

Abstract
Background
Pathological evidence suggests that coronavirus disease 2019 (COVID-19) pulmonary infection involves both alveolar damage (causing shunt) and diffuse microvascular thrombus formation (causing alveolar dead space). We propose that measuring respiratory gas exchange enables detection and quantification of these abnormalities. We aimed to measure shunt and alveolar dead space in moderate COVID-19 during acute illness and recovery.
Methods
We studied 30 patients (22 males; mean±sd age 49.9±13.5 years) 3–15 days from symptom onset and again during recovery, 55±10 days later (n=17). Arterial blood (breathing ambient air) was collected while exhaled oxygen and carbon dioxide concentrations were measured, yielding alveolar–arterial differences for each gas (PA−aO2 and Pa−ACO2, respectively) from which shunt and alveolar dead space were computed.
Results
For acute COVID-19 patients, group mean (range) for PA−aO2 was 41.4 (−3.5–69.3) mmHg and for Pa−ACO2 was 6.0 (−2.3–13.4) mmHg. Both shunt (% cardiac output) at 10.4% (0–22.0%) and alveolar dead space (% tidal volume) at 14.9% (0–32.3%) were elevated (normal: <5% and <10%, respectively), but not correlated (p=0.27). At recovery, shunt was 2.4% (0–6.1%) and alveolar dead space was 8.5% (0–22.4%) (both p<0.05 versus acute). Shunt was marginally elevated for two patients; however, five patients (30%) had elevated alveolar dead space.
Conclusions
We speculate impaired pulmonary gas exchange in early COVID-19 pneumonitis arises from two concurrent, independent and variable processes (alveolar filling and pulmonary vascular obstruction). For most patients these resolve within weeks; however, high alveolar dead space in ∼30% of recovered patients suggests persistent pulmonary vascular pathology.
Short abstract
Hypoxaemia in COVID-19 is due to a variable combination of intrapulmonary shunt and increased dead space, likely from both airspace and vascular pathology. Increased dead space present up to 2 months later suggests persistent pulmonary vascular pathology. https://bit.ly/3ROtboc
Introduction
Since the beginning of 2020, over half a billion people have been diagnosed with coronavirus disease 2019 (COVID-19), a disease caused by infection with the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Many have required hospitalisation, with 10–20% of those requiring intensive care, mainly due to respiratory compromise [1]. Worldwide there have been more than 6 million deaths.
Patients with early COVID-19 respiratory failure present with hypoxaemia and hyperventilation [2–6]. The hypoxaemia is likely related to ventilation/perfusion (V′A/Q′) mismatch and in particular to increased intrapulmonary shunt arising from alveolar filling with fluid or cellular debris. However, pathology reports from COVID-19 infected lungs also frequently demonstrate pulmonary vasculature involvement, including severe endothelial injury, widespread thrombosis with microangiopathy and new vessel growth [7–9]. Pulmonary vessel microembolism reduces capillary blood flow, thus generating areas of high V′A/Q′ and promoting increased alveolar dead space.
We hypothesised that in early COVID-19 pneumonitis, hypoxaemia is associated with both increased intrapulmonary shunt and increased alveolar dead space. Using bedside measurement of exhaled partial pressure of oxygen (PO2) and carbon dioxide (PCO2) combined with arterial blood gas measurements, we measured alveolar–arterial partial pressure differences for both oxygen (PA−aO2) and carbon dioxide (Pa−ACO2), from which both intrapulmonary shunt and alveolar dead space values were then determined using a novel computational model [10].
Methods
Subjects
30 patients admitted to Danderyd Hospital in Stockholm, Sweden who were ≥18 years old and PCR-positive for SARS-CoV-2 were recruited between November and December 2020. Patients were excluded if they were in immediate need of intubation or mechanical ventilation, had advanced pulmonary or cardiac disease, current malignancy, previous thromboembolic disease, current pregnancy, or were unable to tolerate the study protocol, which required spontaneous breathing of ambient air for several minutes. Data were collected within 24 h of presentation to hospital and again 55±10 days after discharge in early recovery (n=17). 13 patients did not return for follow-up. A subset of the acute data has been reported previously [6].
13 healthy volunteers, negative for SARS-CoV-2 on PCR testing and with normal pulmonary function (Vyaire-Vyntus Spiro PC-spirometer with SentrySuite Software; Vyaire, Mettawa, IL, USA), were recruited from hospital staff as methodological controls.
All participants gave written informed consent and the study protocol was approved by the Swedish Ethical Review Authority (diary number 2020-02966).
