

SUMMARY
Mutations affecting isocitrate dehydrogenase (IDH) enzymes are prevalent in glioma, leukemia, and other cancers. Although mutant IDH inhibitors are effective against leukemia, they seem to be less active in aggressive glioma, underscoring the need for alternative treatment strategies. Through a chemical synthetic lethality screen, we discovered that IDH1-mutant glioma cells are hypersensitive to drugs targeting enzymes in the de novo pyrimidine nucleotide synthesis pathway, including dihydroorotate dehydrogenase (DHODH). We developed a genetically engineered mouse model of mutant IDH1-driven astrocytoma and used it and multiple patient-derived models to show that the brain-penetrant DHODH inhibitor BAY 2402234 displays monotherapy efficacy against IDH-mutant gliomas. Mechanistically, this reflects an obligate dependence of glioma cells on the de novo pyrimidine synthesis pathway and mutant IDH’s ability to sensitize to DNA damage upon nucleotide pool imbalance. Our work outlines a tumor-selective, biomarker-guided therapeutic strategy that is poised for clinical translation.
In brief
Shi et al. show that IDH-mutant gliomas are hyperdependent on de novo pyrimidine nucleotide synthesis. Using a newly developed, genetically engineered mouse model of IDH mutant glioma and patient-derived models, they show that blocking pyrimidine synthesis with the DHODH inhibitor BAY 2402234 is effective against these tumors.
Graphical Abstract
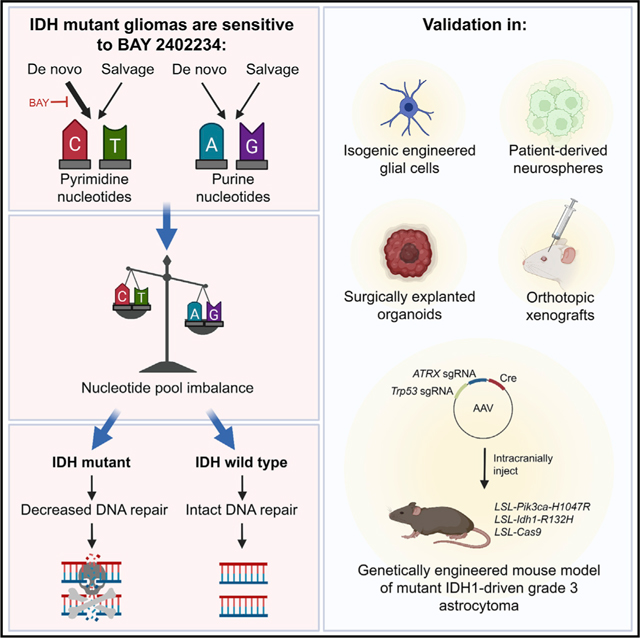
INTRODUCTION
Glioma is the most common primary malignant brain tumor in adults and is usually fatal. Despite intense efforts to develop novel treatments, no new medical therapies have been approved for adult patients with glioma in the last decade. Therefore, there remains a pressing need for better glioma therapies.
Our understanding of the molecular pathogenesis of glioma has grown considerably over the last two decades, driven partly by the advent of high-throughput sequencing. IDH1 and IDH2 mutations, which encode the isocitrate dehydrogenase (IDH) 1 and 2 enzymes (Parsons et al., 2008), are now part of the diagnostic criteria for gliomas (Louis et al., 2021). Most IDH-mutant gliomas are heterozygous for the canonical glioma-associated IDH1-R132H mutation (Losman and Kaelin, 2013). IDH1/2-mutant enzymes gain the neomorphic ability to produce the oncometabolite (R)-2-hydroxyglutarate [(R)-2HG] (Dang et al., 2009). (R)-2HG is structurally similar to 2-oxoglutarate (2OG) and competitively inhibits many 2OG-dependent enzymes (Losman and Kaelin, 2013). (R)-2HG also stimulates the EglN1 prolyl hydroxylase, a 2OG-dependent dioxygenase that promotes degradation of the hypoxia-inducible factor 1 alpha transcription factor (Koivunen et al., 2012; Tarhonskaya et al., 2014). Together, the cumulative biochemical effects of (R)-2HG transform neural cells and initiate glioma formation.
These advances prompted the development of mutant IDH inhibitors that block (R)-2HG synthesis. In contrast with the broad success of this approach in treating IDH-mutant leukemias (Stein et al., 2017), IDH inhibitors have displayed comparably limited antitumor activity against aggressive IDH-mutant gliomas in preclinical (Tateishi et al., 2015) and early clinical studies (Mellinghoff et al., 2021). Among patients with recurrent or progressive IDH-mutant gliomas, the objective response rates after IDH inhibitor treatment in a recent phase I clinical trial were 18% in patients with non-contrast-enhancing tumors and 0% in patients with contrast-enhancing tumors, which are typically more aggressive (Mellinghoff et al., 2021). These results might reflect the fact that dependence on (R)-2HG synthesis is transient in ex vivo models of neural cell transformation by mutant IDH (Johannessen et al., 2016; Turcan et al., 2018). Furthermore, there is emerging clinical evidence that a small fraction of IDH-mutant gliomas select against the mutant IDH allele over time (Favero et al., 2015; Mazor et al., 2017), implying that there is not an ongoing dependence on (R)-2HG in such tumors. Copy number alterations that repress (R)-2HG production also occur spontaneously in cultured IDH1-mutant glioma cells and do not decrease cellular fitness (Luchman et al., 2013). Thus, many IDH-mutant gliomas transition toward (R)-2HG independence over time, indicating that alternative strategies are needed to effectively treat these tumors.
One such strategy is to exploit the collateral vulnerabilities engendered by IDH mutations. Past work from our group and others supports the feasibility of this strategy (Lu et al., 2017; McBrayer et al., 2018; Sulkowski et al., 2017; Tateishi et al., 2015). These efforts have directly translated to new treatments for IDH-mutant gliomas that are currently undergoing clinical testing. Because much of the prior research in this area was borne out of hypothesis-driven approaches, we pursued a complementary, unbiased approach to identify collateral vulnerabilities induced by IDH mutations. Given that metabolic reprogramming is the most proximal consequence of mutant IDH activity in gliomas, we sought to uncover metabolic liabilities conferred by mutant IDH that could nominate new therapeutic targets in glioma.
RESULTS
Mutant IDH1 sensitizes cells to de novo pyrimidine synthesis inhibition
We recently created isogenic IDH1-mutant and IDH1 wild-type (WT) glioma cell culture models that recapitulate (R)-2HG levels in primary brain tumors (McBrayer et al., 2018). Using an endogenous IDH1/2 WT human glioma line, HOG, we expressed the IDH1-R132H oncogene (HOG-R132H) or an empty vector (HOG-EV). We used these isogenic models with a compound screening platform developed by two of us (J.E.E. and I.S.H.), named multifunctional approach to pharmacologic screening (Harris et al., 2019), to identify liabilities conferred by the IDH1-R132H oncogene (Figure 1A). Isogenic HOG stable cell lines were screened against 546 unique metabolic inhibitors and anticancer drugs that target a wide range of cellular processes (Figure S1A). We identified compounds that selectively decreased the fitness of IDH1-mutant cells versus IDH1 WT cells (Figures S1B and S1C). Many of these compounds, hereafter referred to as hits, target enzymes involved in mitochondrial, lipid, or nucleotide metabolism (Figure S1D). Therefore, the IDH1-R132H oncogene induces liabilities related to cellular metabolism.
Figure 1. De novo pyrimidine synthesis inhibitors preferentially kill IDH1-mutant glioma cells.

(A) Schema of the multifunctional approach to pharmacologic screening (MAPS) platform drug screen. Percent difference in area under curve (AUC) values (%AUCDiff) in HOG-EV and HOG-R132H cells were calculated for all drugs.
(B) Top hits and related pathways.
(C) Waterfall plot of all drugs ranked by %AUCDiff.
(D) Schematic of de novo and salvage pyrimidine synthesis pathways and targets of indicated hits.
(E–G) Cell death assays of HOG-EV or HOG-R132H cells treated with the indicated drugs (n = 5).
(H–J) Cell death assays of HOG-R132H cells treated with indicated drugs with or without 100 μM uridine (n = 3).
(K) Cell death assays of GSC lines treated with brequinar for two population doublings (n ≥ 3).
(L) Correlation between 2HG levels and sensitivity of GSC lines to brequinar. Symbols and colors are as in (K). For all panels, data presented are means ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001. For (L), p value was determined by simple linear regression analysis. For all others, two-tailed p values were determined by unpaired t test.
Among the hits were compounds targeting previously described dependencies conferred by IDH mutations, including oxidative stress inducers and nicotinamide phosphoribosyl-transferase, poly adenosine diphosphate-ribose polymerase, and oxidative phosphorylation inhibitors (Figures 1B and Table S1) (Grassian et al., 2014; Lu et al., 2017; McBrayer et al., 2018; Sulkowski et al., 2017; Tateishi et al., 2015). Unexpectedly, multiple de novo pyrimidine synthesis inhibitors scored highly, including three of the top 15 hits (Figures 1B–1D and S1E–S1G). This was specific because inhibitors of purine metabolism, including the de novo purine synthesis inhibitor lometrexol, did not reduce cell fitness in a mutant IDH-dependent manner (Figures S1H and S1I).
To attempt to validate our findings, we measured HOG-EV and HOG-R132H cell death after treatment with three inhibitors of de novo pyrimidine synthesis: the dihydroorotate dehydrogenase (DHODH) inhibitor brequinar and the orotidylate monophosphate decarboxylase inhibitors pyrazofurin and 6-azauridine. Each inhibitor preferentially killed IDH1-mutant glioma cells (Figures 1E–1G), and this effect was likely on-target because cell death could be fully rescued by stimulating pyrimidine nucleotide salvage with supraphysiological uridine (Figures 1H–1J).
We next treated ten patient-derived glioma stem-like cell (GSC) lines, six of which were heterozygous for the IDH1-R132H mutation and four of which were IDH1 WT (Table S2), with brequinar for two population doublings (Figure S1J). Brequinar cytotoxicity was greater in IDH1-mutant GSC lines relative to IDH1 WT lines, except for one line, HK213 (Figure 1K). The 2HG content correlated closely with brequinar sensitivity (Figure 1L), suggesting that it is a better predictor of DHODH inhibitor hypersensitivity than IDH status per se. Notably, HK213 cells displayed the lowest 2HG content of all IDH1-mutant GSCs, possibly explaining their relative insensitivity to this drug (Figures 1K and 1L). Collectively, our studies implicate de novo pyrimidine synthesis as a heretofore unappreciated vulnerability induced by the IDH1-R132H oncogene in glioma.
IDH1-mutant gliomas are sensitive to the brain-penetrant DHODH inhibitor BAY 2402234
We next sought a brain-penetrant inhibitor of de novo pyrimidine synthesis that could be used to target this pathway in vivo. Many classical inhibitors of this pathway are nucleoside analogues with poor CNS penetration. Therefore, we investigated a newly developed DHODH inhibitor, BAY 2402234, that was tested in a clinical trial for treating leukemia (NCT03404726) (Christian et al., 2019). To ask if BAY 2402234 is brain penetrant, we developed a pharmacodynamic (PD) assay by treating non-tumor-bearing mice with BAY 2402234 or vehicle and performing unbiased metabolite profiling on heart and liver samples. BAY 2402234 decreased the levels of orotate (the product of DHODH) and increased carbamoyl aspartate, which is an intermediate in the de novo pyrimidine synthesis pathway upstream of DHODH (Figures 2A and 2B). Therefore, we used the orotate to carbamoyl aspartate ratio (OCAR) as an in vivo PD biomarker for DHODH inhibition. Notably, treatment with BAY 2402234 suppressed OCAR in the brain tissue of tumor-free mice (Figure 2C), indicating that it is brain penetrant. Two weeks of daily BAY 2402234 administration was well tolerated (Figure 2D). Complementary in vitro studies revealed that BAY 2402234 preferentially killed IDH1-mutant HOG cells, as well as late passage, but not early passage, IDH1-mutant normal human astrocyte (NHA) cells, compared with their IDH1 WT counterparts (Figures 2E and 2F and S2A–S2F).
Figure 2. The DHODHi BAY 2402234 is brain-penetrant in mice and selectively kills IDH1-mutant glioma cells.

(A and B) Volcano plot of metabolites in heart (A) and liver (B) of mice treated with BAY 2402234 or vehicle (n = 9 per cohort). Carbamoyl asp = carbamoyl aspartate.
(C) OCAR in tissues of mice treated with BAY 2402234 or vehicle as in (A) (n = 9 per cohort).
(D) Weight changes in mice treated with BAY 2402234 or vehicle.
(E) Cell death assays of HOG-EV or HOG-R132H cells treated with BAY 2402234 with or without 100 μM uridine (n = 3).
(F) Representative photomicrographs of cells in (E). Scale bars, 100 μm.
(G) Cell death assays of GSC lines treated with BAY 2402234 or DMSO for two population doublings (n ≥ 3).
(H) Correlation between 2HG levels and sensitivity of GSC lines to BAY 2402234. Symbols and colors are as in (G).
(I) Cell death assays of HOG-R132H cells expressing EV, DHODH WT, or DHODH A58T treated with BAY 2402234 or DMSO (n = 3).
(J) UTP and dihydroorotate levels in HOG-R132H stable lines treated with 50 nM BAY 2402234 or DMSO (n = 3). For panels (A–E and G), data are means ± SEM; for panels (I and J), data are means ± SD; *p < 0.05, **p < 0.01, ***p < 0.001. For (H), the p value was determined by simple linear regression analysis. For all others, two-tailed p values were determined by unpaired t test.
BAY 2402234 induced greater cell killing in IDH-mutant versus IDH WT GSC lines and sensitivity again correlated with intracellular 2HG content (Figures 2G and 2H). To formally prove that the cytotoxic effects of BAY 2402234 were on-target, we used a BAY 2402234-resistant DHODH variant (DHODH A58T) that was identified in a saturating mutagenesis MITE-seq (mutagenesis by integrated tiles) (Melnikov et al., 2014) screen of human DHODH. Expression of DHODH A58T, but not WT DHODH, fully prevented the killing of IDH1-mutant HOG cells and MGG152 GSCs by BAY 2402234, which correlated with restoration of uridine 5’-triphosphate (UTP) levels, suppression of the DHODH substrate dihydroorotate, and increased OCAR (Figures 2I and 2J and S2G–S2I and S2M–S2O). Inactivating DHODH with CRISPR/Cas9 also preferentially decreased the fitness of HOG-R132H cells versus HOG-EV cells, which was reversed by supraphysiological uridine (Figures S2J–S2L). These findings validate our use of OCAR as a PD biomarker for DHODH inhibition and establish on-target activity of BAY 2402234 in in vitro IDH-mutant glioma models.
We next tested the antitumor activity of BAY 2402234 in mouse models of IDH1-mutant glioma. We used the MGG152 orthotopic xenograft glioma model, which was derived from a recurrent IDH1-mutant grade 4 glioma and is resistant to mutant IDH1 inhibitor treatment (Tateishi et al., 2015; Wakimoto et al., 2014). Treating MGG152 tumor-bearing mice with BAY 2402234 depleted orotate and increased carbamoyl aspartate throughout tumor tissue, as visualized by matrix-assisted laser desorption ionization mass spectrometry imaging (Figures 3A and 3B), thus reducing OCAR. BAY 2402234 accumulated to approximately 150 nM in tumor tissue after oral dosing (Figures 3C and S3A and S3B), exceeding the concentrations of drug required to kill IDH1-mutant GSCs in culture (Figure 2G). BAY 2402234 treatment prolonged survival relative to vehicle-treated mice (Figure 3D), although slightly less so than radiotherapy (Figure 3E), which is a cornerstone of glioma therapy. This effect was specific because BAY 2402234 was not active against TS516 IDH WT orthotopic xenografts (Figure 3F). Importantly, therapy failure in this IDH WT glioma model cannot be explained by poor target engagement, because OCAR and DHODH activity were similarly depressed by BAY 2402234 in MGG152 and TS516 tumors (Figures 3G–3I and S3C). Moreover, DHODH activity was comparably suppressed by BAY 2402234 in both IDH1-mutant and IDH1 WT HOG and GSC lines ex vivo (Figures S3D and S3E). BAY 2402234 also extended the survival of mice bearing orthotopic HOG-R132H xenografts unless those grafts expressed DHODH A58T (Figures 3J and 3K). These data show that BAY 2402234 exerts on-target, monotherapy activity against IDH1-mutant gliomas in vivo.
Figure 3. Treatment with BAY 2402234 improves survival of mice bearing IDH1-mutant glioma orthotopic xenografts.

(A) Brain sections from mice bearing MGG152 xenografts and treated with BAY 2402234 or vehicle (n = 5 per cohort). Top: stain: hematoxylin and eosin. Relative orotate (middle) and carbamoyl aspartate (bottom) levels as determined by matrix-assisted laser desorption ionization mass spectrometry imaging (MALDI-MSI). Scale bar, 3 mm.
(B and C) MALDI-MSI-based quantification of orotate, carbamoyl aspartate, and OCAR (B) and BAY 2402234 (C) in tumor tissues in (A). n.d., not detected.
(D and E) Kaplan-Meier curves of mice bearing MGG152 xenografts treated with BAY 2402234 or vehicle (D) or cranial radiation or sham (E). Red arrows in (E) indicate irradiation.
(F) Kaplan-Meier curves of mice bearing TS516 xenografts treated with BAY 2402234 or vehicle.
(G and H) OCAR in MGG152 (n = 8 per cohort) (G) and TS516 (n = 6 per cohort) (H) tumor tissues. (I) DHODH activity in MGG152 or TS516 tumor tissues after BAY 2402234 or vehicle treatment.
(J and K) Kaplan-Meier curves of mice bearing HOG-R132H xenografts expressing either EV (J) or DHODH-A58T (K). For (B), Tukey plots are shown. For (C), data are means ± SD. For all other panels, data are means ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001. In (D–F), (J), and (K), p values were calculated by log rank test. Grubbs’ test was used to detect and exclude outliers in (J and K) (a = 0.05). In (B and G–I), two-tailed p values were determined by unpaired t test.
Creation of a genetically engineered mouse model of mutant IDH1-driven astrocytoma
Would more indolent IDH-mutant brain tumors also respond to DHODH inhibitors given that nucleotide synthesis dependencies in other cancers have principally been associated with rapidly proliferating cells (Deberardinis et al., 2008)? To address this question, we developed a genetically engineered mouse (GEM) model of mutant IDH1-driven grade 3 astrocytoma to test BAY 2402234 therapy.
Brain-specific activation of the IDH1-R132H oncogene alone is insufficient to cause gliomagenesis in mice (Bardella et al., 2016; Sasaki et al., 2012a). We hypothesized that more fully recreating the mutational landscape of astrocytoma, IDH-mutant, grade 3 in adult mouse brains might cause tumors that recapitulate this disease. A subset of astrocytomas harbor concurrent mutations in IDH1, TP53, and ATRX genes, along with alterations affecting PIK3CA or PIK3R1 genes, which encode the two subunits of PI3-kinase (PI3K) (Figure 4A and Table S3). PI3K mutations define a subset of patients with IDH1-mutant lower grade glioma with poor outcomes (Figure 4B) (Aoki et al., 2018). We hypothesized that mutating Idh1 with Pik3r1 or Pik3ca in neural cells in the setting of astrocytoma-specific Atrx and Trp53 mutations would promote the development of gliomas in mice. To this end, we determined that PIK3R1 (Quayle et al., 2012) and IDH1 (Koivunen et al., 2012) oncogenes cooperate to transform NHA cells that were immortalized via expression of HPV E6 (which phenocopies TP53 mutations) and E7 (which phenocopies RB mutations) proteins as well as hTERT (which phenocopies ATRX loss) (Figures 4C–4G and S4).
Figure 4. IDH1 and PIK3R1 oncogenes cooperate to transform immortalized astrocytes.

(A) Alterations in the indicated genes in the Brain Lower Grade Glioma TCGA dataset.
(B) Kaplan-Meier curves of glioma patients with the indicated genotypes. Note that the apparent tail of the black curve is driven by a single patient.
(C and D) Immunoblot analyses of NHA cell lines expressing HA-tagged IDH1-R132H (or EV) and FLAG-tagged WT or mutant (D560_S565del, R574fs, or T576del) p85 (protein product of PIK3R1 gene) or the corresponding EV.
(E) Representative anchorage independence assays of NHA cells used in (C) and (D) (n = 5). Scale bar, 100 μm.
(F and G) Bioluminescence imaging (F) and Kaplan-Meier curves (G) of mice after intracranial injection of EV/EV (control), IDH1R132H/EV (IDH1mut), EV/p85D560_S565del (PIK3R1mut), IDH1R132H/p85D560_S565del (IDH1mut + PIK3R1mut), or H-Ras-V12 (positive control) NHA lines. *p < 0.05. n.s., not significant. P values were determined by the log rank test.
