

Abstract
Generation of the prototypic second messenger cAMP instigates numerous signaling events. A major intracellular target of cAMP is Protein kinase A (PKA), a Ser/Thr protein kinase. Where and when this enzyme is activated inside the cell has profound implications on the functional impact of PKA. It is now well established that PKA signaling is focused locally into subcellular signaling ‘islands’ or ‘signalosomes.’ The A-Kinase Anchoring Proteins (AKAPs) play a critical role in this process by dictating spatial and temporal aspects of PKA action. Genetically encoded biosensors, small molecule and peptide-based disruptors of PKA signaling are valuable tools for rigorous investigation of local PKA action at the biochemical level. This chapter focuses on approaches to evaluate PKA signaling islands, including a simple assay for monitoring the interaction of an AKAP with a tunable PKA holoenzyme. This latter approach evaluates the composition of PKA holoenzymes, in which regulatory subunits and catalytic subunits can be visualized in the presence of test compounds and small molecule inhibitors.
Keywords: AKAP, cAMP, DSF, Kinase Inhibitor, Mass Spectrometry, Peptide, Protein kinase A (PKA), SDS-PAGE
1. Introduction
The human protein kinome is a remarkable source of signaling enzymes [1]. Investigation of kinase biology requires the exploitation of biochemical and cellular assays that invariably include validated chemical probes [1]. Protein kinase A (PKA) is the textbook example of a second messenger-activated protein kinase, whose regulation by cAMP has been fundamental to the development of the cell signaling field [2]. Recent work raises the prospect of selective intervention in human diseases driven by PKA mutations, such as adrenocortical Cushing’s syndrome [3–5]. The PKA holoenzyme is composed of a tetramer of regulatory (R) and catalytic (C) subunits whose catalytic output is controlled, at least in part, by the reversible binding of cAMP to the R subunits. The A Kinase Anchoring Proteins (AKAPs) are signaling scaffolds that play a fundamental role in the spatial and temporal targeting of the PKA holoenzyme [6, 7]. Although AKAPs differ greatly in primary sequence, subcellular localization and their diverse repertoire of binding partners, they share the defining feature of a high-affinity interaction with the regulatory subunits (RI or RII) of the PKA holoenzyme, at a distinct site to those involved in cAMP binding. PKA anchoring proceeds through an amphipathic helix that inserts into a customized groove formed by the docking and dimerization (D/D) of the R-subunit protomers [8–10]. When tethered to AKAPs, the PKA holoenzyme is spatially restricted with access to appropriate cellular substrates that mediate cellular outcomes [11] (Fig. 1). This offers one mechanism to selectively promote cellular events that proceed through the ubiquitous second messenger molecule cAMP [12, 13]. However, the PKA-binding module is only one facet of AKAP action. Other domains of anchoring proteins can interact independently to allow distinct enzymes to integrate other second messenger signals within multivalent assemblies [14–16]. Diversification of these signaling complexes also occurs, because macromolecular assembles contain other kinases, protein phosphatases, adenylyl cyclases, phosphodiesterases and selected PKA substrates [17–21].
Fig. 1.
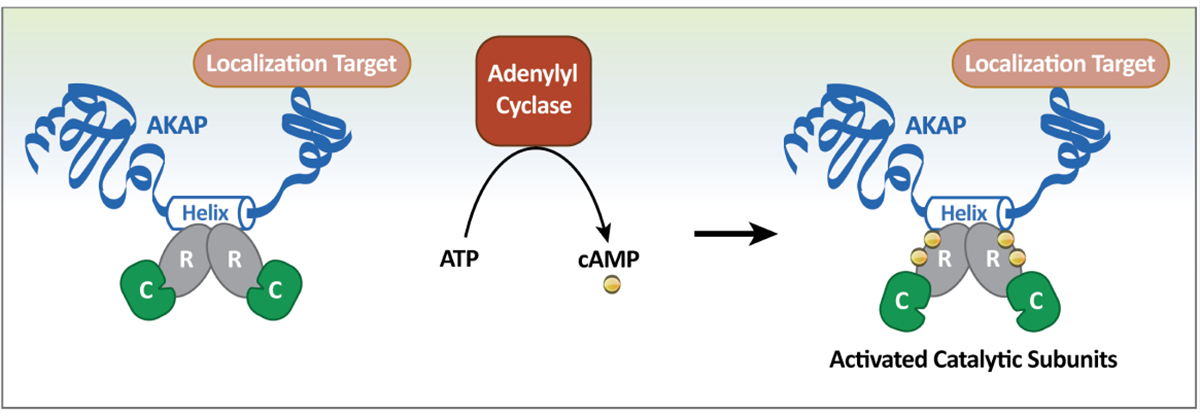
Signaling through AKAP complexes. When intracellular concentrations of cAMP are low, the PKA holoenzyme complex is largely bound to AKAPs. AKAPs are localized to intracellular sites including the plasma membrane and organelles, thereby concentrating PKA to particular locations within the cell. Upon stimulation, intracellular cAMP levels increase. Each R-subunit of PKA binds up to two cAMP molecules and undergoes an allosteric conformational change to loosen contacts with the activated catalytic subunits. Active C subunits are then able to phosphorylate adjacent substrates.
Complexity within PKA signaling is augmented by the utilization of four distinct regulatory subunit isoforms of PKA: RI (RIα and RIβ) and RII (RIIα and RIIβ) which differ in their tissue distribution and concentration, cAMP sensitivity, and affinities for AKAPs. These additional layers finely tune PKA activity [22, 23]. The vast majority of AKAPs selectively bind the RII isoform; however, a limited number of dual-specific AKAPs are thought to interact with RI [9, 24–26]. Moreover, due to the dynamic and compartment-specific nature of interactions with AKAPs and other PKA signaling components, uncovering the intricacies of AKAP-mediated signaling events has proven to be a substantial challenge. To confound matters further, the human genome encodes in excess of fifty AKAP genes and most cell types express at least 10–15 different anchoring proteins [27]. Additionally, the PKA C-subunits are thought to have hundreds of distinct substrates. Likewise, certain AKAPs are expressed as families of alternatively spliced transcripts with distinct biological functions [28, 29]. This level of complexity makes it difficult to definitively elucidate individual roles for each AKAP signaling island. One strategy to study the role of anchoring in signaling events is to selectively displace PKA subtypes from the AKAP platform. Consequently, isoform-selective disruptors have been developed (Fig. 2; Table 1) [30, 31]. Although these reagents are valuable tools to study AKAP–PKA signaling, one major drawback is that these inhibitors nonspecifically occlude the AKAP anchoring site on the regulatory subunits. This results in indiscriminate interruption of AKAP interactions with either the RI or RII isoforms.
Fig. 2.

Some published peptide disruptors of AKAP complexes. Isoform-selective disruptors were developed to have specificity of targeting toward either the RI or RII isoform of PKA. Despite considerable sequence divergence between the different disruptor peptides, they all share the common feature of forming an amphipathic helix with a largely hydrophobic binding interface (shown in gray) that complements the binding surface of the D/D domain of the R-subunits. Asterisks represent incorporation of the unnatural amino acid (S)-2-(4′-pentenyl)alanine to form an all-hydrocarbon bridge within the sequece.
Table 1.
