

Abstract
Protease-targeted chimeras (PROTACs) have been employed as a novel therapeutic approach, utilizing the ubiquitin-proteasome system for targeted protein degradation. PROTACs are heterobifunctional molecules consisting of an E3 ligase ligand and a small-molecule inhibitor for recruiting a protein of interest. After binding, PROTAC molecules recruit E3 ligase for ubiquitination of the protein of interest, which is followed by its proteasome-mediated degradation. PROTAC molecules have several advantages over traditional small-molecule inhibitors. A number of PROTAC molecules based on small-molecule inhibitors have been developed against various diseases, among which cereblon-based PROTAC molecules have received the greatest interest due to their promising clinical use. This article highlights the current trends in the discovery of cereblon-based PROTAC molecules along with their medicinal chemistry, clinical progression and future outlook in cancers, cardiovascular diseases and neurodegenerative disorders.
Keywords: : cereblon, clinical progression, CRBN, E3 ligase, PROTAC, protein degradation, ubiquitination
The landscape of drug discovery has changed significantly in the last decade. The traditional approach to inhibiting various enzymes or proteins, such as kinases and G-protein-coupled receptors, has been moving toward the novel and more challenging approach: targeted protein degradation. Owing to its potential, targeted protein degradation has attracted substantial interest in therapeutically modulating biological proteins or receptors that are difficult to target with traditional approaches or small molecules (i.e., undruggable targets). Some undruggable proteins have been intractable because of their broad or shallow active sites, which are difficult to block with small molecules [1]. Many of these proteins play key roles in various complex diseases like inflammatory diseases, cancer, cardiovascular and neurodegenerative disorders and therefore, have been considered as the important targets for these disease conditions. Molecules that can act on these undruggable proteins or receptors are identified as protease-targeted chimera (PROTAC) protein degraders, which are structurally heterobifunctional compounds. PROTAC molecules consist of two functionalities (moieties) clubbed together with an appropriately sized linker: one moiety binds to E3 ubiquitin ligase while the other binds to the protein of interest (POI). The simultaneous binding of the POI and E3 ubiquitin ligase through a PROTAC begins the ubiquitylation of the POI. After forming a ternary complex with the POI and E3 ligase, PROTACs induce a degradation process. Immediately after forming the complex, ubiquitination occurs followed by the development of a chain of ubiquitin molecules on the POI. These ubiquitylated proteins are then recognized by the specific proteasome and degradation of the POI is begun by the ubiquitin-proteasome system (UPS), even if the POI is not a physiological substrate of E3 ubiquitin ligase (Figure 1) [2–4].
Figure 1. . The mechanism of proteasome degradation mediated by protease-targeted chimera molecules: heterobifunctional protease-targeted chimera molecule brings ubiquitylated ligase and protein of interest in closer proximity and induces formation of ubiquitin chains on protein of interest; these ubiquitylated proteins can be recognized by the specific proteasome.
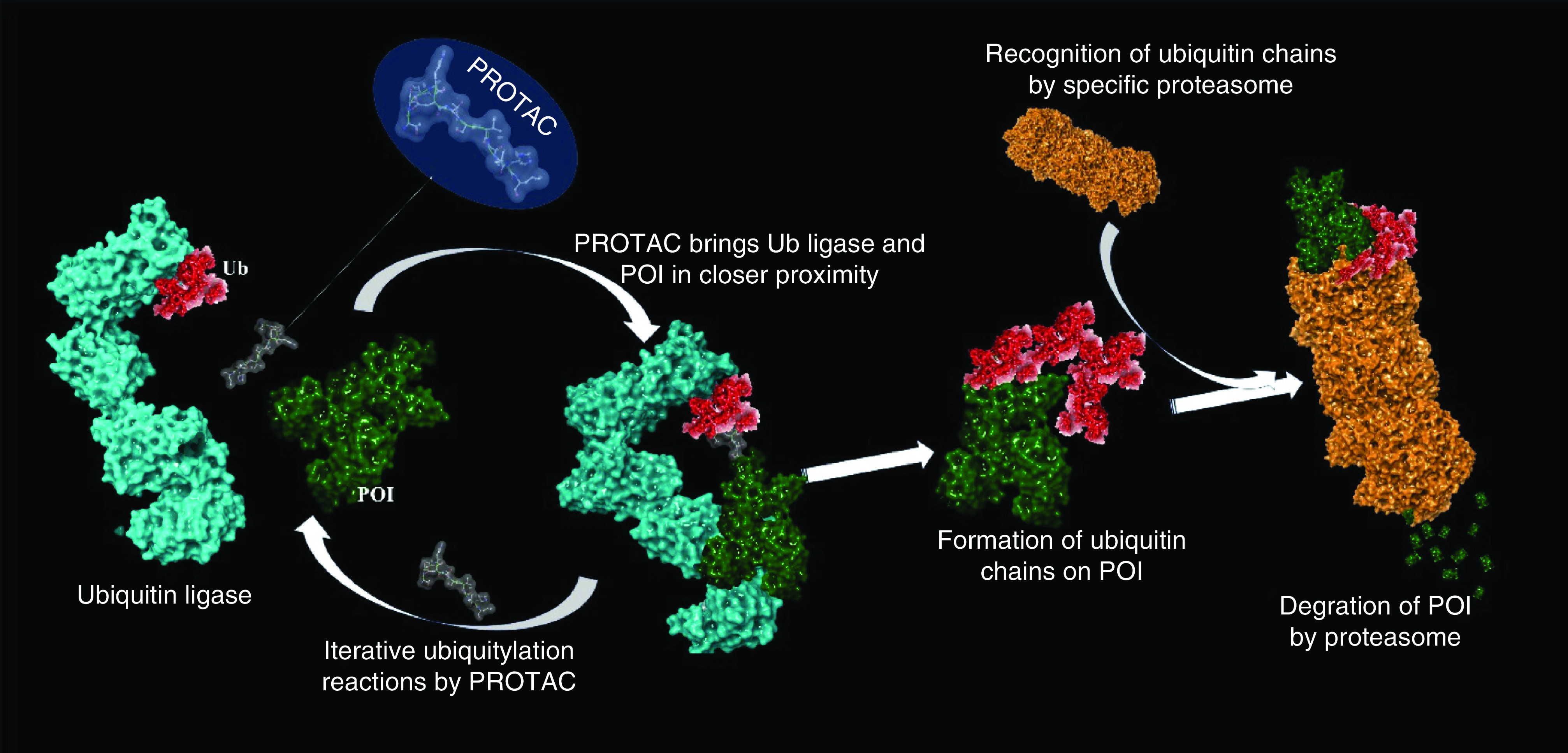
The proteasomal complex then initiates degradation of POI into small peptides.
POI: Protein of interest; PROTAC: Protease-targeted chimera.
PROTAC molecules are potentially advantageous compared with traditional small molecule inhibitors. The most critical advantage is the ability of PROTACs to capture undruggable proteins or receptors like transcription factors, proteins involved in protein–protein interactions or rat sarcoma virus proteins. Additionally, even a small concentration of PROTACs can initiate the catalysis and degradation of the POI in a unique, driven manner that could protect from the toxicity associated with the use of higher concentrations of drugs [5]. Since the PROTAC technology initiates the degradation of the POI, it can thus inhibit drug-resistant mutations or overexpression. A significant number of reports show that PROTAC molecules can induce the degradation of mutated forms of proteins, such as BCR-ABL [6], EGFR [7] and Bruton's tyrosine kinase (BTK) [8]. Furthermore, the selectivity of the targeted protein when using small molecule inhibitors is a major problem for medicinal chemists that can be overcome with the use of PROTACs that are potentially capable of achieving selectivity toward the proteins. For example, some CDKs are difficult to target due to their highly homologous nature. Recently, however, Santanu et al. [9] developed a PROTAC that can specifically degrade CDK2 for the treatment of cancer. Similarly, IL-1 receptor-associated kinases (IRAKs) are membrane-proximal, putative serine-threonine kinases that aid the protective response against pathogens by inducing acute inflammation followed by adaptive immune response through the adaptor protein (MyD88) in the human body [10]. Similarly, cancerous cells containing adaptor proteins activate IRAKs and promote inflammation and growth of the cancerous cells [10]. Therefore, IRAKs are also the therapeutic targets of anti-inflammatory drugs. Some kinases, like IRAK4, not only consist of a catalytic domain that can be targeted by small molecules but also have nonkinase functionality responsible for their signaling pathways [11]. PROTAC molecules could have the ability to target nonfunctional domains of IRAKs [12].
