

Abstract
We identified the BCL11A protein in a proximity-dependent biotinylation screen performed with the DNA glycosylase NTHL1. In vitro, DNA repair assays demonstrate that both BCL11A and a small recombinant BCL11A160–520 protein that is devoid of DNA binding and transcription regulatory domains can stimulate the enzymatic activities of two base excision repair enzymes: NTHL1 and DNA Pol β. Increased DNA repair efficiency, in particular of the base excision repair pathway, is essential for many cancer cells to proliferate in the presence of elevated reactive oxygen species (ROS) produced by cancer-associated metabolic changes. BCL11A is highly expressed in triple-negative breast cancers (TNBC) where its knockdown was reported to reduce clonogenicity and cause tumour regression. We show that BCL11A knockdown in TNBC cells delays repair of oxidative DNA damage, increases the number of oxidized bases and abasic sites in genomic DNA, slows down proliferation and induces cellular senescence. These phenotypes are rescued by ectopic expression of the short BCL11A160–520 protein. We further show that the BCL11A160–520 protein accelerates the repair of oxidative DNA damage and cooperates with RAS in cell transformation assays, thereby enabling cells to avoid senescence and continue to proliferate in the presence of high ROS levels.
Graphical Abstract
Graphical Abstract.
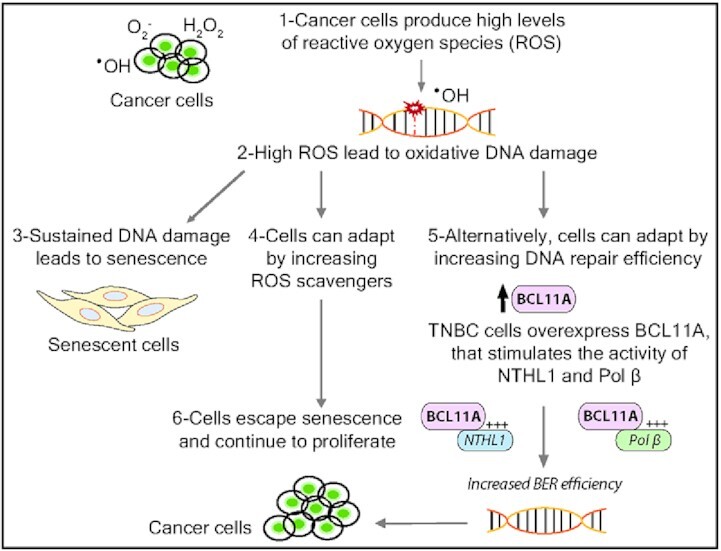
BCL11A expression in triple-negative breast cancer (TNBC) increases base excision repair efficiency and helps cells escape ROS-induced senescence and continue to proliferate.
INTRODUCTION
Base excision repair (BER) is the pathway that repairs single-strand breaks and base lesions including apurinic/apyrimidinic (AP) sites, alkylated, deaminated and oxidized bases as well as uracil residues. In the presence of an altered base, the pathway is initiated by a DNA glycosylase that specifically recognizes and removes the faulty base. UNG, SMUG1 and TDG remove uracil bases, while methyl-purine DNA glycosylase (MPG) removes alkylated purines and MUTYH removes the adenine from an A•8-oxoG base pair to produce an apyrimidinic/apurinic site (AP site) (1,2). In mammals, the AP endonuclease 1, APE1, incises the DNA backbone 5′ to the AP site to generate a single-strand break with a 5'-deoxyribose phosphate (5′-dRP) (3). Oxidized bases are removed by four DNA glycosylases that exhibit a broad substrate range (4). Oxidized purines are primarily removed by OGG1, whereas oxidized pyrimidines are targeted by NTHL1, NEIL1 and NEIL2, the last two enzymes demonstrating affinity for single-stranded or bubble DNA. DNA glycosylases for oxidized bases are bifunctional, being also endowed with an AP/lyase activity that generates a single-strand nick 3' to the AP site. OGG1 and NTH1 generate the single-strand nick through beta elimination, leaving a 3'-phospho-α,β-unsaturated aldehyde (3′PUA), while NEIL1 and NEIL2 proceed by beta delta elimination and produce a 3′-phosphate (3′-P) (reviewed in (4)). As the final ligation step requires a 3′-hydroxyl (3′-OH) and a 5′-phosphate (5′-P), the single-strand breaks produced by BER enzymes or DNA damage must be processed. Pol β carries out a dRP-lyase reaction to convert 5′-dRP into 5′-P, APE1 converts 3′-PUA to 3′-OH, and PNKP changes the 3′-P into 3′-OH. In short-patch repair, Pol β adds a single nucleotide and the single-strand break is then sealed by Ligase 3 in conjunction with XRCC1. In long-patch repair, 2–13 bases are added by Pol β or δ/ϵ, thereby generating a displaced strand that is cleaved by the flap structure-specific endonuclease FEN1 prior to ligation (1,5).
The implication of BER in cancer is complex. On the one hand, somatic mutations in POLB were found in 30% of cancer, while germline mutations in MUTYH and NTHL1 have been associated with colorectal polyposis as well as other cancers including duodenal carcinoma, bladder, ovarian, and skin cancer (6–15). As these mutations were shown to reduce enzymatic activity, one can reasonably conclude that a decrease in BER efficiency increases the risk of cancer. On the other hand, several BER enzymes including Pol β, APE1 and FEN1 were found to be overexpressed in multiple cancers (16–24). Likewise, aberrant or increased expression of the BER accessory factors CUX1, CUX2 and SATB1 were reported in multiple tumours and cancer cell lines (reviewed in (25)). Notably, knockdown of these BER accessory factors was shown to be synthetic lethal in cell lines that exhibit high levels of reactive oxygen species (26–30). In agreement with these findings, a genome-wide RNAi screen to identify synthetic lethal interactions with the KRAS oncogene identified five genes involved in BER: NEIL2, XRCC1, POLβ, LIG3 and CUX1 (31). These studies established that increased activity of BER enzymes accelerates the repair of oxidative DNA damage and enables cancer cells to avoid senescence despite producing excessive levels of reactive oxygen species.
We have previously established that the CUT domain proteins CUX1, CUX2 and SATB1 function as DNA repair accessory proteins that stimulate the enzymatic activities of the 8-oxoguanine DNA glycosylase, OGG1, but no other DNA glycosylases (26–29,32). We therefore hypothesized that other DNA glycosylases may interact with distinct accessory factors. As OGG1 specifically recognizes oxidized purines, we aimed to identify accessory proteins for NTHL1, the main DNA glycosylase for oxidized pyrimidines. Using the proximity-dependent biotinylation screen (BioID) approach, we identified proteins that come into close-proximity to NTHL1. One of these proteins, BCL11A, was found in vitro to stimulate the enzymatic activity of NTHL1.
BCL11A was originally identified as a gene that is amplified or present at a site of chromosomal translocation in B-cell lymphomas (33). BCL11A was quickly found to be the human homolog of Evi9, a mouse gene that was identified as a common site of retroviral integration in murine myeloid leukemias (34,35). These early studies clearly identified BCL11A as an oncogene. Subsequent studies revealed that BCL11A knockdown in triple-negative breast cancer cells (TNBC) decreases tumor development and impedes tumor growth (36). Moreover, cooperation between BCL11A and inactivation of the NF1 GTPase-activating protein in leukemogenesis suggested cooperation between BCL11A and the RAS pathway (37). BCL11A codes for a zinc finger transcription factor that plays a role in B-lymphopoiesis, erythropoiesis, skin development, primary islet cells and neurogenesis (38–42). BCL11A haploinsufficiency and single nucleotide variants have been associated with cerebellar abnormalities (43), intellectual disability (44) and epileptic encephalopathy (45) (reviewed in (46)). It is generally assumed that these disorders result from a deficiency in BCL11A transcriptional activity.
Here, we present the results of in vitro and in cellulo experiments demonstrating that in addition to its role as a transcription factor, BCL11A also functions as an accessory factor in base excision repair. We further show that the DNA repair functions of BCL11A are co-opted by cancer cells to limit oxidative DNA damage below a threshold that would otherwise trigger cellular senescence. Indeed, the synthetic lethality of BCL11A knockdown in some cancer cells is rescued by the expression of a BCL11A fragment that is competent in DNA repair but entirely devoid of transcriptional potential. Moreover, we show that the DNA repair function of BCL11A cooperates with RAS in cellular transformation by preventing senescence triggered by RAS-induced oxidative DNA damage.
MATERIALS AND METHODS
Cell lines and cell culture
293 Flp-In T-REx cells (Invitrogen), HEK293FT, MDA-MB-231, HS578T, MDA-MB-468 and IMR90 cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Wisent). MDA-MB-436 and BT549 were cultured in Roswell Park Memorial Institute medium (RPMI, Wisent), and RPE1 was cultured in Dulbecco's modified Eagle's medium F/12 (DMEM F/12, Wisent). All media were supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin streptomycin (Invitrogen). All cells were maintained at 37°C, 5% CO2 and atmospheric O2. Lentiviruses were produced by co-transfecting HEK293FT cells with plasmids encoding 3xHA-BCL11A160–520, 3xHA-BCL11A-XL, 3xHA- BCL11AΔ160–520, or short hairpin RNA against BCL11A (Mission shRNA pLKO.1 library from Sigma or GipZ shRNA from Dharmacon) with packaging plasmid psPAX2 and envelop plasmid pMD2G. Dicer-substrate siRNA (DsiRNAs) transfections were carried out with two different sequences and cells were collected for the different assays three days after transfection. For doxycline induced knockdown of BCL11A, the sequence of Dicer1 was cloned into a pTRIPZ plasmid. A list of the different shRNA and DsiRNA sequences can be found in Supplementary Table S4. 293 Flp-In T-REX cells were co-transfected with pOG44 (Flp-recombinase expression vector) and a plasmid containing the coding sequence for FLAG-BirA* fusion protein, eGFP-BirA*-FLAG or eGFP-NLS-BirA*-FLAG.
Plasmid construction
Human NTHL1 (GeneCopoeia) was cloned into pcDNA5 FRT/ FLAG-BirA* vector. eGFP-BirA*-FLAG and eGFP-NLS-BirA*-FLAG were provided by Dr Anne-Claude Gingras. For bacterially purified proteins, plasmids expressing histidine-tagged BCL11A small fragments, and the BCL11A160–520 fragment were prepared by inserting gBlocks gene fragments (Integrated DNA Technologies) into pET-30a vector using restriction enzymes. The N-His-BCL11A-XL plasmid was purchased from GeneCopoeia in vector system pReceiver. For the pulldown experiments, GST-NTHL1 recombinant proteins or GST-Pol β were cloned into the pGEX backbone. BCL11A-XL expression plasmid for mammalian cells was purchased from genecopoia (pReceiver-Lv118) and BCL11A160–520 and BCL11AΔ160–520 were cloned into a plenti6 expressing vector using gateway cloning.
Bacterial protein expression
All proteins were expressed in the BL21 strain of Escherichia coli and were induced with Isopropyl ß-d-1-thiogalactopyranoside (ITPG) as described in Ramdzan et al. 2015 (32).
BioID
Human NTHL1 was cloned into pcDNA5 FRT/ FLAGBirA* vector. Using the Flp-In™ T-REx™ system (Thermofisher), we generated 293 TRE Flp-In cells stably expressing NTHL1-BirA*-FLAG, eGFP-NLS-BirA*-FLAG, and eGFP-BirA*-FLAG under a tetracycline inducible system. Cells were treated with 1 μg/ml of tetracycline for 24 h to induce protein expression. Six hours before cell collection, 50 μM of biotin were added and immediately followed by irradiation treatment. BioID purification and mass spectrometry data acquisition and analysis were performed as described (47).
Gene enrichment analysis
Gene enrichment analysis of preys identified by BioID was performed using g:Profiler (48). Analysis is available in Supplementary Tables S2 and S3.
Immunoblotting
Protein extraction and western blotting were performed as described (49). The following antibodies and dilutions were used: FLAG-BirA* fusion proteins were detected using mouse M2 anti-FLAG (1:1000; Sigma-Aldrich). Biotinylated proteins were detected using HRP-conjugated streptavidin (1:1000; BioLegend). Other antibodies used are BCL11A 382A (1:4000, Bethyl), NTHL1 (1:1000, Proteintech), anti-GST (1:1000, Abcam), anti-His (1:3000, Sigma), anti-HA (1:1000 Covance) and anti-γ-tubulin (1:10 000, Sigma). The AF3 BCL11A antibody was made by the McGill Animal Health Facility and raised against a His-tagged peptide of BCL11A (aa 370–509). The V5 antibody for Split Intein-Mediated Protein Ligation (SIMPL) was purchased from Protein Signaling and the Flag antibody was purchased from Sigma. Secondary HRP-conjugated antibodies are from Jackson Laboratories.
