

Abstract
Epigenetic alterations are increasingly recognized as important contributors to the development and progression of pancreatic ductal adenocarcinoma. 5-hydroxymethylcytosine (5hmC) is an epigenetic DNA mark generated through the ten-eleven translocation (TET) enzyme-mediated pathway and is closely linked to gene activation. However, the timing of alterations in epigenetic regulation in the progression of pancreatic neoplasia is not well understood. In this study, we hypothesized that aberrant expression of ten-eleven translocation methylcytosine dioxygenase 1 (TET1) and subsequent global 5hmC alteration are linked to early tumorigenesis in the pancreas. Therefore, we evaluated alterations of 5hmC and TET1 levels using immunohistochemistry in pancreatic neoplasms (n = 380) and normal ducts (n = 118). The study cohort included representation of the full spectrum of precancerous lesions from low- and high-grade pancreatic intraepithelial neoplasia (n = 95), intraductal papillary mucinous neoplasms (all subtypes, n = 129), intraductal oncocytic papillary neoplasms (n = 12), and mucinous cystic neoplasms (n = 144). 5hmC and TET1 were significantly downregulated in all types of precancerous lesion and associated invasive pancreatic ductal adenocarcinomas compared with normal ductal epithelium (all p < 0.001), and expression of 5hmC positively correlated with expression of TET1. Importantly, downregulation of both 5hmC and TET1 was observed in most low-grade precancerous lesions. There were no clear associations between 5hmC levels and clinicopathological factors, thereby suggesting a common epigenetic abnormality across precancerous lesions. We conclude that downregulation of 5hmC and TET1 is an early event in pancreatic tumorigenesis.
Keywords: 5-hydroxymethylcytosine, TET1, epigenetics, immunohistochemistry, pancreatic precancerous lesion, PanIN, IPMN, IOPN, MCN
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a highly lethal malignancy with a 5-year overall survival rate of 9% [1]. A significant fraction of patients develop early recurrence and distant metastasis, even after a histologically complete resection [2,3]. However, PDAC does not originate de novo, but rather through a multistep progression that involves histologically defined precursor lesions [4–7], Three common precursor lesions have been identified in the pancreas; pancreatic intraepithelial neoplasia (PanIN), intraductal papillary mucinous neoplasm (IPMN), and mucinous cystic neoplasm (MCN) [4–7], In addition, intraductal oncocytic papillary neoplasm (IOPN) has been recognized by the World Health Organization (WHO) classification as a unique precancerous neoplasm with some overlapping clinical features with IPMN [8,9]. PanINs are the most prevalent precursor lesions and are microscopic neoplasms, whereas IPMNs, IOPNs, and MCNs occur less frequently and are macroscopic cystic lesions. High-grade precursors are believed to progress to invasive carcinoma; therefore, early detection with prophylactic resection of high-risk precancerous lesions is an important strategy to help prevent invasive cancer growth [10,11].
Recently we carried out multi-region whole exome and targeted sequencing of pancreatic precancerous lesions and showed that low-grade precancerous lesions can arise from multiple genetically independent clones [12–14], suggesting that neoplastic initiation may be driven by non-mutational mechanisms in some lesions. However, the underlying mechanisms that led to this polyclonal neoplastic origin are still largely unexplored. In this study, we investigated epigenetic alterations as one possible early non-mutational alteration. These alterations may be one of the earliest events that predispose to the formation of pancreatic neoplasms.
DNA methylation (5-methylcytosine, 5mC) and hydroxymethylation (5-hydroxymethylcytosine, 5hmC) are epigenetic modifications that can promote neoplastic initiation and progression by altering key cellular mechanisms, such as proliferation and invasiveness of neoplastic cells. When located in a gene promoter, 5mC and 5hmC typically act to repress and activate gene transcription, respectively. Global redistribution of 5mC and 5hmC and preferential enrichment of 5hmC at oncogenic enhancers have been identified as recurrent epigenetic events across multiple cancer types [15–17].
The conversion of 5mC to 5hmC occurs through an oxidative reaction catalyzed by the ten-eleven translocation (TET) protein family of dioxygenases [18]. Notably, ten-eleven translocation methylcytosine dioxygenase 1 (TET1) plays a central role in 5hmC formation, and the depletion of TET1 yields more widespread reduction of 5hmC compared with the depletion of other TET family proteins [19]. In addition, aberrant TET1 expression has been reported to be closely linked to the development and progression of a number of human malignancies [20–32]. However, the role of TET1 in cancer is still controversial because both upregulation and downregulation of TET1 expression can alter the global gene expression profile, leading to phenotypes that could be selected during tumorigenesis. For example, downregulation of TET1 has been reported in esophageal, gastric, prostatic, hepatocellular, and hormone receptor-positive breast cancer [20–25]. Upregulation of TET1 has been reported in bile duct, lung, and ovarian cancer, and triple-negative breast cancer [26–29]. Overall, the results of these studies suggest that the epigenetic abnormalities in neoplastic cells may depend on the tissue environment at the primary site.
It was also recently demonstrated that hyperglycemia can destabilize TET proteins and decrease 5hmC levels, providing a potentially novel mechanism for epigenomic reprogramming by environmental factors, eventually leading to an increased risk of cancer [33]. However, little is known about the molecular basis of the link between such environmental risk factors and PDAC. Also, although previous studies have characterized genome-wide methylation patterns in precancerous pancreatic lesions, identifying genes with distinct methylation patterns [34,35], the change in hydroxymethylation levels in these neoplasms has not been fully investigated. In this study, we hypothesized that aberrant expression of TET1 and subsequent global downregulation or upregulation of 5hmC occur early in human pancreatic tumorigenesis. In order to investigate this hypothesis, we examined alterations of 5hmC and TET1 levels in 498 human pancreatic tissue regions, including normal ducts, low- and high-grade precancerous lesions, and associated invasive carcinomas.
Materials and methods
Case selection
This study was approved by the Institutional Review Board (IRB) of the Johns Hopkins Hospital. We identified patients with precancerous lesions who had undergone pancreatic resection without neoadjuvant chemotherapy. Normal ducts (p = 118) and neoplasms (n = 380), including PanIN, IPMN, IOPN, and MCN, were obtained from anonymized surgical specimens. The sections were derived from individual slides or tissue microarrays containing 70–110 circular samples each. The tissue microarray slides contained two to five distinct cores (1.5–2.0 mm in diameter) from different areas of the tissue blocks in each case. The electronic medical records were reviewed to obtain clinicopathological information (e.g. gender, age, ethnicity, comorbidity, neoplasm location, etc.), as listed in Table 1. In brief, PanIN, IPMN, and IOPN were most common in older males (>60 years old), whereas MCN was commonly observed in middle-aged females (40–50 years old). In addition, PanIN, IPMN, and IOPN arose more frequently in the head of the pancreas, whereas MCN was observed in the body and/or tail.
Table 1.
