

Abstract
The evident genetic, pathological, and clinical heterogeneity of Alzheimer’s disease (AD) poses challenges for traditional drug development. We conducted a computational drug repurposing screen for drugs to treat apolipoprotein (apo) E4-related AD. We first established apoE-genotype-dependent transcriptomic signatures of AD by analyzing publicly-available human brain database. We then queried these signatures against the Connectivity Map database containing transcriptomic perturbations of >1300 drugs to identify those that best reverse apoE-genotype-specific AD signatures. Bumetanide was identified as a top drug for apoE4 AD. Bumetanide treatment of apoE4 mice without or with Aβ accumulation rescued electrophysiological, pathological, or cognitive deficits. Single-nucleus RNA-sequencing revealed transcriptomic reversal of AD signatures in specific cell types in these mice, a finding confirmed in apoE4-iPSC-derived neurons. In humans, bumetanide exposure was associated with a significantly lower AD prevalence in individuals over the age of 65 in two electronic health record databases, suggesting effectiveness of bumetanide in preventing AD.
Introduction
Alzheimer’s disease (AD) is the leading cause of dementia worldwide, and no effective therapies are available1,2. The evident genetic, pathological, and clinical heterogeneity amongst AD patients poses challenges for traditional drug development, with almost all efforts to target individual AD-related pathways having failed in late human trials1,2. These failures together suggest that developing one drug to treat all AD patients may be impossible. Rather, a precision medicine approach, which allows for stratification guided by known disease-associated gene mutations or polymorphisms, might represent a plausible alternative, with the goal of identifying drugs effective for more homogenous subpopulations of AD patients.
The apolipoprotein (apo) E4 genotype, which is the greatest genetic risk factor for late onset AD, incurs a 3–4-fold risk of AD for heterozygote carriers (~25% of the population) and a 12–14-fold risk for homozygotes (~2% of the population) as compared to apoE3 homozygotes2–6. It has been well documented that apoE3 and apoE4 play different roles in AD pathogenesis2–6 and AD patients with different apoE genotypes respond differently to drug treatment7–9. Thus, the apoE genotype represents a unique and untapped genetic guidance tool for precision medicine in AD drug development.
Using a drug repurposing approach to target AD could lower the costs and shorten the drug discovery process by searching amongst well-tolerated drugs with known targets. We utilize a computational drug repurposing algorithm built on the well-validated hypothesis that drugs which reverse or “flip” differentially expressed (DE) genes in a disease state back towards normal levels may be efficacious against the disease10–15. Within this framework for AD drug repurposing, we established apoE-genotype-dependent transcriptomic signatures of AD and then applied the computational drug-repurposing algorithm10,13 to query the Connectivity Map (CMap)16 — a database of transcriptomic perturbation signatures derived from >1300 existing drugs. Each compound receives a prediction score for therapeutic potential in apoE-genotype-dependent AD12,13, where a negative score suggests that a compound might reverse the transcriptomic signature of the disease. Since transcriptomic signatures of AD are compared with those perturbed by each compound, the prediction strategy is driven by the entire DE gene network rather than by isolated receptors, molecules, or pathways, as usually guided in traditional drug development programs. The molecular signature of aging in apoE4 knock-in (apoE4-KI) mice, a mouse model used for studying apoE4 effects in aging and late-onset AD, was used as further validation for predicted efficacy of CMap drugs against apoE4-driven pathophysiology in aging mammalian brain. With this precision drug repurposing approach, one of the top-predicted drugs for apoE4 AD, the loop-diuretic bumetanide, was identified and tested in aged apoE4-KI mice without Aβ accumulation and in J20/E4-KI mice with Aβ accumulation, mouse models of apoE4-driven AD, and its efficacy against physiological, pathological, and behavioral symptoms was validated. The mechanism of action of bumetanide was interrogated via single-nucleus RNA sequencing (snRNA-seq) analysis of the hippocampus of aged apoE4-KI mice and J20/E4-KI mice, where the drug’s predicted transcriptional effects were evident. Furthermore, comparing mouse snRNA-seq data with the transcriptomic perturbation signature of bumetanide in induced pluripotent stem cell (iPSC)-derived human neurons with an apoE4/4 genotype helped to identify translatable mechanisms of action for bumetanide in treating apoE4 AD, warranting further investigation. Importantly, in individuals over 65 years of age, bumetanide exposure was associated with a significantly lower AD prevalence in two independent clinical cohorts containing data on millions of people via electronic health record (EHR) databases, suggesting the potential effectiveness of bumetanide in preventing AD and warranting its further tests in prospective human clinical trials.
Results
ApoE-genotype-dependent transcriptomic signatures of AD
To establish apoE-genotype-dependent transcriptomic signatures of AD, we analyzed the only publicly-available human temporal lobar transcriptomic dataset with AD patients, non-demented controls, and apoE genotype information (GEO accession GSE15222; n = 213, with an n > 3 of non-demented apoE4/4 homozygotes )17 (Fig. 1a and Supplementary Table 1). As in most clinical studies18, the AD group had proportionately more apoE3/4 and apoE4/4 carriers than the control group (Fig. 1b, Chi-Squared test, P < 0.001). The first principal component of the data was significantly correlated with both diagnosis and apoE genotype status, emphasizing the collinearity of these two covariates and underscoring the distinct need for apoE-genotype-specific stratification (Fig. 1c, d). The dataset was then stratified by apoE genotype (Fig. 1a and Supplementary Table 1) and differential expression (DE) of genes was evaluated on each subset of patients (see Methods for details)19. Data were down-sampled in 10 independent permutations to ensure sex-matching (see Methods for details), as sex is also correlated with apoE genotype status in this dataset. Genes with average absolute estimated log fold change (logFC) > 0.4 at P < 0.05 in all ten permutations were used for further analysis (Supplementary Table 1). Age effect was evaluated by ANOVA, which did not reveal a significant difference among various apoE genotype groups. At the selected P-value and fold change thresholds, comparison with apoE-genotype-matched controls resulted in 539, 295, and 1079 DE genes in AD subjects with an apoE4/4, apoE3/4, and apoE3/3 genotype, respectively (Fig. 1e, f, and Supplementary Tables 2–5). Strikingly, only 108 DE genes (5.6% of all DE genes) were shared among all three AD groups (Fig. 1e, f, and Supplementary Tables 2–5), highlighting the differential etiology, at a transcriptomic level, of each apoE genotype in AD pathogenesis.
Fig. 1. ApoE-genotype-dependent transcriptomic signatures of AD.
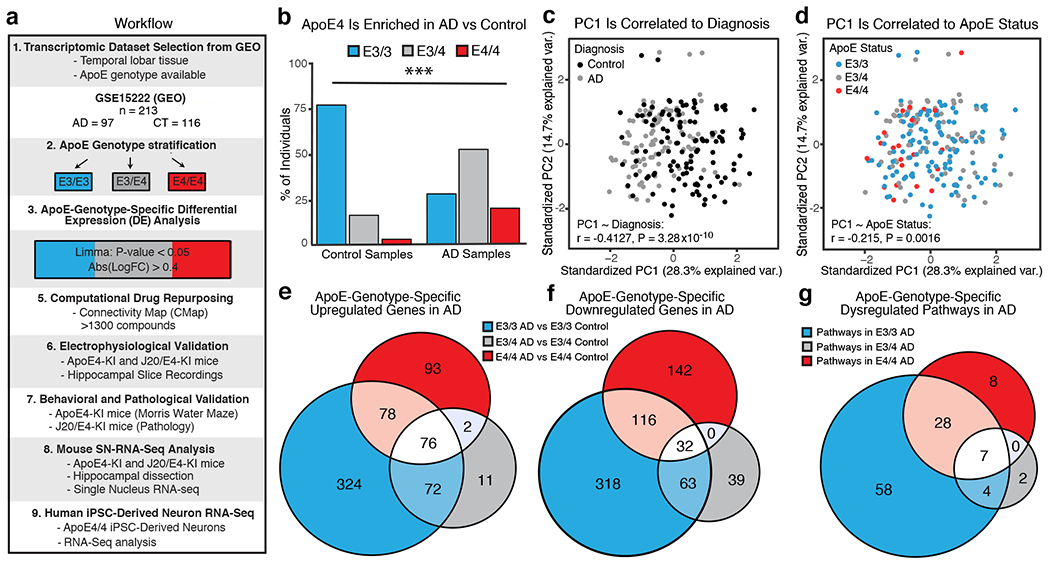
a, Experimental workflow, including dataset selection, apoE genotype stratification, analysis of DE genes, drug repurposing analysis, efficacy validation in apoE4-KI and J20/E4-KI mice, and transcriptomic validation in apoE4-KI and J20/E4-KI mice and apoE4/4-iPSC-derived human neurons. b, ApoE genotype composition of AD (n = 97 individuals) and control (n = 116 individuals) dataset of GSE15222 (total n = 213 individuals). ApoE4 carrier representation (apoE3/4 and apoE4/4) was greater in AD groups (χ2 test of apoE4 carriers versus non-carriers in AD versus control populations, two-sided χ2 = 52.236, df = 2, unadjusted p-value = 4.541e−12). c, PCA of the temporal lobe transcriptomic data showed the first principal component (PC1) is correlated with the diagnosis covariate (two sided Pearson’s correlation, Pearson’s r = −0.4127, unadjusted P = 3.28x10−10). d, PCA of the transcriptomic data showed the first principal component (PC1) is correlated with the apoE genotype covariate (two sided Pearson’s correlation, Pearson’s r = −0.215, unadjusted P = 0.0016). e, f, Venn diagram of overlapping and uniquely upregulated (e) and downregulated (f) DE genes (estimated absolute logFC > 0.4 across 10 sex-matched permutations, P-value < 0.05 in all 10 sex matched permutations) from apoE-genotype-specific DE analysis. 93 genes were uniquely significantly upregulated in apoE4/4 AD, 11 in apoE3/4 AD, and 324 in apoE3/3 AD. Only 76 DE genes were shared across these groups. 142 genes were uniquely significantly downregulated in apoE4/4 AD, 39 in apoE3/4 AD, and 318 in apoE3/3 AD. Only 32 DE genes were shared across all three AD groups. g, Venn diagram of shared and uniquely significantly enriched ontological pathways across apoE-genotype-specific up and down regulated gene groups. Eight pathways were uniquely significantly enriched in apoE4/4 AD, 2 in apoE3/4 AD, and 58 in apoE3/3 AD. Only 7 pathway were shared across all three AD groups.
KEGG pathway analysis of these DE genes identified 43, 13, and 97 perturbed pathways in apoE4/4-, apoE3/4-, and apoE3/3-specific signatures of AD, respectively (Fig. 1g and Supplementary Table 6). Seven pathways were shared among all three AD groups (Fig. 1g and Supplementary Table 6). 8, 2, and 58 pathways were unique to apoE4/4-, apoE3/4-, and apoE3/3-specific signatures of AD (Fig. 1g and Supplementary Table 6). Thus, there are both similarities and differences at the pathway level between the genetically distinct molecular milieus of AD.
Bumetanide is identified as a top predicted drug for apoE4/4 AD
Next, we queried the apoE-genotype-specific transcriptomic signatures of AD against the CMap database to identify potential therapeutic predictions13,16. Previous analyses in studies of cancer drug repurposing indicate that, within the CMap database, compounds found to more dramatically “flip” the transcriptomic signature of the cancer back towards a normal state were more likely to be effective in clinical studies20. We therefore calculated the CMap score of the “flip” for all compounds in the CMap database against the apoE-genotype-specific transcriptomic signatures of AD (Fig. 2a–c, see Methods for details). Of the top five compounds identified as potential therapeutics against apoE4/4 AD, we applied a literature-based search for general pharmacological information and potential mechanism of action to identify one compound to be further evaluated against apoE4/4 AD. The FDA-approved loop-diuretic bumetanide stood out as the highest-ranked drug (Fig. 2a), which had also been investigated for other brain disorders, such as seizure, autism, depression, and schizophrenia, suggesting brain penetration and potential effectiveness in the CNS21–27. We therefore focused our further analysis on the efficacy of bumetanide in apoE4/4-mediated AD.
Fig. 2. ApoE-genotype-dependent drug repurposing analysis identifes bumetanide as a top predicted drug candidate for apoE4 AD.
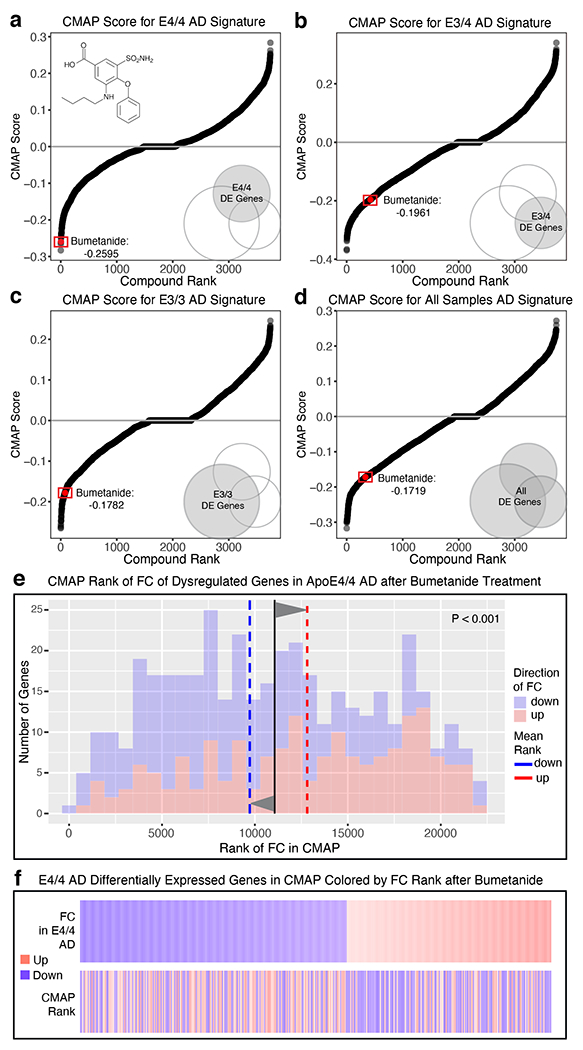
a–d, Graphs of compounds ordered by CMap score against (a) apoE4/4 AD, (b) apoE3/4 AD, (c) apoE3/3 AD, and (d) all AD transcriptomic signatures (see Methods for details). The transcriptomic effects of bumetanide showed apoE4/4 genotype preference (ranked number 4), with a stepwise weaker CMap score (still a negative number toward therapeutic direction) against apoE3/4 AD, apoE3/3 AD, and AD status not controlling for apoE genotype. e, Histogram of the rank of FC of the human apoE4/4-specific transcriptomic signature of AD genes which were also measured in the CMap database after bumetanide treatment. The mean rank of all genes in this gene set is denoted by the black line, the average mean FC rank of up-regulated genes (colored red in histogram) is denoted by the dashed red line and the mean FC rank of the down-regulated genes (colored blue in histogram) is denoted by the blue dashed line. P-value of significance of the “flip” of up- and down-regulated FC rank means away from the rank mean of all genes as calculated by Monte-Carlo simulation is shown (P < 0.001). f, Heatmap of genes from apoE4/4-specific transcriptomic signature of AD, rank ordered and color coded (red for up, blue for down) by estimated FC in human apoE4/4 AD (top) and then re-color-coded by FC rank after bumetanide treatment in the CMap database. Bumetanide flips the expression rank of both up- and down-regulated genes in the human apoE4/4-specific transcriptomic signature of AD.
In bumetanide-treated cells in the CMap database, genes upregulated in apoE4/4 AD were shifted downward, and those downregulated in apoE4/4 AD were shifted upward (P < 0.001 by Monte Carlo Simulation) (Fig. 2e, f). The transcriptomic effects of bumetanide also showed apoE4 genotype preference, with a stepwise weaker CMap score against apoE3/4 AD, apoE3/3 AD, and AD status not controlling for apoE genotype (Fig. 2b–d). However, the overall negative CMap scores suggest that it might also work for apoE3 AD or AD in general at least to some extent.
Bumetanide is predicted to reverse the transcriptomic signature of brain aging in apoE4-KI mice
To be able to test bumetanide’s effects in a mouse model of apoE4-related AD, we analyzed whether bumetanide is also predicted to ameliorate the transcriptomic signature of brain aging in apoE4-KI mice using a publicly available bulk RNA-seq dataset (https://doi.org/10.7303/syn20808171)28. We first analyzed the transcriptomic differences in the cerebral cortex of 12 or 24 versus 3 month-old apoE4-KI female mice. PCA plots of all genes showed clustering between age groups with no distinct outliers (Extended Data Fig. 1a). There were 64 DE genes and 4 DE pathways in common between 12 versus 3 month-old and 24 versus 3 month-old apoE4-KI mouse brains (Extended Data Fig. 1b and Supplementary Tables 7, 8). Analysis of CMap score distribution revealed that bumetanide was in the most efficacious 7th (score: −0.5306) and 8th (score: −0.6217) percentile of drugs predicted against transcriptomic signature of brain aging in 12 versus 3 month-old and 24 versus 3 month-old apoE4-KI mice, respectively (Extended Data Fig. 1c, d). In bumetanide-treated cells in the CMap database, genes upregulated in aged apoE4-KI mouse brains were shifted downward, and those downregulated in aged apoE4-KI mouse brains were shifted upward (Extended Data Fig. 1e, f). This suggests that bumetanide may be efficacious against pathological phenotypes of brain aging in apoE4-KI mice.
Bumetanide treatment rescues neuronal excitability and plasticity deficits in apoE4-KI mice
We then validated the effects of bumetanide treatment on apoE4-induced neuronal excitability and plasticity deficits in 16-month-old female apoE4-KI mice. We first confirmed the known phenotype of hyperexcitability in apoE4-KI mice29, measured by input-output curve analysis30, in the hippocampal CA1 region of aged apoE4-KI mice as compared to age-matched apoE3-KI mice (Fig. 3a). Chronic treatment with bumetanide (0.2 mg/kg, daily intraperitoneal injection for 8 weeks) rescued this pathophysiology (Fig. 3a). We also found that capacity for long term potentiation (LTP) was impaired in these aged apoE4-KI mice compared to apoE3-KI controls (Fig. 3b, c). LTP is an electrophysiological measurement of neuronal plasticity, which is critical for normal memory formation31 and is impaired in animal models of AD32. Strikingly, bumetanide treatment fully rescued the LTP deficit in aged apoE4-KI mice (Fig. 3b, c). Thus, in vivo bumetanide treatment restored normal neuronal excitability and plasticity in the hippocampus of aged apoE4-KI mice.
