

Abstract
Peripheral immune cell infiltration into the brain is a prominent feature in aging and various neurodegenerative diseases such as Alzheimer’s disease (AD). As AD progresses, CD8+ T cells infiltrate into the brain parenchyma, where they tightly associate with neurons and microglia. The functional properties of CD8+ T cells in the brain are largely unknown. To gain further insights into the putative functions of CD8+ T cells in the brain, we explored and compared the transcriptomic profile of CD8+ T cells isolated from the brain and blood of transgenic AD (APPswe/PSEN1dE9, line 85 [APP-PS1]) and age-matched wild-type (WT) mice. Brain CD8+ T cells of APP-PS1 and WT animals had similar transcriptomic profiles and substantially differed from blood circulating CD8+ T cells. The gene signature of brain CD8+ T cells identified them as tissue-resident memory (Trm) T cells. Gene Ontology enrichment and Kyoto Encyclopedia of Genes and Genomes pathway analysis on the significantly upregulated genes revealed overrepresentation of biological processes involved in IFN-β signaling and the response to viral infections. Furthermore, brain CD8+ T cells of APP-PS1 and aged WT mice showed similar differentially regulated genes as brain Trm CD8+ T cells in mouse models with acute virus infection, chronic parasite infection, and tumor growth. In conclusion, our profiling of brain CD8+ T cells suggests that in AD, these cells exhibit similar adaptive immune responses as in other inflammatory diseases of the CNS, potentially opening the door for immunotherapy in AD.
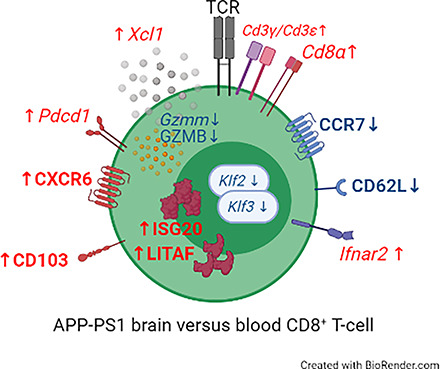
Visual Abstract
Key Points
Brain CD8+ T cells of aged WT and APP-PS1 mice are identified as Trm T cells.
Upregulated genes of brain CD8+ cells are involved in type I IFN signaling.
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disease causing severe cognitive decline in the elderly human population (1, 2). It is the leading cause of dementia and constitutes an enormous and unmet socioeconomic and health burden (3–5). Treatments available do not stop disease progression nor do they regenerate or reinstate functionality (6). AD is historically characterized by its key hallmarks of protein aggregation in the brain, namely the formation of amyloid-β plaques and neurofibrillary tangles, which are assumed as the primary triggers for neuronal cell death (7, 8). However, AD is a very complex pathology with severe structural and functional alterations of the brain, including brain atrophy, neurodegeneration (9), synaptic failure (10), impaired neurogenesis (11), blood–brain barrier disruption (12, 13), and increased neuroinflammation (14–16).
Genome-wide association studies (17, 18) and experimental data illustrated that neuroinflammation, in particular, the innate immune system represented by microglia, is a major contributor to AD pathogenesis (19).
Besides innate immune responses, very little is known about the contribution of adaptive immune responses to the pathophysiology of AD. T cells, as a crucial part of the adaptive immune system, play a major role in the aging brain (20–23) and might also be involved in neurodegenerative diseases such as AD (24–29). In the aged brain, T cells infiltrate into neurogenic niches, where they contribute to reduced levels of neurogenesis (23) and potentiate inflammation and leukocyte recruitment following ischemic brain insult (22). Recently, we and others demonstrated that most brain-infiltrating T cells are CD8+ T cells rather than CD4+ T cells and that the number of CD8+ T cells increases with age and amyloid plaque load in transgenic AD mouse models (30–33). CD8+ T cells seem to form immune synapses with microglia in the mouse brain (33) and associate with neuronal processes in mouse as well as human brains (27).
CD8+ T cells are a heterogeneous cell population of naive cells, central memory T (Tcm) cells, effector memory T (Tem) cells, tissue-resident memory (Trm) T cells, and, at least in humans, terminally tissue-resident memory CD8+ T cells (34, 35).
The molecular identity and profile of brain CD8+ T cells at advanced stages of AD pathology is so far unknown. In this study, we isolated CD8+ T cells from the brain of APPswe/PSEN1dE9, line 85 (APP-PS1) transgenic and age-matched (24- to 25-mo-old) wild-type (WT) mice and performed a detailed analysis on their cellular transcriptome. We compared the gene signature of brain CD8+ T cells with the transcriptome of blood-derived CD8+ T cells. We found that several core signature genes of Trm T cells were significantly regulated in brain CD8+ T cells from APP-PS1 and aged WT mice, and validated the Trm T cell molecular identity of brain CD8+ T cells by immunohistochemistry (IHC) and flow cytometry. Due to the upregulation of genes in brain CD8+ T cells known for their interaction in IFN signaling, we compared our genetic dataset with available genetic data from other studies involving CD8+ T cells in an inflamed brain environment (acute virus infection, chronic parasite infection, and tumor growth). In this study, we demonstrate a unique gene signature of brain CD8+ T cells and the presence of Trm T cells in aging and AD.
Materials and Methods
Animals
Female and male APP-PS1 mice (36, 37) expressing a chimeric mouse/human mutant amyloid precursor protein (Mo/HuAPP695swe) and a mutant human presenilin 1 (PS1-dE9), both directed to CNS neurons under the prion protein promoter, were used (from The Jackson Laboratory, http://www.jax.org/strain/005864). Breeding-derived, age-matched nontransgenic mice were used as WT animals. Mice were housed at the Paracelsus Medical University Salzburg in groups under standard conditions at a temperature of 22°C and a 12-h light/12-h dark cycle with ad libitum access to standard food and water. Animal care, breeding, handling, genotyping, and experiments were approved by Local Ethics Committees (BMWFW-66.019/0011-WF/V/3b/2016). Animal breeding was done under specific pathogen-free conditions.
Perfusion and tissue sectioning
For histology, WT and APP-PS1 mice 24–27 mo of age were used. Mice were anesthetized by i.p. injection of a ketamine (20.5 mg/ml, Richter Pharma), xylazine (5.36 mg/ml, Chanelle Pharma), and acepromazine (0.27 mg/ml, VANA) mixture and animals were perfused (manually or with the help of a perfusion pump) as previously described (31–33, 38). For histology, mice were decapitated and brains were extracted from the skull. Brain hemispheres and spleens were immersed in 4% paraformaldehyde (in 0.1 M sodium phosphate solution, pH 7.4) for fixation overnight before being washed in PBS and transferred into 30% sucrose for cryoprotection. When fully soaked with sucrose, brain hemispheres and spleens were cut into 40-µm slices on dry ice using a sliding microtome (Leica) dividing one brain hemisphere into representative 10ths of the brain. Sections were stored at −20°C in cryoprotectant solution (ethylene glycol, glycerol, 0.1 M sodium phosphate buffer pH 7.4, 1:1:2 by volume).
Flow cytometry and FACS
For flow cytometry analysis and FACS, 24- to 25-mo-old WT and APP-PS1 animals were used (n = 3–5/group). Blood was drawn from the heart with EDTA-coated syringes, filtered through a 40-µm mesh, and collected in 50 µl of PBS containing 0.1 M EDTA. Afterwards, animals were manually perfused through the left ventricle with ice-cold HBSS containing 15 mM HEPES (all from Thermo Fisher Scientific) and 0.5% glucose (Sigma-Aldrich) to wash out the remaining blood in the tissue.
Brain
Brain samples were split in two hemispheres and both hemispheres per mouse were prepared as previously described (32, 33). In short, each brain hemisphere (including brainstem and cerebellum) was mechanically chopped with a razor blade and homogenized in 2 ml of ice-cold HBSS with 15 mM HEPES (all from Thermo Fisher Scientific), 0.5% glucose (Sigma-Aldrich), and RNasin (1:250, Promega) using a glass homogenizer. Cells were passed through a 100-µm cell strainer and rinsed twice with 2 ml of ice-cold HBSS (with 15 mM HEPES and 0.5% glucose). Cell suspensions were centrifuged at 340 × g for 7 min at 4°C. Myelin was removed by resuspending the cell pellet in 30% Percoll (Sigma-Aldrich) solution and centrifuged at 950 × g for 20 min at 4°C. The supernatant was carefully removed and pellets containing cells of interest were diluted in HBSS.
For flow cytometry analysis, left and right brain hemispheres were pooled and centrifuged for 5 min at 350 × g. Cells were stained in 100 µl of PBS with fluorescent-labeled primary Abs (for full Ab list, see Supplemental Table I) for 20 min in the dark, centrifuged for 5 min at 350 × g, resuspended in 600 µl of PBS, filtered using sterile 50-µm CellTrics filters (Sysmex), and measured on a CytoFLEX flow cytometer (Beckman Coulter). Corresponding isotype controls were used for all stainings.
For FACS, the cells were centrifuged at 300 × g for 10 min at 4°C. Pellets were resuspended in PBS containing fixable viability dye eFluor 780 (1:2000, eBioscience, no. 65-0865) and incubated for 3–10 min at room temperature (RT) and transferred to round-bottom polystyrene tubes (Corning). After centrifugation for 5 min at 300 × g, cell pellets were dissolved in FACS buffer (PBS with 2% BSA and 2 mM EDTA) containing rat anti-mouse CD16/CD32 Fc Block (1:100, BD Biosciences, no. 553141) for 5 min at RT. Primary fluorescent-labeled Abs were added and Ab incubation was performed for 45 min at 4°C in the dark. Samples were washed in FACS buffer twice and centrifuged at 400 × g for 5 min. Finally, cell pellets were resuspended in 800 µl of FACS buffer with RNasin (1:250, Promega) and filtered with a 30-µm cell strainer.
Blood
For flow cytometry analysis, 50 µl of whole blood was stained with fluorescent-labeled primary Abs (for full Ab list, see Supplemental Table I) in 50 µl of PBS for 20 min in the dark. RBCs were lysed with 1× FACS lysing solution (BD Biosciences) until the solution was clear and centrifuged for 5 min at 350 × g. Cells were measured in 200 µl of PBS on a CytoFLEX flow cytometer (Beckman Coulter). Corresponding isotype controls were used for all stainings.