Clinicopathological characteristics of patients with pancreatic precancerous lesions.
| PanIN slide | IPMN/IOPN slide | MCN slide | |
|---|---|---|---|
| Age (years); median [range] | 68 [43–88] | 69 [38–87] | 47 [18–78] |
| Gender | |||
| Male | 41 (63%) | 62 (55%) | 2 (1%) |
| Female | 24 (37%) | 51 (45%) | 135 (99%) |
| Ethnicity | |||
| Caucasian | 53 (82%) | 102 (90%) | 15 (11%) |
| African American | 9 (14%) | 6 (5%) | 5 (4%) |
| Asian | 0 (0%) | 2 (2%) | 116 (85%) |
| Other | 3 (5%) | 3 (3%) | 1 (1%) |
| Main neoplasm location | |||
| Head | 40 (62%) | 83 (73%) | 5 (4%) |
| Body or tail | 25 (38%) | 30 (27%) | 132 (96%) |
| Cyst size (cm); median [range] | NA | 2.5 [0.5–10.5] | 4.5 [1.0–8.0] |
| Number of samples | |||
| Normal duct | 45 (32%) | 27 (16%) | 46 (24%) |
| Low grade | 53 (38%) | 80 (48%) | 124 (65%) |
| High grade/carcinoma | 42 (30%) | 61 (36%) | 20 (11%) |
| Histological subtypes | |||
| Gastric | NA | 83 (59%) | NA |
| Intestinal | NA | 24 (17%) | NA |
| Pancreatobiliary | NA | 22 (16%) | NA |
| Oncocytic (IOPN) | NA | 12 (9%) | NA |
NA, not applicable.
Histological examination
All H&E-stained slides were reviewed to assess lesion histology, size, grade of dysplasia, and presence of associated invasive cancer. Four types of precancerous lesion (PanIN, n = 95; IPMN, n = 129; IOPN, n = 12; MCN, n = 144) were diagnosed according to the fifth edition of WHO classification of tumors of the digestive system [36]. Non-invasive precancerous lesions were classified as low or high grade using the two-tiered system [37]. We also examined an associated invasive PDAC observed in 34 PanIN cases and 13 MCN cases, and associated PDACs were grouped with high-grade lesions for statistical analysis because of the limited number of lesions and similar range of H-scores in the specimens. In total, 257 low-grade and 123 high-grade/carcinoma lesions were investigated. The PanIN cohort comprised 53 low-grade and 42 high-grade lesions (see supplementary material. Table S1). IPMNs were histologically divided into three subtypes: gastric (n = 83), intestinal (n = 24), and pancreatobiliary (n = 22) (Table 1). In addition, we also examined IOPNs (n = 12), which were grouped with IPMNs for statistical analysis (referred to as the IPMN/IOPN cohort). In the IPMN/IOPN specimens, the gastric type was the most common (n = 83, 59%) and was usually low grade, whereas the pancreatobiliary type (n = 22. 16%) was more likely to be high grade. Intestinal was the second most common subtype (n = 24, 17%), and low-grade and high-grade lesions were equally observed (see supplementary material, Table S1). All IOPN cases (n = 12, 9%) showed high-grade dysplasia. The majority of MCN cases had low-grade dysplasia (low grade, n = 124; high grade, n = 20) (see supplementary material, Table S1). Normal controls were obtained from histologically normal ducts distant from neoplastic lesions on the same slide (n = 45 on PanIN slides; n = 27 on IPMN slides; n = 46 on MCN slides).
Immunohistochemistry
We performed immunohistochemistry (IHC) analyses for 5hmC and TET1 on all cases with precancerous lesions. Formalin-fixed paraffin-embedded blocks were sectioned at 5 μm, deparaffinized, and subjected to antigen retrieval. Sections were then immunolabeled using antibodies to 5hmC (rabbit polyclonal antibody; 1:20 000 dilution; Active motif, Carlsbad, CA, USA) and TET1 (mouse monoclonal antibody, clone GT1462; 1:400 dilution; Thermo Fisher Scientific, Waltham, MA, USA) using an autostainer (Ventana Benchmark LT: Ventana, Tucson, AZ, USA) or manually. Immunoreactivity was visualized using DAB as a substrate for HRP. After IHC labeling, tissue sections were counterstained with Mayer’s hematoxylin.
To generate positive controls for optimization of staining for TET2, HEK293 cells were transiently transfected with myc-tagged TET2 constructs, or empty vectors, using Lipofectamine 2000 (Thermo Fisher Scientific), as described previously [25]. Cell pellets were fixed in 10% buffered formalin and embedded in paraffin. HEK293 slides and pancreatic tissue slides were stained using a TET2 antibody (mouse monoclonal antibody, clone AMAB91439; 1:50 dilution; Novus Biologicals, Littleton, CO, USA).
IHC results were evaluated using a histological score (H-score; range 0–300). This semiquantitative score was obtained by multiplying the total nuclear immunoreactivity intensity (scored as 0, 1, 2, or 3) and corresponding percentage of labeled cells (0–100%). The intensity score was defined as follows: 0 - no appreciable nuclear labeling (negative); 1 - barely detectable labeling in the nucleus (weak); 2 - readily appreciable brown labeling distinctly marking the nucleus (moderate); 3 - very dark nuclear labeling (strong). The proportion score was evaluated twice by pathologists via counting positive nuclei. When low-grade and high-grade lesions coexisted on a slide, H-scores for these two lesions were separately calculated and were plotted as two different points on the scatter diagram.
Survival analysis
The overall survival of patients was compared using the Kaplan-Meier method, and differences between survival curves were tested using the log-rank test. Median H-scores were used as cutoffs to separate the patients with high and low levels of 5hmC or TET1. An H-score above the median was defined as high and an H-score equal or below the median was defined as low for each marker. A probability of p < 0.05 was considered significant. The software used for the Kaplan-Meier method was MEDCALC (https://www.medcalc.org/).
Statistical analysis
The data were statistically analyzed using the Mann-Whitney U-test or Wilcoxon signed-rank test. A probability of p < 0.05 was considered to be significant. Pearson’s correlation coefficient was used to measure the linear correlation between 5hmC and TET1 levels. Statistical analyses were performed using JMP software (version 10.0; SAS Institute, Cary, NC, USA).
Results
Decreased levels of 5hmC and TET1 in PanINs, IPMNs, IOPNs, and MCNs
IHC labeling for 5hmC revealed nuclear labeling of normal ducts, precancerous lesions, acinar cells, and islet cells (Figure 1). The expression levels of 5hmC were significantly downregulated in all types of precancerous lesion and associated invasive PDACs, compared with normal ductal epithelium (Figures 1,2; Mann-Whitney U-test, all p < 0.001). As TET enzymes can convert 5mC into 5hmC, we next questioned whether TET proteins were also downregulated in these neoplastic lesions. In line with the results of 5hmC IHC labeling, nuclear TET1 expression was significantly lower in precancerous lesions and associated invasive PDACs compared with control normal ductal epithelium (Figures 1,2; Mann-Whitney U-test, all p < 0.001). The Pearson’s correlation coefficients indicated strong correlations between H-scores of 5hmC and TET1 (R2 = 0.55 in PanIN, R2 = 0.52 in IPMN/IOPN, and R2 = 0.49 in MCN) (Figure 2).
Figure 1.
