

ABSTRACT
Receptor for activated C kinase 1 (RACK1) is a small ribosomal subunit protein that is phosphorylated by vaccinia virus (VacV) to maximize translation of postreplicative (PR) mRNAs that harbor 5′ polyA leaders. However, RACK1 is a multifunctional protein that both controls translation directly and acts as a scaffold for signaling to and from the ribosome. This includes stress signaling that is activated by ribosome-associated quality control (RQC) and ribotoxic stress response (RSR) pathways. As VacV infection activates RQC and stress signaling, whether RACK1 influences viral protein synthesis through its effects on translation, signaling, or both remains unclear. Examining the effects of genetic knockout of RACK1 on the phosphorylation of key mitogenic and stress-related kinases, we reveal that loss of RACK1 specifically blunts the activation of c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) at late stages of infection. However, RACK1 was not required for JNK recruitment to ribosomes, and unlike RACK1 knockout, JNK inhibitors had no effect on viral protein synthesis. Moreover, reduced JNK activity during infection in RACK1 knockout cells contrasted with the absolute requirement for RACK1 in RSR-induced JNK phosphorylation. Comparing the effects of RACK1 knockout alongside inhibitors of late stage replication, our data suggest that JNK activation is only indirectly affected by the absence of RACK1 due to reduced viral protein accumulation. Cumulatively, our findings in the context of infection add further support for a model whereby RACK1 plays a specific and direct role in controlling translation of PR viral mRNAs that is independent of its role in ribosome-based stress signaling.
IMPORTANCE Receptor for activated C kinase 1 (RACK1) is a multifunctional ribosomal protein that regulates translation directly and mediates signaling to and from the ribosome. While recent work has shown that RACK1 is phosphorylated by vaccinia virus (VacV) to stimulate translation of postreplicative viral mRNAs, whether RACK1 also contributes to VacV replication through its roles in ribosome-based stress signaling remains unclear. Here, we characterize the role of RACK1 in infected cells. In doing so, we find that RACK1 is essential for stress signal activation by ribotoxic stress responses but not by VacV infection. Moreover, although the loss of RACK1 reduces the level of stress-associated JNK activation in infected cells, this is an indirect consequence of RACK1’s specific requirement for the synthesis of postreplicative viral proteins, the accumulation of which determines the level of cellular stress. Our findings reveal both the specific role of RACK1 and the complex downstream effects of its control of viral protein synthesis in the context of infection.
KEYWORDS: poxvirus, RACK1, ribosome, translation, signaling, stress, ribotoxic stress responses, ribosome quality control, stress signaling
INTRODUCTION
The poxviridae is a family of large double-stranded DNA (dsDNA) viruses that include variola virus (VarV), the causative agent of smallpox, as well as monkeypox virus (1, 2). Although smallpox was declared eradicated in 1980, the unprecedented global spread of monkeypox in 2022 underscores the pandemic potential of zoonotic poxvirus infections and the reasons they remain category A pathogens (3–5). Protection against either smallpox or monkeypox is afforded by vaccination with the closely related orthopoxvirus vaccinia virus (VacV), which has also become the laboratory prototype for poxvirus research (2).
Poxviruses are notable for their exceptional self-sufficiency that includes independently transcribing and replicating their DNA genomes in the cytoplasm rather than the nucleus, where they form DNA-filled structures termed “viral factories” (VFs) (2). This level of independence from the host is enabled by the fact that poxviruses on average encode around 200 genes. Approximately half of their genes are expressed early after entry into the cell and mainly function in regulating uncoating of the viral core, control of host innate immune responses, and progression of the viral replication cycle (2, 6). Progression from early stages involves DNA replication and expression of both intermediate and late viral genes, which are often referred to cumulatively as postreplicative (PR) genes. This is because both intermediate and late genes share a dependence on viral DNA replication for transcriptional activation, as well as promoters that generate mRNAs with 5′-untranslated regions (UTRs) that contain unusual homopolymeric adenosine (polyA) tracts (7, 8). As infection transitions to the PR phase, host protein synthesis is also shut off, and viral proteins are dominantly synthesized. However, despite their ability to both transcribe and fully process their own mRNAs, poxviruses still require the host translational machinery to synthesize their proteins and complete their replication cycle.
Viruses use diverse strategies to take control of the host translation machinery, often revolving around control of translation initiation factors (9). However, VacV also directly targets the ribosome by phosphorylating several subunits due to the activity of the virus-encoded B1 kinase (10–14) (reviewed in reference 8). B1 is expressed as an early protein that phosphorylates unique sites on a number of small ribosomal subunit proteins (RPSs), some of which have been shown to enhance translation of PR mRNAs. These include RPS2, which is located at the mRNA entry channel, and receptor for activated C kinase 1 (RACK1), which is located near the mRNA exit channel (13–16). Of these, RACK1 is best characterized in terms of how poxvirus-induced phosphorylation affects its function. B1 phosphorylates a serine residue (S278) within a linker region of RACK1 that gives rise to electrostatic forces that alter the swivel motion of the 40S head domain (14–16). This in turn facilitates noncanonical modes of cap-independent translation that have been suggested for certain classes of cellular and viral mRNAs. In the case of VacV, this alternate mode of initiation on PR mRNAs is supported by their unusual 5′ polyA leaders, which are not normally found on mammalian transcripts (8, 17–19).
In mammalian cells, homopolymeric stretches of adenosine nucleotides are usually limited to the 3′ polyA tail, in which they play roles in mRNA stability and mRNA circularization that enhance translation initiation efficiency (20, 21). PolyA tracts also play a key role in the cellular mRNA and protein surveillance process called ribosome-associated quality control (RQC) (22, 23). A central aspect of RQC involves ribosome stalling on certain mRNA sequences that are recognized as aberrant, which includes polyA tracts. Under normal conditions, translation has terminated, and the 3′ polyA tail is never decoded. However, if an mRNA is accidentally internally polyadenylated or if a stop codon is absent and leads to readthrough to the polyA tail, the ribosome stalls upon encountering the polyA stretch (24, 25). Trailing ribosomes then collide with stalled ribosomes, which results in regulatory monoubiquitylation of specific RPSs by the E3 ubiquitin-protein ligase ZNF598 (26–30). This in turn promotes disassembly and recycling of stalled ribosomes along with destruction of the aberrant mRNA and nascent peptide. Beyond ZNF598 activation, ribosome collisions that occur during RQC or the related process of the ribotoxic stress response (RSR), which is induced by drugs or conditions that affect ribosome elongation rates, lead to the activation of stress signaling through c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) and p38 mitogen-activated protein kinase (MAPK) (31–33). Notably, RACK1 functions in RQC, and in particular, it plays an essential role in JNK and p38 activation during RSR (16, 26, 34, 35). Indeed, RACK1 was initially discovered and named as a binding partner or receptor for protein kinase C βII (PKCβII), which it binds in response to stress inducers such as phorbol 12-myristate 13-acetate (PMA) or tumor necrosis factor α (TNF-α). Since this discovery, it has become clear that RACK1 is a core ribosomal protein that plays dual roles in regulating translation directly and as a scaffolding protein or hub for signaling to and from the ribosome (36–39). This is due to its structure and position on the ribosome. Structurally, RACK1 is a tryptophan-aspartate (WD) 40 repeat protein that largely consists of seven β-propeller domains that mediate binding to kinases and translation initiation factors. In terms of position, RACK1 lies at the outer edge of the 40S subunit head domain with several of its β-propellers facing outward to the cytosol, which allows it to bridge kinases and initiation factors to the ribosome (39). This also positions RACK1 near the interface of ribosome collisions, also explaining its role in signaling during RSR (29, 40).
Poxvirus infection activates several MAPK pathways, including JNK and p38 (41–45). Separately, VacV has also recently been shown to activate and exploit ZNF598 activity to maximize both early and PR viral protein production (46, 47). Given the centrality of RACK1 in ribosome-based stress signaling and RQC, it therefore remains unclear whether RACK1’s role in poxvirus protein synthesis involves direct effects on translation or by mediating signaling events or a combination of both. Here, we show that unlike its essential role in stress signaling during RSR and unlike the roles of ZNF598 and RQC through all stages of infection (46), RACK1 is specifically required for PR viral protein synthesis. Moreover, RACK1 is not essential for stress signaling during infection and instead only indirectly influences stress activation through effects on PR protein production. Overall, our findings suggest that RACK1’s reported roles in stress signaling have little to do with its role in poxvirus protein synthesis and add additional lines of evidence suggesting that poxvirus-modified RACK1 functions specifically and directly in the regulation of PR mRNA translation.
RESULTS
Characterization of poxvirus infection in HAP1 cells.