Clinical data
Anthropometric and demographic data were collected, along with body temperature for the acutely ill. Pathology test results, pharmacological interventions, respiratory support (including oxygen therapy) and duration of hospitalisation data were also collected.
Study protocol
Subjects were studied in a semirecumbent position, wearing a noseclip and breathing ambient air via a mouthpiece with an antivirus filter (MicroGard II; Vyaire Medical, Hoechberg, Germany; apparatus dead space 75 mL) attached to an inspired/expired gas measurement system (Oxycon Pro; Vyaire Medical). Collected data included PO2, PCO2 and respiratory gas flow at high sampling frequency (100 Hz). After a few minutes of adaptation, participants maintained steady-state breathing (end-tidal PCO2 within ±2 mmHg across several breaths) at a (metronome-facilitated) frequency and tidal volume of their choice. Data were recorded over a steady-state 3–5 min period, during which radial arterial blood was collected over several breaths and then processed immediately (ABL800 Flex Plus; Radiometer Medical, Bronshoj, Denmark) to obtain values for arterial PO2 (PaO2) and PCO2 (PaCO2). For the acute patient studies, arterial blood gas values were expressed at body temperature [11]. Recovered patients and healthy subjects were assumed to have a body temperature of 37°C.
Exhaled gas analysis
Three separate breaths, preceding the arterial blood sample by 35±10 s, were selected, independently analysed and resulting parameters averaged. Following alignment of gas and volume signals, PO2 and PCO2 values for each breath were plotted as a function of expired volume and a linear least-squares fit applied to the alveolar plateau (phase III; see figure 1). Mean alveolar gas values, PAO2 and PACO2 [10], at the mid-point (by volume) of the expired breath were determined from the fitted lines. Exhaled gas measurements in the acute patient studies were corrected for water vapour pressure at body temperature using Antoine's formula [12]. Alveolar–arterial partial pressure differences for both O2 and CO2 were then calculated as PAO2−PaO2 (PA−aO2) and PaCO2−PACO2 (Pa−ACO2).
FIGURE 1.

Expired gas tracings from one patient: exhaled partial pressure of oxygen (PO2; top black line, red symbols and lines) and carbon dioxide (PCO2; bottom black line, blue symbols and lines). The second dotted line indicates the beginning of alveolar emptying (phase III), sloping dashed lines indicate the linear regression fits to phase III. Alveolar partial pressures (PACO2 and PAO2) were measured from the mid-volume of the linear regression fit to phase III (∼225 mL in this example). Filled symbols indicate mean alveolar values; open symbols indicate contemporaneous arterial values; arterial–alveolar differences are indicated by the solid vertical lines connecting the symbols.
Computational analysis
We employed the seven-decade-old Riley and Cournand [13] three-compartment lung model to calculate shunt and alveolar dead space from PA−aO2 and Pa−ACO2. In this model, the lung is considered to have an intrapulmonary shunt compartment with a V′A/Q′ of zero, also encompassing regions of very low V′A/Q′, and an alveolar dead space compartment with V′A/Q′ of infinity, also encompassing regions of very high V′A/Q′. The remainder of the lung is assumed to be normal, with a V′A/Q′ ratio given by non-dead space ventilation divided by non-shunt blood flow. A critical difference exists between the Riley and Cournand model and the current approach: Riley and Cournand used the venous admixture equation for oxygen to calculate shunt and the Bohr equation for carbon dioxide to calculate dead space. However, shunt may increase arterial PCO2 and contribute to calculated alveolar dead space, while dead space (without compensatory hyperventilation) lowers arterial PO2 and contributes to calculated shunt. Our approach [10] recognises this complexity and resolves it, still within the three-compartment framework, by determining the values of shunt and dead space that, present together, predict the measured arterial and expired alveolar PO2 and PCO2 values, and hence their partial pressure differences. In addition, the Bohr equation usually includes anatomical dead space, which commonly dominates total dead space numbers. Our approach eliminates anatomical dead space by using mean alveolar partial pressures as in figure 1 and not those of mixed exhaled gas.
Using the measured V′CO2 (respiratory frequency×volume CO2 per breath), PAO2, fractional alveolar oxygen concentration (FAO2), PACO2 and fractional alveolar carbon dioxide concentration (FACO2), we calculated: 1) total expired alveolar ventilation V′A=V′CO2/FACO2, 2) inspired alveolar ventilation V′I=V′A×((1−FAO2−FACO2)/(1−FIO2)) and 3) V′O2=V′I×FIO2−V′A×FAO2, where FIO2 is the inspiratory oxygen fraction.