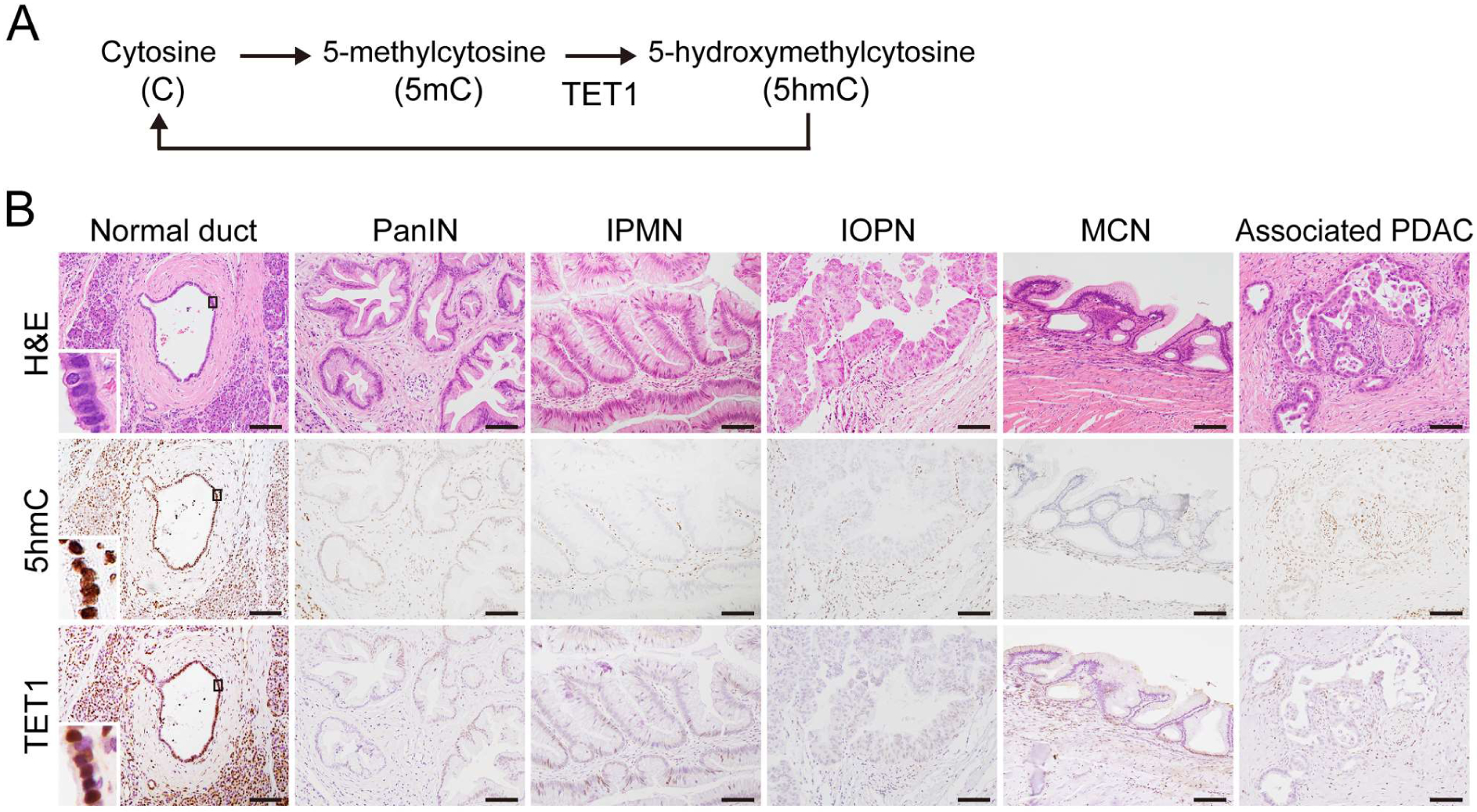
Immunohistological analyses of 5hmC and TET1 on histological slides with pancreatic precancerous lesions. (A) Cycle of DNA methylation and demethylation in mammals. TET enzymes oxidize 5mC to 5hmC. (B) Representative images of H&E and IHC staining in paraffin-embedded pancreatic tissue with any of the four precancerous lesions (PanIN, IPMN, IOPN, and MCN). The normal duct, PanIN, IPMN, IOPN, MCN, and associated invasive PDAC were labeled with anti-5hmC and anti-TET1 antibodies. The enlarged inset shows the nuclear localization of 5hmC and TET1 in the normal duct. Original magnification: × 200. Scale bar = 100 μm.
Figure 2.
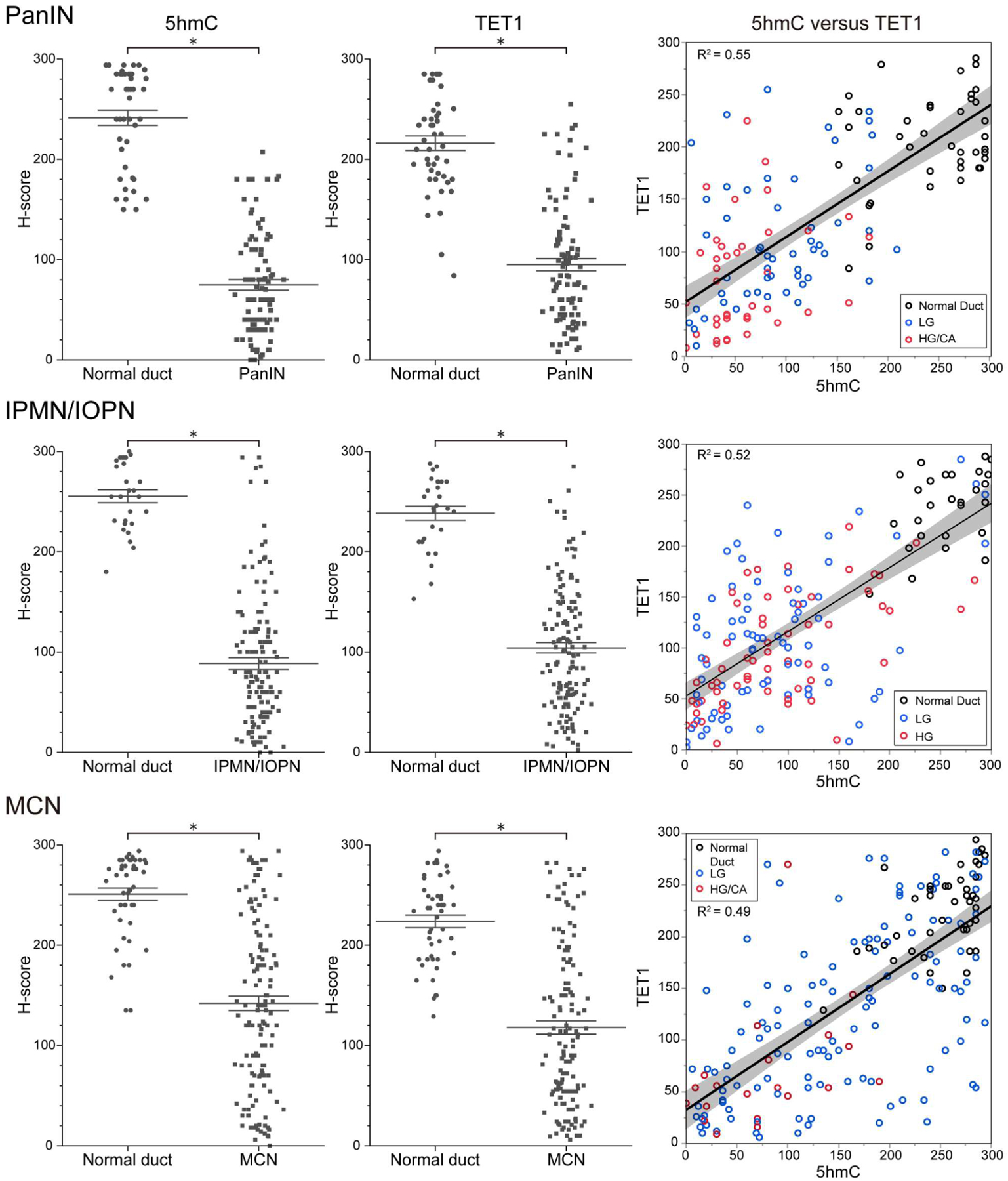
Comparison of the extent of 5hmC and TET1 immunoreactivity in normal ducts versus precancerous lesions. Data are presented as scatter plots of H-scores (0–300) on PanIN slides (normal duct, n = 45; neoplasm, n = 95), IPMN/IOPN slides (normal duct, n = 27; neoplasm, n = 141), or MCN slides (normal duct, n = 46; neoplasm, n = 144). Lines and error bars indicate the mean and SEM, respectively. Correlation plots between 5hmC H-scores and TET1 H-scores are shown in the right-hand panels. Pearson’s correlation coefficients (R2) were calculated to analyze the correlation between TET1 and 5hmC levels. Black dot: normal duct; blue dot: low-grade lesion; red dot: high-grade (/carcinoma) lesion. When low-grade and high-grade lesions coexist on a slide, these two lesions usually have different H-scores and therefore are shown as two separate dots on the graph (see also Figure 4 for further analysis of patients with three matched regions, namely normal duct, low-grade lesion, and high-grade lesion from the same patient). Mann-Whitney U-test, *p < 0.001. H6, high grade; LG, low grade; CA, carcinoma.
