

Abstract
With increasing attention on the essential roles of the tumour microenvironment in recent years, the nervous system has emerged as a novel and crucial facilitator of cancer growth. In this Review, we describe the foundational, translational, and clinical advances illustrating how nerves contribute to tumour proliferation, stress adaptation, immunomodulation, metastasis, electrical hyperactivity and seizures, and neuropathic pain. Collectively, this expanding knowledge base reveals multiple therapeutic avenues for cancer neuroscience that warrant further exploration in clinical studies. we discuss the available clinical data, including ongoing trials investigating novel agents targeting the tumour–nerve axis, and the therapeutic potential for repurposing existing neuroactive drugs as an anti-cancer approach, particularly in combination with established treatment regimens. Lastly, we discuss the clinical challenges of these treatment strategies and highlight unanswered questions and future directions in the burgeoning field of cancer neuroscience.
Introduction
Evidence of nerve involvement in tumours dates back to the 19th century, when Dr Hugh H Young first reported the presence of abundant nerve fibres throughout several peripheral tumours.1 Dr H J Scherer described a similar perineuronal satellitosis of nerves within intracranial gliomas, noting that “the first manifestation of the neoplastic process is a collection of glioma cells about all or a great many of the nerve cells”.2 These foundational observations provided preliminary evidence suggesting an integral role for nerves in malignancy, as opposed to their incidental capture within the developing tumour, and set the stage for what has become known as the field of cancer neuroscience. For more on the parallels between cancer neuroscience and the field of tumour angiogenesis, please see the appendix (p 1).
Perineural invasion, a histopathological finding introduced in 1985, that describes malignant cells surrounding, invading, or within nerves, is a prominent manifestation of tumour–nerve interactions.3,4 Clinically, perineural invasion is a potential indication for adjuvant radiotherapy and is considered in staging and risk stratification of several malignancies, including penile cancer, head and neck cancers, oesophageal cancer, and thyroid cancer.5
Beyond irradiating cancer-infiltrated nerves, molecular targeting of the tumour–nerve axis is potentially poised for a major impact in clinical oncology. In contrast to previous reviews and perspectives that have extensively summarised mechanisms of tumour–nerve crosstalk learned from preclinical models,6–13 we focus herein on providing an integrated clinical and translational framework by examining current and prospective therapeutic avenues within cancer neuroscience.
Therapeutic avenues for cancer neuroscience
A surge of preclinical and clinical evidence in the past two decades has begun to elucidate the role of the nervous system in promoting cancer growth. In light of the many mechanisms of tumour–nerve crosstalk and their accompanying clinicopathological ramifications, we have organised their translational implications into six broad avenues of intervention: inhibiting tumour growth, combating treatment resistance and tumour adaptation to stress, enhancing anti-tumour immunity, preventing dissemination and metastasis, and managing morbidity by dampening electrical hyperactivity and seizures as well as neuropathic pain (figure 1). We will highlight the potential therapeutic opportunities and challenges galvanised by preclinical and preliminary clinical data.
Figure 1: Six therapeutic avenues of cancer neuroscience.
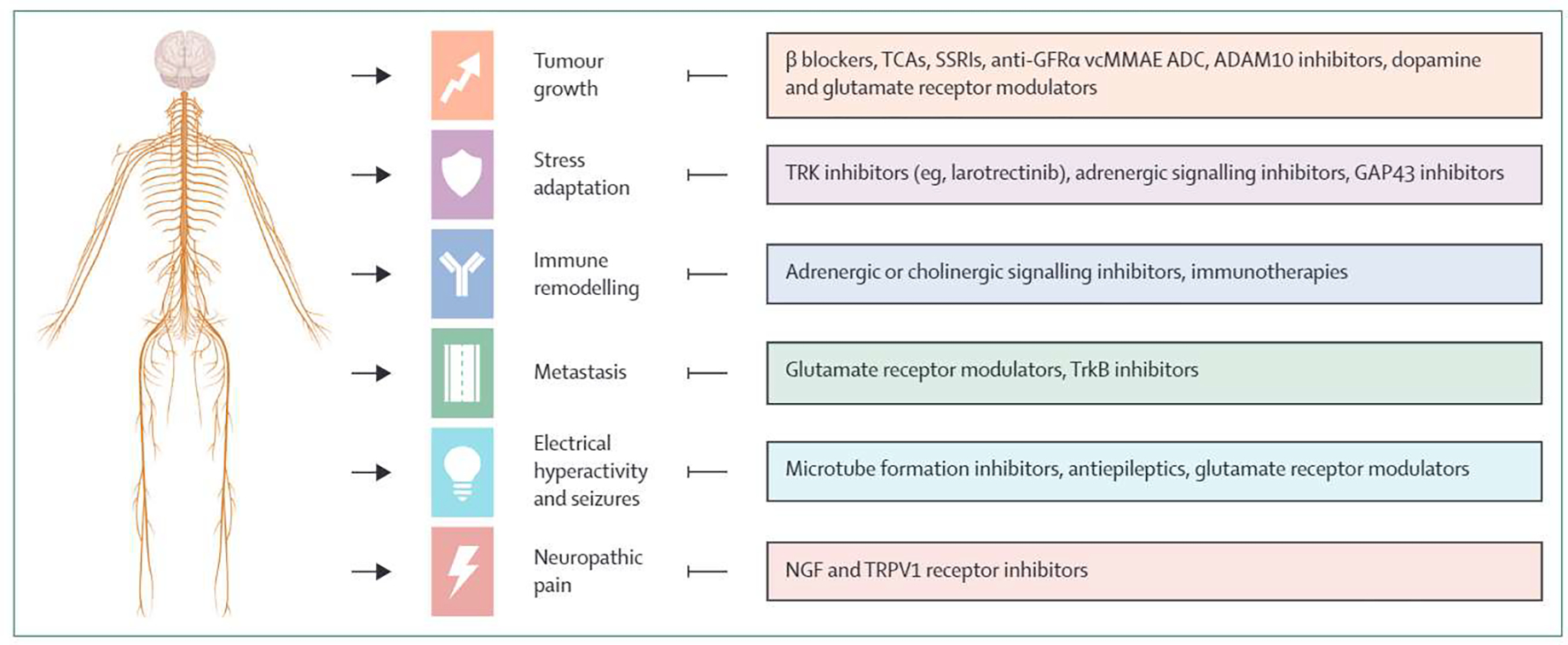
TCA=tricyclic antidepressant. SSRI=selective serotonin reuptake inhibitor. GFRα=glial cell line-derived neurotrophic factor family receptor alpha. ADC=antibody–drug conjugate. BDNF=brain-derived neurotrophic factor. NGF=nerve growth factor.
Inhibiting tumour growth via modulation of the nervous system
Many cancers originate from glandular and ductal epithelial tissues. During normal development of these tissues, cells secrete neurotrophic factors that promote nerve recruitment, axon navigation, and neurite outgrowth.14,15 As reviewed elsewhere,6–13 a wealth of evidence currently suggests that expression of these neurodevelopmental and regenerative processes is aberrantly activated again during tumorigenesis, leading to a marked increase in intratumoural nerve density and subsequent tumour growth.
Multiple mechanisms have been described invoking a clear role of neuronal innervation in tumour growth across a variety of cancers, including gliomas and prostate, pancreas, and gastric cancers (table 1). These data highlight common signalling processes involving neurochemicals (eg, glutamate, norepinephrine, acetylcholine) and secreted factors (NGF, BDNF, GDNF). In addition, neurons can directly communicate with tumour cells by forming tumour–nerve synapses. These signalling mechanisms typically activate a classical oncogenic pathway that promotes tumour growth. For example, seminal work by Venkatesh and colleagues (table 1, row 12) showed that in high-grade gliomas and optic gliomas, the protein neuroligin-3 (NLGN3) is released from surrounding neurons and oligodendroglial precursor cells in response to neuronal activity, undergoes cleavage to its extracellular form by the protease ADAM10, and binds to receptors on the tumour cell surface to activate PI3K-mTOR signalling (figure 2). The net effect is tumour proliferation secondary to increased neural activity. An ADAM 10 inhibitor is now currently undergoing phase 1 testing in paediatric high-grade gliomas (table 2), and trials in adult patients with glioblastoma are planned. Besides the NLGN3–ADAM10 pathway, multiple other mechanisms have similarly delineated the role of neuronal input on promoting tumour growth across cancers (table 1). In addition to molecular inhibition, surgical denervation strategies have also been attempted (appendix p 1).