To recapitulate these mutations in a GEM model, we used recombinant adeno-associated virus (AAV), CRISPR/Cas9 editing, and transgenic mouse lines, including those with the following alleles: LSL-IDH1-R132H, LSL-Pik3ca-H1047R, and LSL-Cas9 (Figure 5A) (Adams et al., 2011; Platt et al., 2014; Sasaki et al., 2012a).We injected AAVs expressing a Cre cDNA and sgRNAs targeting Trp53 and Atrx genes into the brains of adult compound transgenic mice: (1) LSL-Pik3caH1047R/+;Idh1LSL-R132H/+;LSL-Cas9+/−(PIC), (2) Idh1LSL-R132H/+;LSL-Cas9+/− (IC), (3) LSL-Pik3caH1047R/+;LSL-Cas9+/− (PC), and (4) LSL-Cas9+/− (C). Gliomas started forming, as determined by magnetic resonance imaging (MRI), in PIC mice 10 months after AAV injection (Figure 5B). These gliomas displayed 2HG upregulation and the expected genetic mutations (Figures S5A–S5F). However, many AAV-injected PC and PIC mice developed sarcomas at the site of virus injection in the skull that contributed to mortality (Figures 5C and S5G) (Donehower et al., 1992). Nevertheless, sarcoma-free, AAV-injected PIC mice went on to develop grade 3 astrocytomas that were either not (IC and C) or very infrequently (PC) observed in other cohorts (Figure 5D) and exhibited key morphological and lineage marker expression profiles of human astrocytomas (Figures 5E and 5F).
Figure 5. Creation of GEM models of lower grade astrocytoma.

(A) Mouse injection scheme. Indicated AAV was intracranially injected into mouse strains (PIC, IC, PC, and C). LSL, loxP-stop-loxP cassette. Mice were monitored for tumor initiation with serial monthly MRI scans.
(B) Representative MRI images of a PIC mouse after intracranial AAV injection, as in (A).
(C) Kaplan-Meier curves of AAV-injected mice, as in (A). Time 0 = day of injection.
(D) Cumulative glioma incidence in mice (as determined by MRI, histopathologic analysis, or both), as in (A), in the 12 months after AAV injection. Mice that developed injection-site sarcomas were censored.
(E and F) Hematoxylin and eosin (E) and immunohistochemical (F) stained sections of the brain from a representative AAV-injected PIC mouse. In (E), the black box indicates the region in the right panel; in all panels, scale bars, 200 μm.
(G) Representative MRI images from an AAV-injected PIC mouse. Arrows indicate tumor.
(H and I) Photomicrograph (H) of DF-AA27 GEM model (GEMM) GSCs and immunoblot analysis (I) of DF-AA27 GEMM GSCs, human neural stem cells (NSC), and primary human astrocytes. In (H), scale bar, 100 μm. GFAP, glial fibrillary acidic protein.
(J) Kaplan-Meier tumor-free survival curve of mice intracranially injected with DF-AA27 cells. Time 0 = day of injection. (n = 15). For (B and G), MRI images are coronal slices of the entire mouse brain. For all panels, data are means ± SD; *p < 0.05, **p < 0.01. In (C), the p value was determined by log rank test. In (D), the p value was determined by one-way ANOVA.
DHODH inhibition displays monotherapy activity in grades 3 and 4 IDH-mutant gliomas
To circumvent sarcoma formation and protracted tumor latency, we derived a GSC line from an astrocytoma that formed in an AAV-injected PIC mouse (Figures 5G and 5H). This GSC line, DF-AA27, grew as neurospheres, expressed appropriate lineage and stemness (Gfap, Olig2, Sox2, and Nestin) markers, displayed elevated 2HG levels that were depleted by the mutant IDH1 inhibitor AGI-5198, and harbored engineered mutations in Idh1, Pik3ca, Trp53, and Atrx genes (Figures 5H and 5I and S5H–S5L). DF-AA27 cells formed orthotopic allografts within 1 to 4 months after intracranial implantation (Figure 5J and S5M) that retained high 2HG levels (Figure S5N). After testing supraphysiological uridine-sensitive killing of DF-AA27 cells by BAY 2402234 in vitro (Figure 6A), we assessed the efficacy of this drug in DF-AA27 orthotopic allografts. BAY 2402234 treatment decreased OCAR and markedly attenuated tumor growth (Figures 6B–6D). Of the eight DF-AA27 allografts treated with BAY 2402234, three tumors regressed and three displayed less growth relative to vehicle-treated controls (Figure S6). Therefore, de novo pyrimidine synthesis likely represents an oncogene-induced metabolic vulnerability in both grade 3 and grade 4 IDH1-mutant gliomas.
Figure 6. Organoid and genetically engineered allograft models of IDH-mutant glioma respond to DHODH inhibition.

(A) Cell death assay in DF-AA27 GEMM GSCs treated with BAY 2402234 or DMSO with or without 100 μM uridine (n = 3).
(B) OCAR in DF-AA27 orthotopic allografts in mice treated with BAY 2402234 or vehicle (n = 6 per cohort).
(C and D) Tumor volumes (C) and representative MRI images (D) of mice with DF-AA27 orthotopic allografts treated continuously following tumor formation with BAY 2402234 or vehicle.
(E) Histology, immunohistochemistry, and quantification of cleaved caspase 3 (CC3) in glioma SXOs treated with 30 nM BAY 2402234 or DMSO. Scale bars, 50 μm. Gr, tumor grade, A, astrocytoma, O, oligodendroglioma, GBM, glioblastoma. For (A–C), data are means ± SEM. For (D), MRI images are coronal slices of the entire mouse brain. For (E), data are means ± SD. *p < 0.05, ***p < 0.001, n.s., not significant. Two-tailed p values were determined by unpaired t test.
Next, we created a panel of surgically explanted organoid (SXO) models of glioma (five IDH WT and three IDH1 mutant) from primary tissue specimens (Figure 6E) (Abdullah et al., 2022). IDH1-mutant SXOs were derived from both grade 3 and grade 4 gliomas. After confirming that SXOs maintain histological features of the respective parental tumors, we treated them with BAY 2402234 or DMSO and measured apoptosis induction via immunohistochemical quantification of cells expressing cleaved caspase 3 (Figure 6E). BAY 2402234 induced apoptosis in all three IDH1-mutant SXOs, but did so in only one of the five IDH1/2 WT SXOs. As two of the three IDH1-mutant gliomas were grade 3, these data further support our hypothesis that DHODH hyperdependence is not confined to highly proliferative grade 4 tumors.
Mechanisms of de novo pyrimidine synthesis hyperdependence in IDH-mutant glioma
To begin to understand the obligate dependence of IDH1-mutant glioma cells on de novo pyrimidine synthesis, but not de novo purine synthesis, for viability, we cultured IDH1-mutant BT054 GSCs in human plasma-like medium (HPLM) (Cantor et al., 2017) to directly compare the metabolic effects of de novo pyrimidine and de novo purine synthesis inhibition under physiologically relevant conditions. We first confirmed that BAY 2402234 and lometrexol, an inhibitor of glycinamide ribonucleotide transformylase (GART) in the de novo purine synthesis pathway, engaged their respective enzyme targets. BAY 2402234 increased levels of dihydroorotate, the substrate of DHODH, while lometrexol treatment increased levels of glycinamide ribonucleotide, the substrate of GART (Figure 7A). Next, we quantified representative pyrimidine (UMP) and purine (AMP) nucleotides and found that BAY 2402234 decreased pyrimidine nucleotide synthesis, whereas lometrexol did not affect purine nucleotide synthesis. These data suggest that IDH1-mutant glioma cells rely predominantly on the de novo pathway for pyrimidine synthesis and the salvage pathway for purine synthesis (Figure 7B).
Figure 7. Glioma cells use divergent routes for pyrimidine and purine nucleotide synthesis under physiologic conditions.

(A) Steady-state quantification of the indicated metabolites in BT054 GSCs treated with 10 nM BAY 2402234, 5 μM lometrexol, or DMSO (n = 8 per condition).
(B) Pathways targeted by BAY 2402234 (BAY) and lometrexol (Lom).
(C) 15N stable isotope tracing assays in BT054 and TS516 GSC lines (n = 3).
(D) Select pathways using intermediates derived from de novo pyrimidine nucleotide synthesis.
(E and F) Volcano plots of metabolites in HOG-EV (E) or HOG-R132H (F) cells treated with 10 nM BAY 2402234 relative to DMSO (n = 4).
(G) Ratio of pyrimidine (dTTP, dCTP) to purine (dATP) deoxynucleotide triphosphate (dNTP) pools in cells shown in (E and F). For all panels, data are means ± SEM; *p < 0.05, ***p < 0.001. n.s., not significant. Two-tailed p values were determined by unpaired t test.
To test this idea, we conducted parallelized stable nitrogen isotope tracing assays for substrates that feed de novo and salvage nucleotide synthesis pathways and confirmed intracellular accumulation of nitrogen-labeled metabolites (Figure S7A). In support of our model, the de novo pyrimidine synthesis pathway substrate 15N-glutamine robustly labeled the UMP pool, with moderate and no labeling observed from the pyrimidine salvage pathway substrates 15N-uridine and 15N-uracil (Figure 7C). In contrast, the de novo purine synthesis pathway substrate 15N-glutamine failed to label AMP, while the purine salvage pathway substrate 15N-hypoxanthine robustly labeled AMP. Notably, similar labeling patterns were observed in the IDH1/2 WT TS516 GSC line (Figure 7C). Removing hypoxanthine from HPLM triggered induction of de novo purine synthesis and maintenance of the inosine monophosphate pool in BT054 cells (Figures S7B and S7C), indicating that hypoxanthine is a preferred substate for purine synthesis in glioma cells and that they can engage both de novo and salvage purine synthesis pathways. Therefore, GSCs, independent of IDH status, rely primarily on the de novo pathway for pyrimidine synthesis, but display plasticity with respect to purine synthesis, therefore partially explaining the differential impact of de novo pyrimidine and de novo purine synthesis inhibitors in our drug screen.
To illuminate why IDH1-mutant cells are hyperdependent on DHODH for viability relative to WT cells, we performed a metabolomics analysis of HOG-EV and HOG-R132H cells with and without BAY 2402234 treatment. Basal levels of pyrimidine nucleotides were similar in IDH1-mutant and IDH1 WT cells (Figure S7D). Upon drug treatment, we observed alterations in metabolites that link pyrimidine synthesis with key pyrimidine-dependent cellular processes: protein glycosylation, RNA synthesis, phospholipid synthesis, and DNA synthesis (Figures 7D–7F). Although BAY 2402234 treatment depleted some substrates for protein glycosylation (e.g., UDP-hexose) and phospholipid synthesis (e.g., CDP-choline), global levels of dolichol-linked oligosaccharides necessary for N-linked glycosylation and phosphatidylcholine and phosphatidylethanolamine lipids were largely unchanged (Figures S7E–S7H). BAY 2402234 suppressed levels of the RNA precursors UTP and CTP (Figure S7I) irrespective of IDH status, and IDH1-mutant and IDH1 WT glioma cells did not differ in their sensitivity to transcription inhibition (Figure S7J).
Our metabolomics study also revealed depletion of DNA synthesis substrates by DHODH inhibition irrespective of IDH status. The pyrimidine deoxynucleotide triphosphates (dNTPs) dTTP and dCTP were potently suppressed, while the purine dNTP, dATP, was upregulated (Figures 7E–7G and S7K–S7P). Imbalances in the ratio of pyrimidine to purine nucleotides required for DNA synthesis can evoke DNA damage (Kim et al., 2017), raising the possibility that IDH1-mutant cells are hypersensitive to the genotoxic effects of nucleotide imbalances. In support of this idea, the DNA damage marker phospho-histone H2A.X (γH2A.X) was more robustly induced by BAY 2402234 in engineered and patient-derived IDH1-mutant glioma cells compared with IDH WT cells (Figures 8A and 8B). This was specific because lometrexol, which did not create nucleotide imbalance (Figure 7A), was inert in this assay (Figure 8C). As an additional measure of DNA damage, we found that 53BP1 foci were more abundant at baseline in IDH1-mutant versus IDH1 WT HOG cells (Figure 8D), consistent with prior findings from a member of our group (T.W.M.) in long-term hematopoietic stem cells exposed to chronic mutant IDH1 activity (Inoue et al., 2016). Although 53BP1 foci increased acutely in both IDH1-mutant and IDH1 WT cells treated with BAY 2402234, they persisted longer in the IDH1-mutant cells, corroborating the γH2A.X induction data (compare Figures 8D–8A). These data suggest that persistent DNA damage accounts for the preferential cytotoxicity of de novo pyrimidine synthesis inhibitors against IDH1-mutant glioma cells.
Figure 8. The IDH1-R132H mutation enhances DNA damage caused by DHODH inhibition.

(A and B) Immunoblot analysis of HOG-EV and HOG-R132H cells (A) or GSC lines (B) treated with BAY 2402234 (HOGs: 1, 3, or 10 nM; GSCs: 3 or 10 nM) or DMSO.
(C) Immunoblot analysis of HOG-EV and HOG-R132H cells treated with 10 nM BAY 2402234, 5 μM lometrexol, or DMSO.
(D) Quantification of 53BP1 foci formation in HOG-EV and HOG-R132H cells treated with BAY 2402234 (n ≥ 325 cells per condition per time point) and representative images. Scale bar, 5 μm; dashed lines show individual nuclei.
(E–H) Immunoblot analysis and cell death assays in HOG-EV and HOG-R132H cells treated with BAY 2402234, DMSO, 15 μM dC, and/or 15 μM dT (n = 3) (E and F), or pre-treated with 1 μM palbociclib or DMSO (G and H), then treated with BAY 2402234 or DMSO (n = 3).
(I and J) Representative hematoxylin and eosin, anti-γH2A.X IHC staining (I), and γH2A.X quantification (J) of MGG152 orthotopic xenografts treated with BAY 2402234 (n = 5) or vehicle (n = 2). Scale bar, 100 μm.
(K and L) MALDI-MSI ion images (K) and quantified relative changes (L) of brains from mice with MGG152 orthotopic glioma xenografts treated with BAY 2402234 or vehicle. Scale bar, 3 mm.
(M) Schema depicting model by which BAY 2402234 (BAY) treatment preferentially kills IDH-mutant glioma cells. For (D), data are means and variance is not displayed. For (L), Tukey plots are shown. For all other panels, data are means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, n.s., not significant. Two-tailed p values were determined by unpaired t test.
Rescuing pyrimidine dNTP pools by supplementing IDH1-mutant cells with deoxycytidine (dC) and deoxythymidine (dT) decreased γH2A.X and cell death upon DHODH inhibition (Figures 8E and 8F). Importantly, dC and dT provision during BAY 2402234 treatment did not restore pyrimidine substrates for protein glycosylation or phospholipid or RNA synthesis, suggesting this effect was specific for dNTP pool rebalancing (Figure S8A). Blocking cell cycle progression in G1-phase with the CDK4/6 inhibitor palbociclib attenuated DNA damage and cell killing by BAY 2402234 (Figures 8G and 8H), suggesting that suppression of the pyrimidine to purine dNTP ratio by BAY 2402234 elicits DNA damage that depends on replication stress during S-phase. Unlike palbociclib treatment and dC/dT supplementation, blocking 2HG synthesis with a mutant IDH1 inhibitor did not acutely reverse DHODH inhibitor sensitivity in IDH1-mutant glioma cells (Figures S8B and S8C). Moreover, treating IDH1 WT cells with a cell permeable (R)-2HG ester, (R)-2HG-TFMB, did not acutely sensitize them to DHODH inhibition (Figures S8D and S8E). These findings, together with observed passage-dependent sensitization of NHA cells to BAY 2402234 by mutant IDH1 (Figures S2A–S2F), implicate mutant IDH-dependent alterations in chromatin structure and gene expression because they manifest with similarly slow kinetics (Turcan et al., 2012). We evaluated gene expression differences in IDH-mutant and IDH WT human glioma samples from the TCGA dataset. Genes and gene sets related to DNA replication, DNA damage sensing, and DNA repair were downregulated in IDH-mutant gliomas compared with those associated with unrelated cellular processes (Figure S8F–S8I, Table S4). Therefore, we propose that epigenetic and transcriptomic reprogramming by IDH mutations preferentially repress genes that promote genome integrity during nucleotide imbalance, sensitizing them to de novo pyrimidine synthesis inhibition.
To extend our studies in vivo, we quantified γH2A.X in BAY 2402234- or vehicle-treated orthotopic MGG152 IDH1-mutant glioma xenografts and observed robust drug-induced DNA damage (Figures 8I and 8J). Tumoral γH2A.X levels inversely correlated with the abundance of dTTP and dCTP (Figures 8I–8L). The γH2A.X levels in normal brain tissues were not affected by BAY 2402234 (data not shown), despite decreased OCAR (Figure 2C). Collectively, our data indicate that chronic mutant IDH activity sensitizes glioma cells to DHODH inhibition by increasing their susceptibility to replication-dependent DNA damage caused by nucleotide pool imbalances (Figure 8M).
DISCUSSION
Our findings reveal that de novo pyrimidine nucleotide synthesis is a collateral vulnerability induced by IDH oncogenes in glioma. Notably, high DHODH expression is associated with inferior survival in patients with astrocytoma, IDH-mutant, grade 4, but not in IDH WT, glioblastoma (Zhou et al., 2020). Two groups have reported that alternative DHODH inhibitors (DHODHi) are active in IDH WT glioma subcutaneous xenograft models (Echizenya et al., 2020; Lafita-Navarro et al., 2020). However, neither we nor others observed significant monotherapy activity of DHODHi in orthotopic xenografts of IDH WT gliomas (Wang et al., 2019), in contrast with our findings with IDH1-mutant orthotopic xenografts. Therefore, our data suggest that IDH mutations may be sufficient to sensitize gliomas to DHODHi monotherapy. This is further supported by our data using isogenic glioma model systems (Figures 1E–1J and S2A–S2F), as well as independently generated data showing that the introduction of an IDH mutation is sufficient to sensitize leukemia cells to DHODHi (Si and Keenan, 2017).
Our findings that mutant IDH1 expression induces DNA damage resulting from replication stress build on extensive prior research establishing nucleotide pool homeostasis as a critical determinant of DNA replication fidelity (Lee et al., 2018; Meuth, 1989). Moreover, our work extends the idea that certain oncogenic mutations increase pyrimidine synthesis dependence and sensitivity to nucleotide pool imbalance. Lung cancer cells with mutations in KRAS and LKB1 engage a non-canonical pyrimidine biosynthesis program dependent on carbamoyl phosphate synthetase-1 to achieve nucleotide balance and avert DNA damage (Kim et al., 2017). Similar vulnerabilities to nucleotide imbalance driven by IDH1 and KRAS/LKB1 mutations in brain and lung tumors, respectively, suggest a possible paradigm that could be therapeutically exploited for a host of tumors.
IDH mutations have been previously linked with DNA damage in glioma, partly through the repression of homology-directed repair (HDR) (Sulkowski et al., 2017, 2020). However, this process cannot fully explain increased DNA damage sensitivity in our models, because HDR suppression is readily reversed by mutant IDH1 inhibition and (R)-2HG depletion. In contrast, sensitivity of IDH1-mutant glioma cells to DHODHi, although correlating with steady-state (R)-2HG levels (Figures 1L and 2H), is not acutely altered by abrupt changes in (R)-2HG levels. In this regard, Lu and colleagues showed that engineering IDH1 mutations in U251 glioma cells increases their sensitivity to the DNA damaging agent temozolomide via a mechanism that is likewise insensitive to short-term mutant IDH1 inhibition (Lu et al., 2017). IDH1 mutations are known to cause both reversible and irreversible changes in chromatin structure and gene expression (Turcan et al., 2018), the latter of which may underpin DNA damage hypersensitivity in glioma. In support of this idea, we show that the genes involved in DNA damage sensing and repair are globally suppressed in IDH-mutant versus IDH WT gliomas, although the specific gene or genes that control response to DHODH inhibition remain to be identified. During DHODH inhibition, DNA damage may also be reinforced by decreased flux from glucose to pyrimidine nucleotides in IDH-mutant glioma cells (Garrett et al., 2018). Regardless of the underlying mechanism, it will be important to determine whether DHODH inhibition synergizes with other DNA damaging therapies, such as radiation.
We report the development of a GEM model of IDH1-mutant glioma. Both our model and a previously reported GEM model of astrocytoma, IDH-mutant, grade 4 (Philip et al., 2018) show that the IDH1-R132H oncogene drives gliomagenesis when expressed in the presence of clinically relevant co-occurring mutations. These advances circumvent long-standing challenges in developing glioma GEM models driven by mutant IDH. IDH1-R132H expression alone is insufficient to cause glioma formation in mice (Bardella et al., 2016; Sasaki et al., 2012a) and can impede gliomagenesis when coupled with mutations that are infrequently observed in IDH1-mutant human brain tumors (Núñez et al., 2019). The cooperation we observe between the IDH1-R132H oncogene and mutations affecting PI3K signaling reinforces an established association between PIK3R1 mutations and poor outcomes in patients with IDH-mutant astrocytoma (Aoki et al., 2018). Our GEM model opens new avenues to explore mutant IDH1 action in the setting of treatment-naïve, lower grade brain tumors.
