

Abstract
Chlamydia trachomatis is the leading pathogen in sexually transmitted bacterial infections across the globe. The development of a selective treatment against this pathogen could be an attractive therapeutic option that will reduce the overuse of broad-spectrum antibiotics. Previously, we reported some sulfonylpyridine-based compounds that showed selectivity against C. trachomatis. Here, we describe a set of related compounds that display enhanced anti-chlamydial potency when compared to our early leads. We found that the active molecules are bactericidal and have no impact on Staphylococcus aureus or Escherichia coli strains. Importantly, the molecules were not toxic to mammalian cells. Furthermore, a combination of molecule 20 (the most active molecule) and azithromycin at subinhibitory concentrations acted synergistically to inhibit chlamydial growth. Molecule 20 also eradicated Chlamydia in a 3D infection model and accelerated the recovery of Chlamydia-infected mice. This work presents compounds that could be further developed to be used alone or in combination with existing treatment regimens against chlamydial infections.
Keywords: Chlamydia trachomatis, sexually transmitted diseases, 3D culture
Graphical Abstract
INTRODUCTION
Sexually transmitted infections (STIs) are a public health concern, particularly in women, leading to mild and acute health issues that may affect the reproductive system.1,2 According to the World Health Organization (WHO), more than 1 million cases of STIs are reported worldwide every day.3 The 2018 CDC surveillance report shows that ~2.5 million bacterial STIs occur every year in the United States of America.4 These are caused primarily by three bacteria: Chlamydia trachomatis (Ctr), Neisseria gonorrhoeae, and Treponema pallidum (syphilis).5 Ctr is the most common cause of STIs with ~1.8 million reported cases in 2018.6 Furthermore, the WHO estimates that Ctr infections reached 131 million cases globally in the last decade.7 However, given the asymptomatic nature of approximately 70% of the infections, these numbers are likely underrepresented.1
Untreated Ctr infections can lead to long-term sequelae within the reproductive tract with the concomitant increase in Ctr-associated morbidities.8 One such problem is pelvic inflammatory disease, which contributes to ectopic pregnancy.9–11 Furthermore, Ctr can scar the fallopian tubes, ovaries, and endometrial lining, resulting in tubal factor infertility. Also, infection recurrence is common in Ctr, and it is associated with severe complications.9
Ctr is an obligate intracellular bacterium that undergoes a biphasic developmental cycle that alternates between two forms: the elementary body (EB) and the reticulate body (RB).9,12 The EB is the smaller (~0.3 μm diameter) and non-dividing form of the pathogen, which binds to and initiates the infection of the host cell.9,12 The RB is the larger (~1 μm diameter), non-infectious, and replicative form that develops within the host cell in a hybrid vacuole composed of host and bacteria-derived components (termed an inclusion).12 At an unspecified signal, and after multiple rounds of replication, the RBs asynchronously undergo secondary differentiation into EBs, followed by the lysis of the inclusion releasing the bacteria, which infects proximal cells. Most urogenital Chlamydia isolates complete their developmental cycle in 48 to 60 h.
Although a vaccine may be available in the future, such intervention has not yet been approved for use in humans.13–16 Moreover, the efficacy of available treatments, such as azithromycin (AZM) and doxycycline (Doxy), has been recently questioned.17,18 Some studies have shown infection recurrence 28 d after treatment in 10–15% of female patients.19–22 Furthermore, AZM and Doxy can impact the vaginal microbiota, altering its protective function while facilitating the development of antibiotic resistance in other bacterial species.23–28 Also, the widespread utilization of AZM in some countries contributes to macrolide resistance in subsequent or accompanied infections.29,30 Thus, developing new therapies specifically targeting Ctr represents a useful strategy to reduce the pathogen’s prevalence.
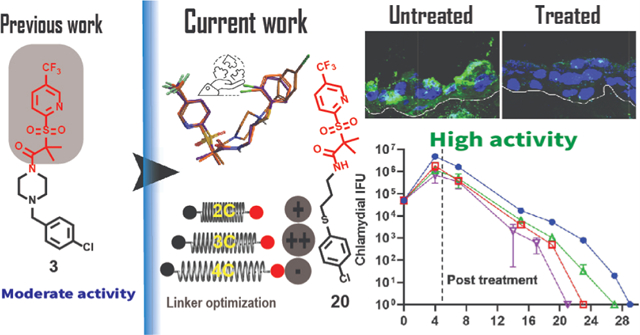
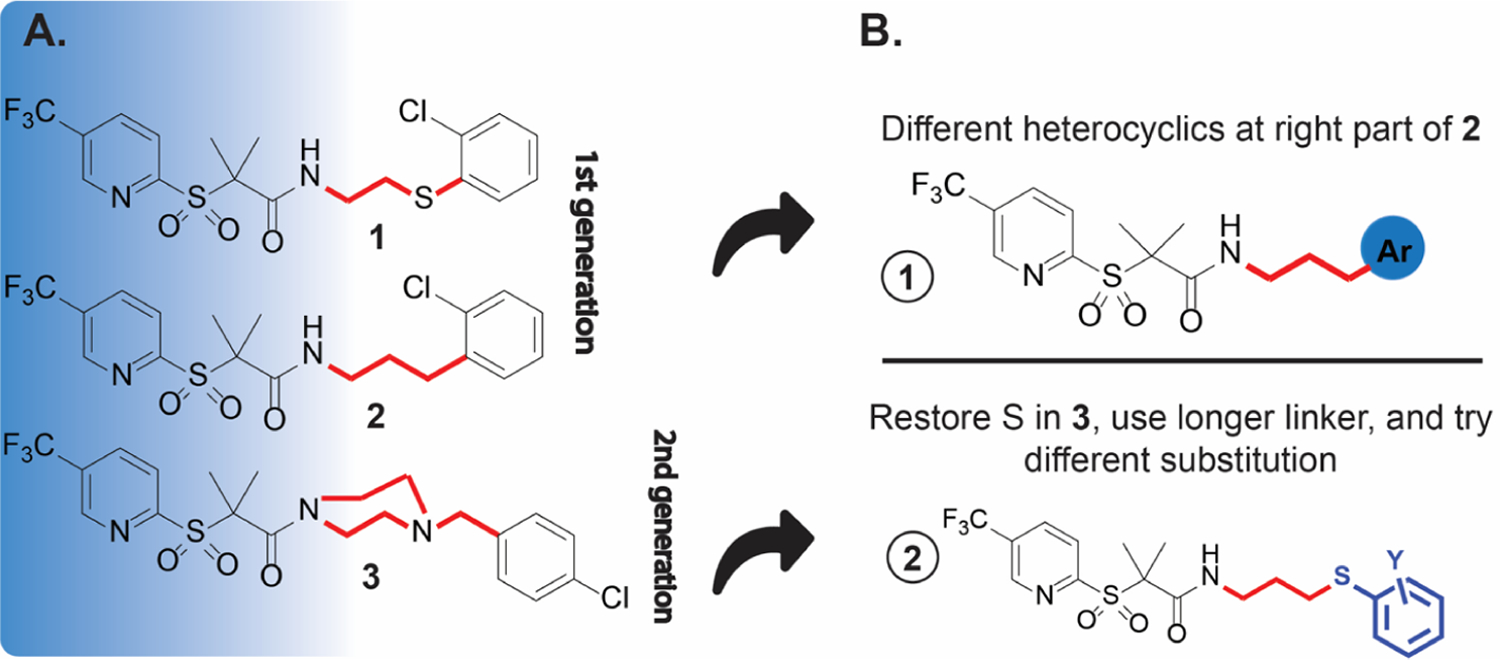
Selective anti-Ctr therapy is a cornerstone to overcome the undesired impact of broad-spectrum antibiotics on commensal flora and to prevent the potential transfer of resistance. In addition, it may block immune escape of the pathogen, which might be a contributing factor in reported treatment failures.1 The unique developmental cycle of Ctr affords an opportunity to create specific treatments against this bacterium instead of broad-spectrum antibiotics.12 Based on the current understanding of the Ctr cycle, several effectors are considered vital targets for treatment intervention. We hypothesize that disrupting the protein turnover process would significantly affect the Ctr development. Previously, we reported the anti-chlamydial activity of sulfonylpyridine derivatives.31,32 Compound 1 (Figure 1A) completely eradicated C. trachomatis at 50 μg/mL, possibly by impacting the chlamydial caseinolytic protease (ClpP) machinery system.31 Meanwhile, derivatives such as 2, which possess a carbon instead of a sulfur atom in the connecting chain, were less potent. We prepared a library of molecules to develop structure–activity relationships (SAR). One of those molecules, compound 3 (Figure 1A), showed good activity at 50 μg/mL with an IC50 of 5.8 μg/mL.33 Even though compound 3 lacks a thioether group, it showed better activity than compound 2 (but less than 1). A comparison between the three molecules shows that 3 has a longer linker than 1 and 2 (Figure 1A) and a chlorine atom in the para position. Although all three compounds shared a similar moiety at the right side, only 1 possesses a thioether linker.
Figure 1.
(A) Chemical structure of lead compounds 1–2, and the most active compound 3 in the second generation; (B) approaches to improving the activity of this scaffold of compounds.
Our previous work suggested that the 3-(trifluoromethyl)-pyridine, the gem dimethyl, and the sulfonyl groups are beneficial for activity (blue shadow, Figure 1A). In this work, we sought to explore how the presence of heterocycles (see 1, Figure 1B), the length of the alkyl amide linker, and the position of the sulfur and chloro atoms (see 2, Figure 1B) affects anti-chlamydial activity. We synthesized 18 compounds carrying the described modifications. One of the prepared molecules (20) was twofold more active than the lead compound 1. The IC50 value of 20 was lower than the previous two generations of this scaffold. A combination of 20 with AZM increased Ctr sensitivity to both drugs. The compound showed activity on a three-dimensional (3D) stratified epithelium cell culture model, which mimics the lower genital tract where Chlamydia first interacts with epithelial cells.34 In addition, the active compounds showed good Ctr inhibition activity in a mouse model. These results suggest that the sulfonylpyridines are promising molecules that can be further developed as anti-chlamydial agents.
RESULTS AND DISCUSSION
Molecular Design.
To rationalize our planned modifications, we performed ligand alignment of the three reference molecules35 (1–3) and evaluated their 3D similarity with some of the designed compounds (Figure 2), utilizing Forge V10 (Cresset Inc., UK). The similarity score parameter relies on calculating specific field points (surface and electrostatic characters) of the tested molecules to compare their chemical structures.36 Figure 2A shows that compounds 2 and 3 possess varying degrees (0.916 vs 0.692) of alignment with the lead compound 1, indicating a high to moderate degree of similarity. Given that 3 was more active than 2, we speculated that either (i) a longer linker could improve the anti-chlamydial activity or (ii) the heterocyclic ring at the right side may provide some hydrogen bonding capabilities that translated into better activity. Thus, we designed two categories of compounds to cover the chemical space. The first set (Figure 1B, top) is like 2 but with heterocycles instead of the o-chlorophenyl group. The second set (Figure 1B, bottom) was built as a blend of compounds 1 and 3 by keeping the same alkyl chain length (three carbons) of 3 and retaining the sulfur atom of 1. The 3D alignment representatives of the new derivatives with the parent compounds (Figure 2B–D) revealed a high superimposition and optimal structure overlap. In addition, compound 20 shows near identical alignment with 1 (Figure 2C) and 3 (Figure 2D).
Figure 2.
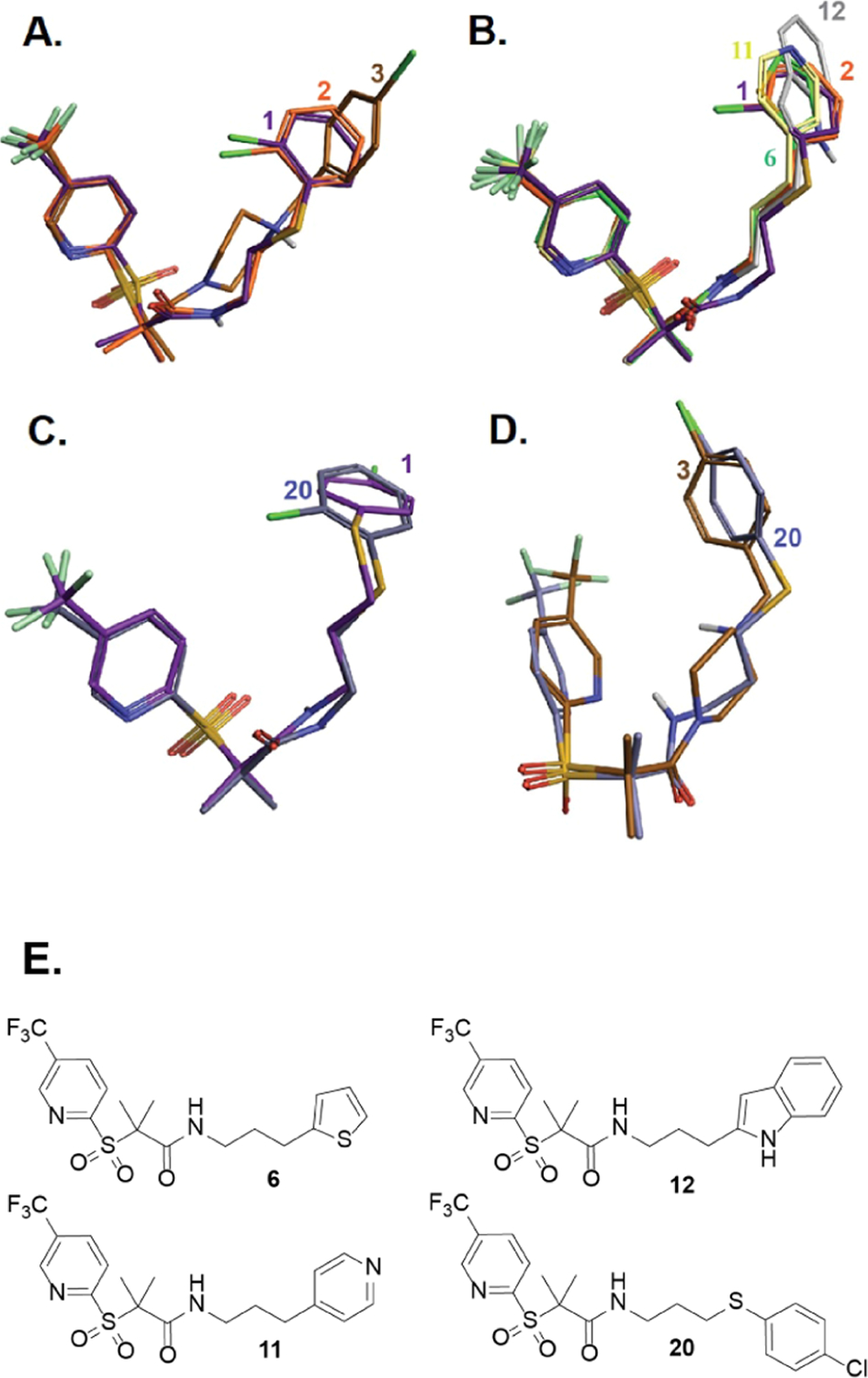
Field alignment of representative derivatives of the new generation with the three parent molecules; colored code and 2D structure are provided for each compound.
Chemistry.
