

ABSTRACT
Zika virus (ZIKV) is unusual among flaviviruses in its ability to spread between humans through sexual contact, as well as by mosquitoes. Sexual transmission has the potential to change the epidemiology and geographic range of ZIKV compared to mosquito-borne transmission and potentially could produce distinct clinical manifestations, so it is important to understand the host mechanisms that control susceptibility to sexually transmitted ZIKV. ZIKV replicates poorly in wild-type mice following subcutaneous inoculation, so most ZIKV pathogenesis studies use mice lacking type I interferon (IFN-αβ) signaling (e.g., Ifnar1−/−). We found that wild-type mice support ZIKV replication following intravaginal infection, consistent with prior studies, although the infection remained localized to the lower female reproductive tract. Vaginal ZIKV infection required a high-progesterone state (pregnancy or pretreatment with depot medroxyprogesterone acetate [DMPA]) even in Ifnar1−/− mice that otherwise are highly susceptible to ZIKV infection. Progesterone-mediated susceptibility did not appear to result from a compromised epithelial barrier, blunted antiviral gene induction, or changes in vaginal leukocyte populations, leaving open the mechanism by which progesterone confers susceptibility to vaginal ZIKV infection. DMPA treatment is a key component of mouse vaginal infection models for herpes simplex virus and Chlamydia, but the mechanisms by which DMPA increases susceptibility to those pathogens also remain poorly defined. Understanding how progesterone mediates susceptibility to ZIKV vaginal infection may provide insights into host mechanisms influencing susceptibility to diverse sexually transmitted pathogens.
IMPORTANCE Zika virus (ZIKV) is transmitted by mosquitoes, similar to other flaviviruses. However, ZIKV is unusual among flaviviruses in its ability also to spread through sexual transmission. We found that ZIKV was able to replicate in the vaginas of wild-type mice, even though these mice do not support ZIKV replication by other routes, suggesting that the vagina is particularly susceptible to ZIKV infection. Vaginal susceptibility was dependent on a high-progesterone state, which is a common feature of mouse vaginal infection models for other pathogens, through mechanisms that have remained poorly defined. Understanding how progesterone mediates susceptibility to ZIKV vaginal infection may provide insights into host mechanisms that influence susceptibility to diverse sexually transmitted pathogens.
KEYWORDS: Zika virus, mice, progesterone, sexually transmitted infection, vagina
INTRODUCTION
The unprecedented size of the 2015–2016 Zika virus (ZIKV) pandemic in the Americas, in which millions of people were infected, revealed new disease manifestations and transmission mechanisms, including congenital infection and sexual transmission (1). Flaviviruses are transmitted to humans by arthropod vectors (mosquitoes and ticks), and ZIKV is the first example of a flavivirus that spreads between humans via sexual transmission (2). The first report of ZIKV sexual transmission predates the 2015–2016 epidemic and resulted from ZIKV infection in Africa (3), suggesting that sexual transmission is a general property of ZIKV, rather than a new trait coincident with its emergence in the Americas. The ability of ZIKV to spread via sexual transmission in addition to mosquito-borne transmission expands the geographic range over which ZIKV transmission can occur, could change the epidemiology of ZIKV even in areas with mosquito-borne transmission, and has the potential to produce distinct pathological outcomes if congenital infection occurs via an ascending route rather than a hematogenous transplacental route. Thus, it is important to understand the antiviral mechanisms that ZIKV may encounter in the vagina that are distinct from antiviral mechanisms present on the skin following mosquito inoculation.
Mouse models of ZIKV vaginal infection involve pretreating mice with progestins (synthetic analogs of progesterone, such as depot medroxyprogesterone acetate [DMPA; Depo-Provera]), based on well-established infection models for herpes simplex virus (HSV) and Chlamydia muridarum (4, 5). The mechanism by which progestins make mice susceptible to ZIKV remains unknown but has been hypothesized to be due to a combination of thinned epithelium, infiltrating immune cells susceptible to ZIKV infection, or deficiencies in antiviral signaling due to decreased expression of antiviral sensing genes (6–8). ZIKV replication is restricted by the type I interferon (IFN-αβ) response in mice because ZIKV is unable to antagonize mouse STAT2 (9, 10). Thus, mouse models of ZIKV pathogenesis, including those investigating vaginal infection, typically use mice deficient in IFN-αβ signaling, usually through genetic loss of the IFN-αβ receptor (Ifnar1−/−) alone or in combination with the IFN-γ receptor, or by treatment of wild-type mice with an IFNAR1-blocking monoclonal antibody (7, 11–15).
Here, we show that although wild-type mice largely are resistant to ZIKV infection via footpad inoculation, vaginal inoculation results in productive local ZIKV replication. Although the ability of ZIKV to replicate in the vaginas of wild-type mice was observed in prior studies (8, 14, 16–18), the mechanisms that affect ZIKV susceptibility specifically at this site were not investigated. We found that permissiveness to vaginal ZIKV replication is regulated by progestins in both wild-type and Ifnar1−/− mice, demonstrating that hormonal status is even more important than IFN-αβ signaling in determining susceptibility to vaginal infection and consistent with prior studies using other IFN-αβ-deficient models of ZIKV vaginal infection (7, 15). Pregnancy, a high-progesterone state, also was sufficient to allow vaginal ZIKV infection in wild-type mice. Vaginal replication was not a unique property of ZIKV, as other flaviviruses that generally are restricted in wild-type mice also were able to replicate in the vagina. Progestin-mediated susceptibility did not appear to result from a compromised epithelial barrier, blunted antiviral gene induction, or changes in vaginal leukocyte populations, leaving open the mechanism by which progesterone confers susceptibility to vaginal ZIKV infection.
RESULTS
Wild-type mice support ZIKV replication after intravaginal inoculation.
Mouse models of ZIKV pathogenesis typically employ mice lacking IFN-αβ signaling (e.g., Ifnar1−/−) to achieve robust infection, as wild-type mice sustain only minimal replication following subcutaneous inoculation (11, 19, 20). Accordingly, in seeking to define host mechanisms that control ZIKV infection in the female reproductive tract, we compared ZIKV replication in wild-type and Ifnar1−/− mice following intravaginal inoculation. We pretreated wild-type and Ifnar1−/− mice with depot medroxyprogesterone acetate (DMPA) (a standard component of mouse vaginal infection models for herpes simplex virus [HSV], Chlamydia, and ZIKV) (4–7) and then 5 days later infected them with 1,000 focus-forming units (FFU) of ZIKV (strain H/PF/2013) via intravaginal instillation (Fig. 1). We assessed viral replication in the vagina by collecting vaginal washes 2, 4, 6, and 8 days postinfection (dpi) and measuring ZIKV RNA by reverse transcription-quantitative PCR (qRT-PCR). We found that viral loads increased from 2 through 8 dpi, indicating productive replication in the vagina. In contrast to minimal viral replication observed in wild-type mice after subcutaneous inoculation in the footpad (11, 19, 20), we observed similar ZIKV replication kinetics and RNA burden in the vaginas of wild-type compared to Ifnar1−/− mice, with the only significant difference being higher viral loads in wild-type mice at 2 dpi (Fig. 1A). Although wild-type mice supported ZIKV replication in the vagina, they did not support systemic infection as viral RNA was detected in serum only in Ifnar1−/− mice (Fig. 1B). Likewise, Ifnar1−/− mice supported ascending infection into the upper female reproductive tract (uterus, ovary, and oviduct), whereas ZIKV infection in wild-type mice was restricted to the lower female reproductive tract (vagina, cervix) (Fig. 1A and C). To confirm that the ZIKV RNA we detected in vaginal washes represented replicating virus, we inoculated wild-type mice with either infectious ZIKV or UV-inactivated virus and measured viral RNA in vaginal washes collected 2 through 8 dpi. No ZIKV RNA was detected in vaginal washes from mice inoculated with UV-inactivated virus, further supporting that the viral RNA detected in vaginal washes results from productive infection (Fig. 1D). Altogether, these results show that ZIKV can replicate in the vagina of wild-type mice but that IFN-αβ signaling restricts systemic spread.
FIG 1.
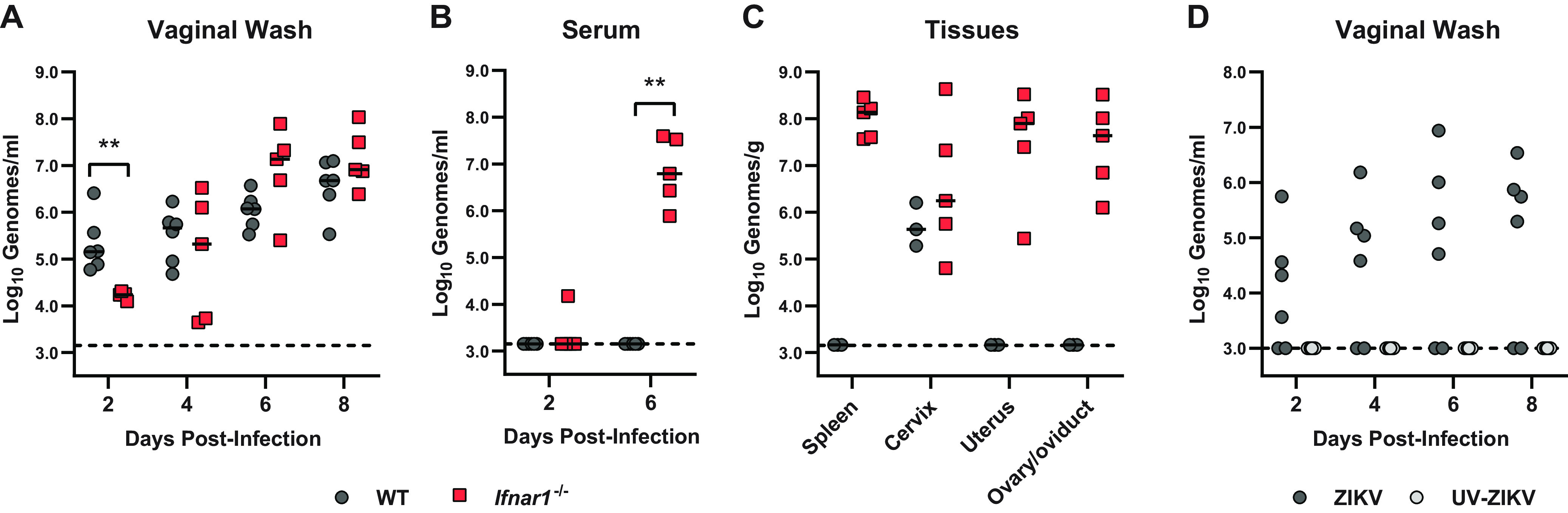
WT mice are susceptible to ZIKV vaginal infection. Six- to 7-week-old mice were pretreated with 2 mg of DMPA and inoculated with 1,000 FFU of ZIKV by intravaginal instillation 5 days later. (A to C) Viral RNA extracted from vaginal washes (A), serum (B), or tissues (C) of wild-type and Ifnar1−/− mice was measured by qRT-PCR. Data represent 5 to 6 (A to B) or 3 to 5 (C) mice per group combined from 2 independent experiments. WT and Ifnar1−/− groups were compared by the Mann-Whitney test with adjustment for multiple comparisons (**, P < 0.01). (D) WT mice were inoculated intravaginally with 1,000 FFU of mock-inactivated or UV-inactivated ZIKV. Viral RNA was extracted from vaginal washes and measured by qRT-PCR. Data represent 6 mice per group combined from 2 independent experiments.
