

Abstract
Fibrolamellar carcinoma (FLC), a rare, lethal hepatic cancer, occurs primarily in adolescents and young adults. Unlike hepatocellular carcinoma, FLC has no known association with viral, metabolic or chemical agents that cause cirrhosis. Currently, surgical resection is the only treatment demonstrated to achieve cure, and no standard of care exists for systemic therapy. Progress in FLC research illuminates a transition from an obscure cancer to one for which an interactive community seems poised to uncover fundamental mechanisms and initiate translation towards novel therapies. In this Roadmap, we review advances since the seminal discovery in 2014 that nearly all FLC tumours express a signature oncogene (DNAJB1–PRKACA) encoding a fusion protein (DNAJ–PKAc) in which the J-domain of a heat shock protein 40 (HSP40) co-chaperone replaces an amino-terminal segment of the catalytic subunit of the cyclic AMP-dependent protein kinase (PKA). Important gains include increased understanding of oncogenic pathways driven by DNAJ–PKAc; identification of potential therapeutic targets; development of research models; elucidation of immune mechanisms with potential for the development of immunotherapies; and completion of the first multicentre clinical trials of targeted therapy for FLC. In each of these key areas we propose a Roadmap for future progress.
Fibrolamellar carcinoma (FLC; also known as fibrolamellar hepatocellular carcinoma (FL-HCC)) is a rare liver cancer that was defined originally by its distinctive histology, especially the abundant lamellar bands of intratumoural fibrosis that are reflected in the tumour’s name1–5 (Fig. 1). FLC primarily affects adolescents and young adults at a median age of approximately 22 years6,7. Cancers in individuals in this age group pose distinct challenges for the understanding of biology, the development of effective therapies, and the provision of care, insurance cover and psychosocial support8,9. Despite the significant advances in FLC research that we discuss in this Roadmap, predisposing factors have not been identified and the population at risk remains poorly defined. Moreover, effective systemic therapy remains elusive5.
Fig. 1 |. Fibrolamellar carcinoma.
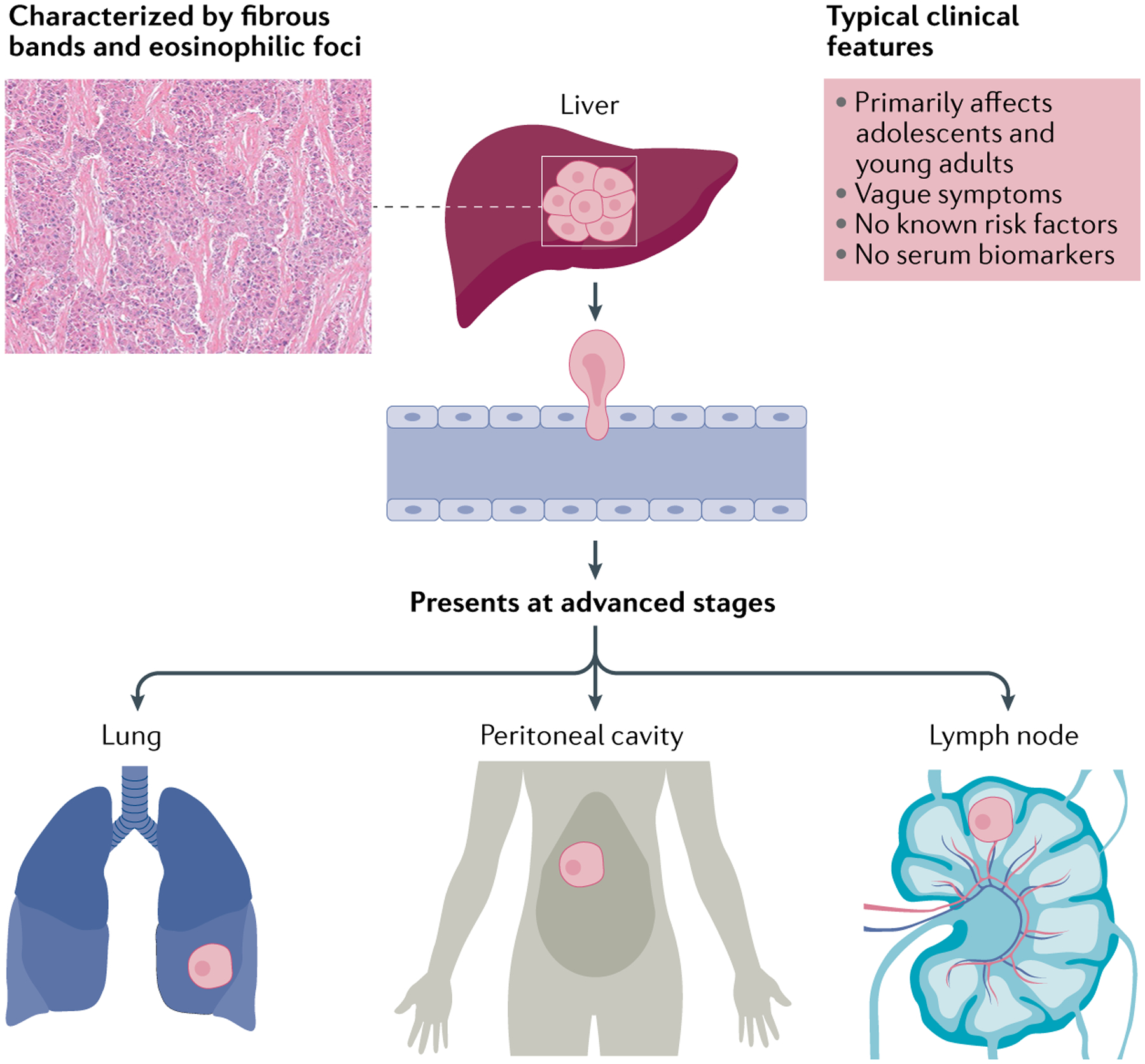
Tumour characteristics (left) and typical clinical features (right). Fibrolamellar carcinoma (FLC) presents, often with vague symptoms, in adolescents and young adults. It is associated neither with liver cirrhosis nor with other factors known to predispose to hepatocellular carcinoma. Primary FLC tumours often grow relatively slowly, but they are highly aggressive and invasive. The cure rate is very low. Common metastatic sites include local lymph nodes, lung and peritoneum. Less frequently observed locations include bone, ovary and intracranial166. Diagnosis often occurs only after extensive tumour growth, due to non-specific symptoms and a lack of non-invasive biomarker assays. Image (top left) courtesy of M. Torbenson, Mayo Clinic, USA.
While FLC cases have been reported around the world, the global incidence of this cancer remains uncertain. Systematic tracking is limited to the USA, where data from the Surveillance, Epidemiology, and End Results (SEER) Program of the National Cancer Institute reveal only 0.2 new diagnoses per million person-years10,11. However, some reports have suggested that FLC comprises a higher fraction of primary liver cancers12–16. The absence of a unique designation for FLC in the International Code of Diseases 10th Revision (ICD-10) might lead to inadvertent under-counting of this cancer. Furthermore, analysis of liver tumours in The Cancer Genome Atlas (TCGA) collection revealed that half (three of six) of those that had both histological and molecular features diagnostic of FLC were inaccurately annotated as having conventional hepatocellular carcinoma (HCC)17. Thus, although FLC undoubtedly is a very rare cancer, its incidence might exceed that indicated by the SEER data.
FLC only infrequently responds to chemotherapy or targeted therapies currently approved for HCC, although these are often used by default without a clear rationale, and therefore have not improved the poor long-term survival rates5,7,18–20. Surgical resection remains the sole available treatment modality with curative intent. However, this option is viable for only a limited number of patients, and tumour recurrence and/or metastasis are common18–20. Orthotopic liver transplantation, utilized for selected patients, gives comparable overall survival (OS) to that reported for HCC21. Radiotherapy and interventional radiology also contribute to current management of FLC, although published data on the effect of these modalities on survival remain scarce5. There is no standard of care for systemic therapy10,11,20. Owing to the lack of specific early warning signs and diagnostic biomarkers, by the time patients present with classic symptoms such as abdominal pain, a palpable mass, anorexia and, rarely, gynaecomastia22 or hyperammonaemia23–25, their disease is typically advanced and curative surgical resection is often not possible (FIG. 1).
Over the past 8 years significant progress has been made towards elucidating the molecular biology of FLC26. However, the challenge in translating knowledge into effective therapies remains unmet. An international group of researchers and physicians focused on FLC met in November 2019 at a Research Summit hosted by the Fibrolamellar Cancer Foundation, Greenwich, USA (for a list of relevant FLC databases, repositories and useful links, see BOX 1). The key goals were to share advances in laboratory and clinical studies, and to consider opportunities for future progress. Participants’ areas of expertise included the clinical care of patients with FLC (both oncological and surgical), biology and genomics, immunotherapy, in vitro and in vivo modelling, and translational research. Patients and family advocates were also represented. All authors of this paper were present at the summit. In this Roadmap, we aim to provide a curated guide to ongoing initiatives. We review developments and unmet needs in four areas: molecular mechanisms, disease models, immunotherapy and clinical trials. For each, we provide a brief background, highlight new research findings and pose important unanswered questions. Finally, we offer a Roadmap for the future, prioritizing next steps towards the development of novel and effective therapeutics.
Box 1 |. Fibrolamellar carcinoma databases and repositories.
FibroLamellar Omics (FLO): https://sethupathy-lab.shinyapps.io/flc_data/. A browser for genome-scale fibrolamellar carcinoma (FLC) data, including small RNA sequencing, RNA sequencing and chromatin run-on sequencing data.
Cancer Dependency Map: https://depmap.org/portal. A systematic effort to catalogue as completely as possible the genetic and pharmacological vulnerabilities in a wide array of established cell lines and 3D organoid cultures from a comprehensive collection of common and rare human cancers.
Kinase Chemogenomic Set (KCGS): https://www.sgc-unc.org/kinase-chemogenomics. A collection of specific small-molecule kinase inhibitors165. At the time of writing, the KCGS includes 645 inhibitors and covers 250 enzymes across all kinase families and includes compounds active against many orphan kinases.
Fibrolamellar Cancer Foundation Biobank (Massachusetts General Hospital): https://fibrofoundation.org/join-the-fight/donate-tissue/. A centralized repository of tumour tissue and blood samples contributed by patients with FLC to help advance research, available to qualified researchers interested in studying FLC.
Fibrolamellar Tissue Repository (Rockefeller university): https://fibrolamellar.rockefeller.edu/repository. A collection of tissue and blood specimens contributed by patients with FLC. Specimens will be distributed only to scientists at Rockefeller University and other approved institutions who have received authorization from their respective Institutional Review Boards. Because tissue samples are rare, investigators must submit a request that justifies their proposed research and use of tissue from the Repository.
The Fibrolamellar Registry: https://fibroregistry.org/. A collection of health data about patients with FLC, stored in a highly secure database. It is a comprehensive, online survey that gathers medical histories and test results. The Registry meets the highest standards for privacy and patient protection and has been approved as a scientific study by the Genetic Alliance Institutional Review Board.
Natural History Study of Rare Solid Tumors: https://www.cancer.gov/pediatric-adult-rare-tumor/participate/natural-history. A natural history trial taking place at the NIH Clinical Center in Bethesda, MD, USA. The purpose of the trial is to collect information and samples from people with rare tumours and their relatives, and to track their health history over a long period of time.
ClinicalTrials.gov, United States National library of Medicine: https://clinicaltrials.gov/. A database of privately and publicly funded clinical studies conducted around the world.
European Union Drug Regulating Authorities Clinical Trials Database (EudraCT): https://eudract.ema.europa.eu/. The European database for all interventional clinical trials on medicinal products authorized in the European Union (EEA) and outside the EU/EEA if they are part of a Paediatric Investigation Plan (PIP) from 1 May 2004 onwards.
Molecular mechanisms
DNAJB1–PRKACA fusion: the molecular hallmark of FLC.