Derivatives 5–13, containing heterocyclic rings, were prepared by reacting the carboxylic acid of 433 with different amines (Scheme 1), as described in the experimental section and in the Supporting Information. Compounds 5–7 carry pyrrolidine, furan, and thiophene rings. Compounds 8–9 possess distinct pyrazoles, enabling us to assess the importance of the hydrogen bonding position in activity (Table 1). To further understand the role of hydrogen bonding, we made compounds 10–11 (bearing pyridine groups) and 12–13 (possessing indole and benzimidazole rings).
Scheme 1.
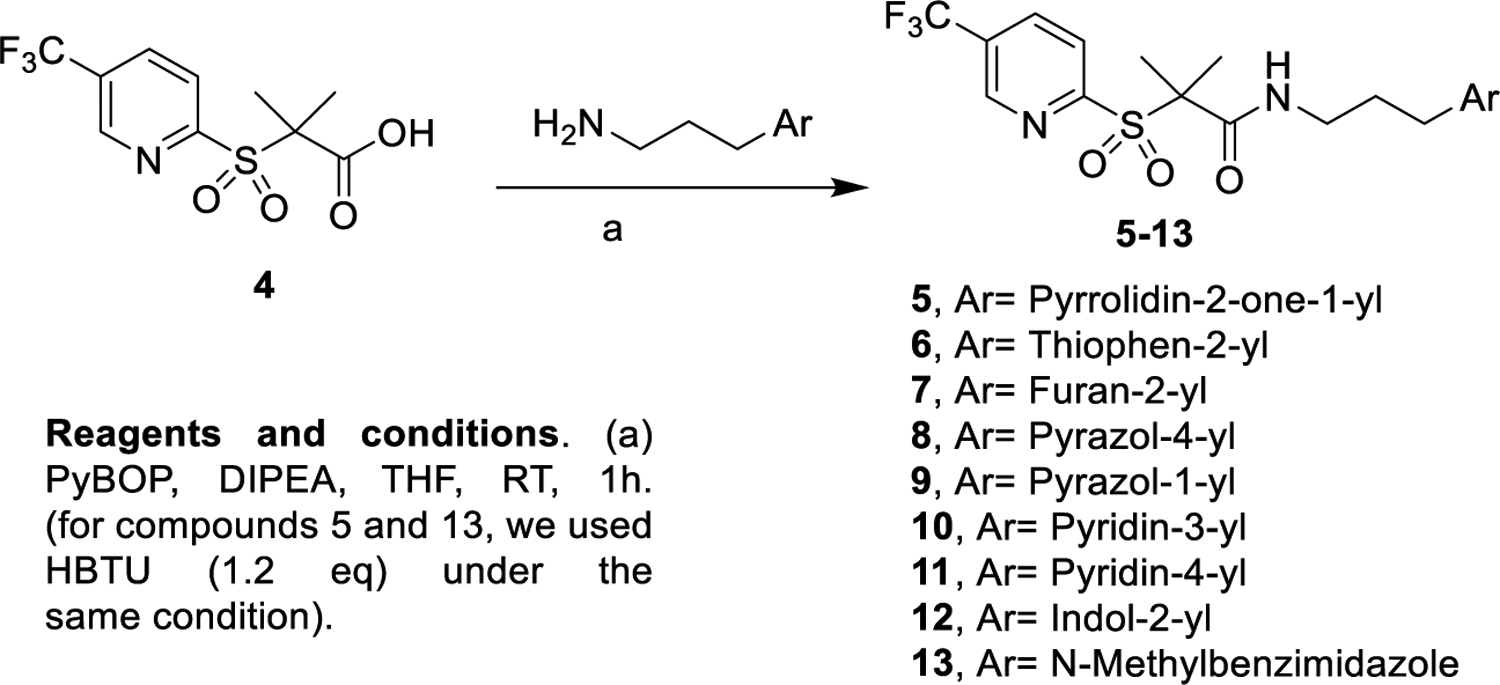
Synthesis of Derivatives with Different Heterocyclics at the Right Part44
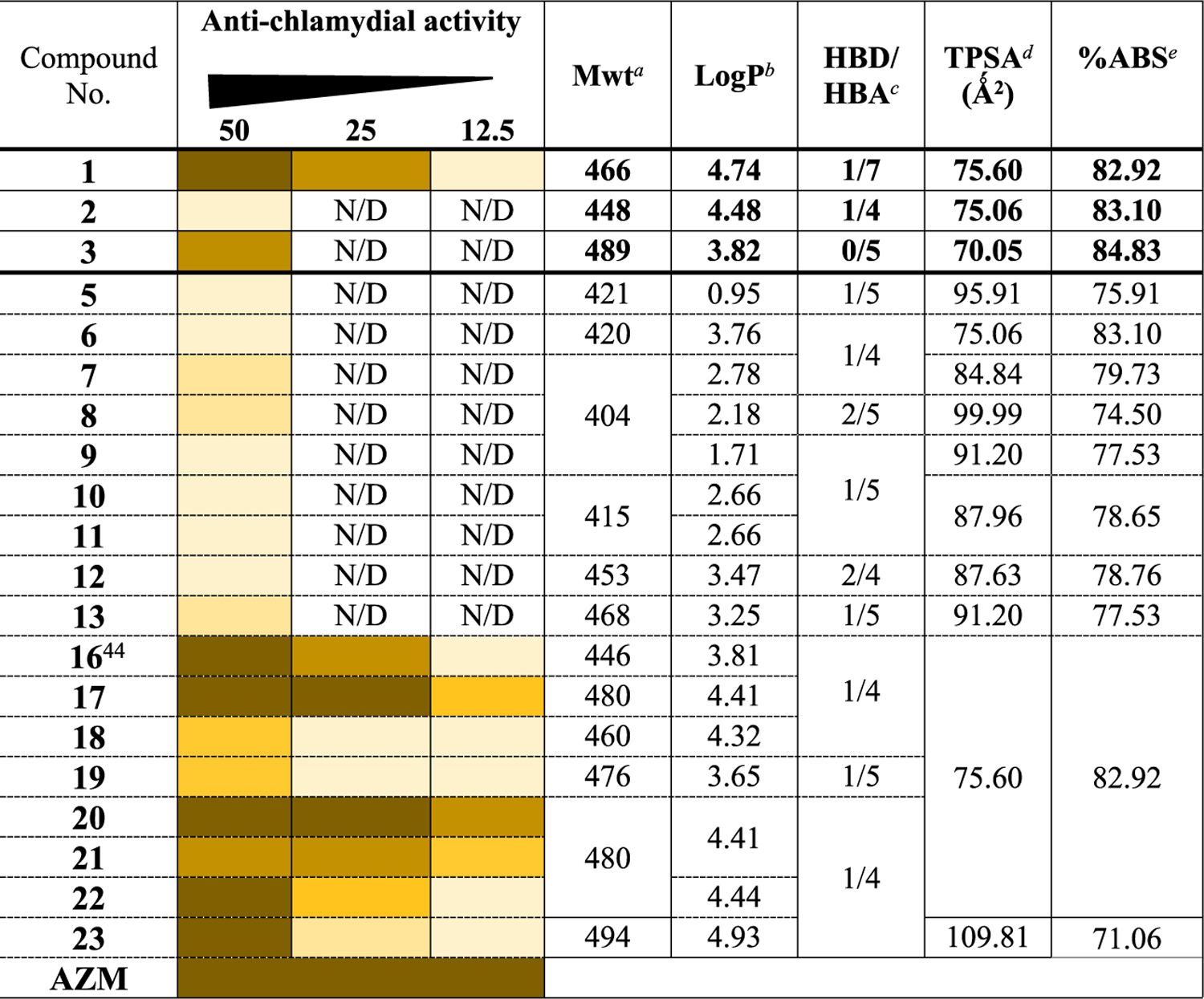
Table 1.
Initial Anti-chlamydial Activity Screening of the New Derivatives against C. trachomatis (Serovar LGV-L2)
|
The second set of modifications possesses diverse substitution patterns and linker size. Molecules 16–24 were obtained by coupling 4 with the appropriate amines 15a–i, as shown in Scheme 2. The amine precursors 15b–g and i were prepared by reacting the N-Boc-protected alkylamines 14a and 14b with the corresponding arylthiols, followed by deprotection under acidic conditions to afford the desired free amines (Scheme 2). On the other hand, molecule 15h was synthesized from 1,4-dibromobutane, as reported (Schemes S1 and S2).37,38 The goal was to understand the effect of substituents at the ortho position on the phenyl group and the effect of translocating the chlorine atom to the para (20) or meta (21) positions. Furthermore, we synthesized 22 with a thioether linker to evaluate the effect of the sulfur atom position; 23 to investigate how linker length affects biological action.
Scheme 2.
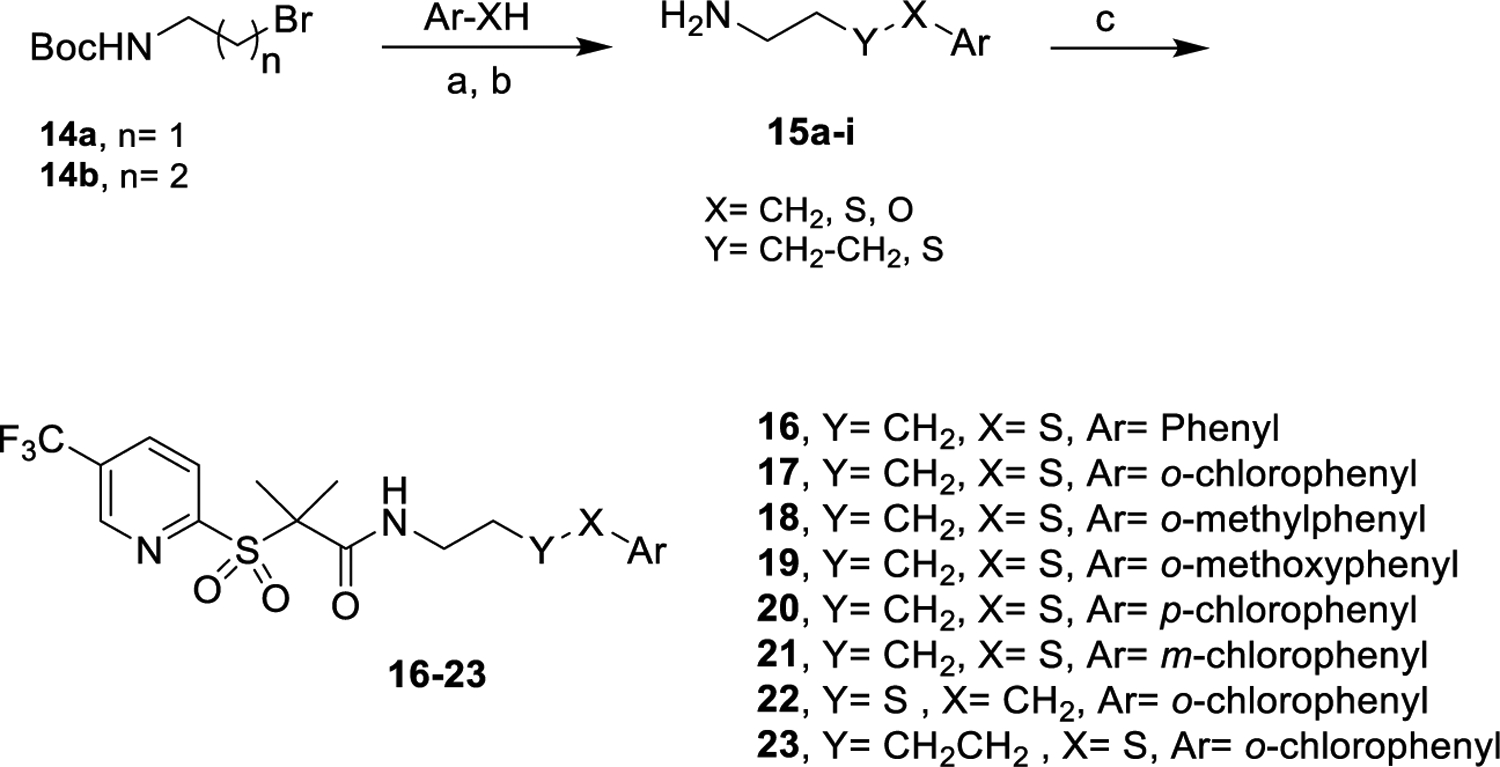
Synthesis of Derivatives with the Longer Alkyl Chain
Drug-Likeness Properties.
We calculated some physicochemical parameters of the compounds (Table 1) to evaluate their drug-likeness.39 All the molecules satisfied the Lipinski and Veber rules.40–42 Their total polar surface area is in the range (60–140) for good intestinal absorption.40 All the compounds are predicted to fall in the accepted range (71–100) for oral bioavailability with minimum blood-brain barrier penetration.43 The % absorption (ABS) of the compounds was 74–81, suggesting high membrane permeability. Finally, all the derivatives are expected to possess moderate to good water solubility, as indicated by their estimated log S values (Table S1). These data indicate the potential of the scaffolds as a starting point to develop anti-chlamydial drugs.
Biological Evaluation.
Anti-chlamydial Investigation.
Due to Chlamydia’s requirement of a host cell to grow,12 we utilized two assays to assess the anti-chlamydial action of the compounds. First, we used an indirect immunofluorescence assay (IFA) to determine the presence of Ctr inclusions. In the assay, we measured the initial potency of the compounds and its impact on the number and size of bacterial inclusions. Then, we used the inclusion-forming unit (IFU) assay to test the infectious progeny generated during the initial primary infection. Here, we harvested infected cells from a primary infection to quantify the IFU per milliliter from infected cultures treated with the tested compounds.
Immunofluorescence Assay (IFA).
We tested the anti-chlamydial activity of the compounds (50 μg/mL, added 6 h post-infection; hpi) against C. trachomatis serovar L2 by analyzing the number and size of Ctr inclusions.31,33 As negative and positive controls, we used media (untreated sample) and azithromycin (2 μg/mL), respectively. Compounds 5–13 did not show meaningful activity at the tested concentration (<50% bacterial inhibition, Table 1), as seen under the microscope. The lack of activity indicated the importance of the arylthiol group on the right part of the molecule. We tested the second set of compounds, which has the arylthiol moiety. Compound 16 (with an unsubstituted phenyl group) and compounds 17 and 20 (with o or p chloro substitutions) presented similar activity to the lead compound 1 at 50 μg/mL. Compounds 18 and 19, carrying methyl and methoxy groups at the o position, showed moderate activity. The m-chloro substitution (21) negatively impacts the anti-chlamydial activity when compared with its isomers (17 and 20). Compound 22, with an “out of place” sulfur, showed 100% inhibition at the tested concentration. Compound 23, with a 4-carbon length linker, did not show improvement in activity when compared to 1 and 17 (shorter linkers). To determine the minimum inhibitory concentration (MIC), we studied the most potent analogues, 16–23, at lower concentrations (25 and 12.5 μg/mL) (Table 1). Bacterial inclusion analysis revealed that compounds 17, 20, and 21 displayed better anti-chlamydial activity than 1, with 20 being the most active. To ensure the activity of compound 20 was not due to a cytotoxic effect, XTT cell proliferation assay was performed at 25 and 50 μg/mL. Compound 20 showed no toxicity toward HEp-2 cells after 48 h of incubation (Figure S1).
IFU Assay to Determine the Effective Inhibitory Concentration.
We next utilized the IFU assay to quantify the production of Ctr infectious progeny (i.e., EBs) from the cultures treated with the tested compounds. This experiment also allowed us to determine the minimum cidal concentration (MCC, the minimum value at which reinfection is not observed). HEp-2 cells were infected and treated with the most active compounds at 50, 25, and 12.5 μg/mL at 6 hpi. The experiment was stopped at 24 hpi, and the cells were harvested. Then, we took the cell lysates (without additional treatment), reinfected a fresh monolayer of HEp-2 cells with them, and counted the number of IFUs in the secondary infection after 24 h (Figure 3).33,45
Figure 3.