PKA inhibitor compounds for modulation of PKA-mediated signaling
| PKA inhibitors | Mechanism of action |
|---|---|
| PKI peptide | Blocks the catalytic site of PKA |
| H89 | ATP-competitive inhibitor of PKA |
| Rp-cAMPS | Prevents cAMP binding to R-subunits |
| AT13148 | Clinical candidate broad PKA/AGC kinase inhibitor |
1.1. RII-Selective Disruptors of AKAP Complexes
The original AKAP disruptor peptide, Ht31, was derived from the PKA-anchoring domain of AKAP-Lbc [32]. The discovery of this peptide set a precedence for investigating docking interactions between AKAPs and the RII subunit. Although Ht31 has limited cell permeability, chemical modification of the peptide increases its overall hydrophobicity[33]. The addition of stearic or myristic acid to the N-terminus of the peptide enhances cellular permeability. However, the conjugation of a lipid moiety contributes to retention of Ht31 in cell membranes. Lipid modified forms of Ht31 and the negative proline analog control (Ht31 and Ht31P) are available as commercial reagents. A bioinformatics approach was subsequently used to identify an RII-specific consensus sequence [34], which was then optimized by peptide array screening to produce a more potent RII inhibitor peptide, AKAP-in silico (AKAP-IS). This peptide was shown to have improved affinity for RII as compared to the Ht31 peptide. The Kd value of AKAP-IS is less than 1 nM for RII, but is in the mid-high nM range for RI. Initially, the AKAP-IS peptide was not cell permeable and had limited solubility in aqueous solution. However, the subsequent addition of a TAT sequence at the N-terminus of AKAP-IS partially improved cell permeability [35]. Despite the enhanced hydrophilicity afforded by the TAT sequence, the conjugated peptide, TAT–AKAP-IS, is still highly hydrophobic and requires solubilization in an aqueous 10 % DMSO solution. Using a structure-guided optimization approach (from the structures of the AKAP docking site on RIIα alone and in complex with AKAP-IS) in combination with peptide screening assays, AKAP-IS was further modified to improve the affinity and selectivity, which resulted in SuperAKAP-IS [9]. This peptide disruptor exhibited superior RII selectivity, with fourfold higher affinity for RII and approximately 12-fold less affinity for RI as compared to AKAP-IS. Based on the observation that AKAP18 has a higher affinity for RIIα and that an N-terminally truncated form, AKAP18δ, has an even higher affinity, a new class of disruptor peptides were generated [36]. This class of peptides possess high-affinity for RIIα with dissociation constants as low as 0.4 nM. Analysis of sequence divergence between these peptides helped to further illuminate important residues for engagement with the RII docking site. Analogous to Ht31, the AKAP18δ peptides were also modified with the addition of a stearate moiety in order to promote cellular uptake. Within the last 10 years, small molecules have also been developed to disrupt AKAP–RII interactions [37, 38]. Very large, and relatively flat surfaced, such as the protein–protein interaction interface between the amphipathic helix of an AKAP and the RII D/D docking site, are notoriously difficult to target using small molecule approaches, which could partially explain the relative lack of progress in this field of research. As such, these compounds currently have limited cellular potency (IC50 = 20–40 μM), but serve as a starting point for compound optimization using a small molecule targeting approach. Despite the current limitations, these small molecule scaffolds remain an exciting research area that merit further investigation.
Another promising development in anchoring disruptor peptides is the recent introduction of Stapled AKAP Disruptor (STAD) peptides. Chemically modified RII-specific AKAP disruptors were developed by incorporating non-natural amino acids into the A-kinase binding (AKB) sequences to bestow small-molecule-like properties onto the peptide sequences [31]. Synthetic libraries were designed based on previously identified AKB or AKB-like sequences, where non-natural olefinic amino acids were incorporated and cyclized so as to conformationally constrain an alpha-helical fold. This chemical modification was previously shown to promote cellular permeability and proteolytic stability to peptides [39]. The STAD peptides developed in this study are highly cell permeable and effectively block interactions between AKAPs and RII inside cells. The incorporation of the peptide “staple” introduced significant hydrophobicity to an already hydrophobic sequence, so the addition of a small PEG-3 linker at the N-terminus was required to improved solubility for cell-based experiments. The rapid cellular uptake, resistance to degradation, and relatively long half-lives in cells of the STAD peptides provide a more flexible platform for studying dynamic AKAP signaling events under a variety of conditions.
All of the PKA-anchoring disruptor reagents discussed have been patterned after an AKAP motif. Recently, a phage selection procedure was employed that exploited high-resolution structural information to engineer RII D/D domain mutants that are selective for a particular AKAP [40]. Competitive selection screening revealed RII sequences (RSelect) that were preferential for interaction with specific AKAP proteins. Biochemical and cell-based experiments validated the efficacy of RSelect mutants for AKAP2 and AKAP18. This new class of engineered proteins based on the reciprocal surface of the AKAP–PKA interaction has the potential to be used to dissect the contributions of different AKAP-targeted pools of PKA and aid in the design of compounds targeting these subset populations. However, the utility of the RSelect mutants is somewhat limited.
Although numerous RII-specific AKAP disruptors have been developed, designing peptides for RI-selective interactions has proven to be more elusive. The first RI-selective peptide inhibitors were identified through peptide array screening nearly a decade after the design of Ht31 and its inactive control Ht31-Pro [41]. The prototype used for the peptide array was derived from the A-kinase binding (AKB) domain of AKAP10 [41]. Although the crystal structure of the AKB-binding domain of RI was not solved at the time, the minimal sequence required, and surface residue interactions involved in docking to RI were described through systematic analysis. Based on this study, the AKB binding site on RI was shown to involve multiple interactions with charged residues, while the analogous binding site on RII was shown to largely provide a hydrophobic patch for AKB binding. A major limitation of the peptides identified in this study, as with many unmodified peptides, is that they lack cell permeability and therefore require transfection or genetic encoding in order to characterize their activity in cells.
Subsequent studies employed a bioinformatics approach coupled with peptide array screening to yield a RI-selective peptide termed RIAD [42]. The binding sequences from several dual-specificity AKAPs were used as the starting point to develop RI specificity. RIAD was found to have a notably improved binding affinity for RI as well as greater specificity for RI over RII. While the RIAD peptide alone was not cell permeable, the C-terminal addition of 11 arginine residues afforded this property. Although transfection can result in artifacts and compensatory expression changes within the cell, the cell-permeable version of RIAD was utilized to illustrate disruption of RI-specific AKAP interactions in intact, non-modified cells. RIAD analogs were later developed that incorporated non-natural and natural amino acids into the sequence to improve proteolytic stability [43]. However, the cell permeability of RIAD analogs remains an issue.
Probing the structure of RIα reveals several unique features that differentiate the D/D domain from that of RII, and which are likely key determinants of AKAP specificity [44]. These changes include an altered depth of the binding groove, the presence of an inter-subunit disulfide bridge within the AKAP binding site, and a unique spatial arrangement of restrictive amino acid lining the AKB binding pocket. These structural insights will undoubtedly lead to the development of optimized peptide-based or synthetic scaffolds that can discriminate against RII interactions while maintaining high-affinity binding with RI. Furthermore, additional AKAP selectivity for RI anchoring may involve a separate interface that is upstream of the amphipathic helix, known as the RI-specifier region (RISR), which is capable of augmenting RI binding [45]. Cellular delivery of a RISR peptide was shown to disrupt RI binding and may serve as an additional targeting site for RI-specific disruption.
1.2. cAMP-Modulatory Effectors
As a means to interrogate AKAP signaling events in cell-based studies, multiple strategies can be applied to stimulate intracellular cAMP production (Table 2). While some reagents stimulate cAMP to physiological levels, many cause inappropriately high concentrations of cAMP. The labdane diterpene, forskolin, is a potent supraphysiological activator of adenylyl cyclase (AC), and is perhaps the most widely used reagent for modulating cAMP signaling and PKA activity. To date, over 10,000 citations describe use the use of forskolin for this purpose. Forskolin is a natural product isolated from Coleus forskohlii that reversibly increases cAMP concentrations in diverse tissue types [46]. Eight of the nine membrane-bound isoforms of AC are stimulated to different extents by forskolin [47], with AC9 being the only major outlier [48] [49]. Since the expression profiles of the AC isoforms are tissue-specific, the potency of forskolin in different cellular contexts can vary considerably, and will often result in cAMP concentrations that are not physiologically relevant [47]. An alternative approach for increasing intracellular cAMP is through inhibition of phosphodiesterase (PDE) activity. A nonspecific PDE inhibitor, 3-isobutyl-1-methylxanthine (IBMX), was identified from a panel of xanthine derivatives to have inhibitory effects on PDEs [50]. IBMX is a moderately potent inhibitor of the majority of PDE isoforms but appears to have no effect on PDE8 or PDE9 [51]. Due to its broad inhibitory activity towards PDEs, IBMX is routinely used in conjunction with an AC-stimulating agent (such as forskolin) to further increase overall intracellular cAMP. However, additional caution must be taken when interpreting results from experiments using a forskolin/IBMX cocktail to stimulate PKA as this combination stimulates ‘unnatural’ cAMP production and prolongs the second messenger response well beyond a ‘typical’ time course.
