal B(act) complex. Cell. 2018;172:454–464.e411. [DOI] [PubMed] [Google Scholar]
- 17. Townsend C, Leelaram MN, Agafonov DE, et al. Mechanism of protein‐guided folding of the active site U2/U6 RNA during spliceosome activation. Science. 2020;370:eabc3753. [DOI] [PubMed] [Google Scholar]
- 18. Zhang Z, Will CL, Bertram K, et al. Molecular architecture of the human 17S U2 snRNP. Nature. 2020;583:310–313. [DOI] [PubMed] [Google Scholar]
- 19. Zhang Z, Rigo N, Dybkov O, et al. Structural insights into how Prp5 proofreads the pre‐mRNA branch site. Nature. 2021;596:296–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. [DOI] [PubMed] [Google Scholar]
- 21. Li H, Koshiba S, Hayashi F, et al. Structure of the C‐terminal phosphotyrosine interaction domain of Fe65L1 complexed with the cytoplasmic tail of amyloid precursor protein reveals a novel peptide binding mode. J Biol Chem. 2008;283:27165–27178. [DOI] [PubMed] [Google Scholar]
- 22. Güntert P. Automated NMR structure calculation with CYANA. Methods Mol Biol. 2004;278:353–378. [DOI] [PubMed] [Google Scholar]
- 23. Fox NK, Brenner SE, Chandonia JM. SCOPe: Structural classification of proteins—Extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic Acids Res. 2014;42:D304–D309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barlow DJ, Thornton JM. Ion‐pairs in proteins. J Mol Biol. 1983;168:867–885. [DOI] [PubMed] [Google Scholar]
- 25. Berndt KD, Beunink J, Schroder W, Wüthrich K. Designed replacement of an internal hydration water molecule in BPTI: Structural and functional implications of a glycine‐to‐serine mutation. Biochemistry. 1993;32:4564–4570. [DOI] [PubMed] [Google Scholar]
- 26. Seviour T, Hansen SH, Yang L, et al. Functional amyloids keep quorum‐sensing molecules in check. J Biol Chem. 2015;290:6457–6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Perkins JR, Diboun I, Dessailly BH, Lees JG, Orengo C. Transient protein‐protein interactions: Structural, functional, and network properties. Structure. 2010;18:1233–1243. [DOI] [PubMed] [Google Scholar]
- 28. Acuner Ozbabacan SE, Engin HB, Gursoy A, Keskin O. Transient protein‐protein interactions. Protein Eng Des Sel. 2011;24:635–648. [DOI] [PubMed] [Google Scholar]
- 29. Provencher SW, Glockner J. Estimation of globular protein secondary structure from circular dichroism. Biochemistry. 1981;20:33–37. [DOI] [PubMed] [Google Scholar]
- 30. van Stokkum IH, Spoelder HJ, Bloemendal M, van Grondelle R, Groen FC. Estimation of protein secondary structure and error analysis from circular dichroism spectra. Anal Biochem. 1990;191:110–118. [DOI] [PubMed] [Google Scholar]
- 31. Ho BK, Brasseur R. The Ramachandran plots of glycine and pre‐proline. BMC Struct Biol. 2005;5:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sampson ND, Hewitt JE. SF4 and SFRS14, two related putative splicing factors on human chromosome 19p13.11. Gene. 2003;305:91–100. [DOI] [PubMed] [Google Scholar]
- 33. Sipiczki M. Phylogenesis of fission yeasts. Contradictions surrounding the origin of a century old genus. Antonie Van Leeuwenhoek. 1995;68:119–149. [DOI] [PubMed] [Google Scholar]
- 34. Sipiczki M. Where does fission yeast sit on the tree of life? Genome Biol. 2000;1:REVIEWS1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Aravind L, Watanabe H, Lipman DJ, Koonin EV. Lineage‐specific loss and divergence of functionally linked genes in eukaryotes. Proc Natl Acad Sci U S A. 2000;97:11319–11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Braun EL, Halpern AL, Nelson MA, Natvig DO. Large‐scale comparison of fungal sequence information: Mechanisms of innovation in Neurospora crassa and gene loss in Saccharomyces cerevisiae . Genome Res. 2000;10:416–430. [DOI] [PubMed] [Google Scholar]
- 37. Tanackovic G, Krämer A. Human splicing factor SF3a, but not SF1, is essential for pre‐mRNA splicing in vivo. Mol Biol Cell. 2005;16:1366–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bax A. Multidimensional nuclear magnetic resonance methods for protein studies. Curr Opin Struct Biol. 1994;4:738–744. [Google Scholar]
- 39. Kay LE. NMR methods for the study of protein structure and dynamics. Biochem Cell Biol. 1997;75:1–15. [DOI] [PubMed] [Google Scholar]