We next investigated whether downregulation of TET2 could also play a role in the alteration of 5hmC in early pancreatic tumorigenesis. To do this, we first carried out IHC for TET2 in a small number of lesions (total lesions, n = 21; normal duct, n = 7; PanIN, n = 10; IOPN, n = 3; MCN, n = 1). In both normal pancreatic ducts and neoplasms, weak nuclear expression of TET2 was observed in a small percentage of cells (usually less than 10%), and there was no significant difference in H-score between normal and neoplastic cells (see supplementary material, Figure S1). There was no correlation between TET2 and 5hmC labeling in this cohort (see supplementary material, Figure S1). Considering the weak TET2 labeling even in normal duct and the lack of difference between normal duct and precancerous neoplasms, we did not expand the TET2 analysis to our larger cohort of precancerous lesions. We focused all subsequent studies on 5hmC and TET1.
When the neoplastic lesions were stratified according to histological grade, higher-grade PanINs and MCNs tended to have lower 5hmC and TET1 H-scores than lower-grade PanINs and MCNs (Mann-Whitney U-test, all p < 0.005), whereas high-grade and low-grade IPMNs/IOPNs had similar H-scores (Figure 3). However, in PanINs and MCNs, the magnitude of the difference in H-score in the normal/low grade comparison was far greater than the difference in H-score in the low grade/high grade comparison. Thus, even in lesion types that had a significant difference between low grade and high grade, the difference in H-score was far greater between normal epithelium and low-grade lesions. Low-grade and high-grade lesions can coexist on a single slide, so we next compared the 5hmC and TET1 H-scores in matched samples of different grades in these cases. We further analyzed 23 PanIN cases with three matched regions (i.e. normal duct - low grade – high grade/carcinoma). As expected, H-scores for 5hmC and TET1 were highest in normal ductal epithelium, then decreased dramatically in low-grade lesions, and further decreased in high-grade/carcinoma lesions, with the greatest difference between normal ductal epithelium and low-grade lesion (Wilcoxon signed-rank test, low grade versus high grade/carcinoma, p = 0.04 in 5hmC and p < 0.001 in TET1) (Figure 4). Similar to the results of comparisons based on grade of dysplasia, there was no significant difference in H-scores among the four histological subtypes in IPMN/IOPN cohort (Figure 5).
Figure 3.
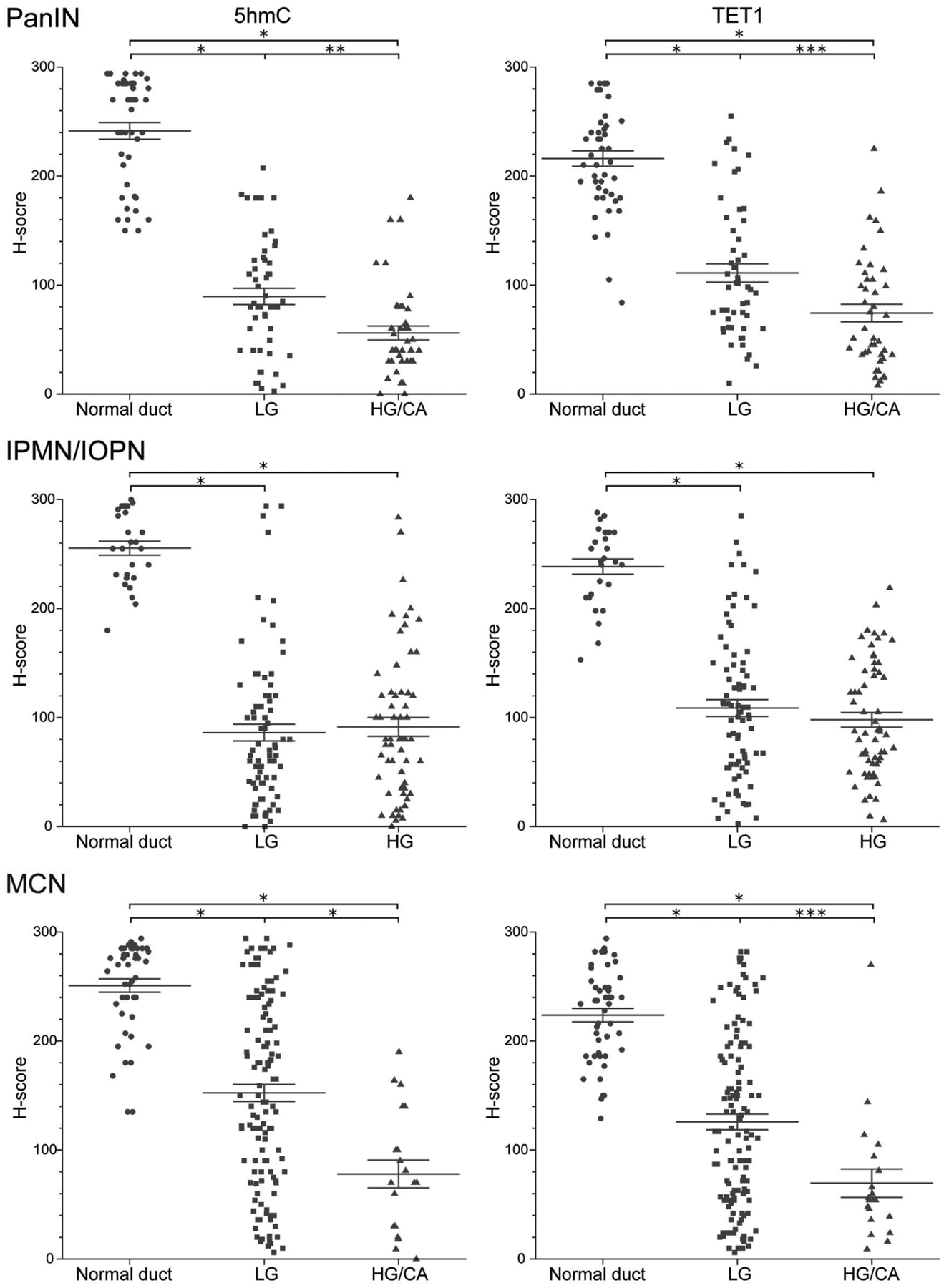
Comparison of H-scores of 5hmC and TETI according to histological grade in three precancerous lesions. Data are presented as scatter plots of H-scores (0–300) on PanIN slides (normal duct, n = 45; low grade, n = 53; high grade/carcinoma, n = 42), IPMN/IOPN slides (normal duct, n = 27; low grade, n = 80; high grade, n = 61), or MCN slides (normal duct, n = 46; low grade, n = 124; high grade/carcinoma, n = 20). Lines and error bars indicate the mean and SEM, respectively. Mann-Whitney U-test, *p < 0.001, **p = 0.001, ***p = 0.002. HG, high grade; LG, low grade; CA, carcinoma.