Fig. 3. Bumetanide treatment rescues AD-like neuronal excitability and plasticity deficits as well as learning impairment in aged apoE4-KI mice.
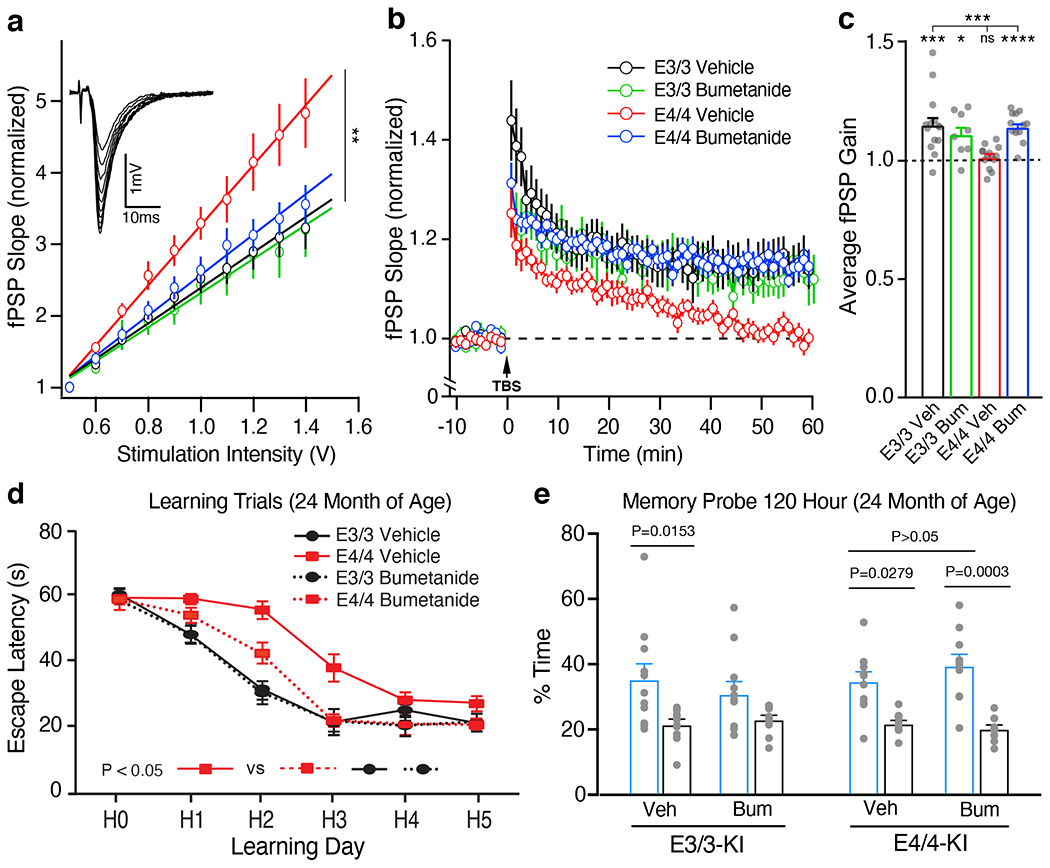
a, Female apoE3-KI and apoE4-KI mice were treated with bumetanide (0.2 mg/kg, daily intraperitoneal injection) for 8 weeks. Input-output relationships in Schaeffer collaterals-CA1 network (inset: example traces) were measured on ex-vivo hippocampal slices from vehicle-treated apoE3-KI (n = 23 slices from 7 mice), bumetanide-treated apoE3-KI (n = 10 slices from 3 mice), vehicle-treated apoE4-KI (n = 11 slices from 3 mice), and bumetanide-treated apoE4-KI (n = 12 slices from 3 mice) mice at 17 months of age. The average fPSP slope values were compared among different groups (Kruskal-Wallis test, P = 0.0011). b, c, LTP was measured on hippocampal slices from vehicle-treated apoE3-KI (n = 14 slices from 6 mice), bumetanide-treated apoE3-KI (n = 8 slices from 3 mice), vehicle-treated apoE4-KI (n = 14 slices from 6 mice), and bumetanide-treated apoE4-KI (n = 12 slices from 3 mice) mice at 17 months of age. Average fPSP slope values were binned to one-minute intervals and normalized to control (b). LTP gain outcomes were summarized as compared to pre-TBS baseline, paired two-sided t test (c). Vehicle-treated (P = 0.0007) and bumetanide-treated (P = 0.0125) apoE3-KI slices and bumetanide-treated apoE4-KI slices (P < 0.0001) showed significant gain while vehicle-treated apoE4-KI slices did not. One way ANOVA, P = 0.0008. d, Escape latency of bumetanide- and vehicle-treated apoE3-KI and apoE4-KI mice (n = 11 for each group) at 24 months of age. One way repeated-measures ANOVA, P = 0.0037 between treatment groups; Tukey’s multiple conparisons test, P = 0.0345 bumetanide-treated apoE4-KI versus vehicle-treated apoE4-KI groups. e, In the 120-hour probe trial, both vehicle-treated (n = 11) and bumetanide-treated (n = 10) apoE4-KI mice as well as vehicle-treated apoE3-KI mice (n = 11), but not bumetanide-treated apoE3-KI mice (n = 11), demonstrated a significant preference for the target quadrant (one way ANOVA p < 0.0001, with Tukey’s multiple comparisons test). Values are mean ± SEM in a-e. * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001 in a-c.
Bumetanide treatment rescues spatial learning deficit in apoE4-KI mice
We then examined the effects of bumetanide treatment (0.2 mg/kg, daily intraperitoneal injection for 8 weeks) on cognitive deficit in 22-month-old female apoE4-KI mice. We used the Morris water maze (MWM)33–35 to test spatial learning over 5 days in hidden platform trials followed by probe trials testing memory. Learning curve analysis showed that vehicle-treated apoE4-KI mice learnt significantly slower than vehicle-treated apoE3-KI mice (Fig. 3d). Bumetanide treatment significantly improved the learning performance of apoE4-KI mice to a level similar to vehicle-treated apoE3-KI mice, while bumetanide treatment had no significant effect on apoE3-KI mice (Fig. 3d). Importantly, bumetanide treatment had no significant effect on swim speed during the hidden trial and did not alter the performance during the visible trials (Extended Data Fig. 2a, b). Likewise, bumetanide treatment also did not significantly alter the learning curve and swim speed of wildtype mice (Extended Data Fig. 2c, d). Furthermore, bumetanide treatment had no significant effect on memory performance in probe trials for either apoE3-KI, apoE4-KI, or wildtype mice (Fig. 3e and Extended Data Fig. 2e, f), probably due to their already good performance on vehicle treatment in this cohort. However, bumetanide treatment appeared to worsen the memory performance of apoE3-KI mice (Fig. 3e). Taken together, these data demonstrate that bumetanide treatment restored normal spatial learning in aged apoE4-KI mice.
Bumetanide treatment flips human apoE4/4 AD transcriptomic signature genes in specific neuron subtypes in apoE4-KI mice
To explore bumetanide’s effects on the transcriptome of apoE4-KI mice in vivo, we performed a snRNA-seq of the hippocampus, a temporal lobe region considered the epicenter of AD pathologies2, from aged apoE4-KI mice treated with vehicle or bumetanide (0.2 mg/kg, daily intraperitoneal injection) for 8 weeks (Fig. 4a). We identified 18 distinct cell clusters of cells that were further analyzed for cell-type specific drug effects (Fig. 4b, c, Extended Data Fig. 3, and Supplementary Table 9). In 12 out of 18 cell types, including all excitatory and mixed neuronal clusters as well as SST/PV interneurons, genes upregulated in apoE4 AD were shifted downward and those downregulated in apoE4 AD were shifted upward after bumetanide treatment (P < 0.05 by Monte Carlo Simulation), confirming that the transcriptomic perturbation signature of bumetanide correlates negatively with that of human apoE4 AD in these neuronal subtypes in aged apoE4-KI mouse hippocampus (Fig. 4d, e, Extended Data Fig. 4a–c, 4e, 4f, 4i, 4k, and 4l, and Supplementary Table 10). When data from all the neuronal subtypes that exhibited a significant “flip” were combined, a significant reversal of apoE4 AD signature genes is clear (P < 0.001 by Monte Carlo Simulation, Fig. 4e and Extended Data Fig. 4o). This shift corroborates the CMap based prediction and the hypothesis that reversal of the disease-specific transcriptomic signature is a rational strategy for computational drug repurposing, even in animal models. The shift was seen across all excitatory neuronal cell types and SST/PV interneurons with various DE gene numbers (P < 0.05, Fig. 4f), suggesting that the drug effect is robust across neuronal cell type regardless of the overall number of affected genes. However, this shift was notably absent from other inhibitory neuron subtypes and almost all non-neuronal cells, including astrocytes and oligodendrocytes, in the hippocampus of apoE4-KI mice treated with bumetanide (Fig. 4f and Extended Data Fig. 4d, 4g, 4h, 4j, 4m, and 4n), suggesting cell-type-selective effects of bumetanide on apoE4 AD signature genes in aged apoE4-KI mouse hippocampus.
Fig. 4. snRNA-seq analysis of the transcriptomic perturbation signature of bumetanide in the hippocampus of aged apoE4-KI mice.
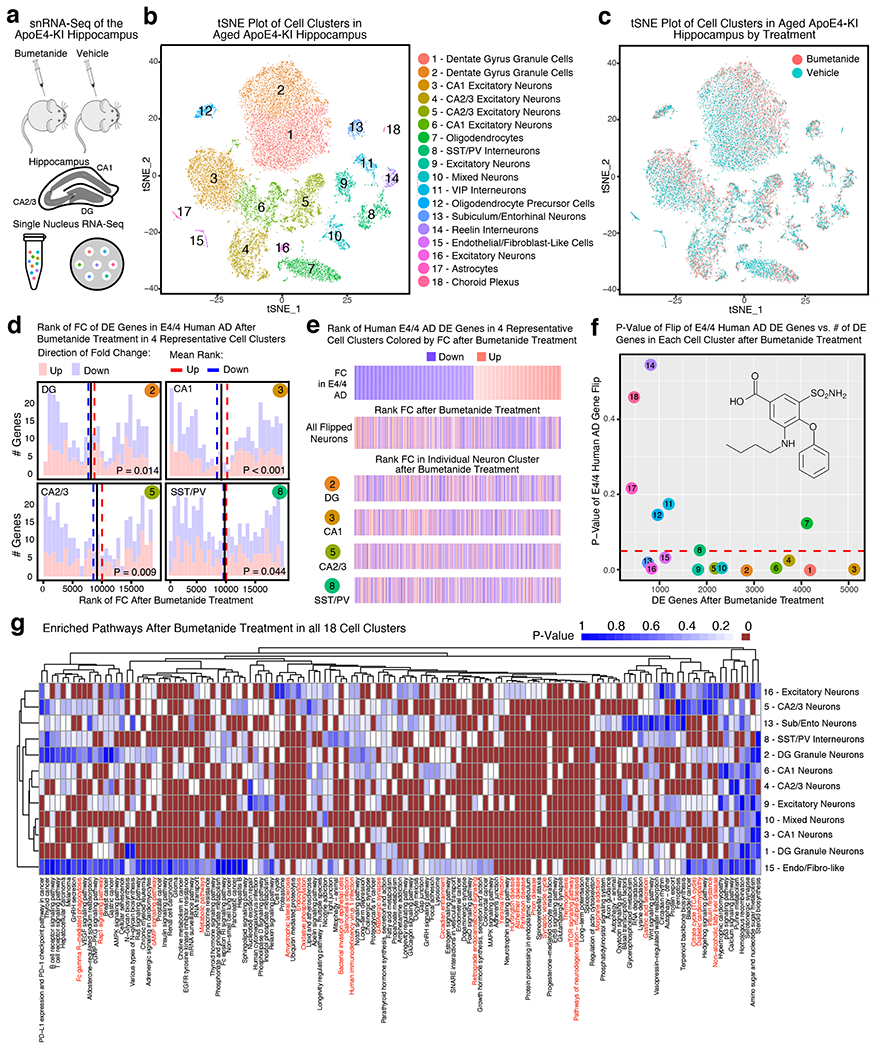
a, Transcripts in ~27,000 single nuclei from the hippocampus of bumetanide- and vehicle-treated female apoE4-KI mice at 17 months (n = 3 per group, 8-week treatment) were sequenced. b, Clustering and visualization by t-SNE identifies 18 distinct cell clusters. c, Cell clusters color-coded by treatment groups. d, Histogram of the rank of FC of apoE4/4-specific transcriptomic signature of AD genes that were also detected by snRNA-seq in vehicle- and bumetanide-treated apoE4-KI mouse hippocampus in four representative cell clusters. The mean rank of all genes in this gene set is denoted by the black line. The average mean FC ranks of up-regulated (red histogram) and down-regulated (blue histogram) genes are denoted by the red and blue dashed lines, respectively. P-value of the “flip” of up- and down-regulated FC rank means away from the rank mean of all genes as calculated by Monte-Carlo simulation is shown (unadjusted P < 0.05 considered significant). e, Heatmap of genes from apoE4/4-specific transcriptomic signature of AD were rank ordered and color coded by estimated FC in human apoE4/4 AD (top) and then re-color-coded by FC rank after bumetanide treatment in combined neuron types exhibiting a significant “flip” of apoE4/4 AD signature genes (clusters 1, 2, 3, 4, 5, 6, 8, 9, 10, 13, 16 combined) and in representative neuron types (clusters 2, 3, 5, 8) in apoE4-KI hippocampus. f, P-value of the “flip” of apoE4/4-specific transcriptomic signatures of AD (y-axis), as calculated by Monte-Carlo simulation, is plotted versus number of DE genes in each cell cluster (x-axis). The red dashed line denotes P = 0.05. g, Heatmap of P-values of enriched pathways for all DE genes from each cell cluster after bumetanide treatment revealed 135 pathways that were affected in at least one of the cell types that had a significant “flip” of human apoE4/4 AD signature genes (unadjusted P < 0.005 by the bespoke enrichment method employed by the kegga function (limma v 3.36.5)). 28 pathways highlighted in red are those shared with apoE4/4-specific signature pathways of AD (P = 0.001 by Monte Carlo Simulation, see Figure 1g and Supplementary Table 6 for human pathways).
The fold change size and directionality of all DE genes after bumetanide treatment in the five major excitatory neuronal cell types in aged apoE4-KI mouse hippocampus (Extended Data Fig. 5a) mimicked the fold change size and directionality after bumetanide treatment in PC3 cells in the CMap database (Extended Data Figure 5b–g). Because of these similarities, bumetanide was in the top 90th percentile of all the CMap scores using DE genes from these cell types in bumetanide-treated apoE4-KI mouse hippocampus, further supporting the finding that the predicted effect of bumetanide in the CMap database is in fact recapitulated in major neuronal cell types in the apoE4-KI mouse hippocampus treated with bumetanide (Extended Data Fig. 5a).
Investigation of enriched pathways in all DE genes from each cell cluster after bumetanide treatment revealed 135 pathways that were enriched in at least one of the cell types that had a significant “flip” of human apoE4 AD signature genes (P < 0.005, Fig. 4g), 28 of which overlapped with those enriched in human apoE4/4 AD signature pathways (overlap enrichment P = 0.001 by Monte Carlo Simulation, highlighted in red in Fig. 4g and Supplementary Table 11).
Bumetanide treatment flips apoE4-mediated transcriptomic signature of brain aging in specific neuron subtypes in apoE4-KI mice
Overlapping upregulated DE genes from 24 vs 3 month-old apoE4-KI mouse brains that were also detected in cell types 1–18 in our apoE4-KI snRNA-seq data were analyzed for whether they were downregulated on average, and P-value of this “flip” was calculated via Monto Carlo simulation. All of the 18 cell types in aged apoE4-KI mouse hippocampus had some overlapping genes that were upregulated (119 genes) in 24 vs 3 month-old apoE4-KI mouse brains, although no cell types had any of the three downregulated genes (Extended Data Fig. 6a, b). Cell types 1, 2 and 4 (DG granule cells and CA2/3 neurons) had a significant flip of these upregulated genes to, on average, a downregulated state (Extended Data Fig. 6c–f). Cell types 3, 5, and 6 as well as cluster 13 trend towards having this “flip” of upregulated genes to a downregulated state as well (Extended Data Fig. 6c). Cell types 1–6 and 13 also experienced a “flip” of DE genes in human apoE4/4 AD, further corroborating the evidence that all major excitatory neuronal clusters in aged apoE4-KI mouse hippocampus may experience a “flip” of both aging DE genes and apoE4/4 AD DE genes in response to bumetanide treatment, as predicted by the CMap database (Extended Data Fig. 6c).
Bumetanide treatment rescues functional deficits and reduces Aβ plaques in brains of J20/E4-KI mice
Next, we validated the effect of bumetanide treatment in apoE4-KI mice expressing mutant human APP related to early onset familial AD (FAD), using mice generated from cross-breeding apoE4-KI mice with mice overexpressing APPFAD (J20 line)36. The J20/E4-KI mice develop significant Aβ plaques, starting from 6 months of age37. We treated J20/E4-KI and J20/E3-KI mice at 10 months of age with bumetanide (0.2 mg/kg, daily intraperitoneal injection) for 12 weeks. After the treatment, we first confirmed the known phenotype of hyperexcitability, electrophysiologically measured by input-output curve analysis30, in the hippocampal CA1 region of both J20/E4-KI and J20/E3-KI mice treated with vehicle (Fig. 5a). Treatment with bumetanide for 12 weeks rescued this pathophysiological phenotype specifically in J20/E4-KI mice (Fig. 5a). We also found that the capacity for LTP was impaired in both J20/E4-KI and J20/E3-KI mice treated with vehicle (Fig. 5b, c). Strikingly, bumetanide treatment rescued the LTP deficit in both J20/E4-KI and J20/E3-KI mice (Fig. 5b, c).
Fig. 5. Bumetanide treatment rescues AD-like neuronal excitability and plasticity deficits and reduces Aβ plaque loads in the hippocampus and cortex in J20/E4-KI mice.
