

Abstract
BACKGROUND
Although hypomethylating agents are currently used to treat patients with cancer, whether they can also reactivate and up-regulate oncogenes is not well elucidated.
METHODS
We examined the effect of hypomethylating agents on SALL4, a known oncogene that plays an important role in myelodysplastic syndrome and other cancers. Paired bone marrow samples that were obtained from two cohorts of patients with myelodysplastic syndrome before and after treatment with a hypomethylating agent were used to explore the relationships among changes in SALL4 expression, treatment response, and clinical outcome. Leukemic cell lines with low or undetectable SALL4 expression were used to study the relationship between SALL4 methylation and expression. A locus-specific demethylation technology, CRISPR–DNMT1-interacting RNA (CRISPR-DiR), was used to identify the CpG island that is critical for SALL4 expression.
RESULTS
SALL4 up-regulation after treatment with hypomethylating agents was observed in 10 of 25 patients (40%) in cohort 1 and in 13 of 43 patients (30%) in cohort 2 and was associated with a worse outcome. Using CRISPR-DiR, we discovered that demethylation of a CpG island within the 5′ untranslated region was critical for SALL4 expression. In cell lines and patients, we confirmed that treatment with a hypomethylating agent led to demethylation of the same CpG region and up-regulation of SALL4 expression.
CONCLUSIONS
By combining analysis of patient samples with CRISPR-DiR technology, we found that demethylation and up-regulation of an oncogene after treatment with a hypomethylating agent can indeed occur and should be further studied. (Funded by Associazione Italiana per la Ricerca sul Cancro and others.)
INTEREST IN EPIGENETIC TARGETING IN cancer treatment is increasing. One example is the use of DNA hypomethylating agents. Two hypomethylating agents, 5-aza-2′-deoxycytidine (decitabine) and 5-azacytidine, are being used clinically. There are many mechanisms of action of these drugs, including irreversibly binding to and inhibiting DNA methyltransferases,1–4 which result in hypomethylation and up-regulation of genes such as tumor suppressors.5 Other major mechanisms of action include incorporation of the hypomethylating agent into newly synthesized DNA, which triggers a DNA-damage response and leads to cytotoxic effects in cancer cells,6,7 as well as alteration of immune responses.8–10
We hypothesized that the global effect of treatment with hypomethylating agents would not only contribute to the demethylation of tumor-suppressor genes but may also induce demethylation of oncogenes. To test this theory, we focused on candidate genes with the following features: they are aberrantly expressed in human cancers; they are functionally proved to be oncogenes with the use of methods such as murine models; and DNA methylation may be important for the expression of these genes. This led to our current study, in which we examined how hypomethylating agents could activate the known oncofetal protein, spalt-like transcription factor 4 (SALL4), in patients with myelodysplastic syndrome, as a model to examine the effects of hypomethylating agents on up-regulation of oncogenes.
SALL4 plays an essential role in myelodysplastic syndrome and acute myeloid leukemia leukemogenesis11 and tumorigenesis in several solid tumors, including germ-cell tumors, hepatocellular carcinoma, breast cancer, and lung cancer.12–14 During development, SALL4 maintains the self-renewal and pluripotency of embryonic stem cells,15 but it is normally repressed in most adult organs with the exception of germ cells and CD34+ hematopoietic stem cells.16 SALL4 is reactivated or aberrantly expressed in a multitude of cancers and has been identified by meta-analysis as a poor prognostic factor. Mechanistically, up-regulation of SALL4 promotes proliferation, metastasis, and drug resistance of cancer cells,17,18 through a number of mechanisms, such as repression of tumor-suppressor genes19 and activation of other oncogenes.20 In hematologic cancers, SALL4 is aberrantly expressed in high-risk myelodysplastic syndrome,21 acute myeloid leukemia,11,20 the blast phase of chronic myeloid leukemia,22 and precursor B-cell lymphoblastic leukemia or lymphoma.23 In a murine model with constitutive SALL4 expression, mice had myelodysplastic syndrome–like features and subsequently leukemic transformation through activation of the Wnt–beta-catenin pathway.24 Methylation is known to regulate SALL4 expression and reactivation in solid tumors.25,26 The expression of SALL4 in hematologic cancers may also be related to its DNA methylation status. The effect of hypomethylating agents on SALL4 expression and its clinical implications are unknown.
Hypomethylating agents are now being used to treat blood cancers as well as solid tumors. Because they were first approved by the Food and Drug Administration (FDA) to treat patients with myelodysplastic syndrome more than a decade ago, we therefore retrospectively analyzed SALL4 expression and clinical survival among 68 patients with myelodysplastic syndrome in two cohorts (25 patients in cohort 1 and 43 patients in cohort 2) with bone marrow samples obtained before and after treatment with a hypomethylating agent. Myelodysplastic syndrome comprises heterogeneous myeloid disorders characterized by cytopenias and dysplasia in peripheral blood and bone marrow, with ineffective hematopoiesis and a variable risk of leukemic transformation.27,28 To define the mechanisms of altered expression of SALL4 in response to treatment with a hypomethylating agent, we used a CRISPR-DiR (clustered regularly interspaced short palindromic repeats–DNA methyltransferase 1 [DNMT1]–interacting RNA) approach. First, we identified and demethylated the CpG island that is critical for SALL4 expression. We then evaluated the effects of treatment with a hypomethylating agent on the methylation status of SALL4 in samples obtained from patients with myelodysplastic syndrome.