Cardiac output (Q′T) was then estimated as Q′T=V′O2+5, with both Q′T and V′O2 expressed in L·min−1 [14, 15]. The oxygen tension at which haemoglobin is 50% saturated (HbP50) was assumed to be 26.8 mmHg. Using these values and each patient's own data for V′O2, V′CO2, V′A, Q′T, base excess, haemoglobin concentration and body temperature, we applied the algorithm first published in 1969 by West [16] to estimate the values of intrapulmonary shunt and alveolar dead space that would result in the measured PaO2 and PaCO2 in each individual to within 0.2 mmHg.
Intrapulmonary shunt was expressed as the percentage of pulmonary blood flow perfusing unventilated regions (shunt %), while alveolar dead space was expressed as the percentage of alveolar ventilation associated with unperfused regions (alveolar dead space %). Based on published measurements using the multiple inert gas elimination technique in normal subjects [17], the 95% upper confidence limit for physiological shunt is 5% and for alveolar dead space is 10%. Further methodological details can be found elsewhere [10].
Statistical analysis
This was an observational study and a sample size was not calculated. Individual data were pooled and reported as group mean with standard deviation or interquartile range. Comparisons were made using paired t-tests. Relationships between variables were examined using Spearman's rank correlation coefficients. p<0.05 was considered significant.
Results
Subject characteristics
See table 1.
TABLE 1.
Anthropometric, demographic and body temperature data for acutely ill COVID-19 patients, recovered COVID-19 patients and healthy subjects
| Acute (n=30) | Recovered (n=17) | Healthy (n=13) | |
| Age (years) | 49.9±13.5 (23–78) | 50.6±12.0 (51–78) | 51.1±17.0 (34–68) |
| Sex | |||
| Female | 8 | 6 | 5 |
| Male | 22 | 11 | 8 |
| Weight (kg) | 87.5±17.0 (46–113) | 87.5±18.2 (46–110) | 80.2±26.1 (58–110) |
| Height (cm) | 177.0±11.3 (150–195) | 176.8±13.2 (150–195) | 177.2±19.5 (159–198) |
| BMI (kg·m−2) | 27.8±4.3 (20.2–38.5) | 27.9±4.7 (20.4–38.5) | 25.5±6.0 (19.6–31.5) |
| Body temperature (°C) | 38.0±1.0 (36.5–40.0) | 37.0# | 37.0# |
Data are presented as mean±sd (range) or n. #: assumed.
Acute COVID-19 patient data
All patients had moderate disease and were studied at hospital presentation and within 3–15 days of symptom onset. 24 were admitted (mean 4.0±2.8 days, range 1–12 days); of these, 18 required supplemental oxygen and one was subsequently admitted to the intensive care unit. There were no deaths.
Pharmacological interventions
22 patients received prophylactic low-molecular-weight heparin (LMWH), 12 of these patients <24 h before the study (tinzaparin 4500 U per day, subcutaneously), while therapeutic LMWH (tinzaparin 175 U·kg−1 twice daily, daily, s.c.) was administered to one patient. Post-study, 14 patients received corticosteroids (betamethasone 6 mg orally), three received remdesivir and convalescent plasma was administered to two patients.
Pathology findings
C-reactive protein levels were elevated in all patients, while almost 40% had elevated levels for D-dimer (table 2).
TABLE 2.
Pathology data at the time of acute COVID-19 infection
| Pathology | Mean±sd | Range | Normal range | Abnormal (n (%)) |
| Haemoglobin (g·L−1) (n=29) | 139.5±11.7 | 110–165 | Female: 117–153; male: 134–170 | 2 (6.8) |
| D-dimer (mg·L−1) (n=28) | 0.8±0.6 | 0.3–3.2 | Female: <0.54; male: <0.50 | 11 (39.2) |
| C-reactive protein (mg·L−1) (n=30) | 79.1±65.7 | 3–256 | <3 | 30 (100) |
Arterial blood gas, oxyhaemoglobin saturation and exhaled alveolar gas data
Arterial blood gas data reflected an acute respiratory alkalosis with hypoxaemia (table 3). PAO2 values were all >102 mmHg, PACO2 values were all <38 mmHg and arterial oxygen saturation (SaO2) values were all >89%.
TABLE 3.