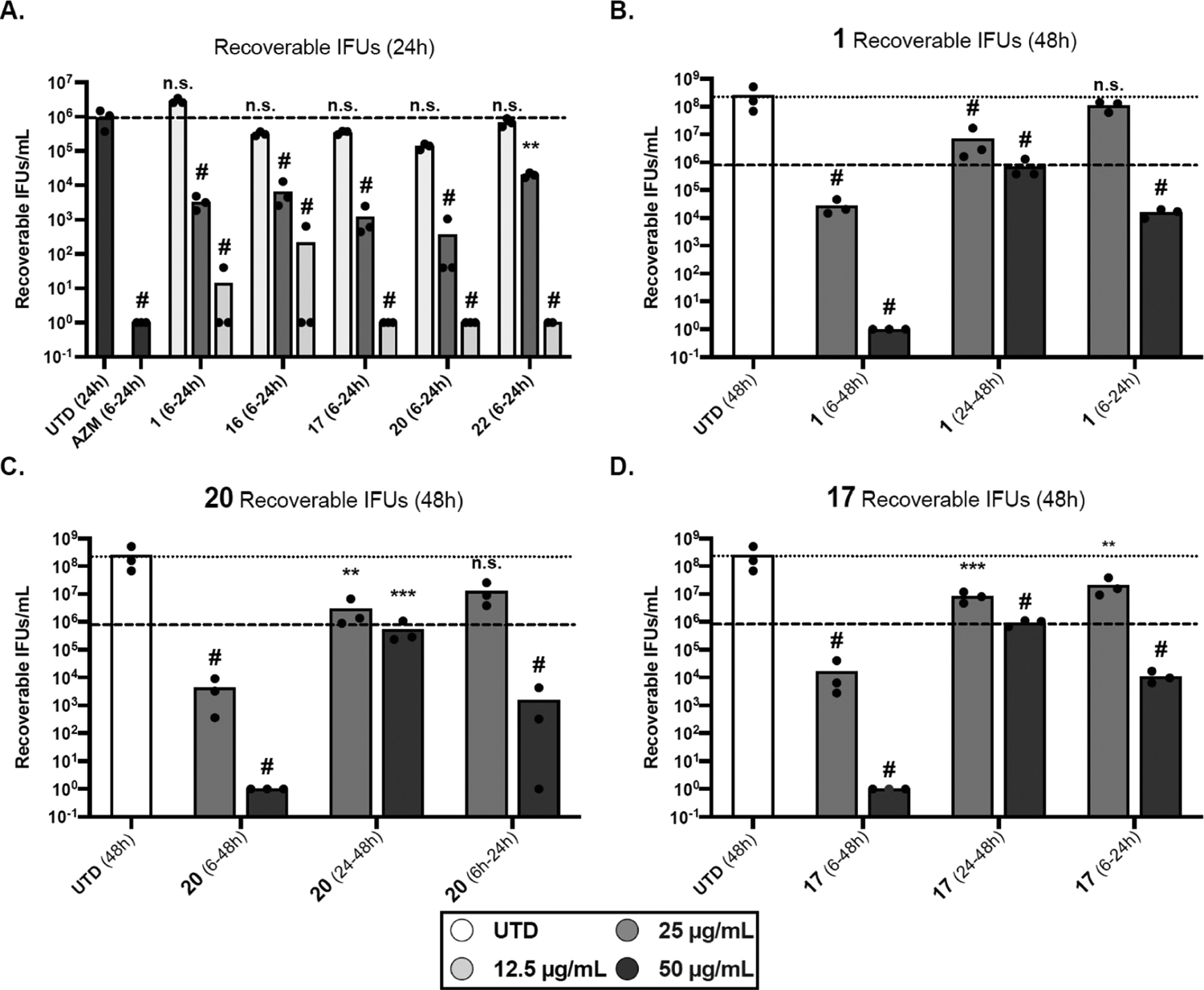
Quantification of infectious progeny yield in the presence of selected derivatives (numbered), as compared to compound 1 and untreated (UTD) control at 24 h (A) and 48 h (B–D) post-infection. Timeframe of treatment is indicated in parentheses. The results are reported on a log10 scale. Symbols indicate individual replicates. The dashed line indicates the average of the 24 h untreated sample, and the dotted line indicates the average of the 48 h untreated sample (same untreated samples for all drug treatments). Data represent three biological replicates. All values were log10 transformed to achieve equal distribution prior to statistical analysis. For each graph, transformed values were analyzed by ordinary two-way analysis of variance (ANOVA) with Dunnet’s post hoc multiple comparisons test. Significance values for each sample compared to the untreated control are shown on the graph. (A) # = p < 0.0001, n.s. = not significant. (B–D) ** = p < 0.01, *** = p < 0.001, and # = p < 0.0001.
As seen in Figure 3A, compounds 17, 20, and 22 blocked Ctr development at 50 μg/mL when cultures were treated at 6 hpi and harvested at 24 hpi. The lead molecule 1 and compound 16 showed a decrease in the bacterial colonies of 5-log10 units at the same concentration. In addition, compounds 17 and 20 were active at 25 μg/mL, showing a roughly 1-log10 decrease in Ctr growth when compared with 1, 16, and 22 (which themselves showed >2-log10 decrease, as compared to UTD). 16, 17, and 20 were moderately active at 12.5 μg/mL, showing ~50–70% inhibition of growth. Conversely, 1 and 22 showed inclusion yield comparable to the untreated sample at that concentration. This observation highlighted the fundamental role of the thiol group position on the right side of the molecule. Immunofluorescence analysis of inclusions (Figure S2A) revealed that both 17 and 20 inhibited the infection at the selected concentrations compared to compound 16. Zoomed images of 1, 17, and 20 (Figure S2B) showed that, at 25 μg/mL, the cells treated with 20 presented minuscule inclusions in comparison with 1 and 17. It was clearly observed that treatment of the infected cells with 20 highly impacted the EB formation. These results are consistent with the IFA work.
Prolonged Treatment to Determine Cidal or Static Mechanism.
Next, we determined at what point the compounds inhibited the developmental cycle of Chlamydia. During this cycle, which lasts 48–60 h for Ctr, the pathogen alternates between the infectious form (EBs) and the replicative form (RBs). Hence, we assessed the long-term inhibition of Ctr caused by 17 and 20 and compared them to 1 at different time points (Figure 3B–D). The compounds were added or removed at a specific timeframe (indicated in parentheses), and the infection yield was calculated to be 48 hpi. We monitored the treated cultures by immunofluorescence microscopy at 10 μm scale at 25 and 50 μg/mL to evaluate the effect of our treatments on inclusion size and morphology as these generally correlate to the number of bacteria (Figure 4).
Figure 4.
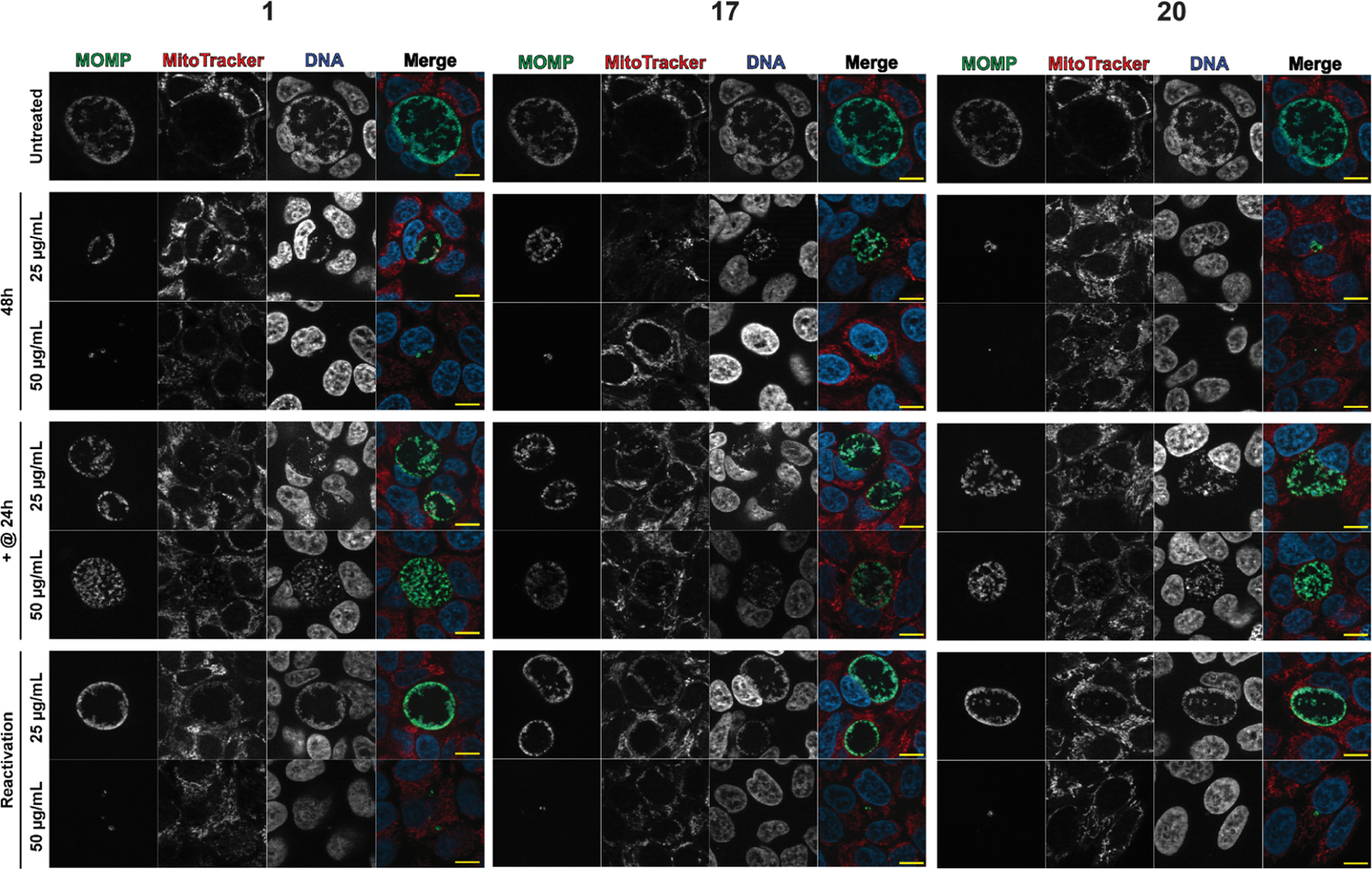
Indirect IFA of 1, 17, and 20 at 50 and 25 μg/mL at 48 hpi. In the upper panels (48 h), cells were treated at 6 hpi, and drug-containing media was maintained throughout the experiment. For the tests for inhibiting developmental cycle progression, the cells were treated with the drugs at 24 hpi (+@24 h). The media was maintained throughout the remainder of the experiment. In the reactivation panels, the cells were treated from 6 to 24hpi, at which point the drug-containing medium was removed and replaced with a drug-free medium. Samples were stained for Ctr L2 using antibodies against the major outer membrane protein (MOMP; green), for mitochondria using MitoTracker (red) and for DNA using DAPI (blue). Scale bar = 10 μm.
We observed that 20 severely attenuated the number of recoverable IFUs following prolonged treatment (6 to 48 hpi, Figure 3C). Complete eradication was observed at 50 μg/mL and a 5-log10 units decrease at 25 μg/mL. Meanwhile, 1 and 17 exhibited complete eradication at 50 μg/mL and reduced activity (effect) at 25 μg/mL, with a 4-log10 units decrease during the same time period (Figure 3B,D). Although the three compounds blocked infectious progeny production at 50 μg/mL, 20 was more active (1-log10 unit) than 1 and 17 at 25 μg/mL at 48 hpi. Immunofluorescence analysis showed that treatment with the molecules for 48 h severely reduced the inclusion size (Figure 4, “48 h”). At 50 μg/mL, inclusions were rarely visible under the effect of our compounds. On the other hand, at 25 μg/mL, 20 exhibited a severe reduction in inclusion sizes in comparison with the other two molecules (see Figure 4, 48 h section). Based on these data, we concluded that compound 20 showed a superior effect among the tested compounds.
Next, we sought to determine the effect of the compounds on preformed EBs [Figure 3B–D, (24–48)]. Infected cells were treated with the molecules at 24 hpi (+@24 h), after a population of EBs has been formed, and incubated for an additional 24 h (total 48 h). We found that the tested compounds led to the rapid growth arrest of Ctr but failed to reduce the titer of infectious progeny already present, as indicated by IFU calculations and immunofluorescence images (Figure 4 “+@24”). The tested compounds showed an IFU yield on par with the 24 h untreated sample and ~1 log10 less than the 48 h untreated sample. Microscopy images (Figure 4 “+@24”) revealed that the tested compounds had little impact on inclusion size in comparison with the untreated control. At both 50 and 25 μg/mL, the tested molecules failed to impact the inclusion size and morphology (see +@24 section). The infection output from the six samples was approximately comparable to the untreated control. This result is similar to what is observed with AZM and Doxy (Figure S3). EBs enter the cell and differentiate into RBs, which then differentiate into EBs; the results from the first two time points seem to indicate that the compounds affect the RBs but not the EBs present when the compounds are introduced. We theorize that these compounds will stop developmental cycle progression (like AZM and Doxy) by inhibiting RB development, with no effect on the metabolically inactive EBs.
Next, we studied whether these compounds are bactericidal or bacteriostatic by allowing a 24 h recovery phase. We utilized the reactivation assay to assess the ability of Chlamydia to recover after treatment withdrawal. Following treatment with 1, 17, or 20 from 6 to 24 h, the drug-containing media was removed, and the samples incubated for an additional 24 h in the absence of the compounds. Although samples treated with 1 recovered to near untreated IFU levels with the 25 μg/mL treatment, both 17 and 20 remained approximately 1-log10 lower, indicating that these inhibitors blocked development more effectively [Figure 3B–D, (24–48)]. This suggests a bacteriostatic effect at 25 μg/mL. Immunofluorescence images of the samples from this concentration showed inclusions of similar size to the untreated ones (see Figure 4, reactivation section at 25 μg/mL).
Conversely, the 50 μg/mL treatment demonstrated a severe attenuation of recoverable infectious progeny, with 20 showing the highest efficacy. In addition, the three compounds showed a dramatic reduction in inclusion sizes, as demonstrated in Figure 4 (reactivation section at 50 μg/mL). Given the severe reduction in IFUs at 50 μg/mL, which was accompanied by an alteration of the inclusion morphology and size, we hypothesized that this concentration is likely cidal. However, we cannot rule out that the residual compound in the sample collected for IFU assay (despite numerous rinses) results in the observed effect; thus, further studies of intracellular drug concentrations may be warranted. The data from prolonged treatment, reactivation, and pre-formed EB indicate that 20 is the most effective of our compounds at blocking Ctr growth and development.
Dose–Response Effect.
The dose–response impact of 1, 17, and 20 on Ctr growth was calculated in a range of concentrations (from 2× to ×/16 the MCC) using IFU assay (Figure 5). The IC50 value of the three molecules, calculated based on the recovered infectious progeny at the indicated concentrations, was 1 = 6.49 μg/mL, 17 = 2.58 μg/mL, and 20 = 1.76 μg/mL. Microscopy of 20 showed a reduction in the number of Ctr inclusions with varying activities in a dose–response manner (Figure S4).