FLC is a distinct disease from HCC26,27. As illustrated histologically, FLC is marked by the presence of thick lamellae of fibrous tissue surrounding clusters of large polygonal and eosinophilic tumour cells2,3,7 (FIG. 1). By immunohistochemistry, FLC tumours consistently co-express cytokeratin 7 (CK7) and CD68 (REFS7,28–30). Global gene expression profiling also clearly distinguishes FLC from HCC17,31–34. Most crucially, FLC tumours characteristically express a fusion gene, DNAJB1–PRKACA, originally identified by Honeyman et al.35 and since confirmed by multiple groups17,28,32,33,36–38 (FIG. 2). The fusion results from a ~400 kb somatic heterozygous deletion on chromosome 19. This genetic alteration, and the lack of consistently recurrent mutations in classic tumour suppressor genes such as PTEN and oncogenes such as CTNNB1, helps distinguish FLC from conventional HCC and other malignancies26,27,32,39. Initially it seemed possible that the gene fusion would be exclusive to FLC17,40. However, in 2020, two groups described some oncocytic neoplasms of the pancreas and bile duct that, like FLC, have eosinophilic cytoplasm due to the accumulation of altered mitochondria, and display recurrent rearrangements involving a PRKAC gene (PRKACB or PRKACA). The genetic alterations include DNAJB1–PRKACA fusions, indistinguishable from those observed in FLC41,42. Thus, the presence of this fusion alone does not uniquely diagnose FLC.
Fig. 2 |. Molecular mechanisms of fibrolamellar carcinoma.
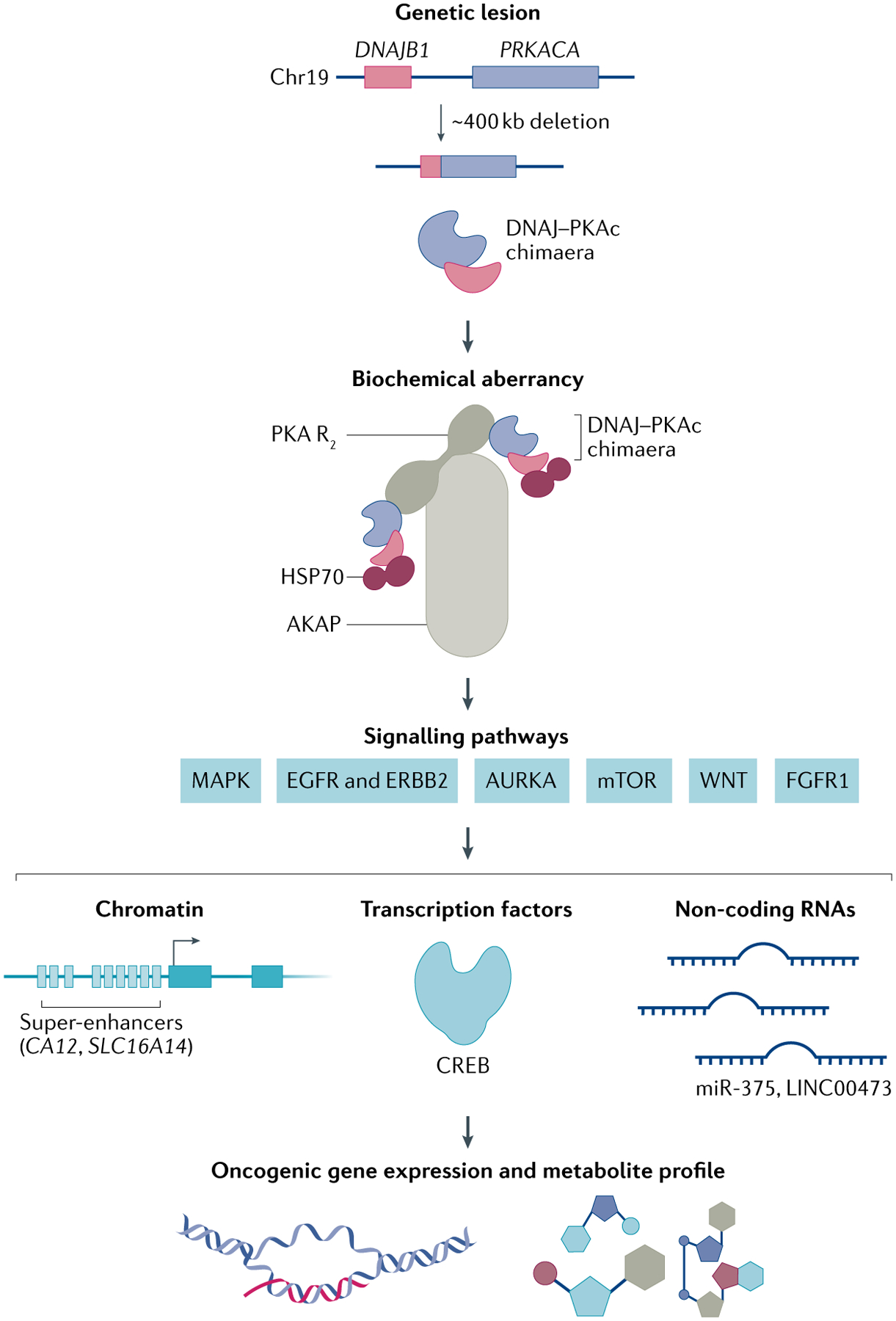
The hallmark genetic lesion of fibrolamellar carcinoma (FLC) is a ~400 kb heterozygous deletion on chromosome 19 that leads to the in-frame fusion of exon 1 of DNAJB1 and exons 2–10 of PRKACA35. The translated protein is referred to in this article as the DNAJ–PKAc chimaera. The chimaera recruits heat shock protein 70 (HSP70), conferring additional protein stability76. This molecular complex is recruited to A-kinase anchoring protein 13 (AKAP13; also called AKAP-Lbc). A key consequence is activation of multiple downstream signalling cascades including the MAPK pathway and a number of others, as shown. It is not yet known which of these are most critical to oncogenesis or most amenable to therapeutic intervention (see the section ‘Additional therapeutic targets?‘). These biochemical alterations lead to notable changes in the wiring of chromatin (enhancers and super-enhancers), as well as in the activity of transcription factors (for example, cAMP response element binding protein (CREB)) and non-coding RNAs (for example, miR-375 and LINC00473), which together shape the aberrant molecular profiles that promote FLC tumour phenotypes. AURKA, aurora kinase A; EGFR, epidermal growth factor receptor; FGFR1, fibroblast growth factor receptor 1; MAPK, mitogen-activated protein kinase; mTOR, mechanistic target of rapamycin; PKA, cyclic AMP-dependent protein kinase; PKA R2, PKA regulatory subunit homodimer.
DNAJB1 encodes a member of the HSP40 family of heat shock proteins43, while PRKACA encodes the α-isoform of the catalytic subunit (Cα) of protein kinase A (PKA), the cyclic AMP-dependent protein kinase44–46. How the fusion of these genes might contribute to oncogenesis is discussed below, in the section ‘Aberrant activation of PKA in FLC’.
Beyond the fusion gene, analyses of FLC have shown a low tumour mutational burden (TMB) and few other consistent genomic changes32,33,39,47. A genomic profiling study did identify activating promoter mutations for the telomerase reverse transcriptase (TERT) gene in 7 of 31 patients with FLC48. In addition, a whole-genome sequencing study in ten patients identified several genes that were mutated recurrently in tumours. For example, genetic lesions in MUC4 were identified in four patients39. This observation is notable because mucin 4 (MUC4) can form a complex with HER2 (also known as ERBB2 or NEU2) to promote cell proliferation and migration49. A separate genome-wide study of 26 FLC samples identified focal deletions present in at least 25% of the patients, including at the LKB1 (also known as STK11) and TAFA5 (also known as FAM19A5) loci32. These findings are concordant with observations that LINC00473, an LKB1-regulated gene, and TAFA5 are among the most aberrantly expressed genes in FLC, and that these loci are associated with FLC-specific chromatin changes50. Although mutations in TP53 are uncommon in FLC, the activated, phosphorylated form of p53 seems to be excluded from its normal site of action in the cell nucleus, while MDM4, a negative regulator of p53, is present in the nucleus at elevated levels51. Dysregulation of the activity or stability of p53, a key tumour suppressor protein, could promote the growth and resistance to therapy of FLC38,51.
Some liver cancers have been reported to present with mixed histological features of FLC and conventional HCC52–54. Although these mixed tumours generally exhibit distinct genomic and transcriptomic profiles relative to canonical or ‘pure’ FLC31, a subset of them do harbour the DNAJB1–PRKACA oncogene37. A genomic study revealed the presence of this hallmark gene fusion in two of ten patients categorized as having mixed FLC–HCC55. Strikingly, six of the remaining eight fusion gene-negative mixed tumours were found to contain biallelic inactivating mutations in the tumour suppressor gene BAP1 (which encodes BRCA1-associated protein 1). Conversely, 2 of 15 tumours classified histologically as pure FLC lacked the DNAJB1–PRKACA fusion and were BAP1-mutated. Most of the patients with fusion-positive pure FLC were relatively young (<40 years), whereas two patients with fusion-negative pure FLC tumours developed FLC later in life (ages 51 and 65 years)55. The mixed tumours bearing BAP1 mutations arose in patients aged 27 to 54 years (median 39 years)55. These observations, along with misdiagnoses of HCC as FLC, might help account for the bimodal age distribution of FLC reported in some epidemiological studies10,11. These intriguing findings merit further validation in follow-up cohorts.
Aberrant activation of PKA in FLC.
Tumours with the DNAJB1–PRKACA gene fusion and BAP1-mutated liver cancers seem to share a crucial functional feature, namely, aberrant activation of PKA. In the genomic study discussed previously, tumours with BAP1-inactivating mutations, regardless of histological classification, generally also displayed amplification of the PRKACA catalytic subunit gene (83%) and/or deletion of a regulatory subunit gene, PRKAR2A (94%)55. Both changes might be expected to deregulate PKA, rendering its activity less dependent on cAMP. When therapeutics are developed for canonical FLC, it will be important to determine whether they are also effective against tumours with a mixed FLC–HCC phenotype and those with BAP1 mutations.
The DNAJB1–PRKACA mRNA transcript found in the vast majority of patients with FLC is translated to a chimeric protein of ~46 kDa (REF.35), designated here as DNAJ–PKAc, in which the J-domain of HSP40 replaces a small segment at the amino terminus of the catalytic subunit of PKA (FIG. 2). This mutant protein retains activity as a serine–threonine-specific protein kinase27,33,35,56. The introduction of DNAJB1–PRKACA into mouse liver via CRISPR–Cas9 or transposon-mediated gene transfer suffices to induce tumorigenesis, implying that DNAJ–PKAc can act as an oncoprotein57,58. Although it has been shown that equally robust expression of wild-type PRKACA fails to drive tumour formation, the kinase activity of DNAJ–PKAc is required for tumorigenesis58. A recent preprint reports data from the preclinical evaluation of a potent, selective active site inhibitor of PKAc that slowed the growth of a human FLC tumour line in a patient-derived xenograft (PDX) model59. However, clinical development of such an inhibitor seems to be highly problematic because of on-target toxicity attributable to inhibition of normal PKAc, in line with the enzyme’s important physiological roles in multiple tissues46,60.
The mechanism by which DNAJ–PKAc transforms cells remains poorly understood and will probably be critical for the development of effective targeted molecular therapies, a central aim of FLC research. The factors that control the level of cAMP in FLC tumour cells are also not well known. As with certain other cancers (for example, lung, colorectal, pancreatic, prostatic and breast cancers)61,62, the peptide neurotensin (NTS) could have a role in oncogenesis by binding to its receptor NTSR1 (NTS receptor type 1) and driving increased production of intracellular cAMP63. Nonetheless, genetic, structural, biochemical and cell biological studies have illuminated key differences in PKA activity between FLC and normal cells.
Wild-type PKA is a heterotetrameric holoenzyme (R2C2) comprising four subunits, namely, two catalytic (C) subunits and a regulatory (R) subunit homodimer that inhibits enzymatic activity45. Cooperative binding of cAMP to the R subunits liberates the kinase function of the C subunits. There are three C isoforms (Cα, Cβ and Cγ), and four functionally non-redundant R isoforms (RIα, RIβ, RIIα and RIIβ). While RIIβ is the predominant isoform in the liver, RIα, a ‘master regulator’ of PKA64, is the only regulatory subunit isoform reported to be upregulated in human FLC tumour cells34,65.