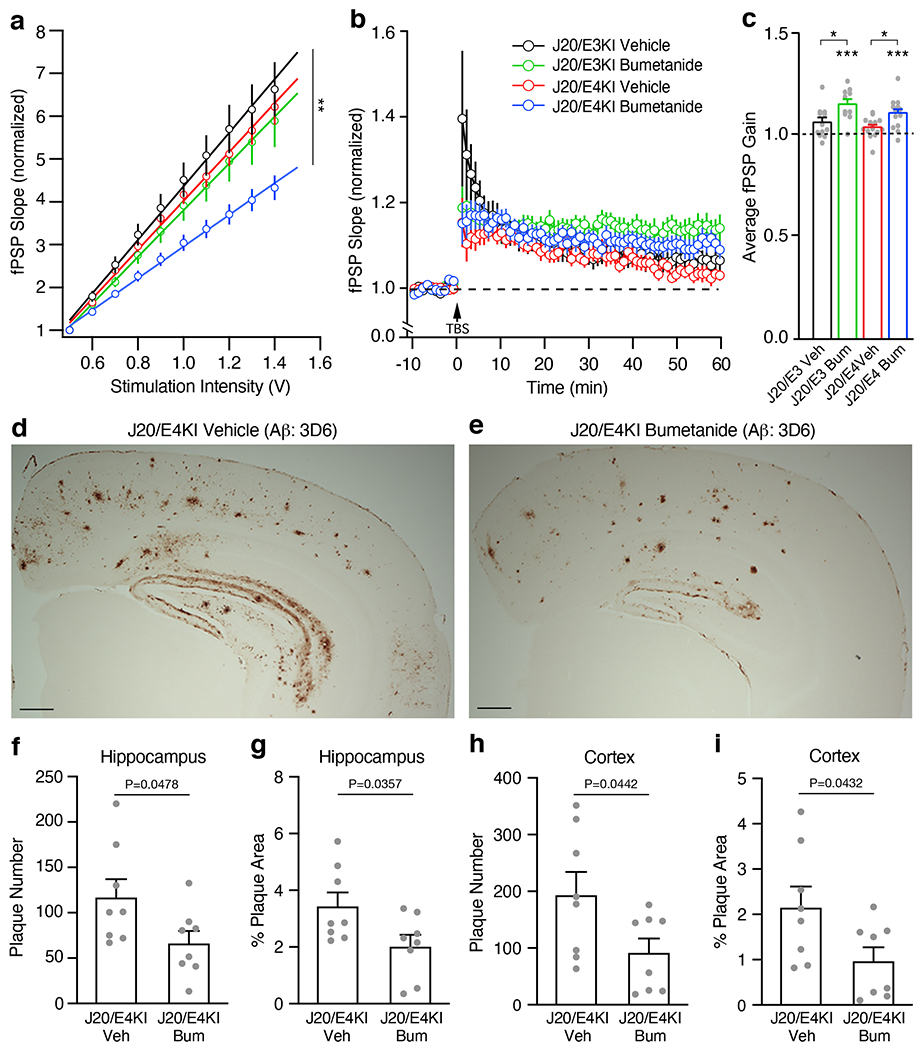
a, Female J20/E-KI mice were treated with bumetanide (0.2 mg/kg, daily intraperitoneal injection) for 12 weeks. Input-output relationships in Schaeffer collaterals-CA1 network were measured on ex-vivo hippocampal slices from vehicle-treated J20/E3-KI (n = 15 slices from 5 mice), bumetanide-treated J20/E3-KI (n = 21 slices from 7 mice), vehicle-treated J20/E4-KI (n = 18 slices from 5 mice), and bumetanide-treated J20/E4-KI (n = 27 slices from 7 mice) mice at 13 months of age. The average fPSP slope values were compared by one way ANOVA (P = 0.0023), with Tukey’s multiple comparisons test P = 0.0205 for bumetanide-treated J20/E4-KI mice versus vehicle-treated J20/E4-KI mice and P = 0.0033 for bumetanide-treated J20/E4-KI mice versus vehicle-treated J20/E3-KI mice. b, c, LTP was measured on ex-vivo hippocampal slices from vehicle-treated J20/E3-KI (n = 12 slices from 5 mice), bumetanide-treated J20/E3-KI (n = 11 slices from 5 mice), vehicle-treated J20/E4-KI (n = 15 slices from 5 mice), and bumetanide-treated J20/E4-KI (n = 15 slices from 5 mice) mice at 13 months of age. Average fPSP slope values were binned to one-minute intervals and normalized to control (b). LTP gain outcomes were summarized as compared to pre-TBS baseline, paired two-sided t test (c). Bumetanide treatment resulted in a significant increase in LTP gain outcome for both treated J20/E3-KI (P = 0.041 versus vehicle-treated J20/E3-KI) and J20/E4-KI (P = 0.0446 versus vehicle-treated J20/E4-KI) mice. d, e, Representative images of Aβ immunostaining from vehicle-treated (d) and bumetanide-treated (e) J20/E4-KI mice at 13 months of age (n = 8 per group, 12-week treatment). Scale bars = 300 μm. f–i, Quantifications of Aβ plaque number (f, h) and area (g, i) in the hippocampus (f, g) and the cortex (h, i) from vehicle-treated or bumetanide-treated J20/E4-KI mice at 13 months (n = 8 per group, 12-week treatment). Unpaired and two-sided t test in f-i. Values are mean ± SEM in a-c and f-i. * P < 0.05; ** P < 0.01; *** P < 0.001 in a-c.
Anti-Aβ immunostaining with the 3D6 monoclonal antibody revealed marked Aβ deposition and amyloid plaque loads in the hippocampus and the cortex of J20/E4-KI mice (Fig. 5d). Strikingly, bumetanide treatment for 12 weeks significantly reduced Aβ plaque numbers and covered areas in both the hippocampus and the cortex of J20/E4-KI mice (Fig. 5e–i). Taken together, these data demonstrated that bumetanide treatment restored normal neuronal excitability and plasticity and reduced Aβ plaque loads in J20/E4-KI mice.
Bumetanide treatment flips human apoE4/4 AD transcriptomic signature genes in neurons and glia in J20/E4-KI mice
To explore bumetanide’s effects on the transcriptome in the presence of both apoE4 and Aβ accumulation, we performed snRNA-seq analysis of the hippocampus from J20/E4-KI mice treated with vehicle or bumetanide (0.2 mg/kg, daily intraperitoneal injection) for 12 weeks. 25 distinct cell clusters were identified and analyzed for cell-type specific drug effects (Extended Data Fig. 7, Extended Data Fig. 8a–c, Supplementary Table 12). In 7 out of 25 cell types, including DG granule cells (cluster 1), subiculum neurons (cluster 5), OPCs (cluster 6), mixed neurons/oligodendrocytes (cluster 9), microglia (cluster 10), astrocytes (cluster 17), and fibroblast-like cells (cluster 20), genes upregulated in apoE4/4 AD were shifted downward and those downregulated in apoE4/4 AD were shifted upward after bumetanide treatment (P < 0.05 by Monte Carlo Simulation) (Extended Data Fig. 8d–f and Supplementary Table 13), confirming that the transcriptomic perturbation signature of bumetanide correlates negatively with that of human apoE4/4 AD in these six subtypes of cells in J20/E4-KI mouse hippocampus.
DE gene pathway analysis revealed 37 pathways that were enriched in at least one of the cell types that had a significant “flip” of human apoE4/4 AD signature genes (Extended Data Fig. 8g and Supplementary Table 14), seven of which overlapped with those enriched in human apoE4/4 AD signature pathways (overlap enrichment P = 0.001 by Monte Carlo Simulation, highlighted in red in Extended Data Fig. 8g).
Bumetanide treatment flips apoE4/4 AD transcriptomic signature genes in apoE4/4-iPSC-derived human neurons.
To evaluate the transcriptional effect of bumetanide on human neurons, we treated apoE4/4-iPSC-derived human neurons (~79% excitatory neurons, ~16% SST/PV inhibitory neurons, and ~5% dopaminergic neurons)38 with bumetanide (10 μM) in culture for 6 hours and then interrogated transcriptional changes by RNA-seq (Fig. 6a). PCA of the top 500 most variable genes did not show clear clustering of samples (Fig. 6b and Supplementary Table 15); however, PCA of the top DE genes from bumetanide versus vehicle treatment did show a distinct clustering pattern between bumetanide and vehicle treated samples (Fig. 6c and Supplementary Table 15). Furthermore, in bumetanide-treated apoE4/4-iPSC-derived human neurons, genes upregulated in apoE4/4 AD were shifted downward and those downregulated in apoE4/4 AD were shifted upward after bumetanide treatment (P < 0.05 by Monte Carlo Simulation) (Fig. 6d, e and Supplementary Table 15), mimicking the neuronal subtype data of snRNA-seq from bumetanide-treated, aged apoE4-KI mouse hippocampus (Fig. 4d, e and Extended Data Fig. 4a–c, 4e, 4f, 4i, 4k, 4l, and 4o) and bumetanide-treated J20/E4-KI mouse hippocampus (Extended Data Fig. 8d, e). These data further corroborate the prediction and underscoring the suitability of human iPSC-derived neurons for further in vitro drug repurposing efforts.
Fig. 6. RNA-seq analysis of the transcriptomic perturbation signature of bumetanide in apoE4/4-iPSC-derived human neurons.
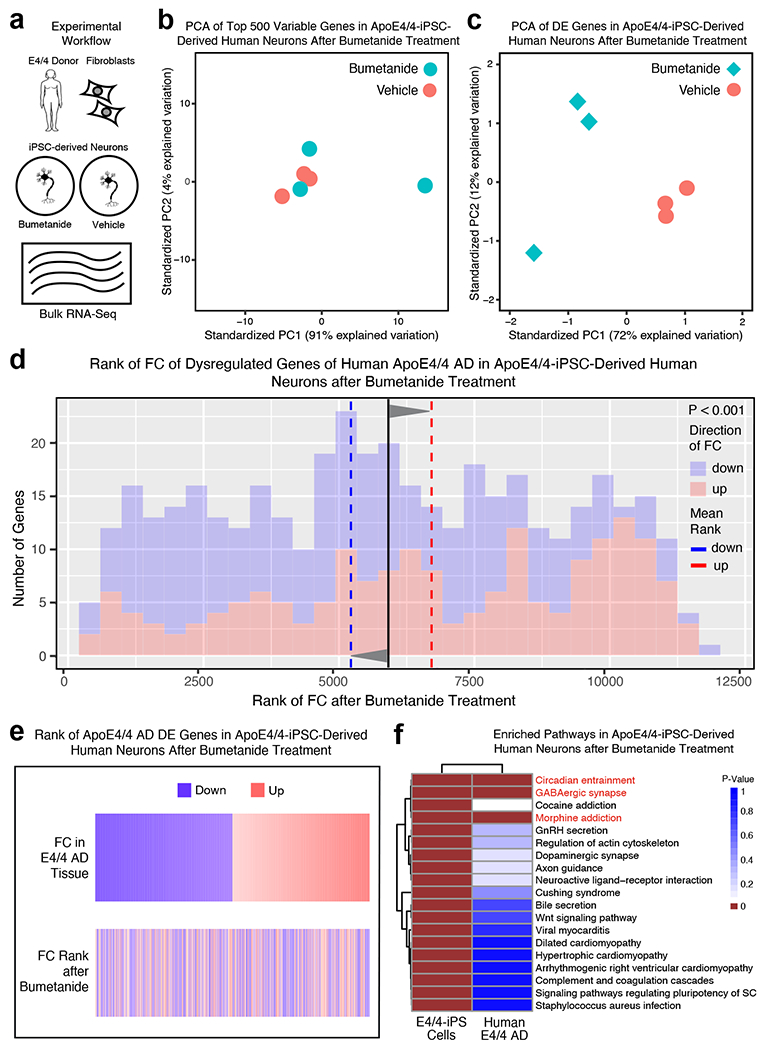
a, ApoE4/4-iPSC-derived human neurons were treated for 6 hours with 10 μM bumetanide or vehicle (n = 3 for each group) and transcriptomic changes were analyzed by RNA-seq. b, PCA of 500 most variale genes in bumetanide treated apoE4/4-iPSC-derived human neurons does not clearly separate bumetanide treated samples from vehicle treated samples. c, PCA of DE genes in bumetanide treated apoE4/4-iPSC-derived human neurons separates bumetanide treated samples from vehicle treated samples. d, Histogram of the human apoE4/4-specific transcriptomic signature of AD geneset that were also detected by RNA-seq in apoE4/4-iPSC-derived human neurons after bumetanide treatment (up and down-regulated gene signatures as denoted in Fig. 1e, f). The rank of the FC of these genes in apoE4/4-iPSC-derived human neurons following bumetanide treatment as compared to vehicle treatment is plotted on the x-axis versus number of genes at that rank on the y-axis. The mean rank of all genes in this geneset is denoted by the black line, the average mean FC rank of up-regulated genes (colored red in histogram) is denoted by the red dashed line and the mean FC rank of the down-regulated genes (colored blue in histogram) is denoted by the blue dashed line. P-value of the significance of the “flip” of up- and down-regulated FC rank means away from the rank mean of all genes as calculated by Monte-Carlo simulation is shown (P < 0.001). e, Heatmap of genes from human apoE4/4-specific transcriptomic signature of AD, rank ordered and color coded (red for up, blue for down) by estimated FC in apoE4/4 AD (top) and then re-color-coded by FC rank in apoE4/4-iPSC-derived human neurons after bumetanide treatment (bottom). f, Heatmap of the p-value of enriched ontological pathways (n = 19) in apoE4/4-iPSC-derived human neurons after bumetanide treatment and their corresponding enrichment p-values in human apoE4/4-specific AD. Pathways highlighted in red (n = 3 pathways) denote those that are shared between apoE4/4-iPSC-derived human neurons and human apoE4/4-specific AD signature pathways (see Figure 1g and Supplementary Table 6 for human pathway information).
Pathway analysis of the DE genes whose expression was affected by bumetanide in apoE4/4-iPSC-derived human neurons identified 19 significantly perturbed pathways (Fig. 6f and Supplementary Table 16). Three of these pathways were also shared with the apoE4/4 AD signature pathways based on human data: GABAergic synapse, circadian entrainment, and morphine addiction (highlighted in red in Fig. 6f and Supplementary Table 16).
Potential mechanisms of bumetanide action targeting apoE4/4 AD signature pathways
To explore the potential mechanisms of bumetanide action targeting apoE4/4 AD signature pathways, we did overlapping-enriched ontological pathway analyses. First, combination of snRNA-seq data from aged apoE4-KI and J20/E4-KI mice revealed 22 shared pathways in bumetanide-“flipped” cell clusters (Extended Data Fig. 9a and Supplementary Table 17). Then, addition of RNA-seq data from E4/4-hiPSC-derived neurons reduced the shared pathways down to 6 (Extended Data Fig. 9b and Supplementary Table 18). Finally, inclusion of human E4/4 AD signature pathways further reduced the shared pathways down to 3, which are GABAergic synapse, circadian entrapment, and morphine addiction pathways (Extended Data Fig. 9c). Taken together, these data suggest that modulation of these three pathways by bumetanide might underlie its beneficial effects against apoE4 AD, which warrants further in-depth studies in the future.
Bumetanide exposure is associated with a significantly lower AD prevalence in individuals over the age of 65.
We hypothesized that if bumetanide is efficacious against AD, we would see a decreased prevalence of AD diagnosis in individuals exposed to bumetanide compared to a matched control cohort in individuals over the age of 65. To test this hypothesis in humans, we analyzed two independent electronic health record (EHR) databases (Fig. 7a). One is an EHR database from the University of California at San Francisco (UCSF), which contains complete medical records for 1.3 million patients from outpatient, inpatient, and emergency room encounters as part of clinical operations from June 2012 through November 2019. The UCSF EHR database was filtered using the medication order table for patients on the drug of interest and found 5,526 patients who had used bumetanide (other names: Bumex or Burinex). Among them, 1,850 patients (1059 men, 57.2% and 791 women, 42.8%) were over the age of 65. The other EHR database was from the Mount Sinai Health System (MSHS), which covers 3.9 million patients with complete medical records from outpatient, inpatient, and emergency room encounters as part of clinical operations from five hospitals in Manhattan, Brooklyn, and Queens boroughs of New York City from January 2003 through February 2020. Of those patients, 806,040 had medication information. After filtering the data, 3,008 patients who had used bumetanide were found, with 1,901 patients (954 men, 50.2% and 947 women, 49.8%) being over the age of 65.
Fig. 7. Bumetanide exposure is associated with a significantly lower AD prevalence in individuals over the age of 65 in two independent EHR databases.
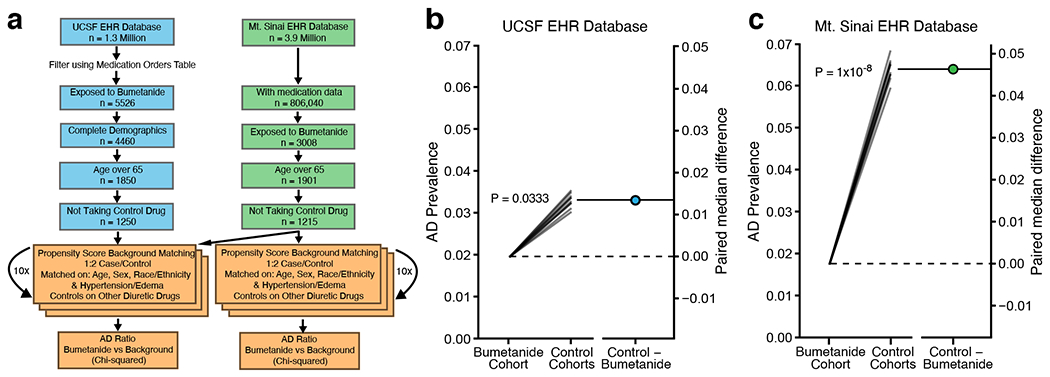
a, Workflow of evaluation of the UCSF EHR database and the Mt. Sinai EHR database for association of bumetanide exposure with AD prevalence. We evaluated two large-scale EHR databases in a cross-sectional manner to test the association of bumetanide exposure with AD prevalence in individuals with the age of 65 or above using a propensity score matching approach to control cohort creation. b, AD prevalence (the left y-axis) in bumetanide-exposed cohort is significantly lower than those in 10 randomly selected non-bumetanide-exposed cohorts controlled for non-bumetanide diuretic drug use for hypertension and edema treatment (Bootstrapped two-sided χ2(1) = 4.530, P = 0.0333, median difference = 0.0133 (95% CI: 0.0123 – 0.0142)) in the UCSF EHR database. The right y-axis represents the paired mean differences in the UCSF EHR database40. c, AD prevalence (the left y-axis) in bumetanide-exposed cohort is significantly lower than those in 10 randomly selected non-bumetanide-exposed cohorts controlled for non-bumetanide diuretic drug use for hypertension and edema treatment (Bootstrapped two-sided χ2(1)= 32.846, P = 1x10−8, median difference = 0.0463 (95% CI: 0.0445–0.0477)) in the Mt. Sinai EHR database. The right y-axis represents the paired mean differences in the Mt. Sinai EHR database.