For FACS, blood samples were incubated for 7–10 min with 1× RBC lysis buffer (eBioscience). Reaction was stopped with FACS buffer and samples were centrifuged for 5 min at 500 × g. The supernatant was removed and cell pellets were resuspended in PBS containing fixable viability dye eFluor 780 (1:2000, eBioscience, no. 65-0865) for 3–10 min at RT. Samples were centrifuged for 5 min at 500 × g and supernatant was removed. Afterwards, cell pellets were dissolved in FACS buffer containing rat anti-mouse CD16/CD32 Fc Block (1:100, BD Biosciences, no. 553141) for 5 min. Primary fluorescent-labeled Abs were directly added to the samples and samples were incubated for 30 min at RT in the dark. Samples were washed with FACS buffer and filtered through a 30-µm cell strainer. After final centrifugation for 5 min at 500 × g, cell pellets were dissolved in 800 µl of FACS buffer with RNasin (1:250, Promega). Samples were placed at 4°C until they were analyzed.
The following primary Abs were used for FACS: CD3 PE (17A2) (1:100, BD Biosciences, no. 555275) and CD8a FITC (1:100, eBioscience, no. 11-0081). Single stains and isotype controls were performed with PBMCs for compensation and verification of the signals. The following isotype control Abs were used for FACS: PE IgG2b, κ (1:100, BD Biosciences, no. 553989), and FITC IgG2a, κ (1:100, eBioscience, no. 11-4321-80). Single living CD3 and CD8 double-positive (CD3+CD8+) cells were gated (see Fig. 1D) and sorted from brain and blood samples using the BD FACSAria III cell sorter (BD Biosciences) with BD FACSDiva software (8.0.1, BD Biosciences). In general, ∼104 CD3+CD8+ T cells were sorted from total brains or blood samples directly in RNAlater (Sigma-Aldrich) and stored at −80°C. Sort purity was analyzed with sorted samples for every experiment day using an LSRFortessa flow cytometer (BD Biosciences). Sorted samples had a purity of ∼95–100% (data not shown). Representative graphs of flow cytometry data were generated with Kaluza analysis software 1.3 (Beckman Coulter).
FIGURE 1.

Isolation of blood and brain CD8+ T cells. (A) Experimental setup. CD3+ CD8+ T cells were isolated from the brain and blood of WT and APP-PS1 mice using FACS and subsequently profiled using mRNA-seq analysis. The image was created with BioRender.com. (B and C) Representative immunohistochemistry images of CD8 (red)- and collagen IV (white)–stained hippocampus brain sections of WT (B) and APP-PS1 mice (C). CD8+ T cells were identified directly in the brain parenchyma (yellow arrows). (D) Representative plots of FACS gating strategy: single, living CD3+CD8+ T cells were isolated from the blood and brain homogenates. Sort purity was regularly analyzed. Scale bars, 25 µm (A, B), 20 µm in inserts (A, B).
Magnetic bead cell sorting of blood CD8+ T cells and analysis of transgene expression
Blood from the hearts of four APP-PS1 and four WT 24- to 25-mo-old animals was collected in 15-ml tubes containing 100 µl of 0.1 M EDTA/PBS. RBCs were lysed by incubation with 10 ml of 1× RBC lysis buffer for 10 min. After stopping the reaction with FACS buffer, cells were pelleted and resuspended in 90 µl of PB buffer (PBS with 0.5% BSA [pH 7.2]). Ten microliters of CD8a MicroBeads (Miltenyi Biotec, no. 130-117-044) was added for 15-min incubation at 4°C. After washing, labeled cells were separated on MS columns using the OctoMACS separator (Miltenyi Biotec). All sorted cells from one genotype were pooled together and RNA was isolated using an RNeasy micro kit (Qiagen) followed by transcription into cDNA (GoScript reverse transcriptase kit, Promega).
Expression of mutant human presenilin 1 (PS1-ΔE9) was tested with PCR using primers specific for PS1 (PS1 reverse, 5′-AGC CTA GAC CAC GAG AAT GC-3′, forward, 5′-CAG GTG GTG GAG CAA GAT G-3′) and housekeeping gene Ywhaz (Mm.PT.39a.22214831, IDT) as an internal control. Reaction mix containing 1× PCR buffer mastermix (GoTaq; M7422, Promega) and 0.5 µM of each primer was denatured at 94°C, followed by 35 cycles of 94°C for 1 min, 63°C for 1 min, and 72°C for 2 min. As a positive control, genomic DNA (gDNA) was isolated from tissue collected during the marking of the animals with ear punches using an NEB Monarch genomic purification kit. Result visualization was performed with an Intas gel documentation system after running PCR samples on a 2% agarose gel. Both cDNA and gDNA of either APP-PS1 or WT animals produced a 147-bp band from amplification of the housekeeping gene Ywhaz, confirming reliability. gDNA of APP-PS1 tissue produced an additional band at 1200 bp for the PS1 transgene, as expected.
Fluorescence IHC
IHC of mouse tissue was performed on free-floating sections as previously described (32, 38, 39). In short, Ag retrieval was performed depending on the used primary Ab by steaming the sections for 15–30 min in 1× citrate buffer (pH 6.0, Sigma-Aldrich). After blocking unspecific Ab binding sites, sections were incubated on a shaker overnight at RT with the following primary Abs: rat anti-CD8a (1:100, eBioscience, clone 4SM16), rabbit anti–collagen IV (1:500, Abcam), goat anti–collagen IV (1:300, Millipore), and Abs for T cell phenotyping (see Supplemental Table I). Sections were extensively washed in PBS and incubated for 4 h at RT in appropriate fluorescent-labeled secondary Abs (Invitrogen or Jackson ImmunoResearch Laboratories, all diluted 1:1000).
Nucleus counterstaining was performed with DAPI (1 mg/ml, 1:2000, Sigma-Aldrich). For amyloid-β plaque staining, Thioflavin S (1 mg/ml, 1:625, Sigma-Aldrich) was added to the secondary Ab solution. Tissue sections were additionally treated with 0.2% Sudan black (Sigma-Aldrich) in 70% ethanol for 30 s to reduce the autofluorescence in tissue from old animals (40). After this treatment, the sections were extensively washed in PBS and mounted onto microscope glass slides (Superfrost Plus, Thermo Scientific). Brain sections were coverslipped semidry in ProLong Gold antifade mountant (Life Technologies).
To verify the different immune cell/T cell markers and their expression in brain tissue, we used spleen tissue as positive controls for our Ab stainings.
Confocal microscopy and image processing
For fluorescence imaging, the confocal laser scanning microscopes LSM 700 and LSM 710 from Zeiss were used and gratefully provided by the microscopy core facility of Spinal Cord Injury and Tissue Regeneration Center Salzburg. Images were taken with the ZEN 2011 SP3 or SP7 (black edition) software (all from Zeiss). For qualitative analysis of CD8+ T cell location and costaining with various other immune markers, we took images as confocal z stacks in ×40 or ×63 oil magnification of cortical and hippocampal brain regions. Images were combined to merged maximum intensity projections and edited as well as processed with the ImageJ/Fiji software (version 1.53c) and Microsoft PowerPoint. For quantification, z stacks of CD8+ T cells in the hippocampus of WT and APP-PS1 mice were taken with ×40 oil magnification. CD8+ T cells were counted on at least nine images (320 × 320 µm) from three or four different sections with three to five images per section using ImageJ’s cell counter plugin. This resulted in an average of ∼50 analyzed cells per animal.
RNA isolation and mRNA sequencing
An RNeasy micro kit (Qiagen) was used to isolate the RNA from the sorted CD3+CD8+ T cells. Isolation procedure was performed according to the manufacturer’s instructions using an elution volume of 14 µl. The elution step was carried out twice to increase the yield.
The mRNA sequencing (mRNA-seq) was performed by Qiagen using QIAseq UPX 3′ transcriptome analysis according to the manufacturer’s protocol. All analyses by the company were carried out using CLC Genomics Workbench (version 20.0.3) and CLC Genomics Server (version 20.0.2).
Statistical analysis
For statistical analysis of CD8+ T cell marker expression (via IHC and flow cytometry), the software GraphPad Prism 9 was used. Differences between two groups were identified using the Student t test, whereas differences between more than two groups were detected via ordinary one-way ANOVA, followed by Šídák’s multiple comparisons test. A p value <0.05 was considered statistically significant.
Bioinformatics analysis
Quality control of raw sequencing data was conducted using the FastQC tool (41). Reads were then mapped to the genome (Mus musculus genome GRCm38) using bowtie2 (version 2.2.2) (42). Reads that overlap with genes were then counted using the HTSeq tool (version 0.11.2, -m intersection-nonempty -s no -i gene_id -t exon) (43). Expression values of protein coding genes were first normalized and differential expression analysis between the different groups was conducted using DESeq2 (44). Genes were considered significantly differentially transcribed with a p value <0.05. Validation of some genes was performed by IHC for respective protein expression. Enrichment analysis for Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were conducted using the clusterProfiler package in R (45). Benjamini and Hochberg multiple testing correction was used to adjust raw p values for multiple testing (an adjusted p value <0.05 was considered significant). To summarize and simplify the GO term enrichment results, we used the rrvgo package (https://ssayols.github.io/rrvgo) (46) to draw treemap plots.
We compared our results with the data from Wakim et al. (47), Landrith et al. (48), and Khalsa et al. (49). For Wakim et al., we downloaded the raw files from the Gene Expression Omnibus (GEO) database (GSE39152) and used the oligo package to conduct RMA (robust multiarray analysis) background correction and quantile normalization. Values were then log2 transformed and differential expression analysis was conducted using the limma package (50). We focused on comparing CD103+ versus CD103− CD8+ T cells in the brain and on comparing brain T cells to splenic T cells. Genes with an adjusted p value <0.05 (Benjamini and Hochberg multiple testing correction) were considered significantly differentially transcribed. For Landrith et al. (48) and Khalsa et al. (49), raw read counts were downloaded from the GEO database (GSE151414 and GSE95105) and DESeq2 was used to normalize and conduct differential expression analysis as explained above. In this study, we compared expression from tumors CT2A and GL261 against control and checked for overlap of differentially expressed genes (DEGs) with our datasets. For Landrith et al. (48) we compared our data to the comparisons CD103+/− brain versus CD103+/− spleen and CD103+ brain versus CD103− brain.