Discovery & development of PROTACs
The concept of PROTACs was first proposed in 2001 by Sakamoto and coworkers [13], specifically in hijacking the cancer-related protein machinery (kinases). They developed the peptide-based PROTAC-1 (Figure 2) to ubiquitinate the protein methionine aminopeptidase (MetAP2) and initiate its degradation. Specifically, PROTAC-1 helps to recruit MetAP2 and β-TRCP, which enables the E3 ligase to ubiquitylate MetAP2 from the unfertilized eggs of Xenopus laevis. In principle, these types of PROTACs have been considered ‘bioPROTACs’ as they contain long peptide-based E3 ligase ligands and are not fully small-molecule structures. Therefore, further advancement in the PROTAC approach employed the ligase ligands having a small molecular structure, leading to the discovery of low-molecular-weight HIF1α peptides [14,15]. The first small-molecule-based PROTAC (MZ1) was designed by incorporating the bromodomain protein inhibitor JQ1 recruit von Hippel–Lindau (VHL; a substrate recognition component of E3 ligase) tumor suppressor to degrade bromodomain-containing protein 4 (BRD4; Figure 2) [16,17]. This research opened the door for the PROTAC approach that produced methods different from the traditional approach to developing small molecule inhibitors.
Figure 2. . Chemical structures of the first reported peptide-based PROTAC-1 against MetAP2 and small inhibitor-based protease-targeted chimera (MZ1) against BRD4.
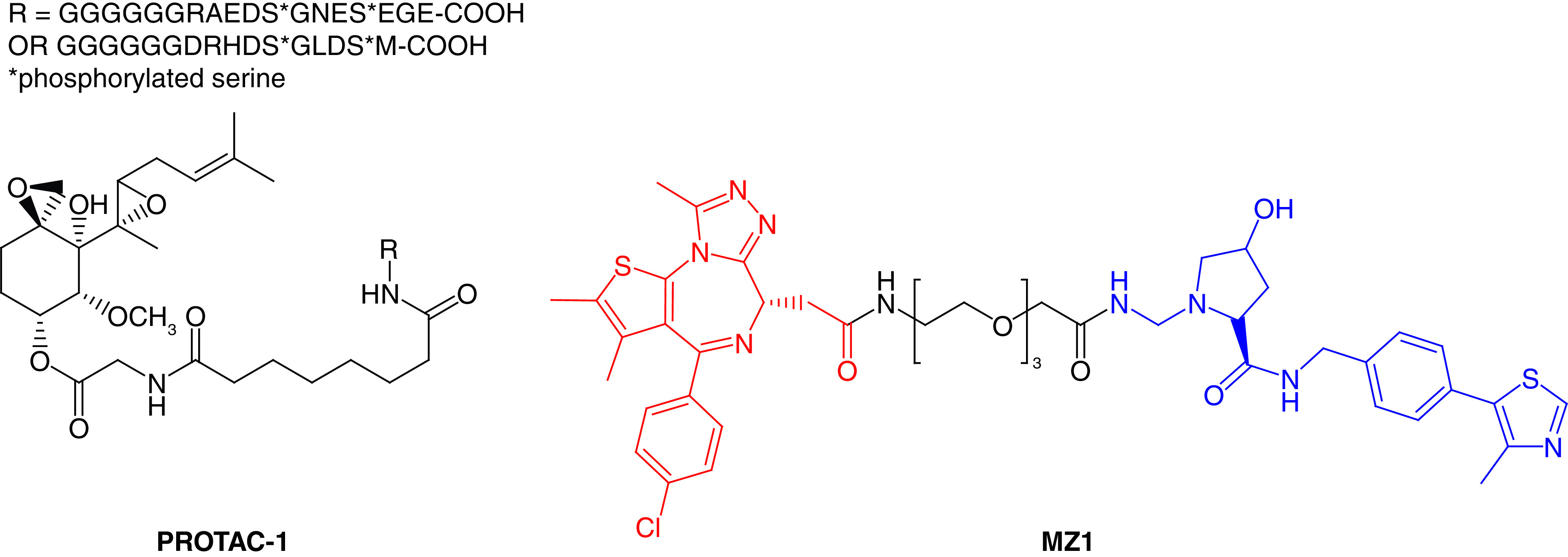
PROTAC: Protease-targeted chimera.
Over the past two decades, the approach has successfully moved from cellular (in vitro) studies toward the animal models of various disease conditions (in vivo) and has progressed fully to clinical trials. Before 2020, it was a general belief in the field of PROTACs that these compounds work safely, have druglike properties and work on the targeted protein as expected. In 2020, the clinical potential of PROTACs was strengthened by the positive results of phase I trials of two PROTAC molecules, ARV-110 (androgen receptor [AR] gene degrader) and ARV-471 (Table 1). The AR degrader PROTAC molecule ARV-110 was clinically tested to treat prostate cancer in humans [18]. The results of a phase I trial demonstrated that PROTAC molecules could be a better option to treat metastatic castration-resistant prostate cancer compared with the insensitive and resistant traditional androgenic therapies. These clinical trials also revealed a safer and tolerable dose of ARV-110 (420 mg). This was the first evidence for the PROTAC approach in humans to elicit significant antitumor activity by degrading the associated proteins, as measured by the reduction in prostate-specific antigen. ARV-110 is currently being assessed in a phase II trial that began in late 2020 with the safer dose of 420 mg [18]. The second PROTAC molecule, ARV-471, is an endoplasmic reticulum protein degrader that entered clinical trials for the treatment of breast cancer. The ARV-471-treated population achieved better results than patients treated with other selective ER degraders at the same stage of development. Currently, ARV-471 is being tested in a phase II clinical trial for the treatment of metastatic breast cancer [19]. After the success of these two PROTAC small molecules and 20 years after the theoretical proposition of this approach, approximately 14 PROTAC molecules had entered into clinical trials by the end of 2021 for the treatment of various disease conditions. The PROTAC molecules currently being tested in clinical trials are summarized in Table 1 [18–29].
Table 1. . Protease-targeted chimera molecules under clinical development.
| PROTAC/structure | E3 ligase & target | Condition | Company | Administration route | Phase | Status/NCT no. | Ref. |
|---|---|---|---|---|---|---|---|
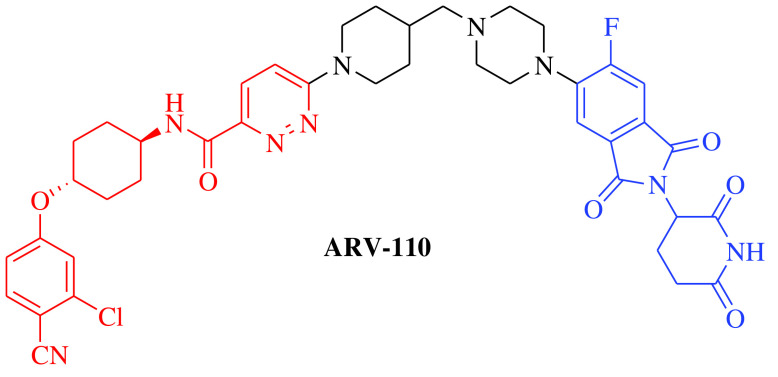
|
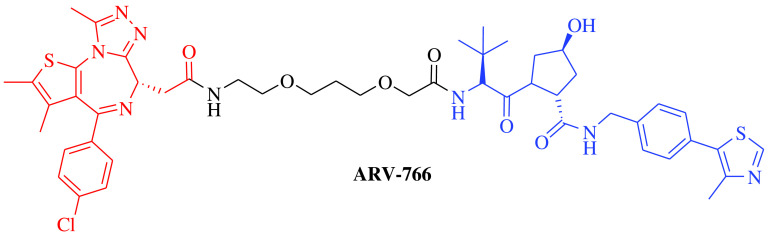
E3 ligase: CRBN Target: AR |
Prostate cancer, metastatic | Arvinas | Oral | Phase II | Recruiting/ NCT03888612 | [18] |