Table 2.
Classical cAMP-stimulating agents for activation of PKA signaling-complexes
| cAMP-stimulating agents | Mechanism of action |
|---|---|
| Forskolin | Activates adenylyl cyclases |
| IBMX | Inhibits PDEs |
| Isoproterenol | Indirectly activates adenylyl cyclases |
| PGE2 | Indirectly activates adenylyl cyclases |
| DB-cAMP | Activates PKA |
A much more physiologically relevant means to stimulate cAMP production is through activation of β1- and β2-adrenergic receptors by isoproterenol (isoprenaline) [52]. Isoproterenol is a synthetic catecholamine that acts as an agonist for this subclass of G protein-coupled receptors (GPCRs). Upon stimulation of β-adrenergic receptors, Gs proteins are activated leading to stimulation of AC activity. After isoproterenol stimulation, cAMP levels rise significantly, but then fall back to near background levelsand are resistant to further stimulation even in the presence of persistent isoproterenol treatment [53]. Although β-adrenergic receptors are widely expressed in a variety of cells and isoproterenol can elicit a notable effect on cAMP levels, isoproterenol-stimulated cAMP production is useful for short time-course studies but is not effective as a cAMP-stimulating agent for sustained periods. A simple competitive chemical strategy to induce cAMP-sensitive signaling was developed using the cell-permeable cAMP analog, dibutyryl cyclic adenosine monophosphate (DB-cAMP). Although the compound enters the cell in an inactive form, hydrolysis of one of the butyrate groups permits the compound to activate PKA [54]. Although there are clear advantaged of using DB-cAMP analogs, it remains unclear whether they are resistant to all of the cAMP PDE subtypes that exist in a typical cell, in particular PDE8, PDE10, and PDE11 [55].
1.3. PKA Inhibitors: From proteins to small molecules
The heat-stable protein inhibitor of PKA was first purified from rabbit skeletal muscle over 50 years ago [56]. Tissue protein kinase inhibitor (PKI) is expressed as three isoforms [57] found in a range of cell types. PKI possesses an affinity for PKA in the sub-micromolar range [58], which can be recapitulated biochemically with purified PKA components [59] and also employed in purified recombinant form for the induction of biological responses in vivo [60]. A short, 20-amino-acid sequence was subsequently identified as the inhibitory component of PKI, and a synthetic peptide spanning this sequence was shown to act as a highly selective, potent inhibitor of PKA [61–63]. At the core of the PKI peptide is a pseudosubstate sequence of RRNAI where the alanine occupies the active site cleft of the C subunit [61–63]. Multiple analogs derived from this 20-mer sequence were synthesized to define the residues that are critical for its inhibitory activity [64, 65]. This sequence was also found to be highly specific for PKA with no inhibitory effect on PKG [64]. PKI not only acts to block the catalytic site on PKA [66], but also serves to mop up any free C subunit inside the cell [67]. A variety of PKI inhibitor peptide analogs and recombinant PKI protein [59] are commercially available that possess a high affinity for PKA and are recognized to have exquisite specificity for PKA at very low concentrations comparable to chemical kinase inhibitors (see below). Recently, very high affinity stapled peptide mimics of the PKI pseudosubstrate with cellular permeability have also been developed [68].
Chemical inhibitors of PKA-C
In addition to targeting by a variety of promiscuous kinases inhibitors, such as staurosporine and K252a [59, 69], several early chemical inhibitors of PKA, such as H89, are widely used in the literature. H89 is an isoquinoline-based small molecule that was derived from an earlier inhibitor, H8 [70]. While H8 targeted both PKA and PKG, H89 was found to be a potent inhibitor of PKA but also had inhibitory activity against several other kinases including PKG, PKC, casein kinases I and II, and CamKII [69, 71]. Further work identified that H89 acts as a competitive inhibitor of ATP binding to occupy and prevent substrate phosphorylation; this binding event can be probed both directly and indirectly, either enzymatically [72] or by using analytical DSF with purified recombinant C-subunits [59]. While H89 is an effective inhibitor of PKA, numerous off-target effects have been documented, including disruption of intracellular signaling pathways and inhibition of a significant number of related (AGC-kinase) protein kinases, some of which are inhibited even more potently than PKA [69, 73, 74]. Although H89 and its analoges are among the most commonly used of all PKA inhibitors in research, caution should be used when interpreting data, due to its numerous off-target effects. The same is likely to be true for multi-targeted AGC kinase inhibitors that inhibit PKA, such as AT13148 [75], which likely exerts anti-cancer effects through the blockade of multiple signaling pathways. Overall, there remains an urgent need to generate new drug-like molecules that can interfere with PKA signaling, although the ubiquity of PKA in cell biology makes this a challenge currently for kinase inhibitors, in contrast to targeted AKAP blockade [5]. Finally, cyclic nucleotide analogs such as Rp-cAMPS (adenosine-3′,5′- cyclic monophosphorothioate Rp-isomer) have also been used as indirect inhibitory agents for PKA signaling. Rp-cAMPS is cell permeable and acts as an antagonist of cAMP to prevent activation of PKA by competing with cAMP at nucleotide-binding sites on the regulatory subunits of PKA [76, 77]. This cAMP analog also demonstrates resistance to hydrolysis by phosphodiesterases. Although Rp-cAMPS has limited cell permeability, newer versions such as Rp-8-Br-cAMPS and Rp-8- Cl-cAMPS are recognized to have improved permeability and greater potency [78]. However, since additional signaling elements bind cAMP, it is possible that these analogs may also have other cellular targets aside from PKA-R and can thereby cause unintended secondary effects.
The versatility of the PKA holoenzyme system for biochemical analysis comes about in part due to a historical ability to purify stable components from biological tissues and recombinant sources for rapid analysis in different experimental systems [60, 79–81]. The ready availability of affinity-tagged recombinant proteins for in vitro study is also central for screening procedures to study the mechanism of PKA signaling in simplified systems (Figure 3). For example, in-frame genetic fusion of the C and RII subunits generates a catalytically-active PKA polypeptide that drives substrate phosphorylation in response to agonists in cells depleted of endogenous subunits [11]. This work also suggested that local PKA signaling in cells can occur without physical separation of the C and RII subunits. The anchored chimeric PKA holoenzyme appears to signal within ~200Å of the AKAP anchor, confirming that spatiotemporal targeting of PKA-based signaling is likely to occur. In addition, we identified that bacterially overexpressed recombinant PKA C-subunit contains at-least 12 autophosphorylation sites not found in a catalytically-dead variant [59, 82], and that these proteins can spontaneously assemble into stoichiometric and sub-stoichiometric complexes in solution that can be analysed by intact MS and DSF [59]. Furthermore, scaffolded cAMP-responsive holoenzyme complexes can be manipulated in vitro for semi-quantitative analysis using gel-based approaches [11].
Fig. 3.