Results
Isolation of blood and brain CD8+ T cells via FACS
To characterize the molecular identity of brain-infiltrating CD8+ T cells at late disease stages in APP-PS1 mice, we isolated CD3 and CD8 double-positive (CD3+CD8+) T cells from the brain and blood of old WT and APP-PS1 mice (24–25 mo old) and performed mRNA-seq analysis on their cellular transcriptome (Fig. 1A). We recently reported that CD8+ T cell infiltration into the brain starts around 10 mo of age in APP-PS1 mice and around 1.5 y of age in WT mice. The number of CD8+ T cells in the brain increases with the age of the animals, peaking at 1.5 y of age in APP-PS1 mice and at 2 y of age in WT mice (32). For the current study, we decided to isolate CD8+ T cells from 24- to 25-mo-old mice, because these animals show the highest number of brain CD8+ T cells (32), allowing higher recovery yields. At this age, WT and APP-PS1 mice showed CD8+ T cells located in the brain parenchyma outside of collagen IV immunolabeled cerebral blood vessels (Fig. 1B, 1C). In APP-PS1 mice, CD8+ T cells were also observed directly at sites of amyloid plaques (Fig. 1C, asterisk).
For cell isolation, we used FACS of immunolabeled T cells and sorted only living, single CD3+CD8+ T cells from the total brain and blood samples of WT and APP-PS1 mice (Fig. 1D). About 1 × 104 CD3+CD8+ T cells (hereafter referred to as CD8+ T cells) were sorted per blood and brain sample, and isolated cells had a purity of ∼95–100% (data not shown).
Transcriptomic profile of blood CD8+ T cells remarkably differs between genotypes whereas brain CD8+ T cells cluster together
To characterize the cellular transcriptome of brain and blood-sorted CD8+ T cells, we performed 3′ mRNA-seq analysis. Unsupervised principal component analysis of gene expression data revealed distinguishable differences between APP-PS1 and WT blood CD8+ T cells and a high degree of similarity between APP-PS1 and WT brain CD8+ T cells. Independent of the genotype, samples clustered by CD8+ T cell origin (i.e., blood versus brain), indicating differences between brain and blood CD8+ T cells, which were more pronounced in APP-PS1 samples (Fig. 2A).
FIGURE 2.

Results of the mRNA-seq analysis from brain- and blood-isolated CD8+ T cells. (A) Principal component analysis of brain and blood samples from WT and APP-PS1 mice (n = 3–5/group). (B) Venn diagram with numbers of significantly regulated genes in all four comparisons. (C–F) Volcano plots showing significantly regulated genes for the comparisons (C) WT brain versus WT blood, (D) APP-PS1 brain versus APP-PS1 blood, (E) APP-PS1 blood versus WT blood, and (F) APP-PS1 brain versus WT brain. Genes with a p value <0.05 were considered as significantly differentially expressed (blue indicates downregulated, red indicates upregulated). PC2, second principal component.
Next, we analyzed the number of significantly DEGs in brain and blood CD8+ T cells from WT and APP-PS1 mice. By comparing gene profiles based on the cell origin (brain versus blood), 442 DEGs were detected on WT CD8+ T cells (Fig. 2B, 2C) and 595 DEGs were detected on APP-PS1 CD8+ T cells (Fig. 2B, 2D). Brain CD8+ T cells from APP-PS1 and WT shared 97 DEGs. By comparing gene profiles based on the animal’s genotype (APP-PS1 versus WT), 488 DEGs were detected on blood CD8+ T cells (Fig. 2B, 2E) and 311 DEGs were detected on brain CD8+ T cells (the smallest number of DEGs across all comparisons) (Fig. 2B, 2F). In APP-PS1 mice, APP and PS1 transgenes are expressed under the prion promoter, which is also present in CD8+ T cells (51). To rule out that genotype-dependent differences in blood CD8+ T cells resulted from T cell transgene expression, we analyzed transgenic PS1 expression in blood CD8+ T cells. Similarly to WT CD8+ T cells, APP-PS1 CD8+ T cells showed no transgene expression (Supplemental Fig. 1A), suggesting that differences in blood CD8+ T cells were likely disease related.
A full list with all significantly DEGs for each comparison was uploaded to the National Center for Biotechnology Information GEO and is accessible through GEO series accession number GSE180746 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE180746) together with the raw data.
Brain-derived CD8+ T cells from aged APP-PS1 and WT mice have a similar transcriptomic profile as Trm CD8+ T cells
Among the most significantly downregulated DEGs in brain versus blood CD8+ T cells of WT and APP-PS1 mice were Klf2 and Klf3 (Krüppel-like factor 2/3) (Fig. 2C, 2D, Tables I, II). These genes are highly expressed in naive T cells but downregulated upon T cell activation (52, 53). The gene Ccr7, an important gene involved in T cell migration and, through downregulation, in the formation of memory T cells (54–56), was significantly downregulated in CD8+ T cells from APP-PS1 and WT brain compared with blood (Fig. 2C, 2D, Table II). Interestingly, Ccr7 was one of the most significantly upregulated DEGs in APP-PS1 blood CD8+ T cells compared with WT blood CD8+ T cells (Fig. 2E). Sell (CD62L), required for lymph node homing and lowly expressed in effector and memory T cells (57), was also downregulated in brain CD8+ T cells (Fig. 2C, 2D). In brain CD8+ T cells, increased expression of the gene Pdcd1 for the programmed cell death protein 1 (PD-1) was observed. PD-1 plays an important role in diminishing the cytotoxic effects of CD8+ T cells and was found to be upregulated in brain CD8+ Trm T cells from viral infection models (58, 59).
Table I.
Top 10 most significantly upregulated and downregulated genes in WT brain versus WT blood CD8+ T cells sorted after p values
| Gene | Description | Fold Change (log2) | p Value (<0.05) |
|---|---|---|---|
| Xcl1 | Chemokine (C motif) ligand 1 | 2.13 | 2.95E−09 |
| Isg20 | IFN-stimulated protein | 2.29 | 8.55E−07 |
| Dusp2 | Dual specificity phosphatase 2 | 2.00 | 1.80E−05 |
| Cxcr6 | CXCR6 | 1.24 | 4.88E−05 |
| Mdfic | MyoD family inhibitor domain containing | 4.73 | 9.32E−05 |
| Itga1 | Integrin α1 | 3.11 | 9.11E−05 |
| Trnt1 | tRNA nucleotidyl transferase, CCA-adding, 1 | 2.97 | 7.63E−05 |
| Pmepa1 | Prostate transmembrane protein, androgen-induced 1 | 2.59 | 1.18E−04 |
| Cish | Cytokine inducible SH2-containing protein | 1.49 | 4.14E−04 |
| Chd3 | Chromodomain helicase DNA binding protein 3 | 1.11 | 4.03E−04 |
| Klf2 | Krüppel-like factor 2 (lung) | −2.34 | 4.40E−14 |
| Hbb-bs | Hemoglobin, β adult S chain | −3.63 | 2.98E−10 |
| Klf3 | Krüppel-like factor 3 (basic) | −2.24 | 1.60E−09 |
| Hba-a2 | Hemoglobin α, adult chain 2 | −3.83 | 3.01E−09 |
| Gzmm | Granzyme M (lymphocyte met-ase 1) | −3.82 | 3.72E−08 |
| Ccl5 | CCL5 | −1.19 | 9.64E−08 |
| Hba-a1 | Hemoglobin α, adult chain 1 | −3.89 | 2.25E−07 |
| Gzma | Granzyme A | −3.44 | 4.27E−05 |
| Arl4c | ADP-ribosylation factor-like 4C | −1.31 | 7.63E−05 |
| Hbb-bt | Hemoglobin, β adult T chain | −3.74 | 8.49E−05 |
Table II.
Top 10 most significantly upregulated and downregulated genes in APP-PS1 brain versus APP-PS1 blood CD8+ T cells sorted after p values
| Gene | Description | Fold Change (log2) | p Value (<0.05) |
|---|---|---|---|
| Cxcr6 | CXCR6 | 1.90 | 1.20E−05 |
| Ccnd2 | Cyclin D2 | 1.40 | 8.22E−05 |
| Id2 | Inhibitor of DNA binding 2 | 1.08 | 1.11E−04 |
| Isg20 | IFN-stimulated protein | 3.61 | 1.35E−04 |
| Efhd2 | EF hand domain containing 2 | 1.74 | 1.58E−04 |
| Litaf | LPS-induced TNF factor | 5.22 | 1.67E−04 |
| Xcl1 | Chemokine (C motif) ligand 1 | 2.27 | 1.70E−04 |
| Ppp1r16b | Protein phosphatase 1, regulatory subunit 16B | 1.55 | 2.43E−04 |
| Casp1 | Caspase 1 | 2.08 | 6.58E−04 |
| Rnf115 | Ring finger protein 115 | 4.41 | 8.36E−04 |
| Klf2 | Krüppel-like factor 2 (lung) | −3.37 | 7.18E−17 |
| Klf3 | Krüppel-like factor 3 (basic) | −3.90 | 6.34E−13 |
| Ccr7 | CCR7 | −4.73 | 1.38E−09 |
| Hbb-bs | Hemoglobin, β adult S chain | −4.22 | 2.84E−08 |
| Hba-a2 | Hemoglobin α, adult chain 2 | −4.36 | 5.78E−08 |
| Ier2 | Immediate early response | −2.17 | 1.94E−07 |
| Hba-a1 | Hemoglobin α, adult chain 1 | −4.98 | 1.96E−07 |
| Limd2 | LIM domain containing 2 | −1.22 | 9.24E−06 |
| Lef1 | Lymphoid enhancer binding factor 1 | −2.20 | 1.17E−05 |
| Junb | Jun B proto-oncogene | −1.39 | 2.46E−05 |
Cxcr6 was the most significantly upregulated gene in the brain CD8+ T cells of APP-PS1 mice and was also among the top 10 upregulated genes in WT brain CD8+ T cells (Tables I, II). Cxcr6 is an important chemokine receptor highly involved in migration and localization of Trm T cells, being highly expressed in Trm CD8+ T cells isolated from other peripheral organs (60, 61). Brain CD8+ T cells also upregulated other genes known to be expressed by memory T cells and related to T cell activation, including Xcl1 (lymphotactin) (62), Isg20 (IFN-stimulated gene) (47), and Litaf (LPS-induced TNF factor) (63) (Fig. 2C, Tables I, II).
In summary, the genetic profile of APP-PS1 and WT brain CD8+ T cells indicates that these cells represent a memory T cell type with a Trm T cell phenotype.