Although IDH inhibitors may display activity in some non-enhancing IDH-mutant gliomas, these agents have thus far not shown benefit in patients with contrast-enhancing brain tumors (Mellinghoff et al., 2021). Mutant IDH oncogenes cause durable changes in gene expression that are insensitive to acute (R)-2HG depletion (Johannessen et al., 2016), and identifying effective therapies independent of an ongoing requirement for (R)-2HG is a key challenge. Our work provides preclinical rationale to initiate clinical studies of BAY 2402234 in glioma by identifying IDH mutational status as a predictive biomarker of response. Furthermore, we outline a new therapeutic strategy that shows promise for treating IDH1-mutant gliomas that display de novo resistance to mutant IDH1 inhibitors.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Samuel K. McBrayer (samuel_mcbrayer@utsouthwestern.edu).
Materials availability
Plasmids generated in this study have been deposited to Addgene.
Data and code availability
Metabolomics data have been deposited to the National Metabolomics Data Repository (NMDR) and will be publicly available as of the date of publication. Accession and identification numbers are listed in the key resources table. All other data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Mouse monoclonal anti-IDH1 R132H antibody (IHC) | Dianova | Cat# DIA-H09, RRID:AB_2335716 |
| Rabbit monoclonal anti-Cleaved Caspase 3 | Cell Signaling Technology | Cat# 9661, RRID:AB_2341188 |
| Mouse monoclonal anti-Akt (pan) | Cell Signaling Technology | Cat# 2920, RRID:AB_1147620 |
| Rabbit monoclonal anti-pAkt | Cell Signaling Technology | Cat# 2965S, RRID:AB_2255933 |
| Rabbit polyclonal anti-GFAP | Abcam | Cat# ab7260, RRID:AB_305808 |
| Rabbit polyclonal anti-Sox2 | Abcam | Cat# ab97959, RRID:AB_2341193 |
| Rabbit polyclonal anti-Olig2 | Ligon et al. (2004) | N/A |
| Mouse monoclonal anti-Nestin | Abcam | Cat# ab6142, RRID:AB_305313 |
| Mouse monoclonal anti-γH2A.X | Cell Signaling Technology | Cat# 9718S, RRID:AB_2118009 |
| Mouse monoclonal anti-FLAG | Sigma-Aldrich | Cat# F1804, RRID:AB_262044 |
| Mouse monoclonal anti-HA | Covance | Cat# MMS-101P RRID: AB_2314672 |
| Mouse monoclonal, anti-Vinculin | Sigma-Aldrich | Cat# V9131, RRID:AB_477629 |
| Anti-53BP1 antibody | Novus | Cat# NB100–304, RRID:AB_10003037 |
| Rabbit polyclonal anti-DHODH | Proteintech | Cat#14877 RRID: AB_2091723 |
| Donkey anti-rabbit IgG, Alexa Fluor 488-conjugated secondary antibody | Invitrogen | Cat# A-21206 RRID:AB_2535792 |
| Goat anti-mouse IgG, HRP-linked secondary antibody | Thermo Fisher | Cat# 31430 RRID:AB_228307 |
| Goat anti-rabbit IgG, HRP-linked secondary antibody | Thermo Fisher | Cat#31460 RRID:AB_228341 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Adeno-associated virus (pAAV2-sgTrp53-sgAtrx-EFS-Cre) | This paper, AAV packaging performed by Vigene Biosciences | https://www.vigenebio.com/aav-packaging/ |
| HB101 Competent Cells | Promega | Cat# L2015 |
|
| ||
| Biological samples | ||
|
| ||
| UTSW6577 glioma specimen | Abdullah et al. (2022) | N/A |
| UTSW1382 glioma specimen | Abdullah et al. (2022) | N/A |
| UTSW9647 glioma specimen | Abdullah et al. (2022) | N/A |
| UTSW2243 glioma specimen | Abdullah et al. (2022) | N/A |
| UTSW2846 glioma specimen | This paper | N/A |
| UTSW2294 glioma specimen | This paper | N/A |
| UTSW9698 glioma specimen | This paper | N/A |
| UTSW9501 glioma specimen | This paper | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Metabolic inhibitor library | This paper | N/A |
| Anticancer drug library | Selleck Chemicals | N/A (purchased in 2015) |
| Brequinar | Sigma-Aldrich | Cat# SML0113 |
| 6-Azauridine | Sigma-Aldrich | Cat# A1882 |
| Pyrazofurin | Sigma-Aldrich | Cat# SML1502 |
| Uridine | Sigma-Aldrich | Cat# U3750 |
| Lometrexol | Cayman Chemical | Cat# 18049 |
| AnnexinV-FITC | Fisher Scientific | Cat# BD556547 |
| AnnexinV-PE | Fisher Scientific | Cat# BD559763 |
| AnnexinV-APC | Fisher Scientific | Cat# BD550475 |
| DAPI | Cayman Chemical | Cat# 14285 |
| AGI-5198 (IDH-C35) | Xcess Bio | Cat# M60068 |
| Deoxythymidine | Sigma-Aldrich | Cat# 3416–05-5 |
| Deoxycytidine | Sigma-Aldrich | Cat# 951–77-9 |
| Palbociclib (PD 0332991) | Cayman Chemical | Cat# 16273 |
| BAY 2402234 | Bayer | N/A |
| (R)-2HG-TFMB | Provided by Ryan E. Looper | N/A |
| amide-15N-glutamine | Cambridge Isotope Laboratories | Cat# NLM-557 |
| 15N-uracil | Cambridge Isotope Laboratories | Cat# NLM-637 |
| 15N-uridine | Cambridge Isotope Laboratories | Cat# NLM-812 |
| 15N-hypoxanthine | Cambridge Isotope Laboratories | Cat# NLM-8500 |
| Tunicamycin | Sigma-Aldrich | Cat# T7765 |
| 4-hydroxytamoxifen | Sigma-Aldrich | Cat# H7904 |
| L-dihydroorotic acid | Sigma-Aldrich | Cat# D7128 |
| Decylubiquinone | Sigma-Aldrich | Cat# D7911 |
| 2,6-dichloroindophenol | Sigma-Aldrich | Cat# D1878 |
| Sodium malonate | Sigma-Aldrich | Cat# 63409 |
| Potassium cyanide | Sigma-Aldrich | Cat# 60178 |
|
| ||
| Critical commercial assays | ||
|
| ||
| KOD Xtreme Hot Start DNA Polymerase Kit | Millipore Sigma | Cat# 71975 |
| DNeasy Blood and Tissue kit | Qiagen | Cat# 695045 |
| TissueLyser | Qiagen | Cat# 85300 |
| Neural Tissue Dissociation Kit | Miltenyi | Cat# 130–095-942 |
| RNeasy Mini Kit | Qiagen | Cat# 74004 |
| AffinityScript qPCR cDNA Synthesis Kit | Agilent | Cat# 600559 |
| NEB Monarch kit | New England BioLabs | Cat# T1030 |
| Qiagen gel extraction kit | Qiagen | Cat# 28706 |
| Mouse genotyping | Transnetyx | https://www.transnetyx.com/ |
| Bond Polymer Refine Detection Kit | Leica Biosystems | Cat# DS9800 |
| BP Clonase | Invitrogen | Cat# 11789100 |
| LR Clonase | Invitrogen | Cat# 11791020 |
|
| ||
| Deposited data | ||
|
| ||
| TCGA Brain Lower Grade Glioma dataset | GDAC Firehose, Broad Institute | http://gdac.broadinstitute.org/runs/analyses__2016_01_28/reports/cancer/LGG-TP/index.html |
| Metabolomics Analysis of HOG-EV and HOG-R132H Cells with and without BAY 2402234 Treatment | National Metabolomics Data Repository | NMDR: https://doi.org/10.21228/M81X41 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| BT054 | Kelly et al. (2010) | N/A |
| BT260 | Koivunen et al. (2012) | N/A |
| TS516 | Rohle et al. (2013) | N/A |
| TS603 | Rohle et al. (2013) | N/A |
| HK213 | Laks et al. (2016) | N/A |
| HK211 | Laks et al. (2016) | N/A |
| HK157 | Laks et al. (2016) | N/A |
| HK252 | Laks et al. (2016) | N/A |
| HK308 | Laks et al. (2016) | N/A |
| MGG152 | Wakimoto et al. (2014) | N/A |
| HOG | Provided by Pablo Paez | N/A |
| NHA (immortalized with HPV E6 and E7 and hTERT) | Provided by Russell Pieper | N/A |
| NHA (EV or IDH1-R132H) | Koivunen et al. (2012) | |
| UTSW6577 glioma organoids | Abdullah et al. (2022) | N/A |
| UTSW1382 glioma organoids | Abdullah et al. (2022) | N/A |
| UTSW9647 glioma organoids | Abdullah et al. (2022) | N/A |
| UTSW2243 glioma organoids | Abdullah et al. (2022) | N/A |
| UTSW2846 glioma organoids | This paper | N/A |
| UTSW2294 glioma organoids | This paper | N/A |
| UTSW9698 glioma organoids | This paper | N/A |
| UTSW9501 glioma organoids | This paper | N/A |
| DF-AA27 GEMM GSC | This paper | N/A |
| MEFs, irradiated | Life Technologies | Cat# A34181 |
| IC (H11LSL-Cas9+/−;Idh1tm1Mak/WT) MEFs | This paper | N/A |
| HEK293T | ATCC | Cat# CRL-11268 |
| Primary human astrocytes | Lonza | Cat# CC-3187 |
| Human neural stem cells | Fisher Scientific | Cat# 10419428 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| NCr Nude mice, 6–10-week-old females | Taconic | Cat# NCRNU-F |
| NCr Nude mice, 4–5-week-old females | Taconic | Cat# NCRNU-F |
| ICR SCID mice, 6–12-week-old females | Taconic | Cat# ICRSC-F |
| R26-Pik3caH1047R (FVB.129S6-Gt(ROSA)26Sortm1(Pik3ca*H1047R)Egan/J mouse | Jackson Laboratories | Cat# 016977 |
| Idh1tm1Mak mouse | Provided by Tak W. Mak | N/A |
| H11LSL-Cas9 (B6; 129-Igs2tm1(CAG-cas9*)Mmw/J mouse | Jackson Laboratories | Cat# 026816 |
| PIC (H11LSL-Cas9+/−;Idh1tm1Mak/WT;R26-Pik3caH1047R+/−) mouse | This paper | N/A |
| PC (H11LSL-Cas9+/−;Idh1WT/WT;R26-Pik3caH1047R+/−) mouse | This paper | N/A |
| IC (H11LSL-Cas9+/−;Idh1tm1Mak/WT) mouse | This paper | N/A |
| C (H11LSL-Cas9+/−;Idh1WT/WT) mouse | This paper | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| sgATRX: ATAAGGACTTTGAGTGAA | This paper | N/A |
| sgTrp53: GTGTAATAGCTCCTGCATG | Platt et al. (2014) | N/A |
| sgControl: GGAGGCTAAGCGTCGCAA | Provided by Feng Zhang | N/A |
| sgDHODH: CATCTTATAAAGTCCGTCCA | This paper | N/A |
| Trp53 outer fwd ATAGAGACGCTGAGTCCGGTTC | Oser et al. (2019) | N/A |
| Trp53 outer rev CCTAAGCCCAAGAGGAAACAGA | Oser et al. (2019) | N/A |
| Trp53 inner fwd TGCAGGTCACCTGTAGTGAGGTAGG | Oser et al. (2019) | N/A |
| Trp53 inner rev GAAACAGGCAGAAGCTGGGGAAGAAAC | Oser et al. (2019) | N/A |
| ATRX outer fwd GCTATCTGAAACTCAATCCACG | This paper | N/A |
| ATRX outer rev GACTTGGTTTCTCCTTTGCCATG | This paper | N/A |
| ATRX inner fwd GCTTCCTGTAAGCTCATAAGTAC | This paper | N/A |
| ATRX inner rev CTAATGCCATATGAGTGTAACTC | This paper | N/A |
| ATRX second inner fwd CTCTTACATAATGGCCATTCTC | This paper | N/A |
| ATRX second inner rev CTGTGAGTCATGATCATTCTTTGC | This paper | N/A |
| Recombined IDH1 fwd GATTGATTCTGCCGCCATGATCCTAGT | Sasaki et al. (2012b) | N/A |
| Recombined IDH1 rev CCTGGTCATTGGTGGCATCACGATTCTC | Sasaki et al. (2012b) | N/A |
| Actb fwd (genomic DNA for Idh1-R132H assay) TGACCCAGGT CAGTATCCCGGGT | This paper | N/A |
| Actb actin rev (genomic DNA for Idh1-R132H assay) GAACACAGCTAAGTTCAGTGTGCTGGGA | This paper | N/A |
| PIK3CA H1047R cDNA fwd CTAGGTAGGGGATCGGGACTCT | This paper | N/A |
| PIK3CA H1047R rev AATTTCTCGATTGAGGATCTTTTCT | This paper | N/A |
| Actb fwd (cDNA for Pik3ca-H1047R assay) TAGGCACCAGGGTGTGATG | This paper | N/A |
| Actb rev (cDNA for Pik3ca-H1047R assay) CATGGCTGGGGTGTTGAAGG | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pLX304-Ubc-GATE-IRES-GFP | Gift from Vidyasagar Koduri | N/A |
| pDONR223 | Invitrogen | N/A |
| pLX304-Ubc-EV-IRES-GFP | EV provided as a gift by Vidyasagar Koduri | N/A |
| pLX304-Ubc-DHODH-WT-IRES-GFP | DHODH WT cDNA provided as a gift by Laura Evans & Julie Losman | N/A |
| pLX304-Ubc-DHODH-A58T-IRES-GFP | DHODH A58T cDNA provided as a gift by Laura Evans & Julie Losman | N/A |
| pLX304-CMV-BFP | Gift from Hilary Nicholson | N/A |
| pLX304-CMV-GFP | Gift from Hilary Nicholson | N/A |
| pLXSN-H-Ras_V12 | Addgene | Cat# 39516 |
| pLenti-Flag-P85 | Addgene | Cat# 40219 |
| pLenti4-Flag-P85-DKRMNS560del | Addgene | Cat# 40225 |
| pLenti-Flag-P85-R574fs | Addgene | Cat# 40227 |
| pLenti4-Flag-T576del | Addgene | Cat# 40228 |
| p1321 HPV-16 E6/E7 | Addgene | Cat# 8641 |
| pAAV2-sgTrp53-sgAtrx-EFS-Cre | This paper | Addgene, Cat# 189977 |
| pLBCX-Large-T-antigenK1 mutant | This paper | Addgene, Cat# 189978 |
| pMSCV-MerCreMer-hygro | This paper | Addgene, Cat# 188982 |
| LeGO-iG2-FLuc | McBrayer et al. (2018) | N/A |
| pBABE-HA-hygro | Koivunen et al. (2012) | N/A |
| pBABE-IDH1-R132H-HA-hygro | Koivunen et al. (2012) | N/A |
| plenti-Ubc-IRES-hygro empty vector | McBrayer et al. (2018) | N/A |
| plenti-Ubc-IDH1-R132H-HA-IRES-hygro | McBrayer et al. (2018) | N/A |
| pMD2.G | Provided by Didier Trono | Addgene, Cat# 12259 |
| Gag/pol | Provided by Tannishtha Reya | Addgene, Cat# 14887 |
| psPAX2 | Provided by Didier Trono | Addgene, Cat# 12260 |
| VSV.G | Provided by Tannishtha Reya | Addgene, Cat# 14888 |
| pX551 | Provided by Feng Zhang | Addgene Cat# 60957 |
|
| ||
| Software and Algorithms | ||
|
| ||
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
| Metran | Yoo et al. (2008) | N/A |
| Multi-Quant 2.1 software | Sciex | https://sciex.com/products/software/multiquant-software |
| GraphPad Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Living Image | PerkinElmer | http://www.perkinelmer.com/product/li-software-for-spectrum-1-seat-add-on-128113 |
| R | The R Project for Statistical Computing | https://www.r-project.org/ |
| AccuCor R package (version 0.2.3) | Su et al. (2017) | https://github.com/lparsons/accucor |
| El-Maven 0.12.0 | Elucidata | https://elucidata.io/el-maven/ |
| TraceFinder 5.1 SP2 | Thermo Fisher | Cat# OPTON-31001 |
| Freestyle 1.7 SP | Thermo Fisher | Cat# XCALI-98282 |
| MetaboAnalyst | Publicly available | https://www.metaboanalyst.ca/ |
| FlexControl, version 4.0 | Bruker | https://www.bruker.com/en/products-and-solutions/mass-spectrometry/maldi-tof/autoflexmax.html |
| FlexImaging, version 5.0 and SCiLS Lab | Bruker Daltonics | https://www.bruker.com/en/products-and-solutions/mass-spectrometry/ms-software/scilslab.html |
| ImageJ | NIH | https://imagej.net/ |
| QuPath software (version 0.2.3) | Publicly available | https://qupath.github.io/ |
|
| ||
| Other | ||
|
| ||
| Geltrex | Life Technologies | Cat# A1413202 |
| AGM SingleQuots Supplements | Lonza | Cat# CC-4123 |
| NeuroCult Basal Medium (Mouse and Rat) | StemCell Technologies | Cat# 05702 |
| NeuroCult NS-A Basal Medium (Human) | StemCell Technologies | Cat# 05750 |
| Neurobasal Medium (MGG152 GSCs) | Gibco | Cat# 21103049 |
| Neurobasal Medium (organoids) | Thermo Fisher | Cat# 21103049 |
| DMEM-F12 Medium | Gibco | Cat# 11320033 |
| DMEM Medium | Gibco | Cat# 11995–065 |
| Accutase | StemCell Technologies | Cat# 07922 |
| RBC lysis buffer | Thermo Fisher | Cat# 00433357 |
| 2-mercaptoethanol | Thermo Fisher | Cat# BP176–100 |
| Glutamax | Thermo Fisher | Cat# 35050061 |
| N-2 supplement | Thermo Fisher | Cat# 17502048 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HOG cells (human oligodendroglioma line from a male) were a gift of P. Paez, SUNY Univ. at Buffalo and were cultured in IMDM medium (Gibco 12440061) with 10% FBS and 1% penicillin/streptomycin. Stable HOG cell lines expressing either EV or IDH1-R132H were generated using plenti-Ubc-IRES-hygro plasmid backbone and cDNA cloning strategies as previously described (McBrayer et al., 2018). Stable cell lines were selected with 500 μg/mL hygromycin and maintained in media the above media supplemented with 200 μg/mL hygromycin and were cultured for at least 5 weeks after selection prior to experimentation. NHA cells (human astrocytes immortalized with HPV E6 and E7 and hTERT) (Sonoda et al., 2001) (sex unknown) were a kind gift of Dr. Russell Pieper (UCSF) or were produced from commercially obtained primary human astrocytes (Lonza CC-3187). NHA cells were cultured in DMEM medium (Gibco 11995–065) containing 10% FBS and 1% penicillin/streptomycin. NHA cells expressing either EV or IDH1-R132H used in this study were described previously (model #1) (Koivunen et al., 2012) or generated de novo (model #2) and maintained in media supplemented with 100 μg/mL hygromycin. Early passage NHA cells (models #1 and #2) were maintained for 5–10 passages prior to use in low passage experiments, and for 25–35 passages prior to use in high passage experiments. Stable NHA cell lines expressing either EV or IDH1-R132H were generated using pBabe-hygro (model #1) or pLenti-EF1α-IRES-hygro (model #2) plasmid backbone and cDNA cloning strategies as previously described (Koivunen et al., 2012; McBrayer et al., 2018). Idh1LSL-R132H/+;LSL-Cas9+/−(IC) MEFs created in this study were cultured in DMEM medium (Gibco 11995–065) containing 10% FBS and 1% penicillin/streptomycin.
All HOG, NHA, and MEFs lines were cultured in the presence of 5% CO2 and ambient oxygen at 37°C. All cell lines were routinely evaluated for mycoplasma contamination and tested negative throughout the study. Cell line authentication was not performed because reference short term tandem repeat profiles have not been established for these cell lines.