- 40. Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. [DOI] [PubMed] [Google Scholar]
- 41. Johnson BA. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol. 2004;278:313–352. [DOI] [PubMed] [Google Scholar]
- 42. Kobayashi N, Iwahara J, Koshiba S, et al. KUJIRA, a package of integrated modules for systematic and interactive analysis of NMR data directed to high‐throughput NMR structure studies. J Biomol NMR. 2007;39:31–52. [DOI] [PubMed] [Google Scholar]
- 43. Herrmann T, Güntert P, Wüthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol. 2002;319:209–227. [DOI] [PubMed] [Google Scholar]
- 44. Güntert P, Mumenthaler C, Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol. 1997;273:283–298. [DOI] [PubMed] [Google Scholar]
- 45. Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. [DOI] [PubMed] [Google Scholar]
- 46. Powers R, Garrett DS, March CJ, Frieden EA, Gronenborn AM, Clore GM. The high‐resolution, three‐dimensional solution structure of human interleukin‐4 determined by multidimensional heteronuclear magnetic resonance spectroscopy. Biochemistry. 1993;32:6744–6762. [DOI] [PubMed] [Google Scholar]
- 47. Case DA, Cheatham TE III, Darden T, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tsuda K, Someya T, Kuwasako K, et al. Structural basis for the dual RNA‐recognition modes of human Tra2‐β RRM. Nucleic Acids Res. 2011;39:1538–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK‐NMR: Programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. [DOI] [PubMed] [Google Scholar]
- 50. Koradi R, Billeter M, Wüthrich K. MOLMOL: A program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. [DOI] [PubMed] [Google Scholar]
- 51. Lobley A, Whitmore L, Wallace BA. DICHROWEB: An interactive website for the analysis of protein secondary structure from circular dichroism spectra. Bioinformatics. 2002;18:211–212. [DOI] [PubMed] [Google Scholar]
- 52. Whitmore L, Wallace BA. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004;32:W668–W673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Whitmore L, Wallace BA. Protein secondary structure analyses from circular dichroism spectroscopy: Methods and reference databases. Biopolymers. 2008;89:392–400. [DOI] [PubMed] [Google Scholar]
- 54. Miles AJ, Ramalli SG, Wallace BA. DichroWeb, a website for calculating protein secondary structure from circular dichroism spectroscopic data. Protein Sci. 2022;31:37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Larkin MA, Blackshields G, Brown NP, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 SURP‐containing proteins and SURP domains in eukaryotic species.
Table S2 CYANA structural statistics for 40 conformers of the chimera of SURP1 and S1BRp (res. 295–327) with the lowest CYANA target function values.
Table S3 Summary of the putative salt bridges and hydrogen bonds between SURP1 and S1BR.
Table S4 Summary of NMR measurements for structural analysis of SURP1 in three forms.
Figure S1 Structure‐based multiple sequence alignment of SURP1 and SURP2 in SF3A1 orthologs, and that of S1BR in SF1 orthologs and S2BR in SF3A3 orthologs.
Figure S2 CD spectroscopy of wild‐type SURP1, the A57S mutant, and the SF1 fragments.
Figure S3 Intermolecular NOEs between SURP1 and S1BR, and comparison of SURP1 structures between free and complex forms.
Figure S4 Comparisons of [1H,15N]‐HSQC spectra between individual SURP1 in free and complex forms, and those between SURP1 of the chimera and individual SURP1 in a complex form.
Figure S5 Equilibrium analysis of binding responses for interactions of SURP1 or mutants to S1BRp using BLI.
Figure S6 A unique topology of α1 and α2 characterized by the G bend.
Figure S7 Comparison of structures among human SURP domains in terms of the G bend.
Data Availability Statement
The atomic coordinates for the ensemble of 20 energy‐refined NMR conformers, representing the solution structure of the SURP1‐Linker‐S1BRp protein, were deposited in the Protein Data Bank under accession code 7VH9. Chemical shift assignments for the SURP1‐Linker‐S1BRp protein were deposited in the BMRB database under accession number 36445.