

After cessation of blood flow or similar ischaemic exposures, deleterious molecular cascades commence in mammalian cells, eventually leading to their death1,2. Yet with targeted interventions, these processes can be mitigated or reversed, even minutes or hours post mortem, as also reported in the isolated porcine brain using BrainEx technology3. To date, translating single-organ interventions to intact, whole-body applications remains hampered by circulatory and multisystem physiological challenges. Here we describe OrganEx, an adaptation of the BrainEx extracorporeal pulsatile-perfusion system and cytoprotective perfusate for porcine whole-body settings. After 1 h of warm ischaemia, OrganEx application preserved tissue integrity, decreased cell death and restored selected molecular and cellular processes across multiple vital organs. Commensurately, single-nucleus transcriptomic analysis revealed organ- and cell-type-specific gene expression patterns that are reflective of specific molecular and cellular repair processes. Our analysis comprises a comprehensive resource of cell-type-specific changes during defined ischaemic intervals and perfusion interventions spanning multiple organs, and it reveals an underappreciated potential for cellular recovery after prolonged whole-body warm ischaemia in a large mammal.
Mammalian cells require oxygen to maintain cellular and tissue viability4. In just minutes after ischaemia, intracellular acidosis and oedema develop and trigger secondary injury to membranes and organelles, often causing cell death1. At the whole-body scale, there is a systemic release of hormones and cytokines, followed by activation of autonomic nervous, immune and coagulation systems, leading to end-organ injury culminating in systemic metabolic acidosis and hyperkalaemia2,5,6.
However, recent studies question the inevitability of cell death even after hours of circulatory interruption. Viable cells can be collected from multiple organs and maintained using in vitro culture after prolonged ischaemia7. Similarly, cellular recovery can be promoted using ex vivo perfusion of isolated whole organs, including heart, liver, kidneys and lungs8–11. Our perfusion-based BrainEx technology also demonstrated restored circulation and cellular activity hours post mortem in isolated porcine brains—the organ that is most vulnerable to ischaemia2,6.
Nevertheless, translating solutions from isolated-organ models for molecular and cellular recovery to whole-body applications after extended ischaemia still presents substantial challenges. Reperfusion of the whole body with autologous blood has several hindrances, including coagulation, microvascular dysfunction, inflammation and blood-intrinsic cellular dysfunction2,5. This has restricted whole-body reperfusion and recovery times to 20 min in large mammals12, although catastrophic/fatal consequences are widespread after several minutes in humans13. A new approach may reinstate systemic circulation while adding targeted molecular and cellular recovery strategies for specific organs in the whole-body setting. From this, achieving recovery after 1 h of warm ischaemia may facilitate the development of opportunities across various clinical disciplines.
Towards these goals, we translated principles from BrainEx technology3 to develop OrganEx, a perfusion system and synthetic, acellular, cytoprotective perfusate, for whole-body use in large mammals. We evaluated OrganEx by perfusing female porcine (Sus scrofa domesticus, 30–35 kg) for 6 h, after 1 h of warm ischaemia induced by the cessation of circulation through cardiac ventricular fibrillation. The non-survival perfusion protocol was implemented in coordination with the Yale University Institutional Animal Care and Use Committee and external advisory and ethics committees and was based on preordained essential principles to ensure humane and compassionate treatment (Methods).
This study demonstrates that targeted interventions can reveal unexpected capacities for molecular and cellular recovery across vital organs in the large mammalian body after prolonged warm ischaemia. These findings shed new light on cell-type- and tissue-specific responses to a range of warm ischaemic intervals and perfusion interventions at the whole-body scale.
Overview of OrganEx technology
The OrganEx technology comprises a perfusion system and synthetic perfusate (Fig. 1b and Methods). The perfusion system circuit includes a custom-made pulse generator that is connected to a centrifugal pump that reproduces physiological pressure waveforms. The circuit is primed with a solution specifically tailored to counter electrolyte and metabolic imbalances after prolonged ischaemia, while automated haemodiafiltration and drug-delivery systems supplement the perfusate (Supplementary Tables 1–3). The perfusate is a modification of our acellular, haemoglobin-based, non-coagulative and cytoprotective BrainEx perfusate3, now optimized for whole-body compatibility. Finally, real-time sensors enable the monitoring of important circulation properties and metabolites.
Fig. 1 |. Overview of the OrganEx technology and the experimental workflow.