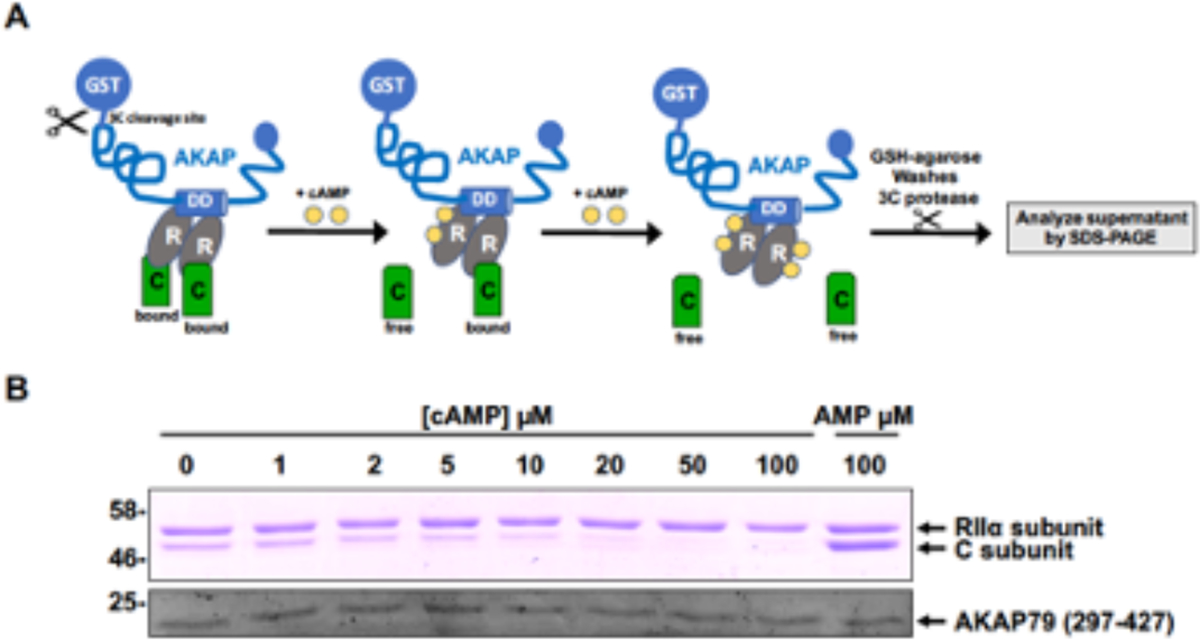
Biochemical analysis of the AKAP:RII:C interaction in the presence and absence of exogenous cAMP titrated over a range of concentrations. (A) Cartoon of the assay procedure, which detects free C-subunits released into solution from previously assembled AKAP:RII complexes (B) SDS-PAGE showing the electrophoretic mobility and staining intensity of 3C-generated cleaved AKAP and the relative amounts of associated RII and C subunits. The amount of 3C-cleaved AKAP and RII remain constant in the assay. A high concentration of 5’AMP serves as an internal control, since it does not lead to C-subunit release.
In this chapter, we describe a simple biochemical assay for the evaluation of cAMP effects on an artificially created AKAP-PKA holoenzyme complex. We also describe, in detail, simple protocols for the generation of highly purified recombinant AKAP-79, PKA RII and C- subunits, which can be reassembled into anchored holoenzyme complex in a test-tube. The formation of these high-order complexes can be investigated by a variety of approaches, including Mass Spectrometry [11, 59]. In particular, complex behavior can be assessed after the addition of exogenous reagents to evaluate multivalent protein binding. This is exemplified in the presented example by the physical separation of RII and C-subunits in the presence of different concentrations of cAMP, which can be readily visualized by SDS-PAGE (Figure 3).
2. Materials
Prepare all solutions in deionised water at room temperature. Store all reagents at room temperature unless otherwise indicated. All binding assays should be performed in chilled tubes kept on ice unless indicated.
2.1. Expression and Purification of AKAP-79, PKA-C and PKA-RII proteins
The following bacterial expression plasmids are validated for the generation of full length recombinant C and RII PKA subunits and a GST-AKAP truncation mutant in BL21 (DE3) pLysS E. coli cells: pOPINJ-human His-GST-AKAP79297–417 (3C cleavable N-terminal 6His, GST tag, amino acids 297–417, ~20 kDa after cleavage of ~30 kDa GST tag), pET-30 Ek/LIC-mouse PKA-C (N-terminal 6His tag, amino acids 1–351, ~40 kDa), pET-30 Ek/LIC-human PKA-RIIα (N-terminal 6His tag, amino acids 1–381, ~50 kDa). See [11, 59] for further details
2.1.1. List of Required Chemicals and Reagents:
Chemically-competent E. coli cells: BL21 (DE3) pLysS cells. Cells can be purchased or made chemically competent in house using standard procedures.
Luria Broth (LB) agar plates: 3.7 g of LB agar in 100 mL water (sufficient to make ~5 × 20 mL agar plates). Sterilise the agar using an autoclave (121 °C under 20 psi for at least 30 min) and allow to cool to 55–60 °C. Under sterile conditions, add the appropriate antibiotics and pour the agar in to the required number of petri dishes. Leave the plates to solidify for ~30 mins.
LB broth: 2.5 g of solid LB is dissolved in 100 mL water. Sterilise using an autoclave.
Antibiotics for selection of transformed E. coli: ampicillin - final concentration 50 μg/mL (pOPINJ); kanamycin-final concentration 50 μg/mL (pET-30 Ek/LIC). Both antibiotics are prepared as 1000x stocks in water. BL21 (DE3) pLysS cells also need to be maintained in chloramphenicol - final concentration 35 μg/mL. Chloramphenicol is prepared as a 1000x stock in 100% (v/v) ethanol. All antibiotic stocks should be stored at −22°C
Isopropyl β-D-1-thiogalactopyranoside (IPTG): 100 mM. 1.19 g of IPTG in 50 mL water. Sterilise using a 0.45 μM syringe and store at −20°C.
Complete™ EDTA-free Protease Inhibitor tablets (Roche).
Ni-NTA agarose resin for purification of His-tagged proteins; reduced glutathione (GSH) sepharose for purification of GST-tagged proteins.
Lysis buffer: 50 mM Tris-HCl, pH 7.4, 300 mM NaCl, 1 mM DTT, 0.1 mM EDTA, 0.1 mM EDTA, 1% (v/v) Triton X-100, 10% (v/v) glycerol, 10 mM imidazole (see Note 1). Pipette 10 mL of glycerol in to a 100 mL graduated measuring cylinder and add 5 mL of 1 M Tris-HCl, pH 7.4, 10 mL of 3 M NaCl, 100 μL of 1 M DTT, 100 μL of 100 mM EDTA, 100 μL of 100 mM EGTA, 1 mL of 1 M imidazole and 1 mL of Triton X-100 (see Note 2). Adjust volume to 100 mL with water and supplement the buffer with 2 × Complete™ EDTA-free Protease Inhibitor tablets just before use. Lysis buffer can be stored at 4°C.
Gel filtration buffer: 50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 1 mM DTT, 10% (v/v) glycerol. Pour 100 mL of glycerol in to a 1 L graduated measuring cylinder and add 50 mL of 1 M Tris-HCl, pH 7.4, 33 mL 3 M NaCl, and 1 mL DTT. Adjust the volume to 1 L with water. (see Note 3).
High salt wash buffer: 50 mM Tris-HCl, pH 7.4, 500 mM NaCl, 20 mM Imidazole. Add 5 mL of 1 M Tris-HCl, pH 7.4, 16.67 mL of 3 M NaCl, 2 mL of 1 M imidazole to a 100 mL graduated measuring cylinder. Add water to a final volume of 100 mL.
Ni-NTA elution buffer: 50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 1 mM DTT, 500 mM imidazole. Add 5 mL of 1 M Tris-HCl, pH 7.4, 3.33 mL of 3 M NaCl, 500 mL of 1 M imidazole, and 100 μL of 1 M DTT to a 100 mL graduated cylinder and add water to a final volume of 100 mL.
GSH elution buffer: Prepare as for Ni-NTA elution buffer, but substitute 500 mM imidazole with 10 mM reduced glutathione (307 mg).
Temperature regulated shaking incubator with capacity for holding 2 and 0.1 L volume glass conical flasks.
HiLoad 16/600 Superdex 200 size exclusion chromatography (SEC) column (GE Healthcare Life Sciences) or equivalent.
Automatic liquid chromatography system (AKTA or similar).
Disposable plastic gravity flow columns (e.g. PD-10 columns).
Ultrasonic cell disruptor.
2.2. In vitro AKAP-RII-C binding assay
List of Reagents:
Purified recombinant PKA-C and RIIα subunits and AKAP79297–417 containing a 3C-protease cleavable GST tag.