METHODS
PATIENTS AND SAMPLE COLLECTION
Bone marrow samples in cohort 1 were obtained from 37 patients with newly diagnosed myelodysplastic syndrome who were enrolled in the BMT-AZA trial.29,30 The current study was overseen and approved by the University of Rome Tor Vergata. Written informed consent was obtained from the patients before inclusion in the study. CD34− bone marrow mononuclear cells obtained from 10 healthy donors and CD34+ cells obtained from 5 healthy donors were used as control cohorts. Of the 37 patients with myelodysplastic syndrome, 25 had paired bone marrow samples collected before and after four cycles of azacytidine. In cohort 2, a total of 43 bone marrow samples were obtained from the Institute of Hematology and Blood Diseases Hospital in Tianjin, China, before or after three to five cycles of treatment with a hypomethylating agent. Patient samples were obtained with written informed consent in accordance with the Declaration of Helsinki and approval of the human research ethics committee at the Institute of Hematology and Blood Disease Hospital, Chinese Academy of Medical Sciences. Bone marrow mononuclear cells were isolated by Ficoll gradient centrifugation with the use of Lympholyte-H (Cedarlane), in accordance with the instructions of the manufacturer. Myelodysplastic syndrome was diagnosed in accordance with the 2016 World Health Organization (WHO) classification.31 Other clinical characteristics, including age at diagnosis, sex, Revised International Prognostic Scoring System risk status, WHO Classification-based Prognostic Scoring System risk status, response after treatment with a hypomethylating agent, peripheral-blood counts, bone marrow blast counts, and cytogenetic features, were also reviewed. The definition of first response follows the International Working Group criteria.32 Patients who had a response included those having complete remission, partial remission, or hematologic improvement, whereas patients who did not have a response included those with stable disease or progressive disease.
STATISTICAL ANALYSIS
Clinical characteristics and mutational profiling are presented as the number and percentage of patients. Data are presented as medians and interquartile ranges for skewed data. Categorical variables were compared with the use of Fisher’s exact test or the chi-square test, as appropriate, and continuous variables were compared with the use of the Wilcoxon rank-sum test. We also used the Wilcoxon rank-sum test for paired observations and factor change in SALL4 messenger RNA (mRNA) levels in patients who had a response as compared with those who did not have a response.
Progression-free survival was defined as the time from treatment with a hypomethylating agent to disease progression or death from the treatment, and overall survival was defined as time from treatment with a hypomethylating agent to death. Overall and progression-free survival were analyzed with the use of the Kaplan–Meier product-limit method with censoring for patients who did not have disease progression or who did not die during the treatment period. A log-rank test was used to compare survival curves for statistical significance. Hazard ratios and 95% confidence intervals were calculated with the use of a Cox proportional-hazards model. All prognostic factors with a P value of less than 0.1 in the univariate model were further entered into the multivariate analysis.
All statistical testing was performed with the use of two-tailed tests; a P value of less than 0.05 was considered to indicate statistical significance. All analyses were performed with the use of SPSS statistical software, version 22 (SPSS). A droplet digital polymerase-chain-reaction (ddPCR) experiment was repeated three times for each group of experiments, and Student’s t-test was used. Additional materials are described in the Supplementary Appendix, available with the full text of this article at NEJM.org.
RESULTS
HYPOMETHYLATING AGENTS AND UP-REGULATION OF SALL4
To evaluate the effect of hypomethylating agents on SALL4 expression after treatment, we first measured the baseline SALL4 expression at diagnosis in bone marrow mononuclear cells obtained from patients with myelodysplastic syndrome before azacytidine treatment in cohort 1. Levels of SALL4 mRNA were significantly higher in 37 patients with myelodysplastic syndrome than in healthy donors (P=0.002) (Fig. S1A in the Supplementary Appendix), a finding similar to what has been reported previously.21 In cohort 1, a total of 25 patients had available paired bone marrow samples at diagnosis and after four cycles of azacytidine. Of these 25 patients, 12 (48%) were classified as having had a response: 9 (36%) had a complete response, 2 (8%) had a partial response, and 1 (4%) had hematologic improvement. The remaining 13 patients (52%) were classified as not having had a response: 10 (40%) had stable disease, and 3 (12%) had progressive disease. Baseline SALL4 expression at diagnosis did not differ substantially among these groups. In cohort 2, of the 43 patients, 20 (47%) were classified as having had response: 15 (35%) had a complete response, and 5 (12%) had hematologic improvement. The remaining 23 patients (53%) were classified as not having had a response: 18 (42%) had stable disease, and 5 (12%) had progressive disease.
These 68 patients were then stratified according to the factor change in SALL4 mRNA expression before and after treatment with a hypomethylating agent. A waterfall plot depicting the factor changes in SALL4 mRNA levels showed that patients can be separated into two groups, one with SALL4 up-regulation and the other with SALL4 down-regulation. In cohort 1, a total of 10 of the 25 patients (40%) had an increase in SALL4 expression and 15 patients (60%) had a decrease in SALL4 expression after four cycles of azacytidine treatment (Fig. 1A). The median log2 factor change was 2.78 (interquartile range, 2.15 to 5.65) in patients with SALL4 up-regulation and −2.25 (interquartile range, −1.26 to −4.45) in those with SALL4 down-regulation. In cohort 2, a total of 13 patients (30%) had SALL4 up-regulation and 30 patients (70%) had SALL4 down-regulation after three to five cycles of treatment with a hypomethylating agent (Fig. 1B). The median log2 factor change was 1.99 (interquartile range, 0.70 to 3.05) in patients with SALL4 up-regulation and −1.99 (interquartile range, −0.84 to −2.71) in those with SALL4 down-regulation. No significant difference in the factor change in SALL4 mRNA was noted between patients who had a response and those who did not have a response in cohort 1 (Fig. S1B) and cohort 2 (Fig. S1C).
Figure 1. Increased SALL4 Expression in Patients with Myelodysplastic Syndrome after Treatment with a Hypomethylating Agent.

Panel A shows a waterfall plot of the log2 factor change in SALL4 expression in 25 patients after four cycles of azacytidine treatment in cohort 1. Panel B shows a waterfall plot of the log2 factor change in SALL4 expression in 43 patients after three to five cycles of treatment with a hypomethylating agent in cohort 2. The term t0 denotes before any cycles of treatment, and t4 after four cycles of treatment. Panel C shows overall survival among patients with SALL4 up-regulation and those with SALL4 down-regulation in cohort 1, and Panel D shows the corresponding data in cohort 2. In Panels C and D, tick marks indicate censored data. A log-rank test was used to compare survival curves for statistical significance in Panels C and D.
When overall survival was compared between the patients with SALL4 up-regulation and those with SALL4 down-regulation in both cohort 1 and cohort 2, those with SALL4 up-regulation had poorer overall survival than those with SALL4 down-regulation (P=0.03 in cohort 1 and P=0.04 in cohort 2) (Fig. 1C and 1D). These findings indicate that SALL4 up-regulation may be associated with worse survival.