The near-haploid human chronic myeloid leukemia cell line HAP1 is becoming increasingly recognized as a powerful genetic system to study host factors required for infection by a variety of viruses (48–55), including modified vaccinia virus (MVA) (56) and monkeypox (57). Indeed, in addition to being adherent and fibroblast-like, HAP1 cells also retain key regulatory features of primary cells that are often lost in other transformed lines. This notably includes the rapid degradation of subunit proteins when they are not part of the ribosome itself (otherwise termed extraribosomal), which serves to limit the activity of RPs such as RACK1 to the ribosome (13, 14, 16, 58–60). Moreover, we previously generated two independent RACK1 knockout HAP1 lines that have been extensively characterized by a number of groups (13, 16, 58, 59). Thus far, these studies have revealed that similar to many other organisms and systems (14, 60–68), there is a limited requirement for RACK1 for global translation and a greater reliance on RACK1 for transcript-specific translation that includes viral mRNAs that harbor internal ribosome entry sites (IRESs) and VacV mRNAs with 5′ polyA leaders (14, 16, 58, 59). However, how closely poxvirus infection in HAP1 cells parallels infection in more established cell systems and precisely how loss of the multifunctional protein RACK1 affects poxvirus protein production remains unclear.
To begin addressing these questions, we first compared VacV infection time courses in parental HAP1 cells and primary normal human dermal fibroblasts (NHDFs). Beginning with immunofluorescence (IF) analysis of viral factory (VF) formation, cells were infected with VacV at a multiplicity of infection (MOI) of 25 and fixed at 0, 6, 16, and 24 h postinfection (hpi). DNA was stained with Hoechst dye, and infection was visualized using antibody against the early-intermediate protein I3, which predominantly localizes to VFs. In HAP1 cells, early VFs were detectable in many but not all cells by 6 hpi (Fig. 1A). By 16 hpi, all cells contained bright VFs, and the size and intensity of VFs increased through 24 hpi. While not readily detected in most NHDF cells at 6 hpi, VFs were evident in all cells at 16 hpi and increased in size through 24 hpi (Fig. 1A). VFs appear more compact and brighter in HAP1 cells compared to NHDFs because HAP1s are smaller in size yet produce more viral protein. Despite this simple difference, the kinetics of VF formation were similar in the two cell types. To extend this comparison, we next measured rates and patterns of protein synthesis at each time point using 35S-methionine/cysteine pulse-labeling. A key feature of the transition from early to PR phases of VacV infection is the shutoff host translation, evident as a robust loss of actively synthesized host proteins and distinctive patterns of PR viral protein synthesis. As would be expected for early stages of infection, host protein synthesis was sustained, while a subset of new bands representing early viral proteins were evident in HAP1 and NHDF cells at 6 hpi (Fig. 1B). Expression of early viral proteins was confirmed by Western blot (WB) analysis, which showed that the early-intermediate protein I3 was produced at 6 hpi and peaked in abundance by 16 hpi in both cell types (Fig. 1B). As infection progressed, robust shutoff host translation and the typical pattern of PR viral protein synthesis was evident in both cell types by 16 hpi and continued through 24 hpi. This was accompanied by ready detection of intermediate (A25) and late (A14) viral proteins by WB, with the abundance of A14 increasing between 16 and 24 hpi in both cell types as the replication cycle progressed. These findings are in line with other studies showing robust host shutoff and late viral protein accumulation spanning 16 to 48 hpi in several cell types (for examples, see references 43 and 69–72). Cumulatively, these findings demonstrate that despite modest differences in how quickly VacV initially establishes infection and begins to form VFs in HAP1 cells, its replication cycle is similar to that in primary NHDFs.
FIG 1.

Comparison of VacV infection in HAP1 and NHDF cells. HAP1 and normal human dermal fibroblasts (NHDF) cells were infected with VacV Western reserve (WR) at a multiplicity of infection (MOI) of 25 for the indicated period of time. (A) Cells were stained with Hoechst dye and I3 (red) antibody. Representative images are shown for each cell type and time point. (B) Cells were labeled with 35S-Met/Cys for 30 min prior to harvesting at the indicated times postinfection. The samples were subjected to polyacrylamide gel analysis and autoradiography to detect nascent protein synthesis (top) or Western blotting for the indicated viral proteins (bottom). The results are representative of at least three independent experiments. hpi, hours postinfection.
MAPK pathway activation during poxvirus infection.
We next examined the phosphorylation status of specific stress and mitogen-activated protein kinases (MAPKs) in HAP1 and other cell types in response to VacV infection, focusing on those reported to be regulated by RACK1. Beginning with HAP1 cells, WB analysis showed that VacV did not alter the levels of phosphorylated PKCβII at any time point examined (Fig. 2A). Indeed, prior studies have already established that PKβII does not function with RACK1 during infection (14). In contrast, infection robustly activated both JNK and p38 specifically at later stages of infection. In addition, VacV infection activated mitogen-associated extracellular signal-regulated kinase (ERK) at late stages of infection in HAP1 cells.
FIG 2.

VacV infection activates mitogenic and stress-related mitogen-activated protein kinase (MAPK) signaling pathways at postreplicative stages of infection. (A) HAP1 cells were infected with VacV at MOI 25 and harvested at 0, 6, and 24 h hpi. The samples were analyzed by Western blotting with the indicated antibodies. (B) NHDF, HEK293A, and MRC5 cells were infected and analyzed as described for HAP1 cells for panel A. The results are representative of at least three independent experiments. JNK, c-Jun N-terminal kinase; ERK, extracellular signal-regulated kinase.
We next determined whether these signaling events in HAP1 cells reflected similar effects of infection in other human cell types, including primary NHDFs and hTERT-immortalized lung fibroblast MRC5 cells (73), as well as HEK293A cells (Fig. 2B). As was seen in HAP1 cells, infection of NHDF, MRC5, or HEK293A cells did not affect the levels of phosphorylated PKCβII, while JNK and p38 also showed the same pattern of phosphorylation; modest JNK phosphorylation occurred at 6 hpi, while a robust activation of both stress kinases occurred at late stages of infection. These kinetics of JNK activation were similar to prior studies in murine fibroblasts and other nonhuman cell lines, wherein the bulk of JNK activation occurs from 24 hpi onward (44, 74). Notably, the pattern of ERK phosphorylation varied between all cell types. There was no change during infection of NHDFs, which contained high basal levels of activated ERK in uninfected cells. Similarly, basal levels of phosphorylated ERK were high in MRC5 cells, and infection caused a decrease at 6 hpi followed by a modest increase again by 24 hpi. In contrast, ERK was highly activated at 6 hpi, and although the level of activation dropped from this peak, it remained activated at later stages of infection in HEK293A cells. This suggests that changes in ERK signaling are unlikely to be directly regulated by VacV or related to stress pathway activation during late stages of infection. Instead, these differences likely reflect how cells of different types or metabolic states respond to viral growth factor homologs in complex ways (75). Notably, however, ERK was active at later stages of infection in all cell types tested, albeit to varying degrees. Collectively, this demonstrated that VacV does not activate the RACK1-associated PKCβII signaling pathway but consistently and robustly activates JNK and p38 stress pathways at later stages of infection in several cell types, including HAP1 cells.
RACK1 specifically regulates postreplicative viral protein synthesis.
To understand the potential role(s) of RACK1 in regulating signaling and/or viral mRNA translation, we first extended upon our earlier analysis of effects on viral protein production. To date, our studies have predominantly focused on RACK1 phosphomimetics to understand its specific effects on translation outside the more complex context of infection (14–16). In terms of the infected cell context, prior studies have shown that knockout of RACK1 in HAP1 cells reduces metabolic labeling of viral proteins at late stages of infection and specifically reduces the accumulation of late viral proteins D8 and A14 but has very little effect on the early-intermediate protein I3 (14). However, whether this is due solely to defects in PR protein synthesis or cumulative feedback effects on viral DNA replication and/or mRNA accumulation is unknown. To address this, we infected parental HAP1 cells or two independent HAP1 RACK1 knockout lines. We then measured viral DNA, RNA, and protein levels at 24 hpi. As a control, cytosine arabinose (AraC) was used to block DNA synthesis, which in turn reduces the transcription of PR viral genes. The results showed that viral DNA levels were at most only modestly reduced in RACK1 knockout cells (Fig. 3A). Moreover, regardless of any modest differences in DNA replication, loss of RACK1 had no detectable effect on the production of representative early-intermediate (I3) or late (A14) viral mRNAs (Fig. 3B). As expected, AraC blocked viral DNA replication (Fig. 3A) and resulted in a 1,000- to 10,000-fold reduction in late (A14) mRNA levels in each cell line (Fig. 3B). Examining an expanded number of viral proteins from different kinetic classes, we found that the levels of the early viral protein E3 and the early-intermediate protein I3 were largely unaffected in RACK1 knockouts compared to parental lines (Fig. 3C). In contrast, the accumulation of PR proteins, including A14, D8, F17, and A25, showed large reductions in abundance in both RACK1 knockouts compared to parental HAP1 cells (Fig. 3C). As expected, AraC treatment selectively blocked the accumulation of these late proteins in each cell line. Cumulatively, this suggests that there is a specific defect in PR viral mRNA translation in the absence of RACK1 in the context of infection, in line with reciprocal approaches demonstrating that a phosphomimetic of poxvirus-modified RACK1 increases translation of mRNA reporters with 5′ polyA leaders (14, 15).