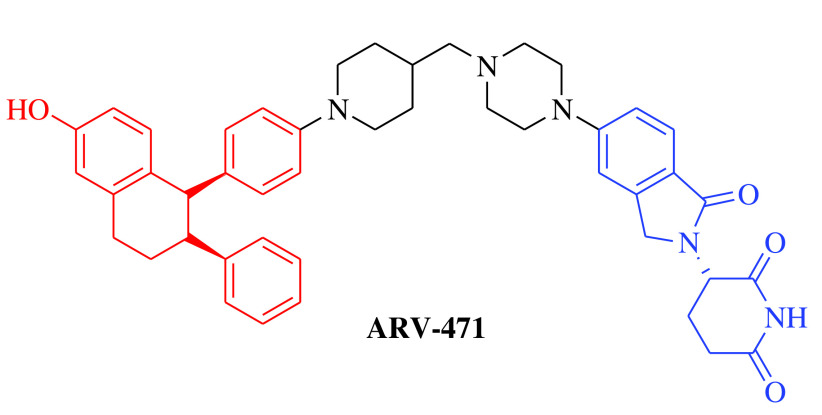
|
E3 ligase: CRBN Target: ER |
Breast cancer | Arvinas/Pfizer | Oral | Phase II | Recruiting/ NCT04072952 | [19] |
|
E3 ligase: VHL Target: AR |
Prostate cancer, metastatic | Arvinas Androgen Receptor, Inc. | Oral | Phase I | Recruiting/ NCT05067140 | [20] |
|
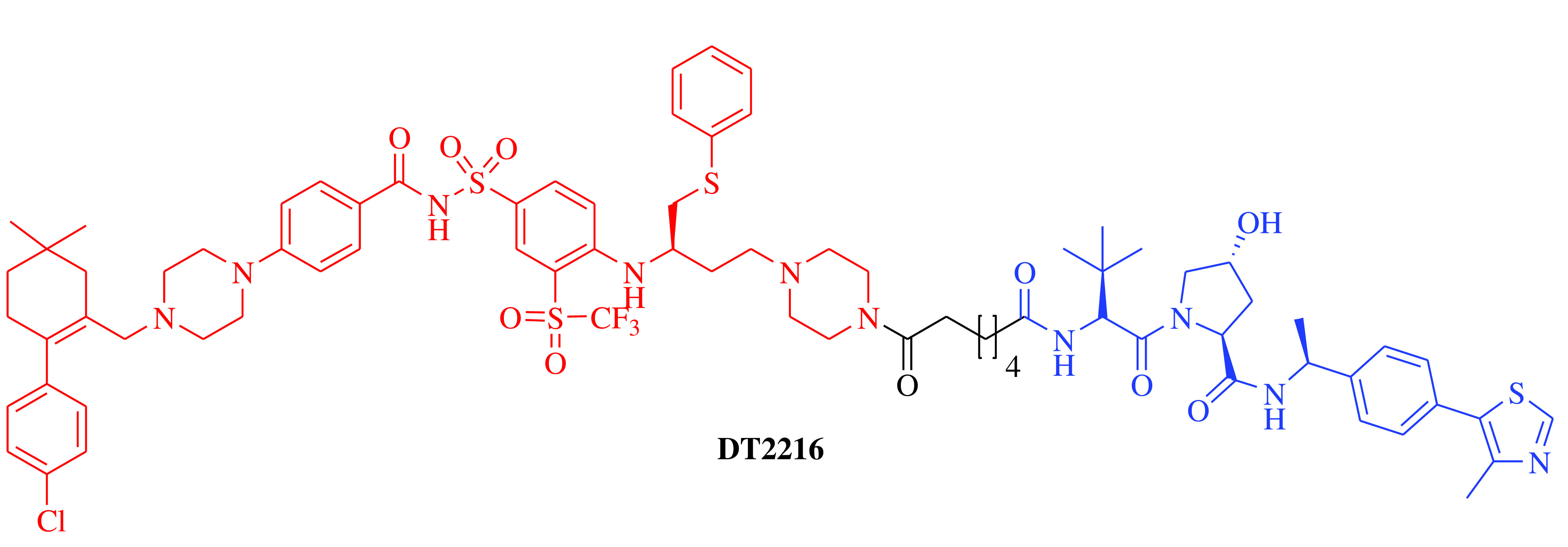
E3 ligase: VHL Target: BCL-XL |
Solid tumor and hematologic malignancy | Dialectic Therapeutics, Inc | Intravenous | Phase I | Recruiting/ NCT04886622 | [21] |
|
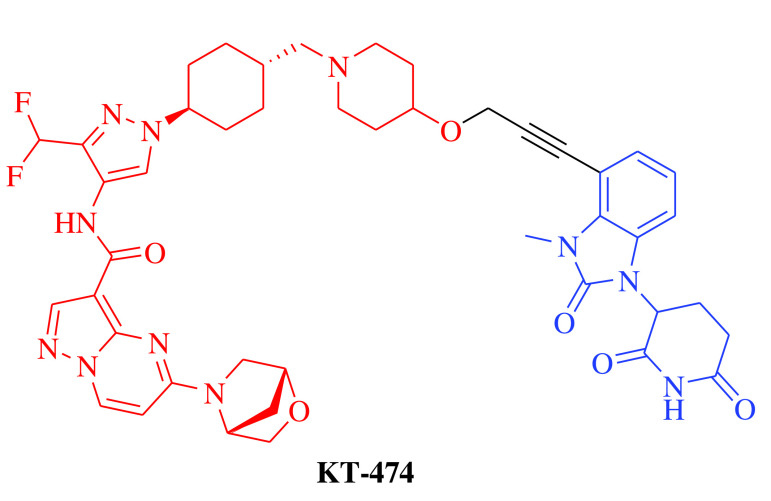
E3 ligase: CRBN Target: IRAK4 |
Atopic dermatitis and hidradenitis suppurativa | Kymera Therapeutics, Inc. | Oral | Phase I | Recruiting/ NCT04772885 | [22] |
| KT-413 | E3 ligase: CRBN Target: IRAK4 |
Non-Hodgkin lymphoma and diffuse large B-cell lymphoma | Kymera Therapeutics, Inc. | Intravenous | Phase I | Recruiting/ NCT05233033 | [23] |
| KT-333 | E3 ligase: Not disclosed Target: STAT3 |
Non-Hodgkin lymphoma, peripheral T-cell lymphoma, cutaneous T-cell lymphoma, large granular lymphocytic leukemia and solid tumors | Kymera Therapeutics, Inc. | Intravenous | Phase I | Recruiting/ NCT05225584 | [24] |
| NX-2127 | E3 ligase: CRBN Target: BTK |
Chronic lymphocytic leukemia, small lymphocytic lymphoma, Waldenstrom macroglobulinemia, mantle cell lymphoma and follicular lymphoma | Nurix Therapeutics, Inc. | Oral | Phase I | Recruiting/ NCT04830137 | [25] |
| NX-5948 | E3 ligase: CRBN Target: BTK |
Chronic lymphocytic leukemia, small lymphocytic lymphoma, Waldenstrom macroglobulinemia, mantle cell lymphoma and follicular lymphoma | Nurix Therapeutics, Inc. | Oral | Phase I | Recruiting/ NCT05131022 | [26] |
| CFT8634 | E3 ligase: CRBN Target: BRD9 |
Synovial sarcoma and soft tissue sarcoma | C4 Therapeutics, Inc. | Oral | Phase II | Recruiting/ NCT05355753 | [27] |
| CC-94676 | E3 ligase: CRBN Target: AR |
Prostate cancer, metastatic | Celgene | Oral | Phase I | Recruiting/ NCT04428788 | [28] |
| AC682 | E3 ligase: CRBN Target: ER |
Breast cancer | Accutar Biotechnology Inc. | Oral | Phase I | Recruiting/ NCT05080842 | [29] |
AR: Androgen receptor; BCL-XL: B-cell lymphoma, extra large; BRD9: Bromodomain-containing protein 9; BTK: Bruton's tyrosine kinase; CRBN: Cereblon protein, ER: Endoplasmic reticulum protein; IRAK: IL-1 receptor-associated kinase; NCT: National clinical trial; PROTAC: Protease-targeted chimera; STAT3: Signal transducer and activator of transcription 3; VHL: von Hippel–Lindau tumor suppressor protein.
Current trends in the discovery & development of PROTACs
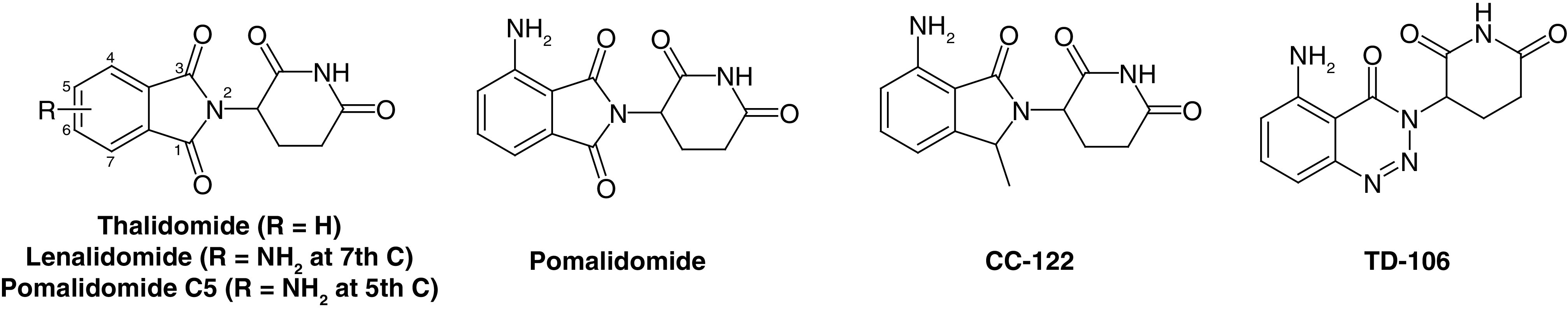
After the discovery of the imine-based ligand thalidomide for cereblon (CRBN) E3 ligase, a transformation in the field of PROTACs occurred [15,30]. Surprisingly, among ∼600 E3 ligases, only 14 PROTAC molecules have successfully reached clinical trials and are currently under investigation for different disease conditions, mainly based on CRBN- and VHL-based PROTACs (Table 1). Indeed, CRBN- and VHL-based mechanisms dominate, likely due to the availability of specific small-molecule binders that are structurally and biophysically well-established and characterized and have suitable physicochemical properties. This molecular profile has so far been difficult to achieve for binders of other E3 ligases [31–34]. CRBN-based PROTACs have been successfully employed in the degradation of different target proteins associated with various diseases. Here, PROTAC molecules discovered based on CRBN as their target E3 ligase published in the last two years (2021–2022) are systematically discussed. A number of reports have recently been published summarizing PROTACs, including CRBN-based PROTAC molecules [35], but may lack detailed clinical trial data as well as perspective on the future of the PROTAC approach. This review describes the development of recently discovered PROTAC molecules as well as current trends and future directions for molecules that have been shown to be active and are currently being tested in clinical trials.