Table 1:
Preclinical and early clinical functional evidence of tumour-nerve interactions
| Tumour type | Therapeutic avenue | Mechanism and target* | Contribution to cancer | |
|---|---|---|---|---|
|
| ||||
| 1 | Prostate cancer | Tumour growth | Adrenergic signalling | Sympathetic neurogenesis and an increase in the density of β-adrenergic nerves promotes early stages of tumorigenesis (eg, low-to-high-grade prostatic intraepithelial neoplasia).16,17 |
| 2 | Prostate cancer | Tumour growth | Adrenergic signalling (ADRB2) | Adrenergic nerves promote a metabolic shift in endothelial cells towards aerobic glycolysis that triggers angiogenesis and malignant growth.16 |
| 3 | Pancreatic cancer | Tumour growth | Sensory neuron input | Dorsal root ganglion sensory neurons convey inflammatory signals from pancreatic cancer tumours to the CNS in a Kras-induced genetically engineered mouse model, contributing to tumour initiation and growth.18 |
| 4 | Pancreatic cancer | Tumour growth | Cholinergic signalling | Cholinergic signalling suppresses pancreatic cancer growth through stimulation of muscarinic receptors.19 |
| 5 | Pancreatic cancer | Tumour growth | Catecholamine signalling (ADRB2, NGF) | Catecholamines bind to ADRB2 and increase nerve density and neurotrophin secretion, creating a feedforward loop that promotes tumour development in a mouse model of pancreatic cancer.20 |
| 6 | Basal cell carcinoma | Tumour growth | Sensory neuron input (Hedgehog pathway) | Cutaneous innervation activates Hedgehog signalling in stem-cell niches within the skin to promote tumorigenesis.21 |
| 7 | Medulloblastoma | Tumour growth | Neurochemical signalling (mGluR4) | Activation of mGluR4 receptors prevents development of medulloblastoma in mouse models.22 |
| 8 | Gastric cancer | Tumour growth | Cholinergic signalling (NGF, Wnt pathway) | Vagal innervation results in NGF secretion via acetylcholine signalling, thereby promoting gastric tumorigenesis through Wnt activation.23 |
| 9 | Pancreatic cancer | Tumour growth | Protein signalling (ARTN) | The neurotrophic factor ARTN contributes to perineural invasion in pancreatic cancer.24 |
| 10 | Glioma | Tumour growth | Protein signalling (BDNF) | BDNF is secreted in an activity-dependent manner and binds to the receptorTrkB, promoting glioma growth.25 |
| 11 | Pancreatic cancer | Tumour growth | Protein signalling (GDNF/RET) | GDNF secretion and its interactions with its receptor RET promote perineural invasion in pancreatic cancer.26 |
| 12 | Glioma | Tumour growth | Protein signalling (NLGN3, ADAM10) | The synaptic adhesion protein NLGN3 is released from neighbouring non-glioma neurons and cleaved by the protease ADAM10 in a neuron activity-dependent manner. The resultant secreted form of NLGN3 promotes growth through a PI3K-mTOR-dependent pathway.27–30 |
| 13 | Multiple myeloma | Tumour growth, immune remodelling | Protein signalling (NNT-1) | NNT-1 is a neurotrophic factor that demonstrates B cell-stimulating capability and promotes growth and survival in myeloma.31 |
| 14 | Multiple myeloma | Tumour growth | Protein signalling (BDNF) | BDNF promotes tumour survival in multiple myeloma through binding to the receptorTrkB, thereby activating MAPK and PI3K/Akt signalling cascades.32 |
| 15 | Thyroid carcinoma (NRTC), NSCLC | Tumour growth | Neurotrophic-tropomyosin receptor kinase (NTRK) rearrangement | NTRK gene fusions can produce chimeric oncoproteins with constitutively activated kinase function (TrkA, TrkB, andTrkC), resulting in proliferation through the RAS–RAF–MAPK pathway.33,34 |
| 16 | Glioma | Tumour growth, electrical hyperactivity and seizures | Direct synapse formation (AMPAR) | Nerves form direct glutamatergic synapses onto glioma cells and modulate tumour microtube-mediated invasion in gliomas.29,35 |
| 17 | Glioma | Electrical hyperactivity and seizures | Glutamate signalling (AMPAR) | Gliomas release high amounts of glutamate to surrounding tissue and also exhibit glutamate-response growth via Ca2+-permeable AMPA receptors, establishing a positive feedback loop of nerve–tumour hyperactivity and tumour proliferation.29,35–37 |
| 18 | Glioma | Tumour growth, electrical hyperactivity and seizures | Neurochemical signalling (GABA) | GABA receptors are present on low-grade astrocytomas and oligodendrogliomas, and GABA currents have also been evoked in glioblastoma.38,39 |
| 19 | Glioma | Tumour growth, electrical hyperactivity and seizures | Neurochemical signalling (DRD4, DRD2) | Blockade of DRD4 has been shown to inhibit glioblastoma proliferation via disruption of autophagy, and blockade of DRD2 alone or in combination with EGFR blockade might have therapeutic potential.40,41 |
| 20 | Glioma | Tumour growth, electrical hyperactivity and seizures | Neurochemical signalling (GABA) | Gliomas downregulate inhibitory GABA currents in the surrounding tumour microenvironment.42 |
| 21 | Brain metastases | Metastasis | Neurochemical signalling (GABA) | Breast cancer brain metastases upregulate GABA receptors and GABA catabolism.43 |
| 22 | Brain metastases | Metastasis, electrical hyperactivity and seizures | Direct synapse formation (NMDAR) | Breast cancer cells form astrocyte-like synapses (pseudo-tripartite synapses) to access glutamate secreted from neurons, which activates the NMDAR pathway and promotes brain colonisation.44 |
| 23 | Metastatic tumours | Metastasis | Anoikis resistance (TrkB) | TrkB is a neurotrophic receptor that is shown to suppress anoikis, contributing to metastatic spread through supporting circulation of cancerous cells.45 |
| 24 | Prostate cancer | Metastasis | Parasympathetic signalling (CHRM1) | Parasympathetic innervation of cholinergic fibres stimulates prostate cancer dissemination at later stages.17 |
| 25 | Brain metastases | Metastasis, immune remodelling | Adrenergic signalling (ADRB2) | β2-adrenergic signalling facilitated breast cancer metastases by recruiting macrophages and inducing a pro-metastatic and immunosuppressive M2 state. Treatment with propranolol reversed this response in animals.46 |
| 26 | Glioma | Tumour growth, electrical hyperactivity and seizures | Neuronal hyperexcitability (GPC3) | Subsets of glioblastoma driven by PIK3CA secrete GPC3, which drives hyperexcitability and glioma growth.47 |
| 27 | Glioma | Electrical hyperactivity and seizures, stress adaptation | Microtube formation (Connexin 43 gap junctions) | Glioma cells form microtubes that facilitate direct communication with surrounding glioma and non-gliomatous cells via Connexin 43-mediated gap junctions, allowing for propagation of calcium waves that have been implicated as a cause of treatment resistance.48 |
| 28 | Pancreatic cancer | Neuropathic pain | Neurotrophin signalling (NGF) | Cancer cells, fibroblasts, and immune cells can release NGF to recruit and activate sensory neurons by way of the receptor TrkA. Elevated expression of NGF–TrkA in pancreatic cancer tissues is thought to be associated with increased perineural invasion and pain.49 |
| 29 | Pancreatic cancer | Neuropathic pain | Neurotrophin signalling (TRPV1) | NGF and other neurotrophins can activate the receptor TRPV1, a cation channel that facilitates nociception in sensory neurons. TRPV1 activation can trigger sensory neuronal membrane depolarisation and the release of substance P or CGRP to transmit pain signals.50 |
| 30 | Glioma | Stress adaptation | Dopamine signalling | Combination treatment with the dopamine antagonist quetiapine, atorvastatin, and radiation prolonged survival in glioblastoma xenografts.51 |
| 31 | Pancreatic cancer | Stress adaptation | Decreased apoptosis (MALT1, TRAF) | In-vitro co-cultures of mouse dorsal root ganglia and human pancreatic cancer cells demonstrated that pro-survival (MALT1, TRAF) genes are upregulated in neoplastic cells.52 |
| 32 | Prostate cancer | Stress adaptation | Decreased apoptosis (NFκB) | Prostate cancer cells exhibiting perineural invasion demonstrate increased proliferation and decreased apoptosis through a NFκB-dependent mechanism.53 |
| 33 | Ovarian cancer | Stress adaptation | Neurotrophin signalling (BDNF) | Activation of the BDNF–TrkB pathway inhibits apoptosis, and knockdown of TrkB enhances apoptosis.54 |
| 34 | Pancreatic cancer | Stress adaptation, tumour growth | Axonal recruitment (NGF) | Starved pancreatic cancer cells in nutrient-poor, serine-deprived desmoplastic environments increase production of NGF, which promotes axonal recruitment as a means of L-serine acquisition. Treatment with theTrk–NGF inhibitor larotrectinib decreased pancreatic cancer tumour growth.55 |
| 35 | Breast cancer | Stress adaptation, metastasis | β-adrenergic signalling | Chronically stressed mice exhibited 38-times higher frequency of distant metastases relative to unstressed mice. Propranolol blocked metastases in chronically stressed animals but had no effect on metastatic burden in unstressed mice.46 |
| 36 | Melanoma | Immune remodelling | β-adrenergic signalling (ADRB2) | Concurrent β-blockade and immunotherapy is associated with improved survival in metastatic melanoma and might be mediated by the ADRB2 receptor.56,57 |
| 37 | Colon cancer, melanoma, and breast cancer | Immune remodelling | β-adrenergic signalling | Addition of β2-adrenergic blockade improved local control and abscopal effects at distant metastatic sites following irradiation.58 |
| 38 | HPV E6+ and E7+ tumours | Immune remodelling | β-adrenergic signalling | Propranolol improved efficacy of an anti-tumour vaccine via enhancement of CD8+ T lymphocyte tumour infiltration.59 |
| 39 | Lung cancer | Immune remodelling | CD8+ T-cell infiltration | Depletion of tumour-associated macrophages in combination with anti-PD-1 immunotherapy restores cytotoxic T-cell migration and infiltration.60,61 |
| 40 | Melanoma | Immune remodelling | Tumour innervation (Schwann cells) | Melanoma cells activate Schwann cells in the tumour microenvironment that promote tumour growth.62 |
| 41 | Pancreatic cancer | Immune remodelling | Cholinergic signalling (acetylcholine) | Acetylcholine impairs the recruitment of CD8+ by pancreatic adenocarcinoma cells and favours T-helper-2 (Th2) cells. Subdiaphragmatic vagotomy in tumour-bearing mice was associated with increased CD8+ concentrations, elevated Th1:Th2 ratio, and improved survival.63 |
GABA=γ-aminobutyric-acid. HPV=human papillomavirus. NGF=nerve growth factor. NSCLC=non-small cell lung cancer. NRTC=NTRK-rearranged thyroid carcinoma. TRAF=TNF receptor associated factors.
Targets are provided in parentheses where available.
Figure 2: Example mechanisms of therapeutic avenues of cancer neuroscience.