Immunoprecipitation
MDA-MB-231 cells were collected, and nuclear proteins were extracted. 500 μg of nuclear extracts were incubated with either 3 μl of anti-NTHL1 antibody (Proteintech), 3 μl of anti-Pol β antibody (Abcam) or 6 μl of AF3 BCL11A antibody and left to spin overnight at 4°C. The next day, 20 μl of magnetic beads (Dynabeads protein-G from Invitrogen) were added for 1 h. Samples were washed 5 times with NP40 buffer and resuspended in 2× Laemmli buffer. Samples were separated by SDS-PAGE followed by immunoblotting with anti-NTHL1 antibody, anti-Pol β antibody or anti-BCL11A antibody.
Split intein-mediated protein ligation (SIMPL)
293T cells were transfected with either BCL11A160–520-V5-IN, IC2-Flag-Pol β, CIC2a-Pol β-Flag or IC2-Flag-LIG3, IC2-Flag-FOXN2, CCNB1-V5-IN4b or CCNE1-V5-IN4b for negative controls. No selection was performed. Cells were treated for 16 h with tetracycline to induce expression of recombinant proteins and total extracts were obtained with RIPA lysis buffer. For each sample, 40 μg were loaded onto an SDS PAGE gel and blotted against V5 (Cell Signaling Technologies) or Flag (Sigma).
GST-pull down assay
Pulldown assays were performed as described in (50), with the following modifications: 1 μg of GST-NTHL1 full length (1–312), GST-NTHL1 peptides (1–116 or 88–312), GST-Pol β or GST-bound beads were incubated with 1 μg of bacterially purified BCL11A peptides.
In vitro fluorescent cleavage assay
The fluorescent cleavage assay was performed as previously described with the following modifications (51,52). We used a 43-mer oligonucleotide (Midland) with a 5,6-dihydrothymidine (DHT) base at its sixth position. The 5′ end of the DNA was conjugated to a FAM fluorophore, and 3′ end conjugated to a Dabcyl quencher. Cleavage reactions were conducted as described in Ramdzan et al. (29).
In vitro radioactive cleavage assay
Double-stranded 32-mer oligonucleotides containing a Tg modification at the 18th position (Midland) were labeled with γ-32P dATPs at the 5′end of the top strand (*) using T4 polynucleotide kinase. Cleavage reactions were conducted in a 20 μl reaction using the indicated concentration of bacterially purified proteins (5, 10 or 15 nM of BCL11A) and enzyme (10 nM of NTHL1) in 20 mM Tris (pH 8), 1 mM EDTA (pH 8.0), 1 mM DTT and 1 pmol (50 nM) of labelled probe. A 5-fold molar excess of DNA over NTHL1 was thus used. Reactions were performed for 30 min at 37°C. Where indicated in Figure 3C and F, 100 mM of NaOH were added and samples were incubated at 70°C for 10 min. Reaction was neutralized by adding 2 μl of 1 M HCl. The reaction was terminated by formamide DNA loading buffer (90% formamide with 0.05% bromophenol blue and 0.05% xylene cyanol). The DNA was loaded on a pre-warmed 20% polyacrylamide-urea gel (19:1) and separated by electrophoresis in Tris-borate and EDTA (TBE; pH 8.0) at 20 mA. The radiolabeled DNA fragments were visualized by storage phosphor screen (GE Healthcare).
Figure 3.

Structure–function analysis of BCL11A defines a region of BCL11A that interacts with NTHL1 and stimulates its enzymatic activity. (A) Diagrammatic representation of the BCL11A-XL and fragments tested in pull-down and DNA repair assays. (B) Diagrammatic representation of the 5,6-dihydrothymidine (DHT) cleavage assay using a fluorophore reporter probe. The assay was performed using 5 nM of NTHL1 and 20 nM of BCL11A-XL, in the presence of 50 nM of BSA. (C) Diagrammatic representation of the thymine glycol (Tg) cleavage assay using a radioactively labeled probe. The assay was performed using purified NTHL1 (10 nM) and the indicated amounts of BSA and/or BCL11A-XL. The reaction in lane 1 was performed with a probe that contains a normal thymidine base instead of thymine glycol. (D) Pull-down assays were performed using His-tagged BCL11A fragments and either GST or GST-NTHL1, followed by immunoblotting with anti-His antibodies. (E) DHT cleavage assays were performed using 20 nM of BCL11A fragments, in the presence of absence of NTHL1 (5 nM), as indicated. An additional assay was performed with 40 nM of BCL11A160–520. All samples included 50 nM of BSA; the sample labeled ‘BSA’ included an additional 20 nM BSA. (F) Thymine-glycol (Tg) cleavage assays were performed using radioactively end-labeled double-stranded oligonucleotides containing a Tg oxidized base, and 250 nM BSA. Where indicated, NTHL1 (2.5 nM), BSA or BCL11A160–520 (15 nM) were included in the reaction. Production of a cleaved product in samples that have not been treated with NaOH (lanes 1–4) requires the glycosylase and AP/lyase activities of NTHL1. In the absence of NaOH, NTHL1 alone cleaves 9.7% of the substrate, whereas BCL11A160–520 cleaves 49%. In the presence of NaOH, NTHL1 alone cleaves 20.3% of the substrate, whereas BCL11A160–520 cleaves 43.8%. (G) Radioactively end-labeled double-stranded oligonucleotides containing a thymine glycol (Tg) base were incubated with the indicated amount of NTHL1, BSA and BCL11A160–520. After incubation at 37°C, 50 mM sodium borohydride was added. The reactions were incubated for another 15 min at 37°C. After termination of the reaction, the trapped complexes were separated from free DNA by 10% SDS-PAGE. (H) Pull-down assays were performed using the His-tagged BCL11A161-366 peptide and either GST or GST-NTHL11-312, GST-NTHL188-312 or GST-NTHL11-116 followed by immunoblotting with anti-His antibodies.
Sodium borohydride trapping of NTHL1
5′-End-labeled 32-mer duplex containing a Tg modification at the 18th position (Midland) was incubated with purified NTHL1, and BCL11A full length protein, short fragment BCL11A160–520 or BSA at the indicated concentrations. After incubation at 37°C for 30 min, 50 mM sodium borohydride was added, and the reactions were incubated for 15 min at 37°C. The trapped complexes were separated from free substrate by 10% SDS-PAGE gel.
Electrophoretic mobility shift assay (EMSA)
EMSAs were performed as previously described (53). For the BCL11A EMSA, the probe carried the consensus sequence from (54).
Clonogenic viability assay
Clonogenic ability of H2O2 treated cells was conducted as described previously (29).
Single cell gel electrophoresis
50 000 cells per condition were plated and 50 μM of H2O2 was used to treat cells on ice for 20 min to induce DNA damage. Immediately after treatment, cells were washed with PBS to eliminate H2O2 residue and allowed to recover at 37°C in fresh medium for the indicated amount of time. For the comet assay performed at pH 10 with Endo III treatment, slides were immersed in lysis solution containing Endo III enzyme for 1h at 37°C prior to electrophoresis. For IMR90, the cells stably carrying BCL11A160–520 were infected with pBABE -HRASG12D or pBABE -empty vector. 48h after infection, cells were selected with 4 μg/μl puromycin for 3 days and pelleted for comet assay. Comet assay was carried out as described in Ramdzan et al. (26).
Abasic site quantification
MDA-MB-231 cells stably carrying a lentivirus expressing a BCL11A short hairpin RNA (shBCL11A) were treated with H2O2 for 20 min or no H2O2. After treatment, cells were washed with PBS and fresh medium was added. In Figure 6C, MDA-MB-231 cells were transfected with Dicer siRNA and pelleted 3 days after transfection. After pelleting, DNA extraction was performed with a DNA extraction kit (Qiagen). Aldehyde-reactive probe labeling and quantification of abasic sites were performed as described by an AP-sites assay kit (Dojindo Molecular Technologies). Briefly, 1 μg of the extracted DNA is incubated with 10 μl of the ARP solution containing the ARP-labelled probe for 1h at 37°C. Labelled DNA is then washed twice with TE buffer on the provided filtration tubes and resuspended from the membrane in 400 μl of TE. ARP-labelled DNA is then bound to the provided 96-well plate overnight by incubation at room temperature with binding buffer. The next day, plates are washed and incubated with HRP-streptavidin solution for 1 h at 37°C, then, washed and incubated for 1 h at 37°C with the substrate solution. The colorimetric reaction is read by the SpectraMax ID3 from Molecular devices.
Figure 6.

The DNA repair and proliferation defects caused by BCL11A knockdown are rescued by ectopic expression of the BCL11A160–520 Fragment. (A, B, E and F) MDA-MB-231 cells were infected with lentiviral vectors as indicated: an empty vector, a vector expressing BCL11A shRNA, and a vector expressing BCL11A160–520. (A) Cells were exposed to 50 μM H2O2 for 20 min and allowed to recover for the indicated time before carrying out single cell gel electrophoresis at pH >13. Comet tail moments were scored for at least 100 cells per condition. Results are a representative of one of three different experiments. Error bars represent standard error. ***P< 0.001; **P< 0.01; *P< 0.05; Student's t-test. (B) Cells were exposed to 50 μM H2O2 for 20 min and allowed to recover for the indicated time before carrying out single cell gel electrophoresis at pH 10 after treatment of cells with the Endo III DNA glycosylase. Comet tail moments were scored for at least 100 cells per condition. Results are a representative of one of three different experiments. Error bars represent standard error. ***P< 0.001; **P< 0.01.; *P< 0.05; Student's t-test. (C) MDA-MB-231 cells stably carrying an empty vector or a vector expressing BCL11A160–520 were transfected with either of two distinct BCL11A dicer RNAs or a control dicer RNA. Genomic DNA was purified and abasic sites were quantified using an aldehyde-reactive probe. Results are an average of three different experiments. Error bars represent standard error. ***P< 0.001; **P< 0.01; *P< 0.05; Student's t-test. (D) MDA-MB-231 cells were stably infected with a vector expressing BCL11A shRNA under the control of a tetracycline-inducible promoter, as well as an empty vector or a vector expressing BCL11A160–520. Cells were treated or not with doxycycline for 3 days before cell extracts were prepared and analyzed in a DNA repair assay using as a substrate double-stranded oligonucleotide containing a thymine glycol base (Tg). Upon incubation in the presence of 32P-dTTP, NTHL1 present in the cell extract removes the thymine glycol and introduces a single-strand break; Pol β adds a radioactively labeled thymidine and ligase III seals the strand-break thereby generating a single-strand that migrates more slowly on a denaturing gel. The amount of repair completion (top band) for each sample was normalized to the value of ‘Vector no dox’. The enzymatic activity of the BCL11A knockdown (‘vector + dox’) is 55.7% whereas the activity of ‘BCL11A160–520 no dox’ is 365% and ‘BCL11A160–520 + dox’ is 361%. (E) Cells were treated with increasing amounts of H2O2 and then submitted to a clonogenic assay. Error bars represent standard error. ***P< 0.001; **P< 0.01; *P< 0.05; Student's t-test. (F) MDA-MB-231 cells carrying BCL11A160–520 or not were infected with lentiviruses expressing either control or BCL11A shRNA. β-Gal associated senescence was measured 5 days after infection. All values are normalized to the control shRNA value. Error bars represent standard error. Results are a representative of one of two different experiments. ***P< 0.001; **P< 0.01; *P< 0.05; Student's t-test. (G) MDA-MB-231 cells were stably infected with a lentiviral vector expressing a BCL11A shRNA under the control of a doxycycline-inducible promoter and either an empty vector or a vector expressing BCL11A160–520. Doxycycline was added to the medium or not and 3 days later cell proliferation was measured by staining with CellTrace™ CFSE. CFSE was added to the medium and a portion of the population was fixed immediately as the ‘0’ generation. The remaining cells were allowed to proliferate for 3 days. Cells were fixed and analyzed by flow cytometry. Small peaks within the CFSE profiles represent successive generations, as indicated above the peaks. The proliferation index is the total number of divisions divided by the number of cells that went into division. The division index is the average number of cell divisions, taking into account the cells that never divided.