Primary cell cultures
GSC lines TS516 and TS603 (sexes unknown) were obtained from I. Mellinghoff at MSKCC (Rohle et al., 2013). BT054 cells (female) were obtained from S. Weiss at Univ. of Calgary (Kelly et al., 2010). BT260 cells (sex unknown) were obtained from K. Ligon at DFCI (Koivunen et al., 2012). HK211 (female), HK213 (male), HK252 (male), HK308 (female), and HK157 (female) cells were obtained from H. Kornblum at UCLA (Laks et al., 2016). TS516, TS603, BT054, and BT260 cells were cultured in NeuroCult NS-A Basal Medium (Human) with Proliferation Supplement (StemCell Technologies 05750) supplemented with EGF (20 ng/mL), bFGF (20 ng/mL), heparin (2 μg/mL), 1% penicillin/streptomycin, amphotericin B (250 ng/mL), and Plasmocin (2.5 μg/mL). HK213, HK211, HK308, and HK157 cells were cultured in DMEM-F12 medium (Gibco 11320033) supplemented with 3 mM glutamine, 1× B27, EGF (20 ng/mL), bFGF (20 ng/mL), heparin (2 μg/mL), 0.5% penicillin/streptomycin, amphotericin B (125 ng/mL), and Plasmocin (2.5 μg/mL). MGG152 cells (Wakimoto et al., 2014) were cultured in Neurobasal Medium (Gibco 21103049) supplemented with 3 mM glutamine, 1× B27, 0.25× N2, EGF (20 ng/mL), bFGF (20 ng/mL), heparin (2 μg/mL), 0.5% penicillin/streptomycin, amphotericin B (125 ng/mL), and Plasmocin (2.5 μg/mL). DF-AA27 GEMM GSCs were cultured in NeuroCult Basal Medium (Mouse and Rat) with Proliferation Supplement (StemCell Technologies 05702) supplemented with EGF (20 ng/mL), bFGF (20 ng/mL), heparin (2 μg/mL), 1% penicillin/streptomycin, amphotericin B (250 ng/mL), and Plasmocin (2.5 μg/mL). Primary human astrocytes (sex unknown) were purchased from Lonza (CC-2565) and cultured in ABM Basal Medium (Lonza CC-3187) supplemented with AGM SingleQuots Supplements (Lonza CC-4123). Human neural stem cells (sex unknown) were purchased from Fisher Scientific (10419428) and cultured in DMEM/F12 medium supplemented with 3 mM glutamine, EGF (20 ng/mL), bFGF (20 ng/mL), heparin (2 μg/mL), 0.5% penicillin/streptomycin, amphotericin B (125 ng/mL), Plasmocin (2.5 μg/mL), and StemPro Neural Supplement (Thermo Fisher A1050801). All GSC lines and human neural stem cells were maintained as neurospheres using ultra low-adherence culture dishes and dissociated 1–2 times per week with Accutase (StemCell Technologies 07922). Primary human astrocytes were cultured adherently on plates coated with Geltrex (Life Technologies A1413202). Sex and source of each line is stated above and listed as unknown if unreported in the original publication describing its derivation.
All human GSC lines were cultured in the presence of 5% CO2 and ambient oxygen at 37°C. DF-AA27 mouse GSCs were cultured in the presence of 5% CO2, 5% O2 at 37°C. All cell lines were routinely evaluated for mycoplasma contamination and tested negative throughout the study. Cell line authentication was not performed because reference short term tandem repeat profiles have not been established for these cell lines. Only low passage GSC lines were used and these cells were discarded after 3 months in culture to prevent genetic and/or phenotypic drift.
Animals
All care and treatment of experimental animals were carried out in strict accordance with Good Animal Practice as defined by the US Office of Laboratory Animal Welfare and approved by the Dana-Farber Cancer Institute (protocol 04–019) or the UT Southwestern Medical Center (protocol 2019–102795) Institutional Animal Care and Use Committee. Animal welfare assessments were carried out daily during treatment periods. Female mice were housed together (2–5 mice per cage) and provided free access to standard diet and water. Mice were randomized to experimental arms prior to cell implantation and/or treatment. For orthotopic glioma cell implantations and intracranial AAV injections, mice were anesthetized via intraperitoneal injection of ketamine (140 mg/kg) and xylazine (12 mg/kg) and immobilized using a stereotactic frame. An incision was made to expose the skull surface, and a hole was drilled into the skull. AAV (1 μL) or cells suspended in 1–3 μL of 2% FBS in PBS (NHA and HOG cells) or cell culture medium (GSC lines) were injected into the brain through the hole using a 5 μL syringe (Hamilton). The skin was closed with surgical clips, and buprenorphine was given for analgesia. Tumor size and survival analyses were performed by researchers who were not blinded to the treatment arms or genotypes of the mice. Mice were euthanized when they either displayed neurological symptoms or became moribund.
Orthotopic xenograft and allograft models
NHA orthotopic xenografts were created by intracranial injection of 3 × 3 105 cells expressing either IDH1-R132H, PIK3R1-D560_S565del, both, or neither or H-Ras-V12 into female NCr nude mice (Taconic). Cells were implanted 1 mm anterior and 2 mm lateral to the lambda, 2.5 mm below the surface of the brain. All injected cells expressed a firefly luciferase-IRES-GFP bicistronic expression cassette (LeGO-iG2-FLuc vector) as previously described (McBrayer et al., 2018) to allow for non-invasive bioluminescence imaging of tumor growth. Mice were imaged once per week following cell injection. In vivo imaging was performed after intraperitoneal injection of luciferin (50 mg/kg). Mice were anesthetized with isoflurane and imaged using an IVIS camera (PerkinElmer). Imaging data were analyzed using Living Image software (PerkinElmer). Tumor growth rates were determined by subtracting the initial signal from the signal at the imaging time point immediately preceding euthanasia or at 16 weeks after cell implantation (for mice that did not require euthanasia during study period) and dividing by the number of intervening weeks.
MGG152 and TS516 orthotopic xenografts were created by intracranial injection of 1 × 105 cells into female ICR SCID mice (Taconic) or Fox Chase SCID mice (Charles River). MGG152 cells were implanted 3 mm anterior and 2 mm lateral to the lambda, 2.5 mm below the surface of the brain. TS516 cells were implanted 1.75 mm posterior and 2 mm lateral to the bregma, 2 mm below the surface of the brain. For survival studies, MGG152 and TS516 tumor-bearing mice were randomized to treatment arms 12 and 9 days after tumor cell implantation, respectively. Immediately following randomization, BAY 2402234 (4 mg/kg PO QD) or vehicle treatments were dosed continuously until mice were euthanized. Radiation (9 Gy in 3 fractions QOD) or sham treatments started two days after randomization. Mice undergoing radiation or sham treatments were anesthetized via intraperitoneal injection of ketamine/xylazine and placed in a lead shield covering their bodies but not their heads. Radiation was delivered using a Gammacell-40 irradiator (Nordion). For MALDI-MSI studies, brains were harvested from MGG152 tumor-bearing mice 33 days after tumor cell implantation.
HOG orthotopic xenografts were created by intracranial injection of 3 × 105 cells into female NCr nude mice (Taconic). HOG-R132H cells expressing LeGO-iG2-FLuc vector and either EV or DHODH A58T were implanted 1 mm anterior and 2 mm lateral to the lambda, 3 mm below the surface of the brain. EV and DHODH A58T tumor-bearing mice were bioluminescently imaged once per week following tumor cell implantation using the IVIS Spectrum In vivo Imaging System (PerkinElmer). Briefly, mice were injected subcutaneously with 75 mg/kg D-luciferin potassium salt (PromegaE1605) in sterile PBS and anesthetized with 2% isoflurane in medical air. Serial bioluminescence images were acquired using the automated exposure set-up. The peak bioluminescence signal intensity within selected regions of interest (ROI) was quantified using the Living Image software (PerkinElmer). To minimize variance in tumor size at randomization, mice with tumor signals ranging from 6 × 105 to 1.5 × 107 photons/sec (unadjusted for background luminescence) were randomized to treatment arms 12 (EV) and 13 (DHODH A58T) days after cell implantation. Immediately following randomization, BAY 2402234 (4 mg/kg PO QD) or vehicle treatments were dosed continuously until mice were euthanized.
DF-AA27 orthotopic allografts were created by intracranial injection of 1 × 105 cells into female ICR SCID mice (Taconic). DF-AA27 cells were implanted 3 mm anterior and 2 mm lateral to the lambda, 2.5 mm below the surface of the brain. For tumor volume measurement studies, DF-AA27 tumor-bearing mice were randomized to BAY 2402234 (4 mg/kg PO QD) or vehicle treatment arms upon tumor detection by MRI. Treatment commenced immediately following randomization and MRIs were acquired again 3 weeks later.
BAY 2402234 in vivo toxicity experiments
Female ICR SCID mice (Taconic) in Figure 2D were treated with BAY 2402234 (4 mg/kg QD PO) or vehicle for 14 days.
Genetically engineered mouse models
Intracranial injection of AAV was performed using 1.5–6-month-old male and female transgenic mice (genotypes indicated in Figure 5A). Strains used were: H11LSL-Cas9 (B6; 129-Igs2tm1(CAG-cas9*)Mmw/J, Jackson stock number 026816) (Chiou et al., 2015), R26-Pik3caH1047R (FVB.129S6-Gt(ROSA)26Sortm1(Pik3ca*H1047R)Egan/J, Jackson stock number 016977) (Adams et al., 2011), Idh1tm1Mak (backcrossed to C57BL/6, provided by T. Mak at University of Toronto) (Sasaki et al., 2012a, 2012b). To generate the mouse strains used for the injection scheme outlined in Figure 5A, we first bred transgenic H11LSL-Cas9+/+ mice with Idh1tm1Mak/+ mice to produce H11LSL-Cas9+/−;Idh1tm1Mak/WT progeny, which were subsequently interbred to produce H11LSL-Cas9+/+;Idh1tm1Mak/WT mice. H11LSL-Cas9+/+;Idh1tm1Mak/WT mice were crossed with R26-Pik3caH1047R+/+ to produce PIC (H11LSL-Cas9+/−;Idh1tm1Mak/WT;R26-Pik3caH1047R+/−) and PC (H11LSL-Cas9+/−;Idh1WT/WT;R26-Pik3caH1047R+/−) mice. Separately, H11LSL-Cas9+/+;Idh1tm1Mak/WT mice were crossed with wild-type FVB mice to produce IC (H11LSL-Cas9+/−;Idh1tm1Mak/WT) and C (H11LSL-Cas9+/−;Idh1WT/WT) mice. Genotyping was performed by Transnetyx. For intracranial AAV injections, mice were first anesthetized via intraperitoneal injection of ketamine (140 mg/kg) and xylazine (12 mg/kg). Mice were immobilized using a stereotactic frame. Following skin incision to expose the skull, a hole was drilled 1 mm posterior and 1 mm lateral to the bregma. 1 μL of virus at 1 × 1013 to 1 × 1014 virions/mL, (1 × 1010 to 1 × 1011 total AAV particles) was injected 2.1 mm below the surface of the brain over 5 minutes. Mice were monitored with serial monthly MRI scans, and glioma incidence was determined by histopathologic evaluation of brain tissues after euthanasia and/or intracranial tumor detection by MRI.
Human subjects
The study was conducted according to the principles of the Declaration of Helsinki. Patient tissue and blood were collected following ethical and technical guidelines on the use of human samples for biomedical research at UT Southwestern Medical Center after informed patient consent under a protocol approved by UT Southwestern Medical Center’s Institutional Review Board. Use of human brain tissue was organized by the Department of Clinical Pathology at UT Southwestern Medical Center. All patient samples were de-identified before processing. All patient samples and organoids were diagnosed and graded according to the 2021 WHO Classification of Tumours of the Central Nervous System (CNS), 5th edition (Louis et al., 2021). Organoid models were created from patients with the following ages and sexes: UTSW6577 (32 male), UTSW1382 (27 male), UTSW2243 (53 female), UTSW9647 (33 male), UTSW2846 (53 male), UTSW2294 (59 male), UTSW9698 (66 female), UTSW9501 (76 male).
Organoid creation from primary glioma tissue was conducted as described previously (Abdullah et al., 2022). Briefly, tumor tissue was collected from the operating room and suspended in ice cold Hibernate A (BrainBits HA). Tumor pieces were exposed to RBC lysis buffer (Thermo Fisher 00433357) and washed with Hibernate A containing Glutamax (final conc. = 2 mM, Thermo Fisher 35050061), penicillin/streptomycin (final conc. = 100 U/mL and 100 μg/mL, respectively, Thermo Fisher 15140122), and Amphotericin B (final conc. = 0.25 μg/mL, Gemini Bio-Products 400104). Tissues were cut using dissection scissors into 1–2 mm3 pieces and suspended in 1 mL Short-Term Glioma Organoid Medium (formulation provided below). One organoid in 1 mL Short-Term Glioma Organoid Medium was plated per well of a 24-well ultra-low adherence plate. Plates were rotated at 120 rpm in a CO2-resistant shaker (Fisher Scientific 88–881-103) in a humidified incubator at 37°C, 5% CO2, and 5% O2. Short-Term Glioma Organoid Medium was refreshed in organoid cultures every 48 hours. All organoids were cultured at least four weeks before treatment. Organoids were randomized to 30 nM BAY 2402234 or vehicle treatments and treated for 14 days before being fixed for IHC analysis. Formulation of Short-Term Glioma Organoid Medium is as follows: 48 mL Long-Term Glioma Organoid Medium, 48 μl 2-mercaptoethanol (BME) (final conc. = 55 μM, Thermo Fisher BP176–100), and 12 μl human insulin (final conc. = 2.375–2.875 μg/mL, Sigma Aldrich I9278). Formulation of Long-Term Glioma Organoid Medium is as follows: 250 mL DMEM:F12 medium (Thermo Fisher 1132033), 250 mL Neurobasal medium (Thermo Fisher 21103049), 5 mL 100X Glutamax (final conc. = 2 mM), 5 mL 100X low-glutamate non-essential amino acids mixture (final conc.: Gly, L-Ala, L-Asn, L-Asp, L-Pro, L-Ser = 100 μM and L-Glu = 300 nM), 5 mL penicillin/streptomycin (final conc. = 100 U/mL and 100 μg/mL, respectively), 10 mL B-27 Supplement without Vitamin A (Thermo Fisher 12587010), 5 mL N-2 Supplement (Thermo Fisher 17502048). Long-Term Glioma Organoid Medium and Short-Term Glioma Organoid Medium stocks were used up to 2 months and 1 week after preparation, respectively.
METHOD DETAILS
Chemicals
TFMB ester of (R)-2-hydroxyglutarate was generated as previously described by R. Looper at University of Utah (Losman et al., 2013). BAY 2402234 was provided by Bayer Pharmaceuticals. Where indicated, cell culture media also contained the following additives: brequinar (Sigma-Aldrich), 6-azauridine (Sigma-Aldrich), pyrazofurin (Sigma-Aldrich), uridine (Sigma-Aldrich), lometrexol (Cayman), AGI-5198 (XcessBio), deoxythymidine (Sigma-Aldrich), deoxycytidine (Sigma-Aldrich), palbociclib (Cayman), tunicamycin (Sigma-Aldrich).
Vectors
Empty vector (plenti-Ubc-IRES-hygro) and IDH1-R132H (plenti-Ubc-IDH1-R132H-HA-IRES-hygro) lentiviral expression plasmids used to create stable HOG cell lines were previously described (McBrayer et al., 2018). Empty vector (pBABE-HA-hygro) and IDH1-R132H (pBABE-IDH1-R132H-HA-hygro) retroviral expression plasmids used to create stable NHA cell lines (model #1) were previously described (Koivunen et al., 2012). Empty vector (pLenti-EF1α-EV-IRES-hygro) and IDH1-R132H (pLenti-EF1α-IDH1-R132H-IRES-hygro) lentiviral expression plasmids used to create stable NHA cell lines (model #2) were previously described (McBrayer et al., 2018). PIK3R1 expression vectors (WT, D560_S565del, R574fs, T576del) used to generate NHA stable cell lines were from Addgene (pLenti-Flag-P85, Addgene 40219; pLenti4-Flag-P85-DKRMNS560del, Addgene 40225; pLenti-Flag-P85-R574fs, Addgene 40227; pLenti4-Flag-P85-T576del, Addgene 40228) (Quayle et al., 2012). H-Ras-V12 expression vector was from Addgene (pLXSN-H-Ras_V12, Addgene 39516). The lentiviral plasmid used to express GFP and firefly luciferase markers in NHA stable cells, LeGO-iG2-FLuc, was previously described (McBrayer et al., 2018). Retroviral vectors used to immortalize (pLBCX-Large-T-antigenK1 mutant) and genetically engineer (pMSCV-MerCreMer-hygro) Idh1LSL-R132H/+;LSL-Cas9+/− (IC) MEFs were previously generated in the Kaelin laboratory. To make pLBCX-Large-T-antigenK1 mutant, Large-T-antigenK1 mutant cDNA was PCR amplified using a 5’ primer that introduced a SalI site and a 3’ primer that introduced a ClaI site. The PCR product and pLBCX empty vector were digested with SalI and ClaI, gel-purified, and ligated. To make pMSCV-MerCreMer-hygro, MerCreMer cDNA was PCR amplified using a 5’ primer that introduced a NotI site and a 3’ primer that introduced a PacI site. The PCR product and pMSCV-hygro empty vector were digested with NotI and PacI, gel-purified, and ligated.
The pAAV2-sgTrp53-sgAtrx-EFS-Cre AAV vector was created using an approach similar to that described previously (Oser et al., 2019). Effective sgRNAs (sequences in Key resources table) targeting mouse Trp53 and Atrx genes were identified from a previous publication (Platt et al., 2014) and empirically, respectively. To generate the AAV vector depicted in Figure 5A, we first generated a destination vector (pAAVGao-DEST-EFS-Cre-spA). This was accomplished by inserting a multiple cloning site sequence between XbaI and NotI sites in pX551 (a gift from F. Zhang at MIT, Addgene 60957). We then inserted the universal gateway cassette between PacI and NheI using restriction enzymes to generate an intermediate vector named pAAVGao-DEST. Next, we performed overlapping PCR to assemble the EFS promoter, Cre, and short poly(A) signal that contained a 5ʼ PacI site and 3ʼ XbaI site. The PCR product was digested with PacI and XbaI and ligated into pAAVGao-DEST cut with these two enzymes. Next, we performed Gibson assembly to generate an entry vector (pENTR223-sgTrp53-sgAtrx) containing sgRNAs targeting Trp53 and Atrx. pENTR223-sgTrp53-sgAtrx was then mixed with pAAVGao-DEST-EFS-Cre-spA to make the final pAAV2-sgTrp53-sgAtrx-EFS-Cre by homologous recombination reaction using LR Clonase II (Life Technologies 11791100) at 25°C for 1 h per the manufacturer’s instructions. The reaction mixtures were then transformed at a ratio of 1:10 (volume recombination reaction:volume competent cells) into HB101 cells and ampicillin-resistant colonies were screened by restriction digestion of miniprep DNA and subsequently validated by whole plasmid DNA sequencing at the MGH CCIB DNA Core. AAV packaging and titering using pAAV2-sgTrp53-sgAtrx-EFS-Cre was performed by Vigene Biosciences.
WT and DHODH A58T mutant human cDNAs were provided by J.A.L. and L.E. pLX304-Ubc-DEST-IRES-GFP lentiviral Gateway destination vector was provided as a gift from V. Koduri (Brigham and Women’s Hospital). A non-coding, control sequence (for the empty vector), DHODH WT cDNA, and the DHODH A58T cDNA were separately PCR amplified using primers designed to append 5’ attB1 and 3’ attB2 sites. PCR products were gel-purified and cloned into the Gateway vector pDONR223 via BP reactions. Resulting pDONR223-EV (empty vector), pDONR223-DHODH-WT, and pDONR-DHODH-A58T plasmids were separately mixed with the pLX304-Ubc-DEST-IRES-GFP lentiviral Gateway destination vector and LR reactions were performed. Resulting pLX304-Ubc-EV-IRES-GFP, pLX304-Ubc-DHODH-WT-IRES-GFP, and pLX304-Ubc-DHODH-A58T-IRES-GFP lentiviral vectors for mammalian cell expression were confirmed by restriction digest analysis and inserts were validated by DNA sequencing.
pLX304-CMV-BFP and pLX304-CMV-GFP constructs used for the sgDHODH competition assay were a gift from H. Nicholson (Tango Therapeutics). Lenti-CRISPR_v2 (Addgene 52961) was digested with Esp3I, and an sgRNA targeting DHODH (CATCTTATAAAGTCCGTCCA) or a non-targeting control sgRNA (GGAGGCTAAGCGTCGCAA) was ligated into this backbone to generate a construct expressing Cas9 and the desired sgRNA. Reaction mixtures were transformed into XL10 gold ultracompetent cells, screened by restriction digestion of miniprep DNA and subsequently validated by Sanger sequencing and whole plasmid sequencing (Plasmidsaurus).