FIG 3.

Loss of RACK1 specifically reduces postreplicative viral protein production. HAP1 parental (par.) or two HAP1 RACK1 knockout cell lines (KO#1, KO#2) were infected with VacV at MOI 25 for 24 h in the presence or absence of AraC (40 μg/mL). (A, B) Total DNA (A) or RNA (B) were extracted and measured by RT-PCR with using I3 (early) and A14 (late) gene primers. The results are representative of at least two independent experiments. (C) The cells were lysed and subjected to Western blotting with the indicated antibodies. The results are representative of at least three independent experiments.
Loss of RACK1 indirectly regulates JNK phosphorylation during VacV infection.
Given that RACK1 is required for JNK and p38 activation as part of the ribotoxic stress response (RSR) (16, 31–35), we next examined whether it was also required for stress signaling during VacV infection. Infection of HAP1 parental or RACK1 knockout cells revealed that loss of RACK1 had little or no effect on VacV-induced increases in p38 activation that occur at late stages (Fig. 4A). In contrast, JNK activation was reduced, while ERK activity increased in both RACK1 knockout lines compared to parental HAP1 cells. Demonstrating that this was directly due to loss of RACK1, expression of FLAG-tagged RACK1 increased both PR viral protein production and JNK phosphorylation in RACK1 knockout cells (Fig. 4B). This could indicate either a direct requirement for RACK1 in JNK activation by VacV or an indirect consequence of reduced PR viral protein accumulation that may thereby impose less stress on infected cells. Indeed, reduced stress may also explain the increased mitogenic ERK signaling in these cells. To test the relationship between stress signal activation and virus replication in these cells, we used either AraC or phosphonoacetic acid (PAA) to inhibit viral DNA replication and PR viral gene expression. The results showed that both inhibitors independently reduced JNK and p38 activation in HAP1 cells, as well as in NHDFs, HEK293A, or MRC5 cells infected with VacV (Fig. 5). This is in line with our earlier kinetic analysis, which suggests that PR stages of infection cause a significant increase in stress signaling, particularly for JNK. Indeed, JNK and p38 activation has been linked to host responses to late stages of VacV infection (45). Furthermore, we noted that ERK phosphorylation increased in inhibitor-treated cells other than NHDFs, which, as discussed above, contain high basal levels of phosphorylated ERK that cannot be further activated upon infection. This suggests that replication stresses in turn reduce the ability of cells to activate ERK, and reductions in virus-induced stress likely allow the cell to better sustain mitogenic signaling. While DNA replication inhibitors block several aspects of late stage replication, the more specific defects in late viral protein accumulation that occur in RACK1 knockout cells (Fig. 3C and 4) likely cause a more limited cellular stress response that is restricted to JNK. Moreover, the fact that ERK activity increases when JNK activity decreases in RACK1 knockout cells suggests that RACK1 does not directly regulate either signaling pathway during infection (Fig. 4A). Instead, these opposing states of JNK and ERK activation likely reflect reduced cellular stress imposed in RACK1 knockout cells due to reduced PR viral protein accumulation.
FIG 4.
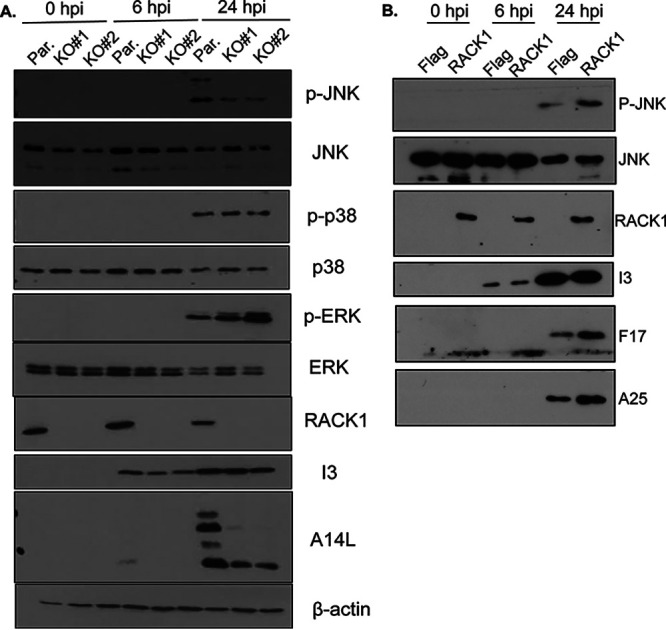
VacV-induced activation of JNK is impaired in RACK1 knockout cells. (A) HAP1 parental (par.) or two HAP1 RACK1 knockout cell lines (KO#1, KO#2) were infected with VacV at MOI 25 and harvested at the indicated time points. The cells were lysed and subjected to Western blotting with the indicated antibodies. (B) HAP1 RACK1 knockout (KO#1) cells expressing either FLAG-control or FLAG-tagged RACK1 were infected as described for panel A. The cells were lysed and subjected to Western blotting with the indicated antibodies. The results are representative of at least three independent experiments.
FIG 5.
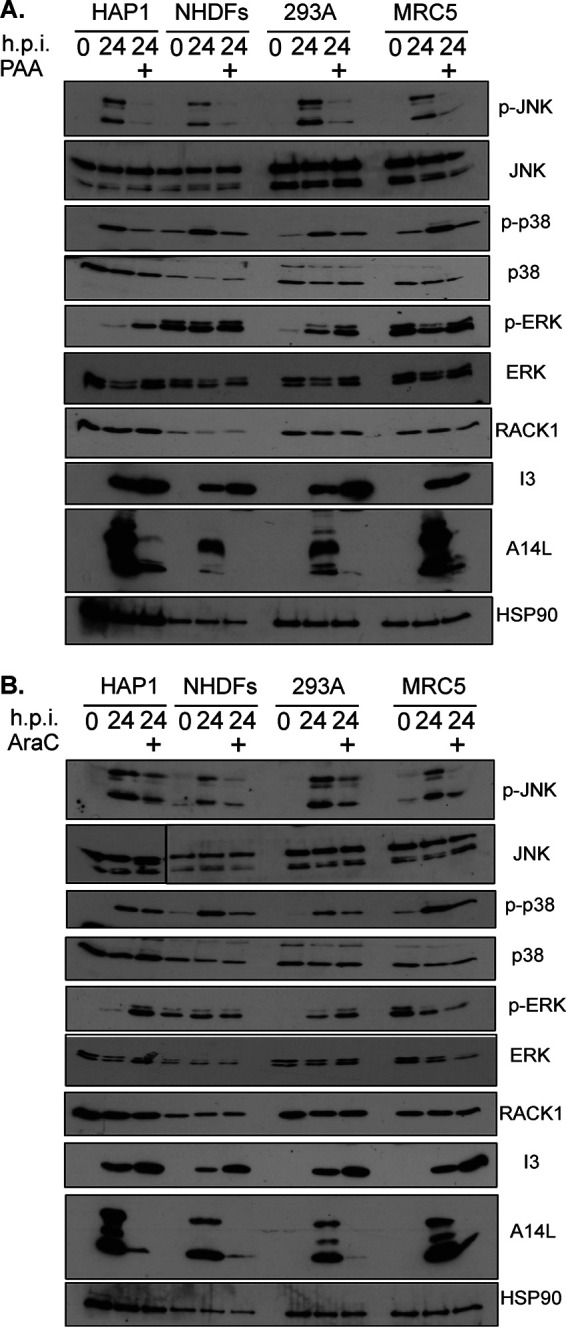
VacV replication is required for maximal JNK and p38 activation. HAP1, NHDFs, HEK 293A, and MRC5 cells were infected with VacV at MOI 25 for 24 h in the presence or absence of phosphonoacetic acid (PAA) (400 μg/mL) (A) and AraC (40 μg/mL) (B). The cells were lysed and subjected to Western blotting with the indicated antibodies. The results are representative of two independent experiments for each inhibitor and cell type.