In addition to the antiviral effects of IFN-αβ, type III IFNs (IFN-λ) contribute to antiviral immunity at epithelial barriers (21). IFN-λ has been reported to restrict HSV infection in the vagina (22) and to restrict ZIKV infection in the vagina when IFN-αβ signaling is inhibited by the administration of an IFNAR1-blocking antibody (15). To test whether IFN-λ controls vaginal ZIKV infection in mice with intact IFN-αβ signaling, we used mice lacking the IFN-λ receptor (Ifnlr1−/−). We treated Ifnlr1+/− and Ifnlr1−/− mice with DMPA and infected them with 1,000 FFU of ZIKV by intravaginal instillation. We measured viral loads in vaginal washes by qRT-PCR and found no significant difference between Ifnlr1+/− and Ifnlr1−/− mice, suggesting that IFN-λ signaling does not restrict ZIKV replication in the vagina in this model (Fig. 2).
FIG 2.

IFN-λ does not restrict ZIKV infection in the vagina. Five- to 6-week-old mice lacking (Ifnlr1−/−) or retaining (Ifnlr1+/−) IFN-λ signaling were pretreated with 2 mg of DMPA and inoculated 5 days later with 1,000 FFU of ZIKV by intravaginal instillation. Viral RNA was measured from vaginal washes by qRT-PCR. Ifnlr1−/− and Ifnlr1+/− groups were compared by Mann-Whitney with adjustment for multiple comparisons. Data are combined from 3 independent experiments.
A high-progesterone state is required for vaginal ZIKV infection.
Pretreatment with DMPA is a standard component of mouse models of vaginal infection with diverse pathogens, including HSV, Chlamydia, and ZIKV (4–7). Since we found that wild-type mice were susceptible to ZIKV infection via an intravaginal but not a subcutaneous inoculation route, we considered whether DMPA treatment rendered mice susceptible to systemic ZIKV infection. We treated wild-type mice with DMPA or PBS, and then 5 days later, we infected them with 1,000 FFU of ZIKV via intravaginal instillation or subcutaneous inoculation in the footpad and measured viral RNA in the vaginal wash and the serum by qRT-PCR. As expected, DMPA treatment increased the permissiveness of wild-type mice to intravaginal infection: ZIKV RNA was detected in the vaginal wash from 10 of 10 DMPA-treated mice compared to only 5 of 10 PBS-treated mice (3 of which were positive on only a single day), and DMPA-treated mice sustained higher viral loads in the vagina than PBS-treated mice (Fig. 3A). Consistent with previous experiments, DMPA-treated wild-type mice supported ZIKV replication in the vagina but no ZIKV RNA was detected in the serum following intravaginal inoculation (Fig. 3B). Furthermore, no ZIKV RNA was detected in the serum of mice inoculated by footpad regardless of DMPA treatment (Fig. 3B), indicating that DMPA treatment was not sufficient to render wild-type mice broadly susceptible to ZIKV infection. Although Ifnar1−/− mice are highly susceptible to ZIKV infection by subcutaneous inoculation, productive vaginal infection required DMPA treatment (1 of 10 PBS-treated mice infected compared to 9 of 9 DMPA-treated mice) (Fig. 3C); all Ifnar1−/− mice with productive vaginal infection subsequently sustained viral RNA in the serum (Fig. 3D). These results demonstrate a key role for progesterone in susceptibility to vaginal ZIKV infection, even in the context of immunodeficient mice that are otherwise highly susceptible to ZIKV infection.
FIG 3.
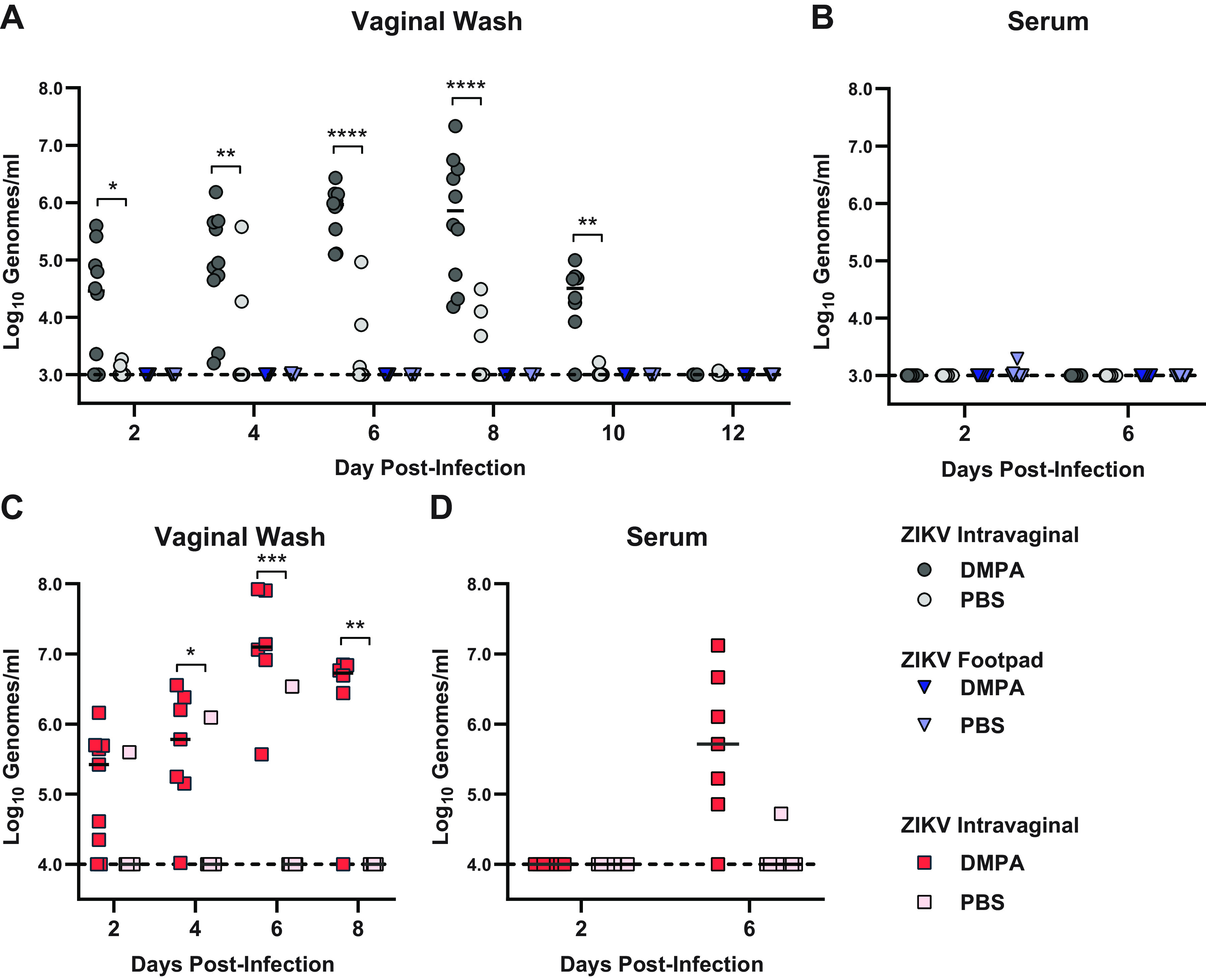
DMPA does not sensitize WT mice to ZIKV infection by footpad inoculation. Six-week-old wild-type (A and B) or Ifnar1−/− mice (C and D) were pretreated with either PBS or 2 mg of DMPA and then infected with 1,000 FFU of ZIKV by intravaginal instillation or subcutaneous inoculation in the footpad. Viral RNA in vaginal washes (A and C) or serum (B and D) was measured by qRT-PCR. Data represent 9 or 10 mice per group combined from 2 independent experiments. PBS and DMPA treated groups were compared by two-way ANOVA with multiple comparison correction (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
Since congenital infection is an important manifestation of ZIKV infection, and pregnancy is a high-progesterone state (23), we evaluated vaginal ZIKV infection in pregnant mice (without DMPA treatment). We mated 7- to 10-week-old wild-type dams with wild-type sires and inoculated them intravaginally with 1,000 FFU of ZIKV at E7. We collected vaginal washes and serum at 2 to 8 dpi and measured ZIKV RNA by qRT-PCR to assess local replication in the vagina and systemic spread, and mice were harvested at 8 dpi to assess congenital infection. A total of 40 fetuses were harvested from 5 pregnant mice; all fetuses had normal morphology and no resorptions were found. Pregnant mice supported vaginal ZIKV replication (viral RNA detected in the vaginal wash from 4 of 5 pregnant mice), but ZIKV RNA was not detected in the vaginal wash of nonpregnant mice (0 of 12 mice) (Fig. 4A). Consistent with our observations in nonpregnant wild-type mice, pregnant wild-type mice did not support systemic ZIKV spread, as ZIKV RNA was not detected in serum, even in the context of robust replication in the vagina (Fig. 4B). Additionally, ZIKV RNA was detected in only 1 of the 40 placentas and none of the corresponding fetuses (Fig. 4C), consistent with the lack of ascending or systemic infection we observed after vaginal ZIKV inoculation in DMPA-treated nonpregnant wild-type mice (Fig. 1B). Altogether these data suggest that a high-progesterone state (DMPA treatment or pregnancy) is required for vaginal permissiveness to ZIKV infection. Furthermore, although vaginal ZIKV infection can cause congenital infection in dams lacking IFN-αβ signaling (7, 13, 14, 24), our data show that viral replication in the vagina is not sufficient for maternal-fetal transmission; additional factors necessary could include maternal viremia or impaired antiviral responses in the uterus or decidua.
FIG 4.

Pregnant WT mice are susceptible to intravaginal ZIKV infection. Seven- to 10-week-old wild-type dams were mated with WT sires and inoculated 7 days afterward intravaginally with 1,000 FFU of ZIKV. Viral RNA was measured by qRT-PCR in vaginal washes (A), serum (B), or fetal tissues harvested at day 8 postinfection (C). Data are combined from 5 pregnant and 12 nonpregnant dams and 40 placentas and fetuses from 2 independent experiments. Pregnant and nonpregnant groups were compared by Mann-Whitney, adjusted for multiple comparisons (**, P < 0.01).
The vagina is permissive to replication of diverse IFN-αβ-restricted flaviviruses.