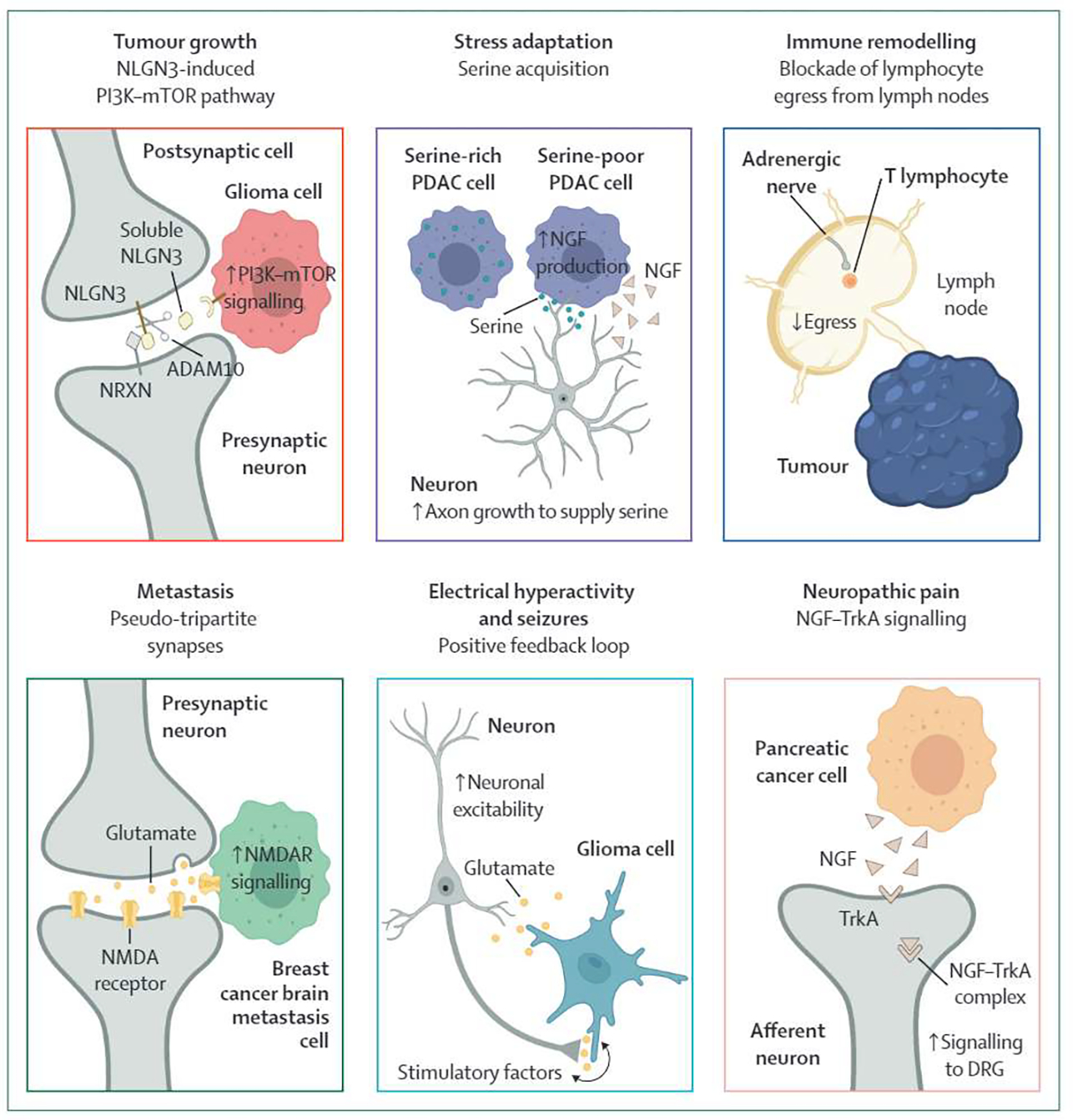
NGF=nerve growth factor. DRG=dorsal root ganglia. PDAC=pancreatic ductal adenocarcinoma.
Table 2:
Select completed and ongoing clinical trials targeting the tumour–nerve axis
| Therapy | Target* | Phase | Actual or target accrual | Primary endpoint | Status | Outcome | Reference | |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| H3K27M spinal cord glioma | ONC201 | DRD2/DRD3 antagonist | Phase 2 | 76 | Progression-free survival | Ongoing | 8 of 12 patients alive at <12 months median follow-up | NCT02525692 |
| H3K27M diffuse midline glioma | ONC201 | DRD2/DRD3 antagonist | Phase 2 | 95 | Objective response rate | Ongoing | No dose-limiting toxicity | NCT03295396 |
| Thalamic H3K27M glioma | ONC201 | DRD2/DRD3 antagonist | Phase 2 | 95 | Objective response rate | Ongoing | 26·3% 6-month progression-free survival | NCT03295396 |
| Recurrent glioblastoma | ONC201 | DRD2/DRD3 antagonist | Phase 2 | 17 | Progression-free survival | Completed | No difference in 6-month progression-free survival | NCT02525692 |
| Neuroendocrine tumours | ONC201 | DRD2/DRD3 antagonist | Phase 2 | 25 | Tumour response | Ongoing | 56% progressed | NCT03034200 |
| Paediatric diffuse intrinsic pontine glioma | ONC201 | DRD2/DRD3 antagonist | Phase 1 | 130 | Recommended phase 2 dose | Ongoing | Not reported | NCT03416530 |
| Endometrial cancer | ONC201 | DRD2/DRD3 antagonist | Phase 2 | 30 | Progression-free survival | Ongoing | Not reported | NCT03485729 |
| Acute myeloid leukaemia | ONC101 | DRD2/DRD3 antagonist | Phase 1 | 20 | Dose-limiting toxicity | Ongoing | Not reported | NCT03932643 |
| Ovarian, fallopian tube, or primary peritoneal cancer | ONC201 | DRD2/DRD3 antagonist | Phase 2 | 62 | Dose-limiting toxicity, adverse events, objective response rate, progression-free survival | Ongoing | Not reported | NCT04055649 |
| Recurrent and rare CNS tumours | ONC206 | DRD2 antagonist | Phase 1 | 102 | Dose-limiting toxicity | Ongoing | Not reported | NCT04541082 |
| Paediatric high-grade glioma | INCB7839 | ADAM10/17 inhibitor | Phase 1 | 28 | Adverse events | Ongoing | Not reported | NCT04295759 |
| Glioblastoma | Talampanel | AMPAR regulator | Phase 2 | 72 | Overall survival | Completed | Improved overall survival compared with historical controls | NCT00943826, NCT00689221, NCT00813943, NCT00884741 |
| Glioblastoma | Memantine, mefloquine, metformin | NMDAR regulator | Phase 1/2 | 85 | Dose-limiting toxicity | Completed | Dose-limiting toxicity: dizziness, gastrointestinal symptoms, lymphopenia | NCT01430351 |
| Glioblastoma | Memantine | NMDAR regulator | Phase 2 | 4 | Overall survival | Terminated | Not reported | NCT01260467 |
| Glioblastoma | Meclofenamic acid | NSAID64 | Phase 1/2 | 72 | Dose-limiting toxicity, progression-free survival | Ongoing | Not reported | EudraCT2021-000708-39 |
| Glioma | Talampanel | AMPAR inhibitor | Phase 2 | 30 | Progression-free survival | Terminated | Early termination due to futility | NCT00062504 |
| Ovarian, primary peritoneal, or fallopian tube cancer | Propranolol, chemotherapy | β-adrenergic antagonist | Phase 1 | 32 | Feasibility | Completed | Not reported | NCT01504126 |
| Ovarian cancer | Propranolol | β-adrenergic antagonist | Phase 1 | 26 | Feasibility | Completed | Improved quality of life | NCT01308944 |
| Colorectal cancer | Propranolol, COX2 inhibitor | β-adrenergic antagonist | Phase 3 | 34 | 3-year recurrence | Completed | No difference in recurrence | NCT00888797 |
| Breast cancer | Propranolol, COX2 inhibitor | β-adrenergic antagonist | Phase 2 | 38 | Pro-metastatic cell markers | Completed | Decreased pro-metastatic and invasive markers | NCT00502684 |
| Breast cancer | Propranolol, chemotherapy | β-adrenergic antagonist | Phase 2 | 10 | Compliance | Ongoing | Not reported | NCT01847001 |
| Breast cancer | Propranolol | β-adrenergic antagonist | Phase 2 | 60 | Gene expression biomarkers | Completed | Reduced metastatic potential | ACTRN12615000889550 |
| Pancreatic cancer | Propranolol etodolac | β-adrenergic antagonist/COX-2 Inhibitor | Phase 2 | 210 | Recurrence | Ongoing | Not reported | NCT03838029 |
| Pancreatic cancer | Bethanechol | Muscarinic agonist | Phase 1 | 18 | Change in Ki-67 | Ongoing | Not reported | NCT03572283 |
| Cancer pain | Tanezumab | Antibody against NGF | Phase 3 | 156 | Change in pain | Completed | Not reported | NCT00545129 NCT02609828 |
| Cancer pain | Resiniferatoxin | Capsaicin analogue | Phase 1 | 45 | Safety | Ongoing | Not reported | NCT00804154 |
| Melanoma | Propranolol, pembrolizumab | β-adrenergic antagonist | Phase 1 | 9 | Safety | Completed | No dose-limiting toxicity; objective response rate 78% | NCT03384836 |
| Bladder cancer | Propranolol, pembrolizumab | β-adrenergic antagonist | Phase 2 | 25 | Objective response rate | Ongoing | Not reported | NCT04848519 |
COX-2=cyclooxygenase 2. DRD2=dopamine receptor D2. DRD3=dopamine receptor D3. NGF=nerve growth factor. NSAID=non-steroidal anti-inflammatory drug.
Where multiple therapies are listed, this column relates only to drugs targeting the tumour–nerve axis.