Figure 4.
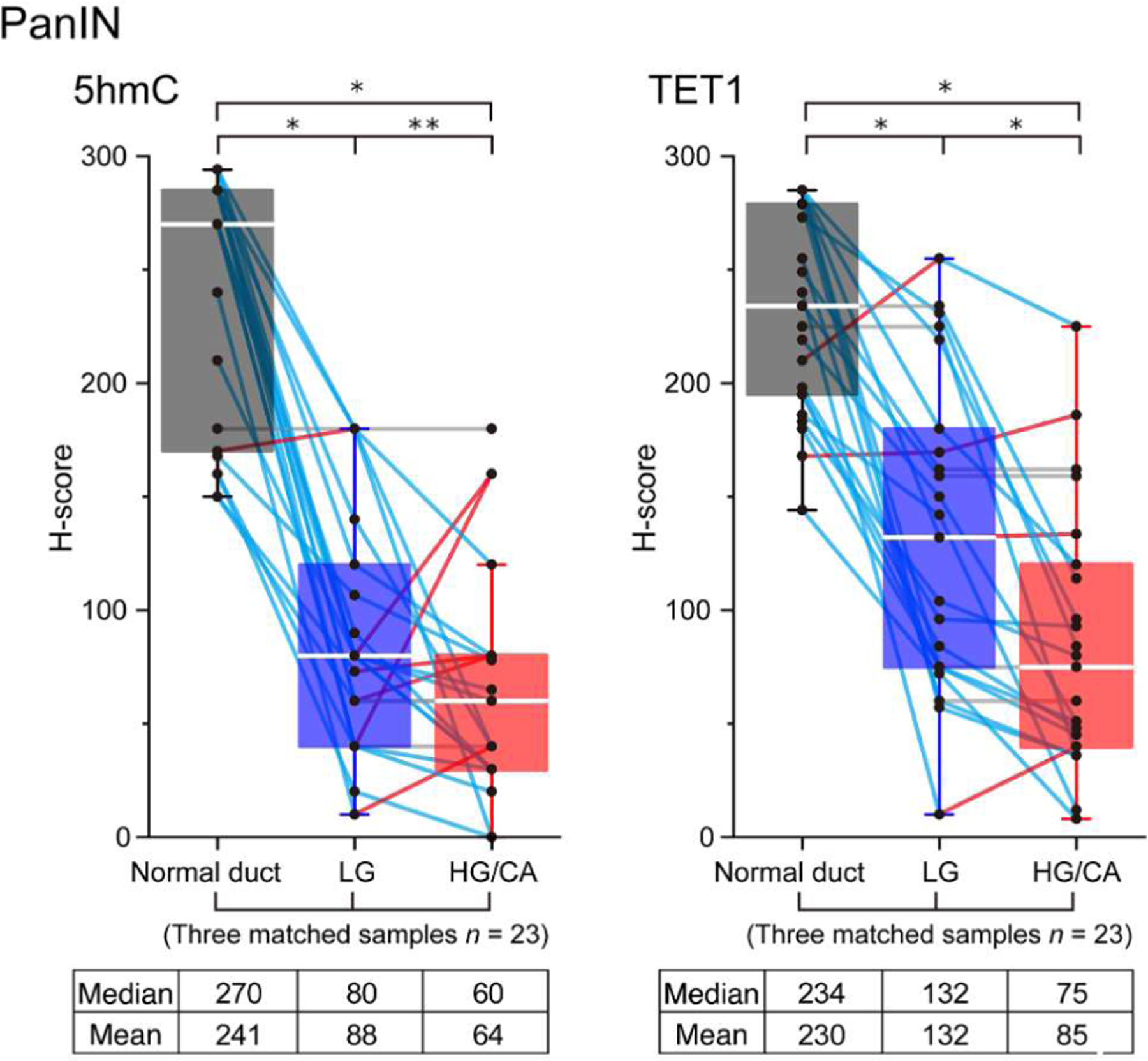
Comparative analysis of H-scores in the matched samples from PanIN cases. Box-plot of H-scores of 5hmC and TET1 on PanIN slides (n = 23) with three matched samples (normal duct, low-grade lesion, and high-grade/carcinoma lesion) at the same time. Each score is represented by a small circle, and the matched data for each individual are joined by a solid line (upregulation in red; downregulation in blue; no change in gray). For all box-plots, the solid center line represents the median, the lower and upper bounds of the box represent the 25th and 75th percentiles, respectively, and the whiskers are Tukey whiskers. Two-sided Wilcoxon signed-rank test, *p < 0.001, **p = 0.04. HG, high grade; LG, low grade; CA, carcinoma.
Figure 5.