The demographic and clinical characteristics of the patients with SALL4 up-regulation and those with SALL4 down-regulation are shown in Tables 1 and 2. The median follow-up after the initiation of therapy was 14.1 months (interquartile range, 9.0 to 20.4) in cohort 1 and 17.0 months (interquartile range, 12.0 to 28.0) in cohort 2. We further conducted a Cox proportional-hazards model analysis of prognostic factors for overall survival on the basis of patient characteristics at diagnosis, mutational profile,30 and SALL4 expression changes. In a multivariate analysis, SALL4 up-regulation was a common independent negative predictor of overall survival (cohort 1: hazard ratio for death, 6.48; 95% confidence interval [CI], 1.06 to 39.67; cohort 2: hazard ratio, 2.74; 95% CI, 0.93 to 8.03) (Tables S1 and S2). In addition, the presence of a RUNX1 mutation in cohort 1 and the lack of a treatment response in cohort 2 were also negative predictors of overall survival in a multivariate analysis.
Table 1.
Baseline Characteristics of 25 Patients Treated with a Hypomethylating Agent in Cohort 1.*
| Characteristic | SALL4 Up-Regulation (N = 10) | SALL4 Down-Regulation (N = 15) |
|---|---|---|
| Median age (IQR) — yr | 59.6 (46.2–61) | 59.5 (45–61.5) |
| Sex — no. (%) | ||
| Male | 9 (90) | 8 (53) |
| Female | 1 (10) | 7 (47) |
| IPSS risk status — no. (%) | ||
| Low or intermediate-1 | 1 (10) | 1 (7) |
| Intermediate-2 or high | 9 (90) | 13 (87) |
| WPSS risk status — no. (%) | ||
| Very low or low | 1 (10) | 0 |
| Intermediate | 1 (10) | 2 (13) |
| High or very high | 8 (80) | 10 (67) |
| IPSS-R risk status — no./total no. (%) | ||
| Very low | 0/9 | 1/13 (8) |
| Low | 0/9 | 1/13 (8) |
| Intermediate | 2/9 (22) | 2/13 (15) |
| High | 3/9 (33) | 2/13 (15) |
| Very high | 4/9 (44) | 7/13 (54) |
| Treatment response — no. (%) | ||
| Yes | 5 (50) | 7 (47) |
| No | 5 (50) | 8 (53) |
| Median white-cell count (IQR) — per mm3 | 2450 (1900–4015) | 3320 (2280–10,410) |
| Median absolute neutrophil count (IQR) — per mm3 | 400(215–1490) | 700 (400–2000) |
| Median hemoglobin level (IQR) — g/dl | 9.9 (8.6–10.6) | 9.4 (8.7–11.5) |
| Median platelet count (IQR) — per mm3 | 49,500 (26,750–164,250) | 77,000 (49,000–129,000) |
| Median percentage of blasts in bone marrow aspirate (IQR) | 13 (11–15) | 12 (3.8–17.8) |
| Cytogenetic features of MDS — no. (%)† | ||
| Good karyotype | 4 (40) | 5 (33) |
| Intermediate karyotype | 2 (20) | 2 (13) |
| Poor karyotype | 3 (30) | 6 (40) |
| Mutational profiling — no. (%)‡ | ||
| TET2 mutation | 1 (10) | 6 (40) |
| ASXL1 mutation | 3 (30) | 6 (40) |
| RUNX1 mutation | 2 (20) | 4 (27) |
| SETBP1 mutation | 2 (20) | 4 (27) |
| TP53 mutation | 3 (30) | 2 (13) |
| ZRSF2 mutation | 4 (40) | 1 (7) |
| DNMT3A mutation | 1 (10) | 3 (20) |
| SRSF2 mutation | 1 (10) | 3 (20) |
| SF3B1 mutation | 1 (10) | 2 (13) |
| U2AF1 mutation | 3 (30) | 0 |
| CEBPA mutation | 0 | 3 (20) |
| CBL mutation | 1 (10) | 1 (7) |
| ETV6 mutation | 0 | 2 (13) |
| IDH2 mutation | 1 (10) | 1 (7) |
| NRAS mutation | 0 | 2 (13) |
| CSF3R mutation | 1 (10) | 0 |
| JAK2 mutation | 1 (10) | 0 |
IPSS denotes International Prognostic Scoring System, IPSS-R Revised IPSS, IQR interquartile range, MDS myelodysplastic syndrome, and WPSS World Health Organization Classification-based Prognostic Scoring System.
Three patients (one with SALL4 up-regulation and two with SALL4 down-regulation) had no metaphase of cytogenetic analysis.
No mutations in ABL1, BRAF, CALR, EZH2, FLT3, HRAS, IDH1, KIT, KRAS, MPL, NPM1, PTPN11, and WT1 were observed in either group.
Table 2.
Baseline Characteristics of 43 Patients Treated with a Hypomethylating Agent in Cohort 2.