ZIKV is unique among flaviviruses in its ability to spread among humans via both vector-borne (mosquito) and vector-independent (sexual) transmission routes. To assess whether this reflects an unusual vaginal tropism specific to ZIKV, we evaluated vaginal infection with 4 additional flaviviruses, Spondweni virus (SPOV), dengue virus (DENV-3), Usutu virus (USUV), and Kunjin virus (KUNV). These flaviviruses were selected because, like ZIKV, they replicate poorly in wild-type mice following subcutaneous inoculation (25–28). Wild-type mice were treated with DMPA 5 days before intravaginal inoculation with 1,000 FFU of ZIKV, SPOV, USUV, and KUNV or 10,000 FFU of DENV-3, and viral RNA was measured by qRT-PCR from vaginal washes at 2, 4, and 6 dpi (Fig. 5). Viral RNA was detected in vaginal washes after infection with ZIKV (5 of 5 mice), USUV (7 of 7 mice), and SPOV (3 of 6 mice), suggesting that these viruses could replicate in the vagina of wild-type mice and at levels similar to ZIKV. In contrast, viral RNA was detected in the vagina of only 1 of 8 mice infected with KUNV and 0 of 3 mice infected with DENV-3. To test whether the vagina is permissive to other positive-sense RNA viruses that generally are restricted by innate antiviral responses in wild-type mice (29, 30), we inoculated wild-type mice intravaginally with 1,000 FFU of rubella virus (Matonaviridae) or 5 × 108 genome equivalents of hepatitis A virus (Picornaviridae) but detected no viral RNA in vaginal washes at any of the time points evaluated through 8 dpi (data not shown). Altogether, these data show that vaginal infection is not a unique property of ZIKV among flaviviruses. Rather, in wild-type mice, the vagina is more permissive to flavivirus replication compared to other inoculation sites but does not allow unrestricted replication of all RNA viruses.
FIG 5.

Diverse flaviviruses replicate in the vagina of WT mice. Six-week-old wild-type mice pretreated with 2 mg of DMPA were inoculated with 1,000 FFU of ZIKV, 1,000 FFU of Spondweni virus (SPOV), 10,000 FFU of dengue virus (DENV-3), 1,000 FFU of Usutu virus (USUV), or 1,000 FFU of Kunjin virus (KUNV) by intravaginal instillation. Viral RNA was measured from vaginal washes by qRT-PCR.
ZIKV infection in the vagina is localized to the epithelium.
To better define the location of the cells targeted by ZIKV in the vagina, we treated wild-type mice with DMPA, infected them intravaginally, and detected ZIKV RNA in vaginal tissue using RNAscope in situ hybridization (Fig. 6). ZIKV-positive cells were infrequent and sporadically distributed in the vagina, but they tended to be clusters of adjacent epithelial cells located along the vaginal lumen, with little staining in the parenchyma. We detected ZIKV staining in 0 of 3 mice at 2 dpi, 1 of 2 at 4 dpi, 3 of 3 at 6 dpi, 2 of 2 at 8 dpi, and 0 of 5 at 10 dpi. The largest clusters of infected cells were detected at 6 dpi. There was no tendency for infected cells to be nearer to the cervix or nearer to the vaginal opening. No sections from infected mice exhibited leukocyte infiltrate into the vaginal tissue relative to uninfected DMPA-treated mice. Altogether, these results indicate that ZIKV infection in the vagina primarily targets epithelial cells, rather than the leukocytes that are the main targets of ZIKV systemic infection (31, 32), and that infected cells are not associated with a pronounced immune infiltrate.
FIG 6.

ZIKV targets vaginal epithelial cells. Five- to 6-week-old wild-type mice were treated with 2 mg of DMPA and 5 days later infected with 1,000 FFU ZIKV intravaginally. Vaginal tissue was harvested 2 to 10 dpi and paraffin embedded, and adjacent sections were stained for ZIKV RNA or H&E. Each image is a single field at 20× (scale bar = 100 μm).
A physically compromised vaginal epithelial barrier is not sufficient to render wild-type mice susceptible to ZIKV infection.
DMPA treatment induces a diestrus-like state in mice, including a vaginal epithelium that is thinned and lacks extensive keratinization (7). Since we found that ZIKV infects epithelial cells in the vagina, we hypothesized that a thinned epithelial barrier is more easily targeted by ZIKV, explaining the DMPA requirement for susceptibility of wild-type and Ifnar1−/− mice to vaginal ZIKV infection. To test whether an impaired epithelial barrier could overcome the requirement for DMPA treatment, we abraded the vaginal epithelium of wild-type mice with an interdental brush before intravaginal inoculation with 1,000 FFU of ZIKV and measured ZIKV RNA in vaginal washes by qRT-PCR. However, vaginal infection was only detected in mice that were treated with DMPA, regardless of vaginal abrasion (Fig. 7A), suggesting that a disrupted epithelial barrier is not sufficient for productive ZIKV infection in the vagina. Vaginal abrasion also did not facilitate ZIKV dissemination, as ZIKV RNA was not detected in serum even from abraded mice (Fig. 7B). In DMPA-treated mice, abrasion did not result in higher viral loads in vaginal washes, altogether suggesting that compromised epithelial barrier integrity is not the mechanism by which DMPA treatment promotes vaginal ZIKV infection.
FIG 7.
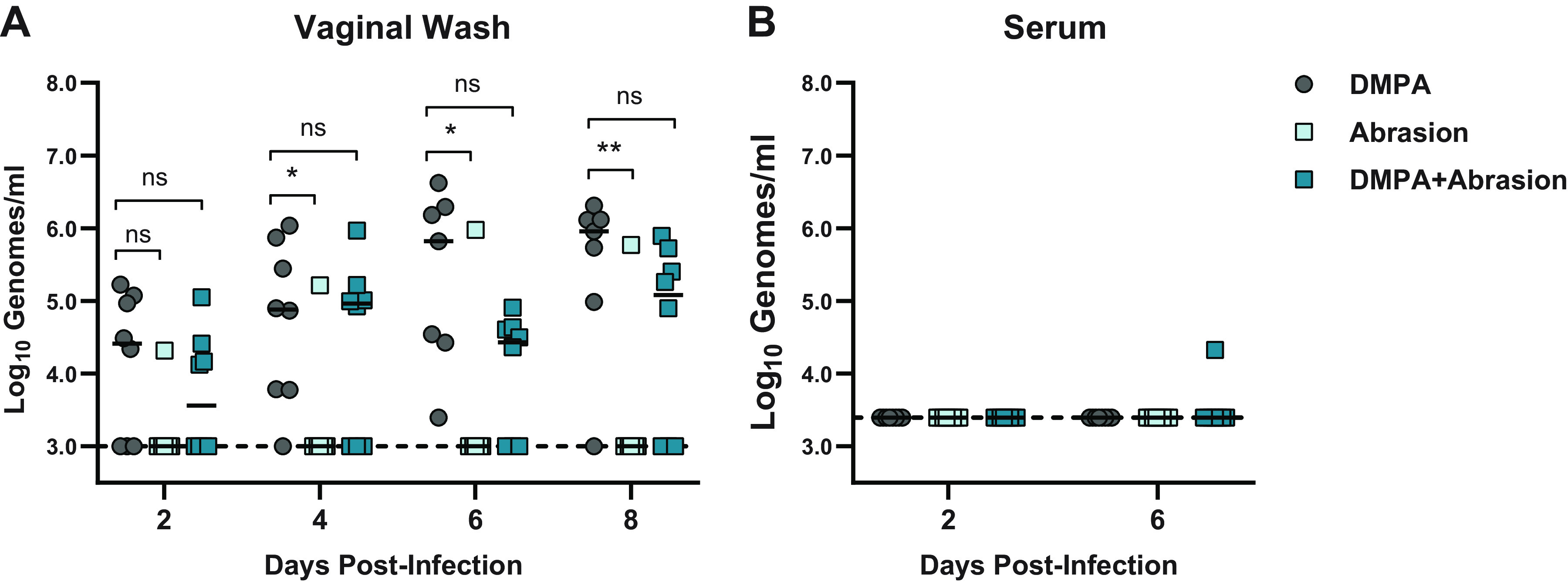
Vaginal abrasion is not sufficient to sensitize WT mice to ZIKV intravaginal infection. Six-week-old wild-type mice were treated with 2 mg of DMPA 5 days before inoculation or vaginally abraded with an interdental brush immediately before inoculation with 1,000 FFU ZIKV via vaginal instillation. Viral RNA in vaginal washes (A) or serum (B) was measured by qRT-PCR. Data represent 8 mice per group combined from 2 independent experiments. Abraded groups were compared to DMPA-only by two-way ANOVA, corrected for multiple comparisons (ns, not significant P > 0.05; *, P < 0.05; **, P < 0.01).
DMPA treatment does not diminish ISG expression or induction.
We next considered whether DMPA treatment might inhibit the basal expression or induction of IFN-stimulated genes (ISGs) in the vagina, thereby permitting ZIKV replication. To test the effect of DMPA treatment on vaginal ISG expression, we treated wild-type mice with DMPA or PBS and then 4 days later administered 50 μg of polyinosinic-polycytidylic acid [poly(I:C)] intravaginally or intraperitoneally. One day after poly(I·C) treatment, we harvested tissues and measured the expression of the canonical ISG Ifit1 by qRT-PCR. We did not observe any DMPA-dependent change in Ifit1 induction in the spleen or lower female reproductive tract (LFRT; vagina and cervix) following intravaginal or intraperitoneal poly(I·C) treatment (Fig. 8A and B). In the upper female reproductive tract (UFRT; uterus and oviduct), DMPA increased Ifit1 expression at baseline and induction in response to intravaginal poly(I·C) treatment (Fig. 8C). DMPA potentially could selectively regulate the expression of some ISGs but not Ifit1 or do so in response to viral infection but not poly(I·C) treatment, but our results do not support a model where DMPA-induced susceptibility to vaginal ZIKV infection is due to a broad inhibition of basal or induced ISG expression.
FIG 8.
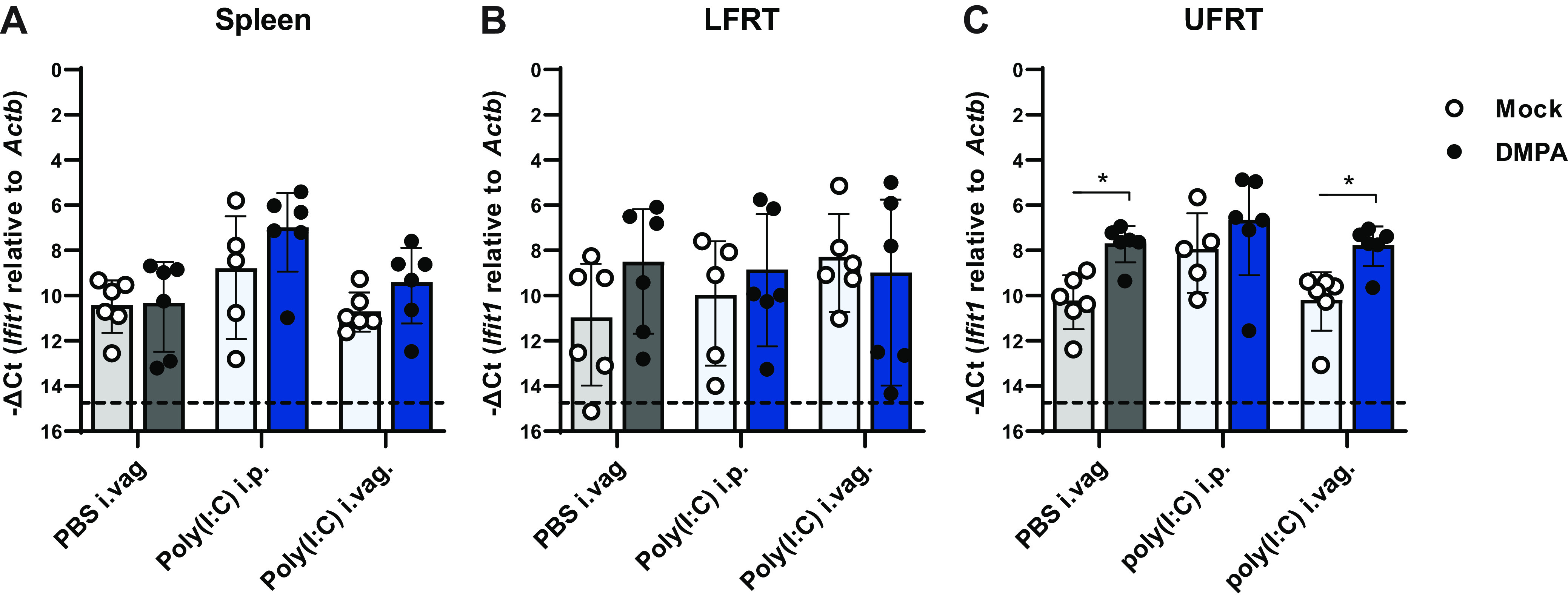
DMPA does not inhibit ISG expression. Five- to 6-week-old wild-type mice were treated with 2 mg of DMPA or PBS (mock). Four days later, mice were treated with 50 μg of poly(I·C) intravaginally (i.vag). or intraperitoneally (i.p.) or PBS i.vag. and tissues were harvested the following day. RNA was extracted from the spleen (A), lower female reproductive tract (LFRT; vagina and cervix) (B), or upper female reproductive tract (UFRT; uterus and oviduct) (C). Ifit1 expression was measured as −ΔCt normalized to Actb. Mock and DMPA-treated groups were compared by Mann-Whitney with adjustment for multiple comparisons (*, P < 0.05).