Figure 5.
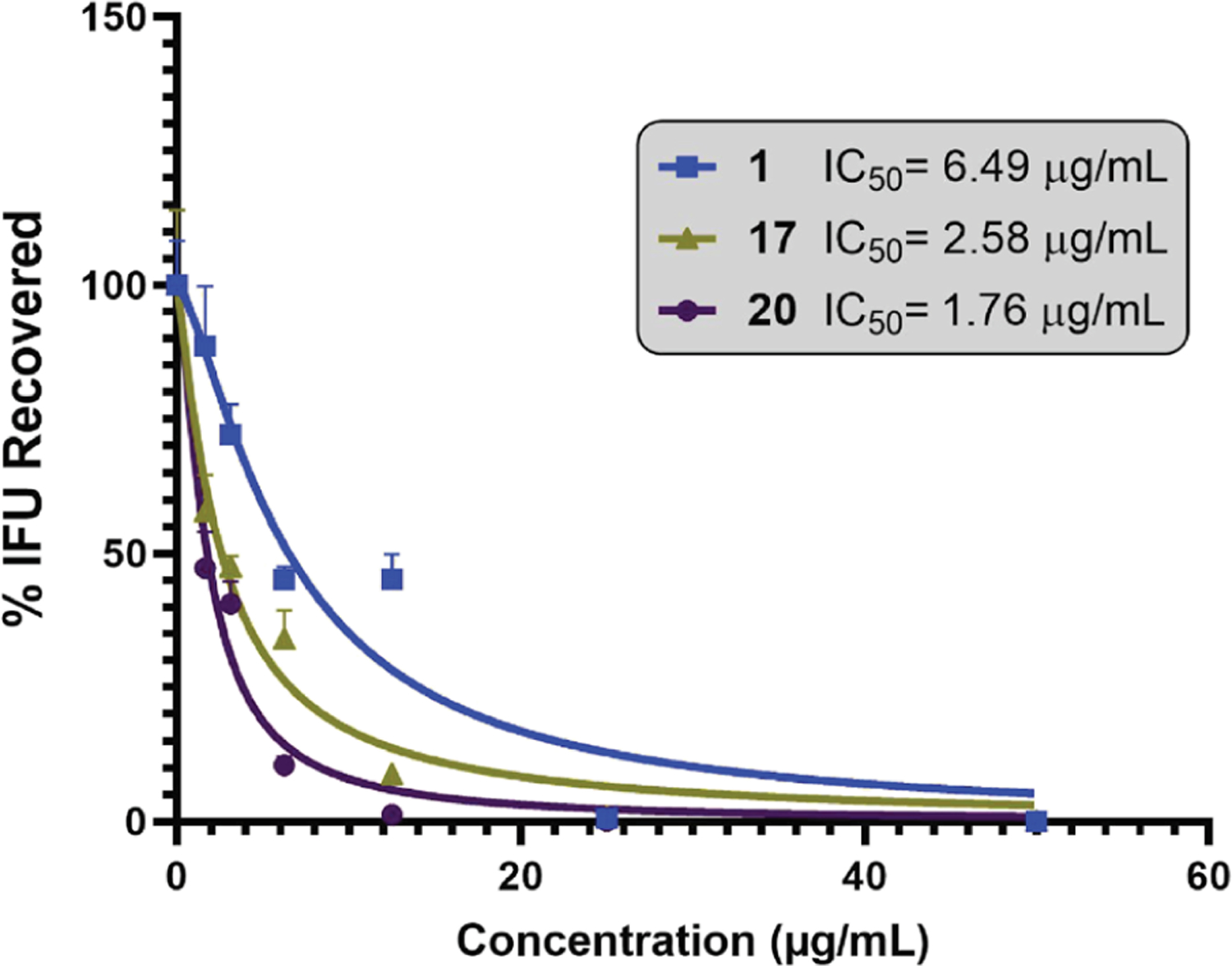
Dose–response curve for the effect of 17 and 20 in comparison with 1 in twofold dilutions starting from the common effective dose of the three derivatives. Data represent two biological replicates [significant difference (P < 0.05, ANOVA)].
Other Antimicrobial Activity.
We then tested 1, 17, and 20 against Escherichia coli K12 and Staphylococcus aureus JE 2. Only compound 20 was able to attenuate E. coli growth at 256 μg/mL (highest tested concentration), while the other molecules did not show any activity up to 256 μg/mL (Figure S5). Based on the current data and the inactivity of our previous generation of compounds against a wide panel of pathogens, we speculated that our compounds might be selective for Chlamydia.
Combination of 20 with Azithromycin.
As discussed, treatment failure in Ctr has been reported.46–48 Two potential issues of C. trachomatis treatments (include AZM and Doxy) are their lack of penetration into certain sites of infection49 and their direct impact on the natural microflora.50,51 Furthermore, AZM may cause adverse effects such as diarrhea and abdominal pain.52 The mass distribution of broad-spectrum antibiotics for trachoma control has led to resistance development by other bacterial pathogens.53,54 Thus, managing Ctr treatment protocols (via adjusting AZM or Doxy doses) may reduce these side effects.52,55 Based on these facts and the reported benefit of multidrug therapies,56,57 we tested the susceptibility of Ctr to combinations of AZM with 20 using a serial dilution titration assay.58,59 First, we used a checkerboard technique to test Ctr growth at different concentrations utilizing the IFA assay.60 Figure 6A represents a growth diagram of Chlamydia in the presence of single or mixed treatments. The combinations kept 100% activity using 0.03 and 3.12 μg/mL of AZM and 20, respectively. Since neither 0.03 μg/mL AZM nor 3.12 μg/mL 20 is an inhibitory concentration when used alone, we concluded that both molecules worked cooperatively to kill Chlamydia.
Figure 6.
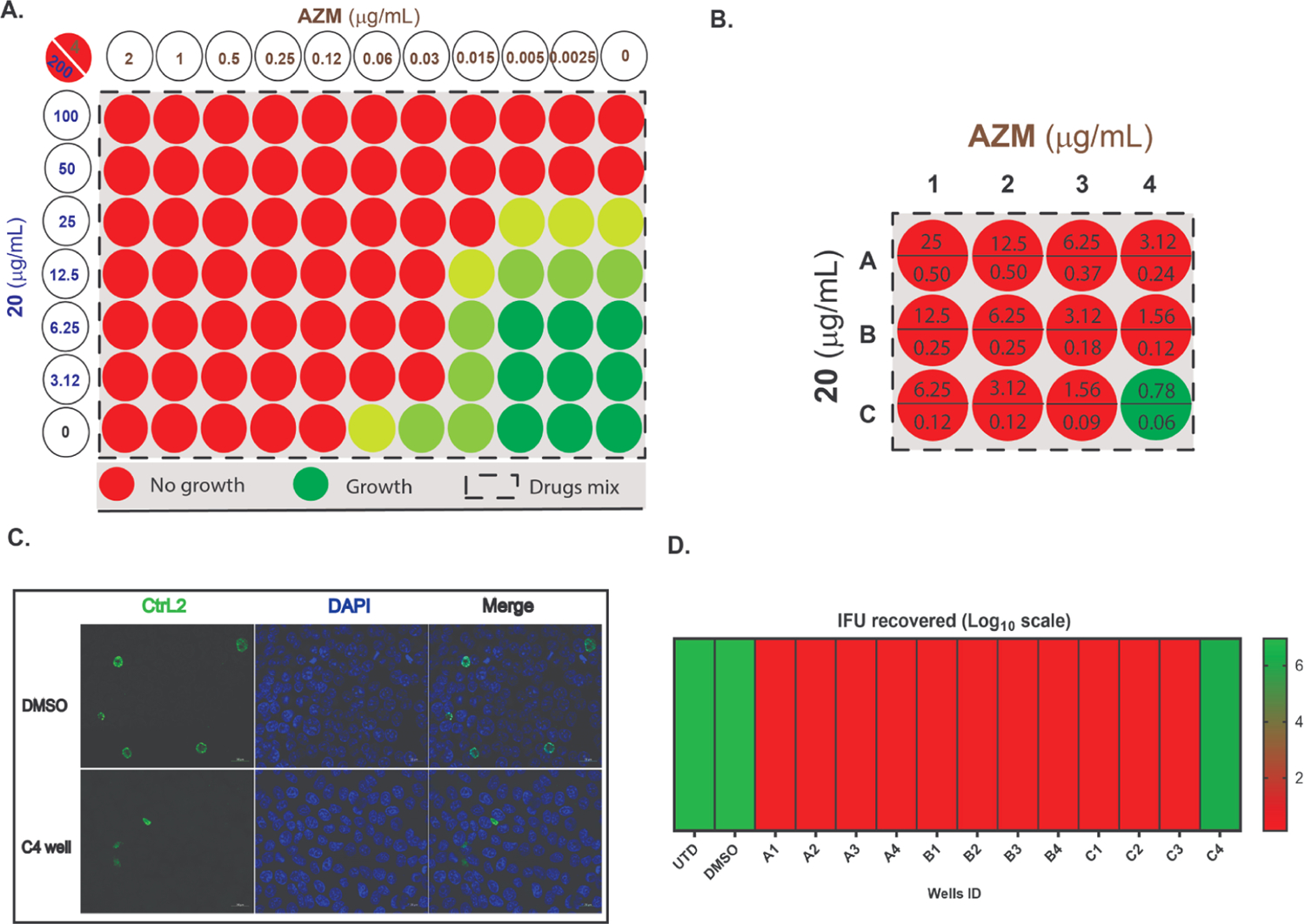
Effect of AZM and 20 combinations on Chlamydia growth showing the degree of growth in color codes; red (no growth); very faint green (<50% growth); light green (50% growth); and dark green (>50% growth) in comparison with an untreated sample; (A) representation of checkerboard assay; columns indicate 20 at different concentrations, rows show AZM at several dilutions, and the box indicates the combinations. For example, the upper left most well has a concentration of 20 at 200 μg/mL and AZM at 4 μg/mL. Data represent three biological replicates. (B) Titration assay diagram of AZM and 20 combinations. Stock solutions of AZM and 20 were mixed, in two directions. Then, 1 μL of each well was transferred into Chlamydia-infected HEp-2 cells (in triplicate) 6 h after infection to obtain the concentrations, as shown in the diagram (μg/mL). For example, well A1 has a concentration of 20 at 25 μg/mL and AZM at 0.5 μg/mL, whereas well B2 has a concentration of 20 at 6.25 μg/mL and AZM at 0.25 μg/mL. (C) Infection output in the case of untreated sample and well C4; samples were stained for MOMP (Ctr L2; green) and DNA (DAPI; blue). (D) Representation of quantified Ctr growth by IFU assay after second round of infection compared to untreated (UTD) and DMSO treatment as controls. Data represent three biological replicates.
To characterize the effect of this drug combination on Ctr growth, we performed an IFU assay. A standard checkerboard technique is difficult to implement due to the obligate intracellular nature of this pathogen and the necessity of propagating each infection onto a secondary cell culture. Hence, we utilized a simplified protocol to work with a manageable number of combinations.59,61–63 We used a two-way serial dilution method (4 × 3), starting with 1 μg/mL of AZM and 50 μg/mL of 20. Employing this approach (serial dilutions from left to right and then from top to bottom) allowed us to test a fewer number of drug combinations. Figure 6B presents the final drug concentrations used and the bacterial growth (A1 contains the highest concentration, and C4 contains the lowest concentration of AZM and 20). The initial IFA study revealed that the combinations blocked Ctr development at low concentrations (red circles, Figure 6B). The AZM/20 mixture gave 100% eradication at 0.09 and 1.56 μg/mL concentration, respectively (C3, Figure 6B). Immunofluorescence (IFA) analysis of C4 revealed that the bacterial inclusion size was dramatically altered compared to the untreated control (Figure 6C). The subsequent IFU titration of the harvested HEp-2 monolayers indicated that the combinations retained the previously detected activity by IFA. Quantification of the AZM/20 harvested samples showed that C4, which represents a combination of 0.06/0.78 μg/mL AZM/20 (Figure 6D), was the only sample in which bacterial growth was detected among all mixtures. The combination of low concentrations of AZM with 1.56 μg/mL of 20 has a great impact on the infectious progeny. The data collectively suggest that our scaffold can be used as co-therapy to improve the MCC value of AZM against Ctr and enhance its efficacy at very low concentrations. The promising activity from these combinations will offer an opportunity to reduce the dose regimen of AZM to get the same anti-chlamydial effect with lower drug concentrations.
3D Stratified Squamous Culture.
C. trachomatis can infect the columnar epithelial cells of the endocervix, the stratified squamous epithelium of the lower genital tract, and the intervening transformation zone where squamous epithelium transitions to columnar epithelium.12 Upon infection, the EBs are attached to non-ciliated columnar or cuboidal epithelial cells in genital and non-genital tract areas.9,12 We decided to study compound 20 using an in vitro HaCaT 3D organotypic culture that mimics the differentiation status and stratification of the lower genital tract. This model provides a conditional approach to study the pathogen’s capability to grow and spread under the inhibitory effect of our compound. The protocol of this assay is summarized in Figure S6 and described in the methods section. The surface of each raft was inoculated with C. trachomatis L2 and kept for a further 24 h to enable bacterial entry and growth. The infected HaCaT cells were then treated with AZM and 20 (below the cell culture inserts, Figure S6), and the infection was allowed to proceed for 8 days. The infected but untreated (termed INF) sample showed high chlamydial growth and cell destruction when compared to the uninfected samples (UIF). On the other hand, both AZM and 20 restricted bacterial growth and prevented the damage and spread associated with infection in the INF sample. In addition, we observed that AZM or 20 as a treatment did not affect cell stratification and development (Figure 7). Thus, we concluded that our compound has a significant effect in retarding the bacterial growth and spread while maintaining tissue integrity.
Figure 7.