Polymerase and strand-displacement activity
Experiments were performed as described (50), with the following modifications: Polymerase and strand-displacement activity was investigated by incubating 15 nM of Pol β and indicated amounts of BCL11A-XL protein or BSA in the presence of 50 mM Tris–HCl, pH 8.0, 10 mM KCl, 1 mM MgCl2, 2 mM DTT, 0.01% Tween-20, 500 ng BSA, 100 μM dTTP and 500 nM fluorescent probe.
Polymerase assays with radiolabeled probes
The gapped probe of Figure 4 was prepared as described (50). Single-nucleotide addition was performed in 50 mM Tris–HCl, pH 8.0, 10 mM KCl, 1 mM MgCl2, 2 mM dithiothreitol, 0.01% Tween-20 with 32P-dCTP in the presence of DNA Pol β (1.25 nM) and either BSA (50 nM) or bacterially purified BCL11A (indicated amounts in the figure).
Figure 4.

BCL11A interacts with Pol β and stimulates its enzymatic activities. (A) Nuclear protein extracts (500 μg) from MDA-MB-231 cells were submitted to co-immunoprecipitation (IP) alternatively with Pol β or BCL11A antibodies and then immunoblotted (IB) with either anti-Polβ or anti-BCL11A antibody. Input (1%) was loaded as a protein expression control. (B) 293 cells were transfected with vectors expressing fusion proteins containing either the N-terminal or C-terminal portion of Intein, as indicated: BCL11A160–520-V5-IN4b with either IC2-Flag-LIG3, IC2-Flag-FOXN2 or IC2-Flag-Pol β. Whole cell extracts were submitted to immunoblotting analysis with the V5 and FLAG antibodies. (C) Pull-down assays were performed using His-tagged BCL11A fragments and either GST or GST-Pol β, followed by immunoblotting with anti-His antibodies. (D) Diagrammatic representation of the strand displacement assay using a fluorophore reporter probe. F: FAM fluorophore; Q: quencher. The strand displacement assay was performed using 2.5 nM of BCL11A and 15 nM of Pol β. 50 nM of BSA were added to each reaction. (E) Double-stranded oligonucleotides containing a uracil residue were incubated with UDG and APE1 to form a gapped substrate for Pol β. The DNA was then incubated with 32P-dCTP in the presence of DNA Pol β and either BSA or BCL11A160–520. (F) Double-stranded oligonucleotides containing a uracil residue were labeled at the 5'-end and incubated with UDG and APE1 to form a gapped substrate for Pol β. The gapped probe was then incubated with all 4 dNTPs in the presence of DNA Pol β and either 25 nM BSA or 25 nM BCL11A160–520. (G) Double-stranded oligonucleotides containing a uracil residue, that were labeled at the 3′ end using Klenow and fluorescent CF 660R dCTP, were incubated with UDG and APE1 to produce a single-strand nick with a 5′-deoxyribose phosphate (dRP). This DNA substrate was incubated with Pol β in the presence of BSA or BCL11A160–520. NaBH4 was added to most samples, except in lane 9, to prevent the spontaneous conversion of dRP into P. From four independent pairs of assays, we calculated an average 1.8 fold (±0.3) stimulation of Pol β dRP-lyase activity by BCL11A160–520.
For the polymerase/elongation assay of Figure 4F, the probe was prepared as described (50). The polymerase reactions were performed with either 1.25 nM or 2.5 nM Pol β and 25 nM of either HOXB3, BCL11A or BSA in the presence of cold dNTPs.
Polymerase β dRP lyase activity
An oligonucleotide containing 32 bp and a uracil at the 18th position (5′CCGGTGCATGACACTGTUACCTATCCTCAGCG-3′) was labeled at the 3′end with Klenow fragment and CF 660R dCTP. The dRP-lyase assay was performed as in Ramdzan et al., with some modifications. Briefly, 200 nM of the CF 660R dCTP labeled oligonucleotide was pre-treated with uracil-DNA glycosylase (UDG) for 15 min and with APE1 for 4 min at 37°C. Next, the oligo was added to the reaction mix, containing 50 mM Tris–HCl pH 8.0, 10 mM KCl, 1 mM MgCl2, 0.01% Tween-20, 2 mM DTT and 10 nM dCTP. The reaction mix was then incubated with 25 nM BCL11A160–520 or BSA as a control. Lastly, 10 nM of purified Pol β (ENZ-168; ProspecBio) was added and the samples were incubated at 37°C for 7.5 or 15 min. To terminate, 50 mM EDTA was added to each sample, and the reaction was stabilized by addition of 340 nM of sodium borohydride (NaBH4) followed by incubation on ice for 30 min. The DNA product was recovered by ethanol precipitation in the presence of 20 μg of glycogen and 300 mM NaOAC. The pelleted DNA was resuspended in 20 μl of 10 mM Tris pH 8.0 + 1 mM EDTA buffer. After addition of formamide loading buffer, samples were incubated at 75°C for 3 min and loaded on a pre-warmed 20% polyacrylamide (19:1)-urea gel. The electrophoresis took place in Tris-Taurine and EDTA (TTE; pH 8.0) at constant 50 W.
Soft agar colony formation assay
IMR90 cells stably expressing BCL11A-XL or BCL11A160–520 were infected with pBABE -HRASG12D or pBABE -empty vector. 48 h after infection, cells were counted and plated for soft agar colony formation assay. Briefly, cells were counted and resuspended to 40 000 cells/ml. A first layer of 1 ml of 0.6% medium-agarose was added to the 6-well plates (for each condition, triplicate wells were plated). Cells at 40 000 cells/ml were then mixed with 0.6% agarose at a ratio of 1:1. Then, 1 ml of agarose-mixed cells was added to the 0.6% agarose layer. Plates were left to solidify for 30 min and 500 μl of fresh DMEM was then added to the top layer. An extra 500 μl of DMEM was added after 4 days at 37°C. After 10 days, 500 μl of thiazolyl blue (at 1 mg/ml) were added to each well and plates were left at 37°C overnight. Colonies were counted the next day.
Senescence quantification
IMR90 cells stably expressing BCL11A-XL or BCL11A160–520 were infected with pBABE -HRASG12V or pBABE-empty vector. 48 h after infection, cells were selected with 4 μg/ml puromycin for 7 days. On day 6, cells were counted and plated to perform the β-galactosidase senescence assay the next day. β-gal senescence was measured by flow cytometry (BD LSR Fortessa) following the instructions of the Spider-βgal senescence kit from Dojindo. MDA-MB-231 cells overexpressing BCL11A160–520 or not were transfected with Dicer siRNA against BCL11A. Senescence was measured by flow cytometry using the Spider-βgal senescence kit from Dojindo.
RT-qPCR
MDA-MB-231 cells were transfected with two different Dicer siRNAs targeting BCL11A or a non-target dicer siRNA. 3 days after transfection, cells were pelleted. IMR90 cells carrying the plenti6-BCL11A160–520 vector or the plenti6-empty vector were infected with pBABE -HRASG12V or pBABE -empty vector as described for the β-galactosidase senescence assay. Three days after selection, cells were pelleted, RNA was extracted using the Qiagen blood & tissue RNA extraction kit which includes a gDNA elimination step. Samples were eluted in 30 μl of RNAse free water. After nanodrop quantification and confirmation of RNA purity (A260/A280 > 2), 1 μg of RNA was reverse transcribed using the Qiagen quantitect reverse transcription kit. Reverse transcription was carried out at 42°C and included an initial gDNA wipe out step. qPCRs were performed in triplicate, using 9 μl of the Biorad SSO advanced sybergreen super mix with 1 μl of cDNA, in the Eppendorf Realplex Mastercycler. The qPCR cycles are the following: 95°C for 30 s–40 cycles (95°C for 15 s–60°C for 45 s)–95°C for 15 s–60°C for 15 s, followed by a melting curve. For each primer pair, standard curves were measured by serial dilution and primer efficiency was measured. Only primer pairs with an efficiency between 0.9 and 1.1 were selected. Analysis was performed by normalizing copy number values of the gene of interest to the copy number values of the housekeeping gene, GAPDH. All primer sequences are available in Supplementary Table S5.
CFSE proliferation
MDA-MB-231 cells were infected with a lentivirus containing a BCL11A shRNA under the control of a doxycycline inducible promoter. On day one, 5 μg/ml doxycline was added or not to the cells. On day 3, cells were stained with CFSE (CellTrace CFSE proliferation kit) according to manufacturer's instructions. Cells were analyzed by Flow cytometry 3 days after CFSE staining.
Total DNA repair assay
MDA-MB-231 cells were infected with a lentivirus containing a BCL11A shRNA under the control of a doxycycline inducible promoter. On day one, 5 μg/ml doxycline was added or not to the cells. On day 3, cells were pelleted. In Figure 7, TNBC cell lines were transfected with two different DsiRNAs and pelleted three days after transfection. Whole cell proteins were extracted with LTGO buffer (50 mM Tris–HCl pH 7.1, 1 mM EDTA, 0.5 mM spermidine, 0.1 mM spermine, 222 mM KCl, 10.2% glycerol). 10 μg of extracts were incubated with a 46 nt probe (at 10 pmol/μl) containing a Tg damaged base in 1× BER buffer (50 mM HEPES (pH 7.5), 10 mM MgCl2, 0.2 mM EDTA, 10% Glycerol, 2 mM DTT, 4 mM ATP (Add fresh)), 400 mM phosphocreatine, 2.5 mg/ml creatine phosphokinase, 100 mM ATP, 10 mM dATP, dGTP, dCTP and 1:50 P32-dTTP (0.25 μCi/μl). Reaction was carried out for 15 min at 37°C. 2 μl of 10 mg/ml Proteinase K and 4 μl of 2.5% SDS and 100 mM EDTA were added, and samples were incubated at 37°C for 60 min. Finally, 2 μl of 1N NaOH were added and incubated for 15 min at 70°C and the reaction was neutralized with 2 μl of 1 M HCl.
Figure 7.

The impact of BCL11A knockdown on DNA repair, genomic DNA damage and clonogenic efficiency is confirmed in other triple-negative breast cancer cells. (A and B) The triple-negative breast cancer cell lines BT549, MDA-MB-468 and Hs578T were transfected with either of two distinct BCL11A dicer RNAs or a control dicer RNA. After 2 days, cell extracts were prepared and in parallel cells were submitted to a comet assay. (A) Cell extracts were prepared and analyzed in a DNA repair assay with a probe containing a thymine glycol, as described in Figure 6D. (B) A fraction of the cells was submitted to single cell gel electrophoresis at pH >13. Comet tail moments were scored for at least 100 cells per condition. Results are a representative of one of three different experiments. Error bars represent standard error. ***P< 0.001; **P< 0.01; *P< 0.05; Student's t-test. (C) Cells were stably infected with a lentiviral vector expressing a BCL11A shRNA under the control of a doxycycline-inducible promoter. Doxycycline was added to the medium or not and 3 days later cells were treated with the indicated concentrations (0, 20, 50) of H2O2 and then submitted to a clonogenic assay. Results are a representative of one of three different experiments. Error bars represent standard error. ***P< 0.001; **P< 0.01; *P< 0.05; Student's t-test.