To further test whether RACK1 might directly affect JNK signaling as part of how it controls viral protein synthesis, we explored whether RACK1 knockout affected JNK recruitment to the ribosome in the context of VacV infection. To do this, we infected HAP1 parental or RACK1 knockout cells followed by sucrose gradient sedimentation and fractionation of cell lysates to isolate free fractions, eukaryotic initiation factor (eIF) complexes, eIF-bound initiating 40S ribosome subunits, 60S subunits, and finally assembled 80S monosomes and polysomes (Fig. 6). Analysis of these fractions showed that beyond subtle differences in the overall distribution of proteins due to the role of RACK1 in regulating ribosome activity at late stages of infection, discussed again directly below, and the expected differences in JNK phosphorylation between parental or RACK1 knockouts, there was no notable difference in the cofractionation of either activated or total JNK with eIFs or initiating 40S subunits. Using I3 to confirm infection, we also observed that it cosediments with eIF4F subunits such as eIF4G, one of I3’s reported binding partners (76). This pattern of cosedimentation was also not affected by the absence of RACK1 (Fig. 6). Cumulatively, our data suggest that RACK1 does not regulate JNK recruitment to initiating or assembled ribosomes in infected cells.
FIG 6.

RACK1 does not regulate JNK recruitment to ribosomes. HAP1 parental or HAP1 RACK1 knockout (KO#1) cells were infected with VacV at MOI 25 and harvested for polysome fractionation. Fractions were trichloroacetic acid (TCA) precipitated and subjected to Western blotting with the indicated antibodies. A sample of total lysate (L) is shown alongside each fraction. Fractions are labeled to illustrate the distribution of initiation complexes (eIF4F and eIF4F+eIF3) followed by 43S (40S subunit bound by eIFs), 60S, and assembled 80S ribosomes (distributed as 80S monosomes, disomes, and polysomes). Representative small (RPS2, RACK1) and large subunit proteins (RPL11, RPL13a) are shown. eIF, eukaryotic initiation factor; SSU, small subunit; LSU, large subunit. The results are representative of three independent experiments.
Stress signal activation has distinct requirements for RACK1 during VacV infection versus ribotoxic stress responses.
It has recently been reported that VacV activates ribosome quality control (RQC) (46), which shares key features of ribosome collisions and stress signal activation that are associated with the broader ribotoxic stress response (RSR). Moreover, RACK1 plays a key role in mediating the sensing of collided ribosomes and the initiation of subsequent stress signals during RQC or RSR (31–35). To explore this further, we first examined the ribosome profiles of uninfected or infected HAP1 parental or RACK1 knockout cells. Notably, the pattern of 40S, 60S, and 80S polysomes was very similar between uninfected parental HAP1 cells and early stages of infection (6 hpi) (Fig. 7A and B), in line with a lack of host shutoff at this point. Moreover, in line with the nonessentiality of RACK1 in overall translation in several systems, including in previous studies of HAP1 cells (16, 59), the loss of RACK1 resulted in only a modest reduction in the levels of polysomes and a slight increase in monosome accumulation in both uninfected cells and cells infected for 6 h. In stark contrast, there was a large reduction in polysomes and a corresponding accumulation of both 80S monosomes and disomes in infected parental HAP1 cells at 24 hpi (Fig. 7C). Further, in line with a more specific requirement for RACK1 in PR viral protein synthesis, there was a more pronounced decrease in polysomes in infected RACK1 knockout cells at 24 hpi, a time point when late viral mRNAs are now the predominant species being translated (Fig. 1B). In addition, we also noted a reduction in disome peaks in infected RACK1 knockout cells at this later time point. This overall phenotype raises two important points. First, while disomes can indicate collided ribosomes, they are usually transient or dynamic and occur at low levels during RQC (22–24, 40). In other words, they are not normally so obvious in polysome traces, which indicate steady-state ribosome levels. Moreover, prior reports suggest that VacV infection induces RQC but similarly, this occurs at a low level, and RQC reaches maximal activity within an hour of infection (46). Accordingly, we do not observe notable disomes at 6 hpi, suggesting that disome formation at late stages is not a reflection of RQC or RSR activity. Second, we recently found that a phosphomimetic of poxvirus-modified RACK1 induces a very similar disome pattern as part of how a negative charge in the RACK1 linker domain remodels ribosome behavior to promote cap-independent translation (16). This suggests that the RACK1-dependent appearance of disomes in infected cells observed at this time point is more likely to be a reflection of viral remodeling of ribosome function and translation of PR viral mRNAs than a reflection of ribosome collisions.
FIG 7.

Ribosome profiles are altered at postreplicative stages of vaccinia virus (VacV) infection. Mock or VacV-infected HAP1 parental and RACK1 KO cells were lysed in polysome buffer and subjected to sucrose gradient centrifugation. Polysome traces were measured by detecting RNA at 254 nm. (A) Polysome profiles in uninfected cells. Note that the absence of RACK1 only modestly reduces the level of polysomes, in particular larger polysomes. (B) Polysome profiles at early stages of the VacV infection (6 hpi) are very similar to those of uninfected cells and exhibit similar effects of RACK1 loss as in panel A. (C) Polysome profiles at postreplicative stages of the VacV infection (24 hpi) illustrating the reduction in heavy polysomes, the increase in 80S monosomes, and the appearance of disome peaks, indicated and marked with red circles. Loss of RACK1 at this stage of infection causes a notable loss of polysomes and the disome shoulder. The results are representative of three independent experiments.
To further explore whether RACK1 might be functionally linked to ribosome collisions during infection, we compared the requirements for RACK1 in VacV-induced versus RSR-induced JNK activation. RSR can be potently activated by moderate doses of anisomycin (ANS), which induces ribosome collisions by slowing translation elongation (16, 31–35). Comparing conditions in parental HAP1 cells, we found that treatment with ANS consistently induced JNK phosphorylation more robustly than VacV infection (Fig. 8A). Moreover, as observed earlier, JNK activation was reduced but not eliminated in VacV-infected RACK1 knockout cells. In contrast, RACK1 was absolutely required for JNK activation by ANS, as reported previously (16, 35). This demonstrates that the essential role of RACK1 in RSR-induced stress signaling is distinct from the effects of RACK1 loss during VacV infection. Furthermore, JNK inhibitor VIII blocked c-Jun phosphorylation by either ANS or VacV (Fig. 8B) but had no detectable effect on the accumulation of representative VacV proteins (Fig. 8C). This was also in line with prior reports that JNK is not required for VacV protein production or replication in murine cells (44). Cumulatively, our data demonstrate that unlike contexts such as RSR in which RACK1 is directly involved in and essential to ribosome-based stress signaling, RACK1 only indirectly affects the levels of JNK activation during infection through its effects on the accumulation of PR viral proteins.
FIG 8.

Differential requirements for RACK1 in JNK activation during poxvirus infection versus ribotoxic stress responses. (A) HAP1 parental and RACK1 KO cells were treated with anisomycin (ANS) (10 μM) for 1 h or infected with VacV at MOI 25 for 24 h. The cells were lysed and subjected to Western blotting with the indicated antibodies. ANS-induced JNK activation is completely dependent on RACK1, while VacV-induced JNK activation is not. (B) Validation of JNK inhibitor VIII efficacy in cells treated with ANS or infected with VacV. JNKVIII blocks JNK activity but not its phosphorylation. To verify inhibitory efficacy, HAP1 cells were treated with dimethyl sulfoxide (DMSO) solvent control or ANS (10 μM) for 1 h or infected with VacV at MOI 25 for 24 h in the presence of JNK inhibitor VIII at the indicated concentrations. Uninfected or infected HAP1 cells were treated with DMSO as a solvent control. Phosphorylated c-Jun was used as a readout for inhibition of JNK activity. (C) JNK activity is not required for VacV protein synthesis. HAP1 cells were infected with VacV as described for panel B. Representative early-intermediate (I3) and late (F17, A14) VacV proteins were analyzed by Western blotting. Early and late viral protein levels were unaffected by JNK inhibition. The results are representative of three independent experiments.
DISCUSSION
The sensing of stalled ribosomes by RQC is an important surveillance mechanism and regulator of stress responses, and it is associated with diseases such as neurodegeneration when the system is not properly functioning (22, 31). Interestingly, factors involved in RQC appear to play a particularly important role in VacV protein synthesis compared to herpes simplex virus 1 (HSV-1) or vesicular stomatitis virus (VSV), but why this is the case remains unclear (47). While our own initial studies revealed that the central RQC factor, ZNF598 is required for efficient accumulation of a limited number of early-intermediate and late viral proteins that were tested, leading us to suggest potential links to the presence of 5′ polyA leaders on their encoding mRNAs (47), subsequent proteomic studies revealed that RQC is activated within 1 h of infection and is more broadly required for the accumulation of all classes of poxvirus proteins (46). Intriguingly, both studies also found that VacV protein synthesis relies upon ubiquitylation of the ZNF598 substrate RPS20 but not RPS10. This contrasts with conventional RQC that occurs on reporters harboring internal polyA stretches in which ubiquitylation of RPS10 appears to play a more prominent role than that of RPS20 (26–28, 40, 77). It remains unclear why these differences arise and how poxviruses activate RPS20 ubiquitylation so rapidly, but it is tempting to suggest that it represents a variation of RQC that is perhaps a host response to infection that the virus ultimately coopts to its benefit. Regardless, the early kinetics of activation and general requirement for ZNF598 and RPS20 ubiquitylation for both early and PR viral protein synthesis contrasts with the more specific requirement for RACK1 at later stages, suggesting that RACK1 has little or no role in the form of RQC that functions during VacV infection.