Arterial blood gases and exhaled alveolar gas measurements
| Acute (n=30)# | Recovered (n=17) | Healthy (n=13) | |
| pH | 7.48±0.04 (7.42–7.60) | 7.44±0.03 (7.39–7.51) | 7.43±0.02 (7.41–7.49) |
| PaCO2 (mmHg) | 34.8±4.2 (26.5–43.0) | 34.4±3.7 (26.7–40.6) | 37.0±2.8 (31.4–40.8) |
| PaO2 (mmHg) | 68.3±12.6 (52.3–105.8) | 100.6±13.6 (84.0–131.3) | 105.0±8.3 (94.5–120.0) |
| PAO2 (mmHg) | 113.3±5.7 (102.2–122.3) | 113.6±6.3 (100.2–125.3) | 119.5±5.1 (112.7–127.3) |
| PACO2 (mmHg) | 30.3±4.0 (23.4–37.3) | 31.4±3.1 (24.9–38.7) | 35.2±2.9 (29.1–39.9) |
| PA−aO2 (mmHg) | 41.4±16.3 (−3.5–69.3) | 13.0±9.7 (−6.0–30.2) | 6.5±6.9 (−5.8–21.4) |
| Pa−ACO2 (mmHg) | 6.0±4.2 (−2.3–13.4) | 3.0±2.3 (−2.2–8.9) | 1.8±1.9 (−1.4–6.0) |
| SaO2 (%) | 94.3±0.2 (89.4–98.7) | 98.7±0.5 (97.7–99.5) | 98.9±0.2 (98.4–99.3) |
Data are presented as mean±sd (range). #: all acute patient gas partial pressures are expressed at body temperature. PaCO2: arterial partial pressure of carbon dioxide; PaO2: arterial partial pressure of oxygen; PAO2: alveolar partial pressure of oxygen; PACO2: alveolar partial pressure of carbon dioxide; PA−aO2: alveolar–arterial partial pressure difference for oxygen; Pa−ACO2: alveolar–arterial partial pressure difference for carbon dioxide; SaO2: arterial oxygen saturation.
Alveolar–arterial differences
There was wide interpatient variance in PA−aO2 and Pa−ACO2 (table 3 and figure 2a). Any negative values (expected from experimental noise) were considered to be zero when calculating shunt and dead space.
FIGURE 2.

a) Alveolar–arterial partial pressure differences for oxygen (PA−aO2) and carbon dioxide (Pa−ACO2). b) Intrapulmonary shunt and alveolar dead space. Acute COVID-19 patients (n=30) and healthy subjects (n=13). Dotted lines in (b) show 95% upper limits for normal [17].
Intrapulmonary shunt and alveolar dead space
Values for intrapulmonary shunt and alveolar dead space varied considerably between patients (table 4 and figure 2b); however, importantly, there was no significant correlation between the shunt and dead space values (p=0.27). Significant positive correlations were detected between shunt and both D-dimer (r=0.36, p=0.03) and CRP (r=0.42, p=0.01), but not for alveolar dead space (p>0.15).
TABLE 4.
Shunt and alveolar dead space in acute COVID-19 patients (including the subgroup of 17 patients studied later in recovery), recovered patients and healthy subjects.
| Acute (n=30) | Acute# (n=17) | Recovered (n=17) | Healthy (n=13) | |
| Shunt (%) | 10.4±5.4 (0–22.0) | 11.7±5.1 (4.1–22.0) | 2.4±1.9** (0–6.1) | 1.2±1.1 (0–4.1) |
| Alveolar dead space (%) | 14.9±9.6 (0–32.3) | 14.4±9.7 (0–29.7) | 8.6±5.5* (0–22.4) | 4.7±4.3 (0–11.8) |
Data are presented as mean±sd (range). #: subgroup studied in recovery. *: p<0.05; **: p<0.0001 (compared with acute subgroup).
Overall, 23 patients had elevated shunt (defined as >5%), while 22 patients had elevated alveolar dead space (defined as >10%). Simultaneously increased shunt and dead space were present in 19 patients, four patients had elevated shunt but normal alveolar dead space and three had elevated alveolar dead space but normal shunt (figure 2b).
Healthy subject data
Healthy subjects were all normoxaemic with normal acid–base status (see table 3). No healthy subject had a shunt value >5% (table 4 and figure 2b); two had slightly elevated values for dead space (table 4 and figure 2b). Overall, shunt and dead space values from the healthy subjects conformed to the previously established upper limits of normal [17].
Transition from acute illness to recovery
Shunt and alveolar dead space fell significantly following recovery (figures 3 and 4). However, shunt was slightly elevated in two patients, while five patients (29.4%) had elevated dead space. There was no correlation with duration after recovery (both p>0.15).