Some relevant insights have come from a heritable condition called Carney complex, which accounts for a small minority (~1%) of FLC cases. Carney complex is marked by multiple, mainly benign tumours of the heart, skin and endocrine system, as well as altered pigmentation66. The disorder is caused by activation of PKA via deleterious mutations in PRKAR1A, which encodes RIα. A report from 2018 describes three individuals with Carney complex who developed histologically typical FLC, but without a DNAJB1–PRKACA gene fusion in their tumours67. In these patients, sequence analysis confirmed a heterozygous mutation in PRKAR1A in germline DNA, although intact RIα was still expressed from the unaffected allele in normal cells. However, the cancer cells lacked detectable RIα protein, consistent with loss of the remaining wild-type PRKAR1A allele as a key event in tumorigenesis. Thus, the absence of this regulatory subunit seems sufficient to deregulate kinase activity and drive elevated phosphorylation of PKA substrates associated with neoplastic growth.
A corresponding mechanism for deregulation of the chimeric catalytic subunit found in the great majority of FLC tumours is less apparent. Enzymological and structural studies indicate that the catalytic core is essentially unaffected in DNAJ–PKAc56,68–70. Similarly, endogenous protein kinase inhibitor peptide (PKI) blocks the activity of the fusion protein and the wild-type kinase with equal efficiency56. Moreover, like wild-type C subunits, the oncogenic DNAJ–PKAc C subunits interact with R subunits to form functional holoenzymes, and PKA activity in FLC cells remains responsive to cAMP65,68,69. Nevertheless, the addition of the J-domain of HSP40 to the C subunit can change biochemical properties when paired with certain regulatory subunits. For example, the holoenzyme formed by the mutant DNAJ–PKAc subunit with RIIβ subunits is activated more readily by cAMP than the corresponding wild-type holoenzyme71.
In 2021, Olivieri and colleagues identified changes in allosteric properties of the hallmark chimeric catalytic subunit that could contribute to deregulating signal transduction72. They noted that normal C subunits (PKAc) exhibit positive binding cooperativity between ATP and the substrate to be phosphorylated. After transfer of a phosphate group, negative binding cooperativity of ADP facilitates release of the phosphoprotein product. However, addition of the flexible J-domain in DNAJ–PKAc strongly attenuates the allosteric binding cooperativity between nucleotide and PKI (a pseudo-substrate). Exactly how this affects the pattern of substrate phosphorylation in FLC cells remains to be determined. Along with other structural studies70, it does offer some hope that subtle differences between DNAJ–PKAc and wild-type PKAc could facilitate the discovery of selective small-molecule inhibitors of the oncogenic chimaera.
Test tube biochemistry over many years suggested that the release of C subunits from R subunits is required for the activation of PKA by cAMP44. In the past 5 years, high-resolution studies of intracellular PKA have revealed, instead, that catalytically active holoenzymes remain intact in the cytoplasm, with some R–C subassemblies constrained to within 150 to 250 Å of target substrates73. This finding implies that a potential mechanism for the oncogenic activity of DNAJ–PKAc could be mis-localization and consequent dysregulation of holoenzymes. PKA binds via the R subunits to A-kinase anchoring proteins (AKAPs), which regulate PKA localization and substrate specificity73–75. Structural analyses support the notion that the J-domain (from HSP40) in DNAJ–PKAc can perturb interaction with AKAPs by altering the position of the dimerization or docking domain of PKA holoenzyme71. Furthermore, Turnham et al. demonstrated that DNAJ–PKAc interacts with heat shock protein 70 (HSP70) via the J-domain76 (FIG. 2). They infer that this interaction creates a pathological AKAP signalling island. Compound screening in a murine hepatocyte cell line engineered to express DNAJ–PKAc suggested that this complex could be targeted pharmacologically by a combination of inhibitors of mitogen-activated protein kinase (MAPK) signalling and HSP70. Clinical development of such drug combinations will first require confirmation of their utility in human FLC cells.
Another aspect of intracellular compartmentation also seems to contribute to oncogenesis in FLC. Cytoplasmic phase separation, the creation of distinct spatial compartments within a solution, has important roles in the formation of membrane-less organelles77,78. In 2020, Zhang et al. discovered that the ubiquitously expressed PKA regulatory subunit RIα normally undergoes liquid–liquid phase separation, thereby spatially constraining cAMP to microdomains within the cytoplasm79. This phase separation leads to the formation of RIα bodies, described by these authors as “biomolecular condensates [that] act as a dynamic ‘sponge’ in recruiting and retaining cAMP and [catalytically] active PKAcat”. Remarkably, they found that the DNAJ–PKAc fusion protein abolishes RIα phase separation, resulting in aberrant cAMP–PKA signalling that promotes cellular transformation. Both the loss of RIα phase separation and the transforming activity depend, at least in part, on the recruitment of HSP70 by DNAJ–PKAc, and also the absence from the fusion protein of a myristoylation site located near the N terminus of the normal C subunit of PKA79.
Additional therapeutic targets?.
Research efforts continue to identify specific substrates and downstream signalling pathways activated by DNAJ–PKAc that are most critical to oncogenesis (FIG. 2). Global analyses of the transcriptomes and focused studies of selected proteins in FLC models and primary tumours have shown elevated copy numbers, expression or activity of genes in several pathways that could contribute to transformation by the fusion kinase. These include the following: the MAPK pathway32,50,76,80; components of the epidermal growth factor receptor (EGFR) pathway and the related receptor tyrosine kinase ERBB231,34,37,38; the mechanistic target of rapamycin (mTOR)81,82; fibroblast growth factor receptor 1 (FGFR1)82,83; the WNT–β-catenin pathway34,58,84; and aurora kinase A (AURKA), a mitotic regulator34,38. However, while multiple reports agree on activation of certain of these pathways (for example, MAPK), the available data on others are more limited and sometimes contradictory. Substantial further work is required to determine which, if any, represent valid targets for FLC therapy. In initial multicentre clinical studies in patients with FLC, neither a pan-HER inhibitor of EGFR and ERBB2 kinase activity, nor an mTOR inhibitor, nor an AURKA inhibitor showed significant benefit as a single agent (see section ‘Clinical trials’). Sporadic responses to other targeted therapies have been observed in various small case series, reported in both meeting abstracts and peer-reviewed publications, but prospective clinical studies are urgently needed85–87.
Genome-scale studies are beginning to reveal additional candidate targets for new FLC treatments. Cataloguing non-coding RNAs in FLC has uncovered RNA species and the pathways they regulate that might have therapeutic significance17,88. One of the most dysregulated small RNAs in FLC is microRNA-375 (miR-375), which is dramatically suppressed in primary human tumours and in several models that express the DNAJ–PKAc chimaera. Restoration of miR-375 in a human FLC cell line suppresses growth and migration of the cancer cells, possibly through the inhibition of expression of proteins in the Hippo signalling pathway, including yes-associated protein 1 (YAP1) and connective tissue growth factor (CTGF)89. The potential therapeutic benefit of this and other miRNAs of interest in FLC, including miR-10b89 and miR-548p88, merits further investigation. Long non-coding RNAs (lncRNAs) have also emerged as key regulators of oncogenesis and metastasis in a growing number of cancer types90–93. The lncRNA LINC00473 is part of a reported gene signature of FLC17. Moreover, the FLC-specific active regulatory elements near LINC00473 are highly enriched in binding motifs for cAMP response element binding protein (CREB)50, an important downstream mediator of PKA signalling that has been implicated as a key transcription factor in FLC33. However, the molecular functions of LINC00473, and other lncRNAs88, in initiation and/or maintenance of this cancer are currently unknown.
Other systematic approaches to target identification follow from the recognition that regulatory alterations might underlie the ‘addiction’ of cancers to specific oncogenic signals94. One study utilized a novel technique called chromatin run-on sequencing (ChRO-seq), which helps define tumour-specific super-enhancers that mark nearby genes that are critical for tumour behaviour. This method also points to aberrant MAPK signalling in FLC tumours. In addition, it reveals several previously unsuspected genes, including CA12 (encoding carbonic anhydrase 12) and SLC16A14 (encoding the orphan transporter known as monocarboxylic acid transporter 14), for proteins that seem to have important roles in the survival of FLC tumour cells50. Strikingly, SLC16A14 is more highly expressed in FLC than in any other tumour type or normal tissue for which data are publicly available50. Studies in FLC cell models suggest that combination therapy with inhibitors of these genes as well as MAPK pathway inhibitors might offer a new therapeutic approach for this cancer50.
Disease models
Model systems are important for the definition of biological processes and vital to the discovery of effective therapeutics. Over the past 6 years, several systems genetically engineered to express a DNAJ–PKAc fusion protein have been described, including a murine cell line (AML12)76, and various transformed human cell lines33,95 as well as in vivo mouse57,58 and zebrafish96 models. In addition, human PDX tumour lines have been generated36,97, and cells from one PDX line have been utilized for in vitro studies50,89. Although the currently available models provide biological insights and facilitate molecular dissection of FLC, they also have important limitations (TABLE 1). Primary human FLC biospecimens are also available from biobanks at Massachusetts General Hospital, Boston, USA, and Rockefeller University, New York, USA (BOX 1).
Table 1 |.
Currently available models for fibrolamellar carcinoma research
| Model | Type | Species | Method | Strengths | Limitations | Refs |
|---|---|---|---|---|---|---|
| Published work | ||||||
| M-DP-1 | In vivo | Mouse | CRISPR–Cas9-based genome editing Transposon-mediated somatic gene transfer |
In vivo tumour growth Mimics many features of FLC |
Slow tumorigenesis Incomplete penetrance Lacks fibrous bands Lacks molecular features of FLC that are primate-specific |
58 |
| M-DP-2 | In vivo | Mouse | CRISPR–Cas9-based genome editing | In vivo tumour growth Mimics many features of FLC |
Slow tumorigenesis Incomplete penetrance Lacks fibrous bands Lacks molecular features of FLC that are primate-specific |
57 |
| ZF-DP-1 | In vivo | Zebrafish | Zebrafish DNAJ–PKAc over-expression | In vivo model | Evolutionarily more distant from humans than mice Lacks molecular features specific to mammals and primates |
96 |
| PDX-1 | In vivo | Human | Xenograft of patient-derived FLC tumour cells in immune-deficient mice | In vivo tumour growth Mimics most/all major features of FLC Can be maintained stably in spheroid (3D) culture |
Generally, tumour grown in host flank, not liver (non-orthotopic model) Not metastatic from flank |
36 |
| PDX-2 to PDX-7 | In vivo | Human | Xenograft of patient-derived FLC tumour cells in immune-deficient mice | In vivo tumour growth Mimics most/all major features of FLC Derived directly from primary Liver tumours and metastases |
Generally, tumour grown in host flank, not liver (non-orthotopic model) Not metastatic from flank |
97 |
| HepG2-DP | In vitro | Human | Lentiviral DP over-expression | All human cells Straightforward to culture and manipulate |
No presence of cells from microenvironment Hepatoblastoma background Transformed cells |
33,50 |
| C3A-DP | In vitro | Human | LentiviraL DP over-expression | All human cells Straightforward to culture and manipulate |
No presence of cells from microenvironment HCC background Transformed cells |
33 |
| AML12-DP | In vitro | Mouse | CRISPR–Cas9-based genome editing | Primary hepatocytes Straightforward to culture and manipulate |
Mouse not human origin Transgenic for human TGFα | 50,76,89 |
| Unpublished or preliminary work | ||||||
| FLX-1 cell line | In vitro | Human | Cell culture from PDX-1 xenograft model | (Nearly) all human tumour cells Established line that retains molecular features of FLC for multiple passages Amenable to treatment and transfection |
Limited (if any) microenvironmental signalling Relatively slow growth |
N. Bardeesy (personal communication) |
| Huh7-DP | In vitro | Human | Lentiviral DP over-expression | All human cells Straightforward to culture and manipulate |
No presence of cells from microenvironment HCC background Transformed cells |
P.S., unpublished work |
| HEK293-DP | In vitro | Human | CRISPR– Cas9-based genome editing |
All human cells Straightforward to culture and manipulate |
No presence of cells from microenvironment Non-hepatic background Transformed cells |
95 (a preprint article) |
All of these 16 in vitro and in vivo models currently in use for fibrolamellar carcinoma (FLC) research offer some strengths but also have key limitations, pointing to the critical need for improved models. DP, DNAJB1–PRKACA; HCC, hepatocellular carcinoma; TGFα, transforming growth factor-α.
Two independent groups described the use of CRISPR–Cas9-mediated deletion, via guide sequences introduced by hydrodynamic injection, to engineer a murine homologue of the DNAJB1–PRKACA fusion gene in hepatocytes of young adult mice57,58. Both groups observed that the chimaera sufficed to induce liver tumorigenesis, albeit slowly and with incomplete penetrance. The tumours in this model resembled those in human FLC, although they lacked fibrolamellar bands. Addition of an activating mutation in the β-catenin gene (CTNNB1), as observed in a small minority of human FLC cases, accelerated tumour initiation and growth58. Exposure to the hepatotoxin 3,5-diethoxycarbonyl-1,4-dihydrocollidine further enhanced tumorigenesis58.