The kinetics and effects on JNK and p38 signaling further support the idea that RACK1 functions distinctly from RQC or RSR during infection. Similar to other studies (44, 74), other than modest JNK activation at 6 hpi, the bulk of JNK and p38 activation occurs at late stages of infection, long after RQC is reported to be maximally activated. Moreover, the fact that RACK1 loss did not coordinately affect both kinases to the same extent further suggests that its function during infection is distinct from any RSR activation that might occur early and persist through the course of infection. Further supporting this idea, we find that while RACK1 is essential for the activation of JNK in response to RSR induction by ANS, loss of RACK1 only blunts JNK activation. Combined with the concurrent increase in mitogenic signaling that occurs, our data suggest that this effect on JNK activation likely reflects reduced cellular stresses as a result of reduced accumulation of PR viral proteins. Indeed, we find no evidence that RACK1 is directly involved in either JNK activation or recruitment to initiating 40S ribosomes during infection. Finally, it is unlikely that JNK activity mediates any of the effects of RACK1 on translation as JNK inhibition had no detectable effect on viral protein production. This is in line with prior reports that also showed that JNK inhibition or knockout had no effect on viral protein production or virus replication, but interestingly, JNK activity appears to be involved in cytoskeletal remodeling and cell motility that subsequently promotes virus spread (44). While it remains possible that RACK1 functions in these later JNK-dependent events, our findings show that RACK1’s role in PR viral protein synthesis does not involve JNK activity.
Our findings in infected cells also align with prior studies that, to date, have largely focused on understanding how poxvirus-modified RACK1 regulates translation using phosphomimetics. These approaches have shown that phosphomimetic RACK1 enhances translation of reporter mRNAs that harbor 5′ polyA mRNAs and more broadly supports cap-independent modes of translation on select subsets of mRNAs (14–16). Mechanistically, phosphomimetic RACK1 alters the swivel motion of the 40S head domain in the same way as type III or IV IRESs (16). While IRESs represent a broad group of structural elements found in the 5′-UTR of many RNA viruses that confer various degrees of independence from host translation initiation factors, type III and IV IRESs interact directly with the 40S ribosome (9). Several of these interactions involve and require RACK1, and they alter the swivel motion of the 40S head domain to control the ribosome and initiate translation independently of the cap-binding eukaryotic translation initiation factor complex, eIF4F (58, 59, 61, 78–83). Interestingly, poxvirus 5′ polyA leaders do not have inherent IRES activity (18) and instead phosphorylated RACK1 mimics the effects of these IRESs (14, 16). In doing so, phosphomimetic RACK1 facilitates a less complex form of initiation with reduced eIF dependency, similar to several IRES elements but on structurally simple leaders such as polyA tracts. In the context of infection, 5′ polyA reporters and PR mRNA translation also have a reduced dependence on the cap-binding eIF4F subunits eIF4E and eIF4G (18, 84). Our findings here, now examining the role of RACK1 in the context of infection in more detail, further support this overall model that RACK1 directly regulates PR viral protein synthesis. Furthermore, the effects of phosphomimetic RACK1 that manifest as a reduction in polysomes and an accumulation of 80S monosomes and disomes (16) were also evident here at late stages of infection. This suggests that these profile changes may not simply reflect host shutoff but instead reflect how poxvirus-modified RACK1 alters ribosome activity.
Cumulatively, our approaches dissect the complex roles of the ribosome-associated regulatory and signaling protein RACK1 in the context of infection. Our findings further support the idea that RACK1 functions directly in regulating translation of PR viral mRNAs yet highlight how this has indirect secondary effects on cellular stress and mitogenic signaling activity in the context of infection.
MATERIALS AND METHODS
Cell lines and viruses.
Stocks of VacV Western reserve (WR) were prepared and titred in BSC40 cells as previously described (73). HAP1 parental cells (Horizon, C859) and HAP1 RACK1 knockout cells, as well as RACK1 knockouts expressing either FLAG control or FLAG-RACK1 (14, 16), were cultured in Iscove’s modified Dulbecco’s Medium (IMDM; Fisher Scientific) containing 5% fetal bovine serum (FBS), 100 IU penicillin-streptomycin, 4 mM l-glutamine, and 25 mM HEPES. Primary normal human dermal fibroblasts (NHDFs) (Lonza, CC-2509), telomerase-expressing MRC5 fibroblasts, human embryonic kidney (HEK) 293A cells (Jeremy Luban, University of Massachusetts School of Medicine), and BSC40 cells (Ian Mohr, New York University School of Medicine) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Fisher Scientific) with 5% FBS, 2 mM l-glutamine, and 100 IU penicillin-streptomycin as described previously (73). All cell lines were cultured at 37°C with 5% CO2 and periodically screened for mycoplasma contamination.
Virus infection and inhibitor treatment.
In 12-well plates, 0.5 × 106 cells were seeded and infected with VacV WR at a MOI 25 for 1 h in 2.5% FBS medium. Then, the medium was replaced with fresh medium containing 5% FBS until the indicated time postinfection. For inhibitors, anisomycin (10 mM) was added for 30 min before cell lysis. AraC (Calbiochem, 251010; 40 μg/mL), PAA (Sigma, 284270; 400 μg/mL), or JNK inhibitor VIII (Selleckchem, S7794) was added 1 h prior to virus infection and maintained throughout the experiment.
Western blot and immunofluorescence analyses.
The cells were harvested with 1× Laemmli buffer and samples were resolved by SDS-polyacrylamide gels and transferred to nitrocellulose membranes (GE Healthcare Life Sciences). The membranes were blocked with 5% milk in TBST, pH 7.4 (20 M Tris, 150 mM NaCl, 0.25% Tween 20), for 1 h. The membranes were probed with the indicated primary antibodies in TBST containing 5% bovine serum albumin (BSA) and 0.02% (wt/vol) sodium azide overnight at 4°C. The antibodies from Cell Signaling Technology are as follows: p-p44/42 (ERK) MAPK (T202/Y204) (catalog no. 4370), p44/42 MAPK (catalog no. 9102, 1:3000), p-SAPK/JNK (T183/Y185) (catalog no. 4668), JNK2 (catalog no. 9258), p-p38 MAPK (T180/Y182) (catalog no. 9211), p38 MAPK (catalog no. 9212), RACK1 (catalog no. 5432 1:3000), eIF4A (catalog no. 2425), eIF3A (catalog no. 3411), RPL13a (catalog no. 2765), p-c-Jun (S63) (catalog no. 9261), β-actin (catalog no. 3700, 1:10,000), and Hsp90 (catalog no. 4877 1:3000). Also used were VacV I3 (David Evans, 1:3000), VacV E3 (Jingxin Cao, 1:5,000), VacV D8 (Paula Traktman, 1:3,000), VacV A14 (Yan Xiang, 1:3000), VacV F17 (Paula Traktman, 1:10,000), VacV A25 (Immune Tech, catalog no. 012-003M1, 1:1000), p-PKCβII (T641) (Millipore Sigma, catalog no. 07-873-I), and RPS2 (Gene Tex, catalog no. GTX114734). eIF4G (1:5,000) and PABP (1:5,000) were described previously (85, 86). All primary antibodies were diluted at 1:1,000 unless otherwise indicated. Secondary antibodies were mouse IgG, horseradish peroxidase (HRP) (GE Healthcare UK, Millipore Sigma, 1:5,000); rabbit IgG, HRP (GE Healthcare UK, Millipore Sigma, 1:5,000) diluted in TBST containing 5% (wt/vol) nonfat milk. Antigens were detected by incubating membranes with Pierce ECL Western blotting substrate or Pierce SuperSignal West Femto maximum sensitivity substrate. For immunofluorescence, HAP1 or NHDF cells were seeded on glass slips in 12-well plates and infected with VacV WR at the indicated MOI for the indicated times. The cells were fixed and stained as described previously using Hoechst dye (Thermo, 33342) and anti-I3 antibody (David Evans, University of Alberta) (71, 87).
Ribosome profiling and TCA precipitation.
We used 1 × 107 uninfected or infected HAP1 cells in a 10-cm dish for lysis and polysome fractionation. Sucrose gradient centrifugation and fractionation followed by trichloroacetic acid (TCA) precipitation for Western blot analysis was performed as previously described (16).
Nucleic acid extraction and qRT-PCR.