| Characteristics | SALL4 Up-Regulation (N = 13) | SALL4 Down-Regulation (N = 30) |
|---|---|---|
| Median age (IQR) — yr | 65 (60–69) | 62.5 (59.8–67.5) |
| Sex — no. (%) | ||
| Male | 11 (85) | 17 (57) |
| Female | 2 (15) | 13 (43) |
| IPSS risk status — no./total no. (%) | ||
| Low or intermediate-1 | 8/11 (73) | 17/26 (65) |
| Intermediate-2 or high | 3/11 (27) | 9/26 (35) |
| WPSS risk status — no./total no. (%) | ||
| Very low or low | 1/11 (9) | 2/26 (8) |
| Intermediate | 2/11 (18) | 9/26 (35) |
| High or very high | 8/11 (73) | 15/26 (58) |
| IPSS-R risk status — no./total no. (%) | ||
| Very low | 0/13 | 1/26 (4) |
| Low | 1/13 (8) | 1/26 (4) |
| Intermediate | 4/13 (31) | 7/26 (27) |
| High | 6/13 (46) | 13/26 (50) |
| Very high | 2/13 (15) | 4/26 (15) |
| Treatment response — no. (%) | ||
| Yes | 5 (38) | 15 (50) |
| No | 8 (62) | 15 (50) |
| Median white-cell count (IQR) — per mm3 | 2220 (1310–4310) | 2180 (1830–2750) |
| Median absolute neutrophil count (IQR) — per mm3 | 780 (330–2240) | 900 (630–1370) |
| Median hemoglobin level (IQR) — g/dl | 7.0 (6.2–10.2) | 8.1 (7.0–9.6) |
| Median platelet count (IQR) — per mm3 | 47,000 (31,000–120,000) | 70,000 (38,000–107,000) |
| Median percentage of blasts in bone marrow aspirate (IQR) | 6 (5.5–10.8) | 7.3 (5.4–11.1) |
| Cytogenetic features of MDS — no./total no. (%)* | ||
| Good karyotype | 10/11 (91) | 22/26 (85) |
| Intermediate karyotype | 1/11 (9) | 0/26 |
| Poor karyotype | 0/11 | 4/26 (15) |
| Mutational profiling — no./total no. (%)† | ||
| TET2 mutation | 1/13 (8) | 2/28 (7) |
| ASXL1 mutation | 4/13 (31) | 5/28 (18) |
| RUNX1 mutation | 3/13 (23) | 3/28 (11) |
| SETBP1 mutation | 0/13 | 2/28 (7) |
| TP53 mutation | 1/13 (8) | 4/28 (14) |
| DNMT3A mutation | 0/13 | 1/28 (4) |
| SRSF2 mutation | 0/13 | 1/28 (4) |
| SF3B1 mutation | 3/13 (23) | 2/28 (7) |
| U2AF1 mutation | 3/13 (23) | 4/28 (14) |
| CEBPA mutation | 0/13 | 1/28 (4) |
| IDH2 mutation | 1/13 (8) | 0/28 |
| NRAS mutation | 0/13 | 3/28 (11) |
| JAK2 mutation | 1/13 (8) | 1/28 (4) |
| EZH2 mutation | 1/13 (8) | 1/28 (4) |
| FLT3 mutation | 1/13 (8) | 0/28 |
| IDH1 mutation | 1/13 (8) | 2/28 (7) |
| KRAS mutation | 1/13 (8) | 0/28 |
| NPM1 mutation | 0/13 | 2/28 (7) |
Two patients with SALL4 down-regulation had no metaphase of cytogenetic analysis. Inadequate Giemsa banding in cytogenetic analysis was seen for one patient with SALL4 up-regulation and two patients with SALL4 down-regulation.
No mutations in ABL1, BRAF, CALR, CBL, CSF3R, ETV6, KIT, MPL, PTPN11, WT1, and ZRSF2 were observed in either group. Two patients with SALL4 down-regulation did not undergo next-generation sequencing.
Because somatic mutations, including epigenetic factors such as DNMT3A, IDH1, IDH2, and TET2, are common in patients with myelodysplastic syndrome,33 we evaluated whether SALL4 up-regulation or down-regulation in patients after treatment was related to their preexisting mutation status. Common somatic mutations in 30 genes, known to be frequently mutated in patients with myelodysplastic syndrome, are reported in Figure S3. At diagnosis, we identified at least one mutation in 22 of 25 patients (88%) in cohort 1 and in 32 of 41 patients (78%) in cohort 2. A total of 19 of 22 patients (86%) in cohort 1 and 16 of 32 patients (50%) in cohort 2 had more than one gene mutation. From these observations, it appears that SALL4 up-regulation or down-regulation in patients after treatment with a hypomethylating agent was not related to their preexisting mutation status.
DEMETHYLATION OF A CRITICAL CPG REGION AND UP-REGULATION OF SALL4
We next investigated the mechanisms of SALL4 up-regulation after treatment with a hypomethylating agent. One possibility is that such treatment can lead to demethylation of a CpG island that is critical for SALL4 expression. The differential methylation of the SALL4 locus has been observed in K562-induced pluripotency reprogrammed cells.34 The major CpG island of SALL4 is located across the 2000-bp locus, including 5′ untranslated region (5′UTR)–Exon 1–Intron 1. Within this CpG island, methylation of 5′UTR–Exon 1–Intron 1, a specific 500-bp DNA segment, was further observed to be negatively correlated with SALL4 expression in patients with hepatocellular carcinoma and in hepatocellular-carcinoma cell lines25 (Fig. 2A). We hypothesized that demethylation of this region could lead to up-regulation of SALL4.
Figure 2. Demethylation of a Critical CpG Island and SALL4 Expression in Leukemic Cells Treated with CRISPR-DiR.

Panel A shows the CpG region within SALL4 5′ untranslated region (5′UTR)–Exon 1–Intron 1. Depicted at the top of the diagram are the 30 CpG residues; depicted below are the four SALL4 exons (in blue) and introns (in purple), with the location of the 30 CpG residues in the Exon 1–Intron 1 region. The location of the target of the guide RNA for CRISPR-DiR (clustered regularly interspaced short palindromic repeats–DNA methyltransferase 1 [DNMT1]–interacting RNA) is shown below, targeting CpG number 11. The term dCas9 denotes nuclease-dead Cas9, and sgDiR single-guide DNMT1-interacting RNA. Panel B shows bisulfite sequencing after CRISPR-DiR of HL-60 cells transduced with either a nontargeting negative control guide RNA (DiR_NT) or a targeting guide RNA (DiR_SALL4), leading to demethylation, with higher methylation indicated by redder shading. Panel C shows up-regulation of SALL4 transcript (copies per cell assessed by droplet digital polymerase-chain-reaction [ddPCR] assay) in HL-60 cells treated with CRISPR-DiR 8 days after transduction of the CRISPR-DiR. A Wilcoxon rank-sum test was used to evaluate the statistical difference in copies per cell between NT (in three patients) and sgSALL4 (in three patients). Data are shown as means, and error bars indicate the standard deviation.
To localize the critical CpG region responsible for regulation of SALL4 expression and to test the causal relationship between DNA methylation of SALL4 and RNA expression, we applied the CRISPR-DiR technique.35 This approach uses induction of locus-specific demethylation by blocking DNMT1 activity36 in cell lines with undetectable or low SALL4 expression.