Bumetanide is usually prescribed for treating hypertension and/or edema. Since hypertension has been identified as a risk factor for AD39, we accordingly matched 10 control cohorts, each was twice the size of the bumetanide exposed group (1:2 match), using a propensity score approach based on age, race, sex, and hypertension and edema diagnosis, out of an overall control group of 252,480 and 402,169 patients over the age of 65 not exposed to bumetanide from the UCSF EHR and the MSHS EHR databases, respectively (Fig. 7a). The control cohort of non-bumetanide-exposed individuals had a similar distribution of hypertension and edema diagnoses to the bumetanide-exposed individuals in both EHR databases (Supplementary Table 19). We then analyzed the number of AD cases in the bumetanide-exposed and control (not-bumetanide-exposed) groups and calculated AD prevalence (the ratio of AD to total patients) for both groups in the UCSF EHR and the MSHS EHR databases (Extended Data Fig. 10a, b). A χ-squared test using 10 permutations of the iterative matching of the control group revealed a significantly lower AD prevalence (~35–75% decrease) in bumetanide-exposed individuals than in non-bumetanide-exposed individuals across all 10 permutations in both EHR databases (Extended Data Fig. 10a, b).
In order to further control for hypertension treatment across the cases and controls, we chose a group of control drugs that represented a comprehensive list of non-loop diuretics with no major co-indications other than hypertension and edema, which were available in the US and were not mixed with bumetanide or a bumetanide analog (Supplementary Table 20). We then filtered our control group to only include patients taking at least one of these drugs, but not bumetanide, for hypertension or edema control. We also excluded, from the bumetanide group, the patients who were on bumetanide treatment together with any one of the non-diuretics in the list (Fig. 7a). A χ-squared test using 10 permutations of the iterative matching of the control group also revealed a significantly lower AD prevalence (~40–70% decrease) in bumetanide-exposed individuals than in non-bumetanide-exposed individuals in 8 out of 10 permutations in the UCSF EHR database and in all 10 permutations in the MSHS EHR database (Extended Data Fig. 10c, d). Bootstrapped χ-squared tests40 confirmed a significantly reduced AD prevalence in bumetanide-exposed individuals, as compared to that in non-bumetanide-exposed individuals, in both EHR databases (Fig. 7b, c). Together these data suggest that bumetanide may be effective in preventing AD in individuals over the age of 65, warranting its further tests in prospective human clinical trials.
DISCUSSION
This study represents the first attempt to apply a precision medicine approach to computational drug repurposing for AD in an apoE-genotype-directed manner. The efficacy of a top predicted drug, bumetanide, for apoE4 AD was validated in vivo in both aged apoE4-KI (without Aβ accumulation) and J20/E4-KI (with Aβ accumulation) mouse models of AD for rescue of electrophysiological, pathological, or behavioral deficits. Importantly, leveraging real world data, bumetanide exposure was associated with a significantly lower AD prevalence in individuals over the age of 65 in two independent EHR databases, suggesting the potential effectiveness of bumetanide in preventing AD in humans. It is impressive and encouraging that, even though EHR data tend to be sparse and are not collected with specific research in mind, EHR analysis across two independent databases corroborated our hypothesis generated from cellular and animal studies, suggesting that these results may translate to human patients. Further studies are warranted to determine whether the observed association between bumetanide exposure and protection against AD in humans is apoE4 genotype dependent. Given the results from our drug repurposing analysis (Fig. 2), we would hypothesize that bumetanide might be beneficial for preventing or treating AD in both apoE4 and apoE3 carriers, with a higher efficacy in apoE4 carriers. Clearly, further testing of this drug in apoE genotype stratified prospective AD clinical trials is warranted.
While studies debate bumetanide’s interaction with canonical target NKCC1 behind the blood brain barrier41–43, the potent and potentially “off-target” effects of bumetanide in the CNS are robust22–27,42,44. Here, we posit that a whole network-level drug effect on the full transcriptome of the temporal lobe, combined with literature knowledge allowing researchers to distinguish between top drug candidates, can be used as both a drug prediction tool and as a roadmap to understand the top candidate bumetanide’s mechanism of action against apoE4 AD. While a comprehensive comparison of a random set of drugs hasn’t been carried out in the context of AD in the current study, we previously have performed a global evaluation of the method for therapeutic discovery in cancer and found that compounds capable of more dramatically “flipping” the transcriptomic signature of the cancer back towards a normal state were more likely to be effective in clinical studies20, leading us to believe that our network-based approach may correctly identify drugs efficacious against AD. Accordingly, we investigate the transcriptomic network perturbation caused by bumetanide in selective cell subtypes in vivo in aged apoE4-KI and J20/E4-KI mouse hippocampus and in vitro in apoE4-iPSC-derived human neurons in culture. Interestingly, based on snRNA-seq analysis, the transcriptomic effects of bumetanide on apoE4 AD signature genes are largely observed in neurons in aged apoE4-KI mouse hippocampus and in both neurons and glial cells in J20/E4-KI mouse hippocampus. This could be due to the fact that gliosis (astrocytosis and microgliosis), which represents a pathological hallmark of AD brains, occurs in J20/E4-KI mouse brains but not in aged apoE4-KI mouse brains. It is conceivable that the AD-related transcriptomic profiles of activated glial cells in response to Aβ accumulation, as reported45–47, make them more responsive to bumetanide’s beneficial effects on transcriptome.
Comparing the RNA-seq data of bumetanide treatment in aged apoE4-KI and J20/E4-KI mouse hippocampus with those from apoE4-iPSC-derived human neurons reveals three shared pathways that also overlap with those enriched in human apoE4 AD signature pathways—GABAergic synapse, circadian entrainment, and morphine addiction, suggesting mechanisms of bumetanide action for preventing or treating apoE4 AD. Importantly, deficits of GABAergic interneurons and synapses have been found in mouse models of AD, including apoE4-KI mice without or with Aβ accumulation2,29, as well as in human AD patients, especially those with apoE42,29. Circadian related impairments, including sleep deficit, have also been reported in AD mouse models and patients, again especially in those with apoE448–51. The morphine addiction pathway shares many key genes with both the GABAergic synapse and the circadian entrainment pathways, and morphine affects both the GABAergic function and circadian rhythms52,53. Importantly, bumetanide treatment rescues transcriptomic deficits of these three pathways in aged apoE4-KI and J20/E4-KI mouse hippocampal neurons and/or glia cells as well as in apoE4-iPSC-derived human neurons, indicating that these rescues likely contribute to the beneficial effects of bumetanide on neuronal physiology, pathology, and cognition in aged apoE4-KI mice without or with Aβ accumulation and possibly also in human apoE4 AD.
The selectivity of bumetanide’s predicted effects on specific neuronal subtypes in vivo in aged apoE4-KI mouse hippocampus and on both neuronal subtypes and glial cells in J20/E4-KI mouse hippocampus, as well as the recapitulation of the predicted effects in apoE4-iPSC-derived human neurons, highlights the necessity of establishing more precise drug repurposing databases created from cell types relevant to neurological diseases, such as subtypes of neurons and glia. While this study made use of data from cancer cell lines included in the CMap database, such a resource in CNS-relevant cell types would enable the large-scale study of drugs that are already FDA approved, providing a faster trajectory to the clinic, dramatically lowering the costs, and shortening the timeline of drug development pipelines for neurological diseases. Similarly, the sample size currently available from AD and control temporal lobe tissues with apoE genotype information is limited, underscoring a need for more human brain region-specific transcriptomic datasets with AD-related genetic information.
There are some limitations in this study. First, the limited availability of human brain transcriptomic databases with clear apoE genotype information restricted us to focus on one dataset with 213 samples in our study. Second, we only validated bumetanide, among the top predicted drugs, in animal models and in real world human EHR databases. Third, since the two EHR databases do not include apoE genotype information, further studies in other EHR databases with apoE genotype information are warranted. Despite these limitations, the current study clearly validates, in several ways, our strategy to develop new therapies for AD and other neurodegenerative disorders with multifactorial etiology, complex mechanisms, and patient heterogeneity. First, combining precision medicine techniques, such as stratification by disease-associated gene mutations or polymorphisms, with computational drug repurposing is a powerful method to identify drugs effective for subpopulations of patients. Second, our data support the theory that perturbing an entire gene-expression network away from a disease state might represent an effective treatment strategy for complex conditions, such as Alzheimer’s disease. Finally, validation of cellular/animal study-generated hypothesis using real world human EHR data can be a powerful approach to bridge preclinical drug development program toward human clinical trials, as exemplified in our study by the potential of bumetanide as a prevention or treatment for apoE4-mediated AD.
Methods
All studies included in this project comply with relevant ethical regulations. Animal protocols and procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of California, San Francisco (UCSF). The protocol of using human iPSC for laboratory research was approved by the Committee on Human Research at the UCSF (10-00234). The two EHR studies were approved by the Institutional Review Board at the UCSF (20-32422) and the Icahn School of Medicine at Mount Sinai (19-02369).
Mice.
All mice were housed under identical conditions from birth through death (12-hour light/dark cycle, housed 5/cage, PicoLab Rodent Diet 20). All mouse lines were maintained on a C57Bl/6J background. ApoE3-KI and apoE4-KI homozygous mice (Taconic)54,55, J20/E3-KI and J20/E4-KI mice generated by cross-breeding apoE-KI mice with J20 mice overexpressing mutant human APP (from Dr. Lennart Mucke’s lab at Gladstone Institutes), as well as wildtype mice were born and aged under normal conditions at the Gladstone Institutes/UCSF animal facility. Female apoE-KI mice were used because of their susceptibility to AD-related neuronal and behavioral deficits. Sex-matched wildtype mice were used as controls. The ages and numbers of the mice are indicated in different Methods sections and in each figure legend.
Differential expression and pathway analysis.
Dataset GSE1522217 was identified as the only available AD brain transcriptomic dataset on GEO Expression Omnibus with apoE genotype information and an n > 3 of apoE4/4 controls. Data had been rank-invariant normalized as described17,56. Negative values were eliminated by adding 0.1 plus the absolute value of the minimum value across the expression matrix and log2 transformation was applied. Since fold change (FC) calculated after this addition is an underestimation, all subsequent FC estimates are conservative relative to the stated thresholds. All samples with an apoE3/3, apoE3/4, or apoE4/4 genotype were used for further analysis except for one sample without a reported sex. Enrichment of apoE4 in AD patients compared to controls was calculated by Chi-squared test. Principal component analysis (PCA) was applied to confirm normalization of technical artifacts, and the first and second PC were regressed against apoE status and AD status (Fig. 1c, d).
Average age did not differ across apoE genotype groups (Supplementary Table 1). To eradicate effects related to sex, 10 permutations of a random down-sampling of each apoE-genotype-specific group was performed to match the sex ratios within each genotype across diagnoses (Supplementary Table 1). Prior to differential expression (DE) analysis, quantile normalization was applied to each genotype-specific cohort to eliminate subsampling bias57,58. Linear modelling (limma v_3.36.5) was applied to each of the 10 permutations to elucidate DE genes. For analysis of the full dataset without genotype stratification, the same normalization procedure and sex permutation analysis described above were applied. Genes with an average absolute logFC greater than 0.4 and a P-value cutoff < 0.05 in all 10 permutations were further analyzed. The kegga function (limma v_3.36.5) was applied to find DE pathways with a background of all genes in the microarray.
Drug repurposing analysis.
The computational drug repurposing algorithm, which was developed by Sirota et al.12, and taken from Chen et al.13, was applied to each of the ten permutations of apoE-genotype-specific gene signatures using the publicly available CMap database (>1300 compounds)16. The drug repurposing algorithm13 takes two inputs: 1) an ordered list of up- and down-regulated genes in a disease, and 2) the data from CMap, consisting of rank FC of each gene after drug treatment. A Kolmogorov-Smirnov test (K-S test) of the gene expression ranks in the disease and drug signatures was used to assign each drug a CMap score, which reflects the degree to which the drug “flips” the signature of disease. The algorithm was modified to use the full DE gene signatures, rather than the top 150 DE genes. Technical replicates, defined as the same drug, concentration, cell line, and treatment, were averaged by CMap score. The CMap score was further averaged over the ten permutations and reported in Figure 2a–d. There are three cell lines (HL60, MCF7, and PC3 cells) used in the CMap database, each of which was treated as a separate experiment with a separate predictive index in candidate drug predictions as we had no prior knowledge as to its relevance as an AD cellular model. Taken from the top five compounds identified as potential therapeutics against apoE4/4 AD, the FDA-approved loop-diuretic bumetanide, evaluated in PC3 cells in the CMap database, was identified as a strong candidate drug (Fig. 2a). Bumetanide had previously been investigated for other brain disorders, such as seizure, autism, depression, and schizophrenia, suggesting brain penetration and potential effectiveness in the CNS21–27. We therefore focused our further analyses on bumetanide. To display how bumetanide “flips” the apoE4/4-specific transcriptomic signature of AD, the CMap data in Figure 2e and f were analyzed by Monte Carlo simulation to calculate the significance of the shift in the average FC rank. Downregulation by bumetanide was defined as a shift of upregulated apoE4/4-specific AD gene signatures to a lower rank than the mean rank of all genes. Upregulation by bumetanide was defined as a shift in average FC rank of the downregulated genes in the apoE4/4-specific AD signature toward a higher rank than the mean rank of all genes.
Bumetanide treatment.
Bumetanide (Sigma-B3023, Lot number BCBR5487) was dissolved in DMSO at 4 mg/ml (stock solution). The working solution was prepared by diluting the stock solution to the final concentration of bumetanide at 0.08 mg/ml in 0.9% sterile saline (2% DMSO), adjusted to pH 8.5 with NaOH for solubility. Bumetanide was given by i.p. injection daily (0.2 mg/kg) to female apoE-KI and female wildtype mice at 22 months of age, starting 6 weeks before and continuing throughout behavioral assessment, or administered to apoE-KI mice at 15 months of age for 8 weeks for electrophysiological and snRNA-Seq assessments. Bumetanide was also given by i.p. injection to female J20/E-KI mice at 10 months of age for 12 weeks for electrophysiological, pathological, and snRNA-Seq assessments. For all bumetanide treatment studies, mouse body weight was measured weekly during bumetanide treatment; injection volume was calculated to achieve a dose of 0.2 mg bumetanide/kg body mass. Control mice were injected with a matched volume of 2% DMSO in 0.9% sterile saline, pH 8.5. Injections were well tolerated and had no adverse effects on health.
Immunohistochemistry and quantitative analysis of Aβ load.
Brains were collected from female J20/E4-KI mice treated with vehicle or bumetanide for 12 weeks (n = 8 for each group). Brain sections were collected (30μm) from paraformaldehyde-fixed right hemibrains on a sliding microtome fitted with a freezing stage as described previously37. Sections from each mouse were immunostained, as reported previously37, with monoclonal anti-Aβ (3D6 from Elan Pharmaceuticals through a Material Transfer Agreement, 1:1000) overnight at 4 °C, Avidin-Biotin Complex (ABCVector Elite) for 60 minutes at room temperature, and development with diaminobenzidine as the substrate for 3 minutes. Stained sections were mounted on Superfrost slides using 1% gelatin, allowed to air dry, and coverslipped. Two sections per mouse were imaged, and the percentage area covered by Aβ deposits in the hippocampus and cortex were quantified and averaged across two sections from each mouse using ImageJ software, as described previously37.
Behavioral testing.
Female mice were singly housed before testing. Each mouse was assigned a random number, so researchers were blinded to genotype and treatment information. Female apoE3-KI and apoE4-KI mice at about 22 months of age were randomly assigned to treatment groups: apoE3-KI vehicle (n = 11, age 22.43 ± 0.26 months), apoE3-KI bumetanide (n = 11, age 22.35 ± 0.23 months), apoE4-KI vehicle (n = 11, age 22.35 ± 0.23 months), apoE4-KI bumetanide (n = 11, age 22.23 ± 0.21 months). Bumetanide or vehicle was administered daily by i.p. injection beginning 6 weeks before and continuing throughout the 14 days of testing in the Morris water maze (MWM); injections were given at the end of the light cycle and after the day’s test.
The MWM test was performed as reported previously33–35. For the probe trials, we analyzed (1) the percent time spent in the target quadrant versus average time spent in the three other quadrants, (2) the number of crossings over the position of the target platform versus the average number of crossings over the equivalent positions in the three other quadrants.
Single-nucleus RNA-seq (snRNA-seq) library preparation and sequencing.