Clinically, the mechanisms by which neurochemicals and proteins promote tumour growth represent attractive therapeutic targets. The potential for utilising existing neuroregulatory medications (eg, β-adrenergic, dopamine, and glutamate receptor modulators) as anti-cancer therapies is appealing and has been explored through a mixture of retrospective work and small prospective trials (table 2). For example, some retrospective studies have found an association between β-blocker usage (especially non-selective) and improved oncological outcomes,65,66 whereas use of tricyclic antidepressants and selective serotonin reuptake inhibitors has been examined in preclinical and retrospective studies in gliomas67,68 with mixed outcomes. Some of these strategies have been tested in small prospective clinical studies, although larger randomised controlled trials are needed to determine whether use of these drugs results in improved clinical outcomes (table 2).
Drugs modulating neuronal paracrine signalling, in contrast to neurotransmitters, are less commonly used, limiting the power of retrospective epidemiological studies. However, some cognate receptors (eg, GFRα1, NTRKs) and their downstream effectors (eg, RET, PI3K) have been known oncogenes for decades, with well-described genetic alterations observed across numerous cancer types.69 Since much of this paracrine signalling converges on classic oncogenic pathways, targeting the upstream neural activators of these pathways might be effective. A 2016 study showed that a small molecule inhibitor of dopamine receptors 2/3 (DRD2/3), ONC201, counteracts the pro-survival Akt–ERK pathway.70 ONC201 and a second generation imipridone targeting DRD2 (ONC206) are currently undergoing phase 1 and 2 testing in multiple tumour types, including H3K27M gliomas, endometrial cancer, and neuroendocrine cancers (table 2).
Although multiple prospective trials are testing novel neurochemical modulators (table 2), repurposing existing medications leverages known safety profiles and could offer an accelerated path towards adjunct use with conventional cancer therapies. This option might be especially attractive for treatment of brain tumours, because many of these neuroregulatory drugs permeate the blood–brain barrier (appendix p 2). However, one major clinical challenge to repurposing neuroregulatory drugs for anti-cancer benefit is the lack of tumour specificity and potential morbidity at effective doses. Furthermore, off-patent strategies are probably less appealing for industry-funded clinical trials. Nonetheless, the recent surge of novel data on neuronal regulation of cancer might reignite interest in this approach. Ongoing prospective trials might also shed light on tolerability and efficacy of neuroregulatory therapeutics (table 2).
Combating treatment resistance by disrupting stress-adaptive mechanisms
A pervasive feature of aggressive cancers is their ability to adapt and proliferate in stressful environments created by surgery, systemic therapy, or radiotherapy, supported by multiple components of the nervous system (table 1, rows 30–35). For example, recent work showed that pancreatic cancer cells deprived of the amino acid serine can selectively increase translation of nerve growth factor (NGF), which in turn recruits axons as an exogenous source of serine (figure 2 and table 1, row 34). Interestingly, it is estimated that approximately 40% of pancreatic tumours cannot synthesise their own serine, highlighting a subpopulation of patients that could benefit from combination of a low-serine diet and larotrectinib, a highly selective TRK inhibitor that has been approved by the US Food and Drug Administration. Such mechanisms are in line with previous predinical findings that dietary restriction of serine (and glycine) can reduce tumour burden in lymphoma and intestinal tumours,71 suggesting that this therapeutic strategy might have broad applicability to densely innervated cancer types.
Beyond serving as a source of essential nutrients, nerves can also directly induce changes in cancer and stromal cells that facilitate survival under microenvironmental stress. For example, adrenergic signalling in prostate cancer promotes a shift in cell metabolism towards aerobic glycolysis to promote angiogenesis (table 1, row 2) and the neurotrophin BDNF inihibits cancer cell apoptosis in ovarian cancer (table 1, row 33).
In the central nervous system (CNS), ultra-long membranous protrusions, known as microtubes, have been described as a mechanism of treatment resistance in gliomas. These microtubes connect cancer cells through gap junctions that protect against cell death from radiotherapy and are associated with high grade tumours and poor clinical outcomes (table 1, row 27). Furthermore, these microtubes are composed of myosin and microtubules, the latter of which might suggest a potential sensitivity to taxanes and vinca alkaloids.72 One of the challenges of taxane-based treatment of CNS malignancies, however, is their limited penetration ofthe blood–brain barrier.73 To this end, it remains to be seen whether novel taxanes with better CNS penetration will become an effective option to combat microtube-mediated resistance.74 There are now industry efforts (eg, Divide and Conquer) focused on small molecule inhibition of GAP43 (neuromodulin), a crucial protein for membrane tube outgrowth, as a therapeutic strategy for glioma.75 As these efforts progress, it will be equally important to identify subsets of patients with glioma that will benefit most from treatment.
Therapeutic strategies targeting pro-survival mechanisms in the context of microenvironmental stress could hold the greatest promise for cancer types known to propagate in nutrient-scarce microenvironments (eg, pancreatic cancer, high-grade gliomas), and treatment of local relapses, where recurrent growth emerges in previously treated tissue with altered vascular and nutrient supply (table 2).
Remodelling the immune microenvironment in combination with immunotherapy
Immune checkpoint blockade has been integrated into standard-of-care management for numerous cancer types. Preclinical studies investigating the neuro–immune axis have shown multiple mechanisms through which the nervous system modulates the activity of tissue-resident as well as lymphatic immune cells.76,77 Given the many neuroactive drugs already used for alternative indications, there might be opportunities to repurpose these drugs in combination with immune checkpoint blockade, cytotoxic therapies, or cancer vaccines to modulate the tumour immune microenvironment.
Among patients with melanoma, use of non-specific β-blockers has been associated with decreased recurrence and improved overall survival in those with metastatic disease who also received immunotherapy (table 1, row 36). As such, early phase clinical trials have begun to explore combination propranolol and pembrolizumab in locally advanced and metastatic melanoma, reporting an initial response rate of 78% (table 2). Similar studies are being conducted for recurrent or metastatic urothelial cancer (table 2).
Emerging evidence also suggests β-blockade might enhance efficacy of cytotoxic therapies through modulation of the immune response. In a preclinical study of multiple cancers, adrenergic signalling suppressed anti-tumour immunity, including the abscopal response following local irradiation (table 1, row 37). When β2-signalling was disrupted, CD8+ T cells exhibited enhanced cytotoxic effector function and migratory capacity. Concurrent irradiation and β-blockade also reduced the abundance of immunosuppressive M2-polarised macrophages and regulatory T cells, both of which have been shown to stymie migration of cytotoxic T cells and their physical interaction with malignant cells in lung cancer (table 1, row 39). These observations are consistent with pervasive adrenergic and sensory innervation in lymph nodes and control of T-cell egress from lymph nodes via β2-adrenergic receptor signalling (figure 2).78,79 Given emerging evidence that a substantial fraction of the response to immune checkpoint blockade occurs outside of the tumour site (eg, draining lymphoid tissue),80–82 modulation of the neural signalling that controls retention or release of T cells can have marked effects on the native as well as the immune checkpoint blockade-induced immune response.
Taken together, these findings could have important implications for immunologically cold tumours for which immune checkpoint inhibitors have been largely unsuccessful, in part due to the dearth of CD8+ T cells within the vicinity of malignant cells.83 The combination of immune checkpoint blockade, radiotherapy, and β2-blockade might therefore warrant further investigation for malignancies of the pancreas, prostate, and other tumours unresponsive to checkpoint inhibition alone. Of note, prostate and pancreatic cancer are also among the most densely innervated tumours, with high rates of perineural invasion4 and profound sympathetic signalling (table 1, rows 1 and 5). We additionally discuss the role of cholinergic signalling, nociceptive sensory neurons, and growth factor signalling (eg, midkine) on the immune microenvironment in the appendix (p 2).
Targeting metastatic spread
Evidence on the role of nerves in promoting metastasis is only beginning to emerge. Nerves can serve not only as a physical conduit, but also as a mediator of vascular or lymphatic dissemination.84 Multiple preclinical and clinical studies have now demonstrated associations of perineural invasion,85 ANXA2–SEMA3D–PLXND1 signalling,86 ADRB2-driven cell signalling (table 1, rows 1 and 5), and NMDA receptor (NMDAR) expression (table 1, row 22) with metastases. Cholinergic signalling has additionally been described as promoting invasion and metastasis in prostate cancer (table 1, row 24). Enhanced metastatic potential can result from cancer cells using nerves as physical channels for transit,85 upregulating neural gene expression to maximise colonisation efficiency in distal neural tissue (eg, the brain; table 1, row 22), or adopting pro-invasive features secondary to dialogue with nerves.87
The association between metastases, angiogenesis, and vascular permeability is well established88 and is regulated by VEGF proteins.89 However, as control of vasculature and nerves are often tightly coupled, neurotrophins (eg, NGF, BDNF) and their receptors are increasingly recognised as regulators of aberrant blood vessel formation as well.90 In preclinical studies, treatment of a breast cancer mouse model with an anti-NGF antibody decreased liver metastases and microvascular density.91 Monoclonal antibodies against NGF (eg, fasinumab, tanezumab) are currently in development and have thus far been clinically tested in non-oncological populations.92
Upon initial escape from the local microenvironment and intravasation into the systemic circulation, malignant cells must resist anoikis, a mechanism of programmed cell death due to detachment from neighbouring cells or extracellular matrix.93 Neurotrophic receptors, and in particular TrkB, are crucial mediators of anoikis resistance, thereby enhancing metastatic spread (table 1, row 23). This might provide a therapeutic rationale for TrkB inhibition (eg, larotrectinib, entrectinib) in locally advanced malignancies as a way of inducing malignant cell death before distant organ colonisation.
The metastatic cascade ends with successful colonisation of a distant organ. Recent work has elucidated the role of the nervous system in breast cancer brain metastases (table 1, row 22). Breast-to-brain metastases were found to exhibit glutamate-dependent signalling through NMDAR by infiltrating synapses between neurons. Breast cancer cells co-opt normal physiological synapse formation, creating so-called pseudo-tripartite synapses to hijack synaptic glutamate from surrounding neurons and feed their own tumour growth (figure 2).44 Glutamate then stimulates NMDAR signalling in breast cancer cells that promotes successful colonisation in the brain. Indeed, mice with knockdown of the GluN2B subunit of NMDAR exhibited significantly longer brain metastasis-free survival and lower brain metastasis burden than mice without this knockdown.44 These findings were supported by correlative clinical data from patients with breast cancer in The Cancer Genome Atlas (table 1, row 22).