FIGURE 3.

Individual (symbols and lines) and grouped data (box-and-whisker plots showing mean, interquartile range and range) for 17 patients during acute COVID-19 infection and again in early recovery, together with 13 healthy individuals: alveolar–arterial partial pressure differences for a) oxygen (PA−aO2) and b) carbon dioxide (Pa−ACO2), and c) shunt and d) alveolar dead space. Horizontal dotted lines in (c) and (d) show the 95% upper confidence intervals for normal. *: p<0.05; **: p<0.0001.
FIGURE 4.
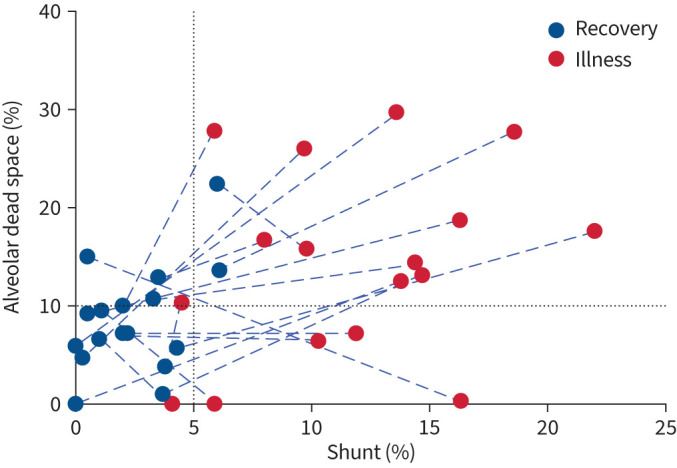
Shunt and dead space trajectories from acute COVID-19 illness to recovery. Blue dashed lines connect paired acute and recovery data (n=17). Black dotted lines show the 95% upper confidence limits of normal.
Discussion
Using simultaneous measurements of arterial and exhaled oxygen and carbon dioxide tensions, interpreted using a three-compartment computational lung model, we developed a bedside methodology for quantifying if there is both intrapulmonary shunt and alveolar dead space [10], and then applied it here in the highly infectious disease setting of COVID-19 pneumonitis. Measurements in a healthy subject cohort (technical controls) conformed to historical normal limits previously established by more sophisticated techniques [17].
Pulmonary gas exchange in early COVID-19 pneumonitis
The main finding from the present study is that in early acute COVID-19 pneumonitis intrapulmonary shunt is elevated for most patients; however, many also have elevated alveolar dead space. Furthermore, intrapulmonary shunt and dead space values were not correlated in moderate COVID-19 pneumonitis. Hence, dead space cannot be predicted from the magnitude of shunt and vice versa. This suggests that in early SARS-CoV-2 pulmonary infection two pathophysiologically distinct process, either separately or together, are in play: 1) patchy alveolar filling/oedema/atelectasis (pneumonia) resulting in lung regions with low or zero V′A/Q′ (i.e. shunt) and 2) patchy pulmonary vascular occlusion (emboli) resulting in high V′A/Q′ regions (i.e. alveolar dead space). However, the relative contribution of these two processes is highly variable from patient to patient. This is similar to findings in intubated patients with severe acute respiratory distress syndrome, where hypoxaemia is due to increased intrapulmonary shunt and increased dead space [18]. This is the first time that this has been demonstrated physiologically in mild/moderate COVID pneumonitis.
While intrapulmonary shunt is perhaps an expected contributor to hypoxaemia in a pulmonary viral infection [18], previous authors have suggested that for COVID-19 patients shunt values can be so elevated as to be considered “excessive” for the degree of lung injury present [2]. This observation has led to the suggestion that SARS-CoV-2 may specifically impair hypoxic pulmonary vasoconstriction, thus preventing hypoxic pulmonary vasoconstriction-driven restriction of pulmonary capillary blood flow to lung regions with poor or no ventilation, increasing shunt [2, 19]. Whether intrapulmonary shunt values detected in the present study are “excessive” remains undetermined.
A major feature of our results was the high dead space values occurring in 73% of patients, with 63% having both elevated intrapulmonary shunt and dead space and 10% having elevated dead space with no evidence of shunt. Increased alveolar dead space is a consequence of ventilation of underperfused alveoli, and we speculate is consistent with reported diffuse and widespread small pulmonary arterial obstruction from thrombus formation, as detected in pulmonary pathological specimens from deceased COVID-19 patients [7, 20]. This interpretation is further supported by the absence of large vessel emboli on contrast computed tomography imaging (performed for clinical purposes) in a subgroup (n=18) of the present patient cohort, although there was no correlation between dead space and D-dimer.