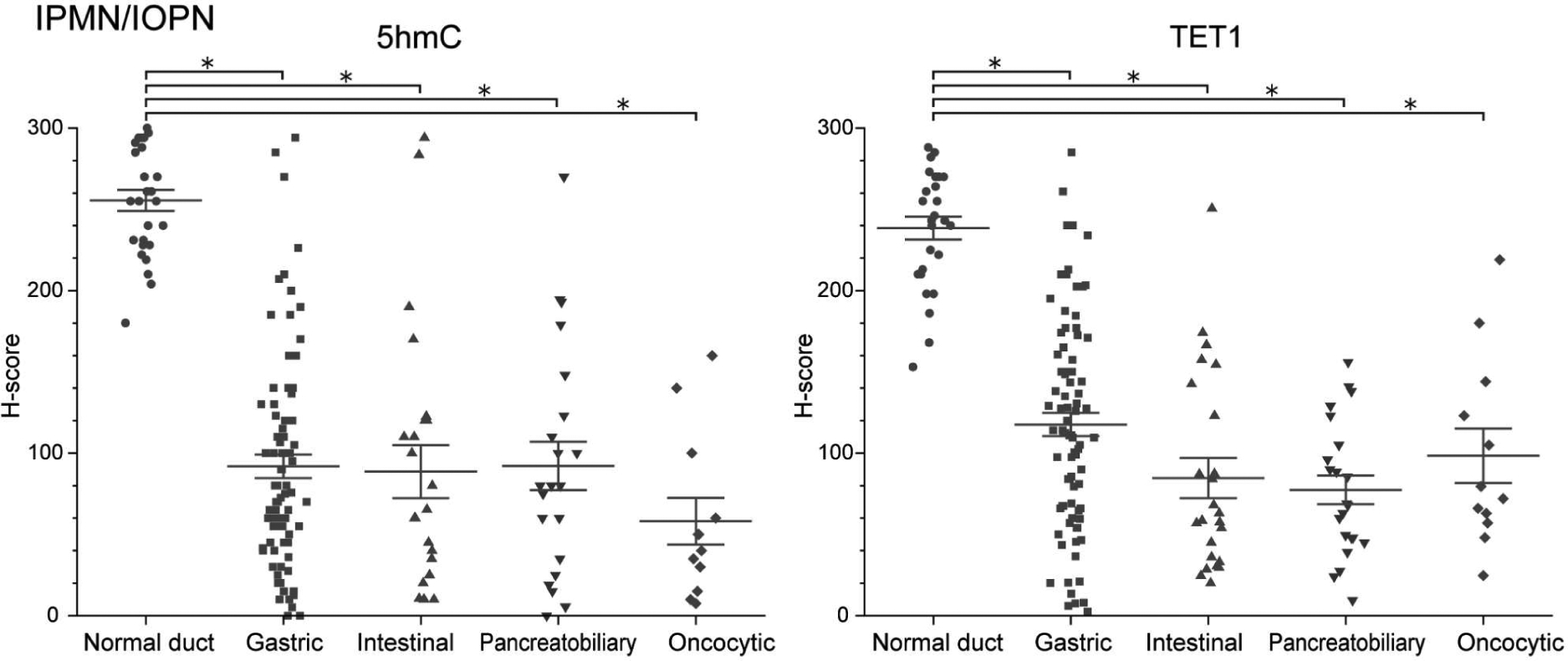
Comparison of H-scores of 5hmC and TET1 according to histological subtype in IPMN/IOPN cohort. Data are presented as scatter plots of H-scores (0–300) on IPMN/IOPN slides (normal duct, n = 27; gastric, n = 83; intestinal, n = 24; pancreatobiliary, n = 22; oncocytic, n = 12). Lines and error bars indicate the mean and SEM, respectively. Mann-Whitney U-test, *p < 0.001.
Relationship between 5hmC levels and diabetes
A previous report suggested that hyperglycemia decreases the levels of 5hmC in blood cells in patients with type 2 diabetes mellitus (T2DM) [33]. As such, we next interrogated whether such a mechanism could apply in the pancreas by determining the correlation of 5hmC levels in our pancreatic tissue samples with a clinical history of T2DM. We observed no significant difference in 5hmC levels in normal or neoplastic epithelial cells between patients with and without T2DM (see supplementary material, Figure S2 and Table S2). There were also no clear associations between 5hmC level and age, gender, cyst size, or neoplasm location (see supplementary material, Tables S2, S3). These results suggest that decreased levels of 5hmC in neoplastic epithelial cells occur across a broad cohort of patients with precancerous pancreatic lesions and are not attributed to a single risk factor.
Relationship between 5hmC/TET1 levels and overall survival
Kaplan-Meier curves and log-rank tests were used to estimate overall survival in relation to 5hmC and TET1 levels in patients with PDAC associated with PanIN. There were no statistically significant differences between the groups with low and high H-scores (5hmC, hazard ratio = 1.18, log-rank test p = 0.72; TET1, hazard ratio = 2.09, log-rank test p = 0.10), although the hazard ratio was slightly higher in the patients with low H-scores in TET1 (see supplementary material, Figure S3).
Discussion
We showed recently that some low-grade pancreatic precancerous lesions contain multiple genetically independent clones [12–14]. The presence of multifocal neoplasia in the pancreas suggests that there may be alternative non-mutational alterations that promote the formation of independent clones in early pancreatic tumorigenesis. In this study, we sought to examine the prevalence of epigenetic alterations in pancreatic precancerous lesions. DNA methylation/hydroxymethylation is one of the most widely studied epigenetic modifications and has been shown to play a significant role in tumorigenesis [38,39]. Previous studies have shown that aberrant DNA methylation/hydroxymethylation is a common event in human malignancy and is associated with the regulation of individual genes and large chromosomal regions [40]. Therefore, epigenetic alterations are a strong candidate mechanism for such early non-genetic alterations. To investigate epigenetic changes in early pancreatic tumorigenesis, we examined 5hmC and its regulator TET1 by IHC in 380 pancreatic precursor lesions and 118 corresponding normal ducts. Our results demonstrate that epigenetic regulation was disrupted in all types and all histological grades of precancerous lesions. In addition, the levels of 5hmC correlated with those of TET1. Importantly, epigenetic alterations were observed in even low-grade PanIN, IPMN, and MCN lesions. These results suggest that global epigenetic alterations might be one of the earliest events in pancreatic neoplasm formation, and 5hmC and TET1 may play an important role in aberrant DNA demethylation processes in the pancreas.
Previous studies have reported the downregulation of TET1 and 5hmC in PDAC [41–43]. We focused our analysis on pancreatic precancerous lesions in order to elucidate the timing of this downregulation in early pancreatic neoplasia. To our knowledge, this is the first comprehensive study investigating four well-characterized human pancreatic precursor lesions. Other previous studies have further investigated 5hmC levels at specific gene loci in either PDAC tissue or cell-free DNA, suggesting that such cancer-associated 5hmC signatures are characteristic of specific cancer types and may serve as potential biomarkers [44–46]. In light of these findings, our results suggest that gene-specific alterations in 5hmC distribution may occur even in the early stages of pancreatic carcinogenesis. As a next step, it will be important to integrate global and gene-specific 5hmC analysis using genome-wide 5hmC profiling approaches in precancerous lesions. In addition to 5hmC dysregulation, tumor development and progression of pancreatic neoplasms also require close coordination between genetic and epigenetic programs [47]. Further experimental data will be required to elucidate the molecular relationship between epigenetic alterations and early driver mutations such as KRAS mutations in pancreatic tumorigenesis.
Previous reports suggest that TET1 is the most abundant among the three TET family proteins and that TET1 plays a dominant role in 5hmC formation [19]. In addition, enrichment of 5hmC at promoters is most likely to be associated with TET1, whereas the 5hmC level at gene bodies and exon boundaries of active genes correlates with TET2 activity [48]. These functional differences between TET enzymes may contribute to the strong correlation of TET1, rather than TET2, with 5hmC. The function of TET1 in epithelial neoplasms is still controversial because both upregulation and downregulation of TET1 expression have been reported in malignant tumors [20–32]. Our results suggest that pancreatic ductal neoplasms including precancerous lesions and associated invasive carcinomas typically have reduced levels of TET1 and 5hmC. Based on our results and previous reports, whether up- or downregulation of TET1 promotes neoplasia may be organ-dependent, and the tissue environment at the primary site may alter the epigenetic status of neoplastic cells.
Activation of TET and restoration of normal 5hmC levels and DNA methylation patterns may provide some value in the treatment and/or prevention of progression in pancreatic ductal neoplasms. Vitamins A and C have recently been shown to remodel the epigenome by enhancing the TET expression and catalytic activity, respectively [49]. The benefit of high doses of vitamins in PDAC treatment is still controversial [50]. Nevertheless, it is possible that vitamin supplementation could reverse global loss of 5hmC in precancerous pancreatic lesions, but the functional impact of such pharmacological epigenetic reprogramming on early pancreatic tumorigenesis remains to be studied in detail in the future. In addition, further clinical studies are needed to investigate the potential efficacy of vitamins as chemo-preventive or therapeutic agents in patients with pancreatic neoplasms.