In the CRISPR-DiR method, two loops of the guide RNA that are not required for guide function have been replaced with sequences that specifically interact with and inhibit DNMT1.35,36 This modified guide RNA can achieve site-specific demethylation.35 We tested several site-specific guide RNAs around the major CpG island of SALL4, and only sgSALL4_1 (named as DiR_SALL4 here), targeting around the 5′UTR CpG 11 region, showed effective demethylation ability.25 We further monitored the methylation of the 5′UTR CpG region in HL-60 cells (with no or low SALL4 expression) after treatment with CRISPR-DiR (Fig. 2A). On transduction of HL-60 cells with DiR_SALL4, substantial demethylation changes were observed after 8 days (Fig. 2B) as well as increased SALL4 transcript levels (Fig. 2C), as compared with a control scrambled guide RNA (DiR-NT). Similar demethylation results by DiR_SALL4 were observed in other SNU387 cells expressing low or no SALL4, whereas CRISPR-DiRs targeting neighboring regions could not demethylate.25 These findings show that demethylation of this region by SALL4 locus-specific CRISPR-DiR can lead to up-regulation of SALL4.
HYPOMETHYLATING AGENTS AND DEMETHYLATION OF SALL4
We then tested whether SALL4 could be up-regulated by hypomethylating agents. Using HL-60 cells, we first evaluated the dynamics of SALL4 mRNA levels through a cycle of decitabine, using a dosage range of 100 to 500 nmol per liter. The number of mRNA copies per cell (assessed by means of ddPCR assay) and protein level (assessed by means of Western blot analysis) for SALL4 was measured at day 5. We noticed a dose-dependent up-regulation of SALL4 mRNA expression at day 5 (Fig. 3A and B).
Figure 3. Demethylation of a Critical CpG Island and SALL4 Expression in Leukemic Cells and Patients with Myelodysplastic Syndrome Treated with a Hypomethylating Agent.
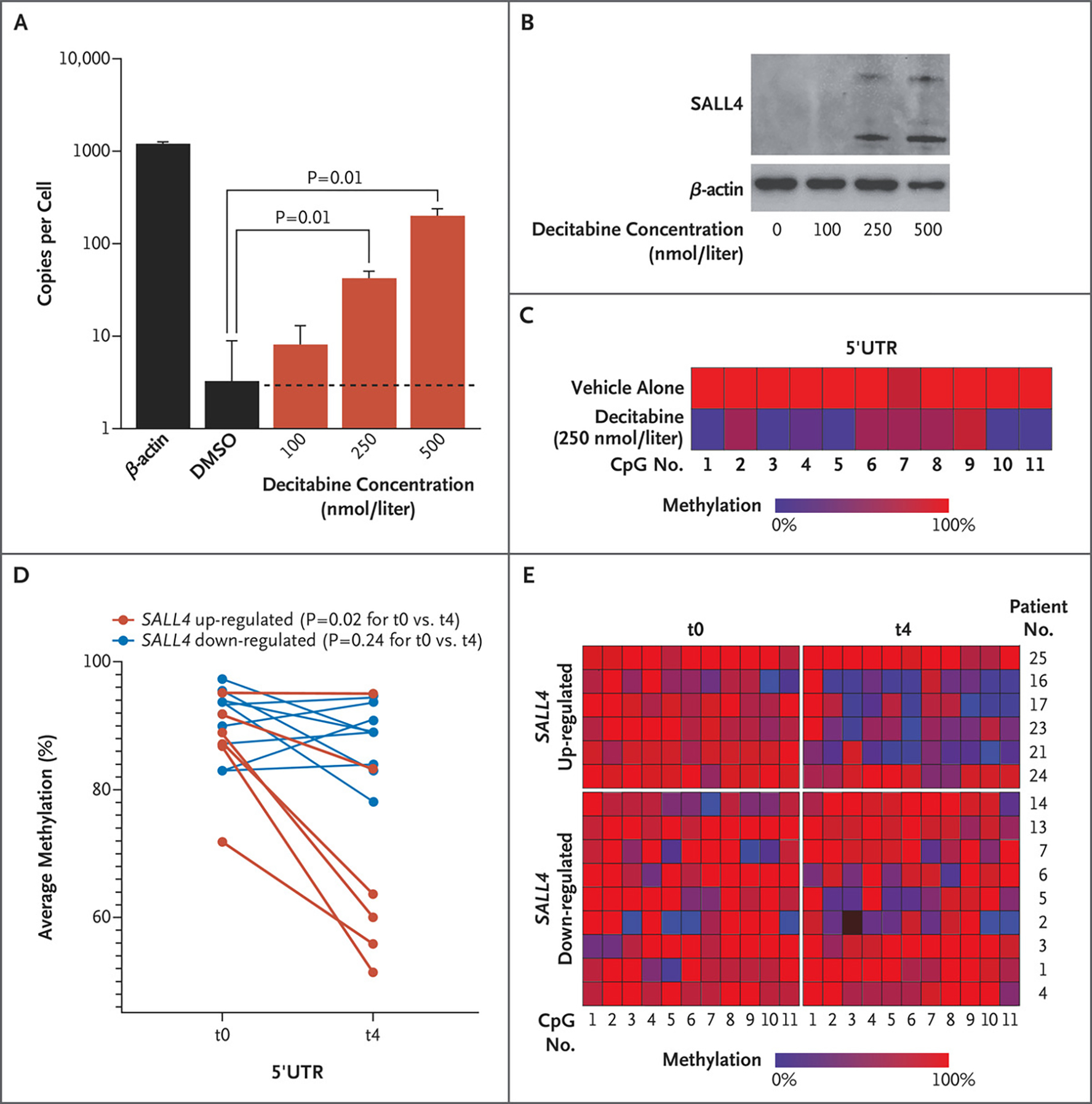
Panel A shows SALL4 copies per cell assessed by ddPCR assay in HL-60 cells treated with decitabine. A Wilcoxon rank-sum test was used to evaluate the statistical difference in copies per cell between dimethylsulfoxide (DMSO) and decitabine treatments. Data are shown as means, and error bars indicate standard deviations of three biologic replicates. Panel B shows Western blot analysis of HL-60 cells treated with decitabine. Panel C shows methylation profiling in HL-60 cells treated with vehicle alone as compared with those treated with decitabine at a dose of 250 nmol per liter. Treatment with a hypomethylating agent leads to demethylation of SALL4 in patients with myelodysplastic syndrome. Panel D shows average methylation across SALL4 5′UTR CpG loci (1 through 11) in patients with myelodysplastic syndrome before any cycles of azacytidine treatment (t0) and after four cycles of such treatment (t4). Red dots indicate patients with up-regulation of SALL4 expression after azacytidine treatments, and blue dots indicate patients with down-regulation of SALL4 after azacytidine treatments. Methylation values at t0 were compared with methylation values at t4 in each up- and down-regulated group separately. The average difference in methylation between the two time points was calculated with the use of a Wilcoxon matched-pairs signed-rank test (one-tailed) for each up- and down-regulated group separately. Panel E shows methylation changes in six patients with SALL4 up-regulation and nine patients with SALL4 down-regulation at t0 and t4.