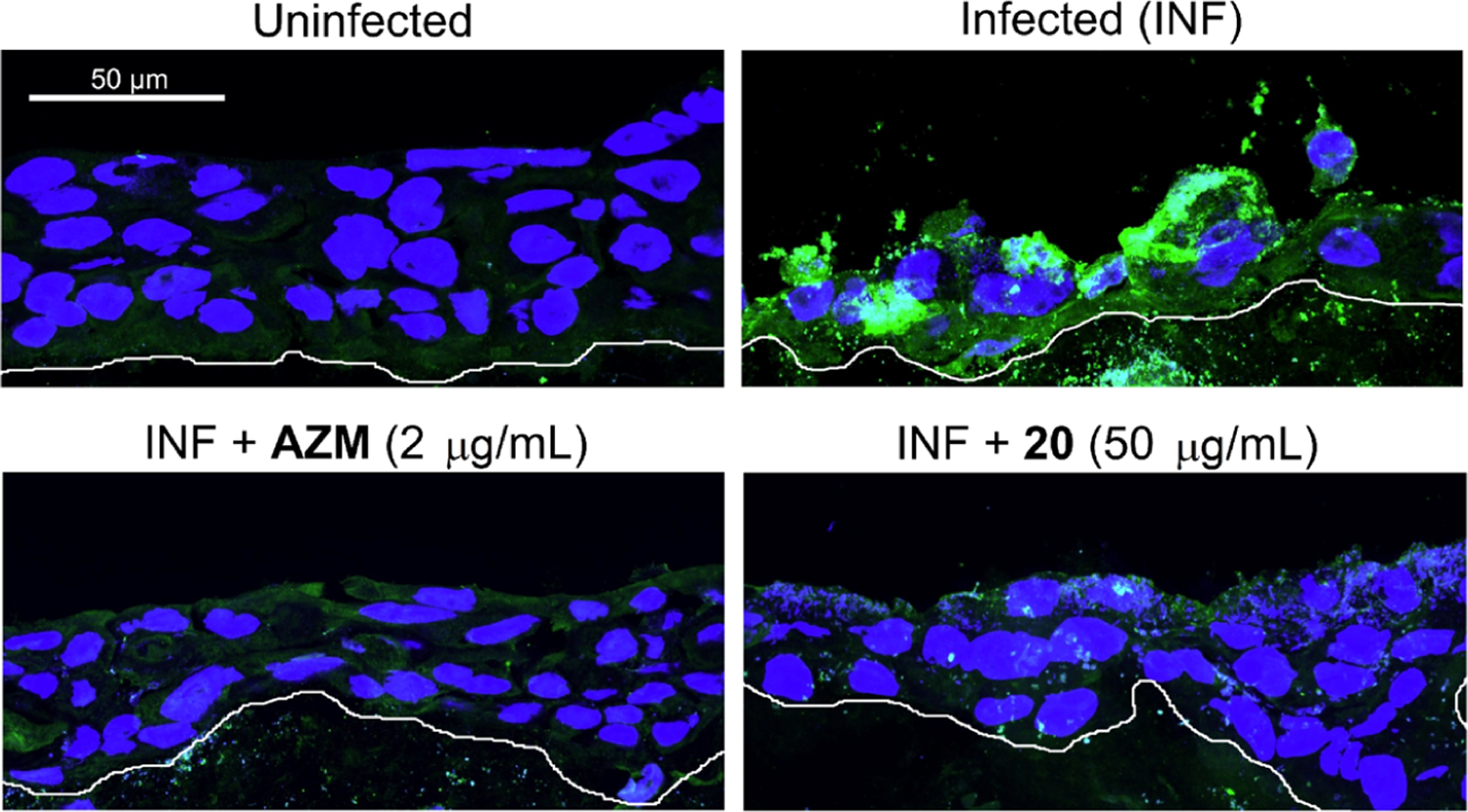
Immunofluorescence images of the fixed tissues; compound 20 was used in a concentration of 50 μg/mL in comparison with azithromycin (2 μg/mL), untreated (INF), and uninfected samples. The infection was detected by human sera staining; in green, chlamydial inclusions and in blue, HaCaT nuclei. The white bottom lines show the bottom of the 3D culture. Data represent three biological replicates.
In Vivo Efficacy.
Finally, we studied the ability of the lead compounds to inhibit the growth of Chlamydia muridarum (previously referred to as C. trachomatis mouse pneumonitis strain), a mouse pathogen that infects the columnar epithelial cells in the genital tracts of female and male mice.64 C. muridarum infection in mice closely mimics acute genital tract Ctr infection in men and women. C. muridarum-infected, dimethyl sulfoxide (DMSO)-treated mice displayed a high level of vaginal chlamydial shedding as early as day 4 after inoculation and displayed progressive reduction followed by the resolution of infection on day 28 after inoculation as a function of innate immune response. We found that mice treated with each of the compounds (100 mg/kg for 5 days) displayed a significant reduction in chlamydial shedding as early as day 4 after inoculation in comparison to DMSO-treated animals (Figure 8). Each of the compound-treated groups displayed a progressive reduction in chlamydial shedding and resolved infection at earlier time periods compared to DMSO alone treated animals. Mice treated with compounds 1 (8.54 × 106 ± 1.18 × 106; mean ± SEM), 17 (6.90 × 106 ± 9.17 × 105), or 20 (3.57 × 106 ± 8.66 × 105) displayed significant (p ≤ 0.001; ANOVA) reduction in area under the curve (AUC) of chlamydial shedding when compared to DMSO-treated animals (2.58 × 107 ± 1.78 × 106). Compound 20 displayed reduced shedding compared to compound 1 or compound 17, although the difference was significant only between 20 and 1. In summary, these data suggest that the compounds are effective in vivo, with compound 20 displaying the highest efficacy, which strongly correlates with our in vitro findings.
Figure 8.
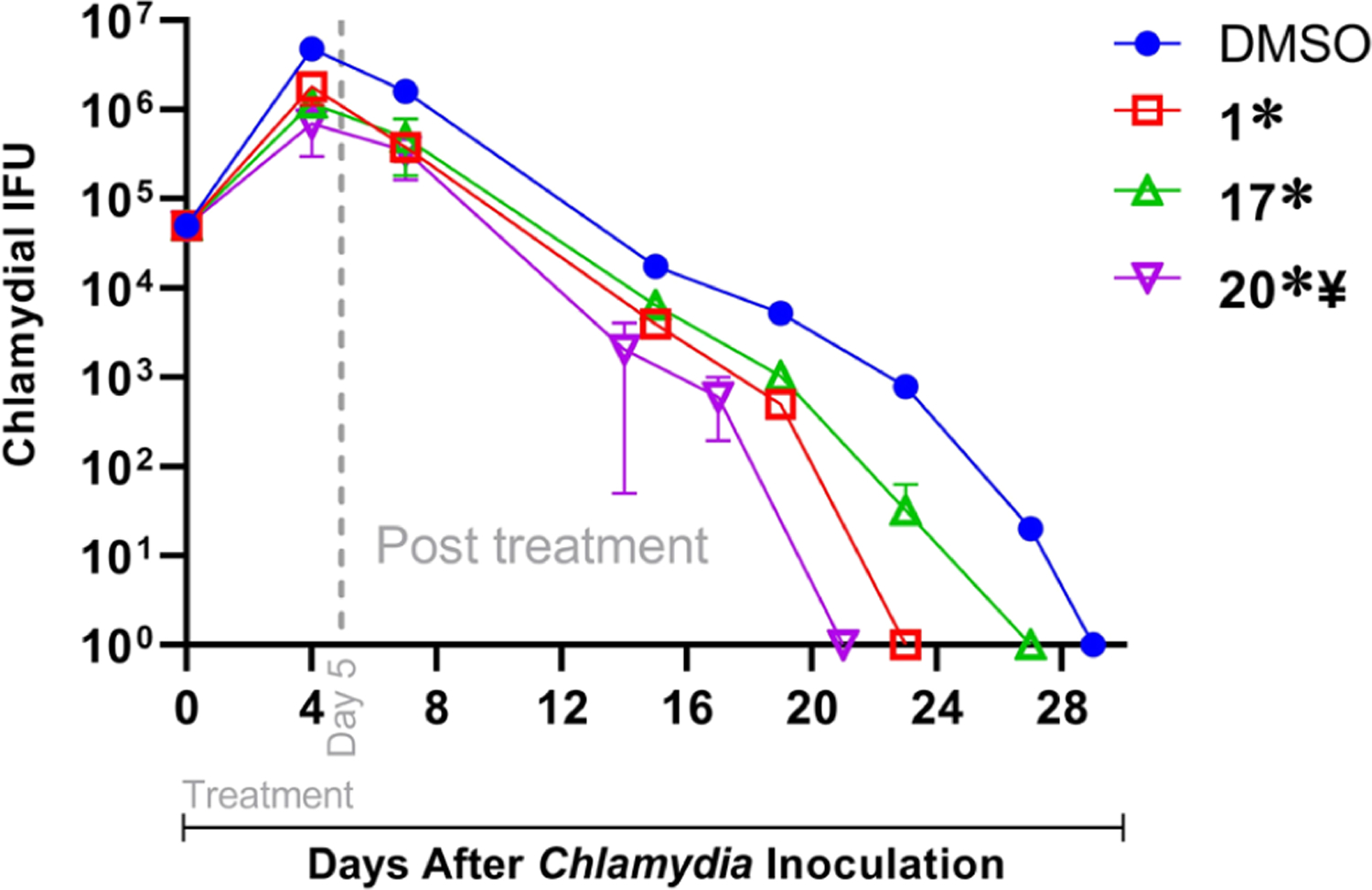
Groups of mice (n = 4–5) were pre-treated with Depo-Provera 5 days before intravaginal infection with 5 × 104 IFU of C. muridarum. Mice were injected intraperitoneally with 100 mg/kg of 1, 17, or 20, in DMSO, or DMSO alone each day from days 0 to 5 after chlamydial inoculation. Chlamydial shedding at indicated time points was measured. Significant difference (p ≤ 0.001, ANOVA) in the AUC of chlamydial shedding is indicated as * between the indicated group and DMSO-alone treated animals and ¥ between 20 and 1.
CONCLUSIONS
In this work, our strategy was to develop a C. trachomatis inhibitor using a field alignment strategy to mix the common features of compounds reported by us31,33 and ClpP activators reported by others.32 Our data confirmed the efficacy of compound 20 against C. trachomatis serovar L2, a fast-growing C. trachomatis strain. Compound 20 inhibited chlamydial growth in infected HEp-2 cells for 24 h, even after removal, indicating a cidal mechanism. This new molecule showed improved IC50 values when compared with our lead molecule 1. Furthermore, the combination of 20 with AZM significantly reduced infectious progeny in cell cultures at sub-MIC concentrations. These molecules maintained efficacy in a HaCaT-stratified 3D culture model that mimics the lower genital tract. A key feature of 20 was its ability to halt the infection and maintain tissue integrity. Furthermore, compound 20, compound 17, or compound 1 displayed significant in vivo efficacy in reduction of chlamydial infection in a mouse model, with the highest efficacy displayed by compound 20. Importantly, the most active compounds did not show activity against S. aureus JE2 and E. coli K12, indicating specificity for C. trachomatis. This result is promising as it suggests that the molecules may be selective for the pathogen (and may not affect the human microflora).
In summary, we describe a new, anti-chlamydial agent generated through SAR-guided alignment. Furthermore, the presented scaffold is an excellent starting point to develop a combination therapy strategy. In future studies, we plan to improve biological activity and to confirm the molecular target of this scaffold to advance to preclinical studies.
METHODS
General.
The commercially available reagents, chemicals, and solvents were used as obtained unless otherwise noted. All the reactions were performed in an oven-dried glassware under an inert atmosphere. Analytical TLC was performed on Merck silica gel IB2-F plates (0.25 mm thickness), and the progression of the reaction was detected using a UV light source at 254 nm. Flash chromatography was performed on a RF 200i Flash Chromatography System from Biotage Isolera. Some compounds were chromatographed on the preparative layer Analtech Uniplate silica gel GF Plates (10 × 20 Cm 250 μm) 02521. All reported yields refer to isolated compounds after purification. The 1H and 13C NMR spectra were performed and recorded at 500 MHz for 1H and 125 MHz for 13C in deuterated solvents on a BRUKER-500 NMR. Chemical shifts were expressed in parts per million on the delta (d) scale and were calibrated relative to the used solvents. Peak multiplicities were represented as s-singlet, d-doublet, t-triplet, q-quartet, p-pentet, m-multiplet, and b rs-broad signal. An Agilent 1200 HPLC system was used to detect the final compound purity with methanol and water as a mobile phase. The accepted purity limit of the final compounds that were tested was 95%, as determined by HPLC of NMR-analyzed samples. High-resolution mass spectrometry (HRMS) was performed on TripleTOF 5600 (SCIEX) using an ESI source. The physicochemical characters of the synthesized compounds were identified using Marvin 20.4, ChemAxon.65
The cell culture materials were purchased from Thermo-Fischer and stored as recommended by the manufacturer. In all states, HEp-2 cells were routinely propagated in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and incubated at 37 °C with 5% CO2. The tested compounds and the reference molecules were dissolved in sterile DMSO, according to the assigned concentrations, and frozen at −20 °C in 5 μL aliquots. The investigational assays were performed in 24-well or 96-well plates in triplicates in duplicate or triplicate biological replicates (specified under each figure). After incubation, the obtained Ctr inclusions were visualized by staining primarily with an anti-L2 goat antibody followed by a secondary donkey anti-goat antibody labeled with Alexa 488. Mitochondria of the live infected cells were stained before fixing, using Mitotracker red fluorescence stain. The cells were stained with DAPI to visualize the nuclei. Fluorescent inclusions were quantified from 15 fields of view at 20× magnification using an Olympus CKX53 fluorescence microscope with EP50 camera. Representative images were captured using a Zeiss Fluoview1000 laser scanning confocal microscope with a 60× objective and 2× digital zoom and used in the raw format without further editing.
Chemistry.
Field Alignment.
The similarity of parent and proposed molecules was evaluated utilizing classic ligand-based alignment calculations in Forge (V10, Cresset, Litlington, Cambridgeshire, UK). The designated structures were drawn and minimized in ChemDraw 16.0.1.4 (MM2 forcefield method). The optimized structures were then imported into Forge, and compound 1 was chosen as the reference molecule. The calculation was carried out in a normal conformation hunt and alignment.
General Method to Synthesize Compounds (5–13 and 16–23).
The acid 4 (0.05 g, 0.17 mmol) was added to an oven-dried round-bottom flask and dissolved in dry THF (10 mL). Then, PyBOP (0.095 g, 0.18 mmol) and Hunig’s base (DIPEA) (87 μL, 0.5 mmol) were added. The mixture was stirred at room temperature for 10 min before adding the appropriate amine derivative (0.17 mmol). Next, the reaction was stirred at optimal temperature for 1 h. Upon completion (as detected by TLC), THF was evaporated in vacuo, and the crude compound was purified by automated flash column chromatography (Biotage Isolera) to afford the desired product (if not purified on the same day, the reaction was stored at −80 °C until the purification time to avoid the previously monitored compounds rearrangement).
Compound 6 was additionally purified using a prep TLC plate.
In the case of compound 8, we utilized HBTU (1 equiv) instead of PyBOP. After 1 h, the organic solvent was evaporated, and EtOAc (20 mL) was added. The organic solution was then transferred to a separating funnel and washed with water (3 × 10 mL) and brine (1 × 10 mL) before the purification.
The physical characters and spectral data of separated products are listed below:
2-Methyl-N-(3-(2-oxopyrrolidin-1-yl)propyl)-2-((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)propanamide (5).
The product was obtained by coupling between 4 and 1-(3-aminopropyl)pyrrolidin-2-one. It gave white crystals (62 mg, 88.5%), purified using DCM/MeOH (0 to 10%) over 15 min and then by paper chromatography to remove tri(pyrrolidin-1-yl)phosphine oxide (TPPO) (see Figure S7); 1H NMR (500 MHz, CDCl3): δ 8.97 (s, 1H), 8.17 (s, 2H), 7.79 (br s, 1H), 3.40 (m, 4H), 3.20 (q, J = 6.0 Hz, 2H), 2.43 (t, J = 8.0 Hz, 2H), 2.06 (p, J = 7.5 Hz, 2H), 1.73 (s, 6H), 2.69 (p, J = 6.0 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 176.3, 167.6, 158.9, 146.8 (q, J = 3.7 Hz), 135.1 (q, J = 3.7 Hz), 129.8 (q, J = 33.7 Hz), 125.1, 123.7, 121.5, 69.2, 47.6, 39.7, 36.3, 30.8, 26.3, 20.3 (2C), 18.0; HPLC purity, % 95.8; HRMS (m/z): [M + Na]+ calcd for C17H22F3N3O4SNa, 444.1175; found, 444.1192.
2 - Methyl - N - (3 - (thiophen - 2 - yl)propyl) - 2 - ((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)propanamide (6).