DMPA treatment does not change vaginal or systemic leukocyte populations.
We next considered whether DMPA might alter leukocyte populations in the vagina or systemically, which could facilitate ZIKV infection by suppressing antiviral immunity or by recruiting susceptible target cells to the site of infection. Wild-type mice were treated with DMPA alone, treated with DMPA then infected with ZIKV intravaginally 5 days later, or left untreated. Six days after ZIKV infection (11 days after DMPA treatment), cells were isolated from the spleen, iliac lymph node, and LFRT. Total cell counts were calculated for T cells, NK cells, B cells, dendritic cells (DCs), eosinophils, monocytes, and neutrophils (markers and gating as defined in methods). ZIKV infection caused an increase in the number of B cells in the spleen and iliac lymph nodes (iLNs) and T cells in the spleen (Fig. 9A and B) and an increase in the number of DCs in the LFRT (Fig. 9C) compared to DMPA treatment alone. However, DMPA treatment alone caused no change in leukocyte populations in any of the tissues analyzed compared to untreated mice. Although DMPA potentially could affect specific leukocyte subsets not analyzed here, or affect activation states independently of cell numbers, our results suggest that DMPA-induced susceptibility to vaginal ZIKV infection does not result from a dramatic change in the immune milieu of the vagina.
FIG 9.
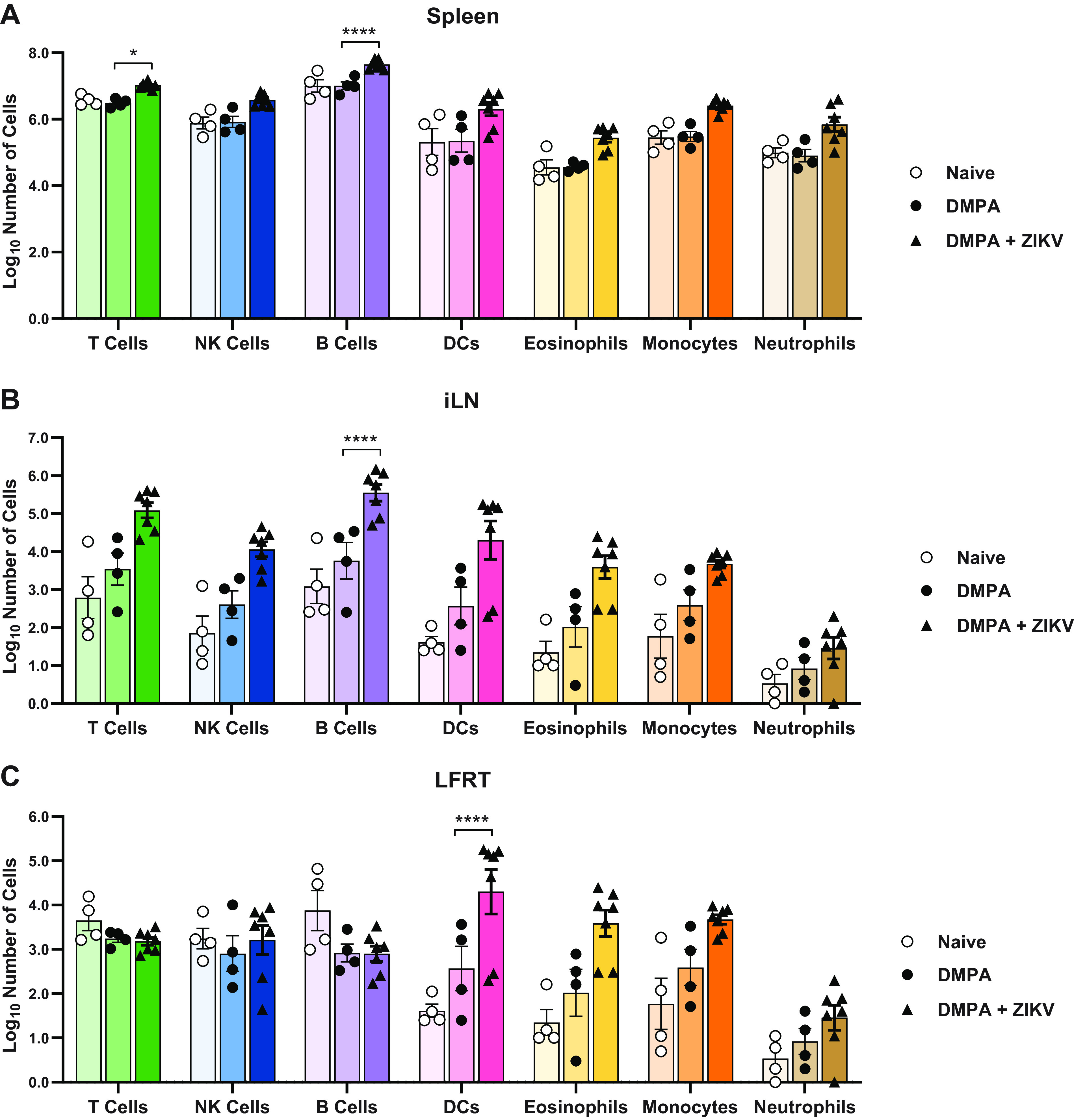
DMPA treatment alone does not impact systemic or vaginal leukocyte populations. Five- to 6-week-old wild-type mice were left untreated, treated with 2 mg of DMPA, or treated with DMPA and 5 days later infected with 1,000 FFU of ZIKV intravaginally. All mice were harvested 6 days after ZIKV infection (11 days after DMPA treatment). Cells were isolated from the spleen, lymph node, and lower female reproductive tract and analyzed by flow cytometry. Total cell counts were calculated for T cells, NK cells, B cells, DCs, eosinophils, monocytes, and neutrophils for the spleen (A), iliac lymph node (B), or lower female reproductive tract (C). Data represent 4 (Naive and DMPA) or 7 (ZIKV) mice per group, combined from 2 independent experiments. Naive and ZIKV-infected groups were compared to DMPA-treated by two-way ANOVA corrected for multiple comparisons (*, P < 0.05; ****, P < 0.00001).
Altogether, our data show that although wild-type mice generally do not support ZIKV replication, the vagina is a unique site that supports the replication of ZIKV as well as certain other flaviviruses. The ability of ZIKV to replicate in the vagina of wild-type mice requires a high-progesterone state (pregnancy or DMPA treatment), but the mechanism by which progesterone promotes ZIKV vaginal infection remains unclear.
DISCUSSION
The emergence of ZIKV in Latin America in 2015–2016 not only revealed new severe disease manifestations but also confirmed a prior report of sexual transmission as an additional mode of transmission for ZIKV, making ZIKV the first arbovirus demonstrated to spread between humans through sexual contact (1, 2). Although most ZIKV cases are presumed to be due to transmission via mosquitoes, it is difficult to estimate the extent to which sexual transmission contributes to ZIKV transmission in areas with frequent and concurrent mosquito-borne transmission. A retrospective study of ZIKV serology in Brazil found that cohabitating with a ZIKV-seropositive sexual partner was associated with a 4-fold greater risk of also being seropositive compared to cohabitating with a ZIKV-seronegative partner, whereas cohabitating with a ZIKV-seropositive nonsexual partner was associated with less than a 2-fold greater risk, supporting a role for sexual transmission even in areas with mosquito-borne transmission (33). Sexual transmission may thus have contributed to the high force of infection of ZIKV in this epidemic even in Latin America where any ZIKV cases were presumed to have been acquired via mosquito.
Sexual transmission among humans appears to be an unusual property of ZIKV compared to other flaviviruses, although the incidence and epidemiology of most flaviviruses preclude certainty about the absence of sexual transmission. The best evidence that ZIKV is sexually transmitted is travel-associated cases in the United States, Europe, and elsewhere, wherein women without mosquito exposure became infected after their male partners returned from areas with ZIKV transmission (2, 34–37). Of 5,399 travel-associated ZIKV cases in the United States from 2015 to 2017, 52 resulted in confirmed transmission to a sexual partner (38, 39). Although this represents only 1% of ZIKV cases in the United States resulting in forward sexual transmission, this is likely an underestimate of the rate at which ZIKV-infected men transmit to their partners, since ~80% of ZIKV infections are asymptomatic and screening has been focused on symptomatic women with travel-related exposure. In contrast, DENV is the most prevalent human flavivirus infection, with an estimated >100 million infections worldwide annually (40), but there have been only two recently described cases of DENV sexual transmission (41, 42) despite tens of thousands of travel-associated DENV cases over the past >40 years (43–50). Our data in mice suggest that the vagina may be a permissive site for replication of other flaviviruses, as we observed replication of other flaviviruses (SPOV and USUV) that do not generally replicate in wild-type mice (25, 26). Since human infections with those flaviviruses are rare (51, 52), it is not known whether they may share with ZIKV the ability to spread through sexual transmission. It may be that there exists a subset of flaviviruses capable of sexual transmission that has not yet been observed because these viruses have not caused a large enough outbreak for sexual transmission to be detected. Sexual transmission would also require these viruses to have tropism for the male reproductive tract as well as secretion into semen. Notably, SPOV has been detected in semen in mice, although it has reduced tropism for the male reproductive tract compared to ZIKV (53). While a lack of DENV-3 replication in the vaginas of wild-type mice was not surprising, since even immunodeficient mice generally are poorly permissive to DENV, the lack of KUNV replication was surprising since we expected this virus to behave similarly to USUV. Altogether, these results suggest that vaginal tropism may vary among even closely related flaviviruses; future studies will assess additional flaviviruses to better define the set of flaviviruses with vaginal tropism and to define viral determinants of this phenotype.