As with other types of cancer, PDAC is known to be closely associated with environmental factors. Previous reports suggested that hyperglycemia, obesity, smoking, and alcohol consumption are linked to an increased risk of PDAC [51–53]. Some of these risk factors, such as tobacco and alcohol exposure, are relatively well understood and have been shown to lead to the accumulation of DNA damage, whereas other factors are still largely unexplored [54,55]. Based on previous data on 5hmC levels in blood cells of diabetic patients [33], we focused our analysis on the correlation between the presence of T2DM and 5hmC expression in precancerous pancreatic neoplasms, but we did not identify a correlation in our cohort. However, patients in our cohort diagnosed with diabetes included those with primary diabetes as well as those with diabetes secondary to their pancreatic neoplasm [56]. Because we cannot distinguish these possibilities based on retrospective clinical data, it is possible that there is a link between primary diabetes and 5hmC in pancreatic cells that cannot be identified using our cohort. Further studies are needed to determine whether predisposing environmental factors contribute to the decline in 5hmC in epithelial cells, as has been reported in blood cells.
In conclusion, our data provide several insights into early non-mutational alterations in pancreatic tumorigenesis. We show that 5hmC and TET1 are significantly downregulated at high prevalence in all types of precancerous lesion compared with normal ductal epithelium. These results suggest that global epigenetic dysregulation via downregulation of 5hmC and TET1 is one of the earliest molecular events in pancreatic tumorigenesis.
Supplementary Material
Figure S1. IHC analyses for TET2
Figure S2. Analysis of 5hmC levels in patients with or without T2DM
Figure S3. Kaplan-Meier survival curves for overall survival after stratification by 5hmC and TET1 levels
Table S1. Number of human samples analyzed in this study
Table S2. Relationship between clinicopathological factors and 5hmC level (Mann-Whitney U-test)
Table S3. Relationship between clinicopathological factors and 5hmC level (Pearson’s correlation coefficients, R2)
Acknowledgements
The authors acknowledge the following sources of funding: NIH/NCI P50 CA62924; NIH/NIDDK K08 DK107781; SKCCC Cancer Center Support Grant (CCSG; P30 CA006973); Sol Goldman Pancreatic Cancer Research Center; Buffone Family Gastrointestinal Cancer Research Fund; Carol S. and Robert M. Long Pancreatic Cancer Research Fund; Kaya Tuncer Career Development Award in Gastrointestinal Cancer Prevention; AGA-Bernard Lee Schwartz Foundation Research Scholar Award in Pancreatic Cancer; Sidney Kimmel Foundation for Cancer Research Kimmel Scholar Award; AACR-Incyte Corporation Career Development Award for Pancreatic Cancer Research; American Cancer Society - The Cornelia T. Bailey Foundation Research Scholar Grant RSG-18-143-01; Emerson Collective Cancer Research Fund; Rolfe Pancreatic Cancer Foundation; Joseph C. Monastra Foundation; The Gerald O. Mann Charitable Foundation (Harriet and Allan Wulfstat, Trustees); Susan Wojcicki and Denis Troper.
Conflict of interest statement LDW receives research funding from Applied Materials unrelated to the studies described in this article and is an Associate Editor of The Journal of Pathology. No other conflicts of interest were declared.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020; 70: 7–30. [DOI] [PubMed] [Google Scholar]
- 2.Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med 2014; 371: 1039–1049. [DOI] [PubMed] [Google Scholar]
- 3.Kamisawa T, Wood LD, Itoi T, et al. Pancreatic cancer. Lancet 2016; 388: 73–85. [DOI] [PubMed] [Google Scholar]
- 4.Hruban RH, Klimstra DS. Adenocarcinoma of the pancreas. Semin Diagn Pathol 2014; 31: 443–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brosens LA, Hackeng WM, Offerhaus GJ, et al. Pancreatic adenocarcinoma pathology: changing “landscape”. J Gastrointest Oncol 2015; 6: 358–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pittman ME, Rao R, Hruban RH. Classification, morphology, molecular pathogenesis, and outcome of premalignant lesions of the pancreas. Arch Pathol Lab Med 2017; 141: 1606–1614. [DOI] [PubMed] [Google Scholar]
- 7.Ren B, Liu X, Suriawinata AA. Pancreatic ductal adenocarcinoma and its precursor lesions: histopathology, cytopathology, and molecular pathology. Am J Pathol 2019; 189: 9–21. [DOI] [PubMed] [Google Scholar]
- 8.Basturk O, Chung SM, Hruban RH, et al. Distinct pathways of pathogenesis of intraductal oncocytic papillary neoplasms and intraductal papillary mucinous neoplasms of the pancreas. Virchows Arch 2016; 469: 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang T, Askan G, Adsay V, et al. Intraductal oncocytic papillary neoplasms: clinical-pathologic characterization of 24 cases, with an emphasis on associated invasive carcinomas. Am J Surg Pathol 2019;43:656–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lennon AM, Wolfgang CL, Canto MI, et al. The early detection of pancreatic cancer: what will it take to diagnose and treat curable pancreatic neoplasia? Cancer Res 2014; 74: 3381–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brugge WR. Diagnosis and management of cystic lesions of the pancreas. J Gastrointest Oncol 2015; 6: 375–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fischer CG, Beleva Guthrie V, Braxton AM, et al. Intraductal papillary mucinous neoplasms arise from multiple independent clones, each with distinct mutations. Gastroenterology 2019; 157: 1123–1137.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noë M, Niknafs N, Fischer CG, et al. Genomic characterization of malignant progression in neoplastic pancreatic cysts. Nat Commun 2020; 11: 4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fujikura K, Hosoda W, Felsenstein M, et al. Multiregion whole-exome sequencing of intraductal papillary mucinous neoplasms reveals frequent somatic KLF4 mutations predominantly in low-grade regions. Gut 2021; 70: 928–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu PY, Hsu HK, Singer GA, et al. Estrogen-mediated epigenetic repression of large chromosomal regions through DNA looping. Genome Res 2010; 20: 733–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stroud H, Feng S, Morey Kinney S, et al. 5-Hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol 2011; 12: R54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kafer GR, Li X, Horii T, et al. 5-Hydroxymethylcytosine marks sites of DNA damage and promotes genome stability. Cell Rep 2016; 14: 1283–1292. [DOI] [PubMed] [Google Scholar]
- 18.Mulholland CB, Traube FR, Ugur E, et al. Distinct and stage-specific contributions of TET1 and TET2 to stepwise cytosine oxidation in the transition from naive to primed pluripotency. Sci Rep 2020; 10: 12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Putiri EL, Tiedemann RL, Thompson JJ, et al. Distinct and overlapping control of 5-methylcytosine and 5-hydroxymethylcytosine by the TET proteins in human cancer cells. Genome Biol 2014; 15: R81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang KC, Kang CH, Tsai CY, et al. Ten-eleven translocation 1 dysfunction reduces 5-hydroxymethylcytosine expression levels in gastric cancer cells. Oncol Lett 2018; 15: 278–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tian X, Sun B, Chen C, et al. Circulating tumor DNA 5-hydroxymethylcytosine as a novel diagnostic biomarker for esophageal cancer. Cell Res 2018; 28: 597–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi X, Yu Y, Luo M, et al. Loss of 5-hydroxymethylcytosine is an independent unfavorable prognostic factor for esophageal squamous cell carcinoma. PLoS One 2016; 11: e0153100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsai KW, Li GC, Chen CH, et al. Reduction of global 5-hydroxymethylcytosine is a poor prognostic factor in breast cancer patients, especially for an ER/PR-negative subtype. Breast Cancer Res Treat 2015; 153: 219–234. [DOI] [PubMed] [Google Scholar]