Purified Rhinovirus 3C protease (cleaves between a glutamine and glycine of targets containing an LEVLFQ/GP consensus sequence, commercially available).
2x AKAP-PKA binding buffer: 100 mM Tris-HCl, pH 7.4, 200 mM NaCl, 10 mM DTT. Add 10 mL 1 M Tris-HCl, pH 7.4, 6.66 mL 3 M NaCl, and 200 μL 1 M DTT to a 100 mL measuring cylinder, and adjust the volume to 100 mL with water. Use at 1x concentration in the assay.
GSH sepharose.
1 mM cAMP and 5’-AMP: Dissolve 3.2 mg cAMP in 10 mL water, 3.2 mg 5’-AMP in 10 mL water. Adjust pH of both solutions to 8.0 with NaOH and freeze prior to storage.
A heating shaking dry bath.
3. Methods
3.1. Expression and purification of recombinant RII, C and AKAP proteins in BL21 (DE3) pLysS E. coli.
For each protein to be purified, prepare 4 × 2 L and 1 × 200 mL glass conical flasks with 750 mL and 100 mL sterile LB broth respectively.
Under sterile conditions, transform 50 μL of competent BL21 (DE3) pLysS cells with 10 ng of plasmid DNA (using standard transformation procedures). Spread the transformed bacteria on to pre-warmed agar plates containing the appropriate selection antibiotics and incubate overnight at 37 °C.
Inoculate 100 mL LB broth (supplemented with the appropriate antibiotics) with a single freshly transformed colony, and incubate overnight at 37 °C in an orbital shaking incubator (180–240 rpm).
Inoculate each of the 4 × 750 mL LB broth flasks (supplemented with the appropriate antibiotics) with 5 mL of the overnight culture and incubate at 37°C (240 rpm) for ~2–3 hours until the OD600nm reaches 0.6 – 0.8. Reduce the temperature of the incubator and the bacterial culture to 18°C and induce protein expression with the addition of 0.4 mM (3 mL 100 mM) IPTG. Incubate overnight (~18 hours) at 240 rpm.
Collect the cells by centrifugation at 5000 × g for 10 mins, 4 °C.
Decant the supernatant and collect and pool the cell pellets. Recombinant proteins can either be immediately harvested (Step 3.2) or the bacterial pellets can be flash frozen in liquid nitrogen and stored at −20°C for ~1 week.
3.2. Lysis of the bacterial cell pellet.
Resuspend the bacterial pellet in ice-cold lysis buffer (~10 mL per gram of bacterial pellet) and transfer to a 100 mL glass beaker.
Use an ultrasonic cell disruptor to lyse the bacterial suspension on ice. Sonicate for 30 seconds and then cool for 1 min to prevent overheating. Repeat for 6–10 cycles or as required (see Note 4).
Centrifuge the lysate in a pre-chilled centrifuge at 43000 × g for 60 min at 4°C.
Collect the supernatant and pass through a 0.45 μM syringe filter to remove any remaining cell debris or aggregated material. Keep the clarified supernatant on ice to maintain protein stability throughout the purification procedure. It is recommended that a sample of the total cell lysate is also taken for further analysis.
3.3. Protein purification.
The following described the procedure for batch affinity purification of PKA and/or AKAP proteins using a ‘column-free’ binding step using a GST-binding resin (such as GSH sepharose) and a 6His-binding resin (such as Ni-NTA agarose). To minimize proteolysis, perform all of the purification steps at 4°C.
Equilibrate 16/600 Superdex 200 pg SEC column with 300 mL gel filtration buffer at a flow rate of 1 mL/min with the pressure limit set to 0.6 MPa.
Transfer 2 mL of 50 % Ni-NTA or GSH sepharose slurry (1 ml resting bead volume) in to a disposable plastic gravity flow column. Wash and equilibrate the settled beads by passing through 10 × column volumes of water followed by 5 × column volumes of cell lysis buffer (see Note 5). Care must be taken to prevent the beads drying.
Resuspend the equilibrated affinity resin in the cell lysate and incubate at 4°C with gentle agitation (using a magnetic stirrer) for 1 h to enable binding of the recombinant proteins. Due to the lower binding efficiency of GST to the GSH resin at 4 °C, it is recommended that the binding stage for GST-AKAP is extended to ~3h.
Sequentially reapply the lysate containing the suspended affinity resin in to the plastic column until all of the beads have been collected. Collect and sample the ‘non-binding’ fraction of the lysate for further analysis.
Wash the beads in with ~ 50 column volumes of high salt wash buffer.
Elute the bound protein by applying 10 column volumes of the appropriate elution buffer and collect as 500 μL fractions as it comes off the column.
Identify protein-containing elution fractions by SDS-PAGE and Coomassie Blue staining (see Note 6). It is recommended that samples of the total lysate and the ‘non-binding’ Ni-NTA fraction are also analysed at this point.
Pool the protein containing fractions and remove any residual aggregated material by centrifugation at 16,000 × g for 20 mins (4 °C) prior to SEC (see Note 7).
If necessary, purify the protein further by SEC, collecting 1.0 mL elution fractions.
Analyse eluted fractions after SEC by SDS-PAGE and Coomassie blue staining.
Pool the protein containing fractions.
The protein can be flash frozen in liquid nitrogen and stored at −80°C (see Note 8).
3.4. AKAP-RII binding assay (Figure 3A)
The following details the specifics of an in vitro GST ‘pull-down’ assay to detect the level of association/dissociation of PKA holoenzyme complexes bound to a purified GST-3C-AKAP79 fragment in the presence of increasing concentrations of cAMP, or the control non-cyclic nucleotide 5’-AMP.
Prepare 6 microcentrifuge tubes containing serial dilution stocks of cAMP at 10x concentration (1 – 0.01 mM). Complete dissociation of the PKA holoenzyme complex is achieved at cAMP concentrations ≥100 μM.
Transfer 320 μl of 50 % glutathione sepharose slurry in to a microcentrifuge tube and centrifuge at 5’000 × g for 1 min. Remove and discard the supernatant, taking care not to disturb the pelleted beads. This will provide a 160 μl packed bead volume which is sufficient to perform 8 individual pull-down assays (20 μl per assay). The volumes can be adjusted proportionally depending on the number assays required.
Equilibrate the beads by resuspending them in 1 mL 1x AKAP-PKA binding buffer before centrifuging them again at 5’000 × g (1 min).
Carefully remove the supernatant and wash the beads an additional two times (see Note 9).
Remove the supernatant and add 200 μL of 1x AKAP-PKA binding buffer containing 40 μg of GST-AKAP79297–417 (~5 μg protein per assay). Incubate for ~3 h at 4 °C with gentle agitation to allow binding of GST-AKAP79297–417 to the resin.
Pellet the beads (5’000 × g, 1 min) and remove the supernatant.
Wash the beads 5 times in 1 mL ice cold AKAP-PKA binding buffer (as for step 3.4.3) to remove residual unbound protein.
Resuspend the pelleted AKAP-bound GSH beads in 1 mL ice cold AKAP-PKA binding buffer and evenly distribute the suspended bead slurry between 8 microcentrifuge tubes (~125 μL per tube). To ensure that all tubes contain an equal volume of AKAP-bound beads, mix the slurry between aliquots by pipetting up and down to prevent bead sedimentation.
Centrifuge the microcentrifuge tubes again and carefully remove as much of the supernatant as possible without disturbing the beads.
Prepare a 450 μL PKA stock solution containing 16 μM PKA-C, 8 μM PKA-RIIα and 250 μL of 2x AKAP-PKA binding buffer. Adjust the volume to 450 μL with water.
Combine the PKA stock solution with the different concentration of 10x cAMP (45 μL PKA mix + 5 μL 10x cAMP). A control assay containing no cAMP (5 μL water) is also required.
As a negative control, prepare a final assay (as above) containing 100 μM 5’-AMP, which will be incapable of dissociating the PKA holoenzyme.