A similar approach in zebrafish has not yet yielded frank malignant tumours. However, the generation of a DNAJ–PKAc chimeric protein from an engineered fusion of the homologous genes in hepatocytes induced hepatomegaly and increased hepatocyte size. Early characterization of this model indicated non-resolving inflammation96. This observation might reflect a species difference in terms of the effects of the fusion protein, because, despite altered expression of inflammatory genes and enhanced cytokine production in a subset of human tumours32, overt inflammation has not been reported as a characteristic feature of FLC.
The first published PDX model sheds light on the potential lineage derivation of human FLC. This tumour line, designated FLC-TD-2010 (listed as PDX-1 in TABLE 1), was developed from cells present in ascitic fluid of a patient at an advanced stage of disease36. Prior to injection into immune-deficient mice, the cells were first cultured briefly in serum-free medium and on a hyaluronan gel substrate, which are conditions known to promote the survival of normal endodermal stem or progenitor cells. The resulting line is strongly enriched for tumour-initiating cells and displays striking transcriptomic and phenotypic similarities to a normal adult population of primitive biliary tree stem cells, which can give rise to hepatic, cholangiocytic and pancreatic cell lineages98,99. These data indicate that the biliary tree stem cell is a potential cell type of origin for FLC30–33,36. The detection of DNAJB1–PRKACA in human intraductal oncocytic papillary neoplasms of the pancreas and bile duct supports the concept that this fusion gene can drive transformation of multipotent endodermal stem or progenitor cells to yield primary cancers outside the liver, in addition to FLC41,42. However, the de-differentiation of more mature liver or pancreatic or biliary ductal cells remains possible100, as suggested by the murine models of FLC in which the fusion gene is inserted into cells of the adult liver. No stable cell lines of human FLC have yet been reported in the literature. However, one line has been established from the original PDX-1 model that is slow-growing and retains the DNAJB1–PRKACA fusion across numerous passages (N. Bardeesy, personal communication; see FLX-1 in TABLE 1).
Lalazar et al. described six new PDX lines derived by direct implantation of tumour specimens from both the liver (one primary and one recurrent tumour) and metastases97 (PDX-2 to PDX-7 in TABLE 1). The new PDXs facilitated the first reported large-scale screening of compounds for the ability to kill dissociated FLC tumour cells in short-term culture (72 h). Active compounds were also assessed on cells obtained directly from patients, taken from lymph node metastases or ascitic fluid, and in vivo in mice bearing the PDX. A library of >5,000 compounds, selected as suitable for drug repurposing, included clinically approved drugs, drugs in preclinical development and other relevant bio-active species. The results revealed promising activity of several classes of inhibitors, including those for topoisomerase I (TOPOI) and histone deacetylase (HDAC). Napabucasin exhibited the most potent activity of the compounds screened. This drug has been touted as a potential blocker of the stem cell activity of cancers via targeting the signal transducer and activator of transcription 3 (STAT3) pathway101,102. Indeed, reports indicate that napabucasin reduces cancer stem cell properties of human HCC and biliary tract cancer cells103,104. The activity against FLC of a drug thought to target cancer stem cells seems to be consistent with both the high proportion of tumour-initiating cells in the PDX-1 model36 and the strong resemblance of the transcriptomes of that line and of multiple primary FLC tumours to normal biliary tree stem cells17,36. However, napabucasin’s anticancer activity does not depend exclusively on the attenuation of STAT3 phosphorylation and signalling. The compound was shown to induce pleiotropic effects through the generation of reactive oxygen species, induction of DNA damage and decreased synthesis of certain proteins via inhibition of eukaryotic initiation factor 4A97,105. Another important observation from the compound screen is that inhibitors of the anti-apoptotic protein BCL-XL, but not BCL-2, synergized with drugs such as the TOPOI inhibitor topotecan and the HDAC inhibitor panobinostat97. Overall, the study may represent a major step towards novel clinical trials of individual drugs and/or combinations possessing documented activity against FLC cells.
Immunotherapy
Immunotherapies have revolutionized cancer treatment and, in some cases, provide robust and lasting protection to patients. Favourable results in clinical trials have led regulators in the USA and Europe to approve immune checkpoint blockade for patients with diseases including, among others, melanoma, renal cell carcinoma, non-small-cell lung cancer and HCC106–108. Can such therapies be developed for FLC? A significant challenge is that the responsiveness of cancers to various immune modulations might correlate positively with TMB, as has been observed with checkpoint blockade immunotherapy109–111. This finding is often attributed to the random generation of large numbers of neoantigens that could be targeted by the immune system110. Indeed, in 2020, the FDA approved the use of the PD1 immune checkpoint inhibitor (ICI) pembrolizumab to treat adult and paediatric patients with TMB-high solid tumours, agnostic of the tissue of origin112,113.
Genomic studies indicate that FLC falls at the low end of the TMB spectrum, suggesting that this cancer would be immunologically ‘cold’32,33,38,39,48. However, the demonstration that some FLC tumours robustly express markers of multiple immune-suppressive mechanisms, including PDL1 (also known as CD274), PD1 (also known as CD279), B7-H3 (also known as CD276) and indoleamine 2,3-dioxygenase, in a pattern consistent with adaptive immune resistance, challenges the assumption that checkpoint blockade therapy necessarily would be ineffective114. No systematic reports have appeared on the efficacy of individual ICIs against FLC, and only a few anecdotal accounts have been published26,86,115. Increasingly, ICIs are being employed in combination, and sometimes also in conjunction with other therapeutic modalities, to expand the range of patients in whom they are effective116. A clinical trial led to the approval by the EC (European Commission) and the FDA of combined ICIs against PD1 and CTLA4 (also known as CD152), nivolumab and ipilimumab, for the treatment of HCC previously treated with sorafenib117. While FLC was excluded from that study, a case report documents a long-lasting, near-complete response to the nivolumab–ipilimumab combination in a patient with advanced, metastatic FLC and low TMB118.
Even for TMB-low tumours, it is possible that the immune system could effectively target a specific antigen common to a particular type of cancer. What neoantigen(s) might be accessible for immune attack in FLC? One obvious candidate is the DNAJ–PKAc oncogenic driver protein itself, in which the fusion junction could comprise a unique epitope not found in normal cells26,119,120. A precedent exists in the robust immune recognition of a comparable junction in the ETV6–RUNX1 driver fusion protein of paediatric acute lymphoblastic leukaemia121. In addition to the use of checkpoint blockade to enhance the immune response to tumours bearing such a neoantigen, therapeutic routes that could be explored include vaccination with a peptide corresponding to the junction (discussed later), or adoptive cell therapy with a cloned T cell receptor specific for a junctional epitope.
Clinical trials
Surgical intervention, when possible, remains the mainstay of FLC treatment, given that there are no effective, standard or agreed-upon systemic therapies for advanced disease5,11. Patients with oligometastatic disease have been shown to benefit from repeated local control interventions with the goal of life prolongation, including resection of the primary tumour, metastasectomy, radiation and embolization with or without the addition of chemotherapy. Advanced surgical approaches include parenchyma-sparing hepatectomies and the associating liver partition and portal vein ligation for staged hepatectomy (ALPPS) procedure122,123. While these methods attempt to radically resect all visible sites of disease, data regarding their effect on OS in patients with FLC are lacking. Therefore, in patients with metastatic disease, systemic therapies remain an important aspect of care, and there is an urgent need for clinical studies to advance new concepts.
Current chemotherapeutic regimens utilized for FLC are often based upon retrospective data analysis and vary among institutions5. Treatment regimens currently in regular use include gemcitabine and oxaliplatin (GEMOX), 5-fluorouracil (5-FU) and oxaliplatin, and 5-FU and interferon124,125. Paediatric patients with FLC have historically been treated with hepatoblastoma-specific regimens, yielding a modest response126. However, assessment of regimens for FLC in clinical trials has been limited, and many clinicians consider this cancer to be generally resistant to chemotherapy6,11,19,48.
Patients with FLC are eligible for a current international, prospective clinical trial led by the Children’s Oncology Group (COG), which is enrolling patients with HCC (with FLC considered as an HCC ‘variant’) to receive cisplatin and doxorubicin postoperatively, in the completely resected context, or cisplatin–doxorubicin–sorafenib versus cisplatin–doxorubicin–sorafenib alternating with GEMOX and sorafenib in the metastatic or unresectable context (Paediatric Hepatic Malignancy International Therapeutic Trial NCT03533582; European Union Drug Regulating Authorities Clinical Trials Database (EudraCT) number 2016-002828-85). The inclusion of sorafenib in this trial is noteworthy. This compound has activity against several kinase activities that conceivably could be relevant in FLC, including RAF serine–threonine kinase phosphorylation in the RAS–RAF–MAPK pathway, and the vascular endothelial growth factor receptor tyrosine kinases (VEGFR2 and VEGFR3)127,128. Sorafenib was the first molecularly targeted drug to receive FDA approval for treatment of HCC, achieving that status in 2007 on the basis of a nearly 3-month improvement in OS in the Sorafenib Hepatocellular Carcinoma Assessment Randomized Protocol (SHARP) study, from which FLC was excluded129. While FLC and HCC are clearly distinct entities, single-centre and individual case reports indicate that a number of oncologists have utilized sorafenib in patients with FLC, both paediatric and adult18,85. For example, in one single-institution experience, four of nine patients with advanced FLC who received sorafenib with palliative intent (median duration 8 months) showed temporary disease stabilization, but progression-free survival (PFS) did not improve86. Newer kinase inhibitors, in particular lenvatinib, have been approved in Europe and in the USA for the treatment of HCC, against which the drug has superior efficacy to sorafenib130. Preliminary data reported in a 2021 meeting abstract show encouraging initial results in patients with FLC treated with lenvatinib in combination with GEMOX or nivolumab, although not yet in the context of a formal, prospective research study131.
Despite the rarity of FLC, studies of several targeted therapies have shown that multicentre clinical trials specific for this cancer can meet recruitment goals. The choices of drugs were based primarily on certain clinical observations, together with results of the initial systematic analyses of gene expression in FLC tumours31,32,34,36. The first example of such a study tested everolimus as a single agent or in combination with an anti-oestrogenic regimen. Everolimus inhibits the serine–threonine protein kinase activity of mTOR complex 1 (mTORC1), which selectively regulates translation of mRNAs encoding key elements of the cell’s protein synthesis machinery132. The anticancer activity of everolimus correlates with its ability to inhibit the phosphorylation of substrates such as ribosomal protein S6 kinase (S6K). The active, phosphorylated forms of mTORC1 and S6K have been observed in biopsy specimens from patients with FLC81. A study in 2015 detected a high level of the phosphorylated form of the key downstream substrate ribosomal protein S6 (P-S6) in 96% of FLC specimens (n = 23), while P-S6 was observed only rarely in other carcinomas of the liver (13% of HCCs and 0% of intrahepatic cholangiocarcinomas) or normal liver (3%)82. Aromatase expression in FLC and its association with the occasional incidence of gynaecomastia in male patients suggested the combined use of everolimus with the aromatase inhibitor letrozole, plus the luteinizing hormone-releasing hormone agonist leuprolide22,133,134. A randomized phase II trial (NCT01642186) evaluated everolimus alone, anti-oestrogen therapy (letrozole plus leuprolide) or the three agents together in FLC; upon progression, patients in the everolimus or anti-oestrogen arms could receive the complete combination. Of 28 patients enrolled, none achieved the primary end point of PFS at 6 months (median PFS was 2.7 months; median OS was 12.4 months), and the study was halted after an interim analysis indicated lack of clinical activity135.
A separate phase II multicentre study (NCT02234986) targeted the serine-threonine kinase AURKA, a cell-cycle regulator for the G2/M transition believed to promote centrosome maturation and mitotic spindle assembly136. Transcriptome and kinome studies point to activation of AURKA as a downstream consequence of both the expression and kinase activity of the DNAJ–PKAc fusion protein34. An open-label study of the oral AURKA inhibitor ENMD-2076, which also inhibits several kinases associated with angiogenesis, enrolled 35 patients with metastatic, advanced FLC137. One patient showed a partial response, and 20 had stable disease. However, the OS (19 months) and PFS (3.9 months) did not meet the study’s primary or secondary end points. The investigators concluded that the results did not support further testing of ENMD-2076 as a single agent for the treatment of FLC138.