Cereblon-based therapeutic PROTAC molecules
CRBN, a component of a cullin-RING ubiquitin ligase complex, is the target of thalidomide [36]. Thalidomide and its analogs directly bind to CRBN and recruit protein targets to the CRL4CRBN E3 ligase [37]. Structurally, CRBN is comprised of 442 amino acid protein sequences that serve as a substrate of the cullin-RING ligase 4 E3 ubiquitin ligase complex that identifies substrates for ubiquitination and consequent proteasomal degradation [38]. CRBN can also directly bind to the cytosolic carboxy-terminus of large conductance Ca2+ and voltage-activated K+ channel subunits [39]. These proteins also participate in glutamine regulation, the dysregulation of which is associated with various disease conditions such as cancer and metabolic abnormalities [40,41]. Glutamine synthetase is a crucial enzyme in the signaling pathway that regulates glutamine levels. This enzyme directly binds to CRBN, leading to its ubiquitination by the E3 ubiquitin ligase complex and an ultimate decrease in glutamine [42]. Based on clinical trial data, CRBN ligands are the most frequently used E3 ligase ligands used in developing new PROTACs [30,43–45]. The major advantages of CRBN ligands include suitable physicochemical properties like molecular weight, lipophilicity, solubility and lack of metabolic sites; strong and specific binding affinities for their E3 ligases and clear and well-characterized binding mechanisms. These characteristics made these ligands successful candidates for the degradation of different types of proteins associated with disease conditions including cancer, neurodegenerative disorders, cardiovascular diseases, immune disorders and various viral infections [46–49]. Figure 3 shows molecule ligands that are specific binders of E3 ligase and used for CRBN PROTACs.
Figure 3. . Small-molecule ligands of E3 ligase used for cereblon protease-targeted chimeras.
Cereblon-based PROTACs for the treatment of cancer
Li et al. designed a series of ALK degraders [50] by conjugating the parent drug LDK378 and CRBN ligands together. In vitro assays revealed that PROTAC molecule B3 (Figure 4) exhibited significant ALK inhibition as well as reductions in cellular levels in the H3122 tumor cell line. The molecule showed promising and potent anticancer effects in vitro as well as in vivo using a xenograft tumor model and PROTAC molecule B3 was better than the parent drug molecule, LDK378. Taking together, the in vitro and in vivo profiles of B3 demonstrate the clinical potential of these PROTACs and warrant further investigation.
Figure 4. . Chemical structures of protease-targeted chimera molecules as degraders of ALK/AR/BCR-ABL for the treatment of cancer (blue indicates the E3 ligase ligand cereblon; red indicates the target inhibitor).
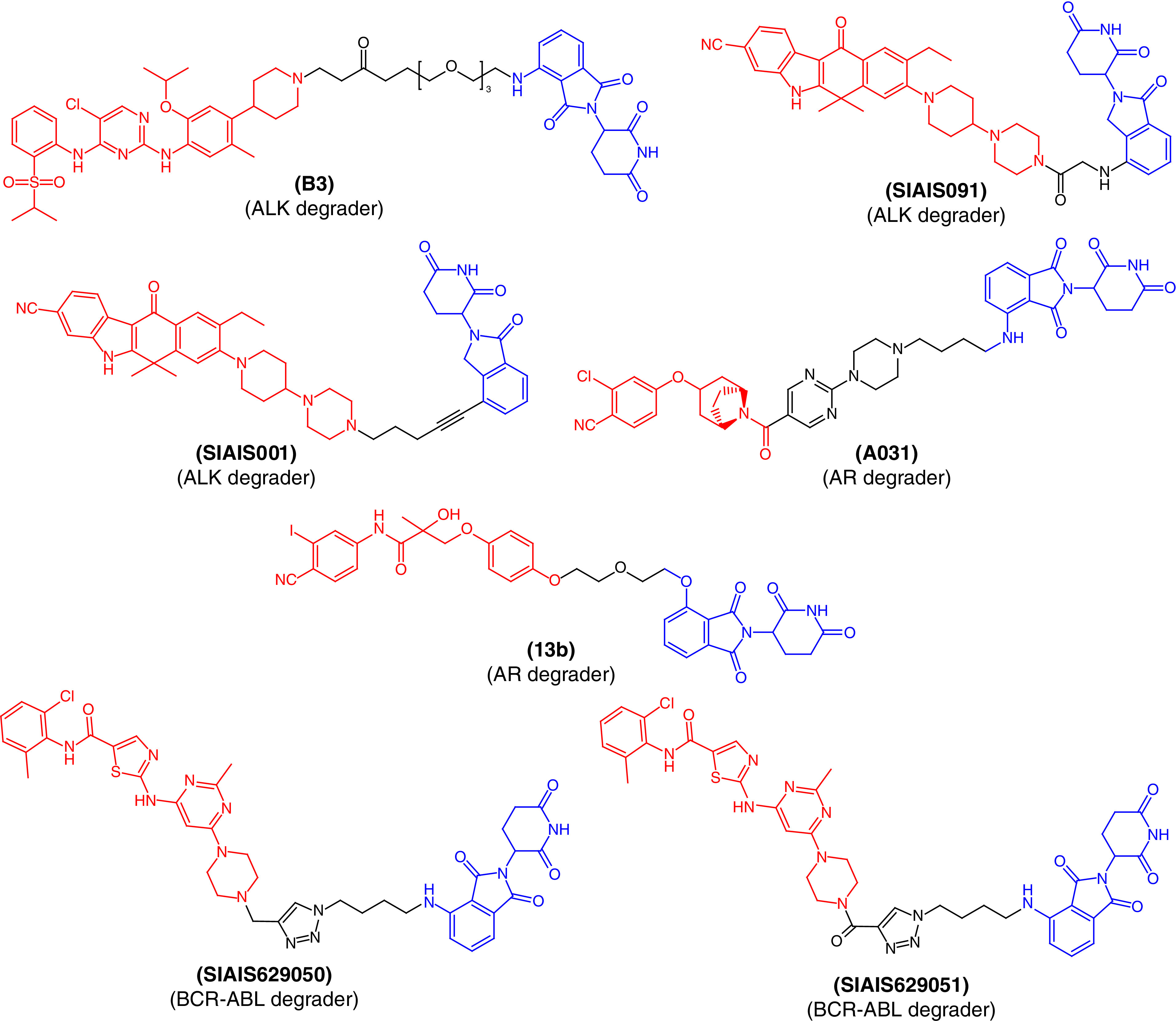
ALK: Anaplastic lymphoma kinase; AR: Androgen receptor.
Jiang et al. also reported a structure-based design, synthesis and evaluation of ALK-degrading PROTACs based on alectinib as the ALK binder [51]. They obtained a series of PROTACs using different CRBN ligands among which two PROTAC molecules, SIAIS091 and SIAIS001 (Figure 4), were found to induce effective degradation of ALK. They also showed anticancer effects that mechanistically arrested the cell cycle in the G1/S phase. Of both ALK degraders, SIAIS001 showed significantly better oral bioavailability and pharmacokinetic profile.
ARs are members of the nuclear hormone superfamily that plays a critical role in the maintenance of sexual characteristics and development of the prostate gland in males. Disruption of this receptor may lead to prostate cancer and long-term treatment with current drug therapies that are prone to produce resistance as well as toxicity. To determine the PROTAC molecule with the lowest toxicity and better binding, Wang et al. synthesized AR degraders consisting of CRBN/VHL E3 ligands and AR antagonists [52]. They found PROTAC molecule A031 (Figure 4) to be the best molecule, inhibiting 55.44% of AR-positive VCaP prostate cancer cells at a 1-μM concentration. Similarly, a series of AR PROTACs having bicalutamide derivatives and thalidomide were designed and synthesized [53]. The synthetic PROTAC molecules were evaluated to examine their primary ability to degrade AR. Among them, 13b (Figure 4) was found to produce the best dose-dependent degradation of AR and could be useful for future drug development and optimization for the treatment of prostate cancer.