Combine all 8 of the 50 μL PKA solutions with the 8 microcentrifuge tubes containing the AKAP-bound GSH beads, and incubate at 30°C for 20 mins in a shaking drybath with enough agitation to maintain the beads in suspension.
Pellet the beads by centrifugation as previously describe and wash three times in 1 mL ice cold 1x AKAP-PKA binding buffer to remove non-bound and dissociated protein. Remove the supernatant.
Resuspend the beads in 30 μL 1x AKAP-PKA binding buffer supplemented with ~200 ng of 3C. Incubate the beads again in a shaking drybath at 30°C for 30 mins (see Note 10).
Pellet the beads by centrifugation and remove 25 μL of the supernatant which will contain the PKA holoenzyme complex and can be analysed by SDS-PAGE (12 % acrylamide gel) and Coomassie blue staining. The order that the proteins will appear on the gel (from top to bottom) will be PKA-RIIα (~50 kDa), PKA-C (~40 kDa) and AKAP79297–417 (~20 kDa). A low intensity band corresponding to 3C protease can sometimes be detected above AKAP79297–417, although this depends upon the amount of 3C protease used
A concomitant reduction in the intensity of the PKA-C band will be observed as a function of the increasing concentration of cAMP. 5’AMP (which does not bind detectably to R subunit) serves as a convenient negative control (See Figure 3B).
3.5. SDS-PAGE and Commassie Blue staining
Resolving gel buffer: 1.5 mM Tris-HCl, pH 8.8: 181.7 g Tris base in 900 mL water. Adjust the pH to 8.8 with HCl and bring the final volume to 1 L with water.
Stacking gel buffer: 0.5 mM Tris-HCl, pH 6.8: 60.6 g Tris base in 900 mL water. Adjust the pH to 6.8 with HCl and bring the final volume to 1 L with water.
Bis-Acrylamide solution: 29.2:0.8 acrylamide:bis ratio.
Ammonium persulfate (APS): 10% (w/v) in water. 0.1 g APS in 1 mL water.
N,N,N,N’-Tetramethyl-ethylenediamine (TEMED).
SDS-PAGE running buffer (10x): 25 mM Tris, 192 mM glycine, 3.5 mM SDS. 60.57 g Tris, 288.3 g glycine and 20 g SDS in 2 L of water and mix. Dilute 100 mL of 10x SDS-PAGE running buffer in 900 mL water for to make a 1x solution for use.
5x SDS-PAGE loading buffer
Pre-stained molecular mass standards.
1 mM glass gel plates, 1 mM 10 well combs, and electrophoresis system for SDS-PAGE.
Coomassie blue: 500 mL methanol, 1 g of Brilliant Blue, 100 mL acetic in 1 L (with water). Mix well.
Destaining solution: 200 mL methanol, 100 mL acetic acid in 1 L (with water). Mix well.
Plastic container for Coomassie blue gel staining.
3.6. Biophysical analysis of purified recombinant PKA signaling components
Related techniques to evaluate biochemical interactions with purified AKAP, RII and C subunits include Differential Scanning Fluorimetry (DSF), circular dichroism and native mass spectrometry, and these are described in more detail in [59, 83]. For example, DSF can be employed for analysis of direct binding of cAMP to purified RII subunits discussed in this chapter (and does not require prior unfolding to strip of endogenous cAMP that can co-purify from bacteria [84]), or for the titration of small molecules such as kinase inhibitors with C-subunits. In addition, the binding of the heat-stable PKI inhibitor protein and derived peptides to C subunits, and the interaction of a variety of non-specific kinase inhibitors with the ATP site of C-subunits is described [59]. These studies also include careful evaluation of a ‘kinase-dead’ PKA catalytic domain [60] that does not bind to ATP or PKI in solution, but can still interact with a panel of chemical PKA inhibitors, as well as an R133A RII mutant that does not interact with RII subunits or PKI [59].
4. Notes
Buffer stock solutions: 1 M Tris-HCl, pH 7.4 – 121.14 g of Tris base in 800 mL water, adjust to pH 7.4 with HCl and make up the volume to 1 L with water; 3 M NaCl – 175.32 g of NaCl in 1 L water; 1 M imidazole – 68.07 g imidazole in 1 L water (adjust pH to 7.4 with HCl); 1 M DTT – 15.43 g DTT in 10 mL water (aliquot and store at −20 °C); 100 mM EGTA – 19 g in 500 mL water; 100 mM EDTA – 14.6 g in 500 mL water (adjust the pH of EDTA and EGTA to 8.0 with NaOH).
Triton X-100 is extremely viscous. To aid pipetting accuracy, remove the end point of the pipette tip with scissors.
Gel filtration buffer must be filtered and degassed for at least 1 hour prior to use in SEC.
Sonication conditions will vary depending on the type of system and the size of the probe used.
Alternatively, a syringe attached to the end of the column with a small stretch of rubber tubing can be used to manually draw through the liquid phase containing the eluate.
We typically use 10% resolving acrylamide gels of 1 mm thickness for SDS-PAGE, loading approximately 5–10 μL of sample per well.
It may be necessary to concentrate the pooled eluted protein in order to load it all on to the SEC column. For improved purification resolution, it is recommended that a ~1 mL protein volume is applied to the Superdex 200 16/60 column.
It is recommended that the purified protein be aliquoted prior to cryo-storage to avoid damage caused repeated freeze/thaw cycles.
GSH beads are provided as a slurry in 20% (v/v) ethanol, which must be removed with successive wash steps to prevent denaturation of the assay proteins.
Proteolytic cleavage of the GST tag (which remains bound to the GSH beads) from the AKAP79 protein results in the elution of the intact PKA holoenzyme complex in the mobile phase, which is then collected for analysis by SDS-PAGE.