RESULTS
Proximity biotinylation with NTHL1-BirA*
To identify proteins that come in close-proximity to the human NTHL1 DNA glycosylase we performed proximity-dependent biotinylation (BioID) in 293 cells by inducing expression of a fusion protein containing NTHL1 fused to the R118G mutant Escherichia coli biotin conjugating enzyme BirA, commonly designated BirA*. The 293 cells have been extensively used for this type of procedure (55). A flow chart of the procedure is presented in Figure 1A. Two negative controls were used in this experiment, BirA*-Flag, a cytoplasmic control, and NLS-BirA*-Flag, a nuclear control. Immediately after adding biotin to the medium, cells were submitted or not to 2 Gy ionizing radiation. Radiation causes DNA damage through direct ionization of DNA and indirectly, through ionization of water to produce hydroxyl radicals that react with DNA to produce single-strand breaks, abasic sites and oxidized DNA base lesions (56). We reasoned that it was likely that the radiation treatment would increase the number of proteins that come in contact with NTHL1. Ectopic expression of the fusion proteins was verified by immunoblotting with NTHL1 and FLAG antibodies (Figure 1B), while biotinylation of cellular proteins following addition of biotin to the medium was confirmed by immunoblotting for streptavidin (Figure 1C). All protein candidates identified in mass spectrometry were given a Bayesian false discovery rate (BFDR), defined as the expected proportion of false positives. After eliminating all candidates with a BFDR greater than 0.2, 223 preys remained: 41 unique to unirradiated cells, 36 unique to irradiated cells, and 146 common to both (Figure 1D). Supplementary Table S1 provides a list of all retained preys together with their BFDR in unirradiated and irradiated cells. The functional enrichment analysis of NTHL1 preys identified in unirradiated and irradiated cells is shown in Supplementary Tables S2 and S3. A literature review of all proteins suspected to function as transcription factors brought our attention to BCL11A because of its reported implication in triple-negative breast cancers and its suspected cooperation with the RAS pathway (36,37,57,58). As a first approach to verify whether these two proteins interact with each other, we performed co-immunoprecipitation assays with endogenous proteins from MDA-MB-231 triple-negative breast cancer cells. Immunoprecipitation with the NTHL1 antibody was able to bring down the BCL11A protein (Figure 1E, top panel, lane 3); while immunoprecipitation with the BCL11A antibody brought down the NTHL1 protein (Figure 1E, bottom panel, lane 4). As a complementary approach to confirm the interaction between NTHL1 and BCL11A, we took advantage of the recently developed method of Split Intein-Mediated Protein Ligation, whereby fusion proteins that respectively carry the N- and C-terminal portion of a modified Intein protein can recombine to produce a third fusion protein (see diagram in Supplementary Figure S1A) (59). Co-expression of BCL11A-V5-IN with IC2-FLAG-NTHL1 led to the production of a third fusion protein, BCL11A-V5-FLAG-NTHL1 that can be detected with both the FLAG and V5 antibodies (Figure 1F, lane 3). In contrast, IC2-Flag-NTHL1 did not recombine with CCNA2-V5-IN4b (Supplementary Figure S1B).
Figure 1.

Proximity Biotinylation with NTHL1-BirA*. (A) Flow chart of manipulations for the BioID experiment. Following treatment with tetracycline and addition of biotin to the medium, cells were treated (2 Gy) or not (0 Gy) with ionizing radiation prior to cell lysis, affinity purification on streptavidin beads and mass spectrometry identification. (B) Immunoblotting analysis of Flp-In™ T-Rex™ 293 cells that were engineered to stably carry vectors that express BirA*-FLAG or NTHL1-BirA*-FLAG upon tetracycline induction. (C) Immunoblotting analysis with streptavidin confirming the expression of BirA* fusion proteins and successful biotinylation by BirA*. (D) The Venn diagram shows the number of NTHL1-BirA* target proteins identified from mass spectrometry after removing all candidates with BFDR above 0.2, and proteins identified in cells expressing BirA*-FLAG and NLS-BirA*-FLAG. 146 preys were identified in both unirradiated and irradiated cells, while 41 and 36 preys were respectively identified only in unirradiated or irradiated cells. (E) Nuclear protein extracts (500 μg) from MDA-MB-231cells were submitted to immunoprecipitation (IP) alternatively with NTHL1 or BCL11A antibodies and then immunoblotted (IB) with either anti-NTHL1 or anti-BCL11A antibody. The control lanes did not include a primary antibody while the input was loaded directly onto the gel. (F) 293 cells were transfected with vectors expressing BCL11A and NTHL1 fusion proteins containing the N-terminal or C-terminal portion of intein, respectively: BCL11A-V5-IN and IC2-FLAG-NTHL1. Whole cell extracts were submitted to immunoblotting analysis with the V5 and FLAG antibodies. The new band detected by both antibodies represents the recombined protein.
BCL11A knockdown causes an increase in genomic DNA damage and a delay in the repair of oxidized bases and abasic sites
Using single-cell gel electrophoresis (comet assay), we monitored the effect of BCL11A knockdown on genomic DNA damage and DNA repair in the MDA-MB-231 breast cancer cells. We observed a significant increase in comet tail moment following BCL11A knockdown (Figure 2A, non-treated cells). To verify the impact of BCL11A knockdown on DNA repair, cells were treated with 50 μM H2O2 and allowed to recover for various periods of time. We observed a drastic increase in comet tail moment immediately after treatment (0 min), and a gradual decrease during recovery (Figure 2A, compare 0 min with 45, 60 and 90 min). Importantly, DNA repair was significantly delayed following BCL11A knockdown. BCL11A knockdown with a different shRNA confirmed these results in MDA-MB-231 cells as well as in another triple-negative breast cancer cell line, MDA-MB-436 (Supplementary Figure S2A and B). Next, we verified whether BCL11A knockdown impacts specifically on the repair of oxidized bases, the repair of which is initiated by NTHL1. To do this, we performed comet assays at pH 10 following treatment of cells with the E. coli Endo III DNA glycosylase, which removes oxidized bases. In alkaline conditions (pH > 13), the comet assay detects many types of DNA damage including double-strand and single-strand breaks (DSBs and SSBs), abasic sites and several types of altered bases that are intrinsically labile at high pH. Comet assay performed at pH 10 only detects DSBs and SSBs; however, pre-treatment with a DNA glycosylase allows the detection of a specific type of altered base, as shown in our previous publications (26–28,32,60). In particular, the E. coli NTH DNA glycosylase allows the detection of a variety of oxidized pyrimidines such as thymine glycol, 5-hydroxycytosine, 5-hydroxyuracil (61). However, it should be noted that this enzyme can also cleave abasic sites and Fapy-purine residues. BCL11A knockdown caused an increase in the level of oxidized bases in non-treated cells, and a delay in the repair of these bases following treatment with H2O2 (Figure 2B, right panel). We conclude that BCL11A is required for efficient repair of oxidized bases in these breast cancer cells.
Figure 2.

BCL11A knockdown causes an increase in genomic DNA damage and a delay in the repair of oxidized bases and abasic sites. MDA-MB-231 cells were infected with lentiviral vectors expressing a BCL11A shRNA or a non-targeting sequence. Knockdown of BCL11A is ∼60%. (A) Cells were exposed to 50 μM H2O2 for 20 min and allowed to recover for the indicated times before carrying out single cell gel electrophoresis at pH > 13. Comet tail moments were scored for at least 100 cells per condition. Results are a representative of one of three independent experiments. Error bars represent standard error. ***P< 0.001; **P< 0.01.; *P< 0.05; Student's t-test. (B) Cells were exposed to 50 μM H2O2 for 20 min and allowed to recover for the indicated time before carrying out single cell gel electrophoresis at pH 10 and pH 10 after treatment of cells with the Endo III DNA glycosylase. Comet tail moments were scored for at least 100 cells per condition. Results are a representative of one of three independent experiments. Error bars represent standard error. ***P< 0.001; **P< 0.01; *P< 0.05; Student's t-test. (C) Cells stably carrying a lentivirus expressing an shRNA against BCL11A or an empty vector were treated or not with 50 μM H2O2. Genomic DNA was purified and abasic sites were quantified using an aldehyde-reactive probe. Results are an average of three independent experiments. Error bars represent standard error. ***P< 0.001; **P< 0.01.; *P< 0.05; Student's t-test.
Measurement of abasic sites revealed that BCL11A knockdown also causes an increase in the number of abasic sites in the genome of MDA-MB-231 cells (Figure 2C, no treatment), and a delay in the repair of abasic sites following treatment with H2O2 (Figure 2C, H2O2 + 45 min). These results suggest that BCL11A knockdown causes a defect in a downstream step of base excision repair. The defects in base excision repair caused by BCL11A knockdown were associated with a decrease in clonogenic efficiency and proliferation potential (Supplementary Figure S2C, 0 μM H2O2 and 2D, CFSE assay). Importantly, BCL11A knockdown did not cause a significant decrease in the expression of NTHL1 nor Pol β, while expression of MDM2, a previously characterized transcriptional target of BCL11A, was markedly decreased (Supplementary Figure S2E) (36).
BCL11A stimulates the enzymatic activities of NTHL1 in vitro
The diagram in Figure 3A shows the longest BCL11A isoform, BCL11A-XL. To verify the effect of BCL11A-XL on the glycosylase and AP-lyase activities of the NTHL1 DNA glycosylase, we performed cleavage assays with purified proteins and two types of probes each containing an oxidized pyrimidine residue and devoid of a BCL11A binding site. The first probe contained a 5,6-dihydrothymidine (DHT) base at position 6 and a FAM-fluorophore next to a dabcyl quencher on the opposite strand (Figure 3B). As indicated in the diagram, increase in fluorescence depends on both NTHL1 glycosylase and AP/lyase activities. The BCL11A-XL protein stimulated the enzymatic activities of NTHL1 (Figure 3B). The second probe was a 5'-radiolabelled double-stranded oligonucleotide that contains a thymine-glycol (Tg) base. In this case, removal of the altered base by NTHL1 produces an abasic site that is incised in the presence of NaOH, thereby producing a shorter radiolabelled product that can be separated by electrophoresis on a denaturing gel (Figure 3C). In this assay, the glycosylase activity of NTHL1 was stimulated by increasing amounts of the BCL11A-XL protein (Figure 3C, compare lane 2 with lanes 3, 4 and 5).
Structure-function analysis of BCL11A defines a region of BCL11A that interacts with NTHL1 and stimulates its enzymatic activity
We performed structure-function analysis to map the region(s) of BCL11A that are involved in the interaction with NTHL1 and the stimulation of its enzymatic activities. Multiple His-tagged BCL11A fragments and a GST-NTHL1 fusion protein were expressed in bacteria and purified by affinity chromatography (Figure 3A and Supplementary Figure S3A and B). In pull-down assays, we observed a strong interaction between GST-NTHL1 and His-BCL11A161–366 and a weaker interaction with BCL11A370–509 (Figure 3D, lanes 4 and 6). Neither of these fragments was able to stimulate NTHL1 enzymatic activity in the cleavage assay with the FAM-probe that contains a DHT base (Figure 3E). However, the BCL11A160–520 fragment exhibited a strong stimulation of NTHL1 activity (Figure 3E). In this assay, production of a cleaved DNA fragment requires both NTHL1 glycosylase and AP-lyase enzymatic activities. This result was confirmed in the cleavage assay with the radioactively labeled probe that contains a Tg base (Figure 3F, compare lanes 3 and 4). As a further confirmation of these results, incubation in the presence of sodium borohydride showed that the BCL11A160–520 fragment stimulates formation of a Schiff base between NTHL1 and the radioactively labeled probe (Figure 3G, lanes 6–8). Of note, as the C-terminal zinc fingers are required for DNA binding, the BCL11A160–520 fragment is unable to bind to DNA containing the optimal BCL11A binding site (Supplementary Figure S3C: C-terminal fragment, lanes 6 and 13; BCL11A160–520, lanes 7 and 14). Pull-down assays with a His- BCL11A161-366 fragment and various GST-tagged NTHL1 peptides showed no interaction with a peptide containing amino acids 88–312 of NTHL1, however, we observed an interaction with a peptide containing the N-terminal amino acids 1–116 (Figure 3H). We conclude that BCL11A interacts with the N-terminal portion of NTHL1.
BCL11A interacts with Pol β
The increase in the number of genomic abasic sites following BCL11A knockdown (Figure 2C) suggested that BCL11A may act on a downstream BER step. Co-immunoprecipitation assays using nuclear extracts from MDA-MB-231 cells show that the BCL11A antibody is able to bring down the Pol β protein (Figure 4A, IB: Pol β, lane 2). In turn, the Pol β antibody was able to bring down BCL11A (Figure 4, IB: BCL11A, lane 2). As an alternative approach to verify the interaction between BCL11A and Pol β, we performed the split intein assay (Supplementary Figure S1A). Co-expression of BCL11A160–520-V5-IN with IC2-FLAG-Pol β led to the production of a third fusion protein, BCL11A160–520-V5-FLAG-Pol β, that is detected with both the V5 and FLAG antibodies (Figure 4B, lane 6). As controls for the specificity of split intein-detected interactions, we observed no recombination between BCL11A160–520-V5-FLAG and IC2-FLAG-LIG3 nor IC2-FLAG-FOXN2 (Figure 4B, lanes 2 and 4). Likewise, IC2-FLAG-Pol β did not recombine with CCNB1-V5-IN4B nor CCNE1-V5-IN4b (Supplementary Figure S1C, lanes 3 and 6). In pull-down assays, we observe an interaction between GST-Pol β and the His-tagged BCL11A161-366 protein, and a very weak signal with His-tagged BCL11A370-509 protein (Figure 4C, lanes 4 and 6).