We next examined the methylation status of the SALL4 5′UTR CpG island region after decitabine treatment at a dose of 250 nmol per liter and found that this region was demethylated in HL-60 cells, in accordance with our CRISPR-DiR result (Fig. 3C). Similar results were also observed in another SALL4-low leukemic cell line, K562 (Fig. S4A, S4B, and S4C).
We next examined the methylation changes in bone marrow samples from patients with myelodysplastic syndrome before and after treatment with a hypomethylating agent. In six patients with SALL4 up-regulation, we noticed decreased methylation at the critical 5′UTR CpG region (Fig. 3D and 3E). Conversely, we did not observe changes in the methylation at the same CpG region in nine patients with SALL4 down-regulation after treatment with a hypomethylating agent (Fig. 3D and 3E).
DISCUSSION
Since its first approval by the FDA to treat myelodysplastic syndrome in 2004, hypomethylating-agent therapy has been used in patients with hematologic cancers and solid tumors. In this study, we were interested in investigating whether hypomethylating agents could activate oncogenes as an unintended consequence. We used myelodysplastic syndrome as the disease model to test our hypothesis because of the long-standing use of hypomethylating-agent therapy in this patient population and because SALL4 has been shown to act as an oncogene in experimental animal models of myelodysplastic syndrome and acute myeloid leukemia. Although continuous hypomethylating-agent therapy in patients with myelodysplastic syndrome who had a response could improve their clinical characteristics, overall survival after such therapy in a “real-world” evaluation of patients with high-risk myelodysplastic syndrome or low-blast-count acute myeloid leukemia was lower than that expected in clinical trials,37 and the average prognosis after failure of a hypomethylating agent was less than 6 months.38 The mechanisms for primary and secondary drug resistance to hypomethylating agents and associated prognostic factors are still under active investigation.
In our two cohorts of patients with myelodysplastic syndrome from distinct ethnic backgrounds, we observed that after treatment with a hypomethylating agent, 23 of 68 patients (34%) had SALL4 up-regulation and 45 (66%) had SALL4 down-regulation. We observed that a poor long-term outcome correlates with SALL4 up-regulation. The important role of SALL4 in myelodysplastic syndrome and acute myeloid leukemia has been shown in previous studies.11 In SALL4 transgenic (Tg) mice, ineffective hematopoiesis and myelodysplastic syndrome–like features were observed: increased apoptosis with decreased complete blood-cell counts and abnormal morphologic features of blood cells. Impairment of DNA-damage repair was also noted in these Tg mice. Therefore, we hypothesize that up-regulation of SALL4 can lead to defective hematopoiesis, accumulation of mutations, and progression to acute myeloid leukemia, which may in part explain the poorer prognosis in patients with myelodysplastic syndrome and SALL4 up-regulation. Identifying other oncogenes that are similarly activated by hypomethylating-agent therapy and evaluation of these and SALL4 expression in larger prospective trial samples in the future will be necessary to validate this notion.
Another unique aspect of SALL4 is that its expression is associated with DNA methylation status. Previously, a major challenge in the field of DNA methylation was to show a causal relationship between the DNA methylation and gene expression at a locus-specific and cellular level. Recent advances in functional genomic approaches such as the use of the CRISPR–Cas9 nuclease system have provided us with additional tools to address this question. Using the locus-specific CRISPR-DiR approach, we have identified a critical CpG island responsible for SALL4 expression and, more importantly, shown that treatment with a hypomethylating agent leads to demethylation of this region and up-regulation of SALL4. Demethylation of this same region was observed in patients with myelodysplastic syndrome having increased SALL4 expression after hypomethylating-agent therapy. These observations support our hypothesis that treatment with a hypomethylating agent can up-regulate oncogenes such as SALL4.