For uninfected or infected samples, 1 × 106 HAP1 cells in 6-well plate were harvested, and total RNA was extracted using TRIzol LS reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. We used 80 ng of extracted RNA for cDNA synthesis using Revert Aid first-strand cDNA synthesis kits (Thermo Fisher Scientific). Total DNA was extracted using a Genomic DNA kit (IBI Scientific). Quantitative reverse transcription (qRT)-PCR was performed using PowerUP SYBR green Master Mix reagent and 7500 Fast real-time PCR system (Applied Biosystems) with following conditions: Heat-labile uracil-DNA glycosylase activation at 95°C for 2 min and Dual-LockDNA polymerase at 95°C for 2 min. There were 40 cycles of denaturing at 95°C for 15 s and annealing/extension at 60°C for 1 min. RNA quantitation was analyzed using comparative Ct methods (ΔΔCt method). Primer sequences were as follows: I3 forward, ccttggccaattgtctttctc; I3 reverse, ccttggccaattgtctttctc; A14 forward, ctttccatgtacgagtgggactg; and A14 reverse, ggacatgatgcttatgattgg.
35S-Methionine/cysteine labeling.
At 30 min prior to harvesting cells, the medium was replaced with methionine/cysteine (Met/Cys)-free DMEM (Corning, 17-204-CL) containing 40 mM HEPES, 2 mM l-glutamine, and 0.035 mCi/mL 35S-l-methionine and 35S-l-cysteine mix (PerkinElmer, NEG072007MC). Cells were harvested in Laemmli buffer and resolved in SDS-PAGE. The gels were then incubated in destaining solution (10% acetic acid, 25% methanol in water) for 30 min. The fixed gels were dried at 80°C for 2 h using a model 583 gel dryer (Bio-Rad) and incubated with audioradiography film at −80°C for detection.
ACKNOWLEDGMENTS
We thank David Evans, Paula Traktman, Jingxin Cao, and Yan Xiang for kindly providing antibodies.
This work was supported by National Institutes of Health grant R01 AI127456 (to D.W.).
We declare no conflict of interest.
Contributor Information
Derek Walsh, Email: derek.walsh@northwestern.edu.
Felicia Goodrum, University of Arizona.
REFERENCES
- 1.McFadden G. 2005. Poxvirus tropism. Nat Rev Microbiol 3:201–213. 10.1038/nrmicro1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moss B. 2007. Poxviridae: the viruses and their replication, p 2849–2883. In Knipe DM, Howley PM (ed), Fields virology. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 3.Rahimi F, Talebi Bezmin Abadi A. 2022. The 2022 monkeypox outbreak: lessons from the 640 cases in 36 countries. Int J Surg 104:106712. 10.1016/j.ijsu.2022.106712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simpson K, Heymann D, Brown CS, Edmunds WJ, Elsgaard J, Fine P, Hochrein H, Hoff NA, Green A, Ihekweazu C, Jones TC, Lule S, Maclennan J, McCollum A, Muhlemann B, Nightingale E, Ogoina D, Ogunleye A, Petersen B, Powell J, Quantick O, Rimoin AW, Ulaeato D, Wapling A. 2020. Human monkeypox—after 40 years, an unintended consequence of smallpox eradication. Vaccine 38:5077–5081. 10.1016/j.vaccine.2020.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shchelkunov SN. 2013. An increasing danger of zoonotic orthopoxvirus infections. PLoS Pathog 9:e1003756. 10.1371/journal.ppat.1003756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang Z, Reynolds SE, Martens CA, Bruno DP, Porcella SF, Moss B. 2011. Expression profiling of the intermediate and late stages of poxvirus replication. J Virol 85:9899–9908. 10.1128/JVI.05446-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Z, Martens CA, Bruno DP, Porcella SF, Moss B. 2012. Pervasive initiation and 3'-end formation of poxvirus postreplicative RNAs. J Biol Chem 287:31050–31060. 10.1074/jbc.M112.390054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meade N, DiGiuseppe S, Walsh D. 2019. Translational control during poxvirus infection. Wiley Interdiscip Rev RNA 10:e1515. 10.1002/wrna.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jan E, Mohr I, Walsh D. 2016. A cap-to-tail guide to mRNA translation strategies in virus-infected cells. Annu Rev Virol 3:283–307. 10.1146/annurev-virology-100114-055014. [DOI] [PubMed] [Google Scholar]
- 10.Banham AH, Leader DP, Smith GL. 1993. Phosphorylation of ribosomal proteins by the vaccinia virus B1R protein kinase. FEBS Lett 321:27–31. 10.1016/0014-5793(93)80614-z. [DOI] [PubMed] [Google Scholar]
- 11.Kaerlein M, Horak I. 1978. Identification and characterization of ribosomal proteins phosphorylated in vaccinia-virus-infected HeLa cells. Eur J Biochem 90:463–469. 10.1111/j.1432-1033.1978.tb12625.x. [DOI] [PubMed] [Google Scholar]
- 12.Buendia B, Person-Fernandez A, Beaud G, Madjar J. 1987. Ribosomal protein phosphorylation in vivo and in vitro by vaccinia virus. Eur J Biochem 162:95–103. 10.1111/j.1432-1033.1987.tb10547.x. [DOI] [PubMed] [Google Scholar]
- 13.DiGiuseppe S, Rollins MG, Astar H, Khalatyan N, Savas JN, Walsh D. 2020. Proteomic and mechanistic dissection of the poxvirus-customized ribosome. J Cell Sci 134:jcs246603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jha S, Rollins MG, Fuchs G, Procter DJ, Hall EA, Cozzolino K, Sarnow P, Savas JN, Walsh D. 2017. Trans-kingdom mimicry underlies ribosome customization by a poxvirus kinase. Nature 546:651–655. 10.1038/nature22814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rollins MG, Jha S, Bartom ET, Walsh D. 2019. RACK1 evolved species-specific multifunctionality in translational control through sequence plasticity within a loop domain. J Cell Sci 132:jcs228908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rollins MG, Shasmal M, Meade N, Astar H, Shen PS, Walsh D. 2021. Negative charge in the RACK1 loop broadens the translational capacity of the human ribosome. Cell Rep 36:109663. 10.1016/j.celrep.2021.109663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cantu F, Cao S, Hernandez C, Dhungel P, Spradlin J, Yang Z. 2020. Poxvirus-encoded decapping enzymes promote selective translation of viral mRNAs. PLoS Pathog 16:e1008926. 10.1371/journal.ppat.1008926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhungel P, Cao S, Yang Z. 2017. The 5’-poly(A) leader of poxvirus mRNA confers a translational advantage that can be achieved in cells with impaired cap-dependent translation. PLoS Pathog 13:e1006602. 10.1371/journal.ppat.1006602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shirokikh NE, Spirin AS. 2008. Poly(A) leader of eukaryotic mRNA bypasses the dependence of translation on initiation factors. Proc Natl Acad Sci USA 105:10738–10743. 10.1073/pnas.0804940105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nicholson AL, Pasquinelli AE. 2019. Tales of detailed poly(A) tails. Trends Cell Biol 29:191–200. 10.1016/j.tcb.2018.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hershey JWB, Sonenberg N, Mathews MB. 2019. Principles of translational control. Cold Spring Harb Perspect Biol 11:a032607. 10.1101/cshperspect.a032607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joazeiro CAP. 2019. Mechanisms and functions of ribosome-associated protein quality control. Nat Rev Mol Cell Biol 20:368–383. 10.1038/s41580-019-0118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.D'Orazio KN, Green R. 2021. Ribosome states signal RNA quality control. Mol Cell 81:1372–1383. 10.1016/j.molcel.2021.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chandrasekaran V, Juszkiewicz S, Choi J, Puglisi JD, Brown A, Shao S, Ramakrishnan V, Hegde RS. 2019. Mechanism of ribosome stalling during translation of a poly(A) tail. Nat Struct Mol Biol 26:1132–1140. 10.1038/s41594-019-0331-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tesina P, Lessen LN, Buschauer R, Cheng J, Wu CC, Berninghausen O, Buskirk AR, Becker T, Beckmann R, Green R. 2020. Molecular mechanism of translational stalling by inhibitory codon combinations and poly(A) tracts. EMBO J 39:e103365. 10.15252/embj.2019103365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sundaramoorthy E, Leonard M, Mak R, Liao J, Fulzele A, Bennett EJ. 2017. ZNF598 and RACK1 regulate mammalian ribosome-associated quality control function by mediating regulatory 40S ribosomal ubiquitylation. Mol Cell 65:751–760.e4. 10.1016/j.molcel.2016.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garzia A, Jafarnejad SM, Meyer C, Chapat C, Gogakos T, Morozov P, Amiri M, Shapiro M, Molina H, Tuschl T, Sonenberg N. 2017. The E3 ubiquitin ligase and RNA-binding protein ZNF598 orchestrates ribosome quality control of premature polyadenylated mRNAs. Nat Commun 8:16056. 