Using the click chemistry approach to synthesize triazole-based molecules, Jiang et al. [54] reported two PROTAC molecules, SIAIS629050 and SIAIS629051, with the potential to inhibit the proliferation of K562 myelogenous leukemia cells (Figure 4). Structurally, the PROTACs were designed to incorporate pomalidomide as a CRBN ligand and dasatinib derivatives as BCR-ABL degraders. These molecules were found to degrade BCR-ABL in a dose-dependent manner and could act as ‘hit’ molecules for further clinical development to treat BCR-ABL-associated disease conditions.
Since the chromatin regulators, CBP and p300, play critical roles in driving enhancer-driven transcription in cancer cells, these proteins became therapeutic targets in cancer treatment [55–57]. Several drug like molecules that inhibit the functionality of these proteins have been developed to treat cancer. However, compounds that act on a single active domain of a target cannot completely block the p300/CBP function in cancerous cells, since p300/CBP consists of eight distinct active domains [58]. Therefore, to target p300/CBP, Vannam et al. [55] designed a series of PROTAC molecules that were surprisingly potent against multiple myeloma cells. dCBP-1 (Figure 5) was the best PROTAC molecule among the series and also abolished the activator of MYC oncogene expression. This molecule could be useful to treat cancer, with its unique class of protein degradation of acetyltransferases.
Figure 5. . Chemical structures of protease-targeted chimera molecules as degraders of p300-CBP/CDK2/CDK4/CDK6 for the treatment of cancer (blue indicates the E3 ligase ligand cereblon; red indicates the target inhibitor).
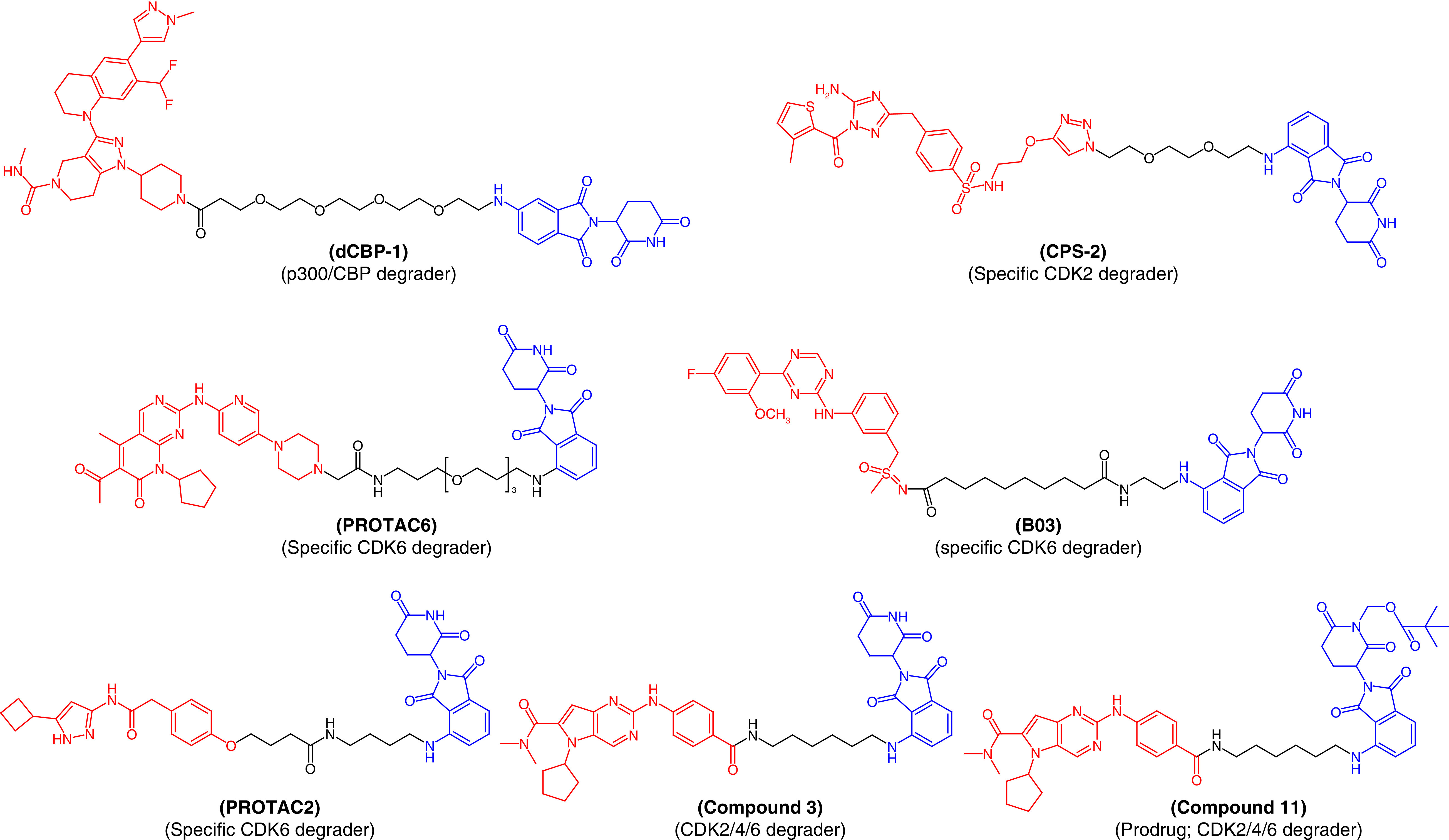
PROTAC: Protease-targeted chimera.
Similarly, cyclin-dependent kinase (CDK) enzymes also play an important role in cancer cell division and their non-selective inhibition lead to unwanted side effects with the current drug therapy associated to these enzymes. However, there are currently no available selective inhibitors that can exhibit specific CDK isoform inhibition. The inactivation of CDK2 is a promising approach to arresting acute myeloid leukemia (AML). Therefore, Rao et al. developed first-in-class specific CDK2 degraders, conjugating a CRBN-based ligand with a nonspecific CDK2 inhibitor, JNJ-7706621 [59]. Among the PROTAC molecules, CPS-2 (Figure 5) initiated rapid and selective CDK2 degradation without affecting other CDK isoforms, inducing remarkable differentiation of AML cell lines. These results exemplify the advantages of PROTAC molecules in specifically targeting sites associated with disease conditions. Similarly, PROTAC6 (Figure 5) was designed to specifically degrade CDK6 without affecting other CDK family proteins [60].
Bian et al. designed a series of CDK9 degraders from its well-known inhibitor, BAY-1143572 [61]. At a very low nanomolar concentration ranging from 7.62 nM to 31.16 nM, they degraded CDK9 in AML cells. PROTAC molecule B03 (Figure 5) demonstrated pronounced efficiency in breaking CDK9, specifically, with a degradation concentration (DC50) value of 7.62 nM against the MV4-11 myelomonocytic leukemia cell line. In vivo experiments also confirmed the degradation of CDK9 resulting from treatment with B03. Therefore, B03 represents a hit molecule that could be valuable in the treatment of AML. Natrajan et al. [62] also reported an aminopyrazole-based CDK9 degrader, PROTAC2, that showed efficiency in degrading CDK9 in the nanomolar range (150 nM). PROTAC2 is a selective CDK9 degrader which was effective against pancreatic cancer and is superior to the BCL2-selective FDA-approved inhibitor, venetoclax. Therefore, PROTAC2 is another example justifying the further development of PROTACs (Figure 5).
Instead of specific degradation, PROTACs can also target multiproteins. Compound 3 (Figure 5), designed by using ribociclib and a CRBN ligand, can simultaneously act on CDK2, CDK4 and CDK6, rapidly halt the progression of the cell cycle and induce apoptosis of cancerous cells [63]. These three CDK isoforms have been considered promising therapeutic targets for solid tumors. PROTAC molecule 3 showed low oral bioavailability, which was improved by designing PROTAC molecule 11 (Figure 5) with the intact efficiency to degrade CDK2/4/6. Oral bioavailability of 68% was found in a mouse model [63].
Cereblon-based PROTACs for the treatment of cardiovascular diseases
HMG-CoA reductase (or HMGCR) is the rate-limiting, eight-pass transmembrane protein for cholesterol synthesis [64]. HMGCR is the main target of statins (antidyslipidemic agents), which have been widely used to treat cardiovascular diseases. Unfortunately, statins are associated with various side effects, including skeletal muscle damage and are not effective in patients with statin intolerance [65]. Therefore, PROTAC molecules could be beneficial for those patients suffering from statin intolerance. To our knowledge, Rao et al. [66] first reported a PROTAC molecule, P22A (Figure 6), that could be useful in cardiovascular diseases. They tethered atorvastatin (a well-known antidyslipidemic agent) to a CRBN ligand using the click chemistry approach. P22A showed potent degradation of HMGCR when tested in Chinese hamster ovary cells, with a DC50 value of 100 nM. The effect on HMGCR by P22A was far better than atorvastatin, indicating that P22A can replace statin therapy in patients with statin intolerance. Interestingly, P22A also activated the sterol regulatory element-binding protein pathway to block the biosynthesis of cholesterol. This first-time targeting of HMGCR by a PROTAC molecule is a potential strategy for the treatment of cardiovascular diseases.