Acknowledgements
This work was supported, in whole or in part, by and BBSRC grants BB/S018514/1, BB/N021703/1 and BB/R000182/1 to D.B. and P.A.E. and National Institutes of Health Grants 1K22CA154600 to EJK, F32DK121415 to MO and NIH DK119186 and DK119192 to J.D.S
References
- 1.Wilson LJ, et al. , New Perspectives, Opportunities, and Challenges in Exploring the Human Protein Kinome. Cancer Res, 2018. 78(1): p. 15–29. [DOI] [PubMed] [Google Scholar]
- 2.Newton AC, Bootman MD, and Scott JD, Second Messengers. Cold Spring Harb Perspect Biol, 2016. 8(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beuschlein F, et al. , Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. N Engl J Med, 2014. 370(11): p. 1019–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Omar MH and Scott JD, AKAP Signaling Islands: Venues for Precision Pharmacology. Trends Pharmacol Sci, 2020. 41(12): p. 933–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bucko PJ and Scott JD, Drugs That Regulate Local Cell Signaling: AKAP Targeting as a Therapeutic Option. Annu Rev Pharmacol Toxicol, 2021. 61: p. 361–379. [DOI] [PubMed] [Google Scholar]
- 6.Carnegie GK, Means CK, and Scott JD, A-kinase anchoring proteins: from protein complexes to physiology and disease. IUBMB Life, 2009. 61(4): p. 394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scott JD and Pawson T, Cell signaling in space and time: where proteins come together and when they’re apart. Science, 2009. 326(5957): p. 1220–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carr DW, et al. , Interaction of the regulatory subunit (RII) of cAMP-dependent protein kinase with RII-anchoring proteins occurs through an amphipathic helix binding motif. J Biol Chem, 1991. 266(22): p. 14188–92. [PubMed] [Google Scholar]
- 9.Gold MG, et al. , Molecular basis of AKAP specificity for PKA regulatory subunits. Mol Cell, 2006. 24(3): p. 383–95. [DOI] [PubMed] [Google Scholar]
- 10.Newlon MG, et al. , A novel mechanism of PKA anchoring revealed by solution structures of anchoring complexes. EMBO J, 2001. 20(7): p. 1651–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith FD, et al. , Local protein kinase A action proceeds through intact holoenzymes. Science, 2017. 356(6344): p. 1288–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Welch EJ, Jones BW, and Scott JD, Networking with AKAPs: context-dependent regulation of anchored enzymes. Mol Interv, 2010. 10(2): p. 86–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skroblin P, et al. , Mechanisms of protein kinase A anchoring. Int Rev Cell Mol Biol, 2010. 283: p. 235–330. [DOI] [PubMed] [Google Scholar]
- 14.Sanderson JL and Dell’Acqua ML, AKAP signaling complexes in regulation of excitatory synaptic plasticity. Neuroscientist, 2011. 17(3): p. 321–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dessauer CW, Adenylyl cyclase--A-kinase anchoring protein complexes: the next dimension in cAMP signaling. Mol Pharmacol, 2009. 76(5): p. 935–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diviani D, et al. , A-kinase anchoring proteins: scaffolding proteins in the heart. Am J Physiol Heart Circ Physiol, 2011. 301(5): p. H1742–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klauck TM, et al. , Coordination of three signaling enzymes by AKAP79, a mammalian scaffold protein. Science, 1996. 271(5255): p. 1589–92. [DOI] [PubMed] [Google Scholar]
- 18.Coghlan VM, Hausken ZE, and Scott JD, Subcellular targeting of kinases and phosphatases by association with bifunctional anchoring proteins. Biochem Soc Trans, 1995. 23(3): p. 592–6. [DOI] [PubMed] [Google Scholar]
- 19.Dodge KL, et al. , mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. EMBO J, 2001. 20(8): p. 1921–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tasken KA, et al. , Phosphodiesterase 4D and protein kinase a type II constitute a signaling unit in the centrosomal area. J Biol Chem, 2001. 276(25): p. 21999–2002. [DOI] [PubMed] [Google Scholar]
- 21.Bauman AL, et al. , Dynamic regulation of cAMP synthesis through anchored PKA-adenylyl cyclase V/VI complexes. Mol Cell, 2006. 23(6): p. 925–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor SS, et al. , PKA: a portrait of protein kinase dynamics. Biochim Biophys Acta, 2004. 1697(1–2): p. 259–69. [DOI] [PubMed] [Google Scholar]
- 23.Gold MG, Swimming regulations for protein kinase A catalytic subunit. Biochem Soc Trans, 2019. 47(5): p. 1355–1366. [DOI] [PubMed] [Google Scholar]
- 24.Herberg FW, et al. , Analysis of A-kinase anchoring protein (AKAP) interaction with protein kinase A (PKA) regulatory subunits: PKA isoform specificity in AKAP binding. J Mol Biol, 2000. 298(2): p. 329–39. [DOI] [PubMed] [Google Scholar]
- 25.Aye TT, et al. , Selectivity in enrichment of cAMP-dependent protein kinase regulatory subunits type I and type II and their interactors using modified cAMP affinity resins. Mol Cell Proteomics, 2009. 8(5): p. 1016–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walker-Gray R, Stengel F, and Gold MG, Mechanisms for restraining cAMP-dependent protein kinase revealed by subunit quantitation and cross-linking approaches. Proc Natl Acad Sci U S A, 2017. 114(39): p. 10414–10419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lester LB, et al. , Cloning and characterization of a novel A-kinase anchoring protein. AKAP 220, association with testicular peroxisomes. J Biol Chem, 1996. 271(16): p. 9460–5. [DOI] [PubMed] [Google Scholar]
- 28.Huang LJ, et al. , Identification of a novel protein kinase A anchoring protein that binds both type I and type II regulatory subunits. J Biol Chem, 1997. 272(12): p. 8057–64. [DOI] [PubMed] [Google Scholar]
- 29.Trotter KW, et al. , Alternative splicing regulates the subcellular localization of A-kinase anchoring protein 18 isoforms. J Cell Biol, 1999. 147(7): p. 1481–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scott JD, Dessauer CW, and Tasken K, Creating order from chaos: cellular regulation by kinase anchoring. Annu Rev Pharmacol Toxicol, 2013. 53: p. 187–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, et al. , Isoform-selective disruption of AKAP-localized PKA using hydrocarbon stapled peptides. ACS Chem Biol, 2014. 9(3): p. 635–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carr DW, et al. , Association of the type II cAMP-dependent protein kinase with a human thyroid RII-anchoring protein. Cloning and characterization of the RII-binding domain. J Biol Chem, 1992. 267(19): p. 13376–82. [PubMed] [Google Scholar]
- 33.Vijayaraghavan S, et al. , Protein kinase A-anchoring inhibitor peptides arrest mammalian sperm motility. J Biol Chem, 1997. 272(8): p. 4747–52. [DOI] [PubMed] [Google Scholar]
- 34.Alto NM, et al. , Bioinformatic design of A-kinase anchoring protein-in silico: a potent and selective peptide antagonist of type II protein kinase A anchoring. Proc Natl Acad Sci U S A, 2003. 100(8): p. 4445–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Faruque OM, et al. , Cell-permeable peptide-based disruption of endogenous PKA-AKAP complexes: a tool for studying the molecular roles of AKAP-mediated PKA subcellular anchoring. Am J Physiol Cell Physiol, 2009. 296(2): p. C306–16. [DOI] [PubMed] [Google Scholar]
- 36.Hundsrucker C, et al. , High-affinity AKAP7delta-protein kinase A interaction yields novel protein kinase A-anchoring disruptor peptides. Biochem J, 2006. 396(2): p. 297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Christian F, et al. , Small molecule AKAP-protein kinase A (PKA) interaction disruptors that activate PKA interfere with compartmentalized cAMP signaling in cardiac myocytes. J Biol Chem, 2011. 286(11): p. 9079–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schafer G, et al. , Highly functionalized terpyridines as competitive inhibitors of AKAP-PKA interactions. Angew Chem Int Ed Engl, 2013. 52(46): p. 12187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Verdine GL and Hilinski GJ, Stapled peptides for intracellular drug targets. Methods Enzymol, 2012. 503: p. 3–33. [DOI] [PubMed] [Google Scholar]
- 40.Gold MG, et al. , Engineering A-kinase anchoring protein (AKAP)-selective regulatory subunits of protein kinase A (PKA) through structure-based phage selection. J Biol Chem, 2013. 288(24): p. 17111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burns-Hamuro LL, et al. , Designing isoform-specific peptide disruptors of protein kinase A localization. Proc Natl Acad Sci U S A, 2003. 100(7): p. 4072–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carlson CR, et al. , Delineation of type I protein kinase A-selective signaling events using an RI anchoring disruptor. J Biol Chem, 2006. 281(30): p. 21535–21545. [DOI] [PubMed] [Google Scholar]