10.1038/ncomms16056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Juszkiewicz S, Hegde RS. 2017. Initiation of quality control during poly(A) translation requires site-specific ribosome ubiquitination. Mol Cell 65:743–750.e4. 10.1016/j.molcel.2016.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ikeuchi K, Tesina P, Matsuo Y, Sugiyama T, Cheng J, Saeki Y, Tanaka K, Becker T, Beckmann R, Inada T. 2019. Collided ribosomes form a unique structural interface to induce Hel2-driven quality control pathways. EMBO J 38:e100276. 10.15252/embj.2018100276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsuo Y, Ikeuchi K, Saeki Y, Iwasaki S, Schmidt C, Udagawa T, Sato F, Tsuchiya H, Becker T, Tanaka K, Ingolia NT, Beckmann R, Inada T. 2017. Ubiquitination of stalled ribosome triggers ribosome-associated quality control. Nat Commun 8:159. 10.1038/s41467-017-00188-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vind AC, Genzor AV, Bekker-Jensen S. 2020. Ribosomal stress-surveillance: three pathways is a magic number. Nucleic Acids Res 48:10648–10661. 10.1093/nar/gkaa757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu CC, Peterson A, Zinshteyn B, Regot S, Green R. 2020. Ribosome collisions trigger general stress responses to regulate cell fate. Cell 182:404–416.e14. 10.1016/j.cell.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iordanov MS, Pribnow D, Magun JL, Dinh TH, Pearson JA, Chen SL, Magun BE. 1997. Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol Cell Biol 17:3373–3381. 10.1128/MCB.17.6.3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gandin V, Gutierrez GJ, Brill LM, Varsano T, Feng Y, Aza-Blanc P, Au Q, McLaughlan S, Ferreira TA, Alain T, Sonenberg N, Topisirovic I, Ronai ZA. 2013. Degradation of newly synthesized polypeptides by ribosome-associated RACK1/c-Jun N-terminal kinase/eukaryotic elongation factor 1A2 complex. Mol Cell Biol 33:2510–2526. 10.1128/MCB.01362-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim TS, Kim HD, Park YJ, Kong E, Yang HW, Jung Y, Kim Y, Kim J. 2019. JNK activation induced by ribotoxic stress is initiated from 80S monosomes but not polysomes. BMB Rep 52:502–507. 10.5483/BMBRep.2019.52.8.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sengupta J, Nilsson J, Gursky R, Spahn CM, Nissen P, Frank J. 2004. Identification of the versatile scaffold protein RACK1 on the eukaryotic ribosome by cryo-EM. Nat Struct Mol Biol 11:957–962. 10.1038/nsmb822. [DOI] [PubMed] [Google Scholar]
- 37.Adams DR, Ron D, Kiely PA. 2011. RACK1, A multifaceted scaffolding protein: structure and function. Cell Commun Signal 9:22. 10.1186/1478-811X-9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmitt K, Smolinski N, Neumann P, Schmaul S, Hofer-Pretz V, Braus GH, Valerius O. 2017. Asc1p/RACK1 connects ribosomes to eukaryotic phospho-signaling. Mol Cell Biol 37:e00279-16. 10.1128/MCB.00279-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nielsen MH, Flygaard RK, Jenner LB. 2017. Structural analysis of ribosomal RACK1 and its role in translational control. Cell Signal 35:272–281. 10.1016/j.cellsig.2017.01.026. [DOI] [PubMed] [Google Scholar]
- 40.Juszkiewicz S, Chandrasekaran V, Lin Z, Kraatz S, Ramakrishnan V, Hegde RS. 2018. ZNF598 is a quality control sensor of collided ribosomes. Mol Cell 72:469–481.e7. 10.1016/j.molcel.2018.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andrade AA, Silva PN, Pereira AC, De Sousa LP, Ferreira PC, Gazzinelli RT, Kroon EG, Ropert C, Bonjardim CA. 2004. The vaccinia virus-stimulated mitogen-activated protein kinase (MAPK) pathway is required for virus multiplication. Biochem J 381:437–446. 10.1042/BJ20031375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stack J, Hurst TP, Flannery SM, Brennan K, Rupp S, Oda SI, Khan AR, Bowie AG. 2013. Poxviral protein A52 stimulates p38 mitogen-activated protein kinase (MAPK) activation by causing tumor necrosis factor receptor-associated factor 6 (TRAF6) self-association leading to transforming growth factor beta-activated kinase 1 (TAK1) recruitment. J Biol Chem 288:33642–33653. 10.1074/jbc.M113.485490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walsh D, Arias C, Perez C, Halladin D, Escandon M, Ueda T, Watanabe-Fukunaga R, Fukunaga R, Mohr I. 2008. Eukaryotic translation initiation factor 4F architectural alterations accompany translation initiation factor redistribution in poxvirus-infected cells. Mol Cell Biol 28:2648–2658. 10.1128/MCB.01631-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pereira AC, Leite FG, Brasil BS, Soares-Martins JA, Torres AA, Pimenta PF, Souto-Padron T, Traktman P, Ferreira PC, Kroon EG, Bonjardim CA. 2012. A vaccinia virus-driven interplay between the MKK4/7-JNK1/2 pathway and cytoskeleton reorganization. J Virol 86:172–184. 10.1128/JVI.05638-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang P, Langland JO, Jacobs BL, Samuel CE. 2009. Protein kinase PKR-dependent activation of mitogen-activated protein kinases occurs through mitochondrial adapter IPS-1 and is antagonized by vaccinia virus E3L. J Virol 83:5718–5725. 10.1128/JVI.00224-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sundaramoorthy E, Ryan AP, Fulzele A, Leonard M, Daugherty MD, Bennett EJ. 2021. Ribosome quality control activity potentiates vaccinia virus protein synthesis during infection. J Cell Sci 134:jcs257188. 10.1242/jcs.257188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DiGiuseppe S, Rollins MG, Bartom ET, Walsh D. 2018. ZNF598 plays distinct roles in interferon-stimulated gene expression and poxvirus protein synthesis. Cell Rep 23:1249–1258. 10.1016/j.celrep.2018.03.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carette JE, Guimaraes CP, Varadarajan M, Park AS, Wuethrich I, Godarova A, Kotecki M, Cochran BH, Spooner E, Ploegh HL, Brummelkamp TR. 2009. Haploid genetic screens in human cells identify host factors used by pathogens. Science 326:1231–1235. 10.1126/science.1178955. [DOI] [PubMed] [Google Scholar]
- 49.Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, Dal Cin P, Dye JM, Whelan SP, Chandran K, Brummelkamp TR. 2011. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477:340–343. 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Acciani MD, Lay Mendoza MF, Havranek KE, Duncan AM, Iyer H, Linn OL, Brindley MA. 2021. Ebola virus requires phosphatidylserine scrambling activity for efficient budding and optimal infectivity. J Virol 95:e0116521. 10.1128/JVI.01165-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fessler E, Jae LT. 2018. Haploid screening for the identification of host factors in virus infection. Methods Mol Biol 1836:121–137. 10.1007/978-1-4939-8678-1_6. [DOI] [PubMed] [Google Scholar]
- 52.Tanaka A, Tumkosit U, Nakamura S, Motooka D, Kishishita N, Priengprom T, Sa-Ngasang A, Kinoshita T, Takeda N, Maeda Y. 2017. Genome-wide screening uncovers the significance of N-sulfation of heparan sulfate as a host cell factor for chikungunya virus infection. J Virol 91:e00432-17. 10.1128/JVI.00432-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Labeau A, Simon-Loriere E, Hafirassou ML, Bonnet-Madin L, Tessier S, Zamborlini A, Dupre T, Seta N, Schwartz O, Chaix ML, Delaugerre C, Amara A, Meertens L. 2020. A genome-wide CRISPR-Cas9 screen identifies the dolichol-phosphate mannose synthase complex as a host dependency factor for dengue virus infection. J Virol 94:e01751-19. 10.1128/JVI.01751-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Riblett AM, Blomen VA, Jae LT, Altamura LA, Doms RW, Brummelkamp TR, Wojcechowskyj JA. 2016. A haploid genetic screen identifies heparan sulfate proteoglycans supporting rift valley fever virus infection. J Virol 90:1414–1423. 10.1128/JVI.02055-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jae LT, Raaben M, Riemersma M, van Beusekom E, Blomen VA, Velds A, Kerkhoven RM, Carette JE, Topaloglu H, Meinecke P, Wessels MW, Lefeber DJ, Whelan SP, van Bokhoven H, Brummelkamp TR. 2013. Deciphering the glycosylome of dystroglycanopathies using haploid screens for lassa virus entry. Science 340:479–483. 10.1126/science.1233675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Luteijn RD, van Diemen F, Blomen VA, Boer IGJ, Manikam Sadasivam S, van Kuppevelt TH, Drexler I, Brummelkamp TR, Lebbink RJ, Wiertz EJ. 2019. A genome-wide haploid genetic screen identifies heparan sulfate-associated genes and the macropinocytosis modulator TMED10 as factors supporting vaccinia virus infection. J Virol 93:e02160-18. 