Some RNA-based studies and/or immunohistochemical analyses of human FLC tumours have indicated at least modestly elevated expression of one or both of the EGFR (encoded by EGFR (also known as ERBB1)), a tyrosine kinase, and its closest relative (encoded by ERBB2 (also known as HER2)), a receptor for which a specific ligand remains to be identified31,32,34,139,140. However, FLC typically does not feature either activating mutations of EGFR, which frequently occur in lung and certain other cancers, or high levels of amplification of HER2, as seen in a subset of breast cancers. Nevertheless, patients with FLC were included in a current clinical trial (NCT01953926) evaluating neratinib, an irreversible pan-HER receptor tyrosine kinase inhibitor which targets both the EGFR and ERBB2 proteins141. A 2021 meeting abstract states that initial results did not show substantial benefit for single-agent neratinib in 15 patients with advanced FLC who had failed other systemic regimens. However, several patients with FLC received neratinib plus pembrolizumab (with or without everolimus) under expanded access regulations, leading to an objective partial response in one and extended disease stabilization in another142. The investigators recommend continued exploration of combinations of neratinib with other anticancer agents.
As FLC PDX lines and other disease models, including cell lines and 3D organoids, become more widely accessible, large-scale screening could reveal compounds and drug combinations with unanticipated potential to revolutionize systemic therapy for this cancer97. In parallel, future efforts at targeted therapy will take advantage of the growing knowledge of FLC mechanisms discussed above. In addition, ‘genetic dependency mapping’, the systematic elucidation of genes and pathways that tumour cells require to survive and grow, has potential to identify an expanded, unbiased set of therapeutic targets and to help elucidate drug mechanisms143–146. These strategies are key components of the proposed Roadmap for future research aimed at translation to novel clinical trials and improved therapy (TABLE 2). At the level of individual patients, assays on cells taken directly from tumour biopsy samples might enable the selection of optimal therapeutic regimens147,148. This could be particularly important for drugs such as neratinib, for which in vitro responses of FLC cells taken from patients have been observed to vary substantially from tumour to tumour97.
Table 2 |.
Fibrolamellar carcinoma research Roadmap
| Unmet need | Actions | Current status or feasibility | Challenges |
|---|---|---|---|
| Define the FLC patient base | |||
| Accurate diagnosis and determination of global incidence of FLC | Standardize diagnostic criteria and establish a unique ICD code | FLC recognized under ICD-10, but included under same numerical code as HCC Identification by pathologists might be inconsistent outside the most experienced groups Molecular testing for fusion gene is feasible to help verify diagnoses28 |
Achieving agreement among pathologists on diagnostic criteria Broader implementation of molecular testing for DNAJB1–PRKACA fusions Clarification of frequency and significance of FLC and mixed FLC/HCC tumours with BAP1 mutations, lacking fusion gene |
| Improve data collection on paediatric and AYA cancers for US SEER database and globally | National Cancer Institute workgroup in place to improve data collection from registries International cooperative group support needed |
AYA demographic is generally under-served, and tracking might be lost in transition between paediatric and adult practices Collection of similar data outside USA |
|
| Source FLC data from patient-reported registries and natural history studies | Fibrolamellar Registry has large sets of patient-reported data Additional registries in development US National Cancer Institute MyPART Natural History Study of Rare Solid Tumors is at early stages of enrolment for FLC |
Increasing participation across full diversity of patient population Collecting and sharing data internationally |
|
| Capture extensive real-world data on FLC incidence and treatment158 | Companies access large percentage of medical records in some areas Seek relevant data from ERNs in EU cross-border health-care initiative159 |
Global coverage beyond USA Search criteria must be defined and tested Who will bear cost? |
|
| Develop targeted molecular therapies | |||
| Experimental systems widely accessible to the research community | Develop a research consortium to generate and distribute models and other resources that are routinely available for common cancers, but scarce for very rare ones154–157 | Biobanks of FLC tumour tissue exist at Rockefeller University and Massachusetts General Hospital; none identified outside USA Multiple PDX models (flank) now reported; orthotopic models could be tried with existing methods No 2D or 3D human FLC cell lines widely available No available animal model accurately replicates human FLC Genomic engineering of murine germline and/or liver cells, and organoids of normal human tissue to generate improved in vivo and culture models should be feasible |
Slow growth of FLC tumours in PDX models and in cell culture Further genomic data on FLC required to better understand its oncogenesis and to determine mutations and/or focal lesions most likely to pair effectively with DNAJB1–PRKACA for FLC experimental models Biobank contribution from treating sites needs to gain momentum Achieving wide distribution of models to FLC research community |
| Drugs selectively targeting DNAJ–PKAc oncoprotein | Apply multiple modalities to target fusion protein but spare normal PKA | Potentially feasible approaches include design or screening for selective small-molecule inhibitors, RNAi143, PROTACs160 | Reluctance of some biopharmaceutical companies to invest in therapeutics development for a very rare disease |
| Identification of additional, druggable therapeutic targets and/or drug sensitivities | Rigorously assess protein complexes through which DNAJ–PKAc acts and define biochemical pathways it activates | Underway in certain cell models | Confirmation needed in human FLC cells, which is limited by availability of PDX models and cell lines |
| Determine comprehensive catalogue of molecular dependencies by genetic knockdown or knockout and testing of compounds161 |
Dependency of FLC on candidate genes identified from genomic and epigenomic analyses can be assessed in low-throughput assays, even with the limited models currently available First systematic drug repurposing screen of >5,000 compounds reported97 Cancer Dependency Maps covering entire human genome are now feasible for all common cancers and some rare ones; large comparator datasets are available144,146,162,163 Available chemical resources include libraries of a large percentage of compounds that have been utilized in human trials Specialized chemical libraries such as protein kinase inhibitors (Kinase Chemogenomic Set (KCGS)) are publicly available164,165 |
For genome-wide studies culture models must grow robustly enough to carry out automated screens of libraries up to 20,000 genetic constructs (RNAi, CRISPR–Cas9) | |
| Develop targeted molecular therapies (cont.) | |||
| Characterization of the tumour ecosystem, including the stromal and immune microenvironment | Leverage state-of-the-art single-cell and/or spatial genomic and proteomic technologies | Rapidly developing technology best implemented at sites with appropriate facilities and expertise | Logistics of rapid collection of rare tumour biospecimens at widely dispersed institutions and transport to centralized study sites without degradation Longitudinal specimen collection to study tumour progression and responses to therapy High cost |
| Develop immune therapies | |||
| Identification of cell surface antigens targetable by antibodies or CAR T cells | Find proteins expressed selectively in FLC tumours at sufficient levels and for which monoclonal antibodies can be obtained | Some candidates already reported from RNA data50 Protein studies with mass spectroscopy and labelling methods, and antibody development are feasible |
Beyond proof of concept, development of therapeutic products will probably require commercial partners |
| Assessment of DNAJ–PKAc as target for immune therapy | Characterize T cells from mouse models, patient biospecimens, clinical trials | Preclinical studies and one clinical trial of a therapeutic vaccine for the fusion protein are in progress | Focuses on a single immune epitope Tumour cells might become independent of DNAJ–PKAc, and escape by losing the antigen |
| Devise effective clinical regimens | |||
| Creation of an international clinical forum of physicians who treat FLC | Share ideas and data towards goal of determining standard(s) of care for systemic therapy | Precedent exists for creation of multi-institutional consortiums for study of rare disease indications | Consensus recommendations might not yet be achievable for FLC in absence of data to support a standard of care for systemic therapy |
| Devise and implement efficient clinical trial designs with strong correlative studies | Lessons from other rare cancers159 | ||
AYA, adolescents and young adults; CAR T cells, chimeric antigen receptor T cells; ERN, European Reference Network; FLC, fibrolamellar carcinoma; HCC, hepatocellular carcinoma; ICD, International Code of Diseases; PDX, patient-derived xenograft; PKA, protein kinase A; PROTAC, proteolysis-targeting chimaera; SEER, Surveillance, Epidemiology, and End Results.
With respect to immunotherapy, an investigator-initiated study of pembrolizumab at four sites in the USA is currently open to enrolment for patients up to age 30 years with HCC or FLC. This trial might shed more light on whether a single-agent PD1 ICI has efficacy against FLC (NCT04134559). A study of another PD1 inhibitor, nivolumab, in conjunction with local yttrium-90 resin microsphere radioembolization therapy149 for liver tumours, in which patients with FLC were eligible, led by investigators at the Clinical University of Navarra, Spain (EudraCT number 2017-000232-34; NCT03380130), was presented at a 2020 meeting150. However, the report did not specifically address whether any patients with FLC were enrolled or responded to therapy.
A clinical trial opened in late 2020 at the MD Anderson Cancer Center, Houston, TX, which combines the frequently used combination of 5-FU and recombinant interferon125 with nivolumab (NCT04380545). A key hypothesis is that the chemotherapy agent and interferon will potentiate the effect of the PD1 ICI. A peer-reviewed paper of initial observations with this triple drug therapy in 14 patients with FLC at Rush University Medical Center (Chicago, IL) was published in 2021 (REF.151). The investigators report that the regimen was well tolerated and suggest the potential for clinical benefit151.
An innovative immunotherapy clinical trial specific to FLC began recruiting patients in March 2020 at Johns Hopkins University, Baltimore, MD. It is designed to focus on the potential attack on cancer cells by T cells with receptors for the signature DNAJ–PKAc driver protein of FLC. The study tests a synthetic peptide vaccine spanning the junction sequence linking the two components of the chimeric protein, corresponding at the mRNA level to the fusion of exon 1 of DNAJB1 to exon 2 of PKACA. The amino acid sequence at the junction should be unique to DNAJ–PKAc and identical in all patients35. The tumour in a few patients also contains a second form of the fusion protein due to a minor RNA splice variant between a donor site within exon 2 of the DNAJB1 sequence and the start of exon 2 of PRKACA. Patients also receive nivolumab and ipilimumab (NCT04248569). Thus, the trial combines a precision immunotherapy directly targeting the FLC oncoprotein with broad immune checkpoint blockade. Observations to date indicate that the regimen has clinical activity (M. Yarchoan, personal communication), but the results have not yet been published.
Additional concepts in immunotherapy remain to be evaluated. For example, a proposed trial at the US National Cancer Institute was designed to test whether oral vancomycin can enhance the immune response against FLC by altering the gut microbiome (NCT04025567). However, the trial was closed prematurely because of slow accrual of patients. Preclinical observations in mice indicate that depletion of commensal bacteria from the gut increases levels of CXCL16, leading to the recruitment of natural killer T cells and a subsequent effect on liver tumours, but this has not yet been tested in an FLC model152.
Looking ahead, emphasis should be placed on multi-agent as opposed to single-agent trials, aimed at targeting multiple protein and pathway alterations driven by the DNAJB1–PRKACA fusion oncogene. Also, it will be important to focus on broad enrolment of clinical trial patients, with specific inclusion of patients at age 12 years and above, per regulatory agency allowances, and consistent with the prevalence of FLC in the adolescent and young adult population.
Roadmap
Many scientists and clinicians working on FLC have forged strong cross-disciplinary and multi-institutional collaborations. These interactions facilitate progress, from understanding of molecular mechanisms to translational findings and clinical trials, all ultimately in the service of developing new treatments and improving outcomes for a cancer that presently lacks a standard of care. This is one of many examples of the role of collaborative networks that include patient-led groups in advancing research in rare and neglected diseases153–155.
Ongoing discussions among the authors of this article and other colleagues for more than 2 years have contributed to a Roadmap for FLC research, which can be categorized into four key goals (TABLE 2):
Define the FLC patient base
Develop targeted molecular therapies
Develop immunotherapies
Devise effective clinical regimens
We appreciate that others in the field might also be working towards the same ends, pursuing similar or superior strategies. While the specific suggestions in TABLE 2 are conditioned by the biology of FLC and the needs of the affected adolescent and young adult population, the overall goals mirror those recognized as essential to improve treatments and discover cures for other rare cancers155–157.