BCL11A stimulates Pol β enzymatic activities
We performed a series of in vitro DNA repair assays to monitor whether BCL11A is able to stimulate the enzymatic activities of Pol β. Using a FAM fluorophore-based probe containing a nick, we observed that BCL11A-XL stimulates strand-displacement by Pol β (Figure 4D). Using double-stranded oligonucleotides containing an abasic site, we observed that the BCL11A160–520 peptide was able to stimulate incorporation of 32P-dCTP in a concentration-dependent manner (Figure 4E, compare lane 2 with lanes 3, 4 and 5). This assay was repeated using the same double-stranded oligonucleotides but this time it was labeled at its 5' end and the four tri-phosphate deoxynucleotides (dNTPs) were added (Figure 4F). As controls, the reactions were performed in the presence of either BSA or HOXB3. The BCL11A160–520 peptide stimulated the addition of several nucleotides by Pol β (Figure 4F, compare lane 5 with lanes 4 and 6, and lane 8 with lanes 7 and 9). Together, the results from the in vitro repair assays with purified proteins indicate that BCL11A-XL and the BCL11A160–520 peptide stimulate the polymerase activity of Pol β.
Another important enzymatic activity of Pol β involves the conversion of a 5′-deoxyribose phosphate (dRP) into a 5′-phosphate (P) to enable the final ligation step. The dRP-lyase activity of Pol β was previously shown to represent a rate-limiting step in base excision repair (62,63). We prepared a fluorescently labeled DNA probe containing a single-strand break with a 5′end dRP residue (Figure 4G, diagram). Note that the dRP residue is a labile chemical group, but its conversion into phosphate can be limited by adding NaBH4 to the reaction (Figure 4G, compare lane 9 with NaCl with lane 10 with NaBH4). The probe was incubated with Pol β in the presence of either BSA or BCL11A160–520. Importantly, in the absence of Pol β, we did not observe any conversion of dRP into P (Figure 4G, compare lanes 1, 2, 7 and 8 with lane 10). The dRP-lyase activity of Pol β was stimulated in the presence of BCL11A160–520 at both the 7.5 min and 15 min time points (Figure 4G, compare lane 5 with 6, and lane 3 with 4). From four independent pairs of assays, we calculated an average 1.8 fold (±0.3) stimulation of Pol β dRP-lyase activity by BCL11A160–520.
Ectopic expression of the BCL11A160–520 fragment accelerates DNA repair and increases resistance to oxidative DNA damage
As a first step to verify whether the BCL11A160–520 peptide can stimulate the repair of oxidative DNA damage in cells, the peptide was ectopically expressed in RPE1 cells, which express low levels of BCL11A. We added a nuclear localization signal to the peptide to ensure that it would localize to the nucleus. Measurement of DNA damage by comet assay revealed a decrease in DNA damage in RPE1 cells expressing BCL11A160–520 as compared to cells harbouring the empty vector (Figure 5A, untreated cells). Moreover, following treatment of cells with H2O2, we observed an acceleration of DNA repair in BCL11A160–520 expressing cells (Figure 5A). In agreement with the comet assay results, clonogenic assays showed that the BCL11A160–520 peptide increased the resistance of RPE1 cells to treatment with 100 μM H2O2 (Figure 5B). In contrast to these results, ectopic expression of a BCL11A protein lacking amino acids 160 to 520, BCL11AΔ160–520, did not reduce genomic DNA damage nor accelerate DNA repair after H2O2 treatment (Supplementary Figure S4A and B).
Figure 5.

Ectopic Expression of BCL11A160–520 Accelerates DNA Repair and Increases Resistance to H2O2. (A) RPE1 cells expressing the BCL11A160–520 peptide were exposed to 100 μM H2O2 for 20 min and allowed to recover for the indicated time before carrying out single cell gel electrophoresis at pH >13. Comet tail moments were scored for at least 100 cells per condition. Results are a representative of one of three different experiments. Error bars represent standard error. ***P< 0.001; **P< 0.01.; *P< 0.05; Student's t-test. (B) RPE1 cells expressing the BCL11A160–520 peptide were treated with 0, 50 or 100 μM H2O2 and submitted to a clonogenic assay. Error bars represent standard error. ***P< 0.001; **P< 0.01; *P< 0.05; Student's t-test.
The DNA repair and proliferation defects caused by BCL11A knockdown are rescued by ectopic expression of the BCL11A160–520 fragment
Using MDA-MB-231 breast cancer cells, we verified whether ectopic expression of the BCL11A160–520 peptide can rescue the DNA repair defect caused by BCL11A knockdown. Comet assays revealed that the increase in DNA damage following BCL11A knockdown was rescued by overexpression of the BCL11A160–520 peptide (Figure 6A, untreated cells). Moreover, following treatment with H2O2, the delay in DNA repair caused by BCL11A knockdown was rescued by the BCL11A160–520 peptide (Figure 6A, recovery after H2O2). In contrast to these results, ectopic expression of the BCL11AΔ160–520 deletion mutant did not rescue the phenotypes caused by BCL11A knockdown but caused a further increase in genomic DNA damage and a slight delay in DNA repair following H2O2 treatment (Supplementary Figure S4C). Rescue of DNA repair by the BCL11A160–520 peptide was confirmed in MDA-MB-436 breast cancer cells (Supplementary Figure S4D). Comet assays at pH 10 following treatment with the Endo III enzyme showed that the BCL11A160–520 peptide rescued the repair of oxidized bases (Figure 6B). In addition, ectopic expression of the BCL11A160–520 peptide was able to prevent the increase in genomic abasic sites resulting from BCL11A knockdown (Figure 6C).
Importantly, expression of NTHL1 and Pol β were not affected by BCL11A knockdown or ectopic expression of BCL11A160–520 (Figure 6C and Supplementary Figures S2E and 5B). RT-PCR analysis further confirmed that BCL11A has no impact on the expression of base excision repair genes and that BCL11A160–520 has no transcriptional activity (Supplementary Figure S5A and B). Notably, expression of MDM2, a reported transcriptional target of BCL11A, was increased following ectopic expression of BCL11A-XL (Supplementary Figure S5A) and reduced by BCL11A knockdown (Supplementary Figure S5B) but was not affected by BCL11A160–520 overexpression (Supplementary Figure S5A and B).
We investigated the effect of BCL11A knockdown on the repair of an oxidized base by cell extracts using double-stranded oligonucleotides containing a thymine glycol base in the presence of 32P-dTTP. As shown in the diagram of Figure 6D, in this assay we expect that NTHL1 present in the extract will remove the oxidized pyrimidine and introduce a single-strand break that will be processed by APE1 to produce a 3'-OH group, and Pol β will add a radiolabeled thymidine and Ligase III will seal the strand-break thereby generating a single-strand that migrates more slowly on a denaturing gel. BCL11A knockdown caused a decrease in thymidine addition and repair completion (Figure 6D, compare lanes 1 and 2), however, ectopic expression of BCL11A160–520 restored the efficiency of both enzymatic reactions (Figure 6D, compare lanes 3 and 4).
In agreement with these results, the decline in clonogenic efficiency caused by BCL11A knockdown was partially rescued by the BCL11A160–520 peptide (Figure 6E). Using the CFSE cell stain assay to investigate cell proliferation, we observed that BCL11A knockdown caused a slight delay in the rate of cell division that was fully rescued by the BCL11A160–520 peptide (Figure 6G). Moreover, the increase in the proportion of senescent cells caused by BCL11A knockdown was also rescued by the BCL11A160–520 peptide (Figure 6F). The significance of these results will be addressed in the discussion.
The impact of BCL11A knockdown on DNA repair, genomic DNA damage and clonogenic efficiency is confirmed in other triple-negative breast cancer cells
Experiments in three other triple-negative breast cancer cell lines confirmed that BCL11A knockdown causes a defect in the DNA repair activity of cell extracts (Figure 7A), an increase in genomic DNA damage (Figure 7B), a decrease in clonogenic efficiency (Figure 7C, 0 μM H2O2), and a decrease in the resistance to H2O2 treatment (Figure 7C, 20 and 50 μM H2O2).
BCL11A cooperates with RAS to transform primary cells and escape senescence
The previously reported cooperation between BCL11A and NF1 inactivation in leukemogenesis raised the possibility that BCL11A could cooperate with RAS oncogene in cellular transformation (37). One obstacle to RAS-transformation is the increased production of reactive oxygen species that causes oxidative DNA damage and ultimately, cellular senescence (64–66). To test whether the DNA repair activity of BCL11A may cooperate with RAS, we performed co-infections into the IMR90 human primary fibroblastic cells to express the HRASG12V oncogene together with BCL11A160–520 or the empty vector (Figure 8A). Co-expression of HRAS with BCL11A160–520 peptide greatly increased the number of colonies in soft agar (Figure 8B). As expected, the HRAS oncogene caused an increase in DNA damage measured by comet assay, but this increase was prevented by co-expression with the BCL11A160–520 fragment (Figure 8C). Importantly, the HRAS oncogene increased the proportion of senescent cells whereas BCL11A160–520 expression was able to tame the increase in senescence as judged by the number of cells exhibiting senescence-associated β-galactosidase activity (Figure 8D and E). These results were confirmed by measuring expression of the senescence markers P16, P21, Il6 and Il8 (Figure 8E). We conclude that the DNA repair function of BCL11A helps RAS-driven cancer cells to avoid cellular senescence and continue to proliferate.
Figure 8.

BCL11A cooperates with RAS to transform primary cells and escape senescence. IMR90 primary fibroblastic cells were infected with retroviruses expressing either HRAS alone or HRAS and BCL11A160–520, as indicated. (A) Total cell extracts were analyzed by immunoblotting with the indicated antibodies. (B) Cells were plated in soft agar 2 days after infection and colonies were counted after 2 weeks Results are an average of three different experiments. Error bars represent standard error. ***P< 0.001; **P< 0.01; *P< 0.05; Student's t-test. (C) IMR90 cells were selected for 5 days with puromycin, cells were then submitted to single cell gel electrophoresis at pH >13. Comet tail moments were scored for at least 100 cells per condition. Error bars represent standard error of three different experiments. ***P< 0.001; **P< 0.01; *P< 0.05; Student's t-test. (D) β-Gal associated senescence was measured 7 days after infection with vectors expressing HRAS or HRAS and BCL11A160–520. Values were normalized to the value of empty vector. Error bars represent standard error of three different experiments. ***P< 0.001; **P< 0.01; *P< 0.05; Student's t-test. (E) Expression of the senescence markers P16, P21, IL6 and IL8 was measured by RT-PCR 5 days following infection of IMR90 cells with vectors expressing either HRAS, BCL11A160–520, HRAS and BCL11A160–520 or nothing. Error bars represent standard error of three different experiments. ***P< 0.001; **P< 0.01; *P< 0.05; Student's t-test.
DISCUSSION
Prior findings demonstrated that CUT domain proteins can stimulate the enzymatic activities of OGG1 but not of other glycosylases of the base excision repair pathway. Hence, we hypothesized that other DNA glycosylases may interact with distinct accessory factors, and therefore aimed to identify accessory factors for NTHL1, the main glycosylase that removes oxidized pyrimidines. Proximity biotinylation with a NTHL1-BirA* protein identified BCL11A, a transcription factor previously characterized as an oncogene. We confirmed that BCL11A interacts with NTHL1 and stimulates its enzymatic activities (Figure 3). In contrast to the situation with CUT domains and OGG1, we did not obtain solid evidence that BCL11A accelerates the binding of NTHL1 to DNA that contains an oxidized base (Supplementary Figure S3D). However, the fact that BCL11A interacts with the N-terminal domain of NTHL1 raises the possibility that it may alleviate the inhibitory effect of this domain as was previously shown for the YB-1 accessory factor (Figure 3F) (67,68). In addition, we found that similarly to CUT domain proteins, BCL11A helps Pol β complete the repair of damaged bases (Figures 4D, E and F; 6D and 7A). In TNBC cells, BCL11A knockdown caused a delay in the repair of oxidative DNA damage and an increase in the number of oxidized bases and abasic sites in genomic DNA (Figures 2B and C; 6A–C and D; 7). The DNA repair defects caused by BCL11A knockdown were rescued by overexpressing BCL11A160–520, a peptide that is devoid of DNA binding and transcriptional activity (Figure 6A–D).