There is extensive pathological evidence of pulmonary microvascular involvement in COVID-19 pneumonia [20]. Histopathology of patients who died from COVID-19, compared with those who died from influenza, showed microvascular changes present in COVID-19 infected lungs that were not present with influenza, including endothelial injury, alveolar capillary microthrombi and new vessel growth [7]. In a study which correlated radiological findings with histopathological inflammation in eight patients who died of COVID-19, there was evidence of vascular damage and thrombosis in regions without concurrent airspace involvement [21]. It has been proposed that the vascular injury may precede the development of frank pneumonia and alveolar filling [22].
High V′A/Q′ regions (dead space) may also be a consequence of redistribution of ventilation from unventilated alveoli (shunt) resulting in relative overventilation of normally perfused alveoli. Our analysis adjusts for any effect of intrapulmonary shunt on Pa−ACO2, so that redistribution of ventilation from obstructed ventilation zones is unlikely to contribute significantly to our measured dead space values.
Pulmonary gas exchange in early recovery from COVID-19 pneumonitis
In recovery, intrapulmonary shunt decreased in all patients and values were within normal expected limits for all but two patients (figures 3 and 4). Consequently, ventilation was restored to lung zones previously identified as having no or very low alveolar ventilation. Dead space also decreased in most patients to within, or close to, normal expected limits (figures 3 and 4). However, in five patients, dead space was >10%, ranging from 10.7% to 22.4%. Consequently, for most patients perfusion was restored to lung zones previously identified as having no or very low perfusion. However, for ∼30% of those studied in recovery (figures 3d and 4), persistent, or even increasing, dead space values suggest persistent or evolving damage to the pulmonary vasculature. This may be a consequence of pre-morbid disease (e.g. COPD or interstitial lung disease). However, if a consequence of COVID-19 infection, this finding is of particular interest, since recently published data suggest that COVID-19 infection poses increased risk for deep vein thrombosis, pulmonary embolism and bleeding episodes, at 3, 6 and 2 months, respectively, after an acute infection [23].
Clinical implications
The finding that there is likely both acute and chronic pulmonary microvascular disease in COVID-19 pneumonitis has implications for the clinical management of patients, both in the acute and recovery phases of the disease process. For example, anticoagulation with LMWH has had mixed outcomes, with benefit in moderate disease but no real improvement found in severe disease [24, 25]. Perhaps a clearer picture might emerge if outcomes were stratified by presence or absence of high V′A/Q′ regions. Targeted therapeutic approaches may then be deployed for patients with increased dead space, who may need anticoagulation, versus those with shunt but low dead space, where anticoagulants may, in theory, have risk without major benefits. Application of this methodology could be used in patients with persistent COVID-19 symptoms to better delineate pulmonary pathology.
Limitations
This study was performed in a small sample of patients with early and mild/moderate disease, early in the pandemic, when few therapeutic interventions were available, and was restricted to those able to tolerate breathing room air. Also, recovery data were collected at only one time-point and we do not have follow-up data on all patients. Consequently, it may not be generalisable to more severe disease. Other (pre-existing) comorbidities such as anaemia may have impacted on our findings, although there was no clinical evidence of these abnormalities. The logistical limitations imposed by an acute airborne infection meant that we were unable to use more sophisticated techniques such as multiple inert gas elimination [26] or imaging tools [27].
We estimated cardiac output and HbP50, required for the calculation of intrapulmonary shunt and dead space. Cardiac output was determined from its well-documented relationship to V′O2 [14, 15], which was measured; HbP50 was taken to be 26.8 mmHg for all participants. Sensitivity analysis for these two uncertain variables shows a minor effect over a wide range of determined intrapulmonary shunt values, with no effect on dead space [10].
Conclusions
Our study shows that in early mild/moderate COVID-19 pneumonitis, there is both increased intrapulmonary shunt and increased alveolar dead space, with marked interpatient variability and lack of correlation. These findings suggest that alveolar filling, resulting in little or no ventilation to some alveoli, and pulmonary microvascular compromise, resulting in little or no blood flow to other alveoli, are both present to varying, but separate, degrees in the early phases of mild/moderate COVID-19 pneumonitis. For essentially all patients, shunt resolved in early recovery; however, for ∼30% of patients, elevated dead space persisted, at least in the early recovery phase. Characterising individual patient pulmonary pathophysiological responses may help inform more personalised approaches to effective treatments in both the acute and recovery phases of infection with SARS-CoV-2.