Flow cytometry and IHC analysis confirmed the presence of Trm CD8+ T cells in the brain of aged WT and APP-PS1 animals
To further investigate the tissue-resident profile of brain CD8+ T cells and validate our mRNA-seq data, we used IHC and flow cytometry for detailed analysis of brain and blood CD8+ T cells of WT and APP-PS1 mice (24–25 mo old) (Figs 3, 4). We analyzed the expression of proteins associated to Trm T cells, which are translated from DEGs of our dataset (i.e., ISG20, LITAF, CXCR6, CCR7, CD62L) (Fig. 2C, 2D), or that are known from the literature to be highly expressed in Trm T cells (i.e., CD103). The list of Abs used for IHC and flow cytometry is provided in Supplemental Table I. The gating strategy for flow cytometry is presented in Supplemental Fig. 1B.
FIGURE 3.

Immunohistochemistry staining and quantitative colocalization analysis of hippocampal CD8+ T cells. (A, C, and E) Representative images of hippocampal CD8+ T cells of APP-PS1 mice. Spleen tissue was used as positive control for Ab stainings. DAPI (blue) was used as a nucleus stain and thioflavin S (white) was used to visualize amyloid plaques. Scale bars, 30 µm, inserts 10 µm. (B, D, and F) Quantitative analysis of ISG20, LITAF, and CXCR6 coexpression on CD8+ T cells in the hippocampal region of WT and APP-PS1 mice (n = 4–5). Statistical differences between two groups were assessed via a Student t test. A p value <0.05 was considered statistically significant.
FIGURE 4.

Immunohistochemistry staining and flow cytometry analysis of blood and brain CD8+ T cells. (A, C, and E) Representative images of hippocampal CD8+ T cells of APP-PS1 mice. Spleen tissue was used as a positive control for Ab stainings. DAPI (blue) was used as a nucleus stain and thioflavin S (white) was used to visualize amyloid plaques. Scale bars, 30 µm, inserts 10 µm. (B, D, and F) Flow cytometry analysis of CCR7, CD62L, and CD103 coexpression on CD8+ T cells in blood and brain of WT and APP-PS1 mice (n = 7–8). Statistical differences between the groups were identified via an ordinary one-way ANOVA, followed by Šídák’s multiple comparisons test. A p value <0.05 was considered statistically significant.
We focused our IHC analysis on brain CD8+ T cells located in the hippocampus, where the amyloid plaque load is high in APP-PS1 mice (Figs. 3, 4) and we included spleen tissue of APP-PS1 mice as a positive control to verify our IHC Ab stainings. Hippocampal CD8+ T cells expressed proteins related to the top upregulated genes identified in our mRNA-seq analysis, including ISG20 and LITAF (Fig. 3A, 3C). ISG20 is an antiviral exoribonuclease and therefore has direct antiviral properties (64), whereas LITAF regulates the expression of various cytokines and TNF (65, 66). Quantification of ISG20+CD8+ T cells in the hippocampus of WT and APP-PS1 mice revealed similar percentages of cells in both genotypes (WT, 21 ± 11%; APP-PS1, 30 ± 9%) (Fig. 3B). The percentage of LITAF+CD8+ T cells was lower (WT, 6 ± 2%; APP-PS1, 8.5 ± 2.5%), but again without a significant difference between genotypes (Fig. 3D).
As expected, based on our mRNA-seq data, we could hardly detect any protein expression for the chemokine receptor CCR7 or the L-selectin CD62L (protein translating from Sell gene) in the brain of APP-PS1 mice (Fig. 4A, 4C). In addition, analysis of brain and blood CD8+ T cells of WT and APP-PS1 animals by flow cytometry confirmed the downregulation of CCR7 and CD62L in the brain in comparison with blood CD8+ T cells (Fig. 4B, 4D). The expression of CCR7 in the brain was so low that we had to rely on the mean fluorescence intensity ratio instead of the percentage of cells expressing this marker. CCR7 and CD62L are both involved in the formation of memory T cells and are not expressed in Tem cells or Trm T cells localized in peripheral tissues (34). Upregulation of CCR7 in blood CD8+ T cells of APP-PS1 compared with WT was not detected by flow cytometry, suggesting that the upregulation may only occur at the RNA level and does not affect protein expression.
In the hippocampus, we observed coexpression of the tissue-resident T cell marker CXCR6 (Fig. 3E), a chemokine receptor also expressed on activated T cells (34, 60, 67). Interestingly, cell counting of CXCR6+CD8+ T cells revealed a high abundance of this marker. In the WT, ∼50% of the CD8+ T cells were positive for CXCR6 (51.6 ± 8.8%) with significantly higher expression levels in APP-PS1 animals (67 ± 9.3%) (Fig. 3F).
CD103 is a commonly used marker for the detection of Trm T cells in peripheral tissues (68). We assessed the number of CD103+CD8+ Trm T cells in the blood and brain of APP-PS1 and WT mice via flow cytometry analysis. We observed increased percentages of CD103+CD8+ T cells in the APP-PS1 brain (16 ± 7.5%) compared with blood (4 ± 6%), but also in comparison with WT brain (8 ± 5%) (Fig. 4F). In the hippocampus, predominantly at sites of amyloid plaques, we observed few single CD103+CD8+ T cells as opposed to CD103−CD8+ T cells (Fig. 4E, arrow).
We additionally characterized brain CD8+ T cells for their cytotoxic activity, which is one of the best known functions of CD8+ T cells. We analyzed the presence of granzyme B (GZMB)–positive vesicles in APP-PS1 brain CD8+ T cells. However, we did not detect GZMB expression in brain CD8+ T cells (Supplemental Fig. 2A). We also investigated the TCR composition of brain CD8+ T cells to exclude the presence of recently discovered rare TCRγδ T cell populations that are involved in synaptic plasticity and short-term memory formation (69). CD8+ T cells in the brain of APP-PS1 mice did not express TCRγδ, concluding the presence of a broad CD8+ T cell population with TCRαβ composition (Supplemental Fig. 2B).
GO enrichment and KEGG pathway analysis demonstrates upregulation of genes associated with type I IFN signaling pathways
To better characterize and understand the phenotype of brain CD8+ T cells in the transgenic AD disease model, we performed GO enrichment analysis on the significantly DEGs. Full tables containing GO enrichment data are available upon request. In APP-PS1 brain CD8+ T cells compared with the blood, the GO terms for the biological processes “response to interferon-beta” and “response to virus” were significantly overrepresented (Fig. 5). Interestingly, the same biological processes were also upregulated in WT brain CD8+ T cells (Supplemental Fig. 2C). Genes enriched in both genotypes included Isg20, Isg15, and genes of the IFN-induced IFIT family (Ifit1, Ifit3 and Ifit3b). Genotype-specific upregulated genes included Ifnar2 and Apobec3 for APP-PS1, and Il10rb as well as Oas3 for WT. All of these genes are involved in either IFN signaling or viral response.
FIGURE 5.

GO enrichment analysis. Overrepresented GO terms for biological processes of significantly upregulated genes in CD8+ T cells from APP-PS1 brain versus APP-PS1 blood samples. All genes with a p value <0.05 were selected for GO enrichment analysis.
In addition, KEGG pathway analysis revealed that significantly upregulated genes in brain versus blood APP-PS1 CD8+ T cells (Table III) are involved in pathways known for virus infections, including “Epstein-Barr virus infection,” “Kaposi sarcoma-associated herpesvirus infection,” and “Human immunodeficiency virus 1 infection.” The same analysis for significantly upregulated genes in WT brain CD8+ T cells compared with blood cells revealed no significant upregulation of KEGG pathways.
Table III.
List of top 10 significantly upregulated KEGG pathways and their corresponding genes in APP-PS1 brain versus APP-PS1 blood CD8+ T cells sorted after adjusted p values
| Pathway | Genes | Adjusted p Value (<0.05) |
|---|---|---|
| EBV infection | Ccnd2/Cd3g/Irf9/Psmd2/Ifnar2/Nfkb2/Cd3e/Nedd4/Isg15/Cdk6/H2-T22/H2-T22/H2-Q7/H2-Q7/H2-Q6 | 3.96E−04 |
| Endocytosis | Ap2a2/Rabep1/Cblb/Agap2/Smap1/Clta/Nedd4/Eea1/Rab11fip2/Sh3kbp1/Arf3/H2-T22/H2-T22/H2-Q7/H2-Q7/H2-Q6 | 3.96E−04 |
| Cellular senescence | Ccnd2/Mapkapk2/Ppp3cc/Ppp3ca/Itpr1/Eif4ebp1/Cdk6/H2-T22/H2-T22/Rbbp4/H2-Q7/H2-Q7/H2-Q6 | 3.96E−04 |
| Kaposi sarcoma−associated herpesvirus infection | Irf9/Mapkapk2/Pik3cg/Ppp3cc/Ifnar2/Ppp3ca/Itpr1/Cdk6/Gng2/H2-T22/H2-T22/H2-Q7/H2-Q7/H2-Q6 | 3.96E−04 |
| HIV1 infection | Cd3g/Apobec3/Ppp3cc/Ppp3ca/Itpr1/Cd3e/Skp1a/Gng2/H2-T22/H2-T22/Trim30d/H2-Q7/H2-Q7/H2-Q6 | 9.00E−04 |
| Human CMV infection | Gna13/Ppp3cc/Ppp3ca/Itpr1/Eif4ebp1/Ptger4/Cdk6/Gng2/ H2-T22/H2-T22/H2-Q7/H2-Q7/H2-Q6 | 6.37E−03 |
| C-type lectin receptor signaling pathway | Irf9/Mapkapk2/Ppp3cc/Cblb/Nfkb2/Casp1/Ppp3ca/Itpr1 | 1.07E−02 |
| Measles | Ccnd2/Cd3g/Irf9/Cblb/Ifnar2/Csnk2b/Ifih1/Cd3e/Cdk6 | 1.15E−02 |
| Human T cell leukemia virus 1 infection | Ccnd2/Cd3g/Ppp3cc/Nfkb2/Xiap/Ppp3ca/Cd3e/H2-T22/H2-T22/H2-Q7/H2-Q7/H2-Q6 | 1.17E−02 |
| Ag processing and presentation | Rfxap/Cd8a/H2-T22/H2-T22/H2-Q7/H2-Q7/H2-Q6 | 1.37E−02 |
In summary, these data indicate that brain CD8+ T cells of aged APP-PS1 and WT mice have an upregulated set of genes that play key functions in CD8+ T cell responses to type I IFN–related pathways such as viral infections, and in view of the KEGG pathways analysis, this upregulation is more pronounced in APP-PS1 than aged WT animals.