10.1128/JVI.02160-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Realegeno S, Puschnik AS, Kumar A, Goldsmith C, Burgado J, Sambhara S, Olson VA, Carroll D, Damon I, Hirata T, Kinoshita T, Carette JE, Satheshkumar PS. 2017. Monkeypox virus host factor screen using haploid cells identifies essential role of GARP complex in extracellular virus formation. J Virol 91:e00011-17. 10.1128/JVI.00011-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johnson AG, Lapointe CP, Wang J, Corsepius NC, Choi J, Fuchs G, Puglisi JD. 2019. RACK1 on and off the ribosome. RNA 25:881–895. 10.1261/rna.071217.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.LaFontaine E, Miller CM, Permaul N, Martin ET, Fuchs G. 2020. Ribosomal protein RACK1 enhances translation of poliovirus and other viral IRESs. Virology 545:53–62. 10.1016/j.virol.2020.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gallo S, Ricciardi S, Manfrini N, Pesce E, Oliveto S, Calamita P, Mancino M, Maffioli E, Moro M, Crosti M, Berno V, Bombaci M, Tedeschi G, Biffo S. 2018. RACK1 specifically regulates translation through its binding to ribosomes. Mol Cell Biol 38:e00230-18. 10.1128/MCB.00230-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Majzoub K, Hafirassou ML, Meignin C, Goto A, Marzi S, Fedorova A, Verdier Y, Vinh J, Hoffmann JA, Martin F, Baumert TF, Schuster C, Imler JL. 2014. RACK1 controls IRES-mediated translation of viruses. Cell 159:1086–1095. 10.1016/j.cell.2014.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ceci M, Welshhans K, Ciotti MT, Brandi R, Parisi C, Paoletti F, Pistillo L, Bassell GJ, Cattaneo A. 2012. RACK1 is a ribosome scaffold protein for beta-actin mRNA/ZBP1 complex. PLoS One 7:e35034. 10.1371/journal.pone.0035034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thompson MK, Rojas-Duran MF, Gangaramani P, Gilbert WV. 2016. The ribosomal protein Asc1/RACK1 is required for efficient translation of short mRNAs. Elife 5:e11154. 10.7554/eLife.11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shor B, Calaycay J, Rushbrook J, McLeod M. 2003. Cpc2/RACK1 is a ribosome-associated protein that promotes efficient translation in Schizosaccharomyces pombe. J Biol Chem 278:49119–49128. 10.1074/jbc.M303968200. [DOI] [PubMed] [Google Scholar]
- 65.Ruan Y, Sun L, Hao Y, Wang L, Xu J, Zhang W, Xie J, Guo L, Zhou L, Yun X, Zhu H, Shen A, Gu J. 2012. Ribosomal RACK1 promotes chemoresistance and growth in human hepatocellular carcinoma. J Clin Invest 122:2554–2566. 10.1172/JCI58488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Romano N, Veronese M, Manfrini N, Zolla L, Ceci M. 2019. Ribosomal RACK1 promotes proliferation of neuroblastoma cells independently of global translation upregulation. Cell Signal 53:102–110. 10.1016/j.cellsig.2018.09.020. [DOI] [PubMed] [Google Scholar]
- 67.Nunez A, Franco A, Madrid M, Soto T, Vicente J, Gacto M, Cansado J. 2009. Role for RACK1 orthologue Cpc2 in the modulation of stress response in fission yeast. Mol Biol Cell 20:3996–4009. 10.1091/mbc.e09-05-0388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rachfall N, Schmitt K, Bandau S, Smolinski N, Ehrenreich A, Valerius O, Braus GH. 2013. RACK1/Asc1p, a ribosomal node in cellular signaling. Mol Cell Proteomics 12:87–105. 10.1074/mcp.M112.017277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu SW, Katsafanas GC, Liu R, Wyatt LS, Moss B. 2015. Poxvirus decapping enzymes enhance virulence by preventing the accumulation of dsRNA and the induction of innate antiviral responses. Cell Host Microbe 17:320–331. 10.1016/j.chom.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Erez N, Wyatt LS, Americo JL, Xiao W, Moss B. 2021. Spontaneous and targeted mutations in the decapping enzyme enhance replication of modified vaccinia virus ankara (MVA) in monkey cells. J Virol 95:e0110421. 10.1128/JVI.01104-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Meade N, Furey C, Li H, Verma R, Chai Q, Rollins MG, DiGiuseppe S, Naghavi MH, Walsh D. 2018. Poxviruses evade cytosolic sensing through disruption of an mTORC1-mTORC2 regulatory circuit. Cell 174:1143–1157.e17. 10.1016/j.cell.2018.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ly M, Burgess HM, Shah SB, Mohr I, Glaunsinger BA. 2022. Vaccinia virus D10 has broad decapping activity that is regulated by mRNA splicing. PLoS Pathog 18:e1010099. 10.1371/journal.ppat.1010099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Meade N, King M, Munger J, Walsh D. 2019. mTOR dysregulation by vaccinia virus F17 controls multiple processes with varying roles in infection. J Virol 93:e00784-19. 10.1128/JVI.00784-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pereira AC, Soares-Martins JA, Leite FG, Da Cruz AF, Torres AA, Souto-Padron T, Kroon EG, Ferreira PC, Bonjardim CA. 2012. SP600125 inhibits orthopoxviruses replication in a JNK1/2-independent manner: implication as a potential antipoxviral. Antiviral Res 93:69–77. 10.1016/j.antiviral.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bonjardim CA. 2017. Viral exploitation of the MEK/ERK pathway—a tale of vaccinia virus and other viruses. Virology 507:267–275. 10.1016/j.virol.2016.12.011. [DOI] [PubMed] [Google Scholar]
- 76.Zaborowska I, Kellner K, Henry M, Meleady P, Walsh D. 2012. Recruitment of host translation initiation factor eIF4G by the vaccinia virus ssDNA-binding protein I3. Virology 425:11–22. 10.1016/j.virol.2011.12.022. [DOI] [PubMed] [Google Scholar]
- 77.Simms CL, Yan LL, Zaher HS. 2017. Ribosome collision is critical for quality control during no-go decay. Mol Cell 68:361–373.e5. 10.1016/j.molcel.2017.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Acosta-Reyes F, Neupane R, Frank J, Fernandez IS. 2019. The Israeli acute paralysis virus IRES captures host ribosomes by mimicking a ribosomal state with hybrid tRNAs. EMBO J 38:e102226. 10.15252/embj.2019102226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Murray J, Savva CG, Shin BS, Dever TE, Ramakrishnan V, Fernandez IS. 2016. Structural characterization of ribosome recruitment and translocation by type IV IRES. Elife 5:e13567. 10.7554/eLife.13567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Quade N, Boehringer D, Leibundgut M, van den Heuvel J, Ban N. 2015. Cryo-EM structure of hepatitis C virus IRES bound to the human ribosome at 3.9-A resolution. Nat Commun 6:7646. 10.1038/ncomms8646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Spahn CM, Kieft JS, Grassucci RA, Penczek PA, Zhou K, Doudna JA, Frank J. 2001. Hepatitis C virus IRES RNA-induced changes in the conformation of the 40s ribosomal subunit. Science 291:1959–1962. 10.1126/science.1058409. [DOI] [PubMed] [Google Scholar]
- 82.Yamamoto H, Collier M, Loerke J, Ismer J, Schmidt A, Hilal T, Sprink T, Yamamoto K, Mielke T, Burger J, Shaikh TR, Dabrowski M, Hildebrand PW, Scheerer P, Spahn CM. 2015. Molecular architecture of the ribosome-bound hepatitis C virus internal ribosomal entry site RNA. EMBO J 34:3042–3058. 10.15252/embj.201592469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Qin X, Sarnow P. 2004. Preferential translation of internal ribosome entry site-containing mRNAs during the mitotic cycle in mammalian cells. J Biol Chem 279:13721–13728. 10.1074/jbc.M312854200. [DOI] [PubMed] [Google Scholar]
- 84.Mulder J, Robertson ME, Seamons RA, Belsham GJ. 1998. Vaccinia virus protein synthesis has a low requirement for the intact translation initiation factor eIF4F, the cap-binding complex, within infected cells. J Virol 72:8813–8819. 10.1128/JVI.72.11.8813-8819.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Walsh D, Mohr I. 2006. Assembly of an active translation initiation factor complex by a viral protein. Genes Dev 20:461–472. 10.1101/gad.1375006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Walsh D, Mohr I. 2004. Phosphorylation of eIF4E by Mnk-1 enhances HSV-1 translation and replication in quiescent cells. Genes Dev 18:660–672. 10.1101/gad.1185304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Procter DJ, Furey C, Garza-Gongora AG, Kosak ST, Walsh D. 2020. Cytoplasmic control of intranuclear polarity by human cytomegalovirus. Nature 587:109–114. 10.1038/s41586-020-2714-x. [DOI] [PMC free article] [PubMed] [Google Scholar]