The product was obtained by coupling between 4 and 3-(thiphen-1yl)propan-1-amine, as a white solid (55 mg, 78.5%), purified using EtOAc/hexanes {0 to 50%} over 10 min; 1H NMR (500 MHz, CDCl3): δ 8.91 (s, 1H), 8.19−8.18 (m, 2H), 7.13 (d, J = 5.0 Hz, 1H), 7.04 (br s, 1H), 6.93−6.91 (m, 1H), 6.82−6.81 (m, 1H), 3.36 (q, J = 6.5 Hz, 2H), 2.93 (t, J = 7.5 Hz, 2H), 1.97 (p, J = 7.0 Hz, 2H), 1.62 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 167.4, 158.2, 147.3 (q, J = 3.7 Hz), 144.0, 135.5 (q, J = 3.3 Hz), 130.4, 130.1, 126.9, 124.5, 124.5, 123.5 (2C), 123.3, 121.3, 67.6, 39.8, 30.9, 27.2, 20.5 (2C); HPLC purity, 95.3%; HRMS (m/z): [M + H]+ calcd for C17H19F3N2O3S2, 421.0862; found, 421.0875.
2-Methyl-N-(3-(furan-2-yl)propyl)-2-((5-(trifluoromethyl)-pyridin-2-yl)sulfonyl)propanamide (7).
The product was obtained by coupling between 4 and 3-(furan-1yl)propan-1-amine, as a light brown solid, (45 mg, 66%), purified using EtOAc/hexanes {0 to 60%} over 15 min; 1H NMR (500 MHz, CDCl3): δ 8.93 (s, 1H), 8.20−8.19 (m, 2H), 7.31 (m, 1H), 7.07 (br s, 1H), 6.29−6.28 (m, 1H), 6.02−6.03 (m, 1H), 3.35 (q, J = 6.5 Hz, 2H), 2.73 (t, J = 7.5 Hz, 2H), 1.93 (p, J = 7.0 Hz, 2H), 1.63 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 167.4, 158.2, 147.3 (q, J = 3.7 Hz), 141.1, 135.5 (q, J = 3.75 Hz), 130.2 (q, J = 33.75 Hz), 124.4, 123.5, 121.3, 110.2, 105.3, 67.6, 39.9, 27.4, 25.4, 20.5 (2C); HPLC purity, 97.3%; HRMS (m/z): [M + H]+ calcd for C17H19F3N2O4S, 405.1202; found, 405.1216; [M + Na]+ calcd for C17H19F3N2O4SNa, 427.0915; found, 427.0928.
N-(3-(1H-Pyrazol-4-yl)propyl)-2-methyl-2-((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)propanamide (8).
The product was obtained by coupling between 4 and 3-(1H-imidazol-1-yl)propan-1-amine, as a white solid, (36 mg, 53%), purified using DCM/MeOH {0 to 5%} over 20 min (two times); 1H NMR (500 MHz, CDCl3): δ 8.91 (s, 1H), 8.19−8.16 (m, 2H), 7.43 (s, 2H), 7.13 (br s, 1H), 3.31 (q, J = 6.5 Hz, 2H), 2.58 (t, J = 7.5 Hz, 2H), 1.85 (p, J = 7.0 Hz, 2H), 1.62 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 167.4, 158.1, 147.2 (q, J = 3.7 Hz), 135.6 (q, J = 3.75 Hz), 132.6, 130.3 (q, J = 33.7 Hz), 124.6 (2C), 123.5, 121.3, 120.0, 67.9, 39.8, 30.0, 21.2, 20.6 (2C); HPLC purity, 95.9%; HRMS (m/z): [M + H]+ calcd for C16H19F3N4O3S, 405.1202; found, 405.1216; [M + Na]+ calcd for C16H19F3N4O3SNa, 427.1028; found, 427.1038.
N-(3-(1H-Pyrazol-1-yl)propyl)-2-methyl-2-((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)propanamide (9).
The product was obtained by coupling between 4 and 3-(pyrrol-1-yl)propan-1-amine, as a white solid, (35 mg, 51.5%), purified using EtOAc/hexanes (25 to 70%) over 15 min; 1H NMR (500 MHz, CDCl3): δ 8.96 (s, 1H), 8.20−8.19 (m, 2H), 7.56 (m, 1H), 7.48 (m, 1H), 7.40 (br s, 1H), 6.28 (m, 1H), 4.31 (q, J = 6.5 Hz, 2H), 3.28 (t, J = 6.1 Hz, 2H), 2.13 (p, J = 6.4 Hz, 2H), 1.64 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 167.7, 158.3, 147.2 (q, J = 3.7 Hz), 138.9, 135.5 (q, J = 3.7 Hz), 130.2 (q, J = 33.7 Hz), 124.8, 123.5, 121.3, 105.9, 68.2, 49.3, 37.6, 29.7, 20.5 (2C); HPLC purity, 96.0%; HRMS (m/z): [M + H]+ calcd for C16H19F3N4O3S, 405.1202; found, 405.1211.
2 - Methyl - N - (3 - (pyridin - 3 - yl)propyl) - 2 - ((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)propanamide (10).
The product was obtained by coupling between 4 and 3-(pyridine-3-yl)propan-1-amine, as a white solid, (60 mg, 87%), purified using EtOAc/hexanes (0 to 60%) over 15 min; 1H NMR (500 MHz, CDCl3): δ 8.90 (s, 1H), 8.56−8.51 (m, 2H), 8.23−8.19 (m, 2H), 7.74 (d, J = 8.0 Hz, 1H), 7.40−7.37 (m, 1H), 7.15 (br s, 1H), 3.36 (q, J = 6.5 Hz, 2H), 2.78 (t, J = 6.1 Hz, 2H), 1.95 (p, J = 6.4 Hz, 2H), 1.63 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 167.5, 158.1, 148.0, 147.2 (q, J = 3.7 Hz), 145.7, 137.9, 135.6 (q, J = 3.7 Hz), 130.2 (q, J = 33.7 Hz), 124.7, 124.6, 124.1, 123.4, 121.3, 67.8, 39.7, 30.3, 30.1, 20.6 (2C); HPLC purity, 96.9%; HRMS (m/z): [M + H]+ calcd for C18H20F3N3O3S, 416.1252; found, 416.1257; ESIMS: calcd for C18H20F3N3O3S, 416.12; found mass [M + H]+, 416.10.
2 - Methyl - N - (3 - (pyridin - 4 - yl)propyl) - 2 - ((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)propanamide (11).
The product was obtained by coupling between 4 and 3-(pyridine-4-yl)propan-1-amine, as a white solid, (56 mg, 81%), purified using EtOAc/hexanes {0 to 100%} over 20 min; 1H NMR (500 MHz, CDCl3): δ 8.91 (s, 1H), 8.53 (d, J = 6 Hz, 2H), 8.23−8.19 (m, 2H), 7.27 (d, J = 6.0 Hz, 2H), 7.11 (br s, 1H), 3.35 (q, J = 6.5 Hz, 2H), 2.77 (t, J = 8.0 Hz, 2H), 1.95 (p, J = 7.5 Hz, 2H), 1.62 (s, 6H); 13C NMR (125 MHz, CDCl3); 167.6, 158.5, 152.7, 147.2 (q, J = 3.7 Hz), 143.9 (2C), 135.7 (q, J = 3.7 Hz), 130.4 (q, J = 35.0 Hz), 126.0 (2C), 124.8, 68.0, 39.4, 32.9, 29.3, 20.7 (2C); HPLC purity, 96.3%; HRMS (m/z): [M + H]+ calcd for C18H20F3N3O3S, 416.1252; found, 416.1259.
N - (3 - (1H - Indol - 2 - yl)propyl) - 2 - methyl - 2 - ((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)propanamide (12).
The product was obtained by coupling between 4 and 3-(1H-indol-2-yl)propan-1-amine, as a brown solid, (69 mg, 91%), purified using EtOAc/hexanes {0 to 50%} over 15 min; 1H NMR (500 MHz, CDCl3): δ 8.871 (s, 1H), 8.14−8.08 (m, 2H), 8.06 (br s, 1H), 7.59 (d, J = 8 Hz, 1H), 7.36 (d, J = 8 Hz, 1H), 7.21−7.17 (m, 1H), 7.12−7.09 (m, 1H), 7.03 (s, 1H), 6.94.11 (br s, 1H), 3.36 (q, J = 6.5 Hz, 2H), 2.87 (t, J = 7 Hz, 2H), 2.01 (p, J = 7.5 Hz, 2H), 1.58 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 167.3, 158.2, 147.2 (q, J = 3.7 Hz), 136.5, 135.5 (q, J = 3.7 Hz), 130.2 (q, J = 33.7 Hz), 127.3, 124.5, 119.3, 118.8, 115.4, 111.2, 67.9, 40.3, 29.1, 22.6, 20.4 (2C); HPLC purity, 96.0%; HRMS (m/z): [M + Na]+ calcd for C21H22F3N3O3SNa, 476.1232; found, 476.1251.
2-Methyl-N-(3-(1-methyl-1H-benzo[d]imidazol-2-yl)-propyl)-2-((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)-propanamide (13).
The products were obtained by coupling between 4 and 3-(1-methyl-1H-benzo[d]imidazol-2-yl)propan-1-amine, as white crystals, (45 mg, 57%), purified using DCM/MeOH {0 to 10%} over 20 min; 1H NMR (500 MHz, CDCl3): δ 8.95 (s, 1H), 8.16−8.12 (m, 2H), 7.80−7.78 (m, 1H), 7.39−7.35 (m, 3H), 3.82 (s, 3H), 3.43 (q, J = 6.5 Hz, 2H), 3.18 (t, J = 6.5 Hz, 2H), 2.22 (p, J = 7 Hz, 2H), 1.70 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 167.9, 158.8, 153.8, 146.9 (q, J = 3.7 Hz), 135.3 (q, J = 3.7 Hz), 134.4, 129.9 (q, J = 33.7 Hz), 125.0, 123.6 (2C), 121.4, 117.6, 109.9, 69.3, 39.6, 30.3, 25.7, 24.1, 20.5 (2C); HPLC purity, 95.3%; HRMS (m/z): [M + H]+ calcd for C21H23F3N4O3S, 469.1512; found, 469.1525.
2-Methyl-N-(3-(phenylthio)propyl)-2-((5-(trifluoromethyl)-pyridin-2-yl)sulfonyl)propanamide (16).
The product was obtained by coupling between 4 and 15a, as a white solid, (48 mg, 64%), purified using EtOAc/hexanes {0 to 50%} over 20 min; 1H NMR (500 MHz, CDCl3): δ 8.94 (s, 1H), 8.19 (m, 2H), 7.38−7.36 (m, 2H), 7.29 (d, J = 7.5 Hz, 2H), 7.21−7.18 (m, 1H), 7.11 (br s, 1H), 3.46 (q, J = 6.5 Hz, 2H), 3.05 (t, J = 7.5 Hz, 2H), 1.95 (p, J = 7.0 Hz, 2H), 1.64 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 167.5, 158.0, 147.3 (q, J = 3.7 Hz), 135.9, 135.5 (q, J = 3.7 Hz), 130.3 (q, J = 33.7 Hz), 129.4, 129.0, 126.2 (2C), 124.5 (2C), 123.5, 121.3, 39.3, 31.1, 28.5, 20.5 (2C); HPLC purity, 97.4%; HRMS (m/z): [M + H]+ calcd for C19H21F3N2O3S2, 447.1022; found, 447.1030.
2-Methyl-N-(3-(2-chlorophenylthio)propyl)-2-((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)propanamide (17).
The product was obtained by coupling between 4 and 15b, as a white solid, (69 mg, 86%), purified using EtOAc/hexanes {0 to 50%} over 20 min; 1H NMR (500 MHz, CDCl3): δ 8.94 (s, 1H), 8.18 (m, 2H), 7.36 (d, J = 7 Hz, 2H), 7.29 (d, J = 6.5 Hz, 2H), 7.19 (t, J = 7.5 Hz, 1H), 7.13 (br s, 1H), 7.12 - (m, 1H), 3.47 (q, J = 6.5 Hz, 2H), 3.05 (t, J = 7.5 Hz, 2H), 1.97 (p, J = 7.0 Hz, 2H), 1.62 (s, 6H); 13C NMR (125 MHz, CDCl3); 167.6, 158.0, 147.3 (q, J = 3.7 Hz), 135.6 (q, J = 3.7 Hz), 133.7, 129.8, 128.6, 127.2, 126.7, 124.5, 67.6, 39.4, 29.9, 28.1, 20.5 (2C); HPLC purity, 96.8%; HRMS (m/z): [M + H]+ calcd for C19H20ClF3N2O3S2, 481.0632; found, 481.0639.
2-Methyl-N-(3-(2-methylphenylthio)propyl)-2-((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)propanamide (18).
The product was obtained by coupling between 4 and 15c, as a white solid, (55 mg, 71%), purified using EtOAc/hexanes {0 to 50%} over 20 min; 1H NMR (500 MHz, CDCl3): δ 8.92 (s, 1H), 8.18−8.14 (m, 2H), 7.27−7.17 (m, 1H), 7.17−7.07 (m, 4H, include the NH br s peak), 3.45 (q, J = 6.5 Hz, 2H), 3.00 (t, J = 7.5 Hz, 2H), 2.37 (s, 3H), 1.94 (p, J = 7.0 Hz, 2H), 1.62 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 167.5, 158.0, 147.3 (q, J = 3.7 Hz), 137.7, 135.5 (q, J = 3.7 Hz), 135.3, 130.2, 127.9, 126.5, 125.8, 124.5, 123.5, 121.3, 67.6, 39.4, 30.2, 28.4, 20.5, 20.4 (2C); HPLC purity, 95.5%; HRMS (m/z): [M + H]+ calcd for C20H23ClF3N2O3S2, 461.1172; found, 461.1185.
2-Methyl-N-(3-(2-methoxyphenylthio)propyl)-2-((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)propanamide (19).
The product was obtained by coupling between 4 and 15d, as a white solid, (57 mg, 71%), purified using EtOAc/hexanes {0 to 50%} over 20 min; 1H NMR (500 MHz, CDCl3): δ 8.93 (s, 1H), 8.16 (m, 2H), 7.29 (d, J = 6.5 Hz, 1H), 7.29 (t, J = 7 Hz, 1H), 7.10 (br s, 1H), 6.90 (t, J = 7.5 Hz, 1H), 6.86 (t, J = 8 Hz, 1H), 3.44 (q, J = 6.5 Hz, 2H), 2.99 (t, J = 7.5 Hz, 2H), 1.91 (p, J = 7.0 Hz, 2H), 1.62 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 167.5, 158.08, 158.07, 157.6 (2C), 147.3 (q, J = 3.7 Hz), 135.5 (q, J = 3.7 Hz), 130.0, 127.5, 124.5, 123.8, 121.1, 110.7, 67.7, 55.8, 39.4, 29.6, 28.3, 20.5 (2C); HPLC purity, 96.3%; HRMS (m/z): [M + Na]+ calcd for C20H23ClF3N2O4S2Na, 499.0960; found, 499.0949.
2-Methyl-N-(3-(4-chlorophenylthio)propyl)-2-((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)propanamide (20).