Brain CD8+ T cells of aged APP-PS1 and WT mice express a gene signature related to brain Trm T cells in acute and chronic inflammation models
Based on the upregulation of Trm T cell–related genes and genes known from type I IFN signaling, we compared the RNA-seq profile from sorted brain CD8+ T cells with the transcriptome of brain CD8+ Trm T cells isolated from inflammation models known to involve CD8+ Trm T cells. We chose two studies in which CD8+ T cells were sorted from the brain after acute infection by vesicular stomatitis virus (47) or chronic parasite infection by Toxoplasma gondii (48) and one study profiling the immune microenvironment of brain tumors (49). In two studies of CNS pathogenic infection, the authors differentiated CD103+CD8+ Trm T cells from CD103−CD8+ T cells for transcriptome analysis. As we did not discriminate CD8+ T cells for CD103 expression in our brain or blood samples, we analyzed our data first in comparison with the mixed populations of CD103+/−CD8+ T cells and afterward to CD103+CD8+ T cells only.
For the systemic vesicular stomatitis virus infection model of Wakim et al. (47), we first reanalyzed the microarray data of Wakim et al. to determine which genes are differentially expressed in CD103+/− brain cells versus CD103− spleen cells (adjusted p value of <0.05). We compared this new dataset with the dataset of APP-PS1 CD8+ brain (versus blood) and WT CD8+ brain (versus blood) T cells. Two hundred twelve genes were similarly differentially expressed in the CD8+ T cells in the brain of APP-PS1 mice (Fig. 6A) as were 203 genes in the brain CD8+ T cells of WT mice (Supplemental Fig. 3A). These included the genes Xcl1, Isg20, Litaf, Pdcd1, CD8a, Cxcr6, Ccr7, Sell, and Klf2/3 for both genotypes. In a second step, we compared our transcriptomic data with the significantly DEGs (adjusted p value of <0.05) of CD103+ brain versus CD103− brain memory CD8+ T cells from Wakim et al. (47). Thereby, we observed that 30 genes overlapped and were similarly differentially expressed in brain CD8+ T cells from APP-PS1 mice (Fig. 6B) and 28 DEGs were shared in WT mice (Supplemental Fig. 3B). Remarkably, the genes Isg20, Litaf, Ccr7, Sell, and Klf2/3 were included for both genotypes.
FIGURE 6.

Comparisons with transcriptomic data of different brain inflammation models. (A and B) Comparison with gene microarray data from brain CD8+ T cells of virus-infected mice (Wakim et al. [47]). (A) Significantly DEGs that are shared in APP-PS1 brain CD8+ T cells and brain CD8+ T cells (CD103+ and CD103−) from an acute virus infection mouse model. (B) Significantly DEGs that are shared in APP-PS1 brain CD8+ T cells and brain CD103+ CD8+ Trm T cells from the virus infection mouse model. The overlapping genes belong to the top regulated genes in the Wakim et al. data and included the genes Klf2, Sell, Cxcr6, Isg20, and Litaf. (C and D) Comparison with transcriptomic data of brain CD8+ T cells of a chronic parasite infection mouse model (Landrith et al. [48]). (C) Significantly DEGs that are shared in APP-PS1 brain CD8+ T cells and brain CD8+ T cells (CD103+ and CD103−) from a chronic parasite infection mouse model. (D) Significantly DEGs that are shared in APP-PS1 brain CD8+ T cells and brain CD103+CD8+ Trm T cells from the parasite infection mouse model. The overlapping genes included Klf2, Sell, Litaf, Cxcr6, and Isg20. (E and F) Comparison with the transcriptomic immune profile of brain tumor (glioblastoma) tissue (Khalsa et al. [49]). (E) Shared DEGs between APP-PS1 brain CD8+ T cells and the immunological inert tumor CT2A. (F) Shared DEGs between APP-PS1 brain CD8+ T cells and the immunological more active tumor GL261. The overlapping genes included Pdcd1, Isg20, Litaf, and Cxcr6 for both tumor types.
For the chronic parasite infection model of Landrith et al. (48), we compared our data with the transcriptomic data of CD103+/−CD8+ T cells from the brain of mice chronically infected with Toxoplasma gondii (48). The analysis revealed 163 overlapping DEGs with APP-PS1 brain CD8+ T cells (Fig. 6C) and 122 DEGs with WT brain CD8+ T cells (Supplemental Fig. 3C), respectively. Comparing our datasets with CD103+ versus CD103− CD8+ T cells, we found 101 similar DEGs in APP-PS1 mice (Fig. 6D) and 76 similar DEGs in the WT mice (Supplemental Fig. 3D), even higher numbers of overlapping genes than with the acute virus infection model. Interestingly, these genes included again Xcl1, Isg20, Litaf, Cxcr6, CD8a, Sell, and Klf2/3.
To clarify, if the upregulation of IFN pathway–related genes in APP-PS1 and WT CD8+ T cells was connected to ongoing inflammation in general, we further compared our data with the transcriptomic profile of two different types of brain tumors from the study of Khalsa et al. (49). Khalsa et al. did not sort for CD8+ T cells but performed RNA-seq on whole brain tissue, either tumor bearing or control. The authors added detailed analysis of cell type populations via CyTOF (cytometry by time of flight), revealing a CD8+ T cell cluster in the tumor tissue in addition to other immune cells, such as macrophages, CD4+ T cells, and microglia. The tumors we selected for bioinformatics comparisons included the tumor CT2A classified as immunologically inert with a high population of exhausted CD8+ T cells, and the tumor GL261 characterized as immunologically active with upregulated pathways such as “response to virus” and “response to interferon-gamma/beta.” We found 262 overlapping genes between APP-PS1 brain CD8+ T cells and tumor CT2A (Fig. 6E) and 209 for the same comparison of WT brain CD8+ T cells (Supplemental Fig. 3E), including the genes Litaf, Pdcd1, Cxcr6, and CD8a. Similar upregulated genes were mainly those involved in pathways for response to IFN-β (e.g., Isg20, Ifit1/3, and Irf9), whereas overlapping downregulated genes were predominantly those involved in cellular and metabolic processes. The comparisons with the immunologically more active tumor GL261 resulted in slightly fewer overlapping genes, with 188 similar DEGs in APP-PS1 brain CD8+ T cells (Fig. 6F) and 158 similar DEGs in WT brain CD8+ T cells (Supplemental Fig. 3F). In both genotypes, the genes Litaf, Isg20, Pdcd1, Cxcr6, CD8a, and Xcl1 were similarly upregulated together with genes involved in response to IFN and activation of innate immune response. Ccr7 and Sell (CD62L) were among the downregulated genes, along with genes that play a role in regulating cellular processes.
In summary, the transcriptomic profiles of brain CD8+ T cells from APP-PS1 and WT mice show similar expression of genes regulated in acute viral and chronic parasitic infections, with a greater number of overlapping genes in the latter. Furthermore, CD8+ T cells from the brain of APP-PS1 and WT mice had similar DEGs as cells from tumor-bearing tissue, indicating regulation of general inflammatory pathways, not limited to pathogen-induced inflammation.
Discussion
The present work reveals major differences in the cellular transcriptome of brain and blood CD8+ T cells isolated from WT and APP-PS1 mice. Brain CD8+ T cells of APP-PS1 and aged WT mice presented a molecular gene signature reminiscent of Trm T cells, which was validated via IHC and flow cytometry analysis. Similar to brain Trm T cells from CNS pathogenic infections and brain tumors, brain CD8+ T cells from APP-PS1 and aged mice upregulated genes associated with type I IFN signaling. Furthermore, APP-PS1 mice showed higher numbers of CD103+CD8+ and CXCR6+CD8+ Trm T cells compared with aged WT mice.
Memory T cells in the AD brain
The presence of brain T cells in AD is well documented in humans (26, 28) and transgenic mouse models (30, 32, 70), but their exact phenotype and function remain poorly characterized (71, 72). In transgenic AD mice, CD8+ T cells clearly outnumber CD4+ T cells in the brain, but their functions are still controversially discussed (30, 33, 73, 74). Our study provides a detailed molecular characterization of brain CD8+ T cells in AD transgenic mice, which is essential to understand the functional role of these cells within the brain.
Memory CD8+ T cells are a very heterogeneous cell population, and subtypes include Tcm cells, Tem cells, and Trm T cells (34, 35). Tcm cells circulate in the blood and migrate back toward secondary lymphoid tissues, whereas Tem cells and Trm T cells are predominantly found in peripheral organs at sites of acute or previous inflammation (34, 54). Memory T cells are long-lived and provide rapid response upon re-encounter with the same Ag and/or pathogen (54, 75). Memory T cells can also undergo self-renewal, and the formation of Trm T cells allows these T cells to reside and survive in various peripheral tissues, including the brain (34, 63, 76, 77).
Several key genes, known to be regulated in Tem cells and Trm CD8+ T cells, were identified by our transcriptome analysis, including Klf2/3, Ccr7, Sell, Xcl1, Isg20, Litaf, Pdcd1, and Cxcr6. The phenotype of Trm T cells is characterized by an increased expression of proteins that promote tissue retention and a reduced expression of surface receptors that support tissue exit (78). Among the most downregulated genes in brain CD8+ T cells from aged APP-PS1 and WT mice was Klf2 that encodes for the transcription factor Klf2. Klf2 drives the expression of S1PR1 and CCR7, proteins involved in lymph node homing and T cell egression from tissue (78). Downregulation of these receptors therefore results in the induction of the Trm T cell phenotype (79). The gene Ccr7 was among the most significantly downregulated genes in brain CD8+ T cells from aged APP-PS1 and WT mice. Downregulation of CCR7 is not only important for Trm T cell formation but also for T cell exiting of the lymphatic system and tissue entrance. Deficiency of CCR7 in meningeal immune cells was previously associated with aging (80) and could be a possible explanation for the increased entry of CD8+ T cells into the parenchymal tissue in our aged mice. Among the most significantly upregulated genes in brain CD8+ T cells from APP-PS1 and WT mice was the gene Cxcr6, involved in the homing of CD8+ T cells to peripheral tissues (60).