In cancer, elevated BCL11A expression has been reported in triple-negative breast cancers (36,57,58), lung squamous cell carcinoma (69), non-small-cell lung cancer (70,71), acute myeloid leukemias (72,73), natural killer/T-cell lymphoma (74) and laryngeal squamous cell carcinoma (75). In breast cancers, BCL11A is highly expressed in the triple-negative breast cancer subtype (TNBC), while its genomic locus is amplified in up to 38% of basal-like breast cancers (36). BCL11A knockdown in three TNBC cell lines suppresses tumor growth in xenografts (36). In the DMBA-induced tumor model, prior deletion of BCL11A greatly decreases tumor formation, whereas deletion of BCL11A after tumor appearance causes tumor regression, indicating that BCL11A represents a potential therapeutic target (36). A retroviral mutagenesis screen in leukemogenesis revealed a strong cooperation between BCL11A overexpression and loss of the NF1 tumor suppressor gene (37). This observation suggested cooperation between BCL11A and RAS pathway activation since NF1 encodes a GTPase-activating protein that negatively regulates RAS signaling (76–78). Indeed, in the RAS-transformation assay, we observed that BCL11A160–520 cooperates with RAS to transform IMR90 primary fibroblast cells in a soft agar assay (Figure 8B). This was associated with a lower level of DNA damage and a reduction in the number of senescent cells (Figure 8C, D and E). These results indicate that the DNA repair function of BCL11A is required for transformation of cells by a RAS oncogene. Moreover, in the fully transformed MDA-MB-231 cells which carry a KRASG13D oncogene, BCL11A knockdown reduced clonogenic efficiency, slowed down cell proliferation and increased the proportion of senescent cells (Figure 6E–G). Similar effects were observed in three other triple-negative breast cancer cells (Figure 7). We conclude that the DNA repair function of BCL11A is required not only for the establishment of the transformed phenotype but also for the continued survival and proliferation of cancer cells.
The role of BCL11A as an accessory factor stimulating both NTHL1 and Pol β is coherent. Indeed, since DNA glycosylases that recognize oxidized bases are also endowed with an AP-lyase activity, it is important that the resulting single-strand break can be rapidly resolved by BER enzymes acting downstream in the pathway. Earlier studies have previously revealed the importance of maintaining a proper balance in the enzymatic activities of BER enzymes (79). For example, cancer cells can be rendered more susceptible to the killing effect of the mono-alkylating agent temozolomide by increasing expression of methylpurine DNA glycosylase or alternatively, by decreasing Pol β expression. The resulting imbalance in base excision repair leads to the accumulation of cytotoxic single-strand breaks with 5'-deoxyribose phosphate (62,80). In a similar manner, the stimulation of OGG1 or NTHL1 enzymatic activities by accessory factors in cancer cells that exhibit high levels of reactive oxygen species could potentially produce cytotoxic single-strand breaks that are deleterious to cell viability unless the increased glycosylase activity is matched by an increase in Pol β activity.
In their seminal study on the role of BCL11A in triple-negative breast cancers, Khaled et al. reported that BCL11A knockdown reduces clonogenic efficiency in several triple-negative breast cancer cell lines but did not have a significant impact on cell viability or apoptosis (36). Results from the present study are entirely consistent with their results, although it should be pointed out that some of the assays performed in the two studies measure distinct cell properties. For example, Khaled et al. observed no difference in cell viability using an MTS assay, however, this metabolic assay is not likely to reveal the increased presence of senescent cells in a population (36). In turn, in the present study subtle differences in cell proliferation were revealed by the CFSE stain assay because this assay measures the proportion of each generation of cells. Following BCL11A knockdown, most cells had only reached generation 2 after 3 days in culture and no cell had reached generation 4, whereas most control cells had reached generation 3, and some cells were at generation 4 (Figure 6G). Strikingly, ectopic expression of BCL11A160–520 restored the ability to progress to generation 4 and even to generation 5 (Figure 6G). The BCL11A160–520 peptide also prevented the increase in senescent cells caused by BCL11A knockdown (Figure 6F). Since the BCL11A160–520 peptide is devoid of transcription regulatory potential, we conclude that the BCL11A DNA repair functions are needed and sufficient for senescence avoidance and efficient proliferation when cells are plated at high density. However, in the clonogenic assay the BCL11A160–520 peptide only partially rescued the decrease in clonogenic efficiency caused by BCL11A knockdown (Figure 6E). We conclude that BCL11A transcriptional function is also needed for efficient proliferation when cells are plated at a very low density. This conclusion is in line with the evidence from Khaled et al. showing that BCL11A provides critical functions in stem and progenitor cells (36).
We note that the TNBC cells that exhibit a dependence on BCL11A all harbor a mutation that leads to the activation of the phosphoinositide 3-kinase and AKT (PI3K/AKT) pathway: KRASG13D in MDA-MB-231, HRASG12D in Hs578T, PIK3R1Y73* in MDA-MB-436, PTENV274* in BT549, PTENA72fs in MDA-MB-468. Such mutations lead to an increase in reactive oxygen species (ROS) production (81–84), which can cause oxidative DNA damage and ultimately, senescence (65,66,85,86). This has been documented in tissue culture, transgenic mouse models and human premalignant lesions (87–93). Cancer cells can reduce ROS levels by increasing the expression of anti-oxidants (94–96), notably following inactivation of the KEAP1 tumor suppressor gene (97). Results from our group and others suggest another mechanism of cancer cell adaptation to elevated ROS production. A genome-wide RNAi screen to identify synthetic lethal interactions with the KRAS oncogene identified 4 enzymes involved in distinct steps of base excision repair (BER): NEIL2, XRCC1, POLβ, LIG3 (31). These results indicated that RAS-driven cancer cells are significantly more dependent on an efficient BER pathway than are normal cells. The CUX1 gene was also identified in the same screen and later studies showed that CUX1 functions as a BER accessory factor that accelerates the repair of oxidative DNA damage, thereby enabling RAS-driven cancer cells to avoid cellular senescence and continue to proliferate in spite of elevated ROS levels (26). Altogether these findings suggest an alternative mechanism of adaptation to heightened ROS production where cancer cells can adapt to elevated ROS and avoid cellular senescence by increasing their capacity to repair oxidative DNA damage. This can be achieved, at least in part, by increasing the expression of BER accessory factors such as CUX1 and BCL11A.
In addition to BCL11A, only a few transcription factors were reported to function as BER accessory factors. YB-1 was reported to stimulate the enzymatic activities of NTHL1 and NEIL2 (68,98,99), XPG was shown to stimulate NTHL1 excision of thymine glycol residue by increasing its DNA binding (100), HMGB1, the functions of the APE1 and FEN1 endonucleases (101), P53, the enzymatic activity of Pol β (102), and CUX1, CUX2 and SATB1, the glycosylase and AP-lyase activities of OGG1 (26–29,32). In addition, CUX1 was shown to stimulate the enzymatic activities of APE1 and Pol β (30,50). While the list of BER accessory factors is still limited, we suspect that a systematic search for such factors would reveal many more. For example, we speculate that the process of DNA demethylation that occurs during differentiation may involve transcription factors that stimulate the DNA binding or enzymatic activity of DNA repair enzymes such as the ten-eleven translocation (TET) proteins, activation-induced deaminase (AID), apolipoprotein B editing complex (APOBEC) proteins, and thymidine-DNA glycosylase (TDG) (103,104). If this notion was confirmed, it would raise the question of why there should be a need for accessory factors in mammalian cells when there does not seem to be any BER accessory factor in bacteria. We suggest that the need for BER accessory factors may be linked to the large genome size of higher organisms. While surveillance of a large genome could be ensured by simply increasing the number of enzymes, one could see benefit in enrolling the proteins that patrol the genome and are active in the regions that are transcribed and thus need better protection from DNA damage.
Supplementary Material
ACKNOWLEDGEMENTS
Image processing and analysis was performed in the McGill University Advanced BioImaging Facility; flow cytometry experiments at the Flow Cytometry and Cell Sorting Facility.
Contributor Information
Elise Vickridge, Goodman Cancer Institute, McGill University, 1160 Pine Avenue West, Montreal, Québec H3A 1A3, Canada.
Camila C F Faraco, Department of Biochemistry, McGill University, 1160 Pine Avenue West, Montreal, Québec H3A 1A3, Canada.
Payman S Tehrani, Lunenfeld-Tanenbaum Research Institute, Mount Sinai Hospital, Department of Molecular Genetics, University of Toronto, Toronto, Canada.
Zubaidah M Ramdzan, Goodman Cancer Institute, McGill University, 1160 Pine Avenue West, Montreal, Québec H3A 1A3, Canada.
Billel Djerir, Centre de Recherche du Centre Hospitalier Universitaire de Sherbrooke, Faculté de médecine et des sciences de la santé, Université de Sherbrooke, Sherbrooke, Québec, J1K 2R1, Canada.
Hedyeh Rahimian, Department of Biochemistry, McGill University, 1160 Pine Avenue West, Montreal, Québec H3A 1A3, Canada.
Lam Leduy, Goodman Cancer Institute, McGill University, 1160 Pine Avenue West, Montreal, Québec H3A 1A3, Canada.
Alexandre Maréchal, Centre de Recherche du Centre Hospitalier Universitaire de Sherbrooke, Faculté de médecine et des sciences de la santé, Université de Sherbrooke, Sherbrooke, Québec, J1K 2R1, Canada.
Anne-Claude Gingras, Lunenfeld-Tanenbaum Research Institute, Mount Sinai Hospital, Department of Molecular Genetics, University of Toronto, Toronto, Canada.
Alain Nepveu, Goodman Cancer Institute, McGill University, 1160 Pine Avenue West, Montreal, Québec H3A 1A3, Canada; Department of Biochemistry, McGill University, 1160 Pine Avenue West, Montreal, Québec H3A 1A3, Canada; Department of Medicine, McGill University, 1160 Pine Avenue West, Montreal, Québec H3A 1A3, Canada; Department of Oncology, McGill University, 1160 Pine Avenue West, Montreal, Québec H3A 1A3, Canada.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Cancer Online.
FUNDING
Canadian Cancer Society [702996]; National Science and Engineering Council [RGPIN-2016-05155]; Canadian Institutes of Health Research [MOP-391532].
Conflict of interest statement. None declared.