Figure 6. . Molecular structures of protease-targeted chimera molecules targeting HMGCR/H-PGDS/IRAK4/GSK-3β for treatment of cardiovascular/immune and neurodegenerative diseases (blue indicates the E3 ligase ligand CRBN; red indicates the target inhibitor).
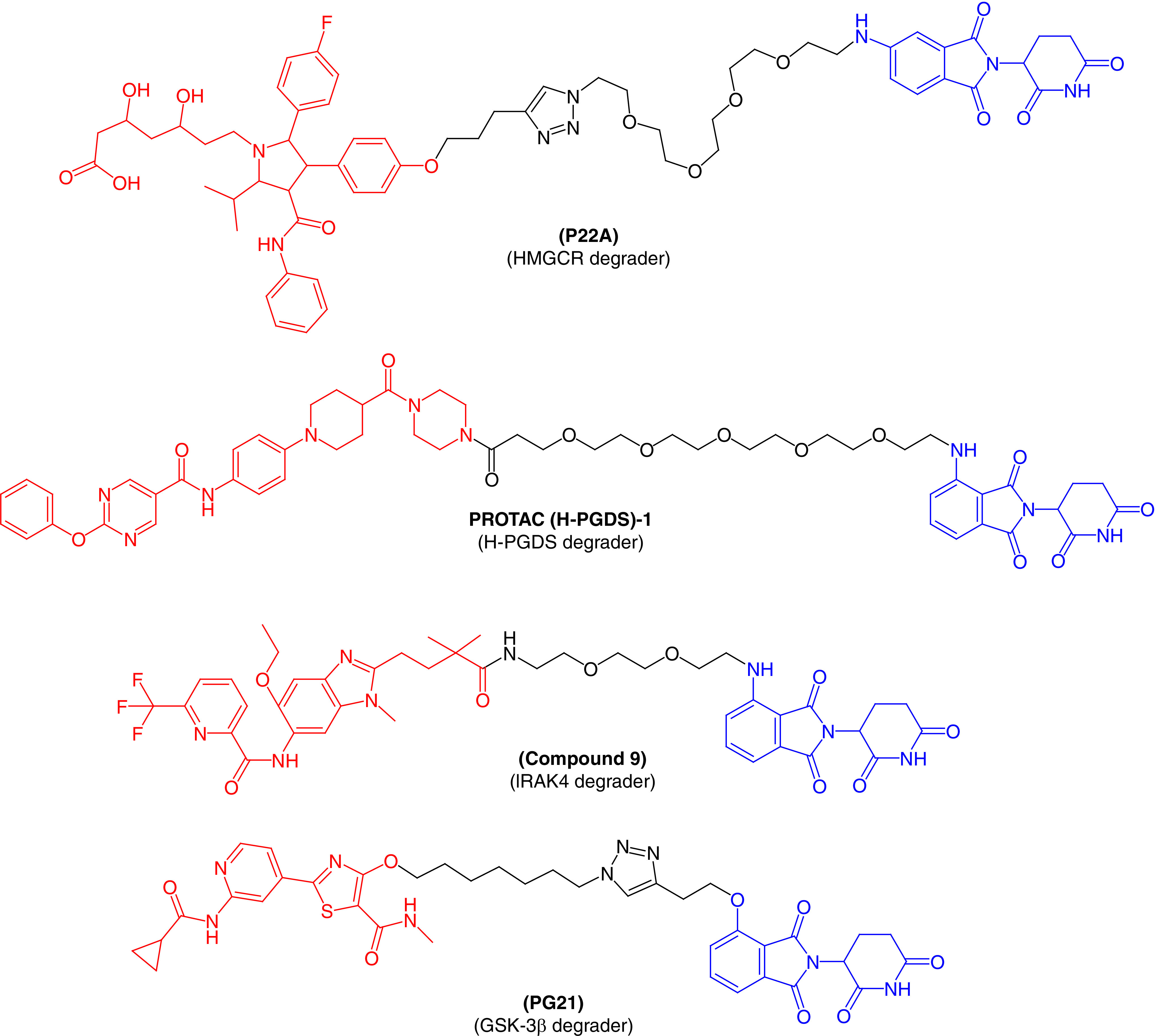
GSK-3β: Glycogen synthase kinase-3β; HMGCR: HMG-CoA reductase; H-PGDS: Hematopoietic prostaglandin-D synthase; IRAK4: IL receptor-associated kinase 4.
Cereblon-based PROTACs for the treatment of immune diseases
Prostaglandin-D2 (PGD2) binds to its receptor and activates downstream signaling pathways to perform its function. This is a major component produced by mast cells in response to allergic reactions. Human prostaglandin synthase (hPGDS) is an enzyme that induces PGD2 synthesis and has been viewed as a therapeutic target for diseases like Duchenne muscular dystrophy, physiological sleep and allergic reactions. There are a number of hPGDS inhibitors described in the published literature, but none are effective for the treatment of these diseases [67]. A chimeric small molecule that can degrade hPGDS via a UPS was recently discovered by Demizu et al. [68] Pamolidomide was used as a CRBN ligand and TFC-007 was used as an hPGDS binder incorporated into a single molecular architecture to produce PROTAC(H-PGDS)-1. Biological results revealed that PROTAC(H-PGDS)-1 effectively and persistently induced the degradation of hPGDS and sustained the suppression of PGD2 production for a longer period than TFC-007. Similarly, Duan et al. [11] also designed and synthesized a series of PROTAC molecules by conjugating an IRAK4 inhibitor with CRBN ligand pomalidomide, with the potential to degrade IRAK4. Of the molecules tested, compound 9 (Figure 6) showed excellent degradation of IRAK4 in lymphoma cell lines (TMD8 and OCILY10). This further blocks the downstream signaling pathway of immune inflammation, including NF-κB signaling. These results suggest a substantial advantage in using PROTAC molecules to inhibit the MYD88 L265P mutant compared with the parent IRAK4 inhibitor.
Cereblon-based PROTAC molecules for the treatment of neurodegenerative disorders
A multifunctional serine/threonine protein kinase, GSK-3, was found to be involved in the pathology of Alzheimer's disease. The proinflammatory action performed by GSK-3β can cause the loss of neuronal functions [69,70]. Therefore, GSK-3β is a major therapeutic target for various neurodegenerative disorders. In an effort to target GSK-3β, Sun et al. [71] first reported a series of PROTACs based on E3 ubiquitin ligase CRBN to treat neuronal abnormalities. They synthesized several PROTAC molecules and most displayed good inhibitory activity against GSK-3β, with IC50 values in the nanomolar range and moderate protein degradation ability against GSK-3β. PG21 (Figure 6) effectively degraded GSK-3β (inducing 44.2% protein degradation at 2.8 mM), which was tested using the western blot technique. Further pharmacological experiments confirmed that the degradation of GSK-3β by PG21 is regulated by the UPS. As the first successful development of a PROTAC to degrade GSK-3β protein, their research could provide candidates for further investigation to treat neurodegenerative disorders.
Conclusion
With the recent increased interest and investment in the development of PROTACs as degraders, this approach could become a therapeutic modality. The first wave of clinical-stage protein degraders is fully aimed at drugged targets. Success against these drugged targets advanced the promise of the approach and underscores the ability of these molecules to become best-in-class drugs by degrading the target instead of its inhibition. In sum, the targeted protein-degradation approach is well defined for novel therapeutic media based on medicinal chemistry and biology, with vast potential in the treatment of various diseases. Therefore, we expect several PROTAC molecules to constitute the next wave of therapeutic drug candidates against various diseases.
Future perspectives
Traditional small-molecule drug discovery for intracellular targets has been focused, thus far, on developing high-affinity inhibitors of the active site or allosteric site binders that can ultimately shut down the function of the POI. No doubt this was a highly effective approach, however, this has left many drug targets undrugged or underdrugged [72]. PROTAC molecules target a POI with existing ligands (available inhibitors). Published reports suggest that the major advantage of PROTACs is having inhibitors as the ligands for POI in their molecular structures that act in an iterative and pseudocatalytic fashion by binding and facilitating the binding of E3 ligase to its target. POIs can be degraded without requiring a large amount of the PROTAC. Intriguingly, the ‘PROTACable’ POI do not require an active site or small-molecule binding site that is approachable by an E3 ligase. Access to the phosphatidylinositol surface in clear proximity to the binding site by a recruited ligase is necessary.
Despite these vast biological applications, the area remains lacking from the E3-ligase point of view. Only a handful of E3 ligases (CRBN, VHL, MDM2, IAPs, DCAF15,16 and RNF114) can be employed to develop POI degraders. Specific ligands for many other ligases remain unknown, thereby limiting extended applications of this approach. Among the available ligands of E3 ligases, ligands of CRBN E3 ligase have been successfully and most widely utilized in the development of PROTACs against several diseases. The majority of the PROTAC molecules currently under clinical translation were designed using CRBN-based E3 ligase ligands. There is a pressing need to further develop new E3 ligases. Some PROTAC molecules examined did not degrade their targeted proteins and the underlying reasons behind their inactivity are unknown. This could be improved by incorporating a computer-aided drug design approach and performing computational modeling. More efforts are warranted to more deeply examine the efficiency and safety profiles of PROTAC molecules based on their clinical utility.