Conclusions
FLC epitomizes both the strengths and challenges of research focused on a very rare cancer. The discovery that nearly all FLC tumours share a unique oncogenic driver, the DNAJ–PKAc fusion protein, shifted the landscape from an obscure variant of HCC to a distinct entity with a unique molecular mechanism centred on a regulatory protein kinase, PKA, from which much of our current understanding of signal transduction evolved. Translation to effective therapies now seems within reach, both to attack the underlying biochemistry and cell biology and to enhance the immune response to a cancer with low mutational burden but an obligate oncogenic driver that might serve as an ideal neoantigen. Yet, as the proposed Roadmap emphasizes, research remains constrained by limitations in basic tools such as cell lines, human tumour xenografts and animal models, along with resources and sufficient patients to support clinical trials. Advances in our fundamental understanding of FLC are likely to bring novel insights to more common cancers that are harder to study because of greater phenotypic and genomic heterogeneity and genetic instability. Progress will continue to depend heavily on active cooperation within the research community, in partnership with patients and caregivers.
Key points.
Fibrolamellar carcinoma (FLC) is a rare but devastating disease that disproportionately affects adolescents and young adults and is characterized by the fusion oncogene DNAJB1–PRKACA.
There is a critical need to establish new cellular and animal models of FLC to perform reliable functional and preclinical studies.
Efforts to directly inhibit cyclic AMP-dependent protein kinase (PKA) catalytic activity for FLC therapy might be constrained by the important role of the normal enzyme in multiple tissues; a deeper understanding of mechanisms underlying cellular transformation by the oncogenic chimaera remains essential to defining tractable pharmacological targets.
Advances in the genetic and molecular underpinnings of FLC, in part through the application of genome-scale techniques, reveal promising avenues for new targeted therapeutic strategies and motivate higher-resolution single-cell and spatial approaches.
Immunotherapy for FLC must overcome the challenge of generating responses against a low-tumour mutational burden cancer, potentially by targeting the specific fusion oncoprotein as a neoantigen.
Clinical trials must overcome obstacles to accruing sufficient patients and should utilize an evidence-based approach and collaborative networks tailored to rare cancers.
Acknowledgements
The authors thank participants at the 2019 Fibrolamellar Research Summit (funded and hosted by the Fibrolamellar Cancer Foundation) and many other members of the FLC research and patient/caregiver communities for their insightful contributions. The authors thank M. Torbenson for providing the histology micrograph in Fig. 1.
Competing interests
E.M.J. is a paid consultant for the following companies: Adaptive Biotech, Cstone Pharmaceuticals, Achilles Therapeutics, DragonFly Therapeutics, Candel Therapeutics and Genocea Biosciences. She receives funding from Lustgarten Foundation and Bristol Myers Squibb. She is the Chief Medical Advisor for Lustgarten and SAB advisor to the Parker Institute for Cancer Immunotherapy (PICI) and for the C3 Cancer Institute. She is a founding member of Abmeta. G.K.A.-A. has had research support from Arcus, Agios, AstraZeneca, Bayer, BioNtech, BMS, Celgene, Flatiron, Genentech/Roche, Genoscience, Incyte, Polaris, Puma, QED, Sillajen, and Yiviva. He has also provided consulting to Agios, AstraZeneca, Alnylam, Autem, Bayer, Beigene, Berry Genomics, Celgene, CytomX, Eisai, Eli Lilly, Exelixis, Flatiron, Genentech/Roche, Genoscience, Helio, Incyte, Ipsen, Legend Biotech, Loxo, Merck, MINA, QED, Redhill, Rafael, Silenseed, Sillajen, Sobi, Surface Oncology, Therabionics, Twoxar, Vector and Yiviva. M.E.F. is employed full-time by the Fibrolamellar Cancer Foundation, a registered non-profit corporation (501c3) in the USA. The following authors serve on the advisory board of the Fibrolamellar Cancer Foundation: E.M.J., J.D.S., J.D.G., G.K.A.-A., A.F.O’N. and J.Z.-R. The following authors are past or current recipients of grant funding from the Fibrolamellar Cancer Foundation: J.D.S., J.D.G., G.K.A.-A., K.C.B. and P.S. T.A.D., A.F.U. and R.M. declare no competing interests.
Footnotes
Cancer dependency Map: https://depmap.org/portal
ClinicalTrials.gov: https://clinicaltrials.gov/
European Union Drug Regulating Authorities Clinical Trials Database (EudraCT): https://eudract.ema.europa.eu/
Fibrolamellar Cancer Foundation Biobank: https://fibrofoundation.org/join-the-fight/donate-tissue/
FibroLamellar Omics (FLo): https://sethupathy-lab.shinyapps.io/flc_data/
Fibrolamellar Registry: https://fibroregistry.org/
Fibrolamellar Tissue Repository: https://fibrolamellar.rockefeller.edu/repository
Kinase Chemogenomic Set (KCGS): https://www.sgc-unc.org/kinase-chemogenomics
Natural History Study of Rare Solid Tumors: https://www.cancer.gov/pediatric-adult-rare-tumor/participate/natural-history
References
- 1.Edmondson HA Differential diagnosis of tumors and tumor-like lesions of liver in infancy and childhood. AMA J. Dis. Child 91, 168–186 (1956). [DOI] [PubMed] [Google Scholar]
- 2.Berman MM, Libbey NP & Foster JH Hepatocellular carcinoma. Polygonal cell type with fibrous stroma–an atypical variant with a favorable prognosis. Cancer 46, 1448–1455 (1980). [DOI] [PubMed] [Google Scholar]
- 3.Craig JR, Peters RL, Edmondson HA & Omata M Fibrolamellar carcinoma of the liver: a tumor of adolescents and young adults with distinctive clinico-pathologic features. Cancer 46, 372–379 (1980). [DOI] [PubMed] [Google Scholar]; This paper names fibrolamellar carcinoma and describes the tumour as a distinct entity.
- 4.Farhi DC, Shikes RH & Silverberg SG Ultrastructure of fibrolamellar oncocytic hepatoma. Cancer 50, 702–709 (1982). [DOI] [PubMed] [Google Scholar]
- 5.O’Neill AF et al. Fibrolamellar carcinoma: an entity all its own. Curr. Probl. Cancer 45, 100770 (2021). [DOI] [PubMed] [Google Scholar]; A current review that emphasizes clinical aspects of FLC.
- 6.Mavros MN, Mayo SC, Hyder O & Pawlik TM A systematic review: treatment and prognosis of patients with fibrolamellar hepatocellular carcinoma. J. Am. Coll. Surg 215, 820–830 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Graham RP & Torbenson MS Fibrolamellar carcinoma: a histologically unique tumor with unique molecular findings. Semin. Diagn. Pathol 34, 146–152 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Close AG, Dreyzin A, Miller KD, Seynnaeve BKN & Rapkin LB Adolescent and young adult oncology–past, present, and future. CA Cancer J. Clin 69, 485–496 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Tricoli JV & Bleyer A Adolescent and young adult cancer biology. Cancer J 24, 267–274 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Eggert T et al. Fibrolamellar hepatocellular carcinoma in the USA, 2000–2010: a detailed report on frequency, treatment and outcome based on the Surveillance, Epidemiology, and End Results database. United European Gastroenterol. J 1, 351–357 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramai D, Ofosu A, Lai JK, Gao ZH & Adler DG Fibrolamellar hepatocellular carcinoma: a population-based observational study. Dig. Dis. Sci 66, 308–314 (2021). [DOI] [PubMed] [Google Scholar]
- 12.Torbenson M Fibrolamellar carcinoma: 2012 update. Scientifica 2012, 743790 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lafaro KJ & Pawlik TM Fibrolamellar hepatocellular carcinoma: current clinical perspectives. J. Hepatocell. Carcinoma 2, 151–157 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graham RP Fibrolamellar carcinoma: what Is new and why it matters. Surg. Pathol. Clin 11, 377–387 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Zakka K et al. Clinical outcomes of rare hepatocellular carcinoma variants compared to pure hepatocellular carcinoma. J. Hepatocell. Carcinoma 6, 119–129 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lemekhova A et al. Clinical features and surgical outcomes of fibrolamellar hepatocellular carcinoma: retrospective analysis of a single-center experience. World J. Surg. Oncol 18, 93 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dinh TA et al. Comprehensive analysis of The Cancer Genome Atlas reveals a unique gene and non-coding RNA signature of fibrolamellar carcinoma. Sci. Rep 7, 44653 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ang CS et al. Clinicopathologic characteristics and survival outcomes of patients with fibrolamellar carcinoma: data from the fibrolamellar carcinoma consortium. Gastrointest. Cancer Res 6, 3–9 (2013). [PMC free article] [PubMed] [Google Scholar]; This article reviews the clinical experience of 95 patients with FLC from a consortium of three institutions over 25 years (1986–2011).
- 19.Stipa F et al. Outcome of patients with fibrolamellar hepatocellular carcinoma. Cancer 106, 1331–1338 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Mayo SC et al. Treatment and prognosis of patients with fibrolamellar hepatocellular carcinoma: a national perspective. J. Am. Coll. Surg 218, 196–205 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Atienza LG et al. Liver transplantation for fibrolamellar hepatocellular carcinoma: a national perspective. J. Surg. Oncol 115, 319–323 (2017). [DOI] [PubMed] [Google Scholar]
- 22.McCloskey JJ, Germain-Lee EL, Perman JA, Plotnick LP & Janoski AH Gynecomastia as a presenting sign of fibrolamellar carcinoma of the liver. Pediatrics 82, 379–382 (1988). [PubMed] [Google Scholar]
- 23.Santiago-Reynoso J et al. Hepatocellular carcinoma of fibrolamellar type in an adolescent: case report and literature review. Gastrointest. Tumors 6, 43–50 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cho J et al. Hyperammonemic encephalopathy in a patient with fibrolamellar hepatocellular carcinoma: case report and literature review. J. Gastrointest. Oncol 10, 582–588 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bartlett AL, Leslie ND, Gupta A & Geller JI Acquired ornithine transcarbamylase deficiency in pediatric and adolescent patients with fibrolamellar hepatocellular carcinoma. Pediatr. Blood Cancer 65, e27392 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Lalazar G & Simon SM Fibrolamellar carcinoma: recent advances and unresolved questions on the molecular mechanisms. Semin. Liver Dis 38, 51–59 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article is a review that emphasizes the molecular aspects of FLC.
- 27.Riggle KM, Turnham R, Scott JD, Yeung RS & Riehle KJ Fibrolamellar hepatocellular carcinoma: mechanistic distinction from adult hepatocellular carcinoma. Pediatr. Blood Cancer 63, 1163–1167 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Graham RP et al. Molecular testing for the clinical diagnosis of fibrolamellar carcinoma. Mod. Pathol 31, 141–149 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article discusses a break-apart FISH assay for DNAJB1–PRKACA gene fusion and reports a correlation between the presence of the fusion and classic diagnostic criteria for FLC.
- 29.Ross HM et al. Fibrolamellar carcinomas are positive for CD68. Mod. Pathol 24, 390–395 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ward SC et al. Fibrolamellar carcinoma of the liver exhibits immunohistochemical evidence of both hepatocyte and bile duct differentiation. Mod. Pathol 23, 1180–1190 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Malouf GG et al. Transcriptional profiling of pure fibrolamellar hepatocellular carcinoma reveals an endocrine signature. Hepatology 59, 2228–2237 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Cornella H et al. Unique genomic profile of fibrolamellar hepatocellular carcinoma. Gastroenterology 148, 806–818.e10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu L et al. Genomic analysis of fibrolamellar hepatocellular carcinoma. Hum. Mol. Genet 24, 50–63 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simon EP et al. Transcriptomic characterization of fibrolamellar hepatocellular carcinoma. Proc. Natl Acad. Sci. USA 112, E5916–E5925 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Honeyman JN et al. Detection of a recurrent DNAJB1-PRKACA chimeric transcript in fibrolamellar hepatocellular carcinoma. Science 343, 1010–1014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article reports the discovery of the fusion oncogene of FLC and the chimeric protein it encodes.
- 36.Oikawa T et al. Model of fibrolamellar hepatocellular carcinomas reveals striking enrichment in cancer stem cells. Nat. Commun 6, 8070 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article describes the first PDX model of FLC and evidence for the cancer’s potential relationship to biliary tree stem cells.