Shareable PDF
Footnotes
Author contributions: The majority of the authors (P. Harbut, G.K. Prisk, C.E. Farrow, G. Hedenstierna, T.C. Amis, A. Malhotra, P.D. Wagner and K. Kairaitis) met on at least a fortnightly basis remotely. The original idea and study design were conceived in these meetings (P. Harbut, G.K. Prisk, C.E. Farrow, G. Hedenstierna, T.C. Amis, A. Malhotra, P.D. Wagner and K. Kairaitis). Ethics approval, subject identification, data collection, including clinical data, and data analysis occurred in Sweden (P. Harbut, R. Lindwall, S. Hamzei and J. Palmgren). Analysis of the raw data signals was primarily performed by G.K. Prisk, with discussion of signal analysis by P. Harbut, G.K. Prisk, C.E. Farrow, G. Hedenstierna, T.C. Amis, A. Malhotra, P.D. Wagner and K. Kairaitis, and further data analysis, including statistical analysis, in Sydney (K. Kairaitis, C.E. Farrow and T.C. Amis). All stages of the study were discussed at fortnightly meetings, including troubleshooting of protocols and quality control of the data. All authors contributed to the preparation of the manuscript and approved the final version.
This article has an editorial commentary: https://doi.org/10.1183/13993003.01962-2022
Data availability: Individual de-identified data and the protocol used in this study will be available to investigators whose proposed use of the data has been approved by an independent review committee identified for this purpose. Data will be available for meta-analysis from 9 to 36 months of publication. After 36 months the data will be available in the Karolinska Institutet (Stockholm, Sweden) data repository but without investigator support other than the deposited data.
Conflict of interest: A. Malhotra has received grants from the National Institutes of Health, and consulting fees for medical education from Livanova, Equilium, Corvus and Jazz. P.D. Wagner has received consulting fees from SMS Biotechnology and Third Pole Inc., and has participated on the data safety monitoring board on Dr Levine's (UT Southwestern, Dallas, TX, USA) PPG. P. Harbut received an unconditional loan of the equipment used in this study from the Djurgården Hockey Club, Stockholm. No other author has a conflict of interest to declare.
Support statement: We would like to thank Djurgården IF Hockey in Stockholm, Sweden for the loan of the equipment used in this study and Vyaire Medical Pty Ltd for data collection software support. A. Malhotra is funded by the National Institutes of Health.
References
- 1.Chew MS, Blixt PJ, Ahman R, et al. National outcomes and characteristics of patients admitted to Swedish intensive care units for COVID-19: a registry-based cohort study. Eur J Anaesthesiol 2021; 38: 335–343. doi: 10.1097/EJA.0000000000001459 [DOI] [PubMed] [Google Scholar]
- 2.Gattinoni L, Coppola S, Cressoni M, et al. COVID-19 does not lead to a “typical” acute respiratory distress syndrome. Am J Respir Crit Care Med 2020; 201: 1299–1300. doi: 10.1164/rccm.202003-0817LE [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ottestad W, Sovik S. COVID-19 patients with respiratory failure: what can we learn from aviation medicine? Br J Anaesth 2020; 125: e280–e281. doi: 10.1016/j.bja.2020.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meng H, Xiong R, He R, et al. CT imaging and clinical course of asymptomatic cases with COVID-19 pneumonia at admission in Wuhan, China. J Infect 2020; 81: e33–e39. doi: 10.1016/j.jinf.2020.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gattinoni L, Chiumello D, Caironi P, et al. COVID-19 pneumonia: different respiratory treatments for different phenotypes? Intensive Care Med 2020; 46: 1099–1102. doi: 10.1007/s00134-020-06033-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kairaitis K, Harbut P, Hedenstierna G, et al. Ventilation is not depressed in patients with hypoxemia and acute COVID-19 infection. Am J Respir Crit Care Med 2022; 205: 1119–1120. doi: 10.1164/rccm.202109-2025LE [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med 2020; 383: 120–128. doi: 10.1056/NEJMoa2015432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leisman DE, Deutschman CS, Legrand M. Facing COVID-19 in the ICU: vascular dysfunction, thrombosis, and dysregulated inflammation. Intensive Care Med 2020; 46: 1105–1108. doi: 10.1007/s00134-020-06059-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Magro C, Mulvey JJ, Berlin D, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res 2020; 220: 1–13. doi: 10.1016/j.trsl.2020.