- 43.Torheim EA, et al. , Design of proteolytically stable RI-anchoring disruptor peptidomimetics for in vivo studies of anchored type I protein kinase A-mediated signalling. Biochem J, 2009. 424(1): p. 69–78. [DOI] [PubMed] [Google Scholar]
- 44.Sarma GN, et al. , Structure of D-AKAP2:PKA RI complex: insights into AKAP specificity and selectivity. Structure, 2010. 18(2): p. 155–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jarnaess E, et al. , Dual specificity A-kinase anchoring proteins (AKAPs) contain an additional binding region that enhances targeting of protein kinase A type I. J Biol Chem, 2008. 283(48): p. 33708–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seamon KB, Padgett W, and Daly JW, Forskolin: unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc Natl Acad Sci U S A, 1981. 78(6): p. 3363–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patel TB, et al. , Molecular biological approaches to unravel adenylyl cyclase signaling and function. Gene, 2001. 269(1–2): p. 13–25. [DOI] [PubMed] [Google Scholar]
- 48.Premont RT, et al. , Identification and characterization of a widely expressed form of adenylyl cyclase. J Biol Chem, 1996. 271(23): p. 13900–7. [DOI] [PubMed] [Google Scholar]
- 49.Dessauer CW, Scully TT, and Gilman AG, Interactions of forskolin and ATP with the cytosolic domains of mammalian adenylyl cyclase. J Biol Chem, 1997. 272(35): p. 22272–7. [DOI] [PubMed] [Google Scholar]
- 50.Beavo JA, et al. , Effects of xanthine derivatives on lipolysis and on adenosine 3’,5’-monophosphate phosphodiesterase activity. Mol Pharmacol, 1970. 6(6): p. 597–603. [PubMed] [Google Scholar]
- 51.Omori K and Kotera J, Overview of PDEs and their regulation. Circ Res, 2007. 100(3): p. 309–27. [DOI] [PubMed] [Google Scholar]
- 52.Robison GA, Butcher RW, and Sutherland EW, Cyclic AMP. Annu Rev Biochem, 1968. 37: p. 149–74. [DOI] [PubMed] [Google Scholar]
- 53.Shear M, et al. , Agonist-specific refractoriness induced by isoproterenol. Studies with mutant cells. J Biol Chem, 1976. 251(23): p. 7572–6. [PubMed] [Google Scholar]
- 54.Schwede F, et al. , Cyclic nucleotide analogs as biochemical tools and prospective drugs. Pharmacol Ther, 2000. 87(2–3): p. 199–226. [DOI] [PubMed] [Google Scholar]
- 55.Beavo JA and Brunton LL, Cyclic nucleotide research -- still expanding after half a century. Nat Rev Mol Cell Biol, 2002. 3(9): p. 710–8. [DOI] [PubMed] [Google Scholar]
- 56.Walsh DA, et al. , Krebs EG: Purification and characterization of a protein inhibitor of adenosine 3’,5’-monophosphate-dependent protein kinases. J Biol Chem, 1971. 246(7): p. 1977–85. [PubMed] [Google Scholar]
- 57.Dalton GD and Dewey WL, Protein kinase inhibitor peptide (PKI): a family of endogenous neuropeptides that modulate neuronal cAMP-dependent protein kinase function. Neuropeptides, 2006. 40(1): p. 23–34. [DOI] [PubMed] [Google Scholar]
- 58.Demaille JG, Peters KA, and Fischer EH, Isolation and properties of the rabbit skeletal muscle protein inhibitor of adenosine 3’,5’-monophosphate dependent protein kinases. Biochemistry, 1977. 16(14): p. 3080–6. [DOI] [PubMed] [Google Scholar]
- 59.Byrne DP, et al. , cAMP-dependent protein kinase (PKA) complexes probed by complementary differential scanning fluorimetry and ion mobility-mass spectrometry. Biochem J, 2016. 473(19): p. 3159–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eyers PA, et al. , Regulation of the G(2)/M transition in Xenopus oocytes by the cAMP-dependent protein kinase. J Biol Chem, 2005. 280(26): p. 24339–46. [DOI] [PubMed] [Google Scholar]
- 61.Scott JD, et al. , Identification of an inhibitory region of the heat-stable protein inhibitor of the cAMP-dependent protein kinase. Proc Natl Acad Sci U S A, 1985. 82(13): p. 4379–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scott JD, et al. , Primary-structure requirements for inhibition by the heat-stable inhibitor of the cAMP-dependent protein kinase. Proc Natl Acad Sci U S A, 1986. 83(6): p. 1613–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cheng HC, et al. , An active twenty-amino-acid-residue peptide derived from the inhibitor protein of the cyclic AMP-dependent protein kinase. Biochem J, 1985. 231(3): p. 655–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Glass DB, et al. , Differential and common recognition of the catalytic sites of the cGMP-dependent and cAMP-dependent protein kinases by inhibitory peptides derived from the heat-stable inhibitor protein. J Biol Chem, 1986. 261(26): p. 12166–71. [PubMed] [Google Scholar]
- 65.Cheng HC, et al. , A potent synthetic peptide inhibitor of the cAMP-dependent protein kinase. J Biol Chem, 1986. 261(3): p. 989–92. [PubMed] [Google Scholar]
- 66.Reed J, et al. , Conformational analysis of PKI(5–22)amide, the active inhibitory fragment of the inhibitor protein of the cyclic AMP-dependent protein kinase. Biochem J, 1989. 264(2): p. 371–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Knighton DR, et al. , Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science, 1991. 253(5018): p. 414–20. [DOI] [PubMed] [Google Scholar]
- 68.Manschwetus JT, et al. , A Stapled Peptide Mimic of the Pseudosubstrate Inhibitor PKI Inhibits Protein Kinase A. Molecules, 2019. 24(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bain J, et al. , The selectivity of protein kinase inhibitors: a further update. Biochem J, 2007. 408(3): p. 297–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hidaka H, et al. , Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide dependent protein kinase and protein kinase C. Biochemistry, 1984. 23(21): p. 5036–41. [DOI] [PubMed] [Google Scholar]
- 71.Chijiwa T, et al. , Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J Biol Chem, 1990. 265(9): p. 5267–72. [PubMed] [Google Scholar]
- 72.Byrne DP, et al. , Aurora A regulation by reversible cysteine oxidation reveals evolutionarily conserved redox control of Ser/Thr protein kinase activity. Sci Signal, 2020. 13(639). [DOI] [PubMed] [Google Scholar]
- 73.Davies SP, et al. , Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J, 2000. 351(Pt 1): p. 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Murray AJ, Pharmacological PKA inhibition: all may not be what it seems. Sci Signal, 2008. 1(22): p. re4. [DOI] [PubMed] [Google Scholar]
- 75.Yap TA, et al. , AT13148 is a novel, oral multi-AGC kinase inhibitor with potent pharmacodynamic and antitumor activity. Clin Cancer Res, 2012. 18(14): p. 3912–23. [DOI] [PubMed] [Google Scholar]
- 76.Rothermel JD, et al. , Inhibition of glycogenolysis in isolated rat hepatocytes by the Rp diastereomer of adenosine cyclic 3’,5’-phosphorothioate. J Biol Chem, 1983. 258(20): p. 12125–8. [PubMed] [Google Scholar]
- 77.Rothermel JD, Jastorff B, and Botelho LH, Inhibition of glucagon-induced glycogenolysis in isolated rat hepatocytes by the Rp diastereomer of adenosine cyclic 3’,5’-phosphorothioate. J Biol Chem, 1984. 259(13): p. 8151–5. [PubMed] [Google Scholar]
- 78.Gjertsen BT, et al. , Novel (Rp)-cAMPS analogs as tools for inhibition of cAMP-kinase in cell culture. Basal cAMP-kinase activity modulates interleukin-1 beta action. J Biol Chem, 1995. 270(35): p. 20599–607. [DOI] [PubMed] [Google Scholar]
- 79.Maller JL, Butcher FR, and Krebs EG, Early effect of progesterone on levels of cyclic adenosine 3’:5’-monophosphate in Xenopus oocytes. J Biol Chem, 1979. 254(3): p. 579–82. [PubMed] [Google Scholar]
- 80.Maller JL and Krebs EG, Progesterone-stimulated meiotic cell division in Xenopus oocytes. Induction by regulatory subunit and inhibition by catalytic subunit of adenosine 3’:5’-monophosphate-dependent protein kinase. J Biol Chem, 1977. 252(5): p. 1712–8. [PubMed] [Google Scholar]
- 81.Mant A, et al. , Protein kinase A is central for forward transport of two-pore domain potassium channels K2P3.1 and K2P9.1. J Biol Chem, 2011. 286(16): p. 14110–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Iyer GH, Moore MJ, and Taylor SS, Consequences of lysine 72 mutation on the phosphorylation and activation state of cAMP-dependent kinase. J Biol Chem, 2005. 280(10): p. 8800–7. [DOI] [PubMed] [Google Scholar]
- 83.Byrne DP, et al. , New tools for evaluating protein tyrosine sulfation: tyrosylprotein sulfotransferases (TPSTs) are novel targets for RAF protein kinase inhibitors. Biochem J, 2018. 475(15): p. 2435–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Buechler YJ, Herberg FW, and Taylor SS, Regulation-defective mutants of type I cAMP-dependent protein kinase. Consequences of replacing arginine 94 and arginine 95. J Biol Chem, 1993. 268(22): p. 16495–503. [PubMed] [Google Scholar]