Taken together, the potential clinical translation of these findings prompts consideration of therapies targeting the tumour–nerve axis to preclude or slow metastatic spread. Preliminary clinical studies are repurposing neuroactive drugs as a strategy for slowing metastases (table 2). Consistent with preclinical evidence (table 1, rows 25 and 35), preoperative propranolol reduced biomarkers associated with metastatic potential in a triple-blind, placebo-controlled, phase 2 trial for primary resectable breast cancer (table 2). However, given that some of the discussed therapeutic rationale presented in this section is geared toward prevention of (as opposed to treatment of) metastatic spread, optimal timing of administration and combination with other therapies needs to be carefully appraised. In situations where metastatic spread has already occurred, therapeutic strategies that target mechanisms of metastatic growth (as opposed to initial seeding)—such as NMDAR regulators (eg, memantine) for patients with brain metastases from basal-like breast cancer (table 1, row 22)—might be viable options.
Disrupting electrical hyperactivity for dual control of tumour growth and seizures
Neuronal electrical activity is crucial for neurodevelopment,94 although evidence of the importance of electrical neuronal input in facilitating cancer proliferation is also emerging, particularly among brain tumours. Analogous to development of the normal human nervous system, glioma cells are affected by electrical activity (table 1, rows 16–17 and 26–27). Gliomas, in turn, release growth stimuli, including glutamate, into the surrounding tissue, increasing neuronal activity in the microenvironment (table 1, row 17). Given the presence of calcium-permeable AM PA receptor-dependent synapses described in both paediatric and adult high-grade gliomas, glioma-derived glutamate contributes a bidirectional, positive feedback loop of tumour–nerve hyperactivity and tumour proliferation (figure 2 and table 1, row 17). However, glutamate represents only one of multiple sources of hyperactivity and glioma growth: recent work has also described the glioma-secreted factor glypican 3 found to drive synaptogenesis and hyperexcitability (table 1, row 26), as well as the ability of gliomas to downregulate inhibitory GABA currents in the surrounding electrical microenvironment (table 1, row 20).29 The precise biological consequences of each source of electrical activity remain to be fully elucidated. For example, glioma–nerve electrical interactions have been described both as potassium currents (table 1, row 12)29 and calcium waves (table 1, row 17). These findings are consistent with existing preclinical data suggesting that multiple mechanisms might be at play, including those involving gap junctions and microtubes (appendix p 2).
Whether electrical networking and complex neural circuits exist in cancer types outside of the CNS remains to be shown. To date, the evidence remains scant for peripheral tumours, although tumour microtubes facilitating intercellular communication have been discovered in pancreatic cancer,95 raising the possibility of electrical processes in non-glial lineages.
The electrical integration of brain tumours with neuronal networks prompts reconsideration of some clinical manifestations of gliomas, including seizures. Although often considered a byproduct of mass effect from the tumour or surrounding oedema, seizures can directly result from glutamate-secreting tumours augmenting neural hyperactivity. Indeed, subgroups of gliomas with higher synaptogenic potential were associated with increased seizure frequency and tumour invasion,96 and recurrent postoperative seizures have been shown to be associated with glioblastoma tumour recurrence.97 These findings raise the question of whether anti-epileptics have oncological therapeutic potential. Although sometimes used in these patients for seizure prevention, anti-epileptics might also confer an additional anti-cancer benefit by dampening neuronal activity; however, the cumulative evidence remains indeterminate, particularly in classes of drugs that modulate postsynaptic signalling. Preliminary work has investigated use of the anti-epileptics perampanel,98 talampanel,99 valproic acid,100 and levetiracetam100 in glioblastoma, although additional work is needed.
There is much that remains unknown regarding how electrical activity from the nervous system affects cancer growth. For instance, although preclinical evidence strongly suggests an oncogenic role of glutamate, glutamate receptor regulators have not yielded positive results in prospective clinical trials thus far (table 2). Although the exact roles of electrical input remain to be fully elucidated, current understanding of electrical networking is consistent with the overall shift in perspective that tumours are driven not only by cell-intrinsic processes but also the intimately connected components of its surrounding microenvironment.
Management of neuropathic pain by targeting the sensory nervous system
Patients with cancer frequently report intense pain at primary and metastatic sites. This has been associated with the presence of perineural invasion and is thought to result in part from the actions of neurotrophins and neuroregulatory proteins (table 1). NGF, for instance, activates sensory afferents from the dorsal root ganglia and has been most directly implicated in pancreatic cancer pain (figure 2 and table 1, rows 3 and 28–29). However, although cancer pain is often treated with the goal of symptom control as opposed to disease eradication, targeting neuropathic pain might simultaneously promote cancer control as well.
Given the morbidity of cancer pain and the fact that the upstream paracrine signalling proteins modulating pain are also known to have pro-tumour growth effects, these neuronal signalling pathways represent attractive therapeutic targets. A subset of patients with advanced cancer experience severe and refractory pain that might in part be explained by these underlying neurotrophic mechanisms, which are not targeted by commonly used analgesics such as opioids. Novel therapeutic strategies tailored to these tumour–nerve signalling dynamics thus have the potential to both control neuropathic cancer pain and confer anti-cancer benefit. Indeed, tanezumab, a monoclonal antibody against NGF, is currently being evaluated in a phase 3 study for patients with painful bone metastases (table 2). Additional ongoing strategies include targeting the TRPV1 receptor, a downstream effector of NGF that facilitates nociception in sensory neurons. Moreover, TRPV1+ sensory nerves promote the growth of pancreatic intraepithelial neoplasia (PanIN) organoids. Denervation of sensory neurons in mice using resiniferatoxin, a high-affinity capsaicin analogue, significantly impaired tumour progression in vivo and is currently in phase 1 testing for patients with severe or refractory pain from advanced cancer (table 2).101
Although cancer itself often causes neuropathic pain, so can treatments. Chemotherapy-induced peripheral neuropathy is a common iatrogenic cause of neuropathic pain most frequently associated with certain platinums, taxanes, and vinca alkaloids.102 The dorsal root ganglion lacks an efficient blood–brain barrier,103 is prone to neurotoxic damage, and is believed to account for many of the sensory symptoms associated with chemotherapy-induced peripheral neuropathy.104 Dorsal root ganglion sensory neurons also play a role in the initiation and growth of certain cancers. In mouse models, previous work revealed perineural invasion, inflammation, and neuronal damage in the nervous system as early as the PanIN-2 stage (table 1, row 3). Ablation of sensory neurons using neonatal capsaicin injections prevented perineural invasion and neuronal damage, suggesting that sensory neurons transmit inflammatory signals from oncogenic Kras-induced neoplasia to the CNS. Taken together, one could postulate that perhaps cytotoxic damage to peripheral sensory nerves has beneficial antineoplastic effects through disruption of tumour–nerve interactions but also results in morbidity through chemotherapy-induced peripheral neuropathy. This hypothesis evokes parallels with the association between immune-related adverse effects caused by immune checkpoint inhibitors and improved treatment response105 and the correlation between mild-to-moderate chronic graft-versus-host disease after allogeneic haematopoietic stem-cell transplantation and lower risk of relapse.106
Future therapeutic perspectives and challenges
As the tumour microenvironment has become increasingly appreciated as a major driver of cancer biology, the nervous system has emerged as a crucial component. The progress in both preclinical and clinical cancer neuroscience has increased exponentially in recent years and enhanced our mechanistic understanding of tumour–nerve crosstalk such that molecularly-targeted strategies are now poised for impactful clinical implementation. Three broad questions that remain incompletely answered include (1) whether the tumour–nerve axis can be successfully targeted with sufficient specificity for broad clinical benefit, (2) how neural regulation can synergistically integrate into existing treatment regimens, and (3) whether histopathological or other companion diagnostics can identify patients who are most likely to benefit from these therapeutic strategies.
This first question is the subject of multiple ongoing prospective clinical trials targeting components of the tumour–nerve axis (table 2). These trials also reflect emerging efforts from joint academic–industry partnerships, including companies such as Divide and Conquer and Cygnal Therapeutics, which are likely to grow as the underpinnings of tumour–nerve crosstalk continue to be illuminated.75 In glioblastoma, for instance, therapeutic strategies are underway to inhibit GAP43, a regulator of synaptic plasticity and membrane tube outgrowth;75 meanwhile, there are also attempts at modulating peripheral nerve signalling to cancer cells and the immune system.75
Beyond therapeutic development, the opportunity for repurposing existing drugs that regulate the nervous system and typically have a tolerable adverse effect profile presents further opportunities. These efforts have taken advantage of both the on-target and off-target effects of these drugs. For example, antiepileptics and neurotransmitter regulators have at times shown promising effects in preclinical and retrospective clinical studies. Perhaps the most clinically vetted neuroregulatory strategy in cancer to date has been β-blockade, which has been evaluated in a range of tumour types including breast, ovarian, and colorectal. It should be noted, however, that the conclusions of retrospective studies in aggregate have been mixed, with some citing benefit and others not. At the same time, this ambiguity should be interpreted with caution, as there might be numerous reasons for a lack of clear improvement, including therapeutic window concerns, inherent limitations of retrospective analyses, and receipt of concurrent therapies in these studies. Importantly, these results also raise questions on how therapeutics targeting the tumour–nerve axis should be optimally utilised with existing treatment strategies. For example, neuroregulatory drugs might have limited efficacy as a monotherapy, but might synergise with treatments such as chemotherapy or radiotherapy. Given the known mechanistic role of nerves, including serving as nutrient sources, immune modulators, and growth stimuli, neural regulation might offer the ability to augment the response to immunotherapy, increase cytotoxic effects with concurrent radiation, or additionally target specific neural signalling pathways that are also commonly altered in cancer (table 1).