- 37.Griffith OL et al. A genomic case study of mixed fibrolamellar hepatocellular carcinoma. Ann. Oncol 27, 1148–1154 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sorenson EC et al. Genome and transcriptome profiling of fibrolamellar hepatocellular carcinoma demonstrates p53 and IGF2BP1 dysregulation. PLoS ONE 12, e0176562 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Darcy DG et al. The genomic landscape of fibrolamellar hepatocellular carcinoma: whole genome sequencing of ten patients. Oncotarget 6, 755–770 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Graham RP et al. DNAJB1-PRKACA is specific for fibrolamellar carcinoma. Mod. Pathol 28, 822–829 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Singhi AD et al. Recurrent rearrangements in PRKACA and PRKACB in intraductal oncocytic papillary neoplasms of the pancreas and bile duct. Gastroenterology 158, 573–582.e2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vyas M et al. DNAJB1-PRKACA fusions occur in oncocytic pancreatic and biliary neoplasms and are not specific for fibrolamellar hepatocellular carcinoma. Mod. Pathol 33, 648–656 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qiu XB, Shao YM, Miao S & Wang L The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol. Life Sci 63, 2560–2570 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krebs EG The Albert Lasker Medical Awards. Role of the cyclic AMP-dependent protein kinase in signal transduction. JAMA 262, 1815–1818 (1989). [DOI] [PubMed] [Google Scholar]
- 45.Taylor SS, Ilouz R, Zhang P & Kornev AP Assembly of allosteric macromolecular switches: lessons from PKA. Nat. Rev. Mol. Cell Biol 13, 646–658 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Turnham RE & Scott JD Protein kinase A catalytic subunit isoform PRKACA; history, function and physiology. Gene 577, 101–108 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ward SC & Waxman S Fibrolamellar carcinoma: a review with focus on genetics and comparison to other malignant primary liver tumors. Semin. Liver Dis 31, 61–70 (2011). [DOI] [PubMed] [Google Scholar]
- 48.El Dika I et al. Molecular profiling and analysis of genetic aberrations aimed at identifying potential therapeutic targets in fibrolamellar carcinoma of the liver. Cancer 126, 4126–4135 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singh AP, Chaturvedi P & Batra SK Emerging roles of MUC4 in cancer: a novel target for diagnosis and therapy. Cancer Res. 67, 433–436 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Dinh TA et al. Hotspots of aberrant enhancer activity in fibrolamellar carcinoma reveal candidate oncogenic pathways and therapeutic vulnerabilities. Cell Rep. 31, 107509 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; A study that describes the identification of the enhancer landscape and genes marked by super-enhancers in FLC tumour tissue.
- 51.Karki A et al. MDM4 expression in fibrolamellar hepatocellular carcinoma. Oncol. Rep 42, 1487–1496 (2019). [DOI] [PubMed] [Google Scholar]
- 52.Malouf GG et al. Pure and mixed fibrolamellar hepatocellular carcinomas differ in natural history and prognosis after complete surgical resection. Cancer 118, 4981–4990 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Chagas AL et al. Clinical and pathological evaluation of fibrolamellar hepatocellular carcinoma: a single center study of 21 cases. Clinics 70, 207–213 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Calderaro J, Ziol M, Paradis V & Zucman-Rossi J Molecular and histological correlations in liver cancer. J. Hepatol 71, 616–630 (2019). [DOI] [PubMed] [Google Scholar]
- 55.Hirsch TZ et al. BAP1 mutations define a homogeneous subgroup of hepatocellular carcinoma with fibrolamellar-like features and activated PKA. J. Hepatol 72, 924–936 (2020). [DOI] [PubMed] [Google Scholar]; A study that describes the identification of frequent biallelic inactivation of BAP1, a tumour suppressor gene, and activation of PKA in liver tumours with mixed features of FLC and HCC.
- 56.Averill AM et al. Inhibition of the chimeric DNAJ-PKAc enzyme by endogenous inhibitor proteins. J. Cell Biochem 120, 13783–13791 (2019). [DOI] [PubMed] [Google Scholar]
- 57.Engelholm LH et al. CRISPR/Cas9 engineering of adult mouse liver demonstrates that the Dnajb1-Prkaca gene fusion is sufficient to induce tumors resembling fibrolamellar hepatocellular carcinoma. Gastroenterology 153, 1662–1673.e10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kastenhuber ER et al. DNAJB1-PRKACA fusion kinase interacts with β-catenin and the liver regenerative response to drive fibrolamellar hepatocellular carcinoma. Proc. Natl Acad. Sci. USA 114, 13076–13084 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; Together with Engelholm et al. (2017), this study shows that the DNAJ–PKAc chimeric protein is oncogenic and can induce liver tumours in mice.
- 59.Schalm SS et al. Evaluation of PRKACA as a therapeutic target for fibrolamellar carcinoma. Preprint at bioRxiv https://doi.org.1101/2022.01.31.477690 (2022). [Google Scholar]
- 60.Sullivan KM, Kenerson HL, Pillarisetty VG, Riehle KJ & Yeung RS Precision oncology in liver cancer. Ann. Transl. Med 6, 285 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qiu S et al. A review of the role of neurotensin and its receptors in colorectal cancer. Gastroenterol. Res. Pract 2017, 6456257 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Myers RM et al. Cancer, chemistry, and the cell: molecules that interact with the neurotensin receptors. ACS Chem. Biol 4, 503–525 (2009). [DOI] [PubMed] [Google Scholar]
- 63.Riehle KJ et al. Neurotensin as a source of cyclic AMP and co-mitogen in fibrolamellar hepatocellular carcinoma. Oncotarget 10, 5092–5102 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lu TW et al. Two PKA RIα holoenzyme states define ATP as an isoform-specific orthosteric inhibitor that competes with the allosteric activator, cAMP. Proc. Natl Acad. Sci. USA 116, 16347–16356 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Riggle KM et al. Enhanced cAMP-stimulated protein kinase A activity in human fibrolamellar hepatocellular carcinoma. Pediatr. Res 80, 110–118 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kamilaris CDC, Faucz FR, Voutetakis A & Stratakis CA Carney complex. Exp. Clin. Endocrinol. Diabetes 127, 156–164 (2019). [DOI] [PubMed] [Google Scholar]
- 67.Graham RP et al. Fibrolamellar carcinoma in the Carney complex: PRKAR1A loss instead of the classic DNAJB1-PRKACA fusion. Hepatology 68, 1441–1447 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cheung J et al. Structural insights into mis-regulation of protein kinase A in human tumors. Proc. Natl Acad. Sci. USA 112, 1374–1379 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tomasini MD et al. Conformational landscape of the PRKACA-DNAJB1 chimeric kinase, the driver for fibrolamellar hepatocellular carcinoma. Sci. Rep 8, 720 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cao B et al. Structures of the PKA RIα holoenzyme with the FLHCC driver J-PKAcα or wild-type PKAcα. Structure 27, 816–828.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lu TW et al. Structural analyses of the PKA RIIβ holoenzyme containing the oncogenic DnaJB1-PKAc fusion protein reveal protomer asymmetry and fusion-induced allosteric perturbations in fibrolamellar hepatocellular carcinoma. PLoS Biol. 18, e3001018 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Olivieri C et al. Defective internal allosteric network imparts dysfunctional ATP/substrate-binding cooperativity in oncogenic chimera of protein kinase A. Commun. Biol 4, 321 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smith FD et al. Local protein kinase A action proceeds through intact holoenzymes. Science 356, 1288–1293 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Scott JD, Dessauer CW & Tasken K Creating order from chaos: cellular regulation by kinase anchoring. Ann. Rev. Pharmacol. Toxicol 53, 187–210 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Soberg K & Skalhegg BS The molecular basis for specificity at the level of the protein kinase A catalytic subunit. Front. Endocrinol 9, 538 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Turnham RE et al. An acquired scaffolding function of the DNAJ-PKAc fusion contributes to oncogenic signaling in fibrolamellar carcinoma. eLife 8, e44187 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article shows that the main FLC oncoprotein interacts with A-kinase anchoring proteins (AKAPs) and that the intracellular organization of PKA signalling is altered in FLC.
- 77.Shin Y & Brangwynne CP Liquid phase condensation in cell physiology and disease. Science 357, eaaf4382 (2017). [DOI] [PubMed] [Google Scholar]
- 78.Boeynaems S et al. Protein phase separation: a new phase in cell biology. Trends Cell Biol. 28, 420–435 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang JZ et al. Phase separation of a PKA regulatory subunit controls cAMP compartmentation and oncogenic signaling. Cell 182, 1531–1544 e1515 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article discusses the possibility that disrupted compartmentation of membrane-less organelles by DNAJ–PKAc is central to its oncogenic activity.
- 80.Kannangai R, Vivekanandan P, Martinez-Murillo F, Choti M & Torbenson M Fibrolamellar carcinomas show overexpression of genes in the RAS, MAPK, PIK3, and xenobiotic degradation pathways. Hum. Pathol 38, 639–644 (2007). [DOI] [PubMed] [Google Scholar]
- 81.Sahin F et al. mTOR and P70 S6 kinase expression in primary liver neoplasms. Clin. Cancer Res 10, 8421–8425 (2004). [DOI] [PubMed] [Google Scholar]
- 82.Riehle KJ et al. mTORC1 and FGFR1 signaling in fibrolamellar hepatocellular carcinoma. Mod. Pathol 28, 103–110 (2015). [DOI] [PubMed] [Google Scholar]
- 83.Graham RP et al. FGFR1 and FGFR2 in fibrolamellar carcinoma. Histopathology 68, 686–692 (2016). [DOI] [PubMed] [Google Scholar]
- 84.Cieply B, Zeng G, Proverbs-Singh T, Geller DA & Monga SP Unique phenotype of hepatocellular cancers with exon-3 mutations in beta-catenin gene. Hepatology 49, 821–831 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Subbiah IM et al. Targeted therapies in early-phase trials for the treatment of advanced fibrolamellar hepatocellular carcinoma. J. Clin. Oncol 31, 232–232 (2013). [Google Scholar]
- 86.Chakrabarti S et al. Clinicopathological features and outcomes of fibrolamellar hepatocellular carcinoma. J. Gastrointest. Oncol 10, 554–561 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jardim DL et al. FBXW7 mutations in patients with advanced cancers: clinical and molecular characteristics and outcomes with mTOR inhibitors. PLoS ONE 9, e89388 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Farber BA et al. Non coding RNA analysis in fibrolamellar hepatocellular carcinoma. Oncotarget 9, 10211–10227 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dinh TA et al. MicroRNA-375 suppresses the growth and invasion of fibrolamellar carcinoma. Cell Mol. Gastroenterol. Hepatol 7, 803–817 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schmitt AM & Chang HY Long noncoding RNAs in cancer pathways. Cancer Cell 29, 452–463 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lin C & Yang L Long noncoding RNA in cancer: wiring signaling circuitry. Trends Cell Biol. 28, 287–301 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jiang MC, Ni JJ, Cui WY, Wang BY & Zhuo W Emerging roles of lncRNA in cancer and therapeutic opportunities. Am. J. Cancer Res 9, 1354–1366 (2019). [PMC free article] [PubMed] [Google Scholar]
- 93.Liu SJ, Dang HX, Lim DA, Feng FY & Maher CA Long noncoding RNAs in cancer metastasis. Nat. Rev. Cancer 21, 446–460 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hnisz D et al. Super-enhancers in the control of cell identity and disease. Cell 155, 934–947 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim SS et al. DNAJB1-PRKACA in HEK293T cells induces LINC00473 overexpression that depends on PKA signaling. Preprint at bioRxiv 10.1101/2021/08.11.455931 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.de Oliveira S, Houseright RA, Korte BG & Huttenlocher A DnaJ-PKAc fusion induces liver inflammation in a zebrafish model of fibrolamellar carcinoma. Dis. Model Mech 13, dmm042564 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lalazar G et al. Identification of novel therapeutic targets for fibrolamellar carcinoma using patient derived xenografts and direct from patient screening. Cancer Discov. 11, 2544–2563 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article describes the first large-scale screening of anti-FLC compounds using new PDX models and cells directly from patients.