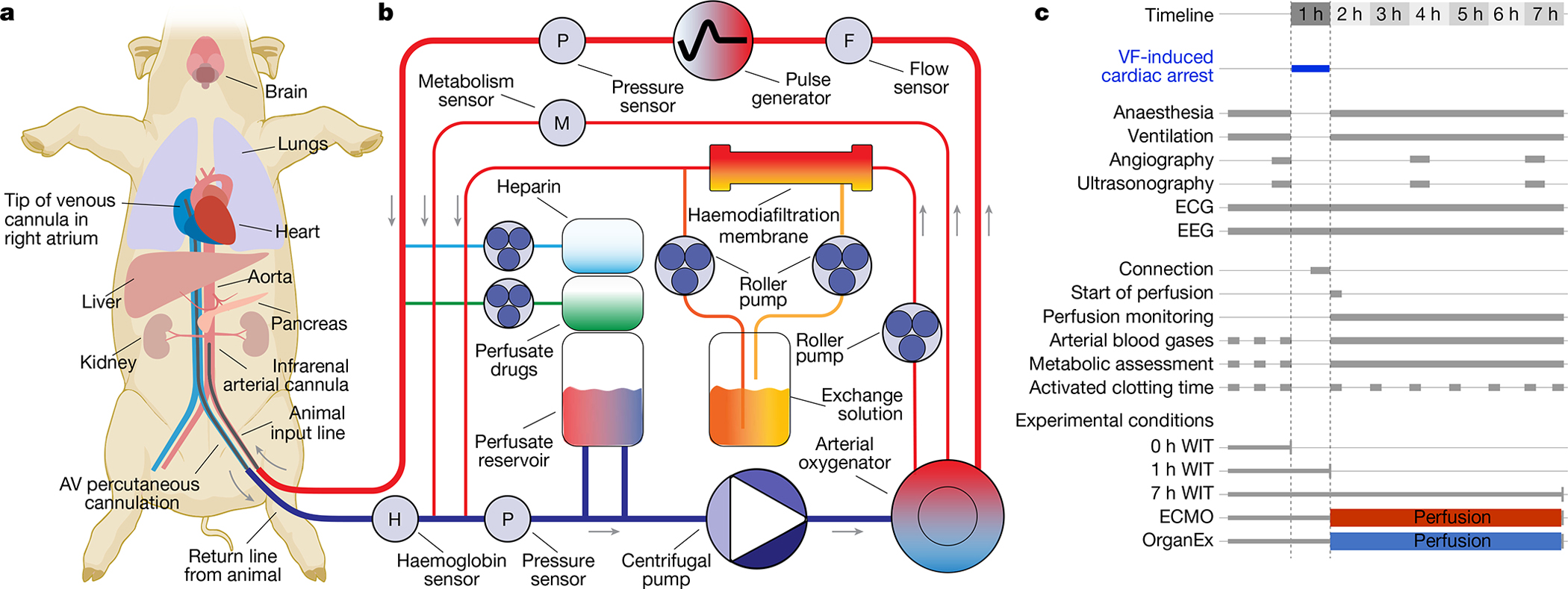
a, Connection of the porcine body to the OrganEx perfusion system (or ECMO, not shown) through cannulation of the femoral artery and vein. b, Simplified schematic of the OrganEx perfusion device. The system is equipped with a centrifugal pump, pulse generator, haemodiafiltration, gas infusion, drug-delivery systems and sensors to measure metabolic and circulation parameters. c, Schematic of the experimental workflow and conditions. VF, ventricular fibrillation.
To evaluate OrganEx technology in large mammals, we implemented a porcine global warm ischaemia model induced by ventricular fibrillation cardiac arrest in anaesthetized and heparinized animals. After cardiac arrest and cessation of the systemic circulation, warm ischaemic damage was allowed to ensue for 1 h, ensuring a core temperature of 36–37 °C. Subsequently, animals were connected to one of the two perfusion systems (see below) through a femoral artery/vein approach to reinstate systemic circulation, without requiring flow to the heart chambers and lung alveoli (Fig. 1a). Our study consisted of five groups: three unperfused control groups corresponding to distinct experimental time points and warm ischaemic times (WIT): (1) control group with minimal/0 h WIT; (2) 1 h WIT to investigate accrued molecular/cellular injury before perfusion interventions; and (3) 7 h WIT to investigate the full extent of the damage that occurs without any intervention. Moreover, in the two perfusion-intervention groups after 1 h of warm ischaemia, we perfused for 6 h under hypothermic conditions (28 °C) either with (4) a clinical standard, heart-and-lung substitution perfusion device—extracorporeal membrane oxygenation system (ECMO)14; or (5) our perfusion technology (OrganEx) (Fig. 1c). The ECMO group was perfused with autologous blood. In the OrganEx group, before initiating the perfusion protocol, autologous blood was drained into the OrganEx system and mixed with the perfusate at an effective 1:1 ratio, which was then used for perfusion.
Systemic circulation parameters
We first examined whether whole-body circulation could be restored with external perfusion after 1 h of warm ischaemia. Perfusion with the ECMO system invariantly resulted in low or no flow states. Using fluoroscopic angiography, ECMO perfusion exhibited limited filling of major conduit arteries and organs, such as the kidneys, liver and brain (Fig. 2a and Extended Data Fig. 1a). ECMO interventions similarly yielded inadequate organ perfusion as indicated by colour Doppler ultrasound analysis (Fig. 2b). Furthermore, systemic perfusion parameters revealed a collapse in circulation as shown by negative venous perfusion pressure and low arterial pressures (Fig. 2c and Extended Data Fig. 1c).
Fig. 2 |. Circulation and blood/perfusate properties during the perfusion protocols.