REFERENCES
- 1. Dianov G.L., Hubscher U.. Mammalian base excision repair: the forgotten archangel. Nucleic Acids Res. 2013; 41:3483–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wilson D.M., Bohr V.A.. the mechanics of base excision repair, and its relationship to aging and disease. DNA Repair (Amst.). 2007; 6:544–559. [DOI] [PubMed] [Google Scholar]
- 3. Demple B., Harrison L.. Repair of oxidative damage to DNA: enzymology and biology. Annu. Rev. Biochem. 1994; 63:915–948. [DOI] [PubMed] [Google Scholar]
- 4. Hegde M.L., Hazra T.K., Mitra S.. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008; 18:27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Horton J.K., Prasad R., Hou E., Wilson S.H.. Protection against methylation-induced cytotoxicity by DNA polymerase beta-dependent long patch base excision repair. J. Biol. Chem. 2000; 275:2211–2218. [DOI] [PubMed] [Google Scholar]
- 6. Starcevic D., Dalal S., Sweasy J.B.. is there a link between DNA polymerase beta and cancer?. Cell Cycle. 2004; 3:998–1001. [PubMed] [Google Scholar]
- 7. Cheadle J.P., Sampson J.R.. MUTYH-associated polyposis–from defect in base excision repair to clinical genetic testing. DNA Repair (Amst.). 2007; 6:274–279. [DOI] [PubMed] [Google Scholar]
- 8. Sampson J.R., Dolwani S., Jones S., Eccles D., Ellis A., Evans D.G., Frayling I., Jordan S., Maher E.R., Mak T.et al.. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet. 2003; 362:39–41. [DOI] [PubMed] [Google Scholar]
- 9. Nielsen M., Morreau H., Vasen H.F., Hes F.J.. MUTYH-associated polyposis (MAP). Crit. Rev. Oncol. Hematol. 2011; 79:1–16. [DOI] [PubMed] [Google Scholar]
- 10. Chow E., Thirlwell C., Macrae F., Lipton L.. Colorectal cancer and inherited mutations in base-excision repair. The Lancet. Oncology. 2004; 5:600–606. [DOI] [PubMed] [Google Scholar]
- 11. Hutchcraft M.L., Gallion H.H., Kolesar J.M.. MUTYH as an emerging predictive biomarker in ovarian cancer. Diagnostics (Basel). 2021; 11:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weren R.D., Ligtenberg M.J., Kets C.M., de Voer R.M., Verwiel E.T., Spruijt L., van Zelst-Stams W.A., Jongmans M.C., Gilissen C., Hehir-Kwa J.Y.et al.. a germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat. Genet. 2015; 47:668–671. [DOI] [PubMed] [Google Scholar]
- 13. Dizdaroglu M. Base-excision repair of oxidative DNA damage by DNA glycosylases. Mutat. Res. 2005; 591:45–59. [DOI] [PubMed] [Google Scholar]
- 14. Weren R.D., Ligtenberg M.J., Geurts van Kessel A., De Voer R.M., Hoogerbrugge N., Kuiper R.P.. NTHL1 and MUTYH polyposis syndromes: two sides of the same coin?. J. Pathol. 2018; 244:135–142. [DOI] [PubMed] [Google Scholar]
- 15. Díaz-Gay M., Alexandrov L.B.. Unraveling the genomic landscape of colorectal cancer through mutational signatures. Adv. Cance. Res. 2021; 151:385–424. [DOI] [PubMed] [Google Scholar]
- 16. Srivastava D.K., Husain I., Arteaga C.L., Wilson S.H.. DNA polymerase beta expression differences in selected human tumors and cell lines. Carcinogenesis. 1999; 20:1049–1054. [DOI] [PubMed] [Google Scholar]
- 17. Albertella M.R., Lau A., O’Connor M.J. the overexpression of specialized DNA polymerases in cancer. DNA Repair (Amst.). 2005; 4:583–593. [DOI] [PubMed] [Google Scholar]
- 18. Canitrot Y., Laurent G., Astarie-Dequeker C., Bordier C., Cazaux C., Hoffmann J.S.. Enhanced expression and activity of DNA polymerase beta in chronic myelogenous leukemia. Anticancer Res. 2006; 26:523–525. [PubMed] [Google Scholar]
- 19. Moore D.H., Michael H., Tritt R., Parsons S.H., Kelley M.R.. Alterations in the expression of the DNA repair/redox enzyme APE/ref-1 in epithelial ovarian cancers. Clin. Cancer Res. 2000; 6:602–609. [PubMed] [Google Scholar]
- 20. Kelley M.R., Cheng L., Foster R., Tritt R., Jiang J., Broshears J., Koch M.. Elevated and altered expression of the multifunctional DNA base excision repair and redox enzyme Ape1/ref-1 in prostate cancer. Clin. Cancer Res. 2001; 7:824–830. [PubMed] [Google Scholar]
- 21. Bobola M.S., Blank A., Berger M.S., Stevens B.A., Silber J.R.. Apurinic/apyrimidinic endonuclease activity is elevated in human adult gliomas. Clin. Cancer Res. 2001; 7:3510–3518. [PubMed] [Google Scholar]
- 22. Wang D., Luo M., Kelley M.R.. Human apurinic endonuclease 1 (APE1) expression and prognostic significance in osteosarcoma: enhanced sensitivity of osteosarcoma to DNA damaging agents using silencing RNA APE1 expression inhibition. Mol. Cancer Ther. 2004; 3:679–686. [PubMed] [Google Scholar]
- 23. Santana T., Sa M.C., de Moura Santos E., Galvao H.C., Coletta R.D., Freitas R.A.. DNA base excision repair proteins APE-1 and XRCC-1 are overexpressed in oral tongue squamous cell carcinoma. J. Oral Pathol. Med. 2017; 46:496–503. [DOI] [PubMed] [Google Scholar]
- 24. Sato M., Girard L., Sekine I., Sunaga N., Ramirez R.D., Kamibayashi C., Minna J.D.. Increased expression and no mutation of the Flap endonuclease (FEN1) gene in human lung cancer. Oncogene. 2003; 22:7243–7246. [DOI] [PubMed] [Google Scholar]
- 25. Ramdzan Z.M., Vickridge E., Faraco C.C.F., Nepveu A.. CUT domain proteins in DNA repair and cancer. Cancers (Basel). 2021; 13:2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ramdzan Z.M., Vadnais C., Pal R., Vandal G., Cadieux C., Leduy L., Davoudi S., Hulea L., Yao L., Karnezis A.N.et al.. RAS Transformation Requires CUX1-Dependent Repair of Oxidative DNA Damage. PLoS Biol. 2014; 12:e1001807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pal R., Ramdzan Z.M., Kaur S., Duquette P.M., Marcotte R., Leduy L., Davoudi S., Lamarche-Vane N., Iulianella A., Nepveu A.. CUX2 Functions As an Accessory Factor in the Repair of Oxidative DNA Damage. J. Biol. Chem. 2015; 290:22520–22531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kaur S., Coulombe Y., Ramdzan Z.M., Leduy L., Masson J.Y., Nepveu A.. Special AT-rich Sequence-binding Protein 1 (SATB1) Functions as an Accessory Factor in Base Excision Repair. J. Biol. Chem. 2016; 291:22769–22780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ramdzan Z.M., Ginjala V., Pinder J.B., Chung D., Donovan C.M., Kaur S., Leduy L., Dellaire G., Ganesan S., Nepveu A.. the DNA repair function of CUX1 contributes to radioresistance. Oncotarget. 2017; 8:19021–19038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kaur S., Ramdzan Z.M., Guiot M.C., Li L., Leduy L., Ramotar D., Sabri S., Abdulkarim B., Nepveu A.. CUX1 Stimulates APE1 Enzymatic Activity and Increases the Resistance of Glioblastoma Cells to the Mono-Alkylating Agent, Temozolomide. Neuro-oncol. 2018; 20:484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Luo J., Emanuele M.J., Li D., Creighton C.J., Schlabach M.R., Westbrook T.F., Wong K.K., Elledge S.J.. a genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009; 137:835–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ramdzan Z.M., Pal R., Kaur S., Leduy L., Berube G., Davoudi S., Vadnais C., Nepveu A.. the function of CUX1 in oxidative DNA damage repair is needed to prevent premature senescence of mouse embryo fibroblasts. Oncotarget. 2015; 6:3613–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Satterwhite E., Sonoki T., Willis T.G., Harder L., Nowak R., Arriola E.L., Liu H., Price H.P., Gesk S., Steinemann D.et al.. the BCL11 gene family: involvement of BCL11A in lymphoid malignancies. Blood. 2001; 98:3413–3420. [DOI] [PubMed] [Google Scholar]
- 34. Nakamura T., Yamazaki Y., Saiki Y., Moriyama M., Largaespada D.A., Jenkins N.A., Copeland N.G.. Evi9 encodes a novel zinc finger protein that physically interacts with BCL6, a known human B-cell proto-oncogene product. Mol. Cell. Biol. 2000; 20:3178–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saiki Y., Yamazaki Y., Yoshida M., Katoh O., Nakamura T.. Human EVI9, a homologue of the mouse myeloid leukemia gene, is expressed in the hematopoietic progenitors and down-regulated during myeloid differentiation of HL60 cells. Genomics. 2000; 70:387–391. [DOI] [PubMed] [Google Scholar]
- 36. Khaled W.T., Choon Lee S., Stingl J., Chen X., Raza Ali H., Rueda O.M., Hadi F., Wang J., Yu Y., Chin S.F.et al.. BCL11A is a triple-negative breast cancer gene with critical functions in stem and progenitor cells. Nat. Commun. 2015; 6:5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yin B., Delwel R., Valk P.J., Wallace M.R., Loh M.L., Shannon K.M., Largaespada D.A.. a retroviral mutagenesis screen reveals strong cooperation between Bcl11a overexpression and loss of the Nf1 tumor suppressor gene. Blood. 2009; 113:1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu P., Keller J.R., Ortiz M., Tessarollo L., Rachel R.A., Nakamura T., Jenkins N.A., Copeland N.G.. Bcl11a is essential for normal lymphoid development. Nat. Immunol. 2003; 4:525–532. [DOI] [PubMed] [Google Scholar]
- 39. Avram D., Fields A., Pretty On Top K., Nevrivy D.J., Ishmael J.E., Leid M.. Isolation of a novel family of C(2)H(2) zinc finger proteins implicated in transcriptional repression mediated by chicken ovalbumin upstream promoter transcription factor (COUP-TF) orphan nuclear receptors. J. Biol. Chem. 2000; 275:10315–10322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sankaran V.G., Menne T.F., Xu J., Akie T.E., Lettre G., Van Handel B., Mikkola H.K., Hirschhorn J.N., Cantor A.B., Orkin S.H.. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008; 322:1839–1842. [DOI] [PubMed] [Google Scholar]
- 41. Li S., Teegarden A., Bauer E.M., Choi J., Messaddeq N., Hendrix D.A., Ganguli-Indra G., Leid M., Indra A.K.. Transcription factor CTIP1/BCL11A regulates epidermal differentiation and lipid metabolism during skin development. Sci. 2017; 7:13427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Peiris H., Park S., Louis S., Gu X., Lam J.Y., Asplund O., Ippolito G.C., Bottino R., Groop L., Tucker H.et al.. Discovering human diabetes-risk gene function with genetics and physiological assays. Nat. Commun. 2018; 9:3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shimbo H., Yokoi T., Aida N., Mizuno S., Suzumura H., Nagai J., Ida K., Enomoto Y., Hatano C., Kurosawa K.. Haploinsufficiency of BCL11A associated with cerebellar abnormalities in 2p15p16.1 deletion syndrome. Mol. Genet. Genomic Med. 2017; 5:429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Soblet J., Dimov I., Graf von Kalckreuth C., Cano-Chervel J., Baijot S., Pelc K., Sottiaux M., Vilain C., Smits G., Deconinck N.. BCL11A frameshift mutation associated with dyspraxia and hypotonia affecting the fine, gross, oral, and speech motor systems. Am. J. Med. Genet. A. 2018; 176:201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yoshida M., Nakashima M., Okanishi T., Kanai S., Fujimoto A., Itomi K., Morimoto M., Saitsu H., Kato M., Matsumoto N.et al.. Identification of novel BCL11A variants in patients with epileptic encephalopathy: expanding the phenotypic spectrum. Clin. Genet. 2018; 93:368–373. [DOI] [PubMed] [Google Scholar]
- 46. Simon R., Wiegreffe C., Britsch S.. Bcl11 transcription factors regulate cortical development and function. Front. Mol. Neurosci. 2020; 13:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lambert J.P., Tucholska M., Go C., Knight J.D., Gingras A.C.. Proximity biotinylation and affinity purification are complementary approaches for the interactome mapping of chromatin-associated protein complexes. J. Proteomics. 2015; 118:81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Raudvere U., Kolberg L., Kuzmin I., Arak T., Adler P., Peterson H., Vilo J.. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019; 47:W191–W198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cadieux C., Kedinger V., Yao L., Vadnais C., Drossos M., Paquet M., Nepveu A.. Mouse mammary tumor virus p75 and p110 CUX1 transgenic mice develop mammary tumors of various histologic types. Cancer Res. 2009; 69:7188–7197. [DOI] [PubMed] [Google Scholar]
- 50. Ramdzan Z.M., Vickridge E., Li L., Faraco C.C.F., Djerir B., Leduy L., Marechal A., Nepveu A.. CUT domains stimulate Pol beta enzymatic activities to accelerate completion of base excision repair. J. Mol. Biol. 2021; 433:166806. [DOI] [PubMed] [Google Scholar]
- 51. Li J., Svilar D., McClellan S., Kim J.H., Ahn E.E., Vens C., Wilson D.M. 3rd, Sobol R.W.. DNA repair molecular beacon assay: a platform for real-time functional analysis of cellular DNA repair capacity. Oncotarget. 2018; 9:31719–31743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Svilar D., Vens C., Sobol R.W.. Quantitative, real-time analysis of base excision repair activity in cell lysates utilizing lesion-specific molecular beacons. J. Vis. Exp. 2012; e4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Moon N.S., Berube G., Nepveu A.. CCAAT displacement activity involves Cut repeats 1 and 2, not the Cut homeodomain. J. Biol. Chem. 2000; 275:31325–31334. [DOI] [PubMed] [Google Scholar]