As we isolated CD8+ T cells from the total mouse brain, without removing the meninges and choroid plexus (structures that harbor CD8+ T cells [81, 82]), we most likely profiled a mix of subpopulations and not only parenchymal brain CD8+ T cells. Thus, we used IHC and flow cytometry to validate the Trm T cell profile of brain CD8+ T cells. In fact, CD8+ T cells located in the hippocampus of APP-PS1 and aged WT mice express proteins associated with tissue residency of Trm T cells (i.e., CXCR6 [60], LITAF [63], and ISG20 [47]), reinforcing the idea that brain CD8+ T cells in APP-PS1 and aged WT mice have a Trm T cell profile.
Interestingly, we detected a heterogeneous population of CD103+ and CD103− CD8+ T cells in the brain of APP-PS1 and aged WT mice, as well as enrichment of CXCR6+CD8+ T cells and CD103+CD8+ T cells in APP-PS1 mice compared with WT mice. The expression of CD103 is associated with tissue residency, but not all Trm T cells automatically express this marker (78, 83). Human brains also show a mixed population of CD103+ and CD103− Trm CD8+ T cells (84), as do mouse brains in aging (22) and after viral infection (85). In our study, most brain CD8+ T cells were CD103− rather than CD103+. Nevertheless, due to the abundant expression of other Trm T cell markers (i.e., CXCR6), we assume the predominance of Trm T cells in contrast to other T cell populations. The presence of Tem cells and Trm CD8+ T cells in the aged mouse brain was already reported by Ritzel et al. (22). In aged mice, memory CD8+ T cells might enter the brain in a stochastic rather than in an Ag-driven way (22). The higher number of Trm CD8+ T cells in APP-PS1 brains raises the question whether AD-specific Ags (e.g., amyloid-β aggregates) might also drive CD8+ T cell brain entrance in transgenic mice. Further experiments addressing T cell clonal expansion and TCR specificity will help to understand these differences between the aged and AD transgenic brain.
The presence and formation of Trm T cell in the CNS is widely known in viral infections (47, 85–88) or in autoimmune diseases such as multiple sclerosis (89), but little is known about Trm T cells in neurodegenerative diseases such as AD (90). In pathogenic CNS infections, brain Trm T cells act as a first line of defense against reinfection (85, 91) and have a distinct transcriptome and marker profile compared with circulating memory T cells (47, 48). To further explore the Trm phenotype of brain CD8+ T cells in AD and aging, we compared significant DEGs of brain CD8+ T cells with two transcriptomic datasets of brain CD8+ T cells isolated from mouse models of CNS pathogenic infections: acute viral infection by systemic vesicular stomatitis virus (47) and chronic parasitic infection by T. gondii (48). Brain CD8+ T cells shared multiple DEGs with brain Trm T cells of virus- and parasite-infected mice, including genes characteristic of Trm T cells (Cxcr6, Klf2, and Sell) and genes involved in inflammatory pathways, particularly in type I IFN signaling. Interestingly, APP-PS1 and WT brain CD8+ T cells shared a higher similarity to brain CD8+ T cells from chronic- than acute-infected mice. A common feature of AD and aging is chronic neuroinflammation (92), leading us to the hypothesis that the inflammatory environment might shape the transcriptomic profile of brain CD8+ T cells. This could explain the similarities of brain CD8+ T cells from APP-PS1 and aged WT mice, and the higher similarity with Trm CD8+ T cells from a chronic than an acute infection model.
Brain inflammation with T cell involvement also occurs in malignant brain tumors such as glioblastoma (49). In fact, several DEGs in glioblastoma tissue (49) were similarly regulated in brain CD8+ T cells of APP-PS1 and aged WT mice, including genes associated to inflammatory processes and IFN-β signaling (Litaf, Isg20, Cd8a, and Irf9). As the glioblastoma dataset we used for this comparative analysis was derived from whole-tumor tissue instead of sorted CD8+ T cells, we need to be careful with our interpretations, as some of the overlapping genes are also expressed by other cell types. However, brain CD8+ T cells might regulate IFN pathway genes such as Isg20, Ifnar2, and Irf9 in response to an increased inflammatory stimulus in their environment, and inflammation might be a potential enhancer of Trm T cell differentiation.
IFN signaling in AD
The induction of type I IFNs was originally identified in responses to viral infection (93). Type I IFNs activate, among others, the canonical STAT1/STAT2/IFN regulatory factor 9 signaling complex by binding to a heterodimeric transmembrane receptor consisting of IFNAR1 and IFNAR2. This leads to the transcription of IFN-stimulated genes (ISGs) (94), primarily known for their antiviral properties.
Brain CD8+ T cells of APP-PS1 and aged WT mice upregulated numerous genes involved in type I IFN signaling (Ifnar2, Irf9, and several ISGs such as Isg20, Isg15, and Ifit1/3). In aged mice, expression of type I IFN–related genes is increased in the choroid plexus (95), and blocking type I IFNs ameliorates age-induced neuroinflammation and improves behavioral deficits (95). In AD mice, type I IFN signaling is increased, resulting in higher protein levels of IFN-α and its downstream transcription factor (96). Likewise, similar upregulation of type I IFN signaling was reported in post-mortem human AD brain tissue (96), suggesting a detrimental role of IFNs in the course of inflammation-induced neurodegeneration. Based on this information, it is not surprising that CD8+ T cells in the brain respond accordingly to the increased levels of type I IFNs. One purpose of type I IFNs is to increase the cytotoxicity of T cells. Although we found downregulation of the gene for cytotoxic granzyme M (Gzmm) and hardly any protein expression of GZMB, it is quite possible that CD8+ T cells contribute to the progression of neurodegeneration via the expression of ISGs. Furthermore, glial cells are able to diminish T cell cytotoxicity via PD-1/programmed cell death ligand 1 interaction (97), and we observed upregulation of Pdcd1, the gene for PD-1, on brain CD8+ T cells.
At present, we can only speculate about the functional consequences of increased numbers of brain CD8+ T cells in aging and AD. In transgenic AD mice and human brain tissue, CD8+ T cells locate close to neuronal structures (27) and might influence neuronal and synaptic function (32). In other mouse models, CD8+ T cells are involved in neuronal apoptosis (86), hippocampal neurogenesis, and synaptic plasticity (98). In the aged mouse brain, increased numbers of CD8+ T cells were associated with a microglia resting phenotype and increased phagocytosis (22). Taken together, these data suggest that brain CD8+ T cells might play an important role in shaping neuronal function and neuropathology during aging and the progression of AD. Based on our present work, a possible mechanism for CD8+ T cells to influence neurodegeneration could by via the expression of ISGs.
Supplementary Material
Acknowledgments
We thank the flow cytometry and microscopy core facility of Spinal Cord Injury and Tissue Regeneration Center Salzburg. Furthermore, we thank Sabine Bernegger for proofreading the manuscript.
This work was supported by the PMU FFF Research Fund (E-20/32/169-UNG) and by the Austrian Science Fund FWF Project P 35417-B. The funding bodies did not influence the design of the study and collection, analysis, interpretation of data, or writing of the manuscript.
M.S.U. planned and designed the study, performed animal and sample preparation, helped with the FACS sort, and wrote the initially submitted manuscript. B.A. helped with the animal sample preparation and performed histology and RNA isolation in the initially submitted manuscript. After M.S.U. had left the institution and changed field, B.A. took over the lead, performed the additional experiments requested, analyzed data, prepared figures, and wrote the revised version of the manuscript. R.P. performed all bioinformatics analyses of the mRNA-seq data and comparison with other genomic datasets. A.H. performed the FACS sort. D.A. performed flow cytometry analysis. H.M. and A.B. were involved in animal work and sample preparation. I.K.G., D.A., R.G., A.E., D.G., and T.W.-C. helped with their T cell expertise and critical revision of the manuscript. D.M.B.d.S. provided critical revision of the manuscript. L.A. is the principal investigator and was involved in the experimental design and critical revision of the manuscript.
The mRNA-seq data presented in this article have been submitted to GEO under accession number GSE180746.
The online version of this article contains supplemental material.
- AD
- Alzheimer’s disease
- APP-PS1
- APPswe/PSEN1dE9, line 85
- DEG
- differentially expressed gene
- gDNA
- genomic DNA
- GEO
- Gene Expression Omnibus
- GZMB
- granzyme B
- IHC
- immunohistochemistry
- ISG
- IFN-stimulated gene
- KEGG
- Kyoto Encyclopedia of Genes and Genomes
- KLF
- Krüppel-like factor
- LITAF
- LPS-induced TNF factor
- mRNA-seq
- mRNA sequencing
- PD-1
- programmed cell death protein 1
- RT
- room temperature
- Tcm
- central memory T
- Tem
- effector memory T
- Trm
- tissue-resident memory
- WT
- wild-type
Disclosures
The authors have no financial conflicts of interest.