Transient transfection and in vitro viral transduction
Lentiviral and retroviral particles were made by Lipofectamine 2000-based cotransfection of HEK293T cells with expression vectors and packaging plasmids psPAX2 (Addgene 12260) and pMD2.G (Addgene 12259) or gag/pol (Addgene 14887) and VSV.G (Addgene 14888), respectively, in a ratio of 4:3:1. Virus-containing media were collected 48 and 72 hours after transfection, passed through a 0.45 μm filter, divided into 1 ml aliquots, and frozen at −80°C until use.
NHA cells or MEFs were plated at a density of 0.3 × 106 cells per well in a 6-well plate. The next day, 2.4 μL polybrene (10 mg/mL) in 0.5 mL media were added to each well in addition to 0.5 mL viral supernatant. Plates were centrifuged at 4,000 × g for 30 minutes at room temperature and incubated overnight. The following day, cells were expanded and replated. NHA cells in Figures 4C and 4D were cultured under low (0.5%) fetal bovine serum conditions prior to lysate preparation. Stable cell lines were selected in 500 μg/mL hygromycin, 2 μg/mL puromycin, 1 mg/mL G418, or 10 μg/mL blasticidin based on the drug selection cassette present in each vector of interest.
Creating genetically engineered MEFs
A dam (LSL-Cas9+/+), impregnated by a male (Idh1LSL-R132H/+) during a timed mating, was euthanized for embryo harvest at day E13.5. Fibroblasts were isolated from an embryo later determined to be heterozygous for both Cas9 and mutant Idh1 alleles (IC genotype). MEFs were immortalized by transduction with retroviral particles produced from the pLBCX-Large-T-antigenK1 mutant vector. Next, immortalized MEFs were transduced with retroviral particles produced from the pMSCV-MerCreMer-hygro vector to constitutively express tamoxifen-inducible Cre recombinase (Sohal et al., 2001) or were mock infected. After selection, MerCreMer-expressing MEFs were treated with 1 μM 4-hydroxytamoxifen for 6 days to activate Cre.
DF-AA27 GSC line creation
After observing brain tumor formation in an AAV-injected PIC mouse by MRI, the mouse was euthanized and brain tissue was harvested. A portion of the right hemisphere of the brain containing tumor tissue was isolated and dissociated to a single cell suspension using the Neural Tissue Dissociation Kit (Miltenyi) and a gentleMACS Dissociator instrument (Miltenyi). Cells were cultured in 5% CO2 and 5% O2 on ultra-low adherence plates to select for neurosphere-forming cells. The resulting murine GSC line was named DF-AA27. Notably, prior attempts to generate murine GSC lines from this GEM model under ambient oxygen conditions were not successful.
Drug screen
The MAPS platform (Harris et al., 2019) was used to test both a commercial anticancer drug library and a custom-curated metabolic inhibitor drug library. Screen was performed at the ICCB-Longwood Screening Facility (https://iccb.med.harvard.edu/small-molecule-screening). HOG-EV or HOG-R132H cells were seeded at a density of 500 cells per well in a final volume of 30 μL per well of 384-well plates. After 24 h, a Seiko Compound Transfer Robot pin transferred 100 nL of each drug library into wells with plated cells. Following pin-transfer, 20 μL of cell culture medium was added to all wells, resulting in each drug being applied at a final 10-point concentration series ranging from 20 μM to 1 nM. After 72 h of drug treatment, the cells were washed with PBS, fixed with 4% formaldehyde, and stained with 5 mg/mL bisbenzimide. An Acumen Cellista plate cytometer was used to image plates and determine the cell numbers in individual wells. XY plots were generated comparing relative numbers of surviving HOG-EV and HOG-R132H cells with concentrations of each drug tested. Area under the curve (AUC) values were calculated for each plot and drugs were ranked based on the difference between the AUCs for HOG-EV and HOG-R132H cells. Hits were defined as drugs that displayed >10 %AUCDiff values, where %AUCDiff = ((AUCEV - AUCR132H)/AUCEV)*100, with positive values indicating preferential activity against HOG-R132H cells. Using these criteria, 56 hits were identified, including the 36 highest ranking hits displayed in Figure 1B.
Cell death quantification
Cells were stained with AnnexinV-FITC (BD Biosciences 556547) for all cell death assays (except for DHODH-A58T drug rescue experiments) according to manufacturer’s instructions and DAPI (100 ng/mL final concentration) to identify early and late apoptotic cells, respectively. For DHODH-A58T drug rescue experiments involving HOG cells, cells were stained with AnnexinV-PE (BD Biosciences 559763) according to manufacturer’s instructions and DAPI as above. For DHODH-A58T drug rescue experiments involving MGG152 cells, were stained with AnnexinV-APC (BD Biosciences 550475) per manufacturer’s instructions and DAPI as above. Cells were analyzed using either an LSR II (BD Biosciences) or LSR Fortessa (BD Biosciences) flow cytometer and data were processed using FCS Express software (De Novo). Dead cells included those that were AnnexinV+/DAPI−, AnnexinV −/DAPI+, or AnnexinV+/DAPI+. In Figures 1K, 2G, and 6A, data are normalized to set cell death = 0% in DMSO-treated cells.
Doubling time calculation
Doubling times of human GSC lines were calculated by plating 2 × 105 cells per well in a 6-well plate. 5 days later, cells were counted using a Vi-Cell XR (Beckman Coulter) cell viability analyzer. Doubling time (Td) was calculated using the following formula:
In vitro pyrimidine synthesis inhibition
Pyrimidine synthesis inhibitor treatments of HOG cells were performed by plating HOG cells at a density of 5,000–10,000 cells per cm2 with or without 100 μM uridine supplementation. 24 h later, the drug being tested was added at the indicated concentrations, and cell death was analyzed at the indicated time points. Cell death was analyzed 48 h (Figures 1E–1J), 72 h (Figures 2E and 2F), 4 days (Figures S2A–S2B and S2E), or 6 days (Figure S2C–S2D and S2F) after drug dosing for adherent cell lines. Uridine rescues were performed by pretreating with either 100 μM uridine or DMSO to cells 24 h prior to drug dosing. For GSC lines, cells were plated in 24-well ultra-low-attachment plates at a density of 50,000 cells per well with 0.25 mL media per well. An additional 0.25 mL medium containing 2× the desired drug concentrations was added at the time of plating to achieve the final concentrations of drug in 0.5 mL media per well. Fresh media (0.5 mL) with drug or vehicle was added every 4 days. In Figures 1 and 2, human GSCs were harvested for cell death quantification after two population doublings (5 days for TS516 and HK308; 6 days for BT260; 7 days for MGG152, BT054 and HK157; 8 days for TS603; 9 days for HK213; 11 days for HK211 and HK252). In Figure 6A, DF-AA27 GSCs were treated with or without 100 μM uridine supplementation, followed by addition of drug 24 h later at the indicated concentrations. Cell death was quantified 4 days after drug dosing.
Metabolite quantification by GC-MS
Quantification and analysis of all metabolite levels by GC-MS (steady state levels from HOG cells, GSCs, and tissues) was performed as previously described (McBrayer et al., 2018). Briefly, to quantify metabolites in HOG cells, cells were plated in 6-well plates (0.5 × 106 cells per well), washed with ice-cold saline, and snap frozen in liquid nitrogen. To extract metabolites, 350 μL 70% methanol at −20°C was added to each well, and adherent HOG cells were scraped from each well into the methanol suspension and transferred to Eppendorf tubes. Chloroform (−20°C, 150 μL) was added, and each sample was vortexed for 20 min at 4°C and centrifuged (17,000 × g for 10 min at 4°C). The upper phase containing polar metabolites in methanol was dried using a vacuum concentrator (CentriVap, Labconco) overnight at 4°C, and dried samples were stored at −80°C if not immediately used for GC-MS analysis.
To quantify metabolites from GSC lines, neurospheres were harvested from 24-well plates, followed by the addition of 4°C saline to quench metabolic activity. Samples were transferred to Eppendorf tubes, centrifuged in an Eppendorf microcentrifuge for 1 min at 4°C, and the resulting supernatant was aspirated, with remaining cell pellets snap frozen in liquid nitrogen and stored at −80°C. Subsequent polar extraction of metabolites from cell pellets was performed as described above for HOG cells.
To quantify metabolites from tumor tissue samples, samples were homogenized using a TissueLyser II (Qiagen) with 70% methanol at −20°C (volume dependent on tissue mass). Chloroform (−20°C, volume dependent on tissue mass) was added to each sample, and subsequent centrifugation, drying, and storage was performed as described above for HOG cells.
GC-MS analysis was performed by derivatizing dried samples and analyzing using an Agilent 7890B GC/5977A MSD system. Peak integration was conducted using the Metran software tool (Yoo et al., 2008). For relative metabolite quantification in tissue culture samples, ion counts were normalized to the total ion counts detected in the sample.
Metabolite quantification by LC-MS/MS or LC-MS
For LC-MS and LC-MS/MS analyses of tissue samples, samples were homogenized using a TissueLyser II (Qiagen) with 80% methanol at −20°C (volume dependent on tissue mass), vortexed for 20 min at 4°C, and centrifuged (21,100 × g at for 10 min at 4°C). Metabolite samples were dried using a CentriVap (Labconco) or SpeedVac (Thermo Fisher) concentrator. In Figures 2A–2C, samples were harvested from the heart and liver of ICR SCID mice treated with BAY 2402234 (4 mg/kg PO QD) or vehicle for 3 days. In Figures 3G and 3H and 6B, mice were treated with BAY 2402234 (4 mg/kg PO QD) or vehicle for 3 days. Tissues were harvested for metabolomics analysis 4 h after the final dose.
For LC-MS and LC-MS/MS analyses of cultured cells, cells were harvested as described above for GC-MS analysis. Metabolites were extracted in 80% methanol, vortexed for 20 min at 4°C, and centrifuged (21,100 × g for 10 min at 4°C). Metabolite samples were dried using a CentriVap (Labconco) or SpeedVac (Thermo Fisher) concentrator. In Figures 2J, S2H, and S2O, cells were harvested 24 h after 50 nM BAY 2402234 or DMSO treatment. In Figure 7A, BT054 cells were grown in HPLM and treated with 10 nM BAY 2402234, 5 μM lometrexol, or DMSO for 24 h. In Figures 7E–7G, HOG-EV or HOG-R132H cells were treated for 24 h with 10 nM BAY 2402234 or DMSO. Note that our LC-MS method cannot distinguish dGTP from ATP; therefore, we report data for dATP only.
For LC-MS/MS analyses, dried metabolites were resuspended in 20 μL HPLC-grade water. 10 μL was injected and analyzed using a 5500 QTRAP hybrid triple quadrupole mass spectrometer (AB/SCIEX) coupled to a Prominence UFLC HPLC system (Shimadzu) with selected reaction monitoring (SRM) with positive/negative polarity switching to detect a total of 263 water-soluble metabolites (Yuan et al., 2012). Amide HILIC chromatography (Waters) at pH 9.0 was used for metabolite separation over a 15-min gradient. Peak areas were integrated using Multi-Quant 2.1 software and metabolite quantification was performed using MATLAB.
For LC-MS analyses, dried metabolites were resuspended in 40–50 μL 80% acetonitrile, vortexed for 20 min at 4°C, and centrifuged (21,100 × g for 10 min at 4°C). 10–20 μL was injected and analyzed with a Q-Exactive HF-X or Orbitrap Exploris hybrid quadrupole-orbitrap mass spectrometer (Thermo Fisher) coupled to a Vanquish Flex UHPLC system (Thermo Fisher). Chromatographic resolution of metabolites was achieved using a Millipore ZIC-pHILIC column using a linear gradient of 10 mM ammonium formate pH 9.8 and acetonitrile. Spectra were acquired with a resolving power of either 120,000 or 240,000 full width at half maximum (FWHM), a scan range set to 80–1,200 m/z, and polarity switching. Data-dependent MS/MS data was acquired on unlabeled pooled samples to confirm metabolite IDs when necessary. Peaks were integrated using El-Maven 0.12.0 software (Elucidata) or TraceFinder 5.1 SP2 software (Thermo Fisher). Total ion counts were quantified using Freestyle 1.7 SP1 software (Thermo Fisher). Peaks were normalized to total ion counts using the R statistical programming language. For stable isotope tracing studies, correction for natural abundance of metabolite labeling was performed using the AccuCor package (version 0.2.3) in the R statistical programming language (Su et al., 2017).
Absolute quantification of 2HG
Absolute 2HG quantification was performed from cell culture and tumor models using GC-MS as previously described (McBrayer et al., 2018). At the time of cell harvest for GC-MS sample preparation, cell counts and average cellular diameter values were determined in parallel cell cultures for each cell line using a Vi-CELL XR cell viability counter (Beckman Coulter). Cell number and average cellular diameter values were then used to calculate total cellular volume using the following formula: , where d is average cellular diameter, n is cell number, and V is total cellular volume. For tissue samples, volumes were calculated using tissue weights and a previously published value of brain tissue density (Bothe et al., 1984). Cell and tissue samples were processed as described above in the GC-MS methods section, with 2HG concentrations calculated by dividing 2HG content by tumor sample volume. 2HG content was determined by comparison to standards containing known quantities of pure (R)-2HG (range: 20 ng–6 μg per vial).
DHODH A58T mutant
The drug resistant DHODH A58T mutant was provided as a gift from L.E. and J.A.L. This drug resistant mutant was identified through a saturating mutagenesis MITE-seq (mutagenesis by integrated tiles) screen of human DHODH (Melnikov et al., 2014). Methods and results of this screen are described in a manuscript under preparation.
BAY 2402234 pharmacokinetics and pharmacodynamics
Naive ICR SCID mice or mice bearing orthotopic xenografts or allografts were treated with BAY 2402234 (4 mg/kg PO QD) for 3 days. 4 h after the last dose, mice were euthanized. Tissues were collected and processed for LC-MS, LC-MS/MS, or MALDI-MSI analysis.
Cell competition assays
For DHODH-A58T competition assays, HOG-R132H stable cell lines expressing either DHODH-A58T-IRES-GFP or an empty vector (EV)-BFP construct were mixed at a ratio of 1:1 and plated at a density of 10,000 cells per cm2. The mixture of cells used for plating was analyzed via flow cytometry to determine baseline GFP:BFP ratio (day −1). 24 h later, cells were dosed with DMSO or the indicated concentrations of BAY 2402234. 3 days after dosing, cells were trypsinized, analyzed via flow cytometry to determine GFP:BFP ratios, and replated at a density of 8,000–10,000 cells per cm2 in drug-containing media. Trypsinization, flow cytometry analysis, and reseeding in drug-containing media were repeated every 3 days at the indicated time points. Flow cytometry was performed using an LSR Fortessa (BD Biosciences), and data were processed using FlowJo (BD Biosciences).
For sgDHODH competition assays, HOG stable lines expressing either EV and BFP or R132H and GFP were mixed 1:1 and plated at a density of 1.5 × 105 cells per well in a 6-well plate. The mixture of cells used for plating was analyzed via flow cytometry using an LSR Fortessa (BD Biosciences) flow cytometer and processed using FlowJo (BD Biosceinces) to determine BFP:GFP ratios (day −1). The next day, 2.4 μL polybrene (10 mg/mL) was added in addition to 1 mL total of media and viral supernatant (lenti-CRISPR-v2 expressing sgControl [sgCntl] or an sgRNA targeting DHODH [sgDHODH]). Plates were centrifuged at 4,000 × g for 30 min at room temperature and incubated overnight. The following day, each well was expanded and divided into a 6 cm plate with IMDM (10% FBS and 1% penicillin/streptomycin) with or without 100 μM uridine supplementation. The following day, 2 μg/mL puromycin was added for selection. Cells were maintained in culture with or without 100 μM uridine supplementation as indicated. Cells were trypsinized, analyzed via flow cytometry to determine BFP:GFP ratios, and reseeded at a density of 10,000 cells per cm2 every 4 days.
MALDI-MSI tissue preparation and microscopy
Intact MGG152 tumor-containing mouse brains were dissected, snap-frozen in liquid nitrogen, and stored at −80°C for MALDI-MSI analysis. The brains were cryo-sectioned in the coronal plane to 10 μm thickness and thaw-mounted onto indium tin oxide (ITO) slides. Serial sections were collected for H&E staining, and imaged using a 10× objective (Zeiss Observer Z.1, Oberkochen, Germany). A tissue drug mimetic was prepared with healthy mouse brain tissue homogenate spiked with BAY 2402234 concentrations ranging from 0.2–50 μM. The spiked homogenate was pipetted into the channels of a tissue microarray array (TMA) mold and frozen. This tissue drug mimetic was cryo-sectioned and thaw mounted adjacent to MGG152 mouse brain tissue sections. In Figures 3A–3C and 8K–8L, brains from MGG152 orthotopic glioma xenografts treated with BAY 2402234 (4 mg/kg PO QD) or vehicle for 3 days were harvested 4 h after the final drug dose.
MALDI matrix preparation
BAY 2402234 quantitation was performed using a 2,5-dihydroxybenzoic acid (160 mg/mL) matrix solution which was dissolved in 70:30 methanol: 0.1% TFA with 1% DMSO. The matrix was applied onto tissue using a TM sprayer (HTX Technologies, Chapel Hill, NC) with a two-pass cycle at a flow rate (0.18 mL/min), spray nozzle velocity (1200 mm/min), nitrogen gas pressure (10 psi), spray nozzle temperature (75°C), and track spacing (2 mm). Matrix was recrystallized by incubation at 85°C in presence of 5% acetic acid solution. Orotate, carbamoyl aspartate, thymidine triphosphate (dTTP) and deoxycytidine triphosphate (dCTP) were imaged using a 1,5-diaminonaphthalene hydrochloride (4.3 mg/mL) matrix solution prepared in 4.5/5/0.5 HPLC grade water/ethanol/1 M HCl (v/v/v). The matrix was applied using a four-pass cycle with 0.09 mL/min flow rate, spray nozzle velocity (1200 mm/min), spray nozzle temperature (75°C), nitrogen gas pressure (10 psi), and track spacing (2 mm).
BAY 2402234 MALDI MRM MSI
BAY 2402234 was quantitatively imaged using a timsTOF fleX mass spectrometer (Bruker Daltonics, Billerica, MA) operating in positive ion mode with a multiple reaction monitoring (MRM) method. The mass range was selected between m/z 100–650. The MRM settings were adjusted using a BAY 2402234-infused solution through the ESI source for the ion transfer funnels, quadrupole, collision cell, and focus pre-TOF parameters. The optimal collision energy for the BAY 2402234 precursor was 35 eV with a 3 m/z isolation width for the precursor to product ion transition 521.101→376.091 corresponding to [C21H18ClF5N4O4+H]+ and [C15H12F4N3O4+H]+ respectively. The optimized ESI method was transferred to a MALDI source method and calibrated using a tune mix solution (Agilent Technologies, Santa Clara, CA). MALDI-MSI conditions included a 10,000 Hz laser repetition rate and a 50 μm pixel size consisting of 1,000 laser shots. SCiLS Lab software (version 2021a premium, Bruker Daltonics, Billerica, MA) was used for data analysis without data normalization. The ion intensity was correlated with BAY 2402234 concentration using a linear regression between 0.2 and 1.0 μM from the tissue drug mimetic resulting in a correlation coefficient of 0.995. A limit of detection (LOD) of 0.10 μM (S/N ratio of >3) and limit of quantification (LOQ) of 0.33 μM (S/N ratio of >10) were calculated.
Metabolite MALDI MRM MSI
Orotate, carbamoyl aspartate, dTTP, and dCTP were imaged from serial tissue sections. The Q-TOF instrument was operated in negative ion mode in full scan mode for m/z 50–1000. Using standards, the orotate and carbamoyl aspartate [M-H]− ions fragmented in MS mode, thus the product ions were directly monitored in MS mode by setting the collision energy to 10 eV. The product ion monitored for orotate was m/z 111.019 and for carbamoyl aspartate m/z 132.029. An MRM approach was used to image dTTP (480.982→158.924) and dCTP (465.982→158.924). Further peak annotation was confirmed by performing on tissue MSMS compared to standards under the same analytical conditions.
DHODH activity assays
For activity assays in HOG cells, HOG stable cell lines expressing either EV or R132H were plated at a density of 0.5 × 106 cells on a 10 cm plate. Cells were allowed to adhere overnight and were dosed with either DMSO or 10 nM BAY 2402234. 24 h later, cells were harvested for mitochondria isolation. For MGG152 and TS516 in vitro activity assays, cells were dosed with 10 nM BAY 2402234 for 24 h before harvesting for mitochondria isolation. For tissues, MGG152 and TS516 orthotopic xenografts were generated as described in the above methods. At tumor formation, mice were dosed with either BAY 2402234 (4 mg/kg PO QD) or vehicle for 3 days. 4 h following the third dose, mice were euthanized, and tumors were harvested for mitochondria isolation.