- 24.Liu C, Liu L, Chen X, et al. Decrease of 5-hydroxymethylcytosine is associated with progression of hepatocellular carcinoma through downregulation of TET1. PLoS One 2013; 8: e62828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haffner MC, Chaux A, Meeker AK, et al. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget 2011; 2: 627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen LY, Huang RL, Chan MW, et al. TET1 reprograms the epithelial ovarian cancer epigenome and reveals casein kinase 2alpha as a therapeutic target. J Pathol 2019; 248: 363–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bai X, Zhang H, Zhou Y, et al. Ten-Eleven Translocation 1 promotes malignant progression of cholangiocarcinoma with IDH1 wild-type. Hepatology 2021; 73: 1747–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Filipczak PT, Leng S, Tellez CS, et al. p53-suppressed oncogene TET1 prevents cellular aging in lung cancer. Cancer Res 2019; 79: 1758–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Good CR, Panjarian S, Kelly AD, et al. TET1-mediated hypomethylation activates oncogenic signaling in triple-negative breast cancer. Cancer Res 2018; 78: 4126–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen YL, Hu CM, Hsu JT, et al. Cellular 5-hydroxylmethylcytosine content determines tumorigenic potential and prognosis of pancreatic ductal adenocarcinoma. Am J Cancer Res 2018; 8: 2548–2563. [PMC free article] [PubMed] [Google Scholar]
- 31.Orr BA, Haffner MC, Nelson WG, et al. Decreased 5-hydroxymethylcytosine is associated with neural progenitor phenotype in normal brain and shorter survival in malignant glioma. PLoS One 2012; 7: e41036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cavalcante GM, Borges DP, de Oliveira RTG, et al. Tissue methylation and demethylation influence translesion synthesis DNA polymerases (TLS) contributing to the genesis of chromosomal abnormalities in myelodysplastic syndrome. J Clin Pathol 2020. 10.1136/jclinpath-2020-207131 [DOI] [PubMed] [Google Scholar]
- 33.Wu D, Hu D, Chen H, et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature 2018; 559: 637–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vincent A, Omura N, Hong SM, et al. Genome-wide analysis of promoter methylation associated with gene expression profile in pancreatic adenocarcinoma. Clin Cancer Res 2011; 17: 4341–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hong SM, Omura N, Vincent A, et al. Genome-wide CpG island profiling of intraductal papillary mucinous neoplasms of the pancreas. Clin Cancer Res 2012; 18: 700–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.WHO Classification of Tumours of the Digestive System (5th edn) edn). IARC Press: Lyon, 2019. [Google Scholar]
- 37.Basturk O, Hong SM, Wood LD, et al. A revised classification system and recommendations from the Baltimore Consensus Meeting for neoplastic precursor lesions in the pancreas. Am J Surg Pathol 2015; 39: 1730–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell 2012; 150: 12–27. [DOI] [PubMed] [Google Scholar]
- 39.Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science 2017; 357: eaal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Do C, Shearer A, Suzuki M, et al. Genetic-epigenetic interactions in cis: a major focus in the post-GWAS era. Genome Biol 2017; 18: 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H, Jiang W, Liu XN, et al. TET1 downregulates epithelial-mesenchymal transition and chemoresistance in PDAC by demethylating CHL1 to inhibit the Hedgehog signaling pathway. Oncogene 2020; 39: 5825–5838. [DOI] [PubMed] [Google Scholar]
- 42.Zeng C, Zhang Z, Wang J, et al. Application of the high-throughput TAB-array for the discovery of novel 5-hydroxymethylcytosine biomarkers in pancreatic ductal adenocarcinoma. Epigenomes 2019; 3: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu J, Li H, Shi M, et al. TET1-mediated DNA hydroxymethylation activates inhibitors of the Wnt/beta-catenin signaling pathway to suppress EMT in pancreatic tumor cells. J Exp Clin Cancer Res 2019; 38: 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cao F, Wei A, Hu X, et al. Integrated epigenetic biomarkers in circulating cell-free DNA as a robust classifier for pancreatic cancer. Clin Epigenetics 2020; 12: 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guler GD, Ning Y, Ku CJ, et al. Detection of early stage pancreatic cancer using 5-hydroxymethylcytosine signatures in circulating cell free DNA. Nat Commun 2020; 11: 5270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li W, Zhang X, Lu X, et al. 5-Hydroxymethylcytosine signatures in circulating cell-free DNA as diagnostic biomarkers for human cancers. Cell Res 2017; 27: 1243–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khan AA, Liu X, Yan X, et al. An overview of genetic mutations and epigenetic signatures in the course of pancreatic cancer progression. Cancer Metastasis Rev 2021; 40: 245–272. [DOI] [PubMed] [Google Scholar]
- 48.Huang Y, Chavez L, Chang X, et al. Distinct roles of the methylcytosine oxidases Tet1 and Tet2 in mouse embryonic stem cells. Proc Natl Acad Sci USA 2014; 111: 1361–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hore TA. Modulating epigenetic memory through vitamins and TET: implications for regenerative medicine and cancer treatment. Epigenomics 2017; 9: 863–871. [DOI] [PubMed] [Google Scholar]
- 50.Cieslak JA, Cullen JJ. Treatment of pancreatic cancer with pharmacological ascorbate. Curr Pharm Biotechnol 2015; 16: 759–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hassan MM, Bondy ML, Wolff RA, et al. Risk factors for pancreatic cancer: case-control study. Am J Gastroenterol 2007; 102: 2696–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abbruzzese JL, Andersen DK, Borrebaeck CAK, et al. The interface of pancreatic cancer with diabetes, obesity, and inflammation: research gaps and opportunities: summary of a National Institute of Diabetes and Digestive and Kidney Diseases Workshop. Pancreas 2018; 47: 516–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stolzenberg-Solomon R, Derkach A, Moore S, et al. Associations between metabolites and pancreatic cancer risk in a large prospective epidemiological study. Gut 2020; 69: 2008–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Garaycoechea JI, Crossan GP, Langevin F, et al. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 2018; 553: 171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alexandrov LB, Ju YS, Haase K, et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016; 354: 618–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sah RP, Nagpal SJ, Mukhopadhyay D, et al. New insights into pancreatic cancer-induced paraneoplastic diabetes. Nat Rev Gastroenterol Hepatol 2013; 10: 423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. IHC analyses for TET2
Figure S2. Analysis of 5hmC levels in patients with or without T2DM
Figure S3. Kaplan-Meier survival curves for overall survival after stratification by 5hmC and TET1 levels
Table S1. Number of human samples analyzed in this study
Table S2. Relationship between clinicopathological factors and 5hmC level (Mann-Whitney U-test)
Table S3. Relationship between clinicopathological factors and 5hmC level (Pearson’s correlation coefficients, R2)