References
- 1. Querfurth H. W., LaFerla F. M.. 2010. Alzheimer’s disease. N. Engl. J. Med. 362: 329–344. [DOI] [PubMed] [Google Scholar]
- 2. Goedert M., Spillantini M. G.. 2006. A century of Alzheimer’s disease. Science 314: 777–781. [DOI] [PubMed] [Google Scholar]
- 3. Reitz C., Mayeux R.. 2014. Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 88: 640–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brookmeyer R., Johnson E., Ziegler-Graham K., Arrighi H. M.. 2007. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 3: 186–191. [DOI] [PubMed] [Google Scholar]
- 5. Alzheimer’s Association . 2015. 2015 Alzheimer’s disease facts and figures. Alzheimers Dement. 11: 332–384. [DOI] [PubMed] [Google Scholar]
- 6. Szeto J. Y. Y., Lewis S. J. G.. 2016. Current treatment options for Alzheimer’s disease and Parkinson’s disease dementia. Curr. Neuropharmacol. 14: 326–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Serrano-Pozo A., Frosch M. P., Masliah E., Hyman B. T.. 2011. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1: a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Selkoe D. J., Hardy J.. 2016. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8: 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pini L., Pievani M., Bocchetta M., Altomare D., Bosco P., Cavedo E., Galluzzi S., Marizzoni M., Frisoni G. B.. 2016. Brain atrophy in Alzheimer’s disease and aging. Ageing Res. Rev. 30: 25–48. [DOI] [PubMed] [Google Scholar]
- 10. Marsh J., Alifragis P.. 2018. Synaptic dysfunction in Alzheimer’s disease: the effects of amyloid beta on synaptic vesicle dynamics as a novel target for therapeutic intervention. Neural Regen. Res. 13: 616–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moreno-Jiménez E. P., Flor-García M., Terreros-Roncal J., Rábano A., Cafini F., Pallas-Bazarra N., Ávila J., Llorens-Martín M.. 2019. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 25: 554–560. [DOI] [PubMed] [Google Scholar]
- 12. Zlokovic B. V. 2008. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57: 178–201. [DOI] [PubMed] [Google Scholar]
- 13. van de Haar H. J., Burgmans S., Jansen J. F. A., van Osch M. J. P., van Buchem M. A., Muller M., Hofman P. A. M., Verhey F. R. J., Backes W. H.. 2016. Blood-Brain barrier leakage in patients with early Alzheimer disease. Radiology 281: 527–535. [DOI] [PubMed] [Google Scholar]
- 14. Wyss-Coray T., Rogers J.. 2012. Inflammation in Alzheimer disease–a brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2: a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heneka M. T., Carson M. J., El Khoury J., Landreth G. E., Brosseron F., Feinstein D. L., Jacobs A. H., Wyss-Coray T., Vitorica J., Ransohoff R. M., et al. 2015. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14: 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Michael J., Marschallinger J., Aigner L.. 2019. The leukotriene signaling pathway: a druggable target in Alzheimer’s disease. Drug Discov. Today 24: 505–516. [DOI] [PubMed] [Google Scholar]
- 17. Jansen I. E., Savage J. E., Watanabe K., Bryois J., Williams D. M., Steinberg S., Sealock J., Karlsson I. K., Hägg S., Athanasiu L., et al. 2019. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. [Published erratum appears in 2020 Nat. Genet. 52: 354.] Nat. Genet. 51: 404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dos Santos L. R., Pimassoni L. H. S., Sena G. G. S., Camporez D., Belcavello L., Trancozo M., Morelato R. L., Errera F. I. V., Bueno M. R. P., de Paula F.. 2017. Validating GWAS variants from microglial genes implicated in Alzheimer’s disease. J. Mol. Neurosci. 62: 215–221. [DOI] [PubMed] [Google Scholar]
- 19. Mosher K. I., Wyss-Coray T.. 2014. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem. Pharmacol. 88: 594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gemechu J. M., Bentivoglio M.. 2012. T cell recruitment in the brain during normal aging. Front. Cell. Neurosci. 6: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Batterman K. V., Cabrera P. E., Moore T. L., Rosene D. L.. 2021. T cells actively infiltrate the white matter of the aging monkey brain in relation to increased microglial reactivity and cognitive decline. Front. Immunol. 12: 607691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ritzel R. M., Crapser J., Patel A. R., Verma R., Grenier J. M., Chauhan A., Jellison E. R., McCullough L. D.. 2016. Age-associated resident memory CD8 T cells in the central nervous system are primed to potentiate inflammation after ischemic brain injury. J. Immunol. 196: 3318–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dulken B. W., Buckley M. T., Navarro Negredo P., Saligrama N., Cayrol R., Leeman D. S., George B. M., Boutet S. C., Hebestreit K., Pluvinage J. V., et al. 2019. Single-cell analysis reveals T cell infiltration in old neurogenic niches. Nature 571: 205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mayne K., White J. A., McMurran C. E., Rivera F. J., de la Fuente A. G.. 2020. Aging and neurodegenerative disease: is the adaptive immune system a friend or foe? Front. Aging Neurosci. 12: 572090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rossi B., Santos-Lima B., Terrabuio E., Zenaro E., Constantin G.. 2021. Common peripheral immunity mechanisms in multiple sclerosis and Alzheimer’s disease. Front. Immunol. 12: 639369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Merlini M., Kirabali T., Kulic L., Nitsch R. M., Ferretti M. T.. 2018. Extravascular CD3+ T cells in brains of Alzheimer disease patients correlate with tau but not with amyloid pathology: an immunohistochemical study. Neurodegener. Dis. 18: 49–56. [DOI] [PubMed] [Google Scholar]
- 27. Gate D., Saligrama N., Leventhal O., Yang A. C., Unger M. S., Middeldorp J., Chen K., Lehallier B., Channappa D., De Los Santos M. B., et al. 2020. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 577: 399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rogers J., Luber-Narod J., Styren S. D., Civin W. H.. 1988. Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer’s disease. Neurobiol. Aging 9: 339–349. [DOI] [PubMed] [Google Scholar]
- 29. Togo T., Akiyama H., Iseki E., Kondo H., Ikeda K., Kato M., Oda T., Tsuchiya K., Kosaka K.. 2002. Occurrence of T cells in the brain of Alzheimer’s disease and other neurological diseases. J. Neuroimmunol. 124: 83–92. [DOI] [PubMed] [Google Scholar]
- 30. Ferretti M. T., Merlini M., Späni C., Gericke C., Schweizer N., Enzmann G., Engelhardt B., Kulic L., Suter T., Nitsch R. M.. 2016. T-cell brain infiltration and immature antigen-presenting cells in transgenic models of Alzheimer’s disease-like cerebral amyloidosis. Brain Behav. Immun. 54: 211–225. [DOI] [PubMed] [Google Scholar]
- 31. Unger M. S., Marschallinger J., Kaindl J., Klein B., Johnson M., Khundakar A. A., Roßner S., Heneka M. T., Couillard-Despres S., Rockenstein E., et al. 2018. Doublecortin expression in CD8+ T-cells and microglia at sites of amyloid-β plaques: a potential role in shaping plaque pathology? Alzheimers Dement. 14: 1022–1037. [DOI] [PubMed] [Google Scholar]
- 32. Unger M. S., Li E., Scharnagl L., Poupardin R., Altendorfer B., Mrowetz H., Hutter-Paier B., Weiger T. M., Heneka M. T., Attems J., Aigner L.. 2020. CD8+ T-cells infiltrate Alzheimer’s disease brains and regulate neuronal- and synapse-related gene expression in APP-PS1 transgenic mice. Brain Behav. Immun. 89: 67–86. [DOI] [PubMed] [Google Scholar]
- 33. Unger M. S., Schernthaner P., Marschallinger J., Mrowetz H., Aigner L.. 2018. Microglia prevent peripheral immune cell invasion and promote an anti-inflammatory environment in the brain of APP-PS1 transgenic mice. J. Neuroinflammation 15: 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martin M. D., Badovinac V. P.. 2018. Defining memory CD8 T cell. Front. Immunol. 9: 2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Woodland D. L., Dutton R. W.. 2003. Heterogeneity of CD4+ and CD8+ T cells. Curr. Opin. Immunol. 15: 336–342. [DOI] [PubMed] [Google Scholar]
- 36. Jankowsky J. L., Slunt H. H., Ratovitski T., Jenkins N. A., Copeland N. G., Borchelt D. R.. 2001. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol. Eng. 17: 157–165. [DOI] [PubMed] [Google Scholar]
- 37. Jankowsky J. L., Fadale D. J., Anderson J., Xu G. M., Gonzales V., Jenkins N. A., Copeland N. G., Lee M. K., Younkin L. H., Wagner S. L., et al. 2004. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum. Mol. Genet. 13: 159–170. [DOI] [PubMed] [Google Scholar]
- 38. Unger M. S., Marschallinger J., Kaindl J., Höfling C., Rossner S., Heneka M. T., Van der Linden A., Aigner L.. 2016. Early changes in hippocampal neurogenesis in transgenic mouse models for Alzheimer’s disease. Mol. Neurobiol. 53: 5796–5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marschallinger J., Sah A., Schmuckermair C., Unger M., Rotheneichner P., Kharitonova M., Waclawiczek A., Gerner P., Jaksch-Bogensperger H., Berger S., et al. 2015. The L-type calcium channel Cav1.3 is required for proper hippocampal neurogenesis and cognitive functions. Cell Calcium 58: 606–616. [DOI] [PubMed] [Google Scholar]
- 40. Schnell S. A., Staines W. A., Wessendorf M. W.. 1999. Reduction of lipofuscin-like autofluorescence in fluorescently labeled tissue. J. Histochem. Cytochem. 47: 719–730. [DOI] [PubMed] [Google Scholar]
- 41. Babraham Bioinformatics . FastQC. A quality control tool for high throughput sequence data. Available at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- 42. Langmead B., Salzberg S. L.. 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Anders S., Pyl P. T., Huber W.. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Love M. I., Huber W., Anders S.. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yu G., Wang L.-G., Han Y., He Q.-Y.. 2012. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16: 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sayols S. 2020. rrvgo: a Bioconductor package to reduce and visualize Gene Ontology terms. Available at: https://ssayols.github.io/rrvgo. [DOI] [PMC free article] [PubMed]