Mitochondria were isolated from cells as previously described (Sun et al., 2021). Briefly, cells were washed with ice-cold SHE buffer (250 mM sucrose, 10 mM HEPES, 0.1 mM EGTA, and pH 7.2). Tissues were washed with ice-cold SHE buffer and ground with a plastic pestle. Protein concentrations of both cells and tissues were quantified and amount of homogenate processed for mitochondria isolation was normalized to equivalent protein amount. Cell or tissue pellets were resuspended in 300 μL complete SHE buffer (SHE buffer supplemented with 2% essentially fatty acid-free BSA [Sigma] and 1× EDTA-free protease inhibitor cocktail [Sigma]) and homogenized with a sonicator at 25% power for 30 s. Homogenates were centrifuged at 8,500 × g for 10 min at 4°C. The pellet was resuspended in 300 μL complete SHE buffer, transferred to a new tube, and centrifuged at 700 × g for 10 min at 4°C. The supernatant was transferred to a clean tube and centrifuged at 8,500 × g for 10 min. The mitochondrial pellet was washed twice with 400 μL complete SHE buffer, followed by centrifugation at 8,500 × g for 10 min at 4°C. Mitochondrial pellets were resuspended in 20 μL complete SHE buffer.
DHODH activity was determined as previously described (Painter et al., 2021). Briefly, DHODH activity was coupled to decylubiquinone and the redox reporter 2,6-dichloroindophenol. Reaction mixtures contained 1 mM L-dihydroorotate (Sigma), 100 μM decylubiquinone (Sigma), 60 μM 2,6-dichloroindophenol (Sigma), 50 mM sodium malonate (Sigma), 2 mM potassium cyanide (Sigma), 100 mM potassium chloride, 0.05% Triton X-100, 10% glycerol, and 0.1 M HEPES (pH 7.6). 200 μL of the reaction mixture was aliquoted into wells of a 96-well plate. 18 μL of resuspended mitochondrial pellet in complete SHE or complete SHE buffer with no mitochondrial protein (control) was added to each reaction mixture, and absorbance at 596 nm was measured every 5 min for 40 (HOG cell samples) or 60 (MGG152, TS516 cell and tumor samples) minutes total using a FLUOstar Omega (BMG Labtech) or SpectraMax iD3 microplate reader (Molecular Devices). For each set of experiments, change in absorbance per minute was calculated during the time frame in which control samples displayed the least change in absorbance in order to minimize background effects (10–40 min for HOG cell samples, 15–60 min for MGG152 and TS516 cell and tumor samples). Change in absorbance per minute in controls was subtracted from change in absorbance in samples and plotted.
15N tracing studies
For 15N stable isotope tracing studies in GSC lines, cells were cultured in human plasma-like medium (HPLM) (Cantor et al., 2017) without glutamate supplemented with 1× B27, 0.25× N2, 1% penicillin/streptomycin, amphotericin B (250 ng/mL), Plasmocin (2.5 μg/mL), EGF (20 ng/mL), bFGF (20 μg/mL), and heparin (2 μg/mL), and lacking glutamine (for amide-15N-glutamine tracing), uracil (for 15N2-uracil tracing), uridine (for 15N2-uridine tracing), or hypoxanthine (for 15N4-hypoxanthine tracing). Cells were acclimated to HPLM over 3 days prior to tracing (100% standard media Day 1, 50% HPLM in standard media Day 2, 100% HPLM Day 3). Tracers were added at their native HPLM concentrations: 550 mM amide-15N-glutamine (Cambridge Isotope Laboratories NLM-557), 3.7 μM 15N2-uracil (Cambridge Isotope Laboratories NLM-637), 3 μM 15N2-uridine (Cambridge Isotope Laboratories NLM-812), or 10 μM 15N4-hypoxanthine (Cambridge Isotope Laboratories NLM-8500). Tracing analysis was performed as previously described (DeVilbiss et al., 2021; Faubert et al., 2021; Tasdogan et al., 2020). In Figure 7C, labeling of representative pyrimidine [uridine monophosphate (UMP)] and purine [adenosine monophosphate (AMP)] nucleotides was measured 18 h after addition of the indicated 15N-labeled metabolites by LC-MS.
For amide-15N-glutamine tracing studies during hypoxanthine deprivation, cells were acclimated as described above. 24 h after completing acclimation, media were refreshed with either HPLM retaining or lacking hypoxanthine every 24 h for 5 days. At this time, media were refreshed with either HPLM containing amide-15N-glutamine and retaining or lacking hypoxanthine. 18 h after tracer addition, cells were harvested for evaluation of 15N labeling patterns by LC-MS.
Metabolomics analysis of GSCs in HPLM
BT054 GSCs were cultured in HPLM without glutamate supplemented with 1× B27, 0.25× N2, 1% penicillin/streptomycin, amphotericin B (250 ng/mL), Plasmocin (2.5 μg/mL), EGF (20 ng/mL), bFGF (20 ng/mL), and heparin (2 μg/mL). Cells were acclimated to HPLM over 3 days prior to treatment (100% standard media Day 1, 50% HPLM in standard media Day 2, 100% HPLM Day 3). For drug treatment experiments, 24 h after completing acclimation, media were refreshed and DMSO, 10 nM of BAY 2402234, or 5 μM lometrexol was added. Cells were harvested 24 h after drug treatment, and metabolites were quantified by LC-MS. For hypoxanthine deprivation experiments, 24 h after completing acclimation, media were refreshed with either HPLM retaining or lacking hypoxanthine every 24 h for 5 days 18 h later, cells were harvested and metabolites were quantified by LC-MS.
TCGA bioinformatics survival analysis
The TCGA Lower Grade Glioma (LGG) and Glioblastoma datasets were used to assess the genetic alterations that co-occur or are mutually exclusive with IDH1 mutations in human gliomas (https://www.cbioportal.org). The LGG dataset was also used to generate the survival curves in patients with IDH1 WT gliomas and patients with IDH1 mutant gliomas with or without PIK3R1/PIK3CA mutations.
Immunoblot analysis of protein expression
Cells were lysed in EBC lysis buffer containing a protease inhibitor cocktail (Sigma Aldrich 11836153001) and, for phosphoprotein immunoblots, a phosphatase inhibitor cocktail (Sigma Aldrich 04906837001). Lysates were resolved by SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad). Primary antibodies used included: anti-Akt (Cell Signaling 2920S, mouse monoclonal), anti-pAkt (Cell signaling 2965S, rabbit monoclonal), anti-GFAP (Abcam 7260, rabbit polyclonal), anti-Sox2 (Abcam 97,959, rabbit polyclonal), anti-Olig2 (DF308, a gift from J. Alberta at DFCI, rabbit polyclonal) (Ligon et al., 2004), anti-nestin (Abcam 6142, mouse monoclonal), anti-γH2A.X (Cell Signaling 9718S, rabbit monoclonal), anti-HA (Covance MMS-101P, mouse monoclonal), anti-FLAG (Sigma F1804, mouse monoclonal), anti-DHODH (Proteintech 14,877), and anti-vinculin (Sigma V9131, mouse monoclonal). Secondary antibodies used included: Goat anti-Mouse IgG (Thermo Fisher 31,430, goat polyclonal) and Goat anti-Rabbit IgG (Thermo Fisher 31,460, goat polyclonal).
Soft agar colony formation assays
Soft agar assays were performed by first plating a base layer (2.5 mL per well of a 6-well plate) of liquefied 1% agarose in DMEM medium with 10% FBS and 1% penicillin/streptomycin. Once solidified, a 1 mL mixture of 5,000 NHA cells and 0.4% agarose in DMEM medium with 10% FBS and 1% penicillin/streptomycin (warmed to 42°C) was added on top of the base layer. Once solidified, 2 mL of DMEM medium with 10% FBS and 1% penicillin/streptomycin was added to each well. Media were exchanged twice weekly. After 3 weeks, cells were stained with 200 μL of 0.1% iodonitrotetrazolium chloride (Sigma-Aldrich) and incubated overnight. Colonies were photographed and analyzed using ImageQuant TL software (GE Healthcare).
Magnetic resonance imaging
MRI was performed using a Bruker BioSpec 7T/30 cm USR horizontal bore Superconducting Magnet System (Bruker Corp.). This system provides a maximum gradient amplitude of 440 mT/m and slew rate of 3,440 T/m/s and uses a 23 mm ID birdcage volume radiofrequency (RF) coil for both RF excitation and receiving. Mice were anesthetized with 1.5% isoflurane mixed with 2 L/min air flow and positioned on the treatment table using the Bruker AutoPac with laser positioning. Body temperature of the mice was maintained at 37°C using a warm air fan while on the treatment table, and respiration and body temperature were monitored and regulated using the SAII (Sa Instruments) monitoring and gating system, model 1025T. T2 weighted images of the brain were obtained using a fast spin echo (RARE) sequence with fat suppression. The following parameters were used for image acquisition: repetition time (TR) = 6,000 ms, echo time (TE) = 36 ms, field of view (FOV) = 19.2 × 19.2 mm2, matrix size = 192 × 192, spatial resolution = 100 × 100 μm2, slice thickness = 0.5 mm, number of slices = 36, rare factor = 16, number of averages = 8, and total acquisition time 7:30 min. Bruker Paravision 6.0.1 software was used for MRI data acquisition, and tumor volume was determined from MRI images processed using a semiautomatic segmentation analysis software (ClinicalVolumes).
Histopathology and immunohistochemistry studies
Histopathological analysis of brain and tumor tissues was performed by harvesting tumor tissues and fixing immediately for 24 h in 10% formalin in PBS. Following fixation, tissues where washed and stored in 70% ethanol. Tissues were embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E). H&E sections of brain tumors were reviewed by a board-certified neuropathologist (K.L.L.) and the Dana-Farber/Harvard Cancer Center Rodent Histopathology Core.
For immunohistochemical (IHC) analyses of GFAP and Olig2, 4 μm-thick tissue sections were prepared from formalin-fixed paraffin-embedded tissue blocks and air dried overnight. Slides were baked in an Isotemp Oven (Fisher Scientific) for 30–60 min at 60 °C to melt excess paraffin and rehydrated. GFAP IHC was performed on the Bond III autostainer (Leica Biosystems) using the Bond Polymer Refine Detection Kit (Leica Biosystems DS9800), according to the manufacturer’s protocol. Antigens were retrieved with Bond Epitope Retrieval Solution 1 (citrate, pH 6.0) for 20 min. Bound antibody was visualized with a 3,3′-diaminobenzidine (DAB) stain, and counterstaining was performed with hematoxylin. Olig2 IHC was performed manually using a humidified chamber. Antigens were retrieved by heating slides with a pressure cooker to 125°C for 30 s and 90°C for 10 s in citrate buffer (pH 6; Life Technologies, 005,000). Slides were then incubated with peroxidase (Dako, S2003) and protein blocking reagents (Dako, X0909), respectively, for 5 min each. Sections were then incubated with primary antibody, washed, and incubated with Envision + System-HRP Labeled Polymer Anti-Rabbit (Dako, K4003) for 30 min. Bound antibody was visualized with a 3,3′-diaminobenzidine (DAB) stain and counterstaining was performed with hematoxylin.
Primary antibodies used were anti-GFAP (Abcam 7260, rabbit polyclonal, 1:3,000) and anti-Olig2 (DF308, a gift from J. Alberta at DFCI, rabbit polyclonal, 1:20,000). The specificity of the GFAP IHC assay was determined by comparing staining in sarcoma (negative control) and normal brain (positive control) mouse tissues. The specificity of the Olig2 IHC assay was determined by comparing staining in liver (negative control) and normal brain (positive control) mouse tissues.
IHC analyses of γH2A.X and cleaved caspase 3 were performed by Histowiz, Inc. (histowiz.com) using a Standard Operating Procedure and fully automated workflow. Samples were processed, embedded in paraffin, and sectioned at 4 μm. Immunohistochemistry was performed on a Bond Rx autostainer (Leica Biosystems) with Heat Induced Epitope Retrieval (HIER). Antibodies used were: anti-γH2A.X (Cell Signaling 9718, rabbit polyclonal, 1:800) and anti-cleaved caspase 3 (Cell Signaling 9661, rabbit monoclonal, 1:300). Chromagen development was done using the Bond Polymer Refine Detection Kit (Leica Biosystems) which was used according to the manufacturer’s protocol. After staining, sections were dehydrated and film coverslipped using a TissueTek-Prisma Coverslipper. Whole slide scanning (40×) was performed on an Aperio AT2 (Leica Biosystems). To quantify cleaved caspase 3 and γH2A.X staining, regions of interest (ROIs) were defined and total and chromogen-positive cells were counted using QuPath software (version 0.2.3). For CC3 quantification, 12–16 ROIs were analyzed from 2 sections per organoid. For γH2A.X quantification, 16 ROIs were analyzed from 2 vehicle-treated tumors and 86 ROIs were analyzed from 5 BAY 2402234-treated tumors. Spectral dissociation was used to separate signals from the chromogen (DAB) and the counterstain (hematoxylin).
Validation of genetic alterations in engineered astrocytomas
For validation of genetic alterations in our GEM model and in DF-AA27 GSCs, RNA and genomic DNA were harvested from astrocytoma tissue, DF-AA27 cells, normal mouse brain tissue, and genetically engineered MEFs. Genomic DNA was extracted using the DNeasy Blood and Tissue kit (Qiagen) per manufacturer’s instructions. To assess CRISPR-based editing of Atrx and Trp53, nested PCR was performed using genomic DNA templates to generate amplicons centered on the PAM sites targeted by Atrx and Trp53 sgRNAs. The KOD Xtreme polymerase kit (EMD Millipore 71975) was used for PCR. For Trp53, the following set of primers were used in sequential PCR reactions to generate amplicons for sequencing:
Outer forward Trp53: ATAGAGACGCTGAGTCCGGTTC
Outer reverse Trp53: CCTAAGCCCAAGAGGAAACAGA
Inner forward Trp53: TGCAGGTCACCTGTAGTGAGGTAGG
Inner reverse Trp53: GAAACAGGCAGAAGCTGGGGAAGAAAC
The inner PCR reaction was repeated using the above inner primers for the third and final PCR reaction for Trp53.
For Atrx, the following set of primers were used in sequential PCR reactions to generate amplicons for sequencing:
Outer forward ATRX: GCTATCTGAAACTCAATCCACG
Outer reverse ATRX: GACTTGGTTTCTCCTTTGCCATG
Inner forward ATRX: GCTTCCTGTAAGCTCATAAGTAC
Inner reverse ATRX: CTAATGCCATATGAGTGTAACTC
Second inner forward ATRX: CTCTTACATAATGGCCATTCTC
Second inner reverse ATRX: CTGTGAGTCATGATCATTCTTTGC
PCR products were purified using the NEB Monarch kit (T1030S) between each PCR reaction. Final PCR products were gel-purified using the Qiagen Gel Extraction Kit (Qiagen 28706) and submitted for next-generation sequencing at the MGH CCIB DNA Core.
Presence of the Idh1-R132H mutation was assessed using a PCR assay specific for the recombined lox-STOP-lox (LSL) element as previously described (Sasaki et al., 2012b). Nested PCR amplification was performed using genomic DNA templates with primers for either the recombined Idh1-R132H allele or Actb (encoding beta actin):
Forward primer recombined Idh1-R132H: GATTGATTCTGCCGCCATGATCCTAGT
Reverse primer recombined Idh1-R132H: CCTGGTCATTGGTGGCATCACGATTCTC
Forward primer Actb: TGACCCAGGTCAGTATCCCGGGT
Reverse primer Actb: GAACACAGCTAAGTTCAGTGTGCTGGGA
Expression of the Pik3ca-H1047R allele was assessed by harvesting RNA and performing RT-PCR as previously described (Adams et al., 2011). RNA was isolated using the RNeasy Mini Kit (Qiagen 74106) prior to cDNA synthesis using the AffinityScript qPCR cDNA Synthesis Kit (Agilent 600559). PCR was performed, and the following primers were used to detect the expression of the Pik3ca-H1047R transgene or Actb:
Forward primer Pik3ca-H1047R: CTAGGTAGGGGATCGGGACTCT
Reverse primer Pik3ca-H1047R: AATTTCTCGATTGAGGATCTTTTCT
Forward primer Actb: TAGGCACCAGGGTGTGATG
Reverse primer Actb: CATGGCTGGGGTGTTGAAGG
γH2A.X immunoblot analysis
HOG-EV or HOG-R132H cells (Figure 8A) were plated and treated with 1 nM, 3 nM, or 10 nM BAY 2402234 or DMSO as described in above in vitro pyrimidine synthesis inhibition methods. Cells were harvested for immunoblot analysis after 48 hours of treatment. GSC lines (MGG152, BT054, BT260, and HK157) were treated with 3 nM or 10 nM BAY2402234 or DMSO. MGG152 cells were treated for 4 days. All other GSCs were treated for 5 days. In Figure 8C, HOG-EV or HOG-R132H cells were treated with 10 nM BAY 2402234, 5 μM lometrexol, or DMSO for 36 hours. In Figures 8I and 8J, mice bearing MGG152 orthotopic glioma xenografts were treated with BAY 2402234 (4 mg/kg PO QD) or vehicle for 3 days before harvesting tissue for γH2A.X analysis.
53BP1 foci formation assays
HOG-EV or HOG-R132H cells were plated at a density of 3 × 104 cells per well onto 18 mm glass coverslips in 12-well plates. The following day, cells were treated with either DMSO or 10 nM BAY 2402234. Cells were fixed at the indicated time points with 4% formaldehyde, permeabilized with 0.3% Triton X-100 in PBS, incubated with Triton Block (0.2 M glycine, 2.5% FBS, 0.1% Triton X-100 in PBS), and immunostained with an anti-53BP1 antibody (1:1,000, Novus Biologicals, NB100–304) diluted in Triton Block. Cells were washed, incubated with a donkey anti-rabbit Alexa Fluor 488 secondary antibody (Invitrogen, A21206), and mounted. Images were acquired using a DeltaVision Ultra (Cytiva) microscope equipped with a 60× objective with 9× 0.5 μm z sections. Images were deconvolved and maximum intensity projections were generated. 53BP1 foci were quantified in individual cells using ImageJ with a macros plugin (n = 325–505 cells per condition per time point).
FACE quantification of lipid-linked oligosaccharides
FACE assays were conducted as previously described (Gao et al., 2013). Briefly, HOG-EV or HOG-R132H cells were plated at a density of 2.5 × 106 cells in 15 cm dishes. 24 h later, 10 nM BAY 2402234, 1 μg/mL tunicamycin, or DMSO was added. 24 h later, plates were transferred to wet ice, medium was aspirated, and cells were rinsed twice with 4°C PBS. 12 mL methanol at room temperature was added to each plate and cells were lifted from plates using cell scrapers and transferred to 15 mL conical tubes. Lipid-linked oligosaccharides (LLOs) were recovered from methanolic cell suspensions and subjected to hydrolysis in weak acid. Oligosaccharides were labeled with the fluorophore 7-amino-1,3-naphthalenedisulfonic acid (ANDS) and resolved by electrophoresis. Fluorophore-labeled LLOs and oligosaccharide standards were detected with a fluorescence imager.
dC and dT supplementation experiments
HOG-EV or HOG-R132H cells were plated at a density of 1 × 105 cells per well in 6-well plates (for cell death analysis), 2.5 × 105 cells per well in 6-well plates (for metabolomics), or at 6 × 105 cells in 10 cm dishes (for γH2A.X immunoblots). 24 h later, BAY 2402234 was added with or without 15 μM dC, 15 μM dT, or both. Cells were harvested after 72 h (cell death analysis), 24 h (for metabolomics), or 48 h (for γH2A.X immunoblots).
Palbociclib rescue experiments
HOG-EV or HOG-R132H cells were pre-treated for 24 h with 1 μM Palbociclib or DMSO in 10 cm dishes. Next, cells were harvested, counted and plated at a density of 1 × 105 cells per well in 6-well plates (for cell death analysis) or at 6 × 105 cells in 10 cm dishes (for γH2A.X immunoblots) and 1 μM Palbociclib or DMSO was re-added. 24 h later, BAY 2402234 was added. Cells were harvested 72 h or 36 h later for cell death analysis or γH2A.X immunoblots, respectively.
Acute (R)-2HG manipulation experiments
Acute manipulation of intracellular (R)-2HG levels during DHODH inhibition involved pre-treatment/concurrent treatment with AGI-5198 or (R)-2HG-TFMB. DF-AA27 GSCs were treated with 3 μM AGI-5198 for 72 h prior to being harvested for 2HG quantification by GC-MS. HOG-R132H cells were pre-treated for 72 h with 3 μM AGI-5198. Then media were exchanged with media containing 3 μM AGI-5198 and 2 mL of IMDM with 500 nM or 1,000 nM brequinar. 48 h later, cells were washed with PBS, trypsinized, and harvested for cell death quantification. 50,000 MGG152 cells were plated per well in a 24-well low-adherent plate in 250 μL media with 110 nM AGI-5198. After 48 h of AGI-5198 pre-treatment, 250 μL media were added to each well with 2× the indicated concentration of brequinar and 110 nM AGI-5198. 96 h later, cells were washed with PBS, trypsinized, and harvested for cell death quantification. HOG-EV cells were plated at a density of 1.5 × 105 cells per well in 6-well plates with 2 mL of IMDM media per well. After 24 h, the media were exchanged with media containing stocks of DMSO or (R)-2HG-TFMB at the indicated concentrations. After 3 h, 5 nM BAY 2402234 was added to each well. After 24 h, media were replenished with (R)-2HG-TFMB and BAY 2402234 at the same doses. 24 h later, cells were washed with PBS, trypsinized, and harvested for cell death quantification.