The product was obtained by coupling between 4 and 15e, as a white solid, (60 mg, 75%), purified using EtOAc: Hexanes {0 to 50%} over 20 min; 1H NMR (500 MHz, CDCl3): δ 8.92 (s, 1H), 8.21−8.17 (m, 2H), 7.27 (d, J = 8.5 Hz, 2H), 7.23 (d, J = 8.5 Hz, 2H), 7.10 (br s, 1H), 3.44 (q, J = 6.5 Hz, 2H), 3.01 (t, J = 7.5 Hz, 2H), 1.91 (p, J = 7.0 Hz, 2H), 1.61 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 167.5, 158.0, 147.3, 147.3 (q, J = 3.2 Hz), 135.7 (q, J = 3.2 Hz), 134.5, 132.2, 130.7, 129.1, 124.5 (2C), 123.5 (2C), 121.3, 67.5, 39.2, 31.3, 28.4, 20.5 (2C); HPLC purity, 96.1%; HRMS (m/z): [M + H]+ calcd for C19H20ClF3N2O3S2, 481.0632; found, 481.0637.
2-Methyl-N-(3-(3-chlorophenylthio)propyl)-2-((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)propanamide (21).
The product was obtained by coupling between 4 and 15f, as a white solid, (33 mg, 41%), purified using EtOAc/hexanes {0 to 50%} over 20 min; 1H NMR (500 MHz, CDCl3): δ 8.93 (s, 1H), 8.19 (m, 2H), 7.29 (s, 1H), 7.20−7.19 (m, 2H), 7.14−7.12 (m, 2H), 7.11 (br s, 1H), 3.45 (q, J = 6.5 Hz, 2H), 3.04 (t, J = 7.5 Hz, 2H), 1.94 (p, J = 7.0 Hz, 2H), 1.62 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 167.6, 158.0, 147.3 (q, J = 3.7 Hz, 2H), 138.3, 135.6 (q, J = 3.7 Hz, 2H), 134.8, 130.0, 128.4 (2C), 126.9, 126.2, 124.5, 67.5, 39.2, 30.7, 28.4, 20.5 (2C); HPLC purity, 95.7%; HRMS (m/z): [M + H]+ calcd for C19H20ClF3N2O3S2, 481.0632; found, 481.0636.
N-(2-((2-Chlorobenzyl)thio)ethyl)-2-methyl-2-((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)propanamide (22).
The product was obtained by coupling between 4 and 15g, as a white solid, (35 mg, 43%), purified using EtOAc/hexanes {0 to 70%} over 15 min; 1H NMR (500 MHz, CDCl3): δ 8.92 (s, 1H), 8.21−8.17 (m, 2H), 7.40−7.36 (m, 2H), 7.29 (br s, 1H), 7.24−7.20 (m, 2H), 3.87 (s, 2H), 3.49 (q, J = 6.5 Hz, 2H), 2.68 (t, J = 6.5 Hz, 2H), 1.65 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 167.6, 158.2, 147.2 (q, J = 3.7 Hz, 2H), 135.9, 135.5 (q, J = 3.7 Hz, 2H), 134.0, 130.8, 129.9, 128.6 (2C), 127.0, 124.5, 123.5, 121.3, 67.8, 39.4, 33.3, 30.8, 20.5 (2C); HPLC purity, 96.8%; HRMS (m/z): [M + H]+ calcd for C19H20ClF3N2O3S2, 481.0632; found, 481.0638.
N-(4-((2-Chlorophenyl)thio)butyl)-2-methyl-2-((5-(trifluoromethyl)pyridin-2-yl)sulfonyl)propanamide (23).
The products were obtained by coupling between 4 and 15i, as white crystals, (56 mg, 67%), purified using EtOAc/hexanes {0 to 40%} over 12 min; 1H NMR (500 MHz, CDCl3): δ 8.98 (s, 1H), 8.20 (s, 1H), 8.19 (s, 1H), 7.35 (dd, J = 1.24 Hz, 1H), 7.26−7.24 (m, 1H), 7.21−7.18 (m, 1H), 7.10−7.07 (m, 1H), 7.01 (s, 1H), 3.36−3.33 (m, 2H), 2.97 (t, J = 6.7 Hz, 2H), 1.78−1.76 (m, 4H), 1.62 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 167.5, 167.4, 158.1, 147.3 (q, J = 3.7 Hz, 2H), 135.8, 135.6, 135.5 (q, J = 3.7 Hz), 133.5, 129.7, 128.3, 127.1, 126.5, 124.5, 67.7, 39.9, 32.1, 28.4, 25.9, 20.6 (2C); HPLC purity, 96.3%; ESIMS: calcd for C20H22ClF3N2O3S2: 494.07; found mass [M + H]+, 495.09.
Biology.
Anti-chlamydial Activity Investigation.
All the IFA assays were initially performed to detect the synthesized compound activity, as reported previously.31,33 To calculate the exact impact of the most active compounds on C. trachomatis, HEp-2 cells were seeded in 24-well plates (2 × 105 cells/well) and then infected with C. trachomatis L2 at a multiplicity of infection of 1. At 24 hpi, cell lysates, which contain C. trachomatis infectious form (EB), were harvested from infected cell cultures (three wells) and stored in a sucrose storage medium (2SP). The resulting mixture was frozen and maintained at −80 °C for the subsequent infection round. The last row in each plate was fixed and stained for imaging. For the IFU work, a fresh HEp-2 cell monolayer was seeded, and the stored cell lysates were thawed and added in a 10-fold dilution series (in 2SP), according to the degree of activity observed in the IFA work. The recoverable EBs from the initial infection were counted at 24 hpi, as mentioned in the general section. The infectious progeny was calculated using the following equation, assuming an inoculum volume of 0.25 mL per well
Titration Assay.
The required concentrations were prepared in a separate plate and then transferred into the corresponding infected HEp-2 cells, as mentioned in the general IFA work. First, the ordinary checkboard assay protocol was adopted, as reported previously.66 We used six concentrations of 20 starting from 100 μg/mL and 10 concentrations of AZM starting from 2 μg/mL. We mixed the concentrations and transferred 1 μL of each well into the corresponding infected HEp-2 cells, 6 hpi sample to achieve the required concentration. After 18 h, The infected cells were then (total 24 hpi), and the chlamydial growth was detected after immunostaining. The MIC value was calculated as the lowest combined concentrations at which no chlamydial growth was detected. For the subsequent IFU work, 50 μg/mL of compound 20 and 1 μg/mL of AZM were diluted in a 3 × 4 way (as indicated in Figure 6). Following, 1 μL of each well was transferred into the corresponding infected cells in triplicate to get the desired concentration indicated. To determine the MCC values, the IFU assay was performed using the harvested samples from the IFA, work as depicted in the IFU work above.
Air–Liquid Interface Model.
Cell Line Cultivation and Media Components.
NIH 3T3 fibroblast cell lines were cultivated in DMEM supplemented with 10% fetal bovine serum, 20 mM L-glutamine, and 10 μg/mL gentamicin. On the other hand, HaCaT cells were propagated in DMEM/F12 1:3 containing 10% FBS, 20 mM L-glutamine, 10 μg/mL gentamicin, 250 μL of insulin, 100 μL of hydrocortisone (final concentration of 10−10 M), 3 μL of cholera toxin (final concentration of 0.5 μg/mL), and 1 μL of the epidermal growth factor (final concentration of 10 ng/mL).
Raft Preparation, Infection, and Treatment.
As described previously,34 collagen solution (RAFT reagent kit, Lonza, cat 016–0R94) was prepared, according to the manufacturer’s protocol (see the Supporting Information for more details). After preparation, NIH 3T3 fibroblast feeder cells were added to the cold collagen solution and transferred to a 24-well plate (1 mL per well). The plate was incubated for 15 min before adsorbers were added to the top of each well for another 15 min. Meanwhile, HaCaT epithelial cells were prepared and transferred to the top of each raft (3 × 105 cells/mL). The plate was then incubated for 48 h until the HaCaT cells reach 100% confluency. After that, the collagen disks were transferred onto 0.4 μm filter inserts in 6-well plates containing 1.5 mL of media in the bottom of each well. The transferred collagen gels were incubated at this air–liquid interface state for 24 h. Then, C. trachomatis serovar L2 in Hanks’ Balanced Salt Solution (HBSS) (1 × 107 IFU per raft) was added to the top of each raft. To allow Ctr to adhere to the cells, the plates were left at room temperature for 1 h. The plates were incubated at 37 °C for 24 h to allow infection to advance into replicative growth before the treatment was applied (the indicated drug concentrations were freshly prepared every day in 1.5 mL of media and applied to the bottom of each insert). After nine doses (11 days post collagen exposure), the rafts were washed with HBSS (1.5 mL × 3) and fixed with 4% paraformaldehyde (PFA) solution in PBS for 30 min. The rafts were then washed with PBS (1.5 mL × 3) and sequentially transferred to PBS with a sucrose gradient (10 to 20 to 30%) to partially dehydrate the samples before processing. Raft optimal cutting temperature (OCT) embedding, freezing, sectioning (10 μm slices), and slide preparation were carried out in the UNMC Tissue Sciences Facility. The resulting slides were gently blocked in PBST with 5% BSA for 2 h, followed by primary staining with human sera and goat-anti-human secondary. The slides were then visualized and imaged using a spinning disk confocal microscope (Nikon Ti-2, CSU-W) with a lambda-S 63× oil objective, and images were processed using ImageJ.67
Minimum Inhibitory Concentration (MIC).
We utilized S. aureus USA 300 JE2 and E. coli K12 in this experiment using the broth microdilution method (ASM Clinical Microbiology Procedures Handbook, 3rd edition). The tested compounds were dissolved in sterile DMSO, and serial 2-fold dilutions were prepared in 96-well plates using 5% DMSO in Muller Hinton broth (Difco BD Diagnostics). The bacterial cultures were then prepared in 0.5 McFarland units. Finally, 10 μL of the suspension was transferred to each well of the previously prepared 96-well plate. The plates were then incubated for 24 h at 37 °C. Data represent three biological replicates.
Cell Proliferation Kit II (XTT).
Human epithelial cells (HEp-2) cell lines were cultured and seeded separately at a density of 5000 cells per well in 96-well plates. After 24 h, each plate of cells was treated with a serial dilution of compound 20 in triplicates and DMSO as control. The plates were incubated for an additional 48 h, followed by adding the premixed XTT solution, as indicated in the manual, and further incubated for 4 h. The absorbance readings were determined at 450 nm using a multiskan FC microplate photometer after subtracting the background absorbance. Data represent two biological replicates.
Animal Model.
In Vivo Infection and Treatment.
All animal experiments reported in this manuscript were approved by the Midwestern University Institutional Animal Use and Care Committee (IAUCC), following the Guide for the Care and Use of Laboratory Animals published by the Institute of Laboratory Animal Research (USA). Mice were administered 2.5 mg of Depo-Provera subcutaneously 5 days before infection in order to render the mice anestrous and receptive to the genital infection. Mice were challenged with 5 × 104 IFUs of C. muridarum in 10 μL of sucrose–phosphate–glutamate (SPG) buffer on day 0. From day 0 to day 5, compounds 1, 17, or 20 in DMSO, or DMSO alone (as a control), were injected intraperitoneally at 100 mg/kg mice once per day. Mice were swabbed once every 3–4 days following infection. Bacterial counts in swabs were measured by plating on HeLa229 cells, followed by immuno-fluorescent staining and enumeration. The mean ± SEM of AUC of chlamydial shedding was calculated for each group and compared using ANOVA. Differences were considered significant for p ≤ 0.01.
Supplementary Material
ACKNOWLEDGMENTS
The authors are thankful to the funding agencies that made this work possible: 1R56AI146062-01A1 (NIH/NIAID, S.P.O. and M.C.-S.); 1R01AI132406 (NIH/NIAID, R.A.C.); and 2R15AI101920-02 and 2R15AI101920-03 (NIH/NIAID to A.K.M.). The funders had no role in designing these studies. We thank Jiachen Feng for assistance. M.A.S. was supported through a graduate fellowship from UNMC.
ABBREVIATIONS
- EB
elementary body
- RB
reticulate body
- STI
sexually transmitted infection
- STDs
sexually transmitted diseases
- Ctr
Chlamydia trachomatis
- PyBOP
benzotriazole-1-yl-oxytris-pyrrolidino-phosphonium hexafluorophosphate
- HBTU
N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- DIPEA
N,N-diisopropylethylamine
- IFA
immunofluorescence assay
- IFU
inclusion forming unit
- Hep-2
human epithelial type 2
- HBD
hydrogen bond donor
- HBA
hydrogen bond acceptor
- TLC
thin-layer chromatography
- DMEM
Dulbecco’s modified Eagle medium
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.1c00553.
Additional synthetic pathways, HPLC traces, MS, Figures S1–S6 and NMR spectra (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acsinfecdis.1c00553
The authors declare no competing financial interest.
Contributor Information
Mohamed A. Seleem, Department of Pharmaceutical Sciences, College of Pharmacy, University of Nebraska Medical Center, Omaha, Nebraska 68198, United States.
Nicholas A. Wood, Department of Pathology and Microbiology, College of Medicine, University of Nebraska Medical Center, Omaha, Nebraska 68198, United States
Amanda J. Brinkworth, Department of Pathology and Microbiology, College of Medicine, University of Nebraska Medical Center, Omaha, Nebraska 68198, United States
Srikanth Manam, Department of Pathology and Population Medicine, Midwestern University, Glendale, Arizona 85308, United States.
Rey A. Carabeo, Department of Pathology and Microbiology, College of Medicine, University of Nebraska Medical Center, Omaha, Nebraska 68198, United States
Ashlesh K. Murthy, Department of Pathology and Population Medicine, Midwestern University, Glendale, Arizona 85308, United States
Scot P. Ouellette, Department of Pathology and Microbiology, College of Medicine, University of Nebraska Medical Center, Omaha, Nebraska 68198, United States
Martin Conda-Sheridan, Department of Pharmaceutical Sciences, College of Pharmacy, University of Nebraska Medical Center, Omaha, Nebraska 68198, United States.
REFERENCES
- (1).Unemo M; Bradshaw CS; Hocking JS; de Vries HJ; Francis SC; Mabey D; Marrazzo JM; Sonder GJ; Schwebke JR; Hoornenborg E Sexually Transmitted Infections: Challenges Ahead. Lancet Infect. Dis 2017, 17, e235–e279. [DOI] [PubMed] [Google Scholar]
- (2).Dallabetta G; Wi T; Nielsen G; Holmes K; Sparling P; Stamm W Prevention and Control of STD and HIV Infection in Developing Countries. Sexually Transmitted Diseases, 4th ed.; McGraw-Hill: New York, NY, 2008. [Google Scholar]
- (3).World Health Organization. Report on Global Sexually Transmitted Infection Surveillance 2018. 2018, https://www.who.int/reproductivehealth/publications/stis-surveillance-2018/en/ (accessed Dec 07, 2021).
- (4).Yoneyama H; Katsumata R Antibiotic Resistance in Bacteria and Its Future for Novel Antibiotic Development. Biosci., Biotechnol., Biochem 2006, 70, 1060–1075. [DOI] [PubMed] [Google Scholar]
- (5).Hopkins Tanne J Sexually Transmitted Diseases Reach Record Highs in US. BMJ [Br. Med. J.] 2018, DOI: 10.1136/bmj.k3747. [DOI] [PubMed] [Google Scholar]
- (6).Centers for Disease Control and Prevention. New CDC Analysis Shows Steep and Sustained Increases in STDs in Recent Years. 2017, https://www.cdc.gov/media/releases/2018/p0828-increases-in-stds.html (accessed Dec 07, 2021).