Fifteen-month-old apoE4-KI mice (n = 3 per condition) received daily i.p. injections of vehicle or bumetanide (0.2 mg/kg body weight) for 8 weeks. 10-month-old J20/E4-KI mice (n = 3 per condition) received daily i.p. injections of vehicle or bumetanide (0.2 mg/kg body weight) for 12 weeks. The snRNA-seq protocol was modified based on 10x Genomics Sample Preparation Demonstrated protocols (Isolation of Nuclei for Single Cell RNA Sequencing). All procedures and samples were harvested and run in randomized order at the same time to minimize any batch effects. Mouse hippocampus was dissected on ice. Dissected hippocampus was placed in 2mL Hibernate A®/B27®/GlutaMAX™ (HEB) medium in a 5mL tube. The HEB medium was removed to a 15mL conical and kept on ice. 2mL of chilled lysis buffer (10 mM Tris-HCl, 10 mM NaCl, 3 mM MgCl2, and 0.1% Nonidet™ P40 Substitute in Nuclease-Free Water) were added to the tissue, and the hippocampus was homogenized by suctioning10x through a 21G needle. After homogenization, the tissue was lysed on ice for 15 min, swirling 2–3 times during this incubation period. The reserved chilled HEB media was then returned to the lysed tissue solution, and the tissue was further triturated with 5–7 passes through a 1mL pipette. A 30μm cell strainer (MACS Smart Strainer; Miltenyi Biotech 130-110-915) was washed with 1mL of PBS, and the lysed tissue solution was filtered through the strainer to remove debris and clumps. Filtered nuclei were centrifuged at 500 rcf for 5min at 4°C. The supernatant was removed, and nuclei were resuspended in 1mL of Nuclei Wash and Resuspension Buffer (1X PBS with 1.0% BSA and 0.2U/µl RNase Inhibitor), then centrifuged at 500 rcf for 5 min at 4°C, and resuspended in 400 µl of Nuclei Wash and Resuspension Buffer. DAPI was added to a final concentration of 0.1µg/mL, and the nuclei solution was filtered through 35µm cell strainer before loaded onto the BD FACSAria-II at the Gladstone Flow Cytometry Core. The DAPI-positive nuclei were selected by gating on DAPI-positive events, excluding debris and doublets. RNA was extracted in accordance with the 10X Genomics isolation of nuclei for single cell RNA sequencing protocol. Sequencing libraries were prepared using the Chromium Single Cell 3′ Library and Gel Bead Kit v2 (10x Genomics, 120267) for apoE4-KI mouse hippocampal samples and using Chromium Next GEM Single Cell 3’ Kit v3.1 (10x Genomics, 1000269) for J20/E4-KI mouse hippocampal samples according to the manufacturer’s instructions. Libraries were sequenced on an Illumina NovaSeq 6000 sequencer at the UCSF Center for Advanced Technology Core. The sequence resulted in detection of a median of 979 genes per nucleus for samples from apoE4-KI mice (Extended Data Fig. 3) and a median of 723 genes per nucleus for samples from J20/E4-KI mice (Extended Data Fig. 7).
snRNA-seq alignment, clustering, and cell type identification.
For the apoE-KI snRNA-seq study, the mouse reference (mm10-3.0.0, Ensembl 97) pre-mRNA genome was created via 10X Genomics Cellranger (v. 2.2.0) mkref function. For the J20/E4-KI snRNA-seq study, reads were aligned to the mm10 reference sequence build version “2020-A”. Reads from each sample were aligned and aggregated using the 10X Cellranger pipeline. Raw data were loaded into the R package Seurat (v. 2.3.4 for apoE4-KI; v3.1.5.9005 for J20/E4-KI)59. Genes with detected expression in at least 3 nuclei, nuclei with 200–2400 detected genes, and mitochondrial percentage of <0.25% were retained for future analysis (Extended Data Fig. 3 and 7). Highly variable genes were calculated via the FindVariableGenes function with the parameters: x.low.cutoff = 0.0123, x.high.cutoff = 3, y.cutoff = 0.5. The expression levels of highly variable genes were scaled and centered via the ScaleData function, and was then fed into the RunPCA function. PCAElbow Plot was used to plot the cumulative standard deviations of each PC, and the significance of the association of each gene with each PC was assessed via the JackStraw and JackStrawPlot functions. The first 15 PCs were selected to feed in to the FindCluster function, with a resolution of 0.6. This method identified 18 distinct cell clusters for apoE4-KI mice and 25 cell clusters for J20/E4-KI mice. We then applied the FindMarkers function to identify marker genes using the default settings to identify the cell type present in each cluster, which were cross referenced with the literature60,61. To better visualize marker gene expression across different cell types, the magic function of the RMagic (Markov Affinity-Based Graph Imputation) package (v.1.4.0 for apoE4-KI mice, k=15; v.2.0.3 for J20/E4-KI mice, k=15) was applied to the data to denoise our count matrix62. The VlnPlot function was used to visualize marker gene expression (Extended Data Fig. 3 and 7)59.
snRNA-seq differential expression and pathway analysis.
To analyze the “flip” of apoE4/4 AD signature genes in different cell clusters in apoE4-KI and J20/E4-KI mice, logFC of all genes between bumetanide and control treated cells in each cell type was calculated via the FindMarkers function using a Wilcoxon rank sum test. In apoE4-KI mice, all neuronal subtypes with significant shift of apoE4/4 AD signature genes (Clusters 1, 2, 3, 4, 5, 6, 8, 9, 10, 13, and 16) were then combined and the logFC of all genes between bumetanide and control treated neuronal cells was calculated in the same manner as above. For apoE4-KI and J20/E4-KI mice, to display how bumetanide “flips” the apoE4/4-specific transcriptomic signature of AD, the FC of all genes present in each cell cluster that also overlapped with those measured in the CMap database were rank transformed, and then were analyzed by Monte Carlo simulation to calculate the significance of the shift in the average FC rank. DE pathways were calculated from all DE genes (defined as p < 0.05) from each cell cluster using the kegga function (limma 3.36.5). The background was all genes in the requisite snRNA-seq dataset that were detected in 3 cells or more. Pathways with an enrichment p < 0.005 were used for further analysis.
To analyze the CMap score of DE genes in apoE4-KI cell types, cell clusters with a median number of cells >50 from individual mice were pseudo-bulkified and analyzed for differential expression using muscat (version 1.0.1). The 300 genes with the lowest P-value were selected for further analysis. Percentile of rank of bumetanide in the CMap as well as the correlation coefficient and correlation p-value between gene changes in the apoE4-KI mouse hippocampus and the CMap were calculated.
RNA-seq analysis of human iPSC-derived neurons.
A human iPSC (hiPSC) line, which was generated from skin fibroblasts of a subject with an apoE4/4 genotype (a written informed consent was obtained from the donor) and published previously38, was used in this study. The apoE4/4-hiPSCs were differentiated into neurons for 6 weeks, as we reported previously38. At 6 weeks in culture, neurons were treated for 6 hours with 10μM bumetanide (Sigma-B3023, Lot number BCBR5487) or vehicle (0.1% DMSO in saline) (n = 3 for each group). Total RNA was extracted and purified with the Qiagen RNeasy Micro kit, which included a DNase treatment. cDNA was generated from full-length RNA (50 ng per sample) with the NuGEN RNA-Seq V2 kit, which uses the single primer isothermal amplification method to deplete ribosomal RNA, and sheared by Covaris to yield fragments of uniform size. The NuGen Ultralow system V2 was used for adding adapters and for barcoding and amplification. The resulting RNA libraries were purified with Agencourt XP magnetic beads, and quantified by qPCR after quality control with an Agilent Bioanalyzer. The libraries were pooled and sequenced with a HiSeq 4000 instrument (Illumina) for single-end (SE50) sequencing. Sequence data were aligned with the STAR short read aligner (v 2.1.3)63 and counts per feature were obtained with the featureCounts function from the Subread package64.
The DESeq pipeline (version 1.20.0) was used to assess DE of the resulting 49,697 features. Genes with a sum of <10 counts across all samples were filtered out for further analysis. Genes with >1 transcript were combined by average logFC and minimum P-value. DE genes with p < 0.05 were considered for future pathway analysis. After count normalization with DESeq, data were regularized log (rld) transformed (vsn package), and PCA was applied to the rld-transformed DE genes as well as the top 500 most variable genes. The kegga function (limma v_3.36.5 ) was used on all DE genes (p < 0.05) to elucidate enriched ontological pathways (p < 0.05), using a background of all genes with a sum of over ten counts across all samples.
To display how bumetanide “flips” the apoE4/4-specific transcriptomic signature of AD, gene symbols were generated from the ENSEMBL ID of each transcript, and the average FC was taken for genes with >1 ENSEMBL ID. The FC of genes which also overlapped with those measured in the CMap database was rank transformed, and then was analyzed by Monte Carlo simulation to calculate the significance of the shift in the average FC rank, as described above. Overlap between enriched pathways in human apoE4/4 AD, apoE4/4-iPSC-derived human neurons after bumetanide, and mouse hippocampal snRNA-Seq analysis after bumetanide was calculated by Monte Carlo simulation. In brief, groups of the same size as the number of enriched pathways from each analysis were chosen at random 1000 times from a pool of the total pathways available in the kegga function (limma v_3.36.5) and the overlap was calculated. P-values of enrichment of the true observed overlap were calculated using this permuted data.
RNA-Seq analysis of the signature of brain aging DE genes in 3-, 12-, and 24-month-old apoE4-KI mouse cerebral cortex
The raw data of the RNA-Seq dataset (https://doi.org/10.7303/syn20808171) from Zhao et al.28 was downloaded, and only female apoE4/4 mice were used as only female mice show electrophysiological and behavioral deficits in this mouse model. The fastq files were pre-processed using the nf-core/rnaseq v1.4.2 pipeline (http://doi.org/10.5281/zenodo.3503887), and the samples were aligned to the mm10 reference genome. PCA of the rlog-transformed top 500 most variable genes was evaluated and found no outliers. DESeq2 (version 1.30.0) was used to analyze DE genes between 3-, 12-, and 24-month-old apoE4-KI female mice, using only genes with a sum of >= 10 counts in all samples. Genes with a logFC >2 and an unadjusted p-value <0.05 were used for further analysis. The kegga function (limma v_3.36.5) was used to determine enriched pathways in DE genes with a background of all genes used in the DESeq pipeline. CMap scores were calculated, as described above, using DE genes from each analysis. Shift in rank of FC in DE genes in 12 and 24 vs 3 month-old apoE4-KI female mice was calculated as described above (the same way as in Figure 2). To measure the “flip” of the apoE4-KI “aging” DE genes in mouse hippocampus, the overlap of DE genes in the 24 vs 3 month-old apoE4-KI mice with genes detected in all 18 cell clusters in the apoE4-KI snRNA-seq dataset was used. P-values of the downregulation of overlapping genes in each of the 18 cell cluster datasets was calculated via Monte Carlo simulation, and rank of FC of the signature DE genes of aging after bumetanide treatment was calculated.
Brain slices electrophysiological recordings and data analyses.
For electrophysiological recording study of apoE-KI mice, female apoE4-KI mice were randomly allocated to vehicle (n = 6, age 15.2 ± 0.56 months) and bumetanide (n=3, age 15.1 ± 0.06) treatment groups. Female apoE3-KI mice were similarly allocated into vehicle (n=6, age 15.5 ± 0.11) and bumetanide (n=3, age 15.57 ± 0.19) treatment groups. All mice were dosed with bumetanide (0.2mg/kg) for 8 weeks. Slice recording day and dosage time were allocated randomly among the groups to allow for equal dosage time while experiments were performed. The final bumetanide injection was performed one hour prior to sacrifice. At the time of recording all mice were about 17 months of age.
For electrophysiological recording study of J20/E-KI mice, the following groups of mice were used after vehicle or bumetanide treatment: female J20/E4-KI mice treated with vehicle (n = 5, age 13.2 ± 0.8 months) or bumetanide (n = 5, age 13.0 ± 0.7 months) and female J20/E3-KI mice treated with vehicle (n = 5, age 13.1 ± 0.4 months) or bumetanide (n = 5, age 13.2 ± 0.5 months). All mice had been dosed with bumetanide (0.2mg/kg) for 12 weeks. Slice recording day and dosage time were allocated randomly among the groups to allow for equal dosage time while experiments were performed. The final bumetanide injection was performed one hour prior to sacrifice. At the time of recording all mice were about 13 months of age.
Animals were anaesthetized with isoflurane and decapitated. Brain was rapidly removed from the skull and placed in the ice-cold (2–5°C) slicing solution. Slicing solution contained (in mM): 110 choline chloride, 2.5 KCl, 26 NaHCO3, 10 MgCl2, 1.25 NaH2PO4, 0.5 CaCl2, 10 glucose, 3 Na Pyruvate, 1 L-Ascorbic acid, pH 7.4. 300 μm-thick sagittal slices were cut from both hemispheres using a vibratome (VT1200, Leica, USA) and transferred to a 95% O2-CO2 vapor interface holding chamber (BSK5, Scientific Systems Design Inc. Canada) containing artificial cerebrospinal fluid (ACSF) where they were allowed to recover at 34°C for one hour and held at room temperature (20–22°C) afterwards. ACSF contained (in mM): 126 NaCl, 2.5 KCl, 1.5 CaCl2, 1.5 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, 10 glucose, and 1.5 L-Ascorbic acid, pH 7.4.
For input/output recording studies, field post-synaptic potentials (fPSPs) were elicited by orthodromic stimulation of Schaffer collaterals by concentric bipolar stimulating electrode (FHC, Inc., USA) connected to a constant voltage isolated stimulator (DS2A-MKII, Digitimer North America) and placed in CA2 stratum radiatum. fPSPs were recorded with a glass borosilicate microelectrode filled with ACSF and placed in CA1 stratum radiatum. Signals were sampled and digitized by MultiClamp 700B amplifier and Digidata 1550B1 acquisition system with pClamp10 software (Molecular Devices, USA), and analyzed using IgorPro6 software (Wavemetrics Inc., USA) running custom macros. fPSP slopes were analyzed as the linear fit slope values between 10% and 90% of fPSP peak. Input-output relationships were recorded as the fPSP slope values in response to increasing stimulation intensity (0.5–1.4V). For long-term potentiation (LTP) recordings, stimulation intensity was adjusted to 50% of saturation and delivered at 0.1Hz. After 10 minutes of control recording, LTP was elicited using two consecutive (5s) theta-frequency burst (HFB) trains of 5 100Hz bursts (4 pulses each) at 0.2Hz. LTP outcome was ascertained as average fPSP gain for a 10-minute period 50 minutes post-induction vs. 10 minutes of control.
Electronic health record (EHR) databases.
UCSF EHR contains electronic health records on 1.3 million unique patients as of November 2019 including diagnosis codes, lab test results, medication orders as well as demographic variables. The database includes male and female patients from July 2012 until November 2019. The Mount Sinai EHR database was obtained dating back from January 2003 until February 2020, which contains 8 million patient records, with 3.9 million male and female patients having clinical data including diagnosis codes, lab test results, medication orders as well as demographic variables, including outpatient, inpatient, and emergency room encounters as part of clinical operations from five hospitals in Manhattan, Brooklyn, and Queens boroughs of New York City.
Permutation and propensity score analyses of EHR databases.
EHR databases were filtered using the medication order table to identify those who had been exposed to bumetanide (other names: Bumex, Burinex) or “control” diuretic compounds as described below. We further focused on patients over the age of 65. Bumetanide is usually prescribed for hypertension and edema (Supplementary Table 19). Hypertension has been identified as a risk factor for AD39. Accordingly, we then matched a 1:2 control cohort with the R package Matchit, using a propensity score based on age, race, sex, and hypertension and edema diagnosis, out of patients over the age of 65. We then calculated the number of AD patients defined by ICD10 codes G30.1, G30.8, G30.9 and ICD9 331.0 in case (bumetanide-exposed) versus control (non-bumetanide-exposed) groups using a Chi-square test on 10 permutations of control group. In order to further control for hypertension across the case and control cohort, we then chose a cohort of control diuretics (Supplementary Table 20) that met the following criteria: a) drugs were not loop diuretics, b) drugs had no other major co-indications other than high blood pressure and edema, c) drugs did not contain a mixture of bumetanide or a bumetanide analog, and d) drugs were available in the US and the UK. We then matched a 1:2 control cohort with the R package Matchit, as described, using the propensity score based on age, sex, race, hypertension and edema diagnosis with control patients taking one of the “control” diuretics. We excluded from the bumetanide cohort anyone who was also taking a “control” diuretic. We then calculated the number of AD patients, as described, in case (bumetanide-exposed) versus control (non-bumetanide-exposed) groups using a Chi-square test on 10 permutations of control group.
General statistics and reproducibility.
Behavior metrics are expressed as mean ± SEM. All values for n are the numbers of mice or biological replicates. Differences between groups were determined by unpaired or paired two-tailed t test. Paired t-tests were used for any analyses which measured the same group of mice across two different metrics. For multiple comparisons, one-way ANOVA and Tukey’s post-test were used. For electrophysiological recording studies, 2-tailed t test was used for statistical analysis of intergroup LTP gain outcomes, while Mann–Whitney U-test (independent samples) was used to analyze differences between animal groups. P < 0.05 was considered significant.
Researchers were blinded to genotypes and treatment information during experiments for all animal studies. For human transcriptomic analysis, sample sizes were determined by the sample sizes of the publicly available datasets. For mouse behavioral, slice electrophysiological, and Aβ plaque quantification studies, sample sizes were determined using effect sizes estimated from pilot cohorts and/or previous studies. For mouse single nucleus RNA-seq studies, sample sizes were determined by a power analysis using effect sizes estimated from literature search. Mice were allocated randomly into different treatment groups within each genotype. No data were excluded from the analyses in animal, electrophysiological, and iPSC studies. One sample was excluded from the publicly available GSE15222 dataset because it had no information regarding its sex.
Data availability.
All data generated or analyzed during this study are included in this published article (or in its supplementary information files) or deposited in the Gene Expression Omnibus (GEO), which are also available from the corresponding authors’ labs. Publicly available datasets used are available in the Gene Expression Omnibus (GEO) under the accession number GSE15222 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE15222) with the associated covariate data found on the Myers Lab website (http://labs.med.miami.edu/myers/LFuN/LFUN/DATA.html) and the associated Google drive (: https://drive.google.com/drive/folders/1ud5F9WN9Xx3oXIkb5xIg1b_zz1nzp3IR) in the “samples.covar.zip” file. The CMap database is available in Sage Synapse in the HCC_NEN project from the Bin Chen lab under the accession number syn6187678, and is linked to the Bin Chen Lab Gitlab repository (also see Code Availability section below). The publicly available RNA-Seq dataset of aging apoE4-KI mouse brains was from Zhao et al. (https://doi.org/10.7303/syn20808171)28. Figure 4, 6 and extended data figure 3, 4, 7, and 8 have associated mouse snRNA-seq data or iPSC-derived human neuron bulk-RNA-seq data generated in this study, which are available in the Gene Expression Omnibus (GEO) under the accession number GSE182765. The UCSF EHR database and the Mt. Sinai EHR database are not yet available to the general public.
Code availability.