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wagner PD, Malhotra A, Prisk GK. Using pulmonary gas exchange to estimate shunt and deadspace in lung disease: theoretical approach and practical basis. J Appl Physiol 2022; 132: 1104–1113. doi: 10.1152/japplphysiol.00621.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bradley AF, Severinghaus JW, Stupfel M. Effect of temperature on Pco2 and Po2 of blood in vitro. J Appl Physiol 1956; 9: 201–204. doi: 10.1152/jappl.1956.9.2.201 [DOI] [PubMed] [Google Scholar]
- 12.Antoine C. Tension des vapeurs: nouvelle relation entre les tension et les temperatures. Comptes Rendus 1888; 107: 681–684, 778–780, 836–837. [Google Scholar]
- 13.Riley RL, Cournand A. ‘Ideal’ alveolar air and the analysis of ventilation–perfusion relationships in the lungs. J Appl Physiol 1949; 1: 825–847. doi: 10.1152/jappl.1949.1.12.825 [DOI] [PubMed] [Google Scholar]
- 14.Astrand PO, Cuddy TE, Saltin B, et al. Cardiac output during submaximal and maximal work. J Appl Physiol 1964; 19: 268–274. doi: 10.1152/jappl.1964.19.2.268 [DOI] [PubMed] [Google Scholar]
- 15.Rowell LB. Human Cardiovascular Control. New York, Oxford University Press, 1993. [Google Scholar]
- 16.West JB. Ventilation–perfusion inequality and overall gas exchange in computer models of the lung. Respir Physiol 1969; 7: 88–110. doi: 10.1016/0034-5687(69)90071-1 [DOI] [PubMed] [Google Scholar]
- 17.Wagner PD, Hedenstierna G, Bylin G. Ventilation–perfusion inequality in chronic asthma. Am Rev Respir Dis 1987; 136: 605–612. doi: 10.1164/ajrccm/136.3.605 [DOI] [PubMed] [Google Scholar]
- 18.Dantzker DR, Brook CJ, Dehart P, et al. Ventilation–perfusion distributions in the adult respiratory distress syndrome. Am Rev Respir Dis 1979; 120: 1039–1052. [DOI] [PubMed] [Google Scholar]
- 19.Herrmann J, Mori V, Bates JHT, et al. Modeling lung perfusion abnormalities to explain early COVID-19 hypoxemia. Nat Commun 2020; 11: 4883. doi: 10.1038/s41467-020-18672-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian S, Hu W, Niu L, et al. Pulmonary pathology of early-phase 2019 novel coronavirus (COVID-19) pneumonia in two patients with lung cancer. J Thorac Oncol 2020; 15: 700–704. doi: 10.1016/j.jtho.2020.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kianzad A, Meijboom LJ, Nossent EJ, et al. COVID-19: histopathological correlates of imaging patterns on chest computed tomography. Respirology 2021; 26: 869–877. doi: 10.1111/resp.14101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Price LC, Ridge C, Wells AU. Pulmonary vascular involvement in COVID-19 pneumonitis: is this the first and final insult? Respirology 2021; 26: 832–834. doi: 10.1111/resp.14123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katsoularis I, Fonseca-Rodriguez O, Farrington P, et al. Risks of deep vein thrombosis, pulmonary embolism, and bleeding after Covid-19: nationwide self-controlled cases series and matched cohort study. BMJ 2022; 377: e069590. doi: 10.1136/bmj-2021-069590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.The ATTACC, ACTIV-4a and REMAP-CAP Investigators . Therapeutic anticoagulation with heparin in noncritically ill patients with Covid-19. N Engl J Med 2021; 385: 790–802. doi: 10.1056/NEJMoa2105911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.The REMAP-CAP, ACTIV-4a and ATTACC Investigators . Therapeutic anticoagulation with heparin in critically ill patients with Covid-19. N Engl J Med 2021: 385: 777–789. doi: 10.1056/NEJMoa2103417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Radermacher P, Herigault R, Teisseire B, et al. Low VA/Q areas: arterial-alveolar N2 difference and multiple inert gas elimination technique. J Appl Physiol 1988; 64: 2224–2229. doi: 10.1152/jappl.1988.64.5.2224 [DOI] [PubMed] [Google Scholar]
- 27.Hopkins SR. Functional magnetic resonance imaging of the lung: a physiological perspective. J Thorac Imaging 2004; 19: 228–234. doi: 10.1097/01.rti.0000142839.37482.c2 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This one-page PDF can be shared freely online.
Shareable PDF ERJ-01117-2022.Shareable (237.4KB, pdf)