ZIKV pathogenesis often is modeled in Ifnar1−/− mice to produce robust disseminated infection, including via vaginal inoculation. Although others previously have observed productive ZIKV vaginal infection in wild-type mice (8, 14, 16–18), these studies did not specifically investigate the mechanisms that make the vagina an unusually susceptible site for ZIKV replication in wild-type mice. We found that ZIKV replicates efficiently in the vagina of wild-type mice as measured by viral RNA detectable in vaginal washes and cervix. Remarkably, wild-type mice not only supported ZIKV replication in the vagina, but they also sustained equivalent viral loads in the vagina compared to Ifnar1−/− mice throughout the course of infection. However, only Ifnar1−/− mice supported systemic infection. These data suggest different roles for IFN-αβ in controlling local ZIKV replication in the vagina versus the disseminated infection. We did not find increased ZIKV replication in the vagina in mice lacking the IFN-λ receptor, contrasting with a prior study reporting that IFN-λ plays a protective role against ZIKV infection in the female reproductive tract (15). The design of the previous study differed from ours in several respects, including using ovariectomized mice supplemented with hormones, treatment with an IFNAR1-blocking antibody, and use of a mouse-adapted ZIKV strain, suggesting that any protective effect of IFN-λ against vaginal ZIKV infection may be context specific.
Importantly, we found that a high-progesterone state confers susceptibility to vaginal ZIKV infection in both wild-type and Ifnar1−/− mice, including high progesterone induced by pregnancy. It is not clear to what extent sex hormones modulate susceptibility to ZIKV infection in humans, though there is precedent for increased HIV susceptibility following progestin treatment (54). Likewise, progestins increase susceptibility to HSV in mice (6, 55). The observation that pregnancy in mice causes susceptibility to vaginal ZIKV infection could be important because the most significant outcome of ZIKV infection is congenital infection after either mosquito-borne or sexual transmission (56). The ability of ZIKV to spread sexually creates the potential for congenital infection via an ascending transvaginal route, which would require the virus to cross distinct anatomic and immunologic barriers compared to hematogenous transplacental transmission. It is not known whether an alternative route of congenital infection would be associated with distinct risks and outcomes for the developing fetus. Studies in nonhuman primates suggest that ZIKV can spread to the placenta and fetus following intravaginal inoculation, but the animals in these studies also developed viremia so the route by which the virus spread to the placenta and fetus is uncertain (57, 58).
We found that most ZIKV-infected cells in the vagina were epithelial cells and that there did not appear to be a pronounced immune infiltrate present near sites of infection. ZIKV infection in vaginal epithelial cells was previously reported in mice with impaired IFN-αβ signaling (15). The observation that epithelial cells appear to be the cells primarily infected in vaginal tissue is notable because ZIKV has a particular tropism for myeloid cells in systemic infection (31, 32). These data suggest a role for vaginal epithelial cells as mediators of host protection at this site of infection. As pregnant wild-type mice also did not exhibit ascending infection or congenital infection, understanding the mechanisms by which epithelial cells and other cell types restrict ZIKV spread will be important for understanding the risks of sexually transmitted ZIKV in the context of congenital infection.
It previously has been reported that the LFRT expresses lower levels of viral RNA pattern recognition receptors than the UFRT, though this expression pattern was not affected by DMPA treatment (8). Accordingly, we found that DMPA did not inhibit baseline or induced expression of Ifit1, an antiviral ISG, either in the vagina or the spleen in response to poly(I:C). Our results suggest that DMPA does not induce a global downregulation of ISG expression that would promote viral infection.
The mechanism by which progesterone confers susceptibility to vaginal ZIKV infection in wild-type mice remains unclear. High-progesterone states such as DMPA treatment and diestrus are associated with a thinner vaginal epithelium (7, 55, 59), but we found that vaginal abrasion was not sufficient to permit ZIKV infection in the absence of DMPA treatment, so a compromised epithelial barrier is unlikely to be the primary mechanism by which the vagina becomes susceptible to ZIKV infection. The vaginal epithelium becomes more permeable to leukocytes and microbiota following administration of exogenous progesterone, neutrophil abundance in the vagina increases during diestrus, and progesterone can skew the immune response away from a Th1 toward a Th2 response (55, 60, 61). However, we did not observe a significant change in leukocyte populations systemically or in vaginal tissue after DMPA treatment. Although DMPA potentially could affect specific leukocyte subsets not analyzed here, or affect activation states independently of cell numbers, our results suggest that DMPA-induced susceptibility to vaginal ZIKV infection does not result from a dramatic change in the immune milieu of the vagina. The lack of immune cell infiltrate after DMPA treatment is consistent with prior observations that sex hormones alone do not modulate large changes in immune cell profiles within the LFRT in the absence of infection (62).
Altogether, our results demonstrate that the vagina is an unusually permissive site for ZIKV replication in wild-type mice, but this susceptibility is dependent on a high-progesterone state, even in immunocompromised mice. The mechanism by which progesterone confers ZIKV susceptibility remains unclear but could include structural changes to the vaginal lumen or epithelial barrier, local or systemic immunomodulatory effects, or direct effects on viral replication in epithelial cells. DMPA treatment is a key component of mouse vaginal infection models for other sexually transmitted pathogens, such as HSV and Chlamydia, but the mechanisms by which DMPA increases susceptibility to those pathogens also remain poorly defined. Thus, understanding how progesterone mediates susceptibility to ZIKV vaginal infection may provide insights into host mechanisms that influence susceptibility to diverse sexually transmitted pathogens.
MATERIALS AND METHODS
Cells and viruses.
Vero cells were maintained in Dulbecco’s modified Eagle’s Media (DMEM) supplemented with 5% heat-inactivated fetal bovine serum (FBS) and l-glutamine at 37°C with 5% CO2. The ZIKV strain H/PF/2013 was obtained from the U.S. Centers for Disease Control and Prevention (63). The SPOV strain SA AR 94 and USUV strain SA AR 1776 were obtained from the World Reference Center for Emerging Viruses and Arboviruses (64, 65). The DENV-3 WHO reference strain (CH54389) was obtained from Aravinda de Silva (University of North Carolina [UNC]), the KUNV strain FLSDX was from Michael Diamond (Washington University in St. Louis) (28, 66), the RUBV strain M33 was from Michael Rossman (Purdue University) (67), and the liver homogenate was from HAV-infected mice from Stanley Lemon (UNC) (29).
Virus stocks were grown in Vero cells in DMEM supplemented with 2% FBS and HEPES and titered by focus forming assay (FFA) (68). The virus was serially diluted in duplicate in DMEM supplemented with 2% FBS and HEPES and added to confluent Vero cells in 96-well plates for 1 to 3 h at 37°C with 5% CO2 before being overlaid with 1% methylcellulose in minimum essential Eagle’s medium (MEM) supplemented with 2% FBS, HEPES, and penicillin and streptomycin. Cells were then incubated for 40 to 45 h at 37°C with 5% CO2 before being fixed with 2% paraformaldehyde for 1 h at room temperature. Cells were then rinsed with 0.05% Tween 20 in PBS and then incubated for 2 h at room temperature or overnight at 4°C with 1 μg/ml of the flavivirus cross-reactive antibody mE60 (69) in 0.1% saponin and 0.1% bovine serum albumin. Following another rinse, cells were then incubated in a 1:5,000 dilution of a horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Sigma). Titration of RUBV was performed similarly but with a polyclonal anti-RUBV goat IgG at 1:4,000 (LifeSpan BioSciences; LS-C103273) and a HRP-conjugated anti-goat IgG at 1:5,000 (Sigma). Color was developed for 30 min in TrueBlue substrate (KPL). Foci were quantified using a CTL Immunospot.
UV-inactivated ZIKV was generated by placing 0.2 ml ZIKV H/PF/2013 at 1 × 106 FFU/ml in a petri dish and exposing it to UV light at 0.9999 J/cm2 in an HL-2000 HybriLinker (UVP Laboratory Products) for 10 min at room temperature. Mock-inactivated ZIKV was generated similarly but placed under a light in a tissue culture hood instead of UV light. Inactivation was confirmed by amplifying UV- and mock-treated virus stocks on Vero cells for 4 days and then titering by FFA.
Mouse infections.
All mouse husbandry and experiments were performed with the approval of the University of North Carolina at Chapel Hill’s Institutional Animal Care and Use Committee. All mice were on a C57BL/6J background. Ifnar1−/− mice were all bred in-house, and wild-type mice were either bred in-house or purchased from The Jackson Laboratory. Unless otherwise indicated, 5- to 10-week-old female mice were subcutaneously injected with 2 mg depot medroxyprogesterone acetate (DMPA) obtained via the UNC pharmacy, diluted in 100 μl of PBS. Five days later, mice were challenged with 1,000 FFU of virus in 5 μl via vaginal instillation or 50 μl via footpad. Vaginal abrasion was accomplished by scrubbing the vagina of anesthetized mice with interdental brushes (GUM Proxabrush Go-Betweens tight-sized cleaners) for a total of 10 combined full rotations and insertions as previously described (70).
Vaginal washes were collected in a total of 100 μl by twice pipetting 50 μl of PBS with 0.4× protease inhibitor (cOmplete, EDTA-free) into the vagina and collecting immediately, every 2 days after infection. Blood was collected into serum collection tubes (BD) on days 2 and 6 after infection via submandibular bleed with a 5-mm Goldenrod lancet or via terminal bleed cardiac puncture. The serum was separated at 8,000 rpm for 5 min. Tissues were collected from mice after euthanasia by isoflurane overdose, cardiac bleed, and perfusion with 5 to 10 ml of PBS. Tissues, vaginal washes, and serum were stored at −80°C until RNA extraction.
Timed pregnancies were set up by exposing females to soiled male cage bedding for 3 days to promote estrus and then housing single pairs of male and female mice in the late afternoon (E0) and separating males and females the next morning (30). All mated females were infected at E7, at which point their pregnancy status was unknown, and then harvested at E15 (8 dpi).
For experiments investigating the responsiveness of tissues to immunogenic RNA, we first treated 5- to 6-week-old mice with either PBS or 2 mg DMPA subcutaneously. Four days later, mice were treated with 50 μg polyinosinic-polycytidylic acid [poly(I:C)], low molecular weight (Invivogen; TLRL-Picw) either intraperitoneally in 100 μl or intravaginally in 20 μl.
Generation of IFN-λ receptor knockout mice.
Mice with a floxed allele of the IFN-λ receptor (Ifnlr1f/f) were received from Herbert Virgin (Washington University in St. Louis). Ifnlr1f/f mice were crossed with mice expressing Cre recombinase under the β-actin promoter (Jackson Laboratory no. 019099, obtained from Jenny Ting, UNC) to generate Ifnlr1f/f mice with ubiquitous Cre recombinase expression from a hemizygous Cre allele (resulting in Ifnlr1−/−). These mice were then crossed with Ifnlr1f/f mice to generate litters in which 50% of pups lacked IFN-λ signaling (Ifnlr1−/−, Cre+) and 50% retained it (Ifnrl1+/−, Cre−). Vaginal infection experiments were conducted in a blinded manner, as genotyping for Cre and Ifnlr1 was performed after the experiment was completed.
qRT-PCR.