The drug repurposing algorithm can be found in the Bin Chen Lab Gitlab Repository (https://github.com/Bin-Chen-Lab/HCC_NEN/). The following packages/softwares were used either as dependencies to downloading or using packages mentioned in the Methods section or in creating the figures in this manuscript: clusterProfiler_3.10.1, pheatmap_1.0.12, vsn_3.48.1, SummarizedExperiment_1.10.1, DelayedArray_0.6.6, BiocParallel_1.14.2, matrixStats_0.54.0, GenomicRanges_1.32.7, GenomeInfoDb_1.16.0, edgeR_3.22.5, mice_3.4.0, lattice_0.20-35, ggbiplot_0.55, scales_1.0.0, plyr_1.8.4, eulerr_5.0.0, VennDiagram_1.6.20, futile.logger_1.4.3, data.table_1.11.8, gridExtra_2.3, GEOquery_2.48.0, qvalue_2.12.0, illuminaHumanv1.db_1.26.0, org.Hs.eg.db_3.6.0 , AnnotationDbi_1.42.1, IRanges_2.14.12, S4Vectors_0.18.3, Biobase_2.40.0, BiocGenerics_0.26.0, nf-core/rnaseq_1.4.2, org.Mm.eg.db_3.6.0, raster_2.8.19, FastQC_0.11.8, featureCounts_1.6.2, Data.table_1.12.8, RMySQL_0.10.17, Dplyr_0.8.3, magrittr_1.5, tidyverse_1.3.1, and biomaRt_2_38_0. All custom codes generated in this study will be made available from the corresponding authors’ labs upon request.
Extended Data
Extended Data Fig. 1. Bumetanide is also predicted to rescue the transcriptomic signature of aging in apoE4-KI mouse hippocampus.
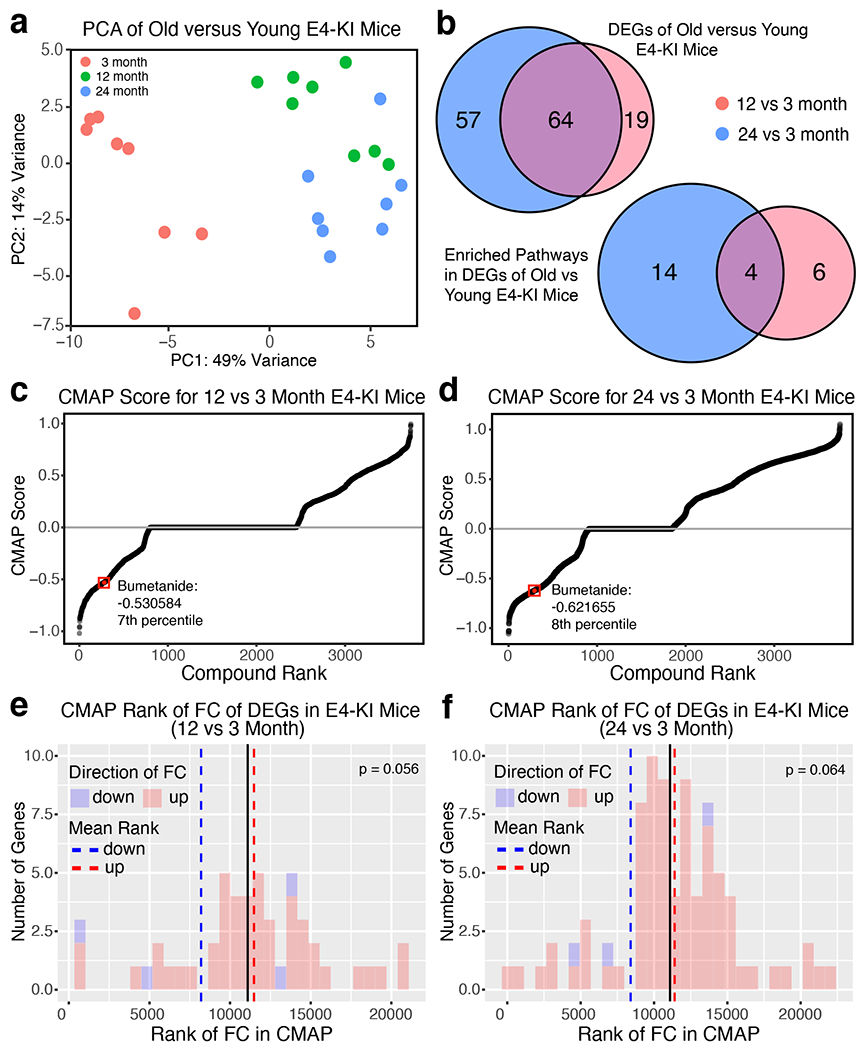
a, PCA plot of top 500 variable genes in apoE4-KI mouse brain shows a distinct effect of age, with 3 month-old brains grouping separately from 12 and 24 month-old brains. b, Venn diagrams of the overlaping DE genes (defined as logFC > 2 and P < 0.05) and DE pathways (defined as P < 0.05) between 12 vs 3 month-old apoE4-KI brains and 24 vs 3 month-old apoE4-KI brains. c, Graphs of compounds ordered by CMap score against DE genes (defined as logFC > 2, P < 0.05) in 12 vs 3 month-old apoE4-KI brains (see Methods for details). Bumetanide has a negative CMap score in the 7th percentile of all drugs in the CMap. d, Graphs of compounds ordered by CMap score against DE genes (defined as logFC > 2, P < 0.05) in 24 vs 3 month-old apoE4-KI brains. Bumetanide has a negative CMap score in the 8th percentile of all drugs in the CMap. e, Histogram of the rank of FC of the DE genes (defined as logFC > 2, P < 0.05) in 12 vs 3 month-old apoE4-KI brains, which were also measured in the CMap database after bumetanide treatment. The mean rank of all genes in this gene set is denoted by the black line, the average mean FC rank of up-regulated genes (colored red in histogram) is denoted by the dashed red line and the mean FC rank of the down-regulated genes (colored blue in histogram) is denoted by the blue dashed line. P-value of significance of the “flip” of up- and down-regulated FC rank means away from the rank mean of all genes as calculated by Monte-Carlo simulation is shown (P = 0.056). This p-value does not reach significance even while the magnitude of the “flip” is quite large. f, Histogram of the rank of FC of the DE genes (defined as logFC > 2, p-value < 0.05) in 24 vs 3 month-old apoE4-KI brains which were also measured in the CMap database after bumetanide treatment. The mean rank of all genes in this gene set is denoted by the black line, the average mean FC rank of up-regulated genes (colored red in histogram) is denoted by the dashed red line and the mean FC rank of the down-regulated genes (colored blue in histogram) is denoted by the blue dashed line. P-value of significance of the “flip” of up- and down-regulated FC rank means away from the rank mean of all genes as calculated by Monte-Carlo simulation is shown (P = 0.064). This p-value does not reach significance even while the magnitude of the “flip” is quite large.
Extended Data Fig. 2. Bumetanide treatment does not affect swim speed or visible trial performance in aged apoE4-KI mice and does not affect behavioral performance in wildtype (WT) mice.
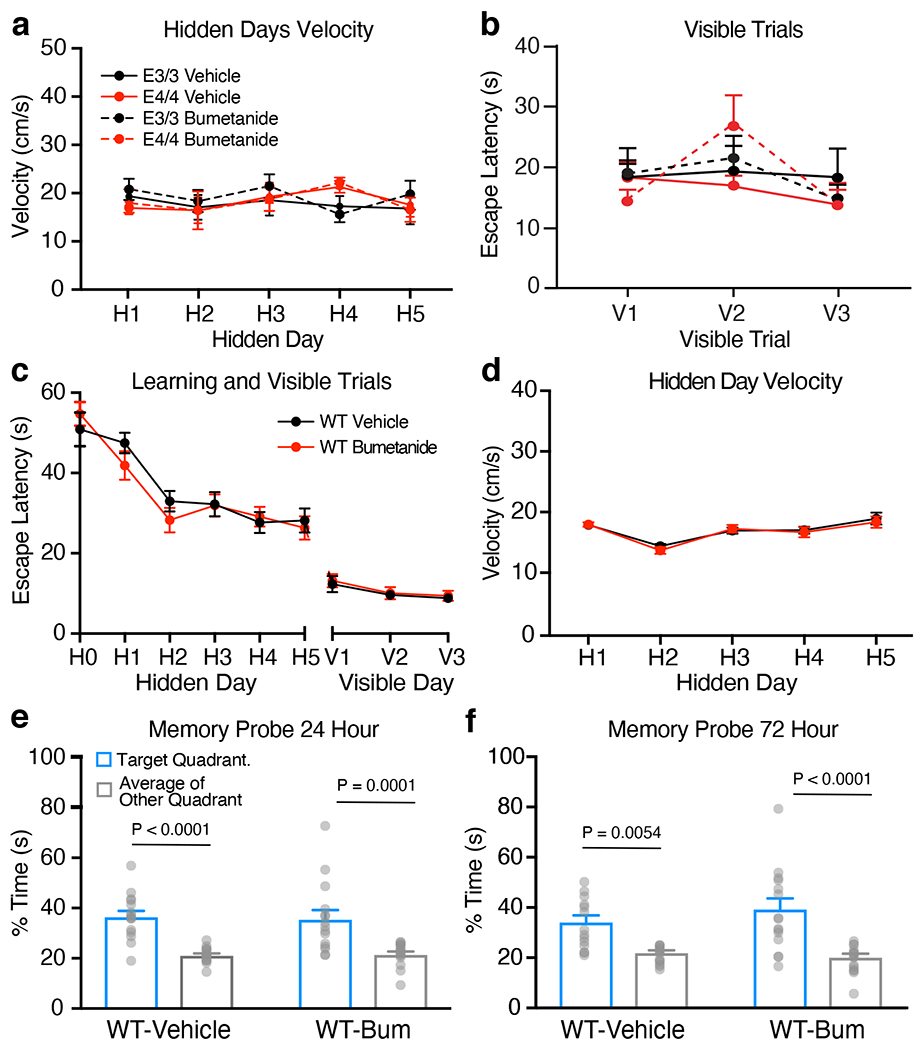
a, Bumetanide did not significantly affect swim speed during hidden platform trials of apoE4-KI and apoE3-KI mice (n = 11 for each group) at 24 month of age over learning days 1–5. b, There was no significant difference between any groups in visible trials (measured by 2-way ANOVA) of apoE4-KI and apoE3-KI mice (n = 11 for each group), indicating there were no motor or vision impairment in any of the groups. c, Escape latency of vehicle (n = 16) and bumetanide (n = 15) treated WT mice during learning days 1–5 did not differ. d, Bumetanide did not significantly affect swim speed during hidden platform trials in WT mice (n = 15) as compared to vehicle treated WT controls (n = 16). e, In the 24-hour probe trial, both vehicle (n = 16, two way ANOVA with Bonferroni’s multiple comparisons test P < 0.0001) and bumetanide (n = 15, two way ANOVA with Bonferroni’s multiple comparisons test P = 0.0001) treated WT mice spent significantly more time in the target quadrant versus average percent time in the other quadrants. f, In the 72-hour probe trial, both vehicle (n = 16, two way ANOVA with Bonferroni’s multiple comparisons test P = 0.0054) and bumetanide (n = 15, two way ANOVA with Bonferroni’s multiple comparisons test P < 0.0001) treated WT mice spent more time in the target quadrant than the other quadrants. Values are mean ± SEM in a-f.
Extended Data Fig. 3. Violin plots of marker genes for 18 cell clusters and their properties identified by snRNA-seq in the hippocampus of aged apoE4-KI mice.
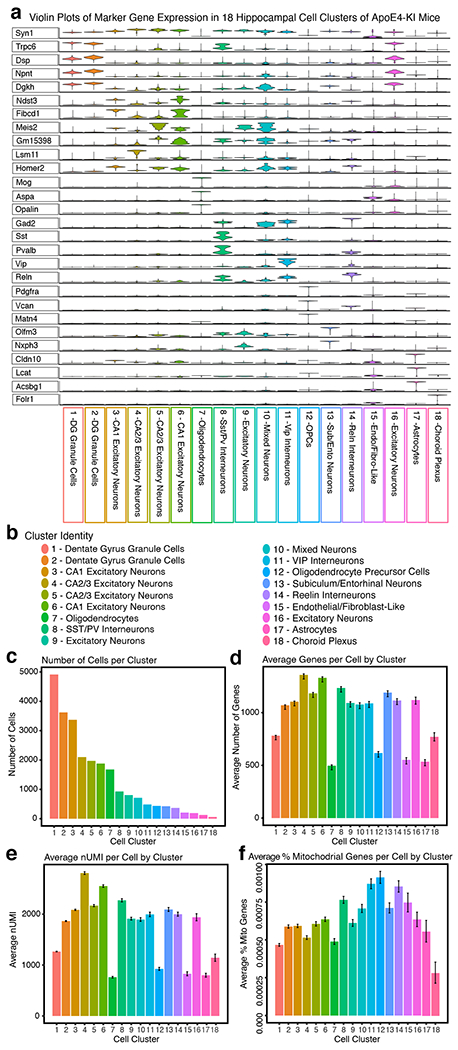
a, Violin plots of expression of marker genes for each of the 18 cell clusters identified by snRNA-seq in the hippocampus of aged apoE4-KI mice with and without bumetanide treatment. Y-axis is average imputed expression of a marker gene across all cells in a cluster (see Methods for details), x-axis denotes each cell cluster. b, snRNA-seq analysis of the hippocampus of aged apoE4-KI mice with and without bumetanide treatment identifies 18 unique cell clusters. c, Number of cells per cluster. d, Average number of genes identified per cell in each cluster (± SEM). Number of cells (n) for each cell cluster can be found in c (> 58 cells in any cluster). e, Average nUMI per cell for each cluster (± SEM). Number of cells (n) for each cell cluster can be found in c (> 58 cells in any cluster). f, Average % mitochondrial genes per cell in each cluster (± SEM). Number of cells (n) for each cell cluster can be found in c (> 58 cells in any cluster).
Extended Data Fig. 4. Histograms of FC rank changes of human apoE4/4-specific AD signature genes in cell clusters 1, 4, 6, 7, 9–18 in aged apoE4-KI mice.
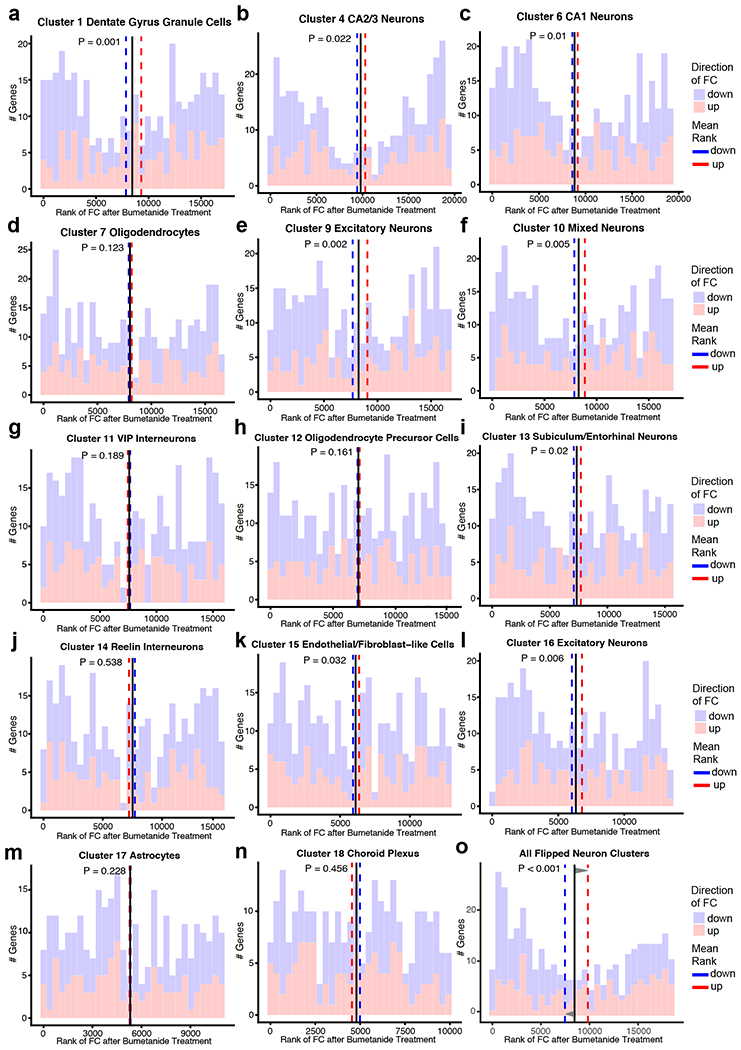
a–n, Histograms of the human apoE4/4-specific transcriptomic signature of AD geneset that was also detected by DE analysis of snRNA-seq in the apoE4-KI mouse hippocampus after bumetanide treatment as compared to controls in cell clusters 1, 4, 6, 7, 9–18. The rank of the FC of these genes in each cluster following bumetanide treatment, as compared to vehicle treatment, is plotted. The mean rank of all genes in this geneset is denoted by the black line, the average mean FC rank of up-regulated genes (colored red in histogram) is denoted by the red dashed line and the mean FC rank of the down-regulated genes (colored blue in histogram) is denoted by the blue dashed line. P-value of the significance of the “flip” of up- and down-regulated FC rank means away from the rank mean of all genes as calculated by Monte-Carlo simulation is shown (P < 0.05 considered significant). Cell clusters 1, 4, 6, 9, 10, 13, 15, and 16, which include all excitatory neurons, mixed neurons, and endothelial/fibroblast-like cells have a significant “flip” of human apoE4/4-specific AD signiture genes, whereas cell clusters 7, 11, 12, 14, 17 and 18, which include oligodendrocytes, VIP-interneurons, OPC’s, RELN-interneurons, astrocytes, choroid plexus, are not significant. o, Histogram of the human apoE4/4-specific transcriptomic signature of AD geneset that was also detected by DE analysis of snRNA-seq in the apoE4-KI mouse hippocampus after bumetanide treatment as compared to controls in combined data from all neuronal clusters that exhibited a significant “flip” of human apoE4/4 AD genes (Clusters 1, 2, 3, 4, 5, 6, 8, 9, 10, 13, and 16 combined). The rank of the FC of these genes in the combined neuronal cells following bumetanide treatment, as compared to vehicle treatment, is plotted. The mean rank of all genes in this geneset is denoted by the black line, the average mean FC rank of up-regulated genes (colored red in histogram) is denoted by the red dashed line and the mean FC rank of the down-regulated genes (colored blue in histogram) is denoted by the blue dashed line. P-value of the significance of the “flip” of up- and down-regulated FC rank means away from the rank mean of all genes as calculated by Monte-Carlo simulation is shown (P < 0.05 considered significant).