- (7).Rowley J; Vander Hoorn S; Korenromp E; Low N; Unemo M; Abu-Raddad LJ; Chico RM; Smolak A; Newman L; Gottlieb S; Thwin SS; Broutet N; Taylor MM Chlamydia, gonorrhoea, trichomoniasis and syphilis: global prevalence and incidence estimates, 2016. Bull. W. H. O 2019. 2019, 97, 548–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Haggerty CL; Gottlieb SL; Taylor BD; Low N; Xu F; Ness RB Risk of Sequelae after Chlamydia trachomatis Genital Infection in Women. J. Infect. Dis 2010, 201, S134–S155. [DOI] [PubMed] [Google Scholar]
- (9).Elwell C; Mirrashidi K; Engel J Chlamydia Cell biology and Pathogenesis. Nat. Rev. Microbiol 2016, 14, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Paavonen J; Eggert-Kruse W Chlamydia trachomatis: Impact on Human Reproduction. Hum. Reprod. Update 1999, 5, 433–447. [DOI] [PubMed] [Google Scholar]
- (11).Darville T; Hiltke TJ Pathogenesis of Genital Tract Disease due to Chlamydia trachomatis. J. Infect. Dis 2010, 201, S114–S125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Abdelrahman YM; Belland RJ The chlamydial developmental cycle: Figure 1. FEMS Microbiol. Rev 2005, 29, 949–959. [DOI] [PubMed] [Google Scholar]
- (13).de la Maza LM; Zhong G; Brunham RC Update in Chlamydia trachomatis Vaccinology. Clin. Vaccine Immunol 2017, 24, No. e00543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Schautteet K; De Clercq E; Vanrompay D Chlamydia trachomatis Vaccine Research Through the Years. Infect. Dis. Obstet. Gynecol 2011, 2011, 963513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kong FYS; Hocking JS Treatment Challenges for Urogenital and Anorectal Chlamydia trachomatis. BMC Infect. Dis 2015, 15, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhou Z; Xie L; Wang L; Xue M; Xu D; Zhong G Effects of Immunomodulatory Drug Fingolimod (FTY720) on Chlamydia Dissemination and Pathogenesis. Infect. Immun 2020, 88, e00281–00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Horner P; Saunders J Should Azithromycin 1 g be Abandoned as a Treatment For Bacterial STIs? The Case for and Against. Sex. Transm. Infect 2017, 93, 85–87. [DOI] [PubMed] [Google Scholar]
- (18).Workowski KA Centers for Disease Control and Prevention Sexually Transmitted Diseases Treatment Guidelines. Clin. Infect. Dis 2015, 61, S759–S762. [DOI] [PubMed] [Google Scholar]
- (19).Sandoz KM; Rockey DD Antibiotic Resistance in Chlamydiae. Future Microbiol. 2010, 5, 1427–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Horner PJ Azithromycin Antimicrobial Resistance and Genital Chlamydia trachomatis Infection: Duration of Therapy May be the Key to Improving Efficacy. Sex. Transm. Infect 2012, 88, 154–156. [DOI] [PubMed] [Google Scholar]
- (21).Somani J; Bhullar VB; Workowski KA; Farshy CE; Black CM Multiple Drug-Resistant Chlamydia trachomatis Associated with Clinical Treatment Failure. J. Infect. Dis 2000, 181, 1421–1427. [DOI] [PubMed] [Google Scholar]
- (22).Wang SA; Papp JR; Stamm WE; Peeling RW; Martin DH; Holmes KK Evaluation of Antimicrobial Resistance and Treatment Failures for Chlamydia trachomatis: A Meeting Report. J. Infect. Dis 2005, 191, 917–923. [DOI] [PubMed] [Google Scholar]
- (23).Jernberg C; Löfmark S; Edlund C; Jansson JK Long-term Impacts of Antibiotic Exposure on the Human Intestinal Microbiota. Microbiology 2010, 156, 3216–3223. [DOI] [PubMed] [Google Scholar]
- (24).Doan T; Arzika AM; Ray KJ; Cotter SY; Kim J; Maliki R; Zhong L; Zhou Z; Porco TC; Vanderschelden B; Keenan JD; Lietman TM Gut Microbial Diversity in Antibiotic-Naive Children After Systemic Antibiotic Exposure: A Randomized Controlled Trial. Clin. Infect. Dis 2017, 64, 1147–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Korpela K; Salonen A; Virta LJ; Kekkonen RA; Forslund K; Bork P; de Vos WM Intestinal Microbiome is Related to Lifetime Antibiotic use in Finnish Pre-school Children. Nat. Commun 2016, 7, 10410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wei S; Mortensen MS; Stokholm J; Brejnrod AD; Thorsen J; Rasmussen MA; Trivedi U; Bisgaard H; Sørensen SJ Short- and Long-term Impacts of Azithromycin Treatment on the Gut Microbiota in Children: A double-blind, Randomized, Placebo-controlled Trial. EBioMedicine 2018, 38, 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Mohammad H; Reddy PVN; Monteleone D; Mayhoub AS; Cushman M; Seleem MN Synthesis and antibacterial evaluation of a novel series of synthetic phenylthiazole compounds against methicillin-resistant Staphylococcus aureus (MRSA). Eur. J. Med. Chem 2015, 94, 306–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Tamarelle J; Ma B; Gajer P; Humphrys MS; Terplan M; Mark KS; Thiébaut ACM; Forney LJ; Brotman RM; Delarocque-Astagneau E; Bavoil PM; Ravel J Nonoptimal Vaginal Microbiota After Azithromycin Treatment for Chlamydia trachomatis Infection. J. Infect. Dis 2019, 221, 627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Chen X-S; Yin Y-P; Wei W-H; Wang H-C; Peng R-R; Zheng H-P; Zhang J-P; Zhu B-Y; Liu Q-Z; Huang S-J High Prevalence of Azithromycin Resistance to Treponema Pallidum in Geographically Different Areas in China. Clin. Microbiol. Infect 2013, 19, 975–979. [DOI] [PubMed] [Google Scholar]
- (30).Read P; Tagg KA; Jeoffreys N; Guy RJ; Gilbert GL; Donovan B Treponema Pallidum Strain Types and Association with Macrolide Resistance in Sydney, Australia: New TP0548 Gene Types Identified. J. Clin. Microbiol 2016, 54, 2172–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wood NA; Chung KY; Blocker AM; Rodrigues de Almeida N; Conda-Sheridan M; Fisher DJ; Ouellette SP Initial Characterization of the Two ClpP Paralogs of Chlamydia trachomatis Suggests Unique Functionality for Each. J. Bacteriol 2019, 201, e00635–e00618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Leung E; Datti A; Cossette M; Goodreid J; McCaw SE; Mah M; Nakhamchik A; Ogata K; El Bakkouri M; Cheng Y-Q; Wodak SJ; Eger BT; Pai EF; Liu J; Gray-Owen S; Batey RA; Houry WA Activators of Cylindrical Proteases as Antimicrobials: Identification and Development of Small Molecule Activators of ClpP Protease. Chem. Biol 2011, 18, 1167–1178. [DOI] [PubMed] [Google Scholar]
- (33).Seleem MA; Rodrigues de Almeida N; Chhonker YS; Murry DJ; Guterres Z. d. R.; Blocker AM; Kuwabara S; Fisher DJ; Leal ES; Martinefski MR; Bollini M; Monge ME; Ouellette SP; Conda-Sheridan M Synthesis and Antichlamydial Activity of Molecules Based on Dysregulators of Cylindrical Proteases. J. Med. Chem 2020, 63, 4370–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Nogueira AT; Braun KM; Carabeo RA Characterization of the Growth of Chlamydia trachomatis in In vitro-generated Stratified Epithelium. Front. Cell. Infect. Microbiol 2017, 7, 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Labute P; Williams C; Feher M; Sourial E; Schmidt JM Flexible Alignment of Small Molecules. J. Med. Chem 2001, 44, 1483–1490. [DOI] [PubMed] [Google Scholar]
- (36).Floresta G; Cilibrizzi A; Abbate V; Spampinato A; Zagni C; Rescifina A FABP4 inhibitors 3D-QSAR model and isosteric replacement of BMS309403 datasets. Data Brief 2019, 22, 471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Sestito S; Pruccoli L; Runfola M; Citi V; Martelli A; Saccomanni G; Calderone V; Tarozzi A; Rapposelli S Design and Synthesis of H2S-donor Hybrids: A new Treatment for Alzheimer’s Disease? Eur. J. Med. Chem 2019, 184, 111745. [DOI] [PubMed] [Google Scholar]
- (38).Du-a-man S; Soorukram D; Kuhakarn C; Tuchinda P; Reutrakul V; Pohmakotr M Synthesis of (+)-Lentiginosine and Its Pyrrolizidine Analogue Based on Intramolecular Cyclization of α-Sulfinyl Carbanions. Eur. J. Org. Chem 2014, 1708–1715. [Google Scholar]
- (39).Saravanakumar A; Sadighi A; Ryu R; Akhlaghi F Physicochemical Properties, Biotransformation, and Transport Pathways of Established and Newly Approved Medications: A systematic Review of the Top 200 Most Prescribed Drugs vs. the FDA-Approved Drugs Between 2005 and 2016. Clin. Pharmacokinet 2019, 58, 1281–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Veber DF; Johnson SR; Cheng H-Y; Smith BR; Ward KW; Kopple KD Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem 2002, 45, 2615–2623. [DOI] [PubMed] [Google Scholar]
- (41).Lipinski CA Drug-like Properties and the Causes of Poor Solubility and Poor Permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [DOI] [PubMed] [Google Scholar]
- (42).Lipinski CA Lead-and drug-like Compounds: the Rule-of-five Revolution. Drug Discovery Today: Technol 2004, 1, 337–341. [DOI] [PubMed] [Google Scholar]
- (43).Zhao YH; Abraham MH; Le J; Hersey A; Luscombe CN; Beck G; Sherborne B; Cooper I Rate-limited Steps of Human Oral Absorption and QSAR Studies. Pharm. Res 2002, 19, 1446–1457. [DOI] [PubMed] [Google Scholar]
- (44).Robert Batey MC; Datti A; Eger BT; Fai EF; Jordan G; Gray-Owen SD; Houry WA; Leung E; Liu J; Nhieu AJ Activators of Cylindrical Proteases. WO 2012079164 A1, 2011. [Google Scholar]
- (45).Ouellette SP; Karimova G; Subtil A; Ladant D Chlamydia Co-Opts the Rod Shape-Determining Proteins MreB and Pbp2 for Cell Division. Mol. Microbiol 2012, 85, 164–178. [DOI] [PubMed] [Google Scholar]
- (46).West SK; Moncada J; Munoz B; Mkocha H; Storey P; Hardick J; Gaydos CA; Quinn TC; Schachter J Is There Evidence for Resistance of Ocular Chlamydia trachomatis to Azithromycin After Mass Treatment for Trachoma Control? J. Infect. Dis 2014, 210, 65–71. [DOI] [PubMed] [Google Scholar]
- (47).O’Brien KS; Emerson P; Hooper P; Reingold AL; Dennis EG; Keenan JD; Lietman TM; Oldenburg CE Antimicrobial Resistance Following Mass Azithromycin Distribution for Trachoma: a Systematic Review. Lancet Infect. Dis 2019, 19, 14–25. [DOI] [PubMed] [Google Scholar]
- (48).Borel N; Leonard C; Slade J; Schoborg RV Chlamydial Antibiotic Resistance and Treatment Failure in Veterinary and Human Medicine. Curr. Clin. Microbiol. Rep 2016, 3, 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Hathorn E; Opie C; Goold P What is the Appropriate Treatment for the Management of Rectal Chlamydia trachomatis in Men and Women? Sex. Transm. Infect 2012, 88, 352–354. [DOI] [PubMed] [Google Scholar]
- (50).Tamarelle J; Ma B; Gajer P; Humphrys MS; Terplan M; Mark KS; Thiébaut ACM; Forney LJ; Brotman RM; Delarocque-Astagneau E; Bavoil PM; Ravel J Nonoptimal Vaginal Microbiota After Azithromycin Treatment for Chlamydia trachomatis Infection. J. Infect. Dis 2020, 221, 627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Chern KC; Shrestha SK; Cevallos V; Dhami HL; Tiwari P; Chern L; Whitcher JP; Lietman TM Alterations in the Conjunctival Bacterial Flora Following a Single Dose of Azithromycin in a Trachoma Endemic Area. Br. J. Ophthalmol 1999, 83, 1332–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Kong FYS; Hocking JS Treatment Challenges for Urogenital and Anorectal Chlamydia trachomatis. BMC Infect. Dis 2015, 15, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Ho DK-H; Sawicki C; Grassly N Antibiotic Resistance in Streptococcus Pneumoniae after Azithromycin Distribution for Trachoma. J. Trop. Med 2015, 2015, 917370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Bojang E; Jafali J; Perreten V; Hart J; Harding-Esch EM; Sillah A; Mabey DCW; Holland MJ; Bailey RL; Roca A; Burr SE Short-term Increase in Prevalence of Nasopharyngeal Carriage of Macrolide-resistant Staphylococcus aureus Following Mass Drug Administration with Azithromycin for Trachoma Control. BMC Microbiol. 2017, 17, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Raymond B Five Rules for Resistance Management in the Antibiotic Apocalypse, A road Map for Integrated Microbial Management. Evol. Appl 2019, 12, 1079–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Ahmed A; Azim A; Gurjar M; Baronia AK Current Concepts in Combination Antibiotic Therapy for Critically Ill Patients. Indian J. Crit. Care Med 2014, 18, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Wambaugh MA; Shakya VPS; Lewis AJ; Mulvey MA; Brown JCS High-throughput Identification and Rational Design of Synergistic Small-molecule Pairs for Combating and Bypassing Antibiotic Resistance. PLoS Biol. 2017, 15, No. e2001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Shang S; Xia L; Zhong M; Zhang J; Zhao J; Gong X; Mabey D; Wang Q In vitro Effects of Spectinomycin and Ceftriaxone Alone or in Combination with Other Antibiotics Against Chlamydia trachomatis. Antimicrob. Agents Chemother 2005, 49, 1584–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Cottarel G; Wierzbowski J Combination Drugs, an Emerging Option for Antibacterial Therapy. Trends Biotechnol. 2007, 25, 547–555. [DOI] [PubMed] [Google Scholar]
- (60).Orhan G; Bayram A; Zer Y; Balci I Synergy tests by E test and Checkerboard Methods of Antimicrobial Combinations Against Brucella melitensis. J. Clin. Microbiol 2005, 43, 140–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Berenbaum MC A Method for Testing for Synergy with any Number of Agents. J. Infect. Dis 1978, 137, 122–130. [DOI] [PubMed] [Google Scholar]
- (62).Foucquier J; Guedj M Analysis of Drug Combinations: Current Methodological Landscape. Pharmacol. Res. Perspect 2015, 3, No. e00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Norden CW; Wentzel H; Keleti E Comparison of Techniques for Measurement of In vitro Antibiotic Synergism. J. Infect. Dis 1979, 140, 629–633. [DOI] [PubMed] [Google Scholar]
- (64).O’Meara CP; Andrew DW; Beagley KW The mouse model of Chlamydia genital tract infection: a review of infection, disease, immunity and vaccine development. Curr. Mol. Med 2014, 14, 396–421. [DOI] [PubMed] [Google Scholar]
- (65).Calculator Plugins Were Used for Structure Property Prediction and Calculation, Marvin 20.4., 2020, ChemAxon. http://www.chemaxon.com (accessed Dec 07, 2021). [Google Scholar]
- (66).Bonapace CR; Bosso JA; Friedrich LV; White RL Comparison of methods of interpretation of checkerboard synergy testing. Diagn. Microbiol. Infect. Dis 2002, 44, 363–366. [DOI] [PubMed] [Google Scholar]
- (67).Schneider CA; Rasband WS; Eliceiri KW NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.