RNA from vaginal washes and serum was extracted with the Qiagen viral RNA minikit. RNA from tissues was extracted with the Qiagen RNeasy minikit after homogenization in a MagNA Lyser instrument (Roche Life Science) with zirconia beads (BioSpec) in 600 μl PBS followed by incubation at room temperature for 10 min in an equal volume RLT buffer for lysis. Viral genomes were quantified by TaqMan one-step qRT-PCR on a CFX96 Touch real-time PCR detection system (Bio-Rad) and were reported on a log10 scale measured against standard curves from either a ZIKV A-plasmid as previously described (71), or from 400 bp gBlock double-stranded DNA fragments (Integrated DNA Technologies [IDT]). ZIKV RNA was quantified as previously published (72), and other viruses were quantified with the indicated primer sets and gBlocks (Table 1 and 2). To measure the expression of Ifit1 in each tissue, the difference in threshold cycle (Ct) values between Ifit1 and ActB as a housekeeping gene was calculated for each tissue sample and plotted as −ΔCt.
TABLE 1.
Primer sets used for qRT-PCR
| Virus or gene target/primer type | Sequence |
|---|---|
| USUV (SA AR 1776) | |
| Forward | TCACAACTAGGAGCATCACAAG |
| Reverse | CCATAGTCACCCATCTTCACAG |
| Probe | TTTACCATCACTCCCAACTCCCCAG |
| SPOV (SA AR 94) | |
| Forward | TGTGCCAATGGTGGGTAAT |
| Reverse | GGAAAGTGGAGCAAACTCAAAG |
| Probe | CGAGATTGACCCTCCGTTTGGTGA |
| DENV3 (CH53489) | |
| Forward | ATTACAGTGCACACAGGAGAC |
| Reverse | CTAGCCCAAGGGTTCCATATTC |
| Probe | TGGGAAATGAAACGCAGGGAGTCA |
| KUNV (FLSDX) | |
| Forward | AGTGGATAGAGGTTGGGGCA |
| Reverse | AGTCCTTCCCGTTGCCTTG |
| Probe | ATCGACACATGCGCCAAATTTGCCTGT |
| RUBV (M33) | |
| Forward | CGAACGAGAAGTGCGGTATATG |
| Reverse | GCGAAACGGCACTGAGAA |
| Probe | ACCTGCTCA/ZEN/ACAAGAATCACACCGA |
| HAV | |
| Forward | GGTAGGCTACGGGTGAAAC |
| Reverse | AACAACTCACCAATATCCGC |
| Probe | AGATGCCTTGGATAGGGTAACAGCG |
| ActB | |
| Forward | GACTCATCGTACTCCTGCTTG |
| Reverse | GATTACTGCTCTGGCTCCTAG |
| Probe | CTGGCCTCACTGTCCACCTTCC |
| Ifit1 | |
| Forward | TGAAGCAGATTCTCCATGACC |
| Reverse | GCAAGAGAGCAGAGAGTCAAG |
| Probe | ACAGCTACCACCTTTACAGCAACCAT |
TABLE 2.
Sequences used for qRT-PCR standard curves
|
Virus (strain) Accession no. |
gBlock sequence |
|---|---|
| USUV (SA AR 1776) AY453412.1 | ACAACTGGGGAGGCCCACAATCCTAAGAGAGCTGAGGACACGTACGTGTGCAAGAGTGGCGTTACTGACAGAGGCTGGGGCAATGGCTGTGGACTATTTGGCAAGGGAAGTATAGACACGTGTGCCAACTTCACCTGCTCCCTGAAAGCGGTGGGCCGAATGATCCAACCGGAAAATGTTAAGTATGAAGTGGGAATCTTCATACATGGTTCCACCAGCTCTGACACTCATGGCAACTATTCTTCACAACTAGGAGCATCACAAGCTGGGCGGTTTACCATCACTCCCAACTCCCCAGCCATCACTGTGAAGATGGGTGACTATGGAGAAATATCAGTTGAGTGTGAACCAAGAAATGGGTTGAACACTGAGGCATACTACATCATGTCAGTGGGCACCA |
| SPOV (SA AR 94) KX227370.1 | TCACCTTCGCTCGCACCCCCTCTGAAACAATTCACGGCACCGCCACAGTGGAGCTGCAATATGCAGGTGAAGATGGGCCGTGCAAAGTTCCCATAGTAATTACCAGTGACACCAATAGCATGGCCTCGACAGGCAGGCTGATCACAGCGAATCCGGTGGTCACGGAAAGTGGAGCAAACTCAAAGATGATGGTCGAGATTGACCCTCCGTTTGGTGATTCTTACATTATTGTGGGCACTGGCACAACAAAAATTACCCACCATTGGCACAGAGCCGGTAGTTCAATTGGACGTGCATTTGAGGCTACCATGAGAGGAGCAAAACGGATGGCGGTCCTCGGCGACACCGCTTGGGACTTTGGCTCTGTTGGGGGCATGTTCAACTCCGTTGGAAAGTTTGTCCACCAGGTGTTTGGATCAGCATTTAAGGCATTGTTTGGAGGCATGTCCTGGTTCACACAGCTCCTGATAGGATTTCT |
| DENV3 (CH53489) DQ863638.1 | CTACGTATGTAAGCATACATACGTGGATAGAGGCTGGGGAAACGGTTGTGGTTTGTTTGGAAAAGGAAGCTTGGTGACATGCGCGAAATTTCAATGCTTAGAATCAATAGAGGGAAAAGTGGTGCAACATGAGAACCTCAAATACACTGTCATCATTACAGTGCACACAGGAGACCAACACCAGGTGGGAAATGAAACGCAGGGAGTCACGGCTGAGATAACACCCCAGGCATCAACCGTTGAAGCTATCTTGCCTGAATATGGAACCCTTGGGCTAGAATGCTCACCACGGACAGGTTTGGATTTCAATGAAATGATCTTATTGACAATGAAGAACAAAGCATGGATGGTACATAGACAATGGTTCTTTGACCTCCCCCTACCATGGACATCAGGAGCT |
| KUNV (FLSDX) AY274504.1 | TACAGCTTTAACTGTCTTGGAATGAGCAACAGAGATTTCCTGGAGGGAGTGTCCGGAGCAACATGGGTGGACTTGGTCCTTGAAGGTGACAGCTGTGTGACCATTATGTCCAAGGACAAGCCCACCATCGATGTGAAGATGATGAACATGGAGGCCGCTAACTTAGCAGAAGTCCGCAGTTATTGCTACTTAGCCACTGTCAGTGAACTCTCCACCAAGGCTGCGTGCCCAACCATGGGGGAAGCCCATAATGACAAGCGGGCTGACCCATCTTTTGTGTGCAAACAAGGAGTAGTGGATAGAGGTTGGGGCAATGGGTGCGGACTTTTTGGTAAAGGAAGCATCGACACATGCGCCAAATTTGCCTGTTCAACCAAGGCAACGGGAAGGACTATCTTAAAGGAGAACATTAAGTATGAGGTGGCTATCTTTGTGCATGGACCAACTACCGTGGAATCGCATGGGAACT |
| RUBV (M33) X72393.1 | CAACCGCGTGACTGAGGGCGAACGAGAAGTGCGGTATATGCGCATCTCGCGTCACCTGCTCAACAAGAATCACACCGAGATGCCCGGAACGGAACGCGTTCTCAGTGCCGTTTCGCCGTGCGGCTACCGCGCG |
| HAV KX343018 | GTTTGGAACGTCACCTTGCAGTGTTAACTTGGCTTTCATGAATCTCTTTGATCTTCCACAAGGGGTAGGCTACGGGTGAAACCTCTTAGGCTAATACTTCTATGAAGAGATGCCTTGGATAGGGTAACAGCGGCGGATATTGGTGAGTTGTTAAGACAAAAACCATTCAACGCCGGAGGACTGACTCTCATCCAGTGGATG |
In situ hybridization.
Tissues were collected from euthanized mice after exsanguination by cardiac puncture and perfusion with 10 ml of PBS followed by 10 ml of 10% neutral buffered formalin (NBF). Tissues were then stored overnight in 1 ml of 10% NBF at 4°C before being transferred to PBS at 4°C for longer-term storage. Tissues were paraffin embedded, and 5-μm sections were stained with a ZIKV-specific RNA probe (Advanced Cell Diagnostics no. 467871) and a hematoxylin counterstain. Positive and negative staining controls for RNA-specific staining were confirmed with probes against peptidyl-prolyl cis-isomerase B (PPIB; no. 321651) and dihydrodicipicolinate reductase (dapB; no. 320751) as recommended by the manufacturer. Tissue processing, histology, and RNAscope were performed by the UNC Histology Research Core Facility.
Flow cytometry.
Spleens and iLNs were mechanically dissociated and red blood cells were lysed using RBC lysis buffer (0.84% NH4Cl in PBS). Cells were pelleted by centrifugation and resuspended in media (RPMI 1640 with 1% FBS). Cells were filtered through a 70-μm cell strainer to make a single-cell suspension. LFRT tissue was excised, minced with scissors, and digested in HBSS (with Ca2+ and Mg2+) containing 1 mg/ml collagenase I and 0.05 mg/ml DNase I for 60 min at 37°C in a shaking incubator. After incubation, 1 ml FBS was added to stop digestion, and cells were serially filtered through a 40- and 70-μm cell strainer and washed with HBSS (with Ca2+ and Mg2+). Cells were resuspended in media at a concentration of 1 × 107 cells/ml for flow cytometric analysis.
Isolated cells were stained in PBS with 1% FBS for 20 to 30 min in the dark on ice. Fc receptor blockade was performed with anti-CD16/32 MAb before surface staining. Dead cells were excluded from analysis using Zombie UV (BioLegend). Cells were fixed in 2% paraformaldehyde, and samples were acquired using an LSRII flow cytometer (BD Biosciences). Data were analyzed using FlowJo software (Tree Star). The following antibodies were used in this study: anti-CD16/32 (clone 2.4G2; BD Biosciences), anti-CD45 AF700 (clone 30-F11; BioLegend), anti-CD3e APC-Fire/750 (clone 17A2; BioLegend), anti-CD19 PE-Cy7 (clone 6D5; BioLegend), anti-NK1.1 PE (clone PK136; BioLegend), anti-CD11b APC (clone M1/70; BioLegend), anti-CD11c BV650 (clone N418; BioLegend), anti-Ly6G FITC (clone IA8; BioLegend), and anti-Ly6C BV605 (clone HK1.4; BioLegend). The following markers were used to identify immune cell populations: T cells (CD45+CD3e+), B cells (CD45+CD19+), NK cells (CD45+NK1.1+), dendritic cells (CD45+CD11c+), neutrophils (CD45+CD11b+Ly6G+), and monocytes (CD45+CD11b+Ly6G-Ly6C+/−).
Statistical analysis.
Statistical tests were performed with GraphPad Prism 9.0. Tests used include unpaired multiple Mann-Whitney analyses with the Holm-Šídák method and two-way ANOVA with matched time points where multiple time points of the same mouse were taken, the Geisser-Greenhouse correction for lack of sphericity, comparison to control cell means, and the Dunnett correction for multiple comparisons.
ACKNOWLEDGMENTS
This work was supported by R21 AI144631 (to H.M.L.) and start-up funds from the UNC Chapel Hill Department of Microbiology and Immunology and the Lineberger Comprehensive Cancer Center. H.M.L. holds an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. C.A.L. was supported by F31 AI143237; and S.J.D. was supported by K12 GM000678. Histology services were provided by the UNC Histology Research Core Facility. The UNC Flow Cytometry Core Facility is supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center.