Extended Data Fig. 5. The fold change size and directionality of all DE genes after bumetanide treatment in the five large excitatory neuronal cell types in aged apoE4-KI mouse hippocampus mimicked the fold change size and directionality after bumetanide treatment in PC3 cells in the CMap database.
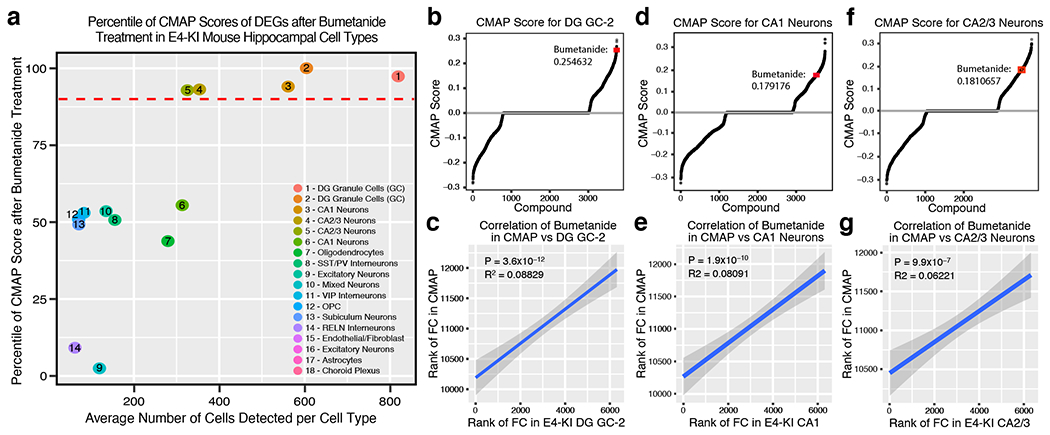
a, Scatterplot of average number of cells per cell cluster (of clusters with > 50 cells) in apoE4-KI hippocampi versus the percentile of CMap score against the DE genes in those clusters after bumetanide treatment (see Methods for details). The top 300 DE genes by p-value of the first five cell clusters (all excitatory neuronal cells) have a CMap score above the top 90 percentile of all drugs in the CMap database. b, Graphs of compounds ordered by CMap score against DE genes in Dentate Gyrus Granule Cells (see Methods for details). Bumetanide has one of the highest positive scores, suggesting that the signature in these cells in vivo is similar to the signature in the CMap database. c, Correlation analysis plot of rank of FC of genes in apoE4-KI Dentate Gyrus Granule Cells after bumetanide treatment versus rank of FC of genes in the CMap database after bumetanide treatment. There is a positive correlation with an R2 = 0.08829 and a P-value = 3.6 × 10−12, indicating that the FC of genes in these two signatures of DE genes after bumetanide treatment mimic each other. The shaded region represents the 95% confidence interval for predictions from the linear model. d, Graphs of compounds ordered by CMap score against DE genes in apoE4-KI CA1 neurons (see Methods for details). Bumetanide has one of the highest positive scores, suggesting that the signature in these cells in vivo is similar to the signature in the CMap database. e, Correlation analysis plot of rank of FC of genes in apoE4-KI CA1 neurons after bumetanide treatment versus rank of FC of genes in the CMap database after bumetanide treatment. There is a positive correlation with an R2 = 0.08091 and a P-value = 1.9 x 10−10, indicating that the FC of genes in these two signatures of DE genes after bumetanide treatment mimic each other. The shaded region represents the 95% confidence interval for predictions from the linear model. f, Graphs of compounds ordered by CMap score against DE genes in apoE4-KI CA 2/3 Neurons (see Methods for details). Bumetanide has one of the highest positive scores, suggesting that the signature in these cells in vivo is similar to the signature in the CMap database. g, Correlation analysis plot of rank of FC of genes in apoE4-KI CA2/3 neurons after bumetanide treatment versus rank of FC of genes in the CMap database after bumetanide treatment. There is a positive correlation with an R2 = 0.06221 and a P-value = 9.9 x 10−7, suggesting that the FC of genes in these two signatures of DE genes after bumetanide treatment mimic each other. The shaded region represents the 95% confidence interval for predictions from the linear model.
Extended Data Fig. 6. Bumetanide treatment flips apoE4-mediated murine transcriptomic signature of aging in specific neuron subtypes in the hippocampus of aged apoE4-KI mice.
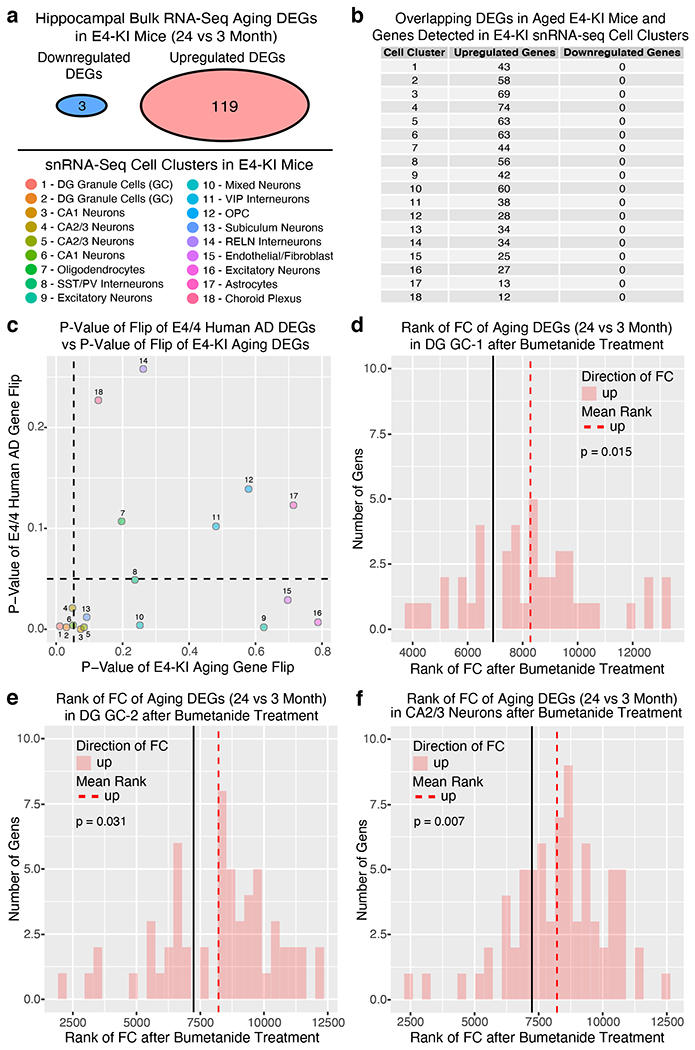
a, Number of upregulated (119) and downregulated (3) genes (defined as logFC > 2, P < 0.05) in 24 vs 3 month-old apoE4-KI cortex. b, Number of overlapping genes that were detected in clusters 1–18 in apoE4-KI hippocampi with and without bumetanide treatment. Due to sequencing depth and gene drop out, none of the 3 downregulated genes were detected in any cell cluster in aged apoE4-KI hippocampi. c, P-value of the “flip” of apoE4/4 specific transcriptomic signatures of AD in humans is plotted on the x-axis versus the P-value of the “flip” of aging signature of upregulated DE genes in 24 vs 3 month-old apoE4-KI hippocampus for each of the 18 cell clusters after bumetanide treatment in apoE4-KI mouse hippocampus. The black dashed lines denotes P = 0.05. Cell clusters 1, 2 and 4 have a significant P-value in each analysis, while Clusters 1–6 and cluster 13 have P-values either trending towards or reaching significance in both analyses, suggesting that most exitatory neuronal clusters experience a “flip” of both aging and apoE4/4 AD signatures when exposed to bumetanide in apoE4-KI hippocampi. d, Histogram of the rank of FC after bumetanide treatment in apoE4-KI Dentate Gyrus Granule Cell (Cluster 1) of the aging DE genes (defined as logFC > 2, P-value < 0.05) in 24 vs 3 month-old apoE4-KI hippocampus. The mean rank of all genes in this gene set is denoted by the black line, the average mean FC rank of up-regulated genes (colored red in histogram) is denoted by the dashed red line. P-value of significance of the “flip” of up-regulated FC rank means away from the rank mean of all genes as calculated by Monte-Carlo simulation is shown (P = 0.015). e, Histogram of the rank of FC after bumetanide treatment in apoE4-KI Dentate Gyrus Granule Cell (Cluster 2) of the aging DE genes (defined as logFC > 2, P-value < 0.05) in 24 vs 3 month-old apoE4-KI hippocampus. The mean rank of all genes in this gene set is denoted by the black line, the average mean FC rank of up-regulated genes (colored red in histogram) is denoted by the dashed red line. P-value of significance of the “flip” of up-regulated FC rank means away from the rank mean of all genes as calculated by Monte-Carlo simulation is shown (P = 0.031). f, Histogram of the rank of FC after bumetanide treatment in apoE4-KI CA 2/3 Neurons (Cluster 4) of the aging DE genes (defined as logFC > 2, P-value < 0.05) in 24 vs 3 month-old apoE4-KI hippocampus. The mean rank of all genes in this gene set is denoted by the black line, the average mean FC rank of up-regulated genes (colored red in histogram) is denoted by the dashed red line. P-value of significance of the “flip” of up-regulated FC rank means away from the rank mean of all genes as calculated by Monte-Carlo simulation is shown (P = 0.007).
Extended Data Fig. 7. Violin plots of marker genes for 25 cell clusters and their properties identified by snRNA-seq in the hippocampus of J20/E4-KI mice.
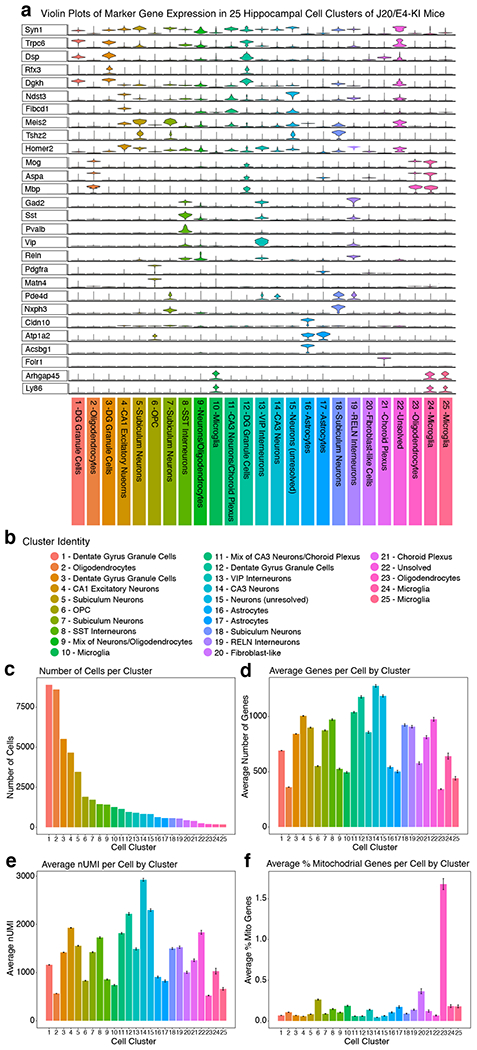
a, Violin plots of expression of marker genes for each of the 25 cell clusters identified by snRNA-Seq in the hippocampus of J20/E4-KI mice with and without bumetanide treatment. Y-axis is average imputed expression of a marker gene across all cells in a cluster (see Methods for details), x-axis denotes each cell cluster. b, snRNA-seq analysis of the hippocampus of aged apoE4-KI mice with and without bumetanide treatment identifies 25 unique cell clusters. c, Number of cells per cluster. d, Average number of genes identified per cell in each cluster (± SEM). Number of cells (n) for each cell cluster can be found in c (> 177 cells in any cluster). e, Average nUMI per cell for each cluster (± SEM). Number of cells (n) for each cell cluster can be found in c (> 177 cells in any cluster). f, Average % mitochondrial genes per cell in each cluster (± SEM). Number of cells (n) for each cell cluster can be found in c (> 177 cells in any cluster).
Extended Data Fig. 8. snRNA-seq analysis of the transcriptomic perturbation signature of bumetanide in the hippocampus of J20/E4-KI mice.
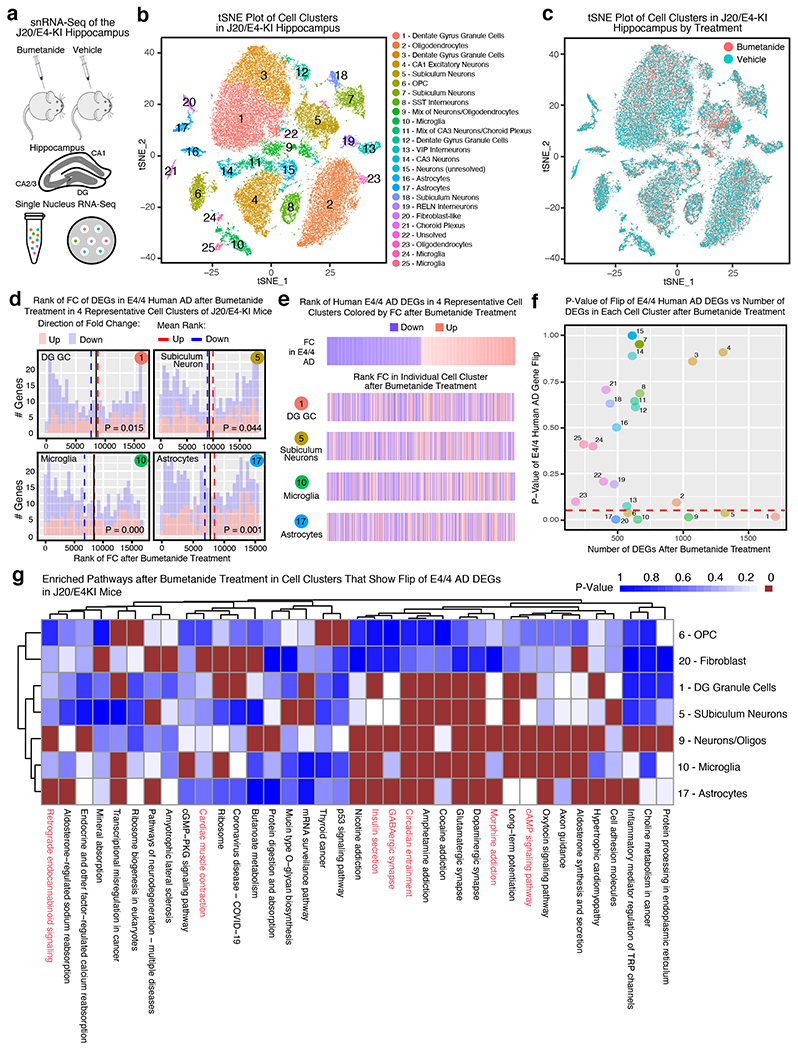
a, Transcripts in 47,619 single nuclei from the hippocampus of bumetanide- and vehicle-treated J20/E4-KI mice (n = 3 mice per group) were sequenced. b, Clustering and visualization by t-SNE identifies 25 distinct cell clusters which are color-coded according to cell-type. c, Cell clusters color-coded by treatment groups. d, Histogram of the rank of FC of the human apoE4/4-specific transcriptomic signature of AD genes that were also detected by snRNA-seq in J20/E4-KI mouse hippocampi in four representative cell clusters. The rank of the FC of these genes in a dentate gyrus granule cell cluster (1), a subiculum neuronal cluster (5), a microglial cluster (10), and an astrocyte cluster (17) in J20/E4-KI mouse hippocampi following bumetanide treatment as compared to vehicle treatment were plotted. The mean rank of all genes in this gene set is denoted by the black line, the average mean FC rank of up-regulated genes (colored red in histogram) is denoted by the red dashed line and the mean FC rank of down-regulated genes (colored blue in histogram) is denoted by the blue dashed line. P-value of significance of the “flip” of up- and down-regulated FC rank means away from the rank mean of all genes as calculated by Monte-Carlo simulation is shown (P < 0.05 considered significant. e, Heatmap of genes from apoE4/4-specific transcriptomic signature of AD, rank ordered and color coded (red for up, blue for down) by estimated FC in human apoE4/4 AD (top), then re-ordered by FC rank after bumetanide treatment in four representative cell types (clusters 1, 5, 10 and 17) in the J20/E4-KI mouse hippocampus. Bumetanide treatment flips the expression rank of both up- and down-regulated genes of the human apoE4/4-specific transcriptomic signature of AD in these four cell types. f, P-value of the “flip” of the apoE4/4-specific transcriptomic signature of AD is plotted on the y-axis versus the number of DE genes in each cell cluster on the x-axis. The red dashed line denotes P = 0.05. Dentate gyrus granule cells, subiculum neurons, OPCs, microglia, and astrocytes exhibit a significant “flip” of the apoE4/4-specific transcriptomic signature of genes in AD despite varying number of DE genes. g, Heatmap of the p-values of enriched ontological pathways in all cell clusters exhibiting the “flip” behavior of the apoE4/4-specific transcriptomic signatures of AD reveals 37 pathways that are affected in at least one of these cell types (P < 0.005). Pathway names highlighted in red (n = 7) are those shared with the apoE4/4-specific signature pathways of AD (see Figure 1g and Supplementary Table 6 for human pathways).
Extended Data Fig. 9. Analyses of overlapping enriched ontological pathways among bumetanide-treated apoE4-KI mice, J20/E4-KI mice, human iPSC-derived neurons, and human apoE4/4-specific transcriptomic signature of AD.