It is unclear why only a subgroup of patients had up-regulation of SALL4 after treatment with a hypomethylating agent, and future studies will be necessary to fully understand the mechanism or mechanisms. It is possible that DNA demethylation is the first step for gene activation, and additional chromatin remodeling and interaction between enhancer and promoter are needed to maintain gene expression. For example, in a recent study, we found that demethylation and subsequent binding of methylation-sensitive regulators could lead to long-range interactions between gene locus and distal regulatory elements, resulting in sustained up-regulation of gene expression.35
Using patient samples and a targeted demethylation assay, we found that monotherapy with a hypomethylating agent can activate or up-regulate oncogenes such as SALL4. Up-regulation of SALL4 probably influences the clinical progression of the disease; similar biologic effects may accompany treatment with a hypomethylating agent in patients with cancers other than myelodysplastic syndrome. Although the up-regulation of SALL4 may be associated with a worse prognosis, it may also provide an additional treatment option on the basis of SALL4-mediated cancer vulnerability. We are currently exploring the concept that if SALL4 expression is up-regulated, a concomitant targeted therapy that directly or indirectly mitigates SALL4 expression, function, or both could be added to the treatment plan.39
Supplementary Material
Acknowledgments
Supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC) 5×1000 MYNERVA (Myeloid Neoplasms Research Venture AIRC) project, #21267 (http://www.progettoagimm.it); a grant (Finalizzata 2018: NET-2018–12365935, to Dr. Voso) from Ministero della Salute, Italy; a Singapore Translational Research Investigator Award (MOH-STaR18nov-0002, MOH-000278, to Dr. Tenen) from the Singapore Ministry of Health National Medical Research Council; the Singapore Ministry of Education under its Research Centers of Excellence initiative; a grant (R35CA197697, to Dr. Tenen) from the National Cancer Institute, National Institutes of Health; a grant (P01HL131477, to Dr. Tenen) from the National Heart, Lung, and Blood Institute, National Institutes of Health; a grant (to Dr. Chai) from the Xiu Research Fund; grants (APP1024364, APP1043934, and APP1102589, to Dr. Pimanda, and APP1163815, to Dr. Unnikrishnan) from the National Health and Medical Research Council of Australia; a Translational Program Grant (6589–210, to Dr. Unnikrishnan) from the Leukemia and Lymphoma Society; a grant (109–2314-B-075–078, to Dr. Y.-C. Liu) from the Ministry of Science and Technology, Taiwan; a grant (to Dr. Y.-C. Liu) from the Chong Hin Loon Memorial Cancer and Biotherapy Research Center, National Yang Ming Chiao Tung University, Taipei, Taiwan; a grant (81870104, to Dr. Xiao) from the National Natural Science Funds, China; a grant (2020-I2M-C&T-A-020, to Dr. Xiao) from the Chinese Academy of Medical Sciences Initiative Fund for Medical Sciences; and a grant (2017RKWNJT, to Dr. Follo) from the Italian Ministry of University and Research–Projects of National Relevance.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
Contributor Information
Yao-Chung Liu, Department of Pathology, Brigham and Women’s Hospital, Boston Division of Hematology, Department of Medicine, Taipei Veterans General Hospital; Faculty of Medicine and the Program in Molecular Medicine, Institute of Biopharmaceutical Sciences, School of Life Science, National Yang Ming Chiao Tung University, Taipei, Taiwan.
Junsu Kwon, Cancer Science Institute of Singapore, Singapore
Emiliano Fabiani, Department of Biomedicine and Prevention, University of Rome Tor Vergata, Italy UniCamillus–Saint Camillus International University of Health Sciences, Italy.
Zhijian Xiao, National Clinical Research Center for Blood Diseases and State Key Laboratory of Experimental Hematology, Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Tianjin, China
Yanjing V. Liu, Cancer Science Institute of Singapore, Singapore
Matilde Y. Follo, Rome, and Cellular Signaling Laboratory, Department of Biomedical and Neuromotor Sciences, University of Bologna, Italy
Jinqin Liu, National Clinical Research Center for Blood Diseases and State Key Laboratory of Experimental Hematology, Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Tianjin, China
Huijun Huang, National Clinical Research Center for Blood Diseases and State Key Laboratory of Experimental Hematology, Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Tianjin, China
Chong Gao, Department of Pathology, Brigham and Women’s Hospital, Boston
Jun Liu, Department of Pathology, Brigham and Women’s Hospital, Boston
Giulia Falconi, Department of Biomedicine and Prevention, University of Rome Tor Vergata, Italy
Lia Valentini, Department of Biomedicine and Prevention, University of Rome Tor Vergata, Italy
Carmelo Gurnari, Department of Biomedicine and Prevention, University of Rome Tor Vergata, Italy
Carlo Finelli, IRCCS Azienda Ospedaliero–Universitaria di Bologna, Istituto di Ematologia "Seràgnoli", Bologna, Italy
Lucio Cocco, Rome, and Cellular Signaling Laboratory, Department of Biomedical and Neuromotor Sciences, University of Bologna, Italy
Jin-Hwang Liu, Faculty of Medicine and the Program in Molecular Medicine, Institute of Biopharmaceutical Sciences, School of Life Science, National Yang Ming Chiao Tung University in Taipei, Taiwan
Adrianna I. Jones, Harvard Stem Cell Institute, Harvard Medical School, Boston
Junyu Yang, Department of Pathology, Brigham and Women’s Hospital, Boston
Henry Yang, Cancer Science Institute of Singapore, Singapore
Julie A.I. Thoms, Harvard Stem Cell Institute, Harvard Medical School, Boston School of Medical Sciences and Lowy Cancer Research Centre, Australia.
Ashwin Unnikrishnan, Prince of Wales Clinical School and Lowy Cancer Research Centre, Australia
John E. Pimanda, School of Medical Sciences and Lowy Cancer Research Centre, Australia Prince of Wales Clinical School and Lowy Cancer Research Centre, Australia; Faculty of Medicine, University of New South Wales, Sydney, and the Department of Hematology, Prince of Wales Hospital, Randwick, NSW, Australia.
Rongqing Pan, Department of Medical Oncology, Dana– Farber Cancer Institute, Boston
Mahmoud A. Bassal, Harvard Stem Cell Institute, Harvard Medical School, Boston Cancer Science Institute of Singapore, Singapore.
Maria T. Voso, Department of Biomedicine and Prevention, University of Rome Tor Vergata, Italy
Daniel G. Tenen, Harvard Stem Cell Institute, Harvard Medical School, Boston Cancer Science Institute of Singapore, Singapore.