TCGA differential expression analysis
Somatic mutations and RNA expression files of TCGA LGG and GBM cohorts were downloaded from FireBrowse data portal (http://firebrowse.org/). All LGG and GBM tumors were labeled either IDH1/2 mutant or IDH1/2 wild type based on protein-altering somatic mutations on either IDH1 or IDH2 coding regions from whole-exome sequencing data of TCGA LGG and GBM cohorts. RNA expression data of TCGA LGG and GBM cohorts was utilized to perform differential gene expression analysis between IDH1/2 mutant and IDH1/2 WT gliomas using the R package limma (Ritchie et al., 2015). Genes with FDR adjusted p < 0.05 and fold change >2 were considered statistically significant. Gene set enrichment analysis (GSEA) was performed on all differentially expressed genes. Genes were determined to be related to the DNA damage response if they were represented in the following gene ontology sets: cell cycle checkpoint signaling, cell cycle DNA replication, DNA damage response, signal transduction by p53 class mediator, DNA damage response, signal transduction by p53 class mediator resulting in transcription of p21 class mediator, DNA damage response, signal transduction resulting in transcription, DNA replication preinitiation complex, intrinsic apoptotic signaling pathway in response to DNA damage, intrinsic apoptotic signaling pathway in response to DNA damage by p53 class mediator, mitotic cell cycle checkpoint signaling, mitotic DNA replication, mitotic spindle assembly checkpoint signaling, mitotic spindle checkpoint signaling, negative regulation of DNA damage response, signal transduction by p53 class mediator, regulation of DNA damage response, signal transduction by p53 class mediator, signal transduction in response to DNA damage, spindle assembly checkpoint signaling, spindle checkpoint signaling.
QUANTIFICATION AND STATISTICAL ANALYSIS
Information related to data presentation and statistical analysis for individual experiments can be found in the corresponding figure legends. Statistical analyses were carried out using GraphPad Prism software or the web-based metabolomics data analysis tool MetaboAnalyst. Significance of all comparisons involving two groups was calculated by unpaired two-tailed t-test. For comparisons of two groups with significantly different variances, Welch’s t-test was used. For comparisons of two groups without significant differences in variances, Student’s t-test was used. Significance of all comparisons involving three or more groups was calculated by one-way ANOVA. Significance of survival data was determined by log rank test. Statistical evaluation of patterns of co-occurrence or mutual exclusivity were performed using one-sided Fisher’s exact tests. Significance of correlations between 2HG content and drug sensitivity was determined by simple linear regression analysis. For all tests, p values < 0.05 were considered statistically significant.
Supplementary Material
Highlights.
IDH-mutant gliomas are hyperdependent on de novo pyrimidine nucleotide synthesis
IDH mutations and 2HG levels predict sensitivity to the DHODH inhibitor BAY 2402234
Allografts from a GEM model of mutant IDH-driven astrocytoma respond to BAY 2402234
IDH mutations enhance DNA damage caused by nucleotide pool imbalance
ACKNOWLEDGMENTS
The authors thank C. Stiles (DFCI), T. Batchelor (BWH), and S. Morrison (UTSW) for insightful feedback; J. Alberta (DFCI) for sharing Olig2 antibody; J. Brugge (HMS) for supporting development of the MAPS platform; Kaelin, McBrayer, Abdullah, DeBerardinis, and Losman laboratory members for helpful discussions; I. Mellinghoff (MSKCC), S. Weiss (Univ. of Calgary), and R. Pieper (UCSF) for sharing cell lines; Bayer Pharmaceuticals for sharing BAY 2402234; M. Yuan (Harvard) for help with mass spectrometry; M. Phillips (UTSW) for guidance on measuring DHODH activity; and DFCI Animal Resources Facility and UTSW Animal Resource Center for help with mouse work. This study was supported by awards from the Broad-Bayer Alliance (Joint Project Proposal Grant 7000062-5500001463) and from NIH/NCI: K22CA237752 to S.K.M., R01CA258586 to S.K.M. and K.G.A., P50CA165962 to N.Y.R.A, D.P.C., K.L.L., and W.G.K. (and related Career Enhancement Project award to S.K.M.), U19CA264504 to N.Y.R.A, D.P.C., K.L.L., W.G.K., and S.K.M., P50CA211015 to H.I.K., 5R35CA210068-02 to W.G.K, F30CA271634 to M.R.S., U54CA210180 to N.Y.R.A, P41EB028741 to N.Y.R.A, R35CA22044901 to R.J.D., and T32EB025823 to S.A.S. N.Y.R.A. is supported by the Pediatric Low-Grade Astrocytoma Program at PBTF. Drug screening using MAPS by J.E.E. and I.S.H. was supported by the Ludwig Center at Harvard and ICCB-L. Organoid modeling work was partly supported by an award from Oligo Nation Foundation to S.K.M. and K.G.A. S.K.M. is supported by a Cancer Prevention and Research Institute of Texas (CPRIT) award (RR190034), a V Scholar Award from the V Foundation for Cancer Research (V2020-006), a Distinguished Scientist Award from the Sontag Foundation, a Moody Medical Research Institute award, and a gift from the Jonesville Foundation. D.D.S. was supported by the HHMI Medical Research Fellows Program, the Scholars in Medicine Program at Harvard Medical School, a Conquer Cancer Young Investigator Award, and the ASTRO Resident Research Biology Seed Grant. W.G.K. and R.J.D. are HHMI Investigators. K.G.A. is supported by the Eugene P. Frenkel, MD, Endowment. C.E.B. is supported by an award from the Burroughs Wellcome Trust.
R.J.D., W.G.K., and S.K.M. have served as paid advisors to Agios Pharmaceuticals. W.G.K. receives compensation for roles as an Eli Lilly and LifeMine Therapeutics Board Director, a founder of Tango Therapeutics and Cedilla Therapeutics, and a scientific advisor for Fibrogen, IconOVir Bio, Circle Pharma, Nextext Invest, and Casdin Capital. K.L.L. receives research support from Eli Lilly and Company via the DFCI. S.K.M. and W.G.K. received research funding from Bayer Pharmaceuticals. Bayer had no influence over the design, execution, or interpretation of studies. N.Y.R.A. is key opinion leader for Bruker Daltonics, scientific advisor to Invicro, and receives support from Thermo Finnegan and EMD Serono. S.G., A.S., M.S., A.J., and L.E. are employees at Bayer. S.G., A.S., and A.J. hold stock in Bayer. D.P.C. has consulted for Lilly, GlaxoSmithKline, and Boston Pharmaceuticals and serves on the advisory board of Pyramid Biosciences, which includes an equity interest.
Footnotes
DECLARATION OF INTERESTS
All other authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.ccell.2022.07.011.
REFERENCES
- Abdullah KG, Bird CE, Buehler JD, Gattie LC, Savani MR, Sternisha AC, Xiao Y, Levitt MM, Hicks WH, Li W, et al. (2022). Establishment of patient-derived organoid models of lower-grade glioma. Neuro Oncol. 24, 612–623. 10.1093/neuonc/noab273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JR, Xu K, Liu JC, Agamez NMR, Loch AJ, Wong RG, Wang W, Wright KL, Lane TF, Zacksenhaus E, and Egan SE (2011). Cooperation between Pik3ca and p53 mutations in mouse mammary tumor formation. Cancer Res. 71, 2706–2717. 10.1158/0008-5472.CAN-10-0738. [DOI] [PubMed] [Google Scholar]
- Aoki K, Nakamura H, Suzuki H, Matsuo K, Kataoka K, Shimamura T, Motomura K, Ohka F, Shiina S, Yamamoto T, et al. (2018). Prognostic relevance of genetic alterations in diffuse lower-grade gliomas. Neuro Oncol. 20, 66–77. 10.1093/neuonc/nox132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardella C, Al-Dalahmah O, Krell D, Brazauskas P, Al-Qahtani K, Tomkova M, Adam J, Serres S, Lockstone H, Freeman-Mills L, et al. (2016). Expression of Idh1R132H in the murine subventricular zone stem cell niche recapitulates features of early gliomagenesis. Cancer Cell 30, 578–594. 10.1016/j.ccell.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothe HW, Bodsch W, and Hossmann KA (1984). Relationship between specific gravity, water content, and serum protein extravasation in various types of vasogenic brain edema. Acta Neuropathol. 64, 37–42. [DOI] [PubMed] [Google Scholar]
- Cantor JR, Abu-Remaileh M, Kanarek N, Freinkman E, Gao X, Louissaint A, Lewis CA, and Sabatini DM (2017). Physiologic medium rewires cellular metabolism and reveals uric acid as an endogenous inhibitor of UMP synthase. Cell 169, 258–272.e17. 10.1016/j.cell.2017.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou SH, Winters IP, Wang J, Naranjo S, Dudgeon C, Tamburini FB, Brady JJ, Yang D, Grüner BM, Chuang CH, et al. (2015). Pancreatic cancer modeling using retrograde viral vector delivery and in vivo CRISPR/Cas9-mediated somatic genome editing. Genes Dev. 29, 1576–1585. 10.1101/gad.264861.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian S, Merz C, Evans L, Gradl S, Seidel H, Friberg A, Eheim A, Lejeune P, Brzezinka K, Zimmermann K, et al. (2019). The novel dihydroorotate dehydrogenase (DHODH) inhibitor BAY 2402234 triggers differentiation and is effective in the treatment of myeloid malignancies. Leukemia 33, 2403–2415. 10.1038/s41375-019-0461-5. [DOI] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. (2009). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744. 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deberardinis RJ, Sayed N, Ditsworth D, and Thompson CB (2008). Brick by brick: metabolism and tumor cell growth. Curr. Opin. Genet. Dev 18, 54–61. 10.1016/j.gde.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVilbiss AW, Zhao Z, Martin-Sandoval MS, Ubellacker JM, Tasdogan A, Agathocleous M, Mathews TP, and Morrison SJ (2021). Metabolomic profiling of rare cell populations isolated by flow cytometry from tissues. Elife 10, e61980. 10.7554/eLife.61980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Butel JS, and Bradley A.(1992). Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356, 215–221. 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Echizenya S, Ishii Y, Kitazawa S, Tanaka T, Matsuda S, Watanabe E, Umekawa M, Terasaka S, Houkin K, Hatta T, et al. (2020). Discovery of a new pyrimidine synthesis inhibitor eradicating glioblastoma-initiating cells. Neuro Oncol. 22, 229–239. 10.1093/neuonc/noz170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Tasdogan A, Morrison SJ, Mathews TP, and DeBerardinis RJ (2021). Stable isotope tracing to assess tumor metabolism in vivo. Nat. Protoc 16, 5123–5145. 10.1038/s41596-021-00605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favero F, Joshi T, Marquard AM, Birkbak NJ, Krzystanek M, Li Q, Szallasi Z, and Eklund AC (2015). Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann. Oncol 26, 64–70. 10.1093/annonc/mdu479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao N, Holmes J, and Lehrman MA (2013). Letter to the glycoforum: improved protocols for preparing lipid-linked and related saccharides for fluorophore-assisted carbohydrate electrophoresis (FACE). Glycobiology 23, 1111. 10.1093/glycob/cwt067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett M, Sperry J, Braas D, Yan W, Le TM, Mottahedeh J, Ludwig K, Eskin A, Qin Y, Levy R, et al. (2018). Metabolic characterization of isocitrate dehydrogenase (IDH) mutant and IDH wildtype gliomaspheres uncovers cell type-specific vulnerabilities. Cancer Metab. 6, 4. 10.1186/s40170-018-0177-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassian AR, Parker SJ, Davidson SM, Divakaruni AS, Green CR, Zhang X, Slocum KL, Pu M, Lin F, Vickers C, et al. (2014). IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 74, 3317–3331. 10.1158/0008-5472.CAN-14-0772-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris IS, Endress JE, Coloff JL, Selfors LM, McBrayer SK, Rosenbluth JM, Takahashi N, Dhakal S, Koduri V, Oser MG, et al. (2019). Deubiquitinases maintain protein homeostasis and survival of cancer cells upon glutathione depletion. Cell Metab. 29, 1166–1181.e6. 10.1016/j.cmet.2019.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue S, Li WY, Tseng A, Beerman I, Elia AJ, Bendall SC, Lemonnier F, Kron KJ, Cescon DW, Hao Z, et al. (2016). Mutant IDH1 downregulates ATM and alters DNA repair and sensitivity to DNA damage independent of TET2. Cancer Cell 30, 337–348. 10.1016/j.ccell.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen T-CA, Mukherjee J, Viswanath P, Ohba S, Ronen SM, Bjerkvig R, and Pieper RO (2016). Rapid conversion of mutant IDH1 from driver to passenger in a model of human gliomagenesis. Mol. Cancer Res 14, 976–983. 10.1158/1541-7786.MCR-16-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly JJP, Blough MD, Stechishin ODM, Chan JAW, Beauchamp D, Perizzolo M, Demetrick DJ, Steele L, Auer RN, Hader WJ, et al. (2010). Oligodendroglioma cell lines containing t(1;19)(q10;p10). Neuro Oncol. 12, 745–755. 10.1093/neuonc/noq031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Hu Z, Cai L, Li K, Choi E, Faubert B, Bezwada D, Rodriguez-Canales J, Villalobos P, Lin Y-F, et al. (2017). CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 546, 168–172. 10.1038/nature22359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, et al. (2012). Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 483, 484–488. 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafita-Navarro MC, Venkateswaran N, Kilgore JA, Kanji S, Han J, Barnes S, Williams NS, Buszczak M, Burma S, and Conacci-Sorrell M.(2020). Inhibition of the de novo pyrimidine biosynthesis pathway limits ribosomal RNA transcription causing nucleolar stress in glioblastoma cells. PLoS Genet. 16, e1009117. 10.1371/journal.pgen.1009117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laks DR, Crisman TJ, Shih MYS, Mottahedeh J, Gao F, Sperry J, Garrett MC, Yong WH, Cloughesy TF, Liau LM, et al. (2016). Largescale assessment of the gliomasphere model system. Neuro Oncol. 18, 1367–1378. 10.1093/neuonc/now045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Adler L, Karathia H, Carmel N, Rabinovich S, Auslander N, Keshet R, Stettner N, Silberman A, Agemy L, et al. (2018). Urea cycle dysregulation generates clinically relevant genomic and biochemical signatures. Cell 174, 1559–1570.e22. 10.1016/j.cell.2018.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon KL, Alberta JA, Kho AT, Weiss J, Kwaan MR, Nutt CL, Louis DN, Stiles CD, Rowitch DH, et al. (2004). The oligodendroglial lineage marker OLIG2 is universally expressed in diffuse gliomas. J. Neuropathol. Exp. Neurol 63, 499–509. 10.1093/jnen/63.5.499. [DOI] [PubMed] [Google Scholar]
- Losman J-A, and Kaelin WG (2013). What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 27, 836–852. 10.1101/gad.217406.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, Cowley GS, Root DE, Ebert BL, and Kaelin WG (2013). (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 339, 1621–1625. 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM, Reifenberger G, et al. (2021). The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 23, 1231–1251. 10.1093/neuonc/noab106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Kwintkiewicz J, Liu Y, Tech K, Frady LN, Su Y-T, Bautista W, Moon SI, MacDonald J, Ewend MG, et al. (2017). Chemosensitivity of IDH1-mutated gliomas due to an impairment in PARP1-mediated DNA repair. Cancer Res. 77, 1709–1718. 10.1158/0008-5472.CAN16-2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchman HA, Chesnelong C, Cairncross JG, and Weiss S.(2013). Spontaneous loss of heterozygosity leading to homozygous R132H in a patient-derived IDH1 mutant cell line. Neuro Oncol. 15, 979–980. 10.1093/neuonc/not064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazor T, Chesnelong C, Pankov A, Jalbert LE, Hong C, Hayes J, Smirnov IV, Marshall R, Souza CF, Shen Y, et al. (2017). Clonal expansion and epigenetic reprogramming following deletion or amplification of mutant IDH1. Proc. Natl. Acad. Sci. USA 114, 10743–10748. 10.1073/pnas.1708914114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBrayer SK, Mayers JR, DiNatale GJ, Shi DD, Khanal J, Chakraborty AA, Sarosiek KA, Briggs KJ, Robbins AK, Sewastianik T, et al. (2018). Transaminase inhibition by 2-hydroxyglutarate impairs glutamate biosynthesis and redox homeostasis in glioma. Cell 175, 101–116.e25. 10.1016/j.cell.2018.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellinghoff IK, Penas-Prado M, Peters KB, Burris HA, Maher EA, Janku F, Cote GM, de la Fuente MI, Clarke JL, Ellingson BM, et al. (2021). Vorasidenib, a dual inhibitor of mutant IDH1/2, in recurrent or progressive glioma; results of a first-in-human phase I trial. Clin. Cancer Res 27, 4491–4499. 10.1158/1078-0432.CCR-21-0611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnikov A, Rogov P, Wang L, Gnirke A, and Mikkelsen TS (2014). Comprehensive mutational scanning of a kinase in vivo reveals substrate-dependent fitness landscapes. Nucleic Acids Res. 42, e112. 10.1093/nar/gku511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuth M.(1989). The molecular basis of mutations induced by deoxyribonucleoside triphosphate pool imbalances in mammalian cells. Exp. Cell Res 181, 305–316. 10.1016/0014-4827(89)90090-6. [DOI] [PubMed] [Google Scholar]