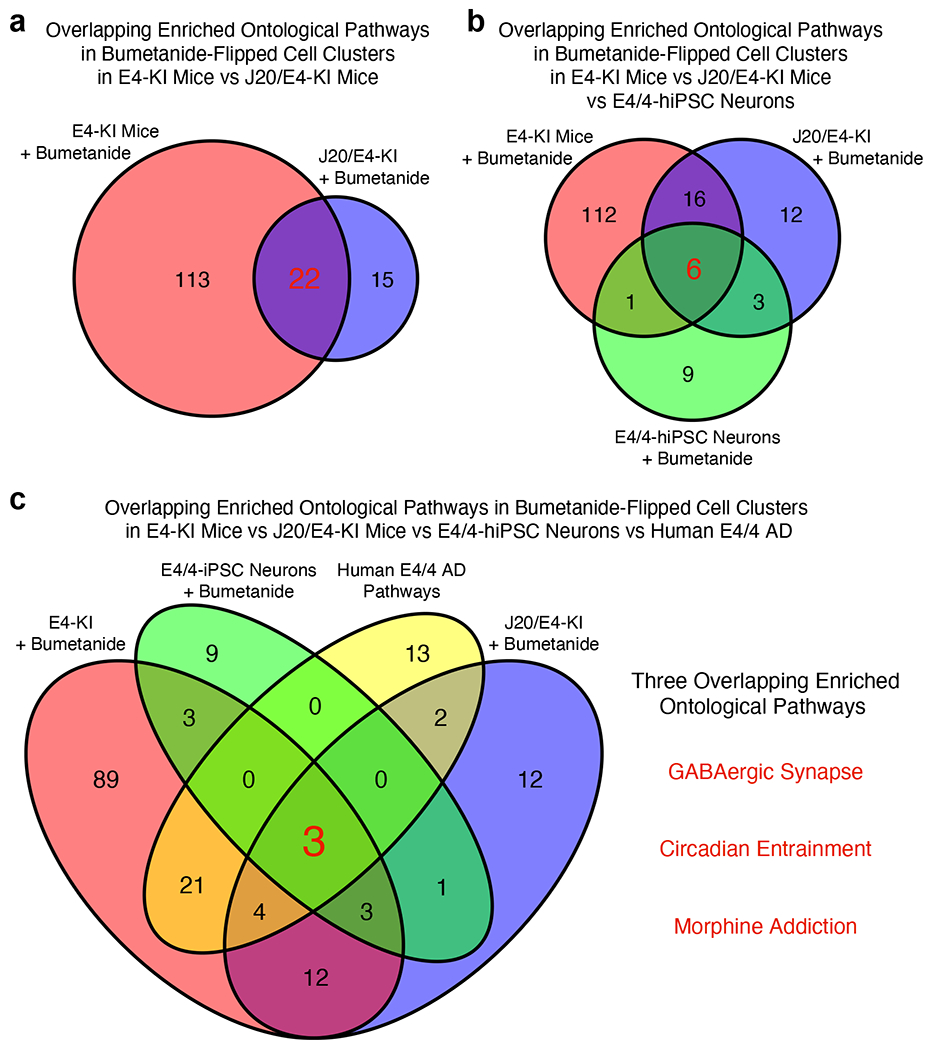
a, 22 overlapping enriched ontological pathways (Supplementary Table 17) in bumetanide-flipped cell clusters in apoE4-KI mice vs J20/E4-KI mice. b, Six overlapping enriched ontological pathways (Supplementary Table 18) in bumetanide-flipped cell clusters in apoE4-KI mice vs J20/E4-KI mice vs E4/4-hiPSC neurons. c, Three overlapping enriched ontological pathways in bumetanide-flipped cell clusters in apoE4-KI mice vs J20/E4-KI mice vs E4/4-hiPSC neurons vs human E4/4 signature of AD, which include GABAergic Synapse, Circadian Entrainment, Morphine Addiction pathways.
Extended Data Fig. 10. Bumetanide exposure is associated with a significantly lower AD prevalence in individuals over the age of 65 in two independent EHR databases.
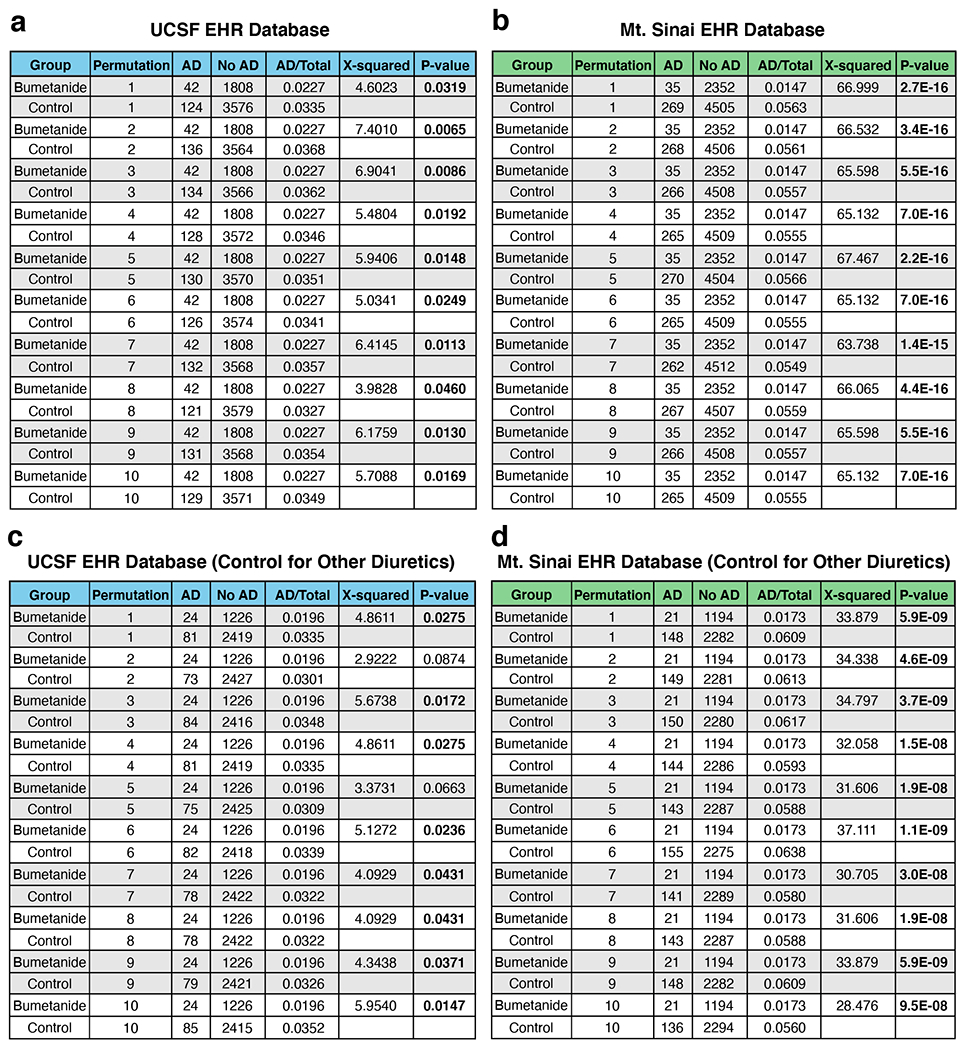
We evaluated two large-scale EHR databases (UCSF EHR and Mt. Sinai EHR) in a cross-sectional manner to test the association of bumetanide exposure with AD prevalence in individuals with the age of 65 or above using a propensity score matching approach to control cohort creation. a, AD prevalence in bumetanide-exposed cohort is significantly lower than those in all 10 randomly selected non-bumetanide-exposed cohorts in the UCSF EHR database. All 10 randomly selected 1:2 control cohorts were matched on propensity score which included age, sex, race, and hypertension and edema diagnosis. χ-squared test, all P < 0.05. b, AD prevalence in bumetanide-exposed cohort is significantly lower than those in all 10 randomly selected non-bumetanide-exposed cohorts in the Mt. Sinai EHR database. All 10 randomly selected 1:2 control cohorts were matched on propensity score which included age, sex, race, and hypertension and edema diagnosis. χ-squared test, all P < 0.0001. c, AD prevalence in bumetanide-exposed cohort is significantly lower than those in 8 out of 10 randomly selected non-bumetanide-exposed cohorts controlled for non-bumetanide diuretic drug use for hypertension and edema treatment in the UCSF EHR database. χ-squared test, 8 out of 10 P < 0.05. d, AD prevalence in bumetanide-exposed cohort is significantly lower than those in all 10 randomly selected non-bumetanide-exposed cohorts controlled for non-bumetanide diuretic drug use for hypertension and edema treatment in the Mt. Sinai EHR database. χ-squared test, all P < 0.0001.
Supplementary Material
Acknowledgments
This work was supported by grants AG057683 to Y.H. and M.S., AG048017 to Y.H., 1F31AG058439 to A.L.T., 1F31AG057150 to E.A.J., and TR001743, ES028047 to B.C., and AG059319 to B.S.G. and M.B. from the National Institutes of Health. The grant funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. GSE15222 data was downloaded from the NCBI GEO database. We thank Reuben Thomas for assistance on snRNA-seq analyses and Theodora Pak for editorial assistance.
Footnotes
Completing Interests Statement
Y. Huang is a cofounder and scientific advisory board member of E-Scape Bio, Inc., GABAeron, Inc., and Mederon Bio, LLC. M.S. is on the advisory board of Aria Pharmaceuticals. A pending patent application related to this work has been filed, on which Y. Huang, AT, MS, and PN were listed as inventors. Other authors declare no competing financial interests.
References
- 1.Golde TE, Schneider LS & Koo EH Anti-Aβ therapeutics in Alzheimer’s disease: the need for a paradigm shift. Neuron 69, 203–213 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang Y & Mucke L Alzheimer mechanisms and therapeutic strategies. Cell 148, 1204–1222 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu CC, Kanekiyo T, Xu H & Bu G Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 9, 106–118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verghese PB, Castellano JM & Holtzman DM Apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol. 10, 241–252 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahley RW & Huang Y Apolipoprotein E sets the stage: response to injury triggers neuropathology. Neuron 76, 871–885 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Corder EH, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923 (1993). [DOI] [PubMed] [Google Scholar]
- 7.Barnes EE & Yaffe K Vitamin E and donepezil for the treatment of mild cognitive impairment. N. Engl. J. Med 353, 951–952 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Marchant NL, King SL, Tabet N & Rusted JM Positive effects of cholinergic stimulation favor young APOE epsilon4 carriers. Neuropsychopharmacology 35, 1090–1096 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salloway S, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med 370, 322–333 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng F, et al. Prediction of drug-target interactions and drug repositioning via network-based inference. PLoS Comput. Biol 8, e1002503 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Csermely P, Korcsmaros T, Kiss HJ, London G & Nussinov R Structure and dynamics of molecular network: a novel paradigm of drug discovery: a comprehensive review. Pharmacol Ther 138, 333–408 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sirota M, et al. Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci. Transl. Med 3, 96ra77 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen B, et al. Computational Discovery of Niclosamide Ethanolamine, a Repurposed Drug Candidate That Reduces Growth of Hepatocellular Carcinoma Cells In Vitro and in Mice by Inhibiting Cell Division Cycle 37 Signaling. Gastroenterology 152, 2022–2036 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen B & Butte AJ Leveraging big data to transform target selection and drug discovery. Clin. Pharmacol. Ther 99, 285–297 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai X, Chen Y, Gao Z & Xu R Explore Small Molecule-induced Genome-wide Transcriptional Profiles for Novel Inflammatory Bowel Disease Drug. AMIA Jt. Summits. Transl. Sci. Proc 2016, 22–31 (2016). [PMC free article] [PubMed] [Google Scholar]
- 16.Lamb J, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Webster JA, et al. Genetic control of human brain transcript expression in Alzheimer disease. Am J Hum Genet 84, 445–458 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farrer LA, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. J. Am. Med. Assoc 278, 1349–1356 (1997). [PubMed] [Google Scholar]
- 19.Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen B, et al. Reversal of cancer gene expression correlates with drug efficacy and reveals therapeutic targets. Nat Commun 8, 16022 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gharaylou Z, et al. A Preliminary Study Evaluating the Safety and Efficacy of Bumetanide, an NKCC1 Inhibitor, in Patients with Drug-Resistant Epilepsy. CNS drugs 33, 283–291 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Goubert E, et al. Bumetanide prevents brain trauma-induced depressive-like behavior. Front. Mol. Neurosci 12, 12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lemonnier E, et al. A randomised controlled trial of bumetanide in the treatment of autism in children. Transl. psychiatry 2, e202 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lemonnier E, Lazartigues A & Ben-Ari Y Treating schizophrenia with the diuretic bumetanide: a case report. Clin. neuropharmacol 39, 115–117 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Lemonnier E, et al. Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl. psychiatry 7, e1056 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rahmanzadeh R, et al. Effect of co-administration of bumetanide and phenobarbital on seizure attacks in temporal lobe epilepsy. Basic Clin. neurosci 9, 408–416 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sivakumaran S & Maguire J Bumetanide reduces seizure progression and the development of pharmacoresistant status epilepticus. Epilepsia 57, 222–232 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao N, et al. Alzheimer’s Risk Factors Age, APOE Genotype, and Sex Drive Distinct Molecular Pathways. Neuron 106, 727–742 e726 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Najm R, Jones EA & Huang Y Apolipoprotein E4, inhibitory network dysfunction, and Alzheimer’s disease. Mol. Neurodegener 14, 24 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Milior G, et al. Electrophysiological properties of CA1 pyramidal neurons along the longitudinal axis of the mouse hippocampus. Sci. Rep 6, 38242 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bliss TV & Collingridge GL A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39 (1993). [DOI] [PubMed] [Google Scholar]
- 32.Selkoe DJ Alzheimer’s disease is a synaptic failure. Science 298, 789–791 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Andrews-Zwilling Y, et al. Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J. Neurosci 30, 13707–13717 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leung L, et al. Apolipoprotein E4 causes age- and sex-dependent impairments of hilar GABAergic interneurons and learning and memory deficits in mice. PLoS ONE 7, e53569 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knoferle J, et al. Apolipoprotein E4 produced in GABAergic interneurons causes learning and memory deficits in mice. J Neurosci 34, 14069–14078 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mucke L, et al. High-level neuronal expression of Ab1–42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci 20, 4050–4058 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bien-Ly N, Gillespie AK, Walker D, Yoon SY & Huang Y Reducing human apolipoprotein E levels attenuates age-dependent Aβ accumulation in mutant human amyloid precursor protein transgenic mice. J. Neurosci 32, 4803–4811 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang C, et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med 24, 647–657 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lennon MJ, Makkar SR, Crawford JD & Sachdev PS Midlife Hypertension and Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J Alzheimers Dis 71, 307–316 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Ho J, Tumkaya T, Aryal S, Choi H & Claridge-Chang A Moving beyond P values: data analysis with estimation graphics. Nat Methods 16, 565–566 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Kharod SC, Kang SK & Kadam SD Off-Label Use of Bumetanide for Brain Disorders: An Overview. Front. Neurosci 13, 310 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Puskarjov M, Kahle KT, Ruusuvuori E & Kaila K Pharmacotherapeutic targeting of cation-chloride cotransporters in neonatal seizures. Epilepsia 55, 806–818 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Töpfer M, et al. Consequences of inhibition of bumetanide metabolism in rodents on brain penetration and effects of bumetanide in chronic models of epilepsy. Eur. J. Neurosci 39, 673–687 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Gharaylou Z, et al. Longitudinal effects of bumetanide on neuro-cognitive functioning in drug-resistant epilepsy. Front. Neurol 10, 483 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sala Frigerio C, et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Abeta Plaques. Cell Rep 27, 1293–1306 e1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nguyen AT, et al. APOE and TREM2 regulate amyloid-responsive microglia in Alzheimer’s disease. Acta Neuropathol 140, 477–493 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Griswold AJ, et al. Increased APOE epsilon4 expression is associated with the difference in Alzheimer’s disease risk from diverse ancestral backgrounds. Alzheimers Dement 17, 1179–1188 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Caselli RJ Obstructive sleep apnea, apolipoprotein E e4, and mild cognitive impairment. Sleep Med. 9, 816–817 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Drogos L, et al. Evidence of association between sleep quality and APOE ε4 in healthy older adults: A pilot study. Neurology 87, 1836–1842 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tranah GJ, et al. APOEε4 and slow wave sleep in older adults. PloS One 13, e0191281 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gozal D, Capdevila OS, Kheirandish-Gozal L & Crabtree VM APOE epsilon 4 allele, cognitive dysfunction, and obstructive sleep apnea in children. Neurology 69, 243–249 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Listos J, et al. The Mechanisms Involved in Morphine Addiction: An Overview. Int J Mol Sci 20(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Falcon E & McClung CA A role for the circadian genes in drug addiction. Neuropharmacology 56 Suppl 1, 91–96 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hamanaka H, et al. Altered cholesterol metabolism in human apolipoprotein E4 knock-in mice. Hum. Mol. Genet 9, 353–361 (2000). [DOI] [PubMed] [Google Scholar]
- 55.Sullivan PM, Mace BE, Maeda N & Schmechel DE Marked regional differences of brain human apolipoprotein E expression in targeted replacement mice. Neuroscience 124, 725–733 (2004). [DOI] [PubMed] [Google Scholar]
- 56.Workman C, et al. A new non-linear normalization method for reducing variability in DNA microarray experiments. Genome Biol 3, research0048.0041–0048.0016 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lui JH, et al. Radial glia require PDGFD-PDGFRbeta signalling in human but not mouse neocortex. Nature 515, 264–268 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raju CS, et al. Secretagogin is expressed by developing neocortical GABAergic neurons in humans but not mice and increases neurite arbor size and complexity. Cereb Cortex 28, 1946–1958 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lein ES, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176 (2007). [DOI] [PubMed] [Google Scholar]
- 61.Rosenberg AB, et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dijk D.v., et al. Recovering gene interactions from single-cell data using data diffusion. Cell 174, 716–729 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article (or in its supplementary information files) or deposited in the Gene Expression Omnibus (GEO), which are also available from the corresponding authors’ labs. Publicly available datasets used are available in the Gene Expression Omnibus (GEO) under the accession number GSE15222 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE15222) with the associated covariate data found on the Myers Lab website (http://labs.med.miami.edu/myers/LFuN/LFUN/DATA.html) and the associated Google drive (: https://drive.google.com/drive/folders/1ud5F9WN9Xx3oXIkb5xIg1b_zz1nzp3IR) in the “samples.covar.zip” file. The CMap database is available in Sage Synapse in the HCC_NEN project from the Bin Chen lab under the accession number syn6187678, and is linked to the Bin Chen Lab Gitlab repository (also see Code Availability section below). The publicly available RNA-Seq dataset of aging apoE4-KI mouse brains was from Zhao et al. (https://doi.org/10.7303/syn20808171)28. Figure 4, 6 and extended data figure 3, 4, 7, and 8 have associated mouse snRNA-seq data or iPSC-derived human neuron bulk-RNA-seq data generated in this study, which are available in the Gene Expression Omnibus (GEO) under the accession number GSE182765. The UCSF EHR database and the Mt. Sinai EHR database are not yet available to the general public.