Li Chai, Department of Pathology, Brigham and Women’s Hospital, Boston
REFERENCES
- 1.Taylor SM, Jones PA. Mechanism of action of eukaryotic DNA methyltransferase: use of 5-azacytosine-containing DNA. J Mol Biol 1982;162:679–92. [DOI] [PubMed] [Google Scholar]
- 2.Leonhardt H, Page AW, Weier HU, Bestor TH. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell 1992;71:865–73. [DOI] [PubMed] [Google Scholar]
- 3.Patel K, Dickson J, Din S, Macleod K, Jodrell D, Ramsahoye B. Targeting of 5-aza-2′-deoxycytidine residues by chromatin-associated DNMT1 induces proteasomal degradation of the free enzyme. Nucleic Acids Res 2010;38:4313–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghoshal K, Datta J, Majumder S, et al. 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol Cell Biol 2005;25:4727–41. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5.Bender CM, Zingg JM, Jones PA. DNA methylation as a target for drug design. Pharm Res 1998;15:175–87. [DOI] [PubMed] [Google Scholar]
- 6.Jüttermann R, Li E, Jaenisch R. Toxicity of 5-aza-2′-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc Natl Acad Sci U S A 1994;91:11797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palii SS, Van Emburgh BO, Sankpal UT, Brown KD, Robertson KD. DNA methylation inhibitor 5-Aza-2′-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol Cell Biol 2008;28:752–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H, Chiappinelli KB, Guzzetta AA, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget 2014;5:587–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fabre C, Grosjean J, Tailler M, et al. A novel effect of DNA methyltransferase and histone deacetylase inhibitors: NFkappaB inhibition in malignant myeloblasts. Cell Cycle 2008;7:2139–45. [DOI] [PubMed] [Google Scholar]
- 10.Chiappinelli KB, Strissel PL, Desrichard A, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 2015;162:974–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma Y, Cui W, Yang J, et al. SALL4, a novel oncogene, is constitutively expressed in human acute myeloid leukemia (AML) and induces AML in transgenic mice. Blood 2006;108:2726–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kobayashi D, Kuribayashi K, Tanaka M, Watanabe N. Overexpression of SALL4 in lung cancer and its importance in cell proliferation. Oncol Rep 2011;26:965–70. [DOI] [PubMed] [Google Scholar]
- 13.Yong KJ, Li A, Ou W-B, et al. Targeting SALL4 by entinostat in lung cancer. Oncotarget 2016;7:75425–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yong KJ, Gao C, Lim JSJ, et al. Oncofetal gene SALL4 in aggressive hepatocellular carcinoma. N Engl J Med 2013;368: 2266–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elling U, Klasen C, Eisenberger T, Anlag K, Treier M. Murine inner cell mass-derived lineages depend on Sall4 function. Proc Natl Acad Sci U S A 2006; 103:16319–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao C, Kong NR, Li A, et al. SALL4 is a key transcription regulator in normal human hematopoiesis. Transfusion 2013; 53:1037–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang L, Xu Z, Xu X, et al. SALL4, a novel marker for human gastric carcinogenesis and metastasis. Oncogene 2014; 33:5491–500. [DOI] [PubMed] [Google Scholar]
- 18.Gao C, Dimitrov T, Yong KJ, et al. Targeting transcription factor SALL4 in acute myeloid leukemia by interrupting its interaction with an epigenetic complex. Blood 2013;121:1413–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu J, Jeong H-W, Kong N, et al. Stem cell factor SALL4 represses the transcriptions of PTEN and SALL1 through an epigenetic repressor complex. PLoS One 2009;4(5):e5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li A, Yang Y, Gao C, et al. A SALL4/MLL/HOXA9 pathway in murine and human myeloid leukemogenesis. J Clin Invest 2013;123:4195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang F, Guo Y, Chen Q, et al. Stem cell factor SALL4, a potential prognostic marker for myelodysplastic syndromes. J Hematol Oncol 2013;6:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu J, Ma Y, Kong N, et al. Dissecting the role of SALL4, a newly identified stem cell factor, in chronic myelogenous leukemia. Leukemia 2011;25:1211–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cui W, Kong NR, Ma Y, Amin HM, Lai R, Chai L. Differential expression of the novel oncogene, SALL4, in lymphoma, plasma cell myeloma, and acute lymphoblastic leukemia. Mod Pathol 2006;19: 1585–92. [DOI] [PubMed] [Google Scholar]
- 24.Shuai X, Zhou D, Shen T, et al. Overexpression of the novel oncogene SALL4 and activation of the Wnt/beta-catenin pathway in myelodysplastic syndromes. Cancer Genet Cytogenet 2009;194:119–24. [DOI] [PubMed] [Google Scholar]
- 25.Kwon J, Liu YV, Gao C, et al. Pseudogene-mediated DNA demethylation leads to oncogene activation. Sci Adv 2021; 7(40):eabg1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fan H, Cui Z, Zhang H, et al. DNA demethylation induces SALL4 gene re-expression in subgroups of hepatocellular carcinoma associated with Hepatitis B or C virus infection. Oncogene 2017;36: 2435–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cazzola M, Malcovati L. Myelodysplastic syndromes — coping with ineffective hematopoiesis. N Engl J Med 2005; 352:536–8. [DOI] [PubMed] [Google Scholar]
- 28.Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015;126: 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Voso MT, Leone G, Piciocchi A, et al. Feasibility of allogeneic stem-cell transplantation after azacitidine bridge in higher-risk myelodysplastic syndromes and low blast count acute myeloid leukemia: results of the BMT-AZA prospective study. Ann Oncol 2017;28:1547–53. [DOI] [PubMed] [Google Scholar]
- 30.Falconi G, Fabiani E, Piciocchi A, et al. Somatic mutations as markers of outcome after azacitidine and allogeneic stem cell transplantation in higher-risk myelodysplastic syndromes. Leukemia 2019;33:785–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016; 127:2391–405. [DOI] [PubMed] [Google Scholar]
- 32.Cheson BD, Greenberg PL, Bennett JM, et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood 2006;108:419–25. [DOI] [PubMed] [Google Scholar]
- 33.Bejar R Implications of molecular genetic diversity in myelodysplastic syndromes. Curr Opin Hematol 2017;24:73–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tatetsu H, Kong NR, Chong G, Amabile G, Tenen DG, Chai L. SALL4, the missing link between stem cells, development and cancer. Gene 2016;584:111–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu YV, Bassal MA, Jayasinghe MK, et al. Targeted intragenic demethylation initiates chromatin rewiring for gene activation. October 31, 2021. ( 10.1101/2020.07.16.205922v2). preprint. [DOI]
- 36.Di Ruscio A, Ebralidze AK, Benoukraf T, et al. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature 2013;503:371–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mozessohn L, Cheung MC, Fallahpour S, et al. Azacitidine in the ‘real-world’: an evaluation of 1101 higher-risk myelodysplastic syndrome/low blast count acute myeloid leukaemia patients in Ontario, Canada. Br J Haematol 2018;181:803–15. [DOI] [PubMed] [Google Scholar]
- 38.Prébet T, Gore SD, Esterni B, et al. Outcome of high-risk myelodysplastic syndrome after azacitidine treatment failure. J Clin Oncol 2011;29:3322–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang J, Gao C, Liu M, et al. Targeting an inducible SALL4-mediated cancer vulnerability with sequential therapy. Cancer Res 2021;81:6018–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.