Several challenges remain in applying cancer neuroscience therapeutics to the clinic, including the need to improve our mechanistic understanding of the processes driving these pathological processes. Improvements to diagnostic and research techniques are helping to achieve this goal, such as use of single cell and spatial omics technologies. Clinically, little is known about how tumour–nerve dialogue affects and is affected by established treatments, including surgery, radiotherapy, chemotherapy, immunotherapy, and other targeted therapies. Furthermore, given that many tumour–nerve interactions invoke signalling pathways that exist in normal tissue function or development, the ability for therapies that target these interactions to exhibit tumour specificity might limit the therapeutic window. Lastly, although treatment strategies targeting the tumour–nerve axis are largely directed at cell-extrinsic processes, they probably act in concert with cell-intrinsic oncogenic events to promote cancer growth. As such, therapeutically targeting tumour–nerve interactions might be best used in combination with other local or systemic treatments. For example, the preclinical finding that adrenergic stress constrains anti-tumour immunity and abscopal responses after focal irradiation (table 1, row 37) suggests that the combination of β blockade and radiotherapy could be worth exploring further.
Effective patient selection for therapeutic targeting of the tumour-nerve axis requires identification of reliable biomarkers and companion diagnostics. Currently, the most histologically apparent feature suggesting a tumour’s direct involvement with the nervous system is perineural invasion, which has variable impact on clinical management depending on cancer type and regional paradigms. Perineural invasion itself is a poor prognostic factor across many cancers.4,107
In practice, perineural invasion status is often integrated with other clinical and histopathological risk factors to determine adjuvant treatment selection, including radiotherapy and or chemotherapy.4 It should be noted, however, that although perineural invasion associates with poorer clinical characteristics in many cancers,4,107 it remains unclear whether perineural invasion itself actively contributes to tumour pathogenesis or whether it simply demarcates an aggressive subset of tumours with a penchant for crosstalk with the nervous system. Furthermore, the relatively limited clinical integration of perineural invasion as a biomarker is in part due to (1) a lack of standardisation in criteria to report perineural invasion, (2) suboptimal tissue sampling and limitations of commonly used staining techniques, and (3) incomplete understanding of the biological and therapeutic implications of perineural invasion. However, akin to how PD-L1 status can guide immunotherapy use, the presence of perineural invasion might eventually serve as a predictive biomarker to guide patient selection for therapies targeting tumour–nerve crosstalk, but the utility of this strategy needs to be rigorously established. Recent efforts to improve detection sensitivity and standardisation of perineural invasion include a pan-cancer perineural invasion gene expression signature developed using machine learning-based algorithms and transcriptomic data.107 Additional diagnostic developments such as an assay for tumour serine biosynthesis and content for pancreatic cancer (table 1, row 34) might also be clinically informative.
The nascent field of cancer neuroscience is growing rapidly, with parallel efforts in uncovering the mechanistic underpinnings of tumour–nerve interactions and developing novel therapies through collaborative academic and commercial efforts. Lessons learned from the tumour microenvironment suggest that disrupting tumour–nerve dialogue could eventually become a mainstay of clinical oncology, akin to anti-angiogenic and immunomodulatory therapies nowadays.
Supplementary Material
Search strategy and selection criteria.
Relevant preclinical studies were identified in PubMed with the search terms “cancer” and specific cancer sites (eg, “prostate”, “pancreatic”, “breast”) in combination with “neuroscience”, “nerve(s)”, or specific neural signalling components such as “glutamate”, “norepinephrine”, “dopamine”, “cholinergic”, and “GABA”. Relevant clinical studies were identified by searching using these same terms on ClinicalTrials.gov. No date restrictions were applied to either search; the last search of both databases was done on Nov 2, 2021. Only articles in English were reviewed. Publications corresponding to trials included in this Review were identified via search of National Clinical Trial numbers on PubMed. The final reference listforthe preclinical studies was generated on the basis of novelty, impact, and relevance to cancer neuroscience mechanisms and therapeutic avenues. The final reference list forthe clinical studies was based on sample size and relevance.
Acknowledgments
We thank Shannon Hoyt, Karen Yee, and Grant Benham for administrative assistance. JetPub Scientific Communications assisted in the preparation of this manuscript, in accordance with Good Publication Practice (GPP3) guidelines.
Footnotes
Declaration of interests
MM-K reports personal fees from H3 Biomedicine and AstraZeneca; grants from Novartis; and royalties from Elsevier, outside the submitted work. TSH reports personal fees from Synthetic Biologies, Novocure, and Merck; equity in PanTher Therapeutics; and grants from Taiho, AstraZeneca, Bristol Myers Squibb, Tesaro, Ipsen, and Puma, outside the submitted work. AJA reports personal fees from Merck, Arrakis Therapeutics, and Oncorus; grants from Mirati Therapeutics, Deerfield, Novo Ventures, National Cancer Institute, Lustgarten Foundation, Pancreatic Cancer Action Network, Doris Duke Charitable Foundation, and Dana-Farber Cancer Institute Hale Center for Pancreatic Cancer Research; and grants and personal fees from Syros Pharmaceuticals, outside the submitted work. TJ reports grants from Lustgarten Foundation; stock in Amgen and Thermo Fisher Scientific; and membership of the Board of Directors of Amgen and Thermo Fisher Scientific, outside the submitted work. LZ reports grants from Merck, during the conduct of the study; and grants from iTeos, Bristol Meyers Squibb, Merck, AstraZeneca, Amgen, NovaRock, Inxmed, and Halozyme; personal fees from Biosion, NovaRock, Akrevia/Xilio, Datarevive, QED, Natera, Ambrx, Snow Lake Capital, and Tempus; personal fees from Alphamab and Mingruizhiyao; and shares in Alphamab and Mingruizhiyao, outside the submitted work. PYW reports research support from Agios, AstraZeneca/Medimmune, Beigene, Celgene, Eli Lily, Genentech/Roche, Kazia, MediciNova, Merck, Novartis, Nuvation Bio, Oncoceutics, Vascular Biogenics, and VBI Vaccines; and membership on the advisory board of Agios, AstraZeneca, Bayer, Boston Pharmaceuticals, CNS Pharmaceuticals, Elevate Bio Immunomic Therapeutics, Imvax, Karyopharm, Merck, Novartis, Nuvation Bio, Vascular Biogenics, VBI Vaccines, and Voyager, outside the submitted work. All other authors declare no competing interests.
See Online for appendix
Contributor Information
Diana D Shi, Department of Radiation, Oncology, Dana-Farber/Brigham and Women’s Cancer, Center, Boston, MA, USA.
Jimmy A Guo, Department of Radiation Oncology, Massachusetts General Hospital, Cancer Center, Boston, MA, USA; School of Medicine, University of California, San Francisco, San Francisco, CA, USA; Broad Institute of MIT and Harvard, Cambridge, MA, USA; Biological and Biomedical Sciences Program, Harvard University, Boston, MA, USA; Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, USA.
Hannah I Hoffman, Department of Radiation Oncology, Massachusetts General Hospital, Cancer Center, Boston, MA, USA; Broad Institute of MIT and Harvard, Cambridge, MA, USA; Department of Biology, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA, USA; Harvard-MIT Health Sciences and Technology Program, Harvard Medical School, Boston, MA, USA.
Jennifer su, Department of Radiation Oncology, Massachusetts General Hospital, Cancer Center, Boston, MA, USA; Broad Institute of MIT and Harvard, Cambridge, MA, USA; Department of Biology, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA, USA.
Mari Mino-Kenudson, Department of Pathology, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA.
Jaimie L Barth, Department of Pathology, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA.
Jason M Schenkel, Department of Biology, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA, USA; Department of Pathology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA.
Jay S Loeffler, Department of Radiation Oncology, Massachusetts General Hospital, Cancer Center, Boston, MA, USA.
Helen A Shih, Department of Radiation Oncology, Massachusetts General Hospital, Cancer Center, Boston, MA, USA.
Theodore S Hong, Department of Radiation Oncology, Massachusetts General Hospital, Cancer Center, Boston, MA, USA.
Jennifer Y Wo, Department of Radiation Oncology, Massachusetts General Hospital, Cancer Center, Boston, MA, USA.
Andrew J Aguirre, Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, USA.
Tyler Jacks, Department of Biology, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA, USA.
Lei Zheng, Department of Oncology, Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD, USA.
Patrick Y Wen, Center for Neuro-Oncology, Dana-Farber Cancer Institute, Boston, MA, USA.
Timothy C Wang, Division of Digestive and Liver Diseases, Columbia University Medical Center, New York, NY, USA.
William L Hwang, Department of Radiation Oncology, Massachusetts General Hospital, Cancer Center, Boston, MA, USA; Broad Institute of MIT and Harvard, Cambridge, MA, USA; Department of Biology, Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA, USA.