- 54. Liu N., Hargreaves V.V., Zhu Q., Kurland J.V., Hong J., Kim W., Sher F., Macias-Trevino C., Rogers J.M., Kurita R.et al.. Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell. 2018; 173:430–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hesketh G.G., Youn J.Y., Samavarchi-Tehrani P., Raught B., Gingras A.C.. Parallel exploration of interaction space by BioID and affinity purification coupled to mass spectrometry. Methods Mol. Biol. 2017; 1550:115–136. [DOI] [PubMed] [Google Scholar]
- 56. Morgan M.A., Randall K., Kaken T., Lawrence T.S.. DeVita V.T. Jr. Cancer: Principles & Practice of Oncology, 10th edition. 2014; Wolter's Kluwer. [Google Scholar]
- 57. Wang X., Xu Y., Xu K., Chen Y., Xiao X., Guan X.. BCL11A confers cell invasion and migration in androgen receptor-positive triple-negative breast cancer. Oncol. 2020; 19:2916–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Seachrist D.D., Hannigan M.M., Ingles N.N., Webb B.M., Weber-Bonk K.L., Yu P., Bebek G., Singh S., Sizemore S.T., Varadan V.et al.. the transcriptional repressor BCL11A promotes breast cancer metastasis. J. Biol. Chem. 2020; 295:11707–11719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yao Z., Aboualizadeh F., Kroll J., Akula I., Snider J., Lyakisheva A., Tang P., Kotlyar M., Jurisica I., Boxem M.et al.. Split intein-mediated protein ligation for detecting protein-protein interactions and their inhibition. Nat. Commun. 2020; 11:2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vadnais C., Davoudi S., Afshin M., Harada R., Dudley R., Clermont P.L., Drobetsky E., Nepveu A.. CUX1 transcription factor is required for optimal ATM/ATR-mediated responses to DNA damage. Nucleic Acids Res. 2012; 40:4483–4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Collins A.R. Investigating oxidative DNA damage and its repair using the comet assay. Mutat. Res. 2009; 681:24–32. [DOI] [PubMed] [Google Scholar]
- 62. Trivedi R.N., Wang X.H., Jelezcova E., Goellner E.M., Tang J.B., Sobol R.W.. Human methyl purine DNA glycosylase and DNA polymerase beta expression collectively predict sensitivity to temozolomide. Mol. Pharmacol. 2008; 74:505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Prasad R., Beard W.A., Strauss P.R., Wilson S.H.. Human DNA polymerase beta deoxyribose phosphate lyase. Substrate specificity and catalytic mechanism. J. Biol. Chem. 1998; 273:15263–15270. [DOI] [PubMed] [Google Scholar]
- 64. Serrano M., Lin A.W., McCurrach M.E., Beach D., Lowe S.W.. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997; 88:593–602. [DOI] [PubMed] [Google Scholar]
- 65. Weyemi U., Lagente-Chevallier O., Boufraqech M., Prenois F., Courtin F., Caillou B., Talbot M., Dardalhon M., Al Ghuzlan A., Bidart J.M.et al.. ROS-generating NADPH oxidase NOX4 is a critical mediator in oncogenic H-Ras-induced DNA damage and subsequent senescence. Oncogene. 2012; 31:1117–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mitsushita J., Lambeth J.D., Kamata T.. the superoxide-generating oxidase Nox1 is functionally required for Ras oncogene transformation. Cancer Res. 2004; 64:3580–3585. [DOI] [PubMed] [Google Scholar]
- 67. Liu X., Roy R.. Truncation of amino-terminal tail stimulates activity of human endonuclease III (hNTH1). J. Mol. Biol. 2002; 321:265–276. [DOI] [PubMed] [Google Scholar]
- 68. Guay D., Garand C., Reddy S., Schmutte C., Lebel M.. the human endonuclease III enzyme is a relevant target to potentiate cisplatin cytotoxicity in Y-box-binding protein-1 overexpressing tumor cells. Cancer Sci. 2008; 99:762–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lazarus K.A., Hadi F., Zambon E., Bach K., Santolla M.F., Watson J.K., Correia L.L., Das M., Ugur R., Pensa S.et al.. BCL11A interacts with SOX2 to control the expression of epigenetic regulators in lung squamous carcinoma. Nat. Commun. 2018; 9:3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jamal-Hanjani M., Wilson G.A., McGranahan N., Birkbak N.J., Watkins T.B.K., Veeriah S., Shafi S., Johnson D.H., Mitter R., Rosenthal R.et al.. Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med. 2017; 376:2109–2121. [DOI] [PubMed] [Google Scholar]
- 71. Jiang B.Y., Zhang X.C., Su J., Meng W., Yang X.N., Yang J.J., Zhou Q., Chen Z.Y., Chen Z.H., Xie Z.et al.. BCL11A overexpression predicts survival and relapse in non-small cell lung cancer and is modulated by microRNA-30a and gene amplification. Mol. Cancer. 2013; 12:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Xutao G., PengCheng S., Yin L., Huijuan D., Yan W., Haiqing Z., Bing X.. BCL11A and MDR1 expressions have prognostic impact in patients with acute myeloid leukemia treated with chemotherapy. Pharmacogenomics. 2018; 19:343–348. [DOI] [PubMed] [Google Scholar]
- 73. Dong H., Shi P., Zhou Y., Yu Y., Guo X., Yao Y., Liu P., Xu B.. High BCL11A expression in adult acute myeloid leukemia patients predicts a worse clinical outcome. Clin. Lab. 2017; 63:85–90. [DOI] [PubMed] [Google Scholar]
- 74. Shi H., Li C., Feng W., Yue J., Song J., Peng A., Wang H.. BCL11A is oncogenic and predicts poor outcomes in natural killer/T-cell lymphoma. Front. Pharmacol. 2020; 11:820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhou J., Zhou L., Zhang D., Tang W.J., Tang D., Shi X.L., Yang Y., Zhou L., Liu F., Yu Y.et al.. BCL11A promotes the progression of laryngeal squamous cell carcinoma. Front. 2020; 10:375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Martin G.A., Viskochil D., Bollag G., McCabe P.C., Crosier W.J., Haubruck H., Conroy L., Clark R., O’Connell P., Cawthon R.M.et al.. the GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell. 1990; 63:843–849. [DOI] [PubMed] [Google Scholar]
- 77. Basu T.N., Gutmann D.H., Fletcher J.A., Glover T.W., Collins F.S., Downward J.. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature. 1992; 356:713–715. [DOI] [PubMed] [Google Scholar]
- 78. DeClue J.E., Papageorge A.G., Fletcher J.A., Diehl S.R., Ratner N., Vass W.C., Lowy D.R.. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell. 1992; 69:265–273. [DOI] [PubMed] [Google Scholar]
- 79. Svilar D., Goellner E.M., Almeida K.H., Sobol R.W.. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid. Redox Signal. 2011; 14:2491–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Trivedi R.N., Almeida K.H., Fornsaglio J.L., Schamus S., Sobol R.W.. the role of base excision repair in the sensitivity and resistance to temozolomide-mediated cell death. Cancer Res. 2005; 65:6394–6400. [DOI] [PubMed] [Google Scholar]
- 81. Cho H.J., Jeong H.G., Lee J.S., Woo E.R., Hyun J.W., Chung M.H., You H.J.. Oncogenic H-Ras enhances DNA repair through the Ras/phosphatidylinositol 3-kinase/Rac1 pathway in NIH3T3 cells. Evidence for association with reactive oxygen species. J. Biol. Chem. 2002; 277:19358–19366. [DOI] [PubMed] [Google Scholar]
- 82. Kim J.H., Chu S.C., Gramlich J.L., Pride Y.B., Babendreier E., Chauhan D., Salgia R., Podar K., Griffin J.D., Sattler M.. Activation of the PI3K/mTOR pathway by BCR-ABL contributes to increased production of reactive oxygen species. Blood. 2005; 105:1717–1723. [DOI] [PubMed] [Google Scholar]
- 83. Koundouros N., Poulogiannis G.. Phosphoinositide 3-kinase/Akt signaling and redox metabolism in cancer. Front. Oncol. 2018; 8:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lim J.K.M., Leprivier G.. the impact of oncogenic RAS on redox balance and implications for cancer development. Cell Death. Dis. 2019; 10:955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lee A.C., Fenster B.E., Ito H., Takeda K., Bae N.S., Hirai T., Yu Z.X., Ferrans V.J., Howard B.H., Finkel T.. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J. Biol. Chem. 1999; 274:7936–7940. [DOI] [PubMed] [Google Scholar]
- 86. Irani K., Xia Y., Zweier J.L., Sollott S.J., Der C.J., Fearon E.R., Sundaresan M., Finkel T., Goldschmidt-Clermont P.J.. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997; 275:1649–1652. [DOI] [PubMed] [Google Scholar]
- 87. Collado M., Serrano M.. the senescent side of tumor suppression. Cell Cycle (Georgetown, Tex). 2005; 4:1722–1724. [DOI] [PubMed] [Google Scholar]
- 88. Dankort D., Filenova E., Collado M., Serrano M., Jones K., McMahon M.. a new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev. 2007; 21:379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bartkova J., Rezaei N., Liontos M., Karakaidos P., Kletsas D., Issaeva N., Vassiliou L.V., Kolettas E., Niforou K., Zoumpourlis V.C.et al.. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006; 444:633–637. [DOI] [PubMed] [Google Scholar]
- 90. Fujita K., Mondal A.M., Horikawa I., Nguyen G.H., Kumamoto K., Sohn J.J., Bowman E.D., Mathe E.A., Schetter A.J., Pine S.R.et al.. p53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence. Nat. Cell Biol. 2009; 11:1135–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kuilman T., Michaloglou C., Vredeveld L.C., Douma S., van Doorn R., Desmet C.J., Aarden L.A., Mooi W.J., Peeper D.S.. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008; 133:1019–1031. [DOI] [PubMed] [Google Scholar]
- 92. Michaloglou C., Vredeveld L.C., Soengas M.S., Denoyelle C., Kuilman T., van der Horst C.M., Majoor D.M., Shay J.W., Mooi W.J., Peeper D.S.. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005; 436:720–724. [DOI] [PubMed] [Google Scholar]
- 93. Collado M., Serrano M.. Senescence in tumours: evidence from mice and humans. Nat. Rev. Cancer. 2010; 10:51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Young T.W., Mei F.C., Yang G., Thompson-Lanza J.A., Liu J., Cheng X.. Activation of antioxidant pathways in ras-mediated oncogenic transformation of human surface ovarian epithelial cells revealed by functional proteomics and mass spectrometry. Cancer Res. 2004; 64:4577–4584. [DOI] [PubMed] [Google Scholar]
- 95. Trachootham D., Zhou Y., Zhang H., Demizu Y., Chen Z., Pelicano H., Chiao P.J., Achanta G., Arlinghaus R.B., Liu J.et al.. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006; 10:241–252. [DOI] [PubMed] [Google Scholar]
- 96. Trachootham D., Alexandre J., Huang P.. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach?. Nat. Rev. Drug Discov. 2009; 8:579–591. [DOI] [PubMed] [Google Scholar]
- 97. Singh A., Misra V., Thimmulappa R.K., Lee H., Ames S., Hoque M.O., Herman J.G., Baylin S.B., Sidransky D., Gabrielson E.et al.. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006; 3:e420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Das S., Chattopadhyay R., Bhakat K.K., Boldogh I., Kohno K., Prasad R., Wilson S.H., Hazra T.K.. Stimulation of NEIL2-mediated oxidized base excision repair via YB-1 interaction during oxidative stress. J. Biol. Chem. 2007; 282:28474–28484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Marenstein D.R., Ocampo M.T., Chan M.K., Altamirano A., Basu A.K., Boorstein R.J., Cunningham R.P., Teebor G.W.. Stimulation of human endonuclease III by Y box-binding protein 1 (DNA-binding protein B). Interaction between a base excision repair enzyme and a transcription factor. J. Biol. Chem. 2001; 276:21242–21249. [DOI] [PubMed] [Google Scholar]
- 100. Klungland A., Hoss M., Gunz D., Constantinou A., Clarkson S.G., Doetsch P.W., Bolton P.H., Wood R.D., Lindahl T.. Base excision repair of oxidative DNA damage activated by XPG protein. Mol. Cell. 1999; 3:33–42. [DOI] [PubMed] [Google Scholar]
- 101. Prasad R., Liu Y., Deterding L.J., Poltoratsky V.P., Kedar P.S., Horton J.K., Kanno S., Asagoshi K., Hou E.W., Khodyreva S.N.et al.. HMGB1 is a cofactor in mammalian base excision repair. Mol. Cell. 2007; 27:829–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zhou J., Ahn J., Wilson S.H., Prives C.. a role for p53 in base excision repair. EMBO J. 2001; 20:914–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Li C.J. DNA demethylation pathways: recent insights. Genet. Epigenet. 2013; 5:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Jost J.P., Oakeley E.J., Zhu B., Benjamin D., Thiry S., Siegmann M., Jost Y.C.. 5-Methylcytosine DNA glycosylase participates in the genome-wide loss of DNA methylation occurring during mouse myoblast differentiation. Nucleic Acids Res. 2001; 29:4452–4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.