Contributor Information
Helen M. Lazear, Email: helen.lazear@med.unc.edu.
Rebecca Ellis Dutch, University of Kentucky College of Medicine.
REFERENCES
- 1.Pierson TC, Diamond MS. 2018. The emergence of Zika virus and its new clinical syndromes. Nature 560:573–581. 10.1038/s41586-018-0446-y. [DOI] [PubMed] [Google Scholar]
- 2.Blitvich BJ, Magalhaes T, Laredo-Tiscareno SV, Foy BD. 2020. Sexual transmission of arboviruses: a systematic review. Viruses 12:933. 10.3390/v12090933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Foy BD, Kobylinski KC, Chilson Foy JL, Blitvich BJ, Travassos da Rosa A, Haddow AD, Lanciotti RS, Tesh RB. 2011. Probable non-vector-borne transmission of Zika virus, Colorado, USA. Emerg Infect Dis 17:880–882. 10.3201/eid1705.101939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaushic C, Ashkar AA, Reid LA, Rosenthal KL. 2003. Progesterone increases susceptibility and decreases immune responses to genital herpes infection. J Virol 77:4558–4565. 10.1128/JVI.77.8.4558-4565.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaushic C, Zhou F, Murdin AD, Wira CR. 2000. Effects of estradiol and progesterone on susceptibility and early immune responses to Chlamydia trachomatis infection in the female reproductive tract. Infect Immun 68:4207–4216. 10.1128/IAI.68.7.4207-4216.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gillgrass AE, Fernandez SA, Rosenthal KL, Kaushic C. 2005. Estradiol regulates susceptibility following primary exposure to genital herpes simplex virus type 2, while progesterone induces inflammation. J Virol 79:3107–3116. 10.1128/JVI.79.5.3107-3116.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang WW, Young MP, Mamidi A, Regla-Nava JA, Kim K, Shresta S. 2016. A mouse model of Zika virus sexual transmission and vaginal viral replication. Cell Rep 17:3091–3098. 10.1016/j.celrep.2016.11.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khan S, Lew I, Wu F, Fritts L, Fontaine KA, Tomar S, Trapecar M, Shehata HM, Ott M, Miller CJ, Sanjabi S. 2019. Low expression of RNA sensors impacts Zika virus infection in the lower female reproductive tract. Nat Commun 10:4344. 10.1038/s41467-019-12371-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, Schwarz MC, Sanchez-Seco MP, Evans MJ, Best SM, Garcia-Sastre A. 2016. Zika virus targets human STAT2 to inhibit type i interferon signaling. Cell Host Microbe 19:882–890. 10.1016/j.chom.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tripathi S, Balasubramaniam VR, Brown JA, Mena I, Grant A, Bardina SV, Maringer K, Schwarz MC, Maestre AM, Sourisseau M, Albrecht RA, Krammer F, Evans MJ, Fernandez-Sesma A, Lim JK, Garcia-Sastre A. 2017. A novel Zika virus mouse model reveals strain specific differences in virus pathogenesis and host inflammatory immune responses. PLoS Pathog 13:e1006258. 10.1371/journal.ppat.1006258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, Miner JJ, Diamond MS. 2016. A mouse model of Zika virus pathogenesis. Cell Host Microbe 19:720–730. 10.1016/j.chom.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uraki R, Jurado KA, Hwang J, Szigeti-Buck K, Horvath TL, Iwasaki A, Fikrig E. 2017. Fetal growth restriction caused by sexual transmission of Zika virus in mice. J Infect Dis 215:1720–1724. 10.1093/infdis/jix204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yockey LJ, Jurado KA, Arora N, Millet A, Rakib T, Milano KM, Hastings AK, Fikrig E, Kong Y, Horvath TL, Weatherbee S, Kliman HJ, Coyne CB, Iwasaki A. 2018. Type I interferons instigate fetal demise after Zika virus infection. Sci Immunol 3:eaao1680. 10.1126/sciimmunol.aao1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yockey LJ, Varela L, Rakib T, Khoury-Hanold W, Fink SL, Stutz B, Szigeti-Buck K, Van den Pol A, Lindenbach BD, Horvath TL, Iwasaki A. 2016. Vaginal exposure to Zika virus during pregnancy leads to fetal brain infection. Cell 166:1247–1256.e4. 10.1016/j.cell.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caine EA, Scheaffer SM, Arora N, Zaitsev K, Artyomov MN, Coyne CB, Moley KH, Diamond MS. 2019. Interferon lambda protects the female reproductive tract against Zika virus infection. Nat Commun 10:280. 10.1038/s41467-018-07993-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scott JM, Lebratti TJ, Richner JM, Jiang X, Fernandez E, Zhao H, Fremont DH, Diamond MS, Shin H. 2018. Cellular and humoral immunity protect against vaginal Zika virus infection in mice. J Virol 92:e00038-18. 10.1128/JVI.00038-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khan S, Woodruff EM, Trapecar M, Fontaine KA, Ezaki A, Borbet TC, Ott M, Sanjabi S. 2016. Dampened antiviral immunity to intravaginal exposure to RNA viral pathogens allows enhanced viral replication. J Exp Med 213:2913–2929. 10.1084/jem.20161289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hastings AK, Yockey LJ, Jagger BW, Hwang J, Uraki R, Gaitsch HF, Parnell LA, Cao B, Mysorekar IU, Rothlin CV, Fikrig E, Diamond MS, Iwasaki A. 2017. TAM receptors are not required for Zika virus infection in mice. Cell Rep 19:558–568. 10.1016/j.celrep.2017.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Winkler CW, Myers LM, Woods TA, Messer RJ, Carmody AB, McNally KL, Scott DP, Hasenkrug KJ, Best SM, Peterson KE. 2017. Adaptive immune responses to Zika virus are important for controlling virus infection and preventing infection in brain and testes. J Immunol 198:3526–3535. 10.4049/jimmunol.1601949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith DR, Hollidge B, Daye S, Zeng X, Blancett C, Kuszpit K, Bocan T, Koehler JW, Coyne S, Minogue T, Kenny T, Chi X, Yim S, Miller L, Schmaljohn C, Bavari S, Golden JW. 2017. Neuropathogenesis of Zika virus in a highly susceptible immunocompetent mouse model after antibody blockade of type i interferon. PLoS Negl Trop Dis 11:e0005296. 10.1371/journal.pntd.0005296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lazear HM, Schoggins JW, Diamond MS. 2019. Shared and distinct functions of type I and type III interferons. Immunity 50:907–923. 10.1016/j.immuni.2019.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ank N, West H, Bartholdy C, Eriksson K, Thomsen AR, Paludan SR. 2006. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J Virol 80:4501–4509. 10.1128/JVI.80.9.4501-4509.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holinka CF, Tseng YC, Finch CE. 1979. Reproductive aging in C57BL/6J mice: plasma progesterone, viable embryos and resorption frequency throughout pregnancy. Biol Reprod 20:1201–1211. 10.1095/biolreprod20.5.1201. [DOI] [PubMed] [Google Scholar]
- 24.Duggal NK, McDonald EM, Ritter JM, Brault AC. 2018. Sexual transmission of Zika virus enhances in utero transmission in a mouse model. Sci Rep 8:4510. 10.1038/s41598-018-22840-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salazar V, Jagger BW, Mongkolsapaya J, Burgomaster KE, Dejnirattisai W, Winkler ES, Fernandez E, Nelson CA, Fremont DH, Pierson TC, Crowe JE, Jr, Screaton GR, Diamond MS. 2019. Dengue and Zika virus cross-reactive human monoclonal antibodies protect against Spondweni virus infection and pathogenesis in mice. Cell Rep 26:1585–1597.e4. 10.1016/j.celrep.2019.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blazquez AB, Escribano-Romero E, Martin-Acebes MA, Petrovic T, Saiz JC. 2015. Limited susceptibility of mice to Usutu virus (USUV) infection and induction of flavivirus cross-protective immunity. Virology 482:67–71. 10.1016/j.virol.2015.03.020. [DOI] [PubMed] [Google Scholar]
- 27.Chen RE, Diamond MS. 2020. Dengue mouse models for evaluating pathogenesis and countermeasures. Curr Opin Virol 43:50–58. 10.1016/j.coviro.2020.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daffis S, Lazear HM, Liu WJ, Audsley M, Engle M, Khromykh AA, Diamond MS. 2011. The naturally attenuated Kunjin strain of West Nile virus shows enhanced sensitivity to the host type I interferon response. J Virol 85:5664–5668. 10.1128/JVI.00232-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirai-Yuki A, Hensley L, McGivern DR, Gonzalez-Lopez O, Das A, Feng H, Sun L, Wilson JE, Hu F, Feng Z, Lovell W, Misumi I, Ting JP, Montgomery S, Cullen J, Whitmire JK, Lemon SM. 2016. MAVS-dependent host species range and pathogenicity of human hepatitis A virus. Science 353:1541–1545. 10.1126/science.aaf8325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Casazza RL, Philip DT, Lazear HM. 2022. Interferon lambda signals in maternal tissues to exert protective and pathogenic effects in a gestational stage-dependent manner. mBio 13:e0385721. 10.1128/mbio.03857-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Michlmayr D, Andrade P, Gonzalez K, Balmaseda A, Harris E. 2017. CD14(+)CD16(+) monocytes are the main target of Zika virus infection in peripheral blood mononuclear cells in a paediatric study in Nicaragua. Nat Microbiol 2:1462–1470. 10.1038/s41564-017-0035-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McDonald EM, Anderson J, Wilusz J, Ebel GD, Brault AC. 2020. Zika virus replication in myeloid cells during acute infection is vital to viral dissemination and pathogenesis in a mouse model. J Virol 94:e00838-20. 10.1128/JVI.00838-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Magalhaes T, Morais CNL, Jacques I, Azevedo EAN, Brito AM, Lima PV, Carvalho GMM, Lima ARS, Castanha PMS, Cordeiro MT, Oliveira ALS, Jaenisch T, Lamb MM, Marques ETA, Foy BD. 2021. Follow-Up household serosurvey in Northeast Brazil for Zika virus: sexual contacts of index patients have the highest risk for seropositivity. J Infect Dis 223:673–685. 10.1093/infdis/jiaa563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hills SL, Russell K, Hennessey M, Williams C, Oster AM, Fischer M, Mead P. 2016. Transmission of Zika virus through sexual contact with travelers to areas of ongoing transmission–continental United States, 2016. MMWR Morb Mortal Wkly Rep 65:215–216. 10.15585/mmwr.mm6508e2. [DOI] [PubMed] [Google Scholar]
- 35.D'Ortenzio E, Matheron S, Yazdanpanah Y, de Lamballerie X, Hubert B, Piorkowski G, Maquart M, Descamps D, Damond F, Leparc-Goffart I. 2016. Evidence of sexual transmission of Zika virus. N Engl J Med 374:2195–2198. 10.1056/NEJMc1604449. [DOI] [PubMed] [Google Scholar]