- 47. Wakim L. M., Woodward-Davis A., Liu R., Hu Y., Villadangos J., Smyth G., Bevan M. J.. 2012. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J. Immunol. 189: 3462–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Landrith T. A., Sureshchandra S., Rivera A., Jang J. C., Rais M., Nair M. G., Messaoudi I., Wilson E. H.. 2017. CD103+ CD8 T cells in the Toxoplasma-infected brain exhibit a tissue-resident memory transcriptional profile. Front. Immunol. 8: 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Khalsa J. K., Cheng N., Keegan J., Chaudry A., Driver J., Bi W. L., Lederer J., Shah K.. 2020. Immune phenotyping of diverse syngeneic murine brain tumors identifies immunologically distinct types. Nat. Commun. 11: 3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., Smyth G. K.. 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43: e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Isaacs J. D., Jackson G. S., Altmann D. M.. 2006. The role of the cellular prion protein in the immune system. Clin. Exp. Immunol. 146: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hart G. T., Hogquist K. A., Jameson S. C.. 2012. Krüppel-like factors in lymphocyte biology. J. Immunol. 188: 521–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Preston G. C., Feijoo-Carnero C., Schurch N., Cowling V. H., Cantrell D. A.. 2013. The impact of KLF2 modulation on the transcriptional program and function of CD8 T cells. PLoS One 8: e77537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Choi H., Song H., Jung Y. W.. 2020. The roles of CCR7 for the homing of memory CD8+ T cells into their survival niches. Immune Netw. 20: e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jung Y. W., Kim H. G., Perry C. J., Kaech S. M.. 2016. CCR7 expression alters memory CD8 T-cell homeostasis by regulating occupancy in IL-7- and IL-15-dependent niches. Proc. Natl. Acad. Sci. USA 113: 8278–8283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Debes G. F., Arnold C. N., Young A. J., Krautwald S., Lipp M., Hay J. B., Butcher E. C.. 2005. Chemokine receptor CCR7 required for T lymphocyte exit from peripheral tissues. Nat. Immunol. 6: 889–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schenkel J. M., Masopust D.. 2014. Tissue-resident memory T cells. Immunity 41: 886–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schøller A. S., Nazerai L., Christensen J. P., Thomsen A. R.. 2021. Functionally competent, PD-1+ CD8+ Trm cells populate the brain following local antigen encounter. Front. Immunol. 11: 595707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shwetank H. A., Abdelsamed H. A., Frost E. L., Schmitz H. M., Mockus T. E., Youngblood B. A., Lukacher A. E.. 2017. Maintenance of PD-1 on brain-resident memory CD8 T cells is antigen independent. Immunol. Cell Biol. 95: 953–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wein A. N., McMaster S. R., Takamura S., Dunbar P. R., Cartwright E. K., Hayward S. L., McManus D. T., Shimaoka T., Ueha S., Tsukui T., et al. 2019. CXCR6 regulates localization of tissue-resident memory CD8 T cells to the airways. J. Exp. Med. 216: 2748–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kumar B. V., Ma W., Miron M., Granot T., Guyer R. S., Carpenter D. J., Senda T., Sun X., Ho S.-H., Lerner H., et al. 2017. Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. 20: 2921–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Boutet M., Benet Z., Guillen E., Koch C., M’Homa Soudja S., Delahaye F., Fooksman D., Lauvau G.. 2021. Memory CD8+ T cells mediate early pathogen-specific protection via localized delivery of chemokines and IFNγ to clusters of monocytes. Sci. Adv. 7: eabf9975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mackay L. K., Rahimpour A., Ma J. Z., Collins N., Stock A. T., Hafon M.-L., Vega-Ramos J., Lauzurica P., Mueller S. N., Stefanovic T., et al. 2013. The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 14: 1294–1301. [DOI] [PubMed] [Google Scholar]
- 64. Weiss C. M., Trobaugh D. W., Sun C., Lucas T. M., Diamond M. S., Ryman K. D., Klimstra W. B.. 2018. The interferon-induced exonuclease ISG20 exerts antiviral activity through upregulation of type I interferon response proteins. mSphere 3: e00209-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tang X., Yang Y., Amar S.. 2011. Novel regulation of CCL2 gene expression by murine LITAF and STAT6B. PLoS One 6: e25083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bolcato-Bellemin A.-L., Mattei M.-G., Fenton M., Amar S.. 2004. Molecular cloning and characterization of mouse LITAF cDNA: role in the regulation of tumor necrosis factor-alpha (TNF-alpha) gene expression. J. Endotoxin Res. 10: 15–23. [DOI] [PubMed] [Google Scholar]
- 67. Heesch K., Raczkowski F., Schumacher V., Hünemörder S., Panzer U., Mittrücker H.-W.. 2014. The function of the chemokine receptor CXCR6 in the T cell response of mice against Listeria monocytogenes. PLoS One 9: e97701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Szabo P. A., Miron M., Farber D. L.. 2019. Location, location, location: tissue resident memory T cells in mice and humans. Sci. Immunol. 4: eaas9673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ribeiro M., Brigas H. C., Temido-Ferreira M., Pousinha P. A., Regen T., Santa C., Coelho J. E., Marques-Morgado I., Valente C. A., Omenetti S., et al. 2019. Meningeal γδ T cell-derived IL-17 controls synaptic plasticity and short-term memory. Sci. Immunol. 4: eaay5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Laurent C., Dorothée G., Hunot S., Martin E., Monnet Y., Duchamp M., Dong Y., Légeron F.-P., Leboucher A., Burnouf S., et al. 2017. Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain 140: 184–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. González H., Pacheco R.. 2014. T-cell-mediated regulation of neuroinflammation involved in neurodegenerative diseases. J. Neuroinflammation 11: 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sommer A., Winner B., Prots I.. 2017. The Trojan horse—neuroinflammatory impact of T cells in neurodegenerative diseases. Mol. Neurodegener. 12: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ní Chasaide C., Lynch M. A.. 2020. The role of the immune system in driving neuroinflammation. Brain Neurosci. Adv. 4: 2398212819901082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. McManus R. M., Mills K. H. G., Lynch M. A.. 2015. T cells—protective or pathogenic in Alzheimer’s disease? J. Neuroimmune Pharmacol. 10: 547–560. [DOI] [PubMed] [Google Scholar]
- 75. Badovinac V. P., Harty J. T.. 2006. Programming, demarcating, and manipulating CD8+ T-cell memory. Immunol. Rev. 211: 67–80. [DOI] [PubMed] [Google Scholar]
- 76. Urban S. L., Jensen I. J., Shan Q., Pewe L. L., Xue H.-H., Badovinac V. P., Harty J. T.. 2020. Peripherally induced brain tissue-resident memory CD8+ T cells mediate protection against CNS infection. Nat. Immunol. 21: 938–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Jiang X., Clark R. A., Liu L., Wagers A. J., Fuhlbrigge R. C., Kupper T. S.. 2012. Skin infection generates non-migratory memory CD8+ TRM cells providing global skin immunity. Nature 483: 227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kok L., Masopust D., Schumacher T. N.. 2022. The precursors of CD8+ tissue resident memory T cells: from lymphoid organs to infected tissues. Nat. Rev. Immunol. 22: 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Skon C. N., Lee J.-Y., Anderson K. G., Masopust D., Hogquist K. A., Jameson S. C.. 2013. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 14: 1285–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Da Mesquita S., Herz J., Wall M., Dykstra T., de Lima K. A., Norris G. T., Dabhi N., Kennedy T., Baker W., Kipnis J.. 2021. Aging-associated deficit in CCR7 is linked to worsened glymphatic function, cognition, neuroinflammation, and β-amyloid pathology. Sci. Adv. 7: eabe4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Korin B., Ben-Shaanan T. L., Schiller M., Dubovik T., Azulay-Debby H., Boshnak N. T., Koren T., Rolls A.. 2017. High-dimensional, single-cell characterization of the brain’s immune compartment. Nat. Neurosci. 20: 1300–1309. [DOI] [PubMed] [Google Scholar]
- 82. Prinz M., Priller J.. 2017. The role of peripheral immune cells in the CNS in steady state and disease. Nat. Neurosci. 20: 136–144. [DOI] [PubMed] [Google Scholar]
- 83. Carbone F. R. 2015. Tissue-resident memory T cells and fixed immune surveillance in nonlymphoid organs. J. Immunol. 195: 17–22. [DOI] [PubMed] [Google Scholar]
- 84. Smolders J., Heutinck K. M., Fransen N. L., Remmerswaal E. B. M., Hombrink P., Ten Berge I. J. M., van Lier R. A. W., Huitinga I., Hamann J.. 2018. Tissue-resident memory T cells populate the human brain. Nat. Commun. 9: 4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Steinbach K., Vincenti I., Kreutzfeldt M., Page N., Muschaweckh A., Wagner I., Drexler I., Pinschewer D., Korn T., Merkler D.. 2016. Brain-resident memory T cells represent an autonomous cytotoxic barrier to viral infection. J. Exp. Med. 213: 1571–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Garber C., Soung A., Vollmer L. L., Kanmogne M., Last A., Brown J., Klein R. S.. 2019. T cells promote microglia-mediated synaptic elimination and cognitive dysfunction during recovery from neuropathogenic flaviviruses. Nat. Neurosci. 22: 1276–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wakim L. M., Woodward-Davis A., Bevan M. J.. 2010. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc. Natl. Acad. Sci. USA 107: 17872–17879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lebrun A., Kean R. B., Hooper D. C.. 2020. Brain tissue-resident immune memory cells are required for long-term protection against CNS infection with rabies virus. Future Virol. 15: 755–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Fransen N. L., Hsiao C.-C., van der Poel M., Engelenburg H. J., Verdaasdonk K., Vincenten M. C. J., Remmerswaal E. B. M., Kuhlmann T., Mason M. R. J., Hamann J., et al. 2020. Tissue-resident memory T cells invade the brain parenchyma in multiple sclerosis white matter lesions. Brain 143: 1714–1730. [DOI] [PubMed] [Google Scholar]
- 90. Stojić-Vukanić Z., Hadžibegović S., Nicole O., Nacka-Aleksić M., Leštarević S., Leposavić G.. 2020. CD8+ T cell-mediated mechanisms contribute to the progression of neurocognitive impairment in both multiple sclerosis and Alzheimer’s disease? Front. Immunol. 11: 566225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Steinbach K., Vincenti I., Merkler D.. 2018. Resident-memory t cells in tissue-restricted immune responses: for better or worse? Front. Immunol. 9: 2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Giunta B., Fernandez F., Nikolic W. V., Obregon D., Rrapo E., Town T., Tan J.. 2008. Inflammaging as a prodrome to Alzheimer’s disease. J. Neuroinflammation 5: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lazear H. M., Schoggins J. W., Diamond M. S.. 2019. Shared and distinct functions of type I and type III interferons. Immunity 50: 907–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. McNab F., Mayer-Barber K., Sher A., Wack A., O’Garra A.. 2015. Type I interferons in infectious disease. Nat. Rev. Immunol. 15: 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Baruch K., Deczkowska A., David E., Castellano J. M., Miller O., Kertser A., Berkutzki T., Barnett-Itzhaki Z., Bezalel D., Wyss-Coray T., et al. 2014. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 346: 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Taylor J. M., Minter M. R., Newman A. G., Zhang M., Adlard P. A., Crack P. J.. 2014. Type-1 interferon signaling mediates neuro-inflammatory events in models of Alzheimer’s disease. Neurobiol. Aging 35: 1012–1023. [DOI] [PubMed] [Google Scholar]
- 97. Schachtele S. J., Hu S., Sheng W. S., Mutnal M. B., Lokensgard J. R.. 2014. Glial cells suppress postencephalitic CD8+ T lymphocytes through PD-L1. Glia 62: 1582–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zarif H., Nicolas S., Guyot M., Hosseiny S., Lazzari A., Canali M. M., Cazareth J., Brau F., Golzné V., Dourneau E., et al. 2018. CD8+ T cells are essential for the effects of enriched environment on hippocampus-dependent behavior, hippocampal neurogenesis and synaptic plasticity. Brain Behav. Immun. 69: 235–254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.