Executive summary.
Protease-targeted chimeras (PROTACs) have been developed as a useful therapeutic approach for targeted protein degradation and have a number of advantages over the traditional drug-discovery approach to small molecule inhibitors.
For the degradation of a protein of interest (POI), this approach mainly utilizes the ubiquitin-proteasome system.
Recent literature has focused on the foremost advantage of PROTACs, having inhibitors as ligands for POIs in the molecular skeleton of PROTACs. This allows them to act in an iterative and pseudocatalytic fashion by interacting and facilitating the binding of E3 ligase to its target. A large amount of PROTACs is not required to degrade the POI.
Among the available ligands of E3 ligases, cereblon E3 ligase ligands have been successfully and widely utilized in the development of PROTACs against several diseases. A majority of the PROTAC molecules currently under clinical translation have been designed using cereblon-based E3 ligase ligands.
Few papers employing computational modeling have been reported. More efforts are needed to deeply assess the crystal structures of POIs and E3 ligases to determine exact binding relations and develop novel PROTAC molecules through computer-aided drug design.
Footnotes
Financial & competing interest disclosure
The research of DK Agrawal is supported by research grants R01 HL144125 and R01 HL147662 from the National Institutes of Health, USA. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Sawyer A. Developing drugs for the ‘undruggable’. BioTechniques 69(4), 239–241 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Goracci L, Desantis J, Valeri A, Castellani B, Eleuteri M, Cruciani G. Understanding the metabolism of proteolysis targeting chimeras (PROTACs): the next step toward pharmaceutical applications. J. Med. Chem. 63(20), 11615–11638 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.An S, Fu L. Small-molecule PROTACs: an emerging and promising approach for the development of targeted therapy drugs. EBioMedicine 36, 553–562 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paiva SL, Crews CM. Targeted protein degradation: elements of PROTAC design. Curr. Opin. Chem. Biol. 50, 111–119 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khan S, He Y, Zhang X et al. PROteolysis TArgeting Chimeras (PROTACs) as emerging anticancer therapeutics. Oncogene 39(26), 4909–4924 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu H, Ding X, Liu L et al. Discovery of novel BCR-ABL PROTACs based on the cereblon E3 ligase design, synthesis, and biological evaluation. Eur. J. Med. Chem. 223, 113645 (2021). [DOI] [PubMed] [Google Scholar]
- 7.Jang J, To C, De Clercq DJH et al. Mutant-selective allosteric EGFR degraders are effective against a broad range of drug-resistant mutations. Angew Chem. Int. Ed. Engl. 59(34), 14481–14489 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buhimschi AD, Armstrong HA, Toure M et al. Targeting the C481S ibrutinib-resistance mutation in Bruton's tyrosine kinase using PROTAC-mediated degradation. Biochemistry 57(26), 3564–3575 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Hati S, Zallocchi M, Hazlitt R et al. AZD5438-PROTAC: a selective CDK2 degrader that protects against cisplatin- and noise-induced hearing loss. Eur. J. Med. Chem. 226, 113849 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; • References 5–9 highlight the advantages of the proteolysis targeting chimera approach over traditional small molecule inhibitors.
- 10.Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z. MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity 7(6), 837–847 (1997). [DOI] [PubMed] [Google Scholar]
- 11.Chen Y, Ning Y, Bai G et al. Design, synthesis, and biological evaluation of IRAK4-targeting PROTACs. ACS Med. Chem. Lett. 12(1), 82–87 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ishida T, Ciulli A. E3 ligase ligands for PROTACs: how they were found and how to discover new ones. SLAS Discov. 26(4), 484–502 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. PROTACs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl Acad. Sci. USA 98(15), 8554–8559 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study provided the first example and discovery of a peptide-based proteolysis targeting chimera molecule.
- 14.Buckley DL, Gustafson JL, Van Molle I et al. Small-molecule inhibitors of the interaction between the E3 ligase VHL and HIF1alpha. Angew Chem. Int. Ed. Engl. 51(46), 11463–11467 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buckley DL, Van Molle I, Gareiss PC et al. Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1alpha interaction. J. Am. Chem. Soc. 134(10), 4465–4468 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bondeson DP, Mares A, Smith IE et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 11(8), 611–617 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zengerle M, Chan KH, Ciulli A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem. Biol. 10(8), 1770–1777 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• References 16 and 17 paved the way for the discovery of further proteolysis targeting chimera molecules based on small molecule inhibitors instead of peptide-based long proteolysis targeting chimeras.
- 18.Clinicaltrials.gov NCT03888612. Trial of ARV-110 in Patients with Metastatic Castration-Resistant Prostate Cancer. Available from: www.clinicaltrials.gov/ct2/show/NCT03888612?term=NCT03888612&draw=2&rank=1
- 19.Clinicaltrials.gov NCT04072952. A Phase 1/2 Trial of ARV-471 Alone and in Combination with Palbociclib (IBRANCE®) in Patients with ER+/HER2- Locally Advanced or Metastatic Breast Cancer. Available from: www.clinicaltrials.gov/ct2/show/NCT04072952?term=NCT04072952&draw=2&rank=1
- 20.Clinicaltrials.gov NCT05067140. Clinical Trial to Evaluate ARV-766 in Patients with Metastatic Castration-Resistant Prostate Cancer. Available from: www.clinicaltrials.gov/ct2/show/NCT05067140?term=NCT05067140&draw=2&rank=1
- 21.Clinicaltrials.gov NCT04886622. A Study of DT2216 in Relapsed/Refractory Malignancies. Available from: www.clinicaltrials.gov/ct2/show/NCT04886622?term=NCT04886622&draw=2&rank=1
- 22.Clinicaltrials.gov NCT04772885. A Single and Multiple Ascending Dose Trial of KT-474 in Healthy Adult Volunteers and Patients with Atopic Dermatitis (AD) or Hidradenitis Suppurativa (HS). Available from: www.clinicaltrials.gov/ct2/show/NCT04772885?term=NCT04772885&draw=2&rank=1
- 23.Clinicaltrials.gov NCT05233033. Safety, PK/PD, and Clinical Activity of KT-413 in Adult Patients with Relapsed or Refractory B-cell NHL. Available from: www.clinicaltrials.gov/ct2/show/NCT05233033?term=NCT05233033&draw=2&rank=1
- 24.Clinicaltrials.gov NCT05225584. Safety, PK, PD, Clinical Activity of KT-333 in Adult Patients with Refractory Lymphoma, Large Granular Lymphocytic Leukemia, Solid Tumors. Available from: www.clinicaltrials.gov/ct2/show/NCT05225584?term=NCT05225584&draw=2&rank=1
- 25.Clinicaltrials.gov NCT04830137. A Study of NX-2127 in Adults with Relapsed/Refractory B-cell Malignancies. Available from: www.clinicaltrials.gov/ct2/show/NCT04830137?term=NCT04830137&draw=2&rank=1
- 26.Clinicaltrials.gov NCT05131022. A Study of NX-5948 in Adults with Relapsed/Refractory B-cell Malignancies. Available from: www.clinicaltrials.gov/ct2/show/NCT05131022?term=NCT05131022&draw=2&rank=1
- 27.Clinicaltrials.gov NCT05355753. A Study to Assess the Safety and Tolerability of CFT8634 in Locally Advanced or Metastatic SMARCB1-Perturbed Cancers, Including Synovial Sarcoma and SMARCB1-Null Tumors. Available from: www.clinicaltrials.gov/ct2/show/NCT05355753?term=NCT05355753&draw=2&rank=1
- 28.Clinicaltrials.gov NCT04428788. Study to Evaluate the Safety and Tolerability of CC-94676 in Participants with Metastatic Castration-Resistant Prostate Cancer. Available from: www.clinicaltrials.gov/ct2/show/NCT04428788?term=NCT04428788&draw=2&rank=1
- 29.Clinicaltrials.gov NCT05080842. A Study of AC682 for the Treatment of Locally Advanced or Metastatic ER+ Breast Cancer. Available from: www.clinicaltrials.gov/ct2/show/NCT05080842?term=NCT05080842&draw=2&rank=1 ; • References 18–28 justify the clinical importance of proteolysis targeting chimera molecules against various disease conditions.