- Núñez FJ, Mendez FM, Kadiyala P, Alghamri MS, Savelieff MG, Garcia-Fabiani MB, Haase S, Koschmann C, Calinescu A-A, Kamran N, et al. (2019). IDH1-R132H acts as a tumor suppressor in glioma via epigenetic upregulation of the DNA damage response. Sci. Transl. Med 11, eaaq1427. 10.1126/scitranslmed.aaq1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oser MG, Sabet AH, Gao W, Chakraborty AA, Schinzel AC, Jennings RB, Fonseca R, Bonal DM, Booker MA, Flaifel A, et al. (2019). The KDM5A/RBP2 histone demethylase represses NOTCH signaling to sustain neuroendocrine differentiation and promote small cell lung cancer tumorigenesis. Genes Dev. 33, 1718–1738. 10.1101/gad.328336.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter HJ, Morrisey JM, Mather MW, Orchard LM, Luck C, Smilkstein MJ, Riscoe MK, Vaidya AB, and Llinás M.(2021). Atypical molecular basis for drug resistance to mitochondrial function inhibitors in plasmodium falciparum. Antimicrob. Agents Chemother 65. e02143–20. 10.1128/AAC.02143-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC-H, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu I-M, Gallia GL, et al. (2008). An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812. 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip B, Yu DX, Silvis MR, Shin CH, Robinson JP, Robinson GL, Welker AE, Angel SN, Tripp SR, Sonnen JA, et al. (2018). Mutant IDH1 promotes glioma formation in vivo. Cell Rep. 23, 1553–1564. 10.1016/j.celrep.2018.03.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, et al. (2014). CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455. 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quayle SN, Lee JY, Cheung LWT, Ding L, Wiedemeyer R, Dewan RW, Huang-Hobbs E, Zhuang L, Wilson RK, Ligon KL, et al. (2012). Somatic mutations of PIK3R1 promote gliomagenesis. PLoS One 7, e49466. 10.1371/journal.pone.0049466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, et al. (2013). An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 340, 626–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Knobbe CB, Itsumi M, Elia AJ, Harris IS, Chio IIC, Cairns RA, McCracken S, Wakeham A, Haight J, et al. (2012a). D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev. 26, 2038–2049. 10.1101/gad.198200.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brüstle A, Harris IS, Holmes R, Wakeham A, Haight J, et al. (2012b). IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 488, 656–659. 10.1038/nature11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si Y, and Keenan MC (2017). Treatment of tumors incorporating mutant isocitrate dehydrogenase. Agios Pharmaceuticals. Patent Number: WO2017117372. Filing date: 12/29/2016. Issue date: 06/07/2017. [Google Scholar]
- Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, and Molkentin JD (2001). Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ. Res 89, 20–25. [DOI] [PubMed] [Google Scholar]
- Sonoda Y, Ozawa T, Hirose Y, Aldape KD, McMahon M, Berger MS, and Pieper RO (2001). Formation of intracranial tumors by genetically modified human astrocytes defines four pathways critical in the development of human anaplastic astrocytoma. Cancer Res. 61, 4956–4960. [PubMed] [Google Scholar]
- Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, Stone RM, DeAngelo DJ, Levine RL, Flinn IW, et al. (2017). Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 130, 722–731. 10.1182/blood-2017-04-779405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Lu W, and Rabinowitz JD (2017). Metabolite spectral accuracy on orbitraps. Anal. Chem 89, 5940–5948. 10.1021/acs.analchem.7b00396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulkowski PL, Corso CD, Robinson ND, Scanlon SE, Purshouse KR, Bai H, Liu Y, Sundaram RK, Hegan DC, Fons NR, et al. (2017). 2Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med 9, eaal2463. 10.1126/scitranslmed.aal2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulkowski PL, Oeck S, Dow J, Economos NG, Mirfakhraie L, Liu Y, Noronha K, Bao X, Li J, Shuch BM, et al. (2020). Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature 582, 586–591. 10.1038/s41586-020-2363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Rahbani JF, Jedrychowski MP, Riley CL, Vidoni S, Bogoslavski D, Hu B, Dumesic PA, Zeng X, Wang AB, et al. (2021). Mitochondrial TNAP controls thermogenesis by hydrolysis of phosphocreatine. Nature 593, 580–585. 10.1038/s41586-021-03533-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarhonskaya H, Rydzik AM, Leung IKH, Loik ND, Chan MC, Kawamura A, McCullagh JSO, Claridge TDW, Flashman E, and Schofield CJ (2014). Non-enzymatic chemistry enables 2-hydroxyglutaratemediated activation of 2-oxoglutarate oxygenases. Nat. Commun 5, 3423. 10.1038/ncomms4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasdogan A, Faubert B, Ramesh V, Ubellacker JM, Shen B, Solmonson A, Murphy MM, Gu Z, Gu W, Martin M, et al. (2020). Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 577, 115–120. 10.1038/s41586-019-1847-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateishi K, Wakimoto H, Iafrate AJ, Tanaka S, Loebel F, Lelic N, Wiederschain D, Bedel O, Deng G, Zhang B, et al. (2015). Extreme vulnerability of IDH1 mutant cancers to NAD+ depletion. Cancer Cell 28, 773–784. 10.1016/j.ccell.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcan S, Makarov V, Taranda J, Wang Y, Fabius AWM, Wu W, Zheng Y, El-Amine N, Haddock S, Nanjangud G, et al. (2018). MutantIDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat. Genet 50, 62–72. 10.1038/s41588-017-0001-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AWM, Lu C, Ward PS, et al. (2012). IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483. 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakimoto H, Tanaka S, Curry WT, Loebel F, Zhao D, Tateishi K, Chen J, Klofas LK, Lelic N, Kim JC, et al. (2014). Targetable signaling pathway mutations are associated with malignant phenotype in IDH-mutant gliomas. Clin. Cancer Res 20, 2898–2909. 10.1158/1078-0432.CCR13-3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Yang K, Wu Q, Kim LJY, Morton AR, Gimple RC, Prager BC, Shi Y, Zhou W, Bhargava S, et al. (2019). Targeting pyrimidine synthesis accentuates molecular therapy response in glioblastoma stem cells. Sci. Transl. Med 11, eaau4972. 10.1126/scitranslmed.aau4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo H, Antoniewicz MR, Stephanopoulos G, and Kelleher JK (2008). Quantifying reductive carboxylation flux of glutamine to lipid in a brown adipocyte cell line. J. Biol. Chem 283, 20621–20627. 10.1074/jbc.M706494200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Breitkopf SB, Yang X, and Asara JM (2012). A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat. Protoc 7, 872–881. 10.1038/nprot.2012.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Yao Y, Scott AJ, Wilder-Romans K, Dresser JJ, Werner CK, Sun H, Pratt D, Sajjakulnukit P, Zhao SG, et al. (2020). Purine metabolism regulates DNA repair and therapy resistance in glioblastoma. Nat. Commun 11, 3811. 10.1038/s41467-02017512-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Metabolomics data have been deposited to the National Metabolomics Data Repository (NMDR) and will be publicly available as of the date of publication. Accession and identification numbers are listed in the key resources table. All other data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Mouse monoclonal anti-IDH1 R132H antibody (IHC) | Dianova | Cat# DIA-H09, RRID:AB_2335716 |
| Rabbit monoclonal anti-Cleaved Caspase 3 | Cell Signaling Technology | Cat# 9661, RRID:AB_2341188 |
| Mouse monoclonal anti-Akt (pan) | Cell Signaling Technology | Cat# 2920, RRID:AB_1147620 |
| Rabbit monoclonal anti-pAkt | Cell Signaling Technology | Cat# 2965S, RRID:AB_2255933 |
| Rabbit polyclonal anti-GFAP | Abcam | Cat# ab7260, RRID:AB_305808 |
| Rabbit polyclonal anti-Sox2 | Abcam | Cat# ab97959, RRID:AB_2341193 |
| Rabbit polyclonal anti-Olig2 | Ligon et al. (2004) | N/A |
| Mouse monoclonal anti-Nestin | Abcam | Cat# ab6142, RRID:AB_305313 |
| Mouse monoclonal anti-γH2A.X | Cell Signaling Technology | Cat# 9718S, RRID:AB_2118009 |
| Mouse monoclonal anti-FLAG | Sigma-Aldrich | Cat# F1804, RRID:AB_262044 |
| Mouse monoclonal anti-HA | Covance | Cat# MMS-101P RRID: AB_2314672 |
| Mouse monoclonal, anti-Vinculin | Sigma-Aldrich | Cat# V9131, RRID:AB_477629 |
| Anti-53BP1 antibody | Novus | Cat# NB100–304, RRID:AB_10003037 |
| Rabbit polyclonal anti-DHODH | Proteintech | Cat#14877 RRID: AB_2091723 |
| Donkey anti-rabbit IgG, Alexa Fluor 488-conjugated secondary antibody | Invitrogen | Cat# A-21206 RRID:AB_2535792 |
| Goat anti-mouse IgG, HRP-linked secondary antibody | Thermo Fisher | Cat# 31430 RRID:AB_228307 |
| Goat anti-rabbit IgG, HRP-linked secondary antibody | Thermo Fisher | Cat#31460 RRID:AB_228341 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Adeno-associated virus (pAAV2-sgTrp53-sgAtrx-EFS-Cre) | This paper, AAV packaging performed by Vigene Biosciences | https://www.vigenebio.com/aav-packaging/ |
| HB101 Competent Cells | Promega | Cat# L2015 |
|
| ||
| Biological samples | ||
|
| ||
| UTSW6577 glioma specimen | Abdullah et al. (2022) | N/A |
| UTSW1382 glioma specimen | Abdullah et al. (2022) | N/A |
| UTSW9647 glioma specimen | Abdullah et al. (2022) | N/A |
| UTSW2243 glioma specimen | Abdullah et al. (2022) | N/A |
| UTSW2846 glioma specimen | This paper | N/A |
| UTSW2294 glioma specimen | This paper | N/A |
| UTSW9698 glioma specimen | This paper | N/A |
| UTSW9501 glioma specimen | This paper | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Metabolic inhibitor library | This paper | N/A |
| Anticancer drug library | Selleck Chemicals | N/A (purchased in 2015) |
| Brequinar | Sigma-Aldrich | Cat# SML0113 |
| 6-Azauridine | Sigma-Aldrich | Cat# A1882 |
| Pyrazofurin | Sigma-Aldrich | Cat# SML1502 |
| Uridine | Sigma-Aldrich | Cat# U3750 |
| Lometrexol | Cayman Chemical | Cat# 18049 |
| AnnexinV-FITC | Fisher Scientific | Cat# BD556547 |
| AnnexinV-PE | Fisher Scientific | Cat# BD559763 |
| AnnexinV-APC | Fisher Scientific | Cat# BD550475 |
| DAPI | Cayman Chemical | Cat# 14285 |
| AGI-5198 (IDH-C35) | Xcess Bio | Cat# M60068 |
| Deoxythymidine | Sigma-Aldrich | Cat# 3416–05-5 |
| Deoxycytidine | Sigma-Aldrich | Cat# 951–77-9 |
| Palbociclib (PD 0332991) | Cayman Chemical | Cat# 16273 |
| BAY 2402234 | Bayer | N/A |
| (R)-2HG-TFMB | Provided by Ryan E. Looper | N/A |
| amide-15N-glutamine | Cambridge Isotope Laboratories | Cat# NLM-557 |
| 15N-uracil | Cambridge Isotope Laboratories | Cat# NLM-637 |
| 15N-uridine | Cambridge Isotope Laboratories | Cat# NLM-812 |
| 15N-hypoxanthine | Cambridge Isotope Laboratories | Cat# NLM-8500 |
| Tunicamycin | Sigma-Aldrich | Cat# T7765 |
| 4-hydroxytamoxifen | Sigma-Aldrich | Cat# H7904 |
| L-dihydroorotic acid | Sigma-Aldrich | Cat# D7128 |
| Decylubiquinone | Sigma-Aldrich | Cat# D7911 |
| 2,6-dichloroindophenol | Sigma-Aldrich | Cat# D1878 |
| Sodium malonate | Sigma-Aldrich | Cat# 63409 |
| Potassium cyanide | Sigma-Aldrich | Cat# 60178 |
|
| ||
| Critical commercial assays | ||
|
| ||
| KOD Xtreme Hot Start DNA Polymerase Kit | Millipore Sigma | Cat# 71975 |
| DNeasy Blood and Tissue kit | Qiagen | Cat# 695045 |
| TissueLyser | Qiagen | Cat# 85300 |
| Neural Tissue Dissociation Kit | Miltenyi | Cat# 130–095-942 |
| RNeasy Mini Kit | Qiagen | Cat# 74004 |
| AffinityScript qPCR cDNA Synthesis Kit | Agilent | Cat# 600559 |
| NEB Monarch kit | New England BioLabs | Cat# T1030 |
| Qiagen gel extraction kit | Qiagen | Cat# 28706 |
| Mouse genotyping | Transnetyx | https://www.transnetyx.com/ |
| Bond Polymer Refine Detection Kit | Leica Biosystems | Cat# DS9800 |
| BP Clonase | Invitrogen | Cat# 11789100 |
| LR Clonase | Invitrogen | Cat# 11791020 |
|
| ||
| Deposited data | ||
|
| ||
| TCGA Brain Lower Grade Glioma dataset | GDAC Firehose, Broad Institute | http://gdac.broadinstitute.org/runs/analyses__2016_01_28/reports/cancer/LGG-TP/index.html |
| Metabolomics Analysis of HOG-EV and HOG-R132H Cells with and without BAY 2402234 Treatment | National Metabolomics Data Repository | NMDR: https://doi.org/10.21228/M81X41 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| BT054 | Kelly et al. (2010) | N/A |
| BT260 | Koivunen et al. (2012) | N/A |
| TS516 | Rohle et al. (2013) | N/A |
| TS603 | Rohle et al. (2013) | N/A |
| HK213 | Laks et al. (2016) | N/A |
| HK211 | Laks et al. (2016) | N/A |
| HK157 | Laks et al. (2016) | N/A |
| HK252 | Laks et al. (2016) | N/A |
| HK308 | Laks et al. (2016) | N/A |
| MGG152 | Wakimoto et al. (2014) | N/A |
| HOG | Provided by Pablo Paez | N/A |
| NHA (immortalized with HPV E6 and E7 and hTERT) | Provided by Russell Pieper | N/A |
| NHA (EV or IDH1-R132H) | Koivunen et al. (2012) | |
| UTSW6577 glioma organoids | Abdullah et al. (2022) | N/A |
| UTSW1382 glioma organoids | Abdullah et al. (2022) | N/A |
| UTSW9647 glioma organoids | Abdullah et al. (2022) | N/A |
| UTSW2243 glioma organoids | Abdullah et al. (2022) | N/A |
| UTSW2846 glioma organoids | This paper | N/A |
| UTSW2294 glioma organoids | This paper | N/A |
| UTSW9698 glioma organoids | This paper | N/A |
| UTSW9501 glioma organoids | This paper | N/A |
| DF-AA27 GEMM GSC | This paper | N/A |
| MEFs, irradiated | Life Technologies | Cat# A34181 |
| IC (H11LSL-Cas9+/−;Idh1tm1Mak/WT) MEFs | This paper | N/A |
| HEK293T | ATCC | Cat# CRL-11268 |
| Primary human astrocytes | Lonza | Cat# CC-3187 |
| Human neural stem cells | Fisher Scientific | Cat# 10419428 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| NCr Nude mice, 6–10-week-old females | Taconic | Cat# NCRNU-F |
| NCr Nude mice, 4–5-week-old females | Taconic | Cat# NCRNU-F |
| ICR SCID mice, 6–12-week-old females | Taconic | Cat# ICRSC-F |
| R26-Pik3caH1047R (FVB.129S6-Gt(ROSA)26Sortm1(Pik3ca*H1047R)Egan/J mouse | Jackson Laboratories | Cat# 016977 |
| Idh1tm1Mak mouse | Provided by Tak W. Mak | N/A |
| H11LSL-Cas9 (B6; 129-Igs2tm1(CAG-cas9*)Mmw/J mouse | Jackson Laboratories | Cat# 026816 |
| PIC (H11LSL-Cas9+/−;Idh1tm1Mak/WT;R26-Pik3caH1047R+/−) mouse | This paper | N/A |
| PC (H11LSL-Cas9+/−;Idh1WT/WT;R26-Pik3caH1047R+/−) mouse | This paper | N/A |
| IC (H11LSL-Cas9+/−;Idh1tm1Mak/WT) mouse | This paper | N/A |
| C (H11LSL-Cas9+/−;Idh1WT/WT) mouse | This paper | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| sgATRX: ATAAGGACTTTGAGTGAA | This paper | N/A |
| sgTrp53: GTGTAATAGCTCCTGCATG | Platt et al. (2014) | N/A |
| sgControl: GGAGGCTAAGCGTCGCAA | Provided by Feng Zhang | N/A |
| sgDHODH: CATCTTATAAAGTCCGTCCA | This paper | N/A |
| Trp53 outer fwd ATAGAGACGCTGAGTCCGGTTC | Oser et al. (2019) | N/A |
| Trp53 outer rev CCTAAGCCCAAGAGGAAACAGA | Oser et al. (2019) | N/A |
| Trp53 inner fwd TGCAGGTCACCTGTAGTGAGGTAGG | Oser et al. (2019) | N/A |
| Trp53 inner rev GAAACAGGCAGAAGCTGGGGAAGAAAC | Oser et al. (2019) | N/A |
| ATRX outer fwd GCTATCTGAAACTCAATCCACG | This paper | N/A |
| ATRX outer rev GACTTGGTTTCTCCTTTGCCATG | This paper | N/A |
| ATRX inner fwd GCTTCCTGTAAGCTCATAAGTAC | This paper | N/A |
| ATRX inner rev CTAATGCCATATGAGTGTAACTC | This paper | N/A |
| ATRX second inner fwd CTCTTACATAATGGCCATTCTC | This paper | N/A |
| ATRX second inner rev CTGTGAGTCATGATCATTCTTTGC | This paper | N/A |
| Recombined IDH1 fwd GATTGATTCTGCCGCCATGATCCTAGT | Sasaki et al. (2012b) | N/A |
| Recombined IDH1 rev CCTGGTCATTGGTGGCATCACGATTCTC | Sasaki et al. (2012b) | N/A |
| Actb fwd (genomic DNA for Idh1-R132H assay) TGACCCAGGT CAGTATCCCGGGT | This paper | N/A |
| Actb actin rev (genomic DNA for Idh1-R132H assay) GAACACAGCTAAGTTCAGTGTGCTGGGA | This paper | N/A |
| PIK3CA H1047R cDNA fwd CTAGGTAGGGGATCGGGACTCT | This paper | N/A |
| PIK3CA H1047R rev AATTTCTCGATTGAGGATCTTTTCT | This paper | N/A |
| Actb fwd (cDNA for Pik3ca-H1047R assay) TAGGCACCAGGGTGTGATG | This paper | N/A |
| Actb rev (cDNA for Pik3ca-H1047R assay) CATGGCTGGGGTGTTGAAGG | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pLX304-Ubc-GATE-IRES-GFP | Gift from Vidyasagar Koduri | N/A |
| pDONR223 | Invitrogen | N/A |
| pLX304-Ubc-EV-IRES-GFP | EV provided as a gift by Vidyasagar Koduri | N/A |
| pLX304-Ubc-DHODH-WT-IRES-GFP | DHODH WT cDNA provided as a gift by Laura Evans & Julie Losman | N/A |
| pLX304-Ubc-DHODH-A58T-IRES-GFP | DHODH A58T cDNA provided as a gift by Laura Evans & Julie Losman | N/A |
| pLX304-CMV-BFP | Gift from Hilary Nicholson | N/A |
| pLX304-CMV-GFP | Gift from Hilary Nicholson | N/A |
| pLXSN-H-Ras_V12 | Addgene | Cat# 39516 |
| pLenti-Flag-P85 | Addgene | Cat# 40219 |
| pLenti4-Flag-P85-DKRMNS560del | Addgene | Cat# 40225 |
| pLenti-Flag-P85-R574fs | Addgene | Cat# 40227 |
| pLenti4-Flag-T576del | Addgene | Cat# 40228 |
| p1321 HPV-16 E6/E7 | Addgene | Cat# 8641 |
| pAAV2-sgTrp53-sgAtrx-EFS-Cre | This paper | Addgene, Cat# 189977 |
| pLBCX-Large-T-antigenK1 mutant | This paper | Addgene, Cat# 189978 |
| pMSCV-MerCreMer-hygro | This paper | Addgene, Cat# 188982 |
| LeGO-iG2-FLuc | McBrayer et al. (2018) | N/A |
| pBABE-HA-hygro | Koivunen et al. (2012) | N/A |
| pBABE-IDH1-R132H-HA-hygro | Koivunen et al. (2012) | N/A |
| plenti-Ubc-IRES-hygro empty vector | McBrayer et al. (2018) | N/A |
| plenti-Ubc-IDH1-R132H-HA-IRES-hygro | McBrayer et al. (2018) | N/A |
| pMD2.G | Provided by Didier Trono | Addgene, Cat# 12259 |
| Gag/pol | Provided by Tannishtha Reya | Addgene, Cat# 14887 |
| psPAX2 | Provided by Didier Trono | Addgene, Cat# 12260 |
| VSV.G | Provided by Tannishtha Reya | Addgene, Cat# 14888 |
| pX551 | Provided by Feng Zhang | Addgene Cat# 60957 |
|
| ||
| Software and Algorithms | ||
|
| ||
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
| Metran | Yoo et al. (2008) | N/A |
| Multi-Quant 2.1 software | Sciex | https://sciex.com/products/software/multiquant-software |
| GraphPad Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Living Image | PerkinElmer | http://www.perkinelmer.com/product/li-software-for-spectrum-1-seat-add-on-128113 |
| R | The R Project for Statistical Computing | https://www.r-project.org/ |
| AccuCor R package (version 0.2.3) | Su et al. (2017) | https://github.com/lparsons/accucor |
| El-Maven 0.12.0 | Elucidata | https://elucidata.io/el-maven/ |
| TraceFinder 5.1 SP2 | Thermo Fisher | Cat# OPTON-31001 |
| Freestyle 1.7 SP | Thermo Fisher | Cat# XCALI-98282 |
| MetaboAnalyst | Publicly available | https://www.metaboanalyst.ca/ |
| FlexControl, version 4.0 | Bruker | https://www.bruker.com/en/products-and-solutions/mass-spectrometry/maldi-tof/autoflexmax.html |
| FlexImaging, version 5.0 and SCiLS Lab | Bruker Daltonics | https://www.bruker.com/en/products-and-solutions/mass-spectrometry/ms-software/scilslab.html |
| ImageJ | NIH | https://imagej.net/ |
| QuPath software (version 0.2.3) | Publicly available | https://qupath.github.io/ |
|
| ||
| Other | ||
|
| ||
| Geltrex | Life Technologies | Cat# A1413202 |
| AGM SingleQuots Supplements | Lonza | Cat# CC-4123 |
| NeuroCult Basal Medium (Mouse and Rat) | StemCell Technologies | Cat# 05702 |
| NeuroCult NS-A Basal Medium (Human) | StemCell Technologies | Cat# 05750 |
| Neurobasal Medium (MGG152 GSCs) | Gibco | Cat# 21103049 |
| Neurobasal Medium (organoids) | Thermo Fisher | Cat# 21103049 |
| DMEM-F12 Medium | Gibco | Cat# 11320033 |
| DMEM Medium | Gibco | Cat# 11995–065 |
| Accutase | StemCell Technologies | Cat# 07922 |
| RBC lysis buffer | Thermo Fisher | Cat# 00433357 |
| 2-mercaptoethanol | Thermo Fisher | Cat# BP176–100 |
| Glutamax | Thermo Fisher | Cat# 35050061 |
| N-2 supplement | Thermo Fisher | Cat# 17502048 |