- 36.Armstrong P, Hennessey M, Adams M, Cherry C, Chiu S, Harrist A, Kwit N, Lewis L, McGuire DO, Oduyebo T, Russell K, Talley P, Tanner M, Williams C, Zika Virus Response E, Laboratory T . 2016. Travel-associated Zika virus disease cases among U.S. residents–United States, January 2015–February 2016. MMWR Morb Mortal Wkly Rep 65:286–289. 10.15585/mmwr.mm6511e1. [DOI] [PubMed] [Google Scholar]
- 37.Venturi G, Zammarchi L, Fortuna C, Remoli ME, Benedetti E, Fiorentini C, Trotta M, Rizzo C, Mantella A, Rezza G, Bartoloni A. 2016. An autochthonous case of Zika due to possible sexual transmission, Florence, Italy, 2014. Euro Surveill 21:30148. 10.2807/1560-7917.ES.2016.21.8.30148. [DOI] [PubMed] [Google Scholar]
- 38.Wilder-Smith A, Chang CR, Leong WY. 2018. Zika in travellers 1947–2017: a systematic review. J Travel Med 25:tay044. 10.1093/jtm/tay044. [DOI] [PubMed] [Google Scholar]
- 39.Centers for Disease Control and Prevention. 2021. December 22. Zika cases in the United States. Centers for Disease Control and Prevention, Atlanta, GA. https://www.cdc.gov/zika/reporting/index.html. Accessed 3 February 2022. [Google Scholar]
- 40.Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GR, Simmons CP, Scott TW, Farrar JJ, Hay SI. 2013. The global distribution and burden of dengue. Nature 496:504–507. 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee C, Lee H. 2019. Probable female to male sexual transmission of dengue virus infection. Infect Dis (Lond) 51:150–152. 10.1080/23744235.2018.1521004. [DOI] [PubMed] [Google Scholar]
- 42.Liew CH. 2020. The first case of sexual transmission of dengue in Spain. J Travel Med 27:taz087. 10.1093/jtm/taz087. [DOI] [PubMed] [Google Scholar]
- 43.Mohammed HP, Ramos MM, Rivera A, Johansson M, Munoz-Jordan JL, Sun W, Tomashek KM. 2010. Travel-associated dengue infections in the United States, 1996 to 2005. J Travel Med 17:8–14. 10.1111/j.1708-8305.2009.00374.x. [DOI] [PubMed] [Google Scholar]
- 44.Centers for Disease Control and Prevention. 2010. Travel-associated Dengue surveillance–United States, 2006–2008. MMWR Morb Mortal Wkly Rep 59:715–719. [PubMed] [Google Scholar]
- 45.Wichmann O, Gascon J, Schunk M, Puente S, Siikamaki H, Gjorup I, Lopez-Velez R, Clerinx J, Peyerl-Hoffmann G, Sundoy A, Genton B, Kern P, Calleri G, de Gorgolas M, Muhlberger N, Jelinek T, European Network on Surveillance of Imported Infectious D, European Network on Surveillance of Imported Infectious Diseases . 2007. Severe dengue virus infection in travelers: risk factors and laboratory indicators. J Infect Dis 195:1089–1096. 10.1086/512680. [DOI] [PubMed] [Google Scholar]
- 46.Wilder-Smith A, Schwartz E. 2005. Dengue in travelers. N Engl J Med 353:924–932. 10.1056/NEJMra041927. [DOI] [PubMed] [Google Scholar]
- 47.Jelinek T, Muhlberger N, Harms G, Corachan M, Grobusch MP, Knobloch J, Bronner U, Laferl H, Kapaun A, Bisoffi Z, Clerinx J, Puente S, Fry G, Schulze M, Hellgren U, Gjorup I, Chalupa P, Hatz C, Matteelli A, Schmid M, Nielsen LN, da Cunha S, Atouguia J, Myrvang B, Fleischer K, European Network on Imported Infectious Disease S . 2002. Epidemiology and clinical features of imported dengue fever in Europe: sentinel surveillance data from TropNetEurop. Clin Infect Dis 35:1047–1052. 10.1086/342906. [DOI] [PubMed] [Google Scholar]
- 48.Rigau-Perez JG, Gubler DJ, Vorndam AV, Clark GG. 1997. Dengue: a literature review and case study of travelers from the United States, 1986–1994. J Travel Med 4:65–71. 10.1111/j.1708-8305.1997.tb00782.x. [DOI] [PubMed] [Google Scholar]
- 49.Centers for Disease Control and Prevention. 1987. Imported and indigenous dengue fever–United States, 1986. MMWR Morb Mortal Wkly Rep 36:551–554. [PubMed] [Google Scholar]
- 50.Gossner CM, Fournet N, Frank C, Fernandez-Martinez B, Del Manso M, Gomes Dias J, de Valk H. 2022. Dengue virus infections among European travellers, 2015 to 2019. Euro Surveill 27:2001937. 10.2807/1560-7917.es.2022.27.2.2001937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haddow AD, Woodall JP. 2016. Distinguishing between Zika and Spondweni viruses. Bull World Health Organ 94:711–711A. 10.2471/BLT.16.181503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cle M, Beck C, Salinas S, Lecollinet S, Gutierrez S, Van de Perre P, Baldet T, Foulongne V, Simonin Y. 2019. Usutu virus: a new threat? Epidemiol Infect 147:e232. 10.1017/S0950268819001213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McDonald EM, Duggal NK, Brault AC. 2017. Pathogenesis and sexual transmission of Spondweni and Zika viruses. PLoS Negl Trop Dis 11:e0005990. 10.1371/journal.pntd.0005990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ferreira VH, Dizzell S, Nazli A, Kafka JK, Mueller K, Nguyen PV, Tremblay MJ, Cochrane A, Kaushic C. 2015. Medroxyprogesterone acetate regulates HIV-1 uptake and transcytosis but not replication in primary genital epithelial cells, resulting in enhanced T-cell infection. J Infect Dis 211:1745–1756. 10.1093/infdis/jiu832. [DOI] [PubMed] [Google Scholar]
- 55.Quispe Calla NE, Vicetti Miguel RD, Boyaka PN, Hall-Stoodley L, Kaur B, Trout W, Pavelko SD, Cherpes TL. 2016. Medroxyprogesterone acetate and levonorgestrel increase genital mucosal permeability and enhance susceptibility to genital herpes simplex virus type 2 infection. Mucosal Immunol 9:1571–1583. 10.1038/mi.2016.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yarrington CD, Hamer DH, Kuohung W, Lee-Parritz A. 2019. Congenital Zika syndrome arising from sexual transmission of Zika virus, a case report. Fertil Res Pract 5:1. 10.1186/s40738-018-0053-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gurung S, Nadeau H, Maxted M, Peregrine J, Reuter D, Norris A, Edwards R, Hyatt K, Singleton K, papin jf, myers da. 2020. maternal zika virus (zikv) infection following Vaginal Inoculation with ZIKV-infected semen in timed-pregnant olive baboons. J Virol 94:e00058-20. 10.1128/JVI.00058-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Newman CM, Tarantal AF, Martinez ML, Simmons HA, Morgan TK, Zeng X, Rosinski JR, Bliss MI, Bohm EK, Dudley DM, Aliota MT, Friedrich TC, Miller CJ, O'Connor DH. 2021. Early embryonic loss following intravaginal Zika virus challenge in Rhesus macaques. Front Immunol 12:686437. 10.3389/fimmu.2021.686437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pessina MA, Hoyt RF, Jr, Goldstein I, Traish AM. 2006. Differential effects of estradiol, progesterone, and testosterone on vaginal structural integrity. Endocrinology 147:61–69. 10.1210/en.2005-0870. [DOI] [PubMed] [Google Scholar]
- 60.Miyaura H, Iwata M. 2002. Direct and indirect inhibition of Th1 development by progesterone and glucocorticoids. J Immunol 168:1087–1094. 10.4049/jimmunol.168.3.1087. [DOI] [PubMed] [Google Scholar]
- 61.Piccinni MP, Giudizi MG, Biagiotti R, Beloni L, Giannarini L, Sampognaro S, Parronchi P, Manetti R, Annunziato F, Livi C, Romagnani S, Maggi E. 1995. Progesterone favors the development of human T helper cells producing Th2-type cytokines and promotes both IL-4 production and membrane CD30 expression in established Th1 cell clones. J Immunol 155:128–133. [PubMed] [Google Scholar]
- 62.Patton DL, Thwin SS, Meier A, Hooton TM, Stapleton AE, Eschenbach DA. 2000. Epithelial cell layer thickness and immune cell populations in the normal human vagina at different stages of the menstrual cycle. Am J Obstet Gynecol 183:967–973. 10.1067/mob.2000.108857. [DOI] [PubMed] [Google Scholar]
- 63.Baronti C, Piorkowski G, Charrel RN, Boubis L, Leparc-Goffart I, de Lamballerie X. 2014. Complete coding sequence of zika virus from a French polynesia outbreak in 2013. Genome Announc 2:e00500-14. 10.1128/genomeA.00500-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kokernot RH, Smithburn KC, Muspratt J, Hodgson B. 1957. Studies on arthropod-borne viruses of Tongaland. VIII. Spondweni virus, an agent previously unknown, isolated from Taeniorhynchus (Mansonioides) uniformis. S Afr J Med Sci 22:103–112. [PubMed] [Google Scholar]
- 65.Williams MC, Simpson DI, Haddow AJ, Knight EM. 1964. The isolation of West Nile Virus from man and of usutu virus from the bird-biting mosquito Mansonia Aurites (Theobald) in the Entebbe Area of Uganda. Ann Trop Med Parasitol 58:367–374. 10.1080/00034983.1964.11686258. [DOI] [PubMed] [Google Scholar]
- 66.Khromykh AA, Kenney MT, Westaway EG. 1998. trans-Complementation of flavivirus RNA polymerase gene NS5 by using Kunjin virus replicon-expressing BHK cells. J Virol 72:7270–7279. 10.1128/JVI.72.9.7270-7279.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mangala Prasad V, Klose T, Rossmann MG. 2017. Assembly, maturation and three-dimensional helical structure of the teratogenic rubella virus. PLoS Pathog 13:e1006377. 10.1371/journal.ppat.1006377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brien JD, Lazear HM, Diamond MS. 2013. Propagation, quantification, detection, and storage of West Nile virus. Curr Protoc Microbiol 31:15D.3.1–15D.3.18. 10.1002/9780471729259.mc15d03s31. [DOI] [PubMed] [Google Scholar]
- 69.Oliphant T, Nybakken GE, Engle M, Xu Q, Nelson CA, Sukupolvi-Petty S, Marri A, Lachmi BE, Olshevsky U, Fremont DH, Pierson TC, Diamond MS. 2006. Antibody recognition and neutralization determinants on domains I and II of West Nile Virus envelope protein. J Virol 80:12149–12159. 10.1128/JVI.01732-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Oh JE, Iijima N, Song E, Lu P, Klein J, Jiang R, Kleinstein SH, Iwasaki A. 2019. Migrant memory B cells secrete luminal antibody in the vagina. Nature 571:122–126. 10.1038/s41586-019-1285-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Widman DG, Young E, Yount BL, Plante KS, Gallichotte EN, Carbaugh DL, Peck KM, Plante J, Swanstrom J, Heise MT, Lazear HM, Baric RS. 2017. A reverse genetics platform that spans the Aika virus family tree. mBio 8:e02014-16. 10.1128/mBio.02014-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carbaugh DL, Baric RS, Lazear HM. 2019. Envelope protein glycosylation mediates Zika virus pathogenesis. J Virol 93:e00113-19. 10.1128/JVI.00113-19. [DOI] [PMC free article] [PubMed] [Google Scholar]