- 98.Cardinale V et al. Multipotent stem/progenitor cells in human biliary tree give rise to hepatocytes, cholangiocytes and pancreatic islets. Hepatology 54, 2159–2172 (2011). [DOI] [PubMed] [Google Scholar]
- 99.Cardinale V et al. The biliary tree–a reservoir of multipotent stem cells. Nat. Rev. Gastroenterol. Hepatol 9, 231–240 (2012). [DOI] [PubMed] [Google Scholar]
- 100.Alison MR The cellular origins of cancer with particular reference to the gastrointestinal tract. Int. J. Exp. Pathol 101, 132–151 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hubbard JM & Grothey A Napabucasin: an update on the first-in-class cancer stemness inhibitor. Drugs 77, 1091–1103 (2017). [DOI] [PubMed] [Google Scholar]
- 102.Locken H, Clamor C & Muller K Napabucasin and related heterocycle-fused naphthoquinones as STAT3 inhibitors with antiproliferative activity against cancer cells. J. Nat. Prod 81, 1636–1644 (2018). [DOI] [PubMed] [Google Scholar]
- 103.Li Y, Han Q, Zhao H, Guo Q & Zhang J Napabucasin reduces cancer stem cell characteristics in hepatocellular carcinoma. Front. Pharmacol 11, 597520 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Beyreis M et al. The cancer stem cell inhibitor napabucasin (BBI608) shows general cytotoxicity in biliary tract cancer cells and reduces cancer stem cell characteristics. Cancers 11, 276 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Froeling FEM et al. Bioactivation of napabucasin triggers reactive oxygen species-mediated cancer cell death. Clin. Cancer Res 25, 7162–7174 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Boutros C et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat. Rev. Clin. Oncol 13, 473–486 (2016). [DOI] [PubMed] [Google Scholar]
- 107.Kourie HR, Awada G & Awada A The second wave of immune checkpoint inhibitor tsunami: advance, challenges and perspectives. Immunotherapy 9, 647–657 (2017). [DOI] [PubMed] [Google Scholar]
- 108.Hargadon KM, Johnson CE & Williams CJ Immune checkpoint blockade therapy for cancer: an overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol 62, 29–39 (2018). [DOI] [PubMed] [Google Scholar]
- 109.Goodman AM et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol. Cancer Ther 16, 2598–2608 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yarchoan M, Hopkins A & Jaffee EM Tumor mutational burden and response rate to PD-1 inhibition. N. Engl. J. Med 377, 2500–2501 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Riviere P et al. High tumor mutational burden correlates with longer survival in immunotherapy-naive patients with diverse cancers. Mol. Cancer Ther 19, 2139–2145 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Subbiah V, Solit DB, Chan TA & Kurzrock R The FDA approval of pembrolizumab for adult and pediatric patients with tumor mutational burden (TMB) ≥10: a decision centered on empowering patients and their physicians. Ann. Oncol 31, 1115–1118 (2020). [DOI] [PubMed] [Google Scholar]
- 113.Marabelle A et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 21, 1353–1365 (2020). [DOI] [PubMed] [Google Scholar]
- 114.Kim AK et al. Multiple immune-suppressive mechanisms in fibrolamellar carcinoma. Cancer Immunol. Res 7, 805–812 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that immune checkpoints are expressed in FLC tumours, suggesting the potential therapeutic value of ICIs.
- 115.Bauer U et al. Progression after immunotherapy for fibrolamellar carcinoma. Visc. Med 35, 39–42 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zappasodi R, Merghoub T & Wolchok JD Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell 33, 581–598 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yau T et al. Efficacy and safety of nivolumab plus ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: the CheckMate 040 randomized clinical trial. JAMA Oncol. 6, e204564 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.De Toni EN & Roessler D Using dual checkpoint blockade to treat fibrolamellar hepatocellular carcinoma. Gut 69, 2056–2058 (2020). [DOI] [PubMed] [Google Scholar]
- 119.Yarchoan M, Johnson BA 3rd, Lutz ER, Laheru DA & Jaffee EM Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 17, 569 (2017). [DOI] [PubMed] [Google Scholar]
- 120.Chang TC et al. The neoepitope landscape in pediatric cancers. Genome Med. 9, 78 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zamora AE et al. Pediatric patients with acute lymphoblastic leukemia generate abundant and functional neoantigen-specific CD8(+) T cell responses. Sci. Transl. Med 11, eaat8549 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; A study that sets the precedent for targeted immune therapy against a junctional epitope in an oncogenic fusion protein.
- 122.Schnitzbauer AA et al. Right portal vein ligation combined with in situ splitting induces rapid left lateral liver lobe hypertrophy enabling 2-staged extended right hepatic resection in small-for-size settings. Ann. Surg 255, 405–414 (2012). [DOI] [PubMed] [Google Scholar]
- 123.Aloia TA & Vauthey JN Associating liver partition and portal vein ligation for staged hepatectomy (ALPPS): what is gained and what is lost? Ann. Surg 256, e9 (2012). author reply e16–19. [DOI] [PubMed] [Google Scholar]
- 124.Lim II, Farber BA & LaQuaglia MP Advances in fibrolamellar hepatocellular carcinoma: a review. Eur. J. Pediatr. Surg 24, 461–466 (2014). [DOI] [PubMed] [Google Scholar]
- 125.Kaseb AO et al. Prognostic indicators and treatment outcome in 94 cases of fibrolamellar hepatocellular carcinoma. Oncology 85, 197–203 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Katzenstein HM et al. Fibrolamellar hepatocellular carcinoma in children and adolescents. Cancer 97, 2006–2012 (2003). [DOI] [PubMed] [Google Scholar]
- 127.Liu L et al. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 66, 11851–11858 (2006). [DOI] [PubMed] [Google Scholar]
- 128.Iyer R, Fetterly G, Lugade A & Thanavala Y Sorafenib: a clinical and pharmacologic review. Expert Opin. Pharmacother 11, 1943–1955 (2010). [DOI] [PubMed] [Google Scholar]
- 129.Keating GM Sorafenib: a review in hepatocellular carcinoma. Target. Oncol 12, 243–253 (2017). [DOI] [PubMed] [Google Scholar]
- 130.Nair A et al. FDA supplemental approval summary: Lenvatinib for the treatment of unresectable hepatocellular carcinoma. Oncologist 26, e484–e491 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gottlieb S, Gliksberg A, Schadde E & Kent P Novel systemic therapies in the treatment of fibrolamellar carcinoma [abstract]. J. Clin. Oncol 39, e16161 (2021). [Google Scholar]
- 132.Thoreen CC et al. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485, 109–113 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Agarwal VR et al. Molecular basis of severe gynecomastia associated with aromatase expression in a fibrolamellar hepatocellular carcinoma. J. Clin. Endocrinol. Metab 83, 1797–1800 (1998). [DOI] [PubMed] [Google Scholar]
- 134.Muramori K et al. High aromatase activity and overexpression of epidermal growth factor receptor in fibrolamellar hepatocellular carcinoma in a child. J. Pediatr. Hematol. Oncol 33, e195–e197 (2011). [DOI] [PubMed] [Google Scholar]
- 135.El Dika I et al. A multicenter randomized three-arm phase II study of (1) everolimus, (2) estrogen deprivation therapy (EDT) with leuprolide + letrozole, and (3) everolimus + EDT in patients with unresectable fibrolamellar carcinoma. Oncologist 25, 925–e1603 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bavetsias V & Linardopoulos S Aurora kinase inhibitors: current status and outlook. Front. Oncol 5, 278 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Fletcher GC et al. ENMD-2076 is an orally active kinase inhibitor with antiangiogenic and antiproliferative mechanisms of action. Mol. Cancer Ther 10, 126–137 (2011). [DOI] [PubMed] [Google Scholar]
- 138.Abou-Alfa GK et al. Phase II multicenter, open-label study of oral ENMD-2076 for the treatment of patients with advanced fibrolamellar carcinoma. Oncologist 25, e1837–e1845 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Buckley AF, Burgart LJ & Kakar S Epidermal growth factor receptor expression and gene copy number in fibrolamellar hepatocellular carcinoma. Hum. Pathol 37, 410–414 (2006). [DOI] [PubMed] [Google Scholar]
- 140.Patonai A et al. Molecular characteristics of fibrolamellar hepatocellular carcinoma. Pathol. Oncol. Res 19, 63–70 (2013). [DOI] [PubMed] [Google Scholar]
- 141.Bose P & Ozer H Neratinib: an oral, irreversible dual EGFR/HER2 inhibitor for breast and non-small cell lung cancer. Expert Opin. Investig. Drugs 18, 1735–1751 (2009). [DOI] [PubMed] [Google Scholar]
- 142.Abou-Alfa GK et al. Evaluation of neratinib (N), pembrolizumab (P), everolimus (E), and nivolumab (V) in patients (pts) with fibrolamellar carcinoma (FLC) [abstract]. J. Clin. Oncol 39 (3 Suppl.), 310 (2021). [Google Scholar]
- 143.McFarland JM et al. Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat. Commun 9, 4610 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tseng YY & Boehm JS From cell lines to living biosensors: new opportunities to prioritize cancer dependencies using ex vivo tumor cultures. Curr. Opin. Genet. Dev 54, 33–40 (2019). [DOI] [PubMed] [Google Scholar]
- 145.Ghandi M et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Vazquez F & Boehm JS The Cancer Dependency Map enables drug mechanism-of-action investigations. Mol. Syst. Biol 16, e9757 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nagourney RA Ex vivo programmed cell death and the prediction of response to chemotherapy. Curr. Treat. Options Oncol 7, 103–110 (2006). [DOI] [PubMed] [Google Scholar]
- 148.Letai A Functional precision cancer medicine-moving beyond pure genomics. Nat. Med 23, 1028–1035 (2017). [DOI] [PubMed] [Google Scholar]
- 149.Kennedy A et al. Safety of selective internal radiation therapy (SIRT) with yttrium-90 microspheres combined with systemic anticancer agents: expert consensus. J. Gastrointest. Oncol 8, 1079–1099 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.De La Torre M et al. Abstract O-27. in ILCA Annual Conference, 2020: Book of Abstracts 16 (ILCA, 2020). [Google Scholar]
- 151.Gottlieb S, O’Grady C, Gliksberg A & Kent P Early experiences with triple immunochemotherapy in adolescents and young adults with high-risk fibrolamellar carcinoma. Oncology 99, 310–317 (2021). [DOI] [PubMed] [Google Scholar]
- 152.Ma C et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 360, eaan5931 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Fajgenbaum DC, Ruth JR, Kelleher D & Rubenstein AH The collaborative network approach: a new framework to accelerate Castleman’s disease and other rare disease research. Lancet Haematol. 3, e150–e152 (2016). [DOI] [PubMed] [Google Scholar]
- 154.Blay JY, Coindre JM, Ducimetiere F & Ray-Coquard I The value of research collaborations and consortia in rare cancers. Lancet Oncol. 17, e62–e69 (2016). [DOI] [PubMed] [Google Scholar]
- 155.Painter CA et al. The Angiosarcoma Project: enabling genomic and clinical discoveries in a rare cancer through patient-partnered research. Nat. Med 26, 181–187 (2020). [DOI] [PubMed] [Google Scholar]
- 156.Komatsubara KM & Carvajal RD The promise and challenges of rare cancer research. Lancet Oncol. 17, 136–138 (2016). [DOI] [PubMed] [Google Scholar]
- 157.Boyd N, Dancey JE, Gilks CB & Huntsman DG Rare cancers: a sea of opportunity. Lancet Oncol. 17, e52–e61 (2016). [DOI] [PubMed] [Google Scholar]
- 158.Sherman RE et al. Real-world evidence–what is it and what can it tell us? N. Engl. J. Med 375, 2293–2297 (2016). [DOI] [PubMed] [Google Scholar]
- 159.Mathoulin-Pelissier S & Pritchard-Jones K Evidence-based data and rare cancers: the need for a new methodological approach in research and investigation. Eur. J. Surg. Oncol 45, 22–30 (2019). [DOI] [PubMed] [Google Scholar]
- 160.Bondeson DP et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol 11, 611–617 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sharifnia T, Hong AL, Painter CA & Boehm JS Emerging opportunities for target discovery in rare cancers. Cell Chem. Biol 24, 1075–1091 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tsherniak A et al. Defining a cancer dependency map. Cell 170, 564–576.e16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Shimada K, Bachman JA, Muhlich JL & Mitchison TJ shinyDepMap, a tool to identify targetable cancer genes and their functional connections from Cancer Dependency Map data. eLife 10, e57116 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Matossian MD et al. Novel application of the published kinase inhibitor set to identify therapeutic targets and pathways in triple negative breast cancer subtypes. PLoS ONE 12, e0177802 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Elkins JM et al. Comprehensive characterization of the Published Kinase Inhibitor Set. Nat. Biotechnol 34, 95–103 (2016). [DOI] [PubMed] [Google Scholar]
- 166.Hammond WJ et al. Intracranial metastasis in fibrolamellar hepatocellular carcinoma. Pediatr. Blood Cancer 65, e26919 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]