- 30.Itoh Y, Ishikawa M, Naito M, Hashimoto Y. Protein knockdown using methyl bestatin-ligand hybrid molecules: design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J. Am. Chem. Soc. 132(16), 5820–5826 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Van Molle I, Thomann A, Buckley DL et al. Dissecting fragment-based lead discovery at the von Hippel-Lindau protein: hypoxia-inducible factor 1alpha protein-protein interface. Chem. Biol. 19(10), 1300–1312 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Galdeano C, Gadd MS, Soares P et al. Structure-guided design and optimization of small molecules targeting the protein-protein interaction between the von Hippel-Lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. J. Med. Chem. 57(20), 8657–8663 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soares P, Gadd MS, Frost J et al. Group-based optimization of potent and cell-active inhibitors of the von Hippel-Lindau (VHL) E3 ubiquitin ligase: structure-activity relationships leading to the chemical probe (2S,4R)-1-((S)-2-(1-cyanocyclopropanecarboxamido)-3,3-dimethylbutanoyl)-4-hydroxy -N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (VH298). J. Med. Chem. 61(2), 599–618 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frost J, Galdeano C, Soares P et al. Potent and selective chemical probe of hypoxic signaling downstream of HIF-alpha hydroxylation via VHL inhibition. Nat. Commun. 7, 13312 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang C, Zhang Y, Wu Y, Xing D. Developments of CRBN-based PROTACs as potential therapeutic agents. Eur. J. Med. Chem. 225, 113749 (2021). [DOI] [PubMed] [Google Scholar]
- 36.Girardini M, Maniaci C, Hughes SJ, Testa A, Ciulli A. Cereblon versus VHL: hijacking E3 ligases against each other using PROTACs. Bioorg. Med. Chem. 27(12), 2466–2479 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ito T, Handa H. Molecular mechanisms of thalidomide and its derivatives. Proc. Jpn Acad. Ser. B Phys. Biol. Sci. 96(6), 189–203 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ito T, Ando H, Suzuki T et al. Identification of a primary target of thalidomide teratogenicity. Science 327(5971), 1345–1350 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Jo S, Lee KH, Song S, Jung YK, Park CS. Identification and functional characterization of cereblon as a binding protein for large-conductance calcium-activated potassium channel in rat brain. J. Neurochem. 94(5), 1212–1224 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Hensley CT, Wasti AT, Deberardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J. Clin. Invest. 123(9), 3678–3684 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deberardinis RJ, Cheng T. Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 29(3), 313–324 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nguyen TV, Lee JE, Sweredoski MJ et al. Glutamine triggers acetylation-dependent degradation of glutamine synthetase via the thalidomide receptor cereblon. Mol. Cell 61(6), 809–820 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schneekloth AR, Pucheault M, Tae HS, Crews CM. Targeted intracellular protein degradation induced by a small molecule: en route to chemical proteomics. Bioorg. Med. Chem. Lett. 18(22), 5904–5908 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu J, Ma J, Liu Y et al. PROTACs: a novel strategy for cancer therapy. Semin. Cancer Biol. 67(Pt 2), 171–179 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Testa A, Lucas X, Castro GV et al. 3-Fluoro-4-hydroxyprolines: synthesis, conformational analysis, and stereoselective recognition by the VHL E3 ubiquitin ligase for targeted protein degradation. J. Am. Chem. Soc. 140(29), 9299–9313 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou X, Dong R, Zhang JY, Zheng X, Sun LP. PROTAC: a promising technology for cancer treatment. Eur. J. Med. Chem. 203, 112539 (2020). [DOI] [PubMed] [Google Scholar]
- 47.He M, Cao C, Ni Z et al. PROTACs: great opportunities for academia and industry (an update from 2020 to 2021). Signal Transduct. Target Ther. 7(1), 181 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liang Y, Nandakumar KS, Cheng K. Design and pharmaceutical applications of proteolysis-targeting chimeric molecules. Biochem. Pharmacol. 182, 114211 (2020). [DOI] [PubMed] [Google Scholar]
- 49.Huang W, Wang B, Zhang Z, Zhang C, Zeng S, Shen Z. Progress on small-molecule proteolysis-targeting chimeras. Future Med. Chem. 11(20), 2715–2734 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Yan G, Zhong X, Yue L et al. Discovery of a PROTAC targeting ALK with in vivo activity. Eur. J. Med. Chem. 212, 113150 (2021). [DOI] [PubMed] [Google Scholar]
- 51.Ren C, Sun N, Kong Y et al. Structure-based discovery of SIAIS001 as an oral bioavailability ALK degrader constructed from Alectinib. Eur. J. Med. Chem. 217, 113335 (2021). [DOI] [PubMed] [Google Scholar]
- 52.Chen L, Han L, Mao S et al. Discovery of A031 as effective proteolysis targeting chimera (PROTAC) androgen receptor (AR) degrader for the treatment of prostate cancer. Eur. J. Med. Chem. 216, 113307 (2021). [DOI] [PubMed] [Google Scholar]
- 53.Kim GY, Song CW, Yang YS et al. Chemical degradation of androgen receptor (AR) using bicalutamide analog-thalidomide PROTACs. Molecules 26(9), 2525 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu H, Sun R, Ren C, Qiu X, Yang X, Jiang B. Construction of an IMiD-based azide library as a kit for PROTAC research. Org. Biomol. Chem. 19(1), 166–170 (2021). [DOI] [PubMed] [Google Scholar]
- 55.Vannam R, Sayilgan J, Ojeda S et al. Targeted degradation of the enhancer lysine acetyltransferases CBP and p300. Cell. Chem. Biol. 28(4), 503–514 e512 (2021). [DOI] [PubMed] [Google Scholar]
- 56.Kitabayashi I, Aikawa Y, Yokoyama A et al. Fusion of MOZ and p300 histone acetyltransferases in acute monocytic leukemia with a t(8;22)(p11;q13) chromosome translocation. Leukemia 15(1), 89–94 (2001). [DOI] [PubMed] [Google Scholar]
- 57.Wang L, Gural A, Sun XJ et al. The leukemogenicity of AML1-ETO is dependent on site-specific lysine acetylation. Science 333(6043), 765–769 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Attar N, Kurdistani SK. Exploitation of EP300 and CREBBP lysine acetyltransferases by cancer. Cold Spring Harb. Perspect. Med. 7(3), a026534 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang L, Shao X, Zhong T et al. Discovery of a first-in-class CDK2 selective degrader for AML differentiation therapy. Nat Chem. Biol. 17(5), 567–575 (2021). [DOI] [PubMed] [Google Scholar]
- 60.Rana S, Bendjennat M, Kour S et al. Selective degradation of CDK6 by a palbociclib based PROTAC. Bioorg. Med. Chem. Lett. 29(11), 1375–1379 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qiu X, Li Y, Yu B et al. Discovery of selective CDK9 degraders with enhancing antiproliferative activity through PROTAC conversion. Eur. J. Med. Chem. 211, 113091 (2021). [DOI] [PubMed] [Google Scholar]
- 62.King HM, Rana S, Kubica SP et al. Aminopyrazole-based CDK9 PROTAC sensitizes pancreatic cancer cells to venetoclax. Bioorg. Med. Chem. Lett. 43, 128061 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wei M, Zhao R, Cao Y et al. First orally bioavailable prodrug of proteolysis targeting chimera (PROTAC) degrades cyclin-dependent kinases 2/4/6 in vivo. Eur. J. Med. Chem. 209, 112903 (2021). [DOI] [PubMed] [Google Scholar]
- 64.Brown MS, Goldstein JL. Multivalent feedback regulation of HMG CoA reductase, a control mechanism coordinating isoprenoid synthesis and cell growth. J. Lipid Res. 21(5), 505–517 (1980). [PubMed] [Google Scholar]
- 65.Brown AS, Watson KE. Statin intolerance. Rev. Cardiovasc. Med. 19(S1), S9–S19 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Li MX, Yang Y, Zhao Q et al. Degradation versus inhibition: development of proteolysis-targeting chimeras for overcoming statin-induced compensatory upregulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase. J. Med. Chem. 63(9), 4908–4928 (2020). [DOI] [PubMed] [Google Scholar]
- 67.Aritake K, Kado Y, Inoue T, Miyano M, Urade Y. Structural and functional characterization of HQL-79, an orally selective inhibitor of human hematopoietic prostaglandin D synthase. J. Biol. Chem. 281(22), 15277–15286 (2006). [DOI] [PubMed] [Google Scholar]
- 68.Yokoo H, Shibata N, Naganuma M et al. Development of a hematopoietic prostaglandin D synthase-degradation inducer. ACS Med. Chem. Lett. 12(2), 236–241 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol. Ther. 148, 114–131 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Golpich M, Amini E, Hemmati F et al. Glycogen synthase kinase-3 beta (GSK-3beta) signaling: Implications for Parkinson's disease. Pharmacol. Res. 97, 16–26 (2015). [DOI] [PubMed] [Google Scholar]
- 71.Jiang X, Zhou J, Wang Y et al. PROTACs suppression of GSK-3beta, a crucial kinase in neurodegenerative diseases. Eur. J. Med. Chem. 210, 112949 (2021). [DOI] [PubMed] [Google Scholar]
- 72.Hopkins AL, Groom CR. The druggable genome. Nat. Rev. Drug Discov. 1(9), 727–730 (2002). [DOI] [PubMed] [Google Scholar]