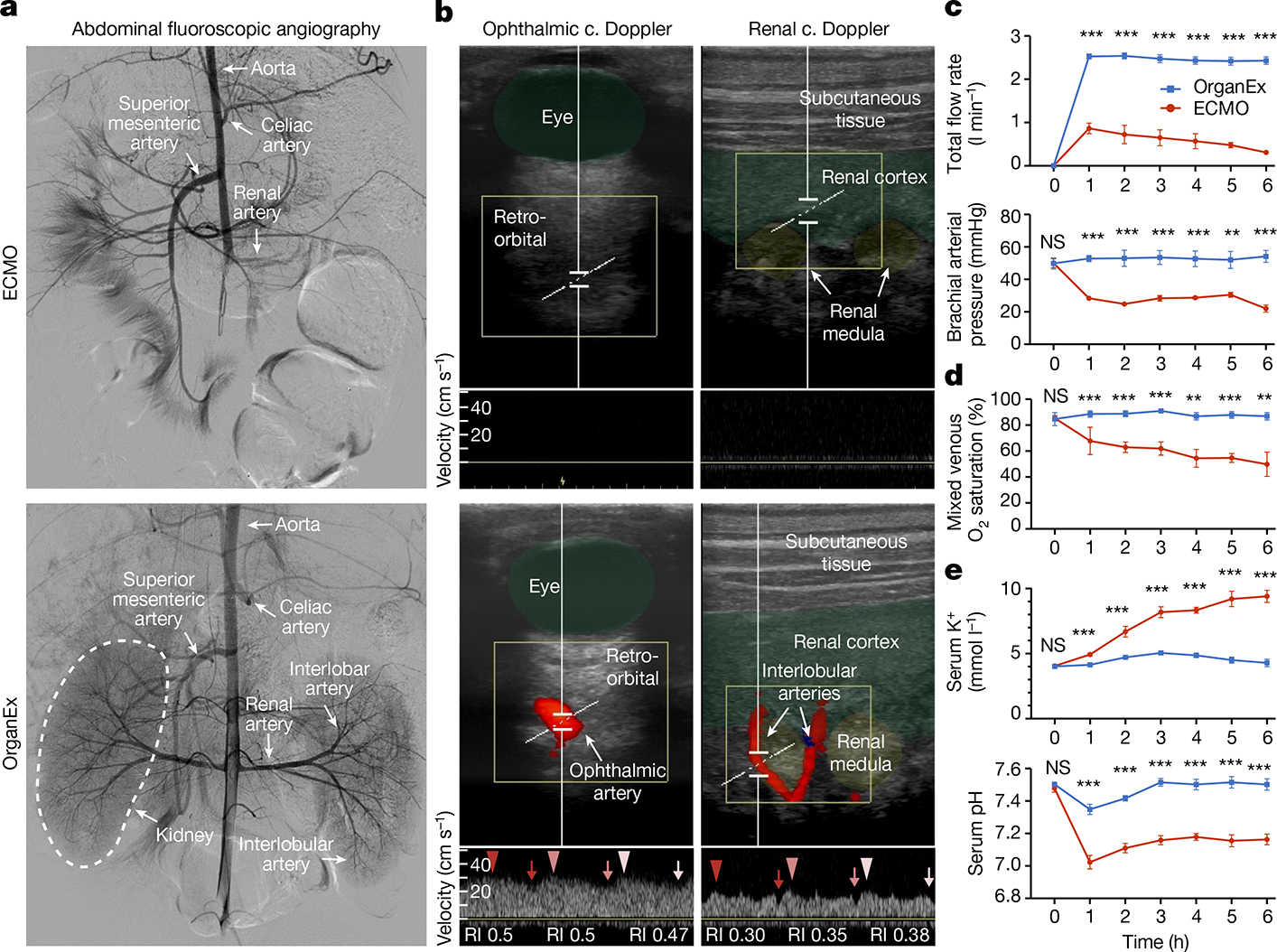
a,b, Representative images of abdominal fluoroscopy (a; n = 9) and ophthalmic and renal ultrasound (b; n = 6) after 3 h of perfusion. ECMO is shown at the top and OrganEx is shown at the bottom. c., colour; RI, resistive index. c–e, Changes in the total flow rate and brachial arterial pressure (c), the percentage of venous O2 saturation (d) and K+ concentration and pH in the serum (e) throughout the perfusion protocols. n = 6. For c–e, data are mean ± s.e.m. Statistical analysis was performed using unpaired two-tailed t-tests; **P < 0.01, ***P < 0.001; NS, not significant. Further detailed information on statistics and reproducibility are provided in the Methods.
By contrast, we observed robust whole-body perfusion in the OrganEx group, as indicated by contrast enhancement of major conduit arteries and organs, and colour Doppler analysis showing whole-body pulsatile flow (Fig. 2a,b and Extended Data Fig. 1a). In particular, flow in the ophthalmic artery—a proxy indicator of cerebral perfusion—was present in the OrganEx group but not in the ECMO perfusion group when analysed at hour three of the perfusion protocols (Fig. 2b). Similar patterns were observed in renal intralobular arteries (Fig. 2b), whereas flow was reduced, but not absent, in the carotid arteries of animals in the ECMO group compared with in the OrganEx group (Extended Data Fig. 1b). These findings were further supported by the system’s sensors, which showed restoration of physiological flow rates and arterial pressures (Fig. 2c).