References
- 1.Young HH. On the presence of nerves in tumours and of other structures in them as revealed by a modification of Ehrlich’s method of “ vital staining “ with methylene blue. J Exp Med 1897; 2: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scherer HJ. Structural development in gliomas. Am J Cancer 1938; 34: 333–51. [Google Scholar]
- 3.Batsakis JG. Nerves and neurotropic carcinomas. Ann Otol Rhinol Laryngol 1985; 94: 426–27. [PubMed] [Google Scholar]
- 4.Liebig C, Ayala G, Wilks JA, Berger DH, Albo D. Perineural invasion in cancer, a review of the literature. Cancer 2009; 115: 3379–91. [DOI] [PubMed] [Google Scholar]
- 5.Amin MB, Greene FL, Edge SB, et al. The eighth edition AJCC cancer staging manual: continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J Clin 2017; 67: 93–99. [DOI] [PubMed] [Google Scholar]
- 6.Monje M, Borniger JC, D’Silva NJ, et al. Roadmap for the emerging field of cancer neuroscience. Cell 2020; 181: 219–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boilly B, Faulkner S, Jobling P, Hondermarck H. Nerve dependence: from regeneration to cancer. Cancer Cell 2017; 31: 342–54. [DOI] [PubMed] [Google Scholar]
- 8.Zahalka AH, Frenette PS. Nerves in cancer. Nat Rev Cancer 2020; 20:143–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Venkatesh H, Monje M. Neuronal activity in ontogeny and oncology. Trends Cancer 2017; 3: 89–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faulkner S, Jobling P, March B, Jiang CC, Hondermarck H. Tumor neurobiology and the war of nerves in cancer. Cancer Discov 2019; 9: 702–10. [DOI] [PubMed] [Google Scholar]
- 11.Demir IE, Friess H, Ceyhan GO. Nerve-cancer interactions in the stromal biology of pancreatic cancer. Front Physiol 2012; 3: 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reavis HD, Chen HI, Drapkin R. Tumor innervation: cancer has some nerve. Trends Cancer 2020; 6:1059–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jurcak N, Zheng L. Signaling in the microenvironment of pancreatic cancer: transmitting along the nerve. Pharmacol Ther 2019; 200:126–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 2001; 24: 677–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knox SM, Lombaert IMA, Reed X, Vitale-Cross L, Gutkind JS, Hoffman MP. Parasympathetic innervation maintains epithelial progenitor cells during salivary organogenesis. Science 2010; 329:1645–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zahalka AH, Arnal-Estapé A, Maryanovich M, et al. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science 2017; 358: 321–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Magnon C, Hall SJ, Lin J, et al. Autonomic nerve development contributes to prostate cancer progression. Science 2013; 341:1236361. [DOI] [PubMed] [Google Scholar]
- 18.Saloman JL, Albers KM, Li D, et al. Ablation of sensory neurons in a genetic model of pancreatic ductal adenocarcinoma slows initiation and progression of cancer. Proc Natl Acad Sci USA 2016; 113: 3078–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Renz BW, Tanaka T, Sunagawa M, et al. Cholinergic signalling via muscarinic receptors directly and indirectly suppresses pancreatic tumourigenesis and cancer sternness. Cancer Discov 2018; 8:1458–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Renz BW, Takahashi R, Tanaka T, et al. (β2 adrenergic-neurotrophin feedforward loop promotes pancreatic cancer. Cancer Cell 2018; 33: 75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peterson SC, Eberl M, Vagnozzi AN, et al. Basal cell carcinoma preferentially arises from stem cells within hair follicle and mechanosensory niches. Cell Stem Cell 2015; 16: 400–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iacovelli L, Arcella A, Battaglia G, et al. Pharmacological activation of mGlu4 metabotropic glutamate receptors inhibits the growth of medulloblastomas. J Neurosci 2006; 26: 8388–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayakawa Y, Sakitani K, Konishi M, et al. Nerve growth factor promotes gastric tumourigenesis through aberrant cholinergic signalling. Cancer Cell 2017; 31: 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ceyhan GO, Giese NA, Erkan M, et al. The neurotrophic factor artemin promotes pancreatic cancer invasion. Ann Surg 2006; 244: 274–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lawn S, Krishna N, Pisklakova A, et al. Neurotrophin signaling via TrkB and TrkC receptors promotes the growth of brain tumor-initiating cells. j Biol Chem 2015; 290: 3814–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He S, Chen CH, Chernichenko N, et al. GFRal released by nerves enhances cancer cell perineural invasion through GDNF-RET signaling. Proc Natl Acad Sci USA 2014; 111: E2008–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Venkatesh HS, Johung TB, Caretti V, et al. Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell 2015; 161: 803–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Venkatesh HS, Tam LT, Woo PJ, et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 2017; 549: 533–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Venkatesh HS, Morishita W, Geraghty AC, et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019; 573: 539–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pan Y, Hysinger JD, Barron T, et al. NF1 mutation drives neuronal activity-dependent initiation of optic glioma. Nature 2021; 594: 277–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Senaldi G, Varnum BC, Sarmiento U, et al. Novel neurotrophin-l/B cell-stimulating factor-3: a cytokine of the IL-6 family. Proc Natl Acad Sci USA 1999; 96:11458–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pearse RN, Swendeman SL, Li Y, Rafii D, Hempstead BL. A neurotrophin axis in myeloma: TrkB and BDNF promote tumor-cell survival. Blood 2005; 105: 4429–36. [DOI] [PubMed] [Google Scholar]
- 33.Chu Y-H, Dias-Santagata D, Farahani AA, et al. Clinicopathologic and molecular characterization of NTRK-rearranged thyroid carcinoma (NRTC). Mod Pathol 2020; 33: 2186–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Farago AF, Taylor MS, Doebele RC, et al. Clinicopathologic features of non-small-cell lung cancer harboring an NTRK gene fusion. JCO Precis Oncol 2018; 2018:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Venkataramani V, Tanev DI, Strahle C, et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019; 573: 532–38. [DOI] [PubMed] [Google Scholar]
- 36.Ishiuchi S, Tsuzuki K, Yoshida Y, et al. Blockage of Ca(2+)-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat Med 2002; 8: 971–78. [DOI] [PubMed] [Google Scholar]
- 37.Ye ZC, Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res 1999; 59: 4383–91. [PubMed] [Google Scholar]
- 38.Babateen O, Jin Z, Bhandage A, et al. Etomidate, propofol and diazepam potentiate GABA-evoked GABAA currents in a cell line derived from human glioblastoma. Eur J Pharmacol 2015; 748:101–07. [DOI] [PubMed] [Google Scholar]
- 39.Labrakakis C, Patt S, Hartmann J, Kettenmann H. Functional GABA(A) receptors on human glioma cells. Eur J Neurosci 1998; 10: 231–38. [DOI] [PubMed] [Google Scholar]
- 40.Dolma S, Selvadurai HJ, Lan X, et al. Inhibition of dopamine receptor D4 impedes autophagic flux, proliferation, and survival of glioblastoma stem cells. Cancer Cell 2016; 29: 859–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li J, Zhu S, Kozono D, et al. Genome-wide shRNA screen revealed integrated mitogenic signaling between dopamine receptor D2 (DRD2) and epidermal growth factor receptor (EGFR) in glioblastoma. Oncotargel 2014; 5: 882–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Campbell SL, Robel S, Cuddapah VA, et al. GABAergic disinhibition and impaired KCC2 cotransporter activity underlie tumor-associated epilepsy. Glia 2015; 63: 23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neman J, Termini J, Wilczynski S, et al. Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc Natl Acad Sci USA 2014; 111: 984–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeng Q, Michael IP, Zhang P, et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 2019; 573: 526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Douma S, Van Laar T, Zevenhoven J, Meuwissen R, Van Garderen E, Peeper DS. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature 2004; 430:1034–39. [DOI] [PubMed] [Google Scholar]
- 46.Sloan EK, Priceman SJ, Cox BF, et al. The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res 2010; 70: 7042–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu K, Lin CJ, Hatcher A, et al. PIK3CA variants selectively initiate brain hyperactivity during gliomagenesis. Nature 2020; 578:166–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Osswald M, Jung E, Sahm F, et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015; 528: 93–98. [DOI] [PubMed] [Google Scholar]
- 49.Zhu Z, Friess H, diMola FF, et al. Nerve growth factor expression correlates with perineural invasion and pain in human pancreatic cancer. J Clin Oncol 1999; 17: 2419–28. [DOI] [PubMed] [Google Scholar]
- 50.Anand U, Otto WR, Casula MA, et al. The effect of neurotrophic factors on morphology, TRPV1 expression and capsaicin responses of cultured human DRG sensory neurons. Neurosci Lett 2006; 399: 51–56. [DOI] [PubMed] [Google Scholar]
- 51.Bhat K, Saki M, Cheng F, et al. Dopamine receptor antagonists, radiation, and cholesterol biosynthesis in mouse models of glioblastoma. J Natl Cancer Inst 2021; 113:1094–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dai H, Li R, Wheeler T, et al. Enhanced survival in perineural invasion of pancreatic cancer: an in vitro approach. Hum Pathol 2007; 38: 299–307. [DOI] [PubMed] [Google Scholar]
- 53.Ayala GE, Dai H, Ittmann M, et al. Growth and survival mechanisms associated with perineural invasion in prostate cancer. Cancer Res 2004; 64: 6082–90. [DOI] [PubMed] [Google Scholar]
- 54.Au CWH, Siu MKY, Liao X, et al. Tyrosine kinase B receptor and BDNF expression in ovarian cancers - effect on cell migration, angiogenesis and clinical outcome. Cancer Lett 2009; 281:151–61. [DOI] [PubMed] [Google Scholar]
- 55.Banh RS, Biancur DE, Yamamoto K, et al. Neurons release serine to support mRNA translation in pancreatic cancer. Cell 2020; 183:1202–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De Giorgi V, Grazzini M, Benemei S, et al. Propranolol for off-label treatment of patients with melanoma: results from a cohort study. JAMA Oncol 2018; 4: el72908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kokolus KM, Zhang Y, Sivik JM, et al. Beta blocker use correlates with better overall survival in metastatic melanoma patients and improves the efficacy of immunotherapies in mice. Oncolmmunology 2017; 7: el405205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen M, Qiao G, Hylander BL, et al. Adrenergic stress constrains the development of anti-tumor immunity and abscopal responses following local radiation. Nat Commun 2020; 11:1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Daher C, Vimeux L, Stoeva R, et al. Blockade of (3-adrenergic receptors improves CD8* T-cell priming and cancer vaccine efficacy. Cancer Immunol Res 2019; 7:1849–63. [DOI] [PubMed] [Google Scholar]
- 60.Peranzoni E, Lemoine J, Vimeux L, et al. Macrophages impede CD8 T cells from reaching tumour cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci USA 2018; 115: E4041–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leclerc M, Voilin E, Gros G, et al. Regulation of antitumour CD8 T-cell immunity and checkpoint blockade immunotherapy by Neuropilin-1. Nat Commun 2019; 10: 3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shurin GV, Kruglov O, Ding F, et al. Melanoma-induced reprogramming of schwann cell signaling aids tumor growth. Cancer Res 2019; 79: 27362–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang M-W, Tao L-Y, Jiang Y-S, et al. Perineural invasion reprograms the immune microenvironment through cholinergic signaling in pancreatic ductal adenocarcinoma. Cancer Res 2020; 80:1991–2003. [DOI] [PubMed] [Google Scholar]
- 64.Schneider M, Vollmer L, Potthoff A-L, et al. Meclofenamate causes loss of cellular tethering and decoupling of functional networks in glioblastoma. Neuro-oncol 2021; 23:1885–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Melhem-Bertrandt A, Chavez-Macgregor M, Lei X, et al. Beta-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer. J Clin Oncol 2011; 29: 2645–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Udumyan R, Montgomery S, Fang F, et al. Beta-blocker drug use and survival among patients with pancreatic adenocarcinoma. Cancer Res 2017; 77: 3700–07. [DOI] [PubMed] [Google Scholar]
- 67.Shchors K, Massaras A, Hanahan D. Dual targeting of the autophagic regulatory circuitry in gliomas with repurposed drugs elicits cell-lethal autophagy and therapeutic benefit. Cancer Cell 2015; 28: 456–71. [DOI] [PubMed] [Google Scholar]
- 68.Caudill JS, Brown PD, Cerhan JH, Rummans TA. Selective serotonin reuptake inhibitors, glioblastoma multiforme, and impact on toxicities and overall survival: the Mayo clinic experience. Am J Clin Oncol 2011; 34: 385–87. [DOI] [PubMed] [Google Scholar]
- 69.Mulligan LM. GDNF and the RET receptor in cancer: new insights and therapeutic potential. Front Physiol 2019; 9:1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kline CLB, Van den Heuvel APJ, Allen JE, Prabhu W, Dicker DT, El-Deiry WS. ONC201 kills solid tumor cells by triggering an integrated stress response dependent on ATF4 activation by specific eIF2a kinases. Sci Signal 2016; 9: ral8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maddocks ODK, Athineos D, Cheung EC, et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 2017; 544: 372–76. [DOI] [PubMed] [Google Scholar]
- 72.Perez EA. Microtubule inhibitors: differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol Cancer Ther 2009; 8: 2086–95. [DOI] [PubMed] [Google Scholar]
- 73.Rice A, Michaelis ML, Georg G, Liu Y, Turunen B, Audus KL. Overcoming the blood-brain barrier to taxane delivery for neurodegenerative diseases and brain tumors. J Mol Neurosci 2003; 20: 339–43. [DOI] [PubMed] [Google Scholar]
- 74.Kumfhekar P, Tang S-C, Brenner AJ, et al. ANG1005, a brain-penetrating peptide-drug conjugate, shows activity in patients with breast cancer with leptomeningeal carcinomatosis and recurrent brain metastases. Clin Cancer Res 2020; 26: 2789–99. [DOI] [PubMed] [Google Scholar]
- 75.Dolgin E Cancer-neuronal crosstalk and the startups working to silence it. Nat Biotechnol 2020; 38:115–17. [DOI] [PubMed] [Google Scholar]
- 76.Huh JR, Veiga-Fernandes H. Neuroimmune circuits in inter-organ communication. Nat Rev Immunol 2020; 20: 217–28. [DOI] [PubMed] [Google Scholar]
- 77.Shurin MR, Shurin GV, Zlotnikov SB, Bunimovich YL. The neuroimmune axis in the tumor microenvironment. J Immunol 2020; 204: 280–85. [DOI] [PubMed] [Google Scholar]
- 78.Panuncio AL, De La Peña S, Gualco G, Reissenweber N, Reissenweber N. Adrenergic innervation in reactive human lymph nodes. J Anat 1999; 194:143–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nakai A, Hayano Y, Furuta F, Noda M, Suzuki K. Control of lymphocyte egress from lymph nodes through β2-adrenergic receptors. j Exp Med 2014; 211: 2583–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Valpione S, Galvani E, Tweedy J, et al. Immune awakening revealed by peripheral T cell dynamics after one cycle of immunotherapy. Nat Cancer 2020; 1: 210–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yost KE, Satpathy AT, Wells DK, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med 2019. 25:1251–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wu TD, Madireddi S, de Almeida PE, et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 2020; 579: 274–78. [DOI] [PubMed] [Google Scholar]
- 83.Bonaventura P, Shekarian T, Alcazer V, et al. Cold tumors: a therapeutic challenge for immunotherapy. Front Immunol 2019; 10:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Le CP, Nowell CJ, Kim-Fuchs C, et al. Chronic stress in mice remodels lymph vasculature to promote tumour cell dissemination. Nat Commun 2016; 7:10634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fowler BZ, Crocker IR, Johnstone PAS. Perineural spread of cutaneous malignancy to the brain: a review of the literature and five patients treated with stereotactic radiotherapy. Cancer 2005; 103: 2143–53. [DOI] [PubMed] [Google Scholar]
- 86.Jurcak NR, Rucki AA, Muth S, et al. Axon guidance molecules promote perineural invasion and metastasis of orthotopic pancreatic tumors in mice. Gastroenterology 2019; 157: 838–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Amit M, Na’ara S, Gil Z. Mechanisms of cancer dissemination along nerves. Nat Rev Cancer 2016; 16: 399–408. [DOI] [PubMed] [Google Scholar]
- 88.Zetter BR. Angiogenesis and tumor metastasis. Annu Rev Med 1998; 49: 407–24. [DOI] [PubMed] [Google Scholar]
- 89.Ferrara N VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer 2002; 2: 795–803. [DOI] [PubMed] [Google Scholar]
- 90.Lam C-T, Yang Z-F, Lau C-K, Tam K-H, Fan S-T, Poon RTP. Brain-derived neurotrophic factor promotes tumorigenesis via induction of neovascularization: implication in hepatocellular carcinoma. Clin Cancer Res 2011; 17: 3123–33. [DOI] [PubMed] [Google Scholar]
- 91.Adriaenssens E, Vanhecke E, Saule P, et al. Nerve growth factor is a potential therapeutic target in breast cancer. Cancer Res 2008; 68: 346–51. [DOI] [PubMed] [Google Scholar]
- 92.Hochberg MC, Carrino JA, Schnitzer TJ, et al. Long-term safety and efficacy of subcutaneous tanezumab versus nonsteroidal antiinflammatory drugs for hip or knee osteoarthritis: a randomized trial. Arthritis Rheumatol 2021; 73:1167–77. [DOI] [PubMed] [Google Scholar]
- 93.Kim YN, Koo KH, Sung JY, Yun UJ, Kim H. Anoikis resistance: an essential prerequisite for tumor metastasis. Int J Cell Biol 2012; 2012: 306879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Barres BA, Raff MC. Proliferation of oligodendrocyte precursor cells depends on electrical activity in axons. Nature 1993; 361: 258–60. [DOI] [PubMed] [Google Scholar]
- 95.Latario CJ, Schoenfeld LW, Howarth CL, et al. Tumor microtubes connect pancreatic cancer cells in an Arp2/3 complex-dependent manner. Mol Biol Cell 2020; 31:1259–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.John Lin CC, Yu K, Hatcher A, et al. Identification of diverse astrocyte populations and their malignant analogs. Nat Neurosci 2017; 20: 396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chaichana KL, Parker SL, Olivi A, Quiñones-Hinojosa A. Long-term seizure outcomes in adult patients undergoing primary resection of malignant brain astrocytomas. Clinical article. J Neurosurg 2009; 111: 282–92. [DOI] [PubMed] [Google Scholar]
- 98.Lange F, Wefilau K, Porath K, et al. AMPA receptor antagonist perampanel affects glioblastoma cell growth and glutamate release in vitro. PLoS One 2019; 14: e0211644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Grossman SA, Ye X, Chamberlain M, et al. Talampanel with standard radiation and temozolomide in patients with newly diagnosed glioblastoma: a multicenter phase II trial. J Clin Oncol 2009; 27:4155–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Happold C, Gorlia T, Chinot O, et al. Does valproic acid or levetiracetam improve survival in glioblastoma? A pooled analysis of prospective clinical trials in newly diagnosed glioblastoma. J Clin Oncol 2016; 34: 731–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sinha S, Fu Y-Y, Grimont A, et al. PanIN neuroendocrine cells promote tumorigenesis via neuronal cross-talk. Cancer Res 2017; 77: 1868–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Loprinzi CL, Lacchetti C, Bleeker J, et al. Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: ASCO guideline update. J Clin Oncol 2020; 38: 3325–48. [DOI] [PubMed] [Google Scholar]
- 103.Allen DT, Kiernan JA. Permeation of proteins from the blood into peripheral nerves and ganglia. Neuroscience 1994; 59: 755–64. [DOI] [PubMed] [Google Scholar]
- 104.Theiss C, Meller K. Taxol impairs anterograde axonal transport of microinjected horseradish peroxidase in dorsal root ganglia neurons in vitro. Cell Tissue Res 2000; 299: 213–24. [DOI] [PubMed] [Google Scholar]
- 105.Eggermont AMM, Kicinski M, Blank CU, et al. Association between immune-related adverse events and recurrence-free survival among patients with stage III melanoma randomized to receive pembrolizumab or placebo: a secondary analysis of a randomized clinical trial. JAMA Oncol 2020; 6: 519–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Weiden PL, Sullivan KM, Flournoy N, Storb R, Thomas ED. Antileukemic effect of chronic graft-versus-host disease: contribution to improved survival after allogeneic marrow transplantation. N Engl J Med 1981; 304:1529–33. [DOI] [PubMed] [Google Scholar]
- 107.Guo JA, Hoffman HI, Shroff SG, et al. Pan-cancer transcriptomic predictors of perineural invasion improve occult histopathologic detection. Clin Cancer Res 2021; 27: 2807–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.