After successful restoration of circulation, we assessed the potential for OrganEx to normalize related metabolic parameters. By measuring the oxygen saturation of venous blood, we confirmed that the OrganEx technology was able to deliver adequate levels of oxygen to the whole body during the perfusion (Fig. 2d). This was coupled with the stabilization of tissue metabolic expenditures (Extended Data Fig. 1d) and correction of physiological imbalances that occur during prolonged ischaemia, most notably hyperkalaemia and metabolic acidosis (Fig. 2e). Furthermore, the post mortem rigidity and lividity observed in the ECMO group was absent after OrganEx perfusion (Extended Data Fig. 1e). Taken together, these observations indicate that, after 1 h of warm ischaemia, OrganEx could reinstate circulation and restore observed physiological and metabolic parameters at the whole-body scale.
Histological analysis
After restoration of systemic circulation and certain key metabolic parameters using OrganEx perfusion, we next investigated cytoarchitectural changes of multiple organs using unbiased sampling and histological analysis. We analysed brain cytoarchitecture using Nissl staining in the hippocampal CA1 region and the prefrontal cortex (PFC)–two regions that are particularly susceptible to ischaemia15,16. CA1 and PFC cell numbers both diminished as ischaemic durations increased, with comparable depletion in the 7 h and ECMO groups. However, OrganEx exposure restored counts nearly to the levels of the 0 h WIT group (Extended Data Fig. 2a,b,d). As an indicator of early cytotoxic oedema from ischaemic stress, ellipsoid cell morphologies increased with longer WIT in the PFC but experienced a ceiling effect at all time points in the CA1. Advanced injury morphologies, with fewer ellipsoid cells, predominated in the ECMO group, but OrganEx cell counts approximated those of the 0 h WIT group in both regions (Extended Data Fig. 2a,c,e).
We next assessed the brain’s three major cell types: neurons, astrocytes and microglia. Immunolabelling intensity of the pan-neuronal marker NeuN (also known as RBFOX3), which has previously been shown to decrease in hypoxia17, was lower in the ECMO and 7 h WIT groups in both regions, as compared to the OrganEx group (Fig. 3a,b and Extended Data Fig. 2h,i). Although immunolabelling of NeuN in the OrganEx and 0 h and 1 h WIT groups did not differ in the CA1, the levels were reduced for the 1 h WIT and OrganEx groups in the PFC compared with the 0 h WIT controls (Extended Data Fig. 2h,i). Similarly, the numbers of astrocytes immunolabelled for GFAP were comparable between the OrganEx and 0 h WIT groups but were decreased in the other groups in both the PFC and CA1 (Extended Data Fig. 2f,g,h,k). Within the CA1, analysis of GFAP immunolabelling revealed increased fragmentation, suggesting impaired astrocytic integrity, after 7 h WIT and ECMO exposure, but the OrganEx, 1 h and 0 h WIT groups had similarly low GFAP fragmentation (Fig. 3a,c). GFAP fragmentation levels in the PFC were also increased in the 7 h WIT and ECMO groups, but higher variance meant that some differences were not significant (Extended Data Fig. 2h,j). Microglial populations marked by IBA1 immunolabelling likewise were similar in both the CA1 and PFC of the OrganEx and 1 h and 0 h WIT groups, while in the ECMO group, a distinct increase in CA1 (Fig. 3a,d) and a decrease in PFC (Extended Data Fig. 2h,l) were observed. Collectively, these findings indicate that, across the brain regions and cell types analysed, cellular and tissue integrity was preserved when assessed immediately after perfusion with OrganEx, consistent with our previous study in the isolated porcine brain3.
Fig. 3 |. Analysis of tissue integrity across experimental conditions and organs.
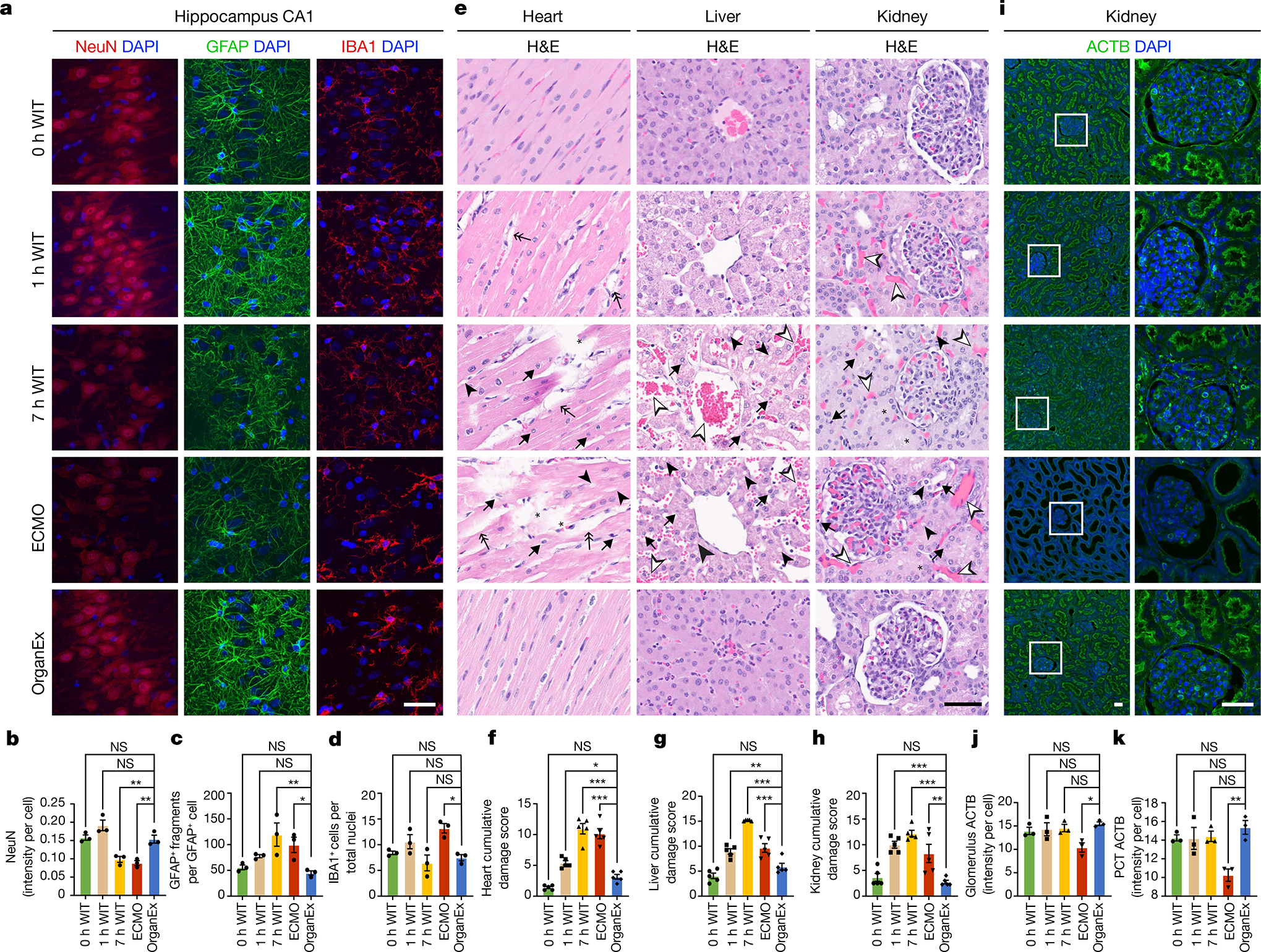
a, Representative confocal images of immunofluorescence staining for neurons (NeuN), astrocytes (GFAP) and microglia (IBA1) counterstained with DAPI nuclear stain in the hippocampal CA1 region. b–d, Quantification of NeuN immunoreactivity intensity (b), the number of GFAP fragments (c) and the microglia number (d) in the CA1. n = 3. e, Representative images of H&E staining in the heart, liver and kidneys. f–h, Evaluation of histopathological criteria, including nuclear pyknosis (arrow), tissue integrity (asterisk), haemorrhage/congestion (empty arrowhead), cell vacuolization (full arrowhead) and tissue oedema (double arrow) in the heart (f), liver (g) and kidneys (h). n = 5. i–k, Representative confocal images of immunofluorescence staining for ACTB in the kidneys (i) and its quantification in the glomerulus (j) and proximal convoluted tubule (PCT) (k). n = 3. Scale bars, 40 μm. For b–d, f–h, j and k, data are mean ± s.e.m. Statistical analysis was performed using one-way analysis of variance (ANOVA) with post hoc Dunnett adjustment; *P < 0.05, **P < 0.01, ***P < 0.001. Further detailed information on statistics and reproducibility are provided in the Methods.
After assessing selected oxygen-sensitive brain regions, we next investigated the effects of OrganEx on tissue and cellular integrity in essential peripheral organs, including the heart, lungs, liver, kidneys and pancreas. We evaluated patterns of haemorrhage, tissue oedema, nuclear pyknosis, cell vacuolization and cellular morphology using haematoxylin and eosin (H&E) staining, and combined them into a cumulative damage score according to standard pathologic criteria18. Notably, the OrganEx group showed a decrease in the H&E damage score, as compared to the 7 h WIT and ECMO groups (Fig. 3e–h and Extended Data Fig. 3a–c). Furthermore, organs that were treated with OrganEx perfusion exhibited fewer signs of haemorrhage or tissue oedema compared with the 1 h WIT group. These results are indicative of the absence of injury promotion and cytoarchitectural damage as compared to ECMO and, importantly, a reduction of H&E damage scores, approaching the 0 h WIT state after OrganEx perfusion.
To examine indications of cytoarchitectural recovery in the OrganEx group and apparent injury progression in the ECMO group, we investigated renal cellular criteria that are well characterized for stratifying injury, including periodic acid–Schiff (PAS) and immunohistochemistry staining for cytoskeletal β-actin (ACTB), kidney injury molecule-1 (KIM-1 also known as HAVCR1)19 and proliferative marker Ki-67 (MKI67)20. The combined PAS damage score, evaluating tubular and Bowman space dilatation, casts and disrupted brush border, indicated greater damage in the ECMO group compared with in the OrganEx group (Extended Data Fig. 3d,e). These results were corroborated by a decrease in ACTB immunolabelling in the ECMO group compared with the OrganEx group (Fig. 3i–k). Similarly, the percentages of renal tubules immunolabelled for HAVCR1 was comparable between the 0 h WIT, 1 h WIT and OrganEx groups. By contrast, the 7 h WIT and ECMO groups had diminished HAVCR1 immunolabelling compared with the OrganEx group consistent with the staining patterns in other organs, possibly indicating protein degradation (Extended Data Fig. 3f,g). Similarly, the numbers of Ki-67+ immunolabelled nuclei were comparable in the 0 h WIT, 1 h WIT and OrganEx groups, but significantly decreased in the 7 h WIT and ECMO groups. This indicated a preserved proliferative response of the kidney tubular epithelial cells in the OrganEx group (Extended Data Fig. 3h–j).
Analysis of cell death processes
As the OrganEx perfusate contains pharmacological suppressors of cell death and we observed decreased cellular demise based on the histopathological analysis in the OrganEx group (Fig. 3e–h and Extended Data Fig. 3), we next investigated key proteins of key cell death pathways using immunohistochemistry analysis18. For apoptosis, we measured the immunolabelling intensity of activated caspase-3 (actCASP3) and performed a TUNEL assay, and observed their increase across peripheral organs such as the heart, liver, kidneys and pancreas in the ECMO compared with the OrganEx group. Furthermore, the respective intensities of actCASP3 and TUNEL in the OrganEx group were comparable to the 0 h WIT group, which did not sustain ischaemic injury, indicating that OrganEx perfusion diminished caspase-3 activation and decreased apoptosis (Extended Data Fig. 4a–j).
The analysis of the CA1 and PFC revealed that the intensity of actCASP3 immunolabelling in the OrganEx group was lower than in the 0 h and 1 h WIT groups (Extended Data Fig. 4k–m). Conversely, the intensity of the TUNEL assay had a lower trend in the OrganEx group compared with the 7 h WIT and ECMO groups (Extended Data Fig. 4n–p). Thus, it is conceivable that the weak brain immunolabelling of actCASP3 in the OrganEx group can be explained by active suppression of actCAPS3 by the pharmacological compounds in the perfusate as previously reported3, rather than cellular or protein destruction, as also supported by our other evaluations (Figs. 3a–d and 5a and Extended Data Figs. 2, 6h–k and 9f,g).
Fig. 5 |. Organ- and cell-type-specific transcriptomic changes assessed by snRNA-seq across various warm ischaemia intervals and different perfusion interventions.
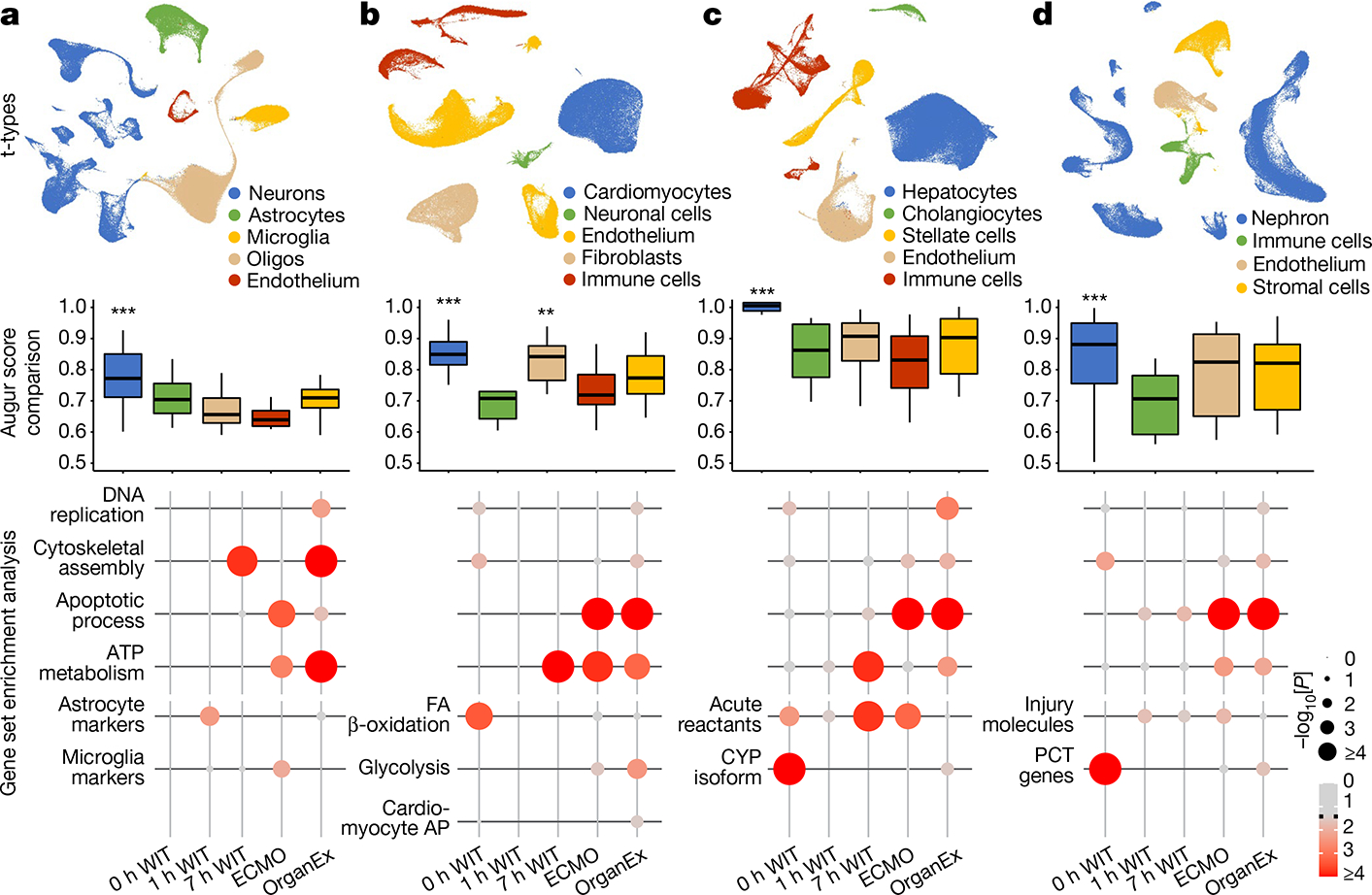
a–d, Uniform manifold approximation and projection (UMAP) layout showing major t-types in the hippocampus (a), heart (b), liver (c) and kidneys (d) (top). Middle, comparison of averaged Augur area under the curve (AUC) scores across the t-types indicating which cell type underwent the most transcriptomic changes. Bottom, P values of gene set enrichment of gene sets that are important in cellular recovery and specific cellular functions in major respective t-types. **P < 0.01, ***P < 0.001. FA, fatty acid.
Next, to investigate pyroptosis, the cell death pathway triggered by proinflammatory signals, we performed interleukin-1β (IL-1B) immunohistochemistry analysis. Across all of the investigated peripheral organs such as the heart, liver and kidneys, IL-1B immunolabelling intensity was comparable in the 0 h WIT group and increased in the ECMO group compared with the OrganEx group. In the brain, immunolabelling intensity was decreased in the ECMO group compared with in the OrganEx group (Extended Data Fig. 5a–e). The trends in both of the peripheral organs and the brain were similar to the observed actCASP3 results (Extended Data Fig. 4).
Finally, we investigated necroptosis and ferroptosis, two distinct cell death pathways, using immunohistochemistry analysis of the important proteins in these pathways—receptor-interacting serine/threonine kinase 3 (RIPK3) and the glutathione peroxidase 4 (GPX4), respectively. The results were consistent between the two cell death pathways and between all of the organs evaluated, such as the brain, heart, liver and kidneys. Compared with the OrganEx group, immunolabelling intensity was comparable in the 0 h WIT group and significantly decreased in the 7 h WIT and ECMO groups (Extended Data Fig. 5f–o).
Organ metabolic and functional analysis
After observing improvements in metabolic function, tissue cytoarchitecture and cell death outcomes using OrganEx, we next investigated cellular energy balance in detail. We measured glucose uptake in highly metabolic organs (brain, heart, kidneys) using the fluorescent glucose analogue 2-NBDG21. This showed comparable levels of glucose uptake in the OrganEx and 0 h WIT groups in all of the assessed organs and reduced cellular glucose capture in the ECMO group, which may indicate impaired glucose utilization or cellular leakage (Fig. 4a–c). Such findings imply that the recovery of cellular metabolism may have a reciprocal relationship with the restoration of systemic metabolic parameters (Fig. 2d,e and Extended Data Fig. 1d).
Fig. 4. Functional characterization and metabolic activity of selected organs.
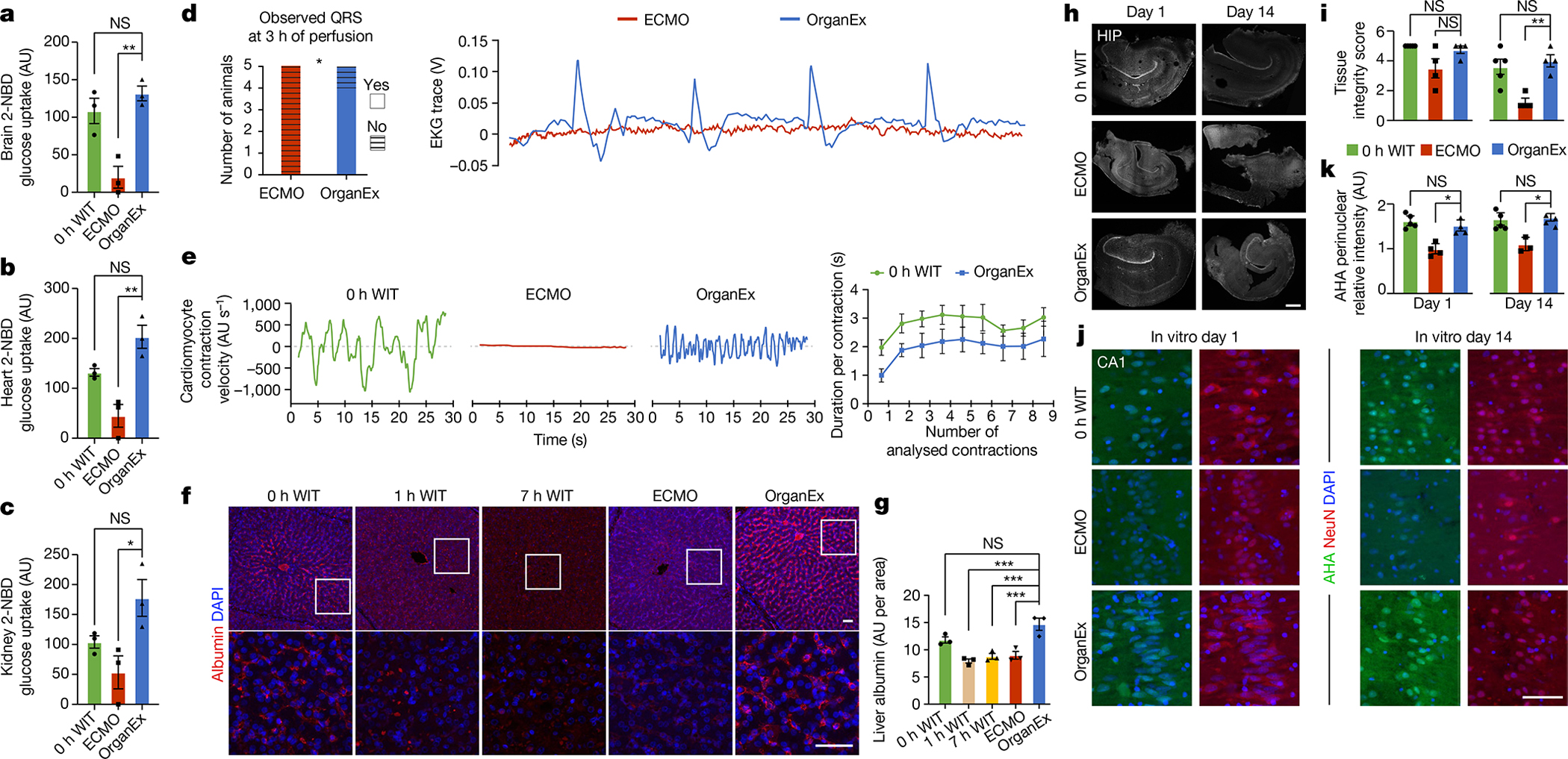
| a–c, Measurement of 2-NBDG uptake in the brain (a), heart (b) and kidney (c). n = 3. d, Observed QRS complexes in ECMO- and OrganEx-treated animals 3 h into the perfusion period (left). Statistical analysis was performed using χ2 tests. Right, representative EKG trances in the OrganEx and ECMO groups 3 h into the perfusion period. e, Measurement of cardiomyocyte contraction velocity in acute heart slices in the 0 h WIT, OrganEx and ECMO groups (left), and cardiomyocyte contraction duration (right). n = 5. f, Representative confocal images of immunolabelling for albumin in the liver. Scale bars, 50 μm. g, Measurement of normalized immunolabelling signal intensity of albumin in the liver demonstrating similar expression between the OrganEx and 0 h WIT groups. n = 3. h, Representative images of organotypic hippocampal slices after 1 and 14 days in culture. Scale bar, 500 μm. i, Quantification of hippocampal slice integrity. n = 4–5. j, Representative confocal images of newly synthesized proteins (AHA, Click-iT Chemistry) with DAPI counterstaining in the long-term organotypic hippocampal slice culture. Scale bar, 100 μm. k, Quantification of AHA relative intensity in the CA1. n = 3–5. For a–c, g, i and k, data are mean ± s.e.m. Statistical analysis was performed using one-way ANOVA with post hoc Dunnett adjustment; *P < 0.05, **P < 0.01, ***P < 0.001. AU, arbitrary units. HIP, hippocampus.
We next proceeded to test indicators of cell- and tissue-level recovery in relevant organs. Cardiac assessment using electrocardiography demonstrated spontaneous re-emergence of QRS complexes during OrganEx perfusion, indicating ventricular depolarization (Fig. 4d). However, no QRS re-emergence was observed in the ECMO group. To further evaluate recovery of ventricular activity, we examined cardiomyocyte contractility using bright-field microscopy analysis of left ventricle tissue slices acquired at the experimental end point. We observed contractions in OrganEx and 0 h WIT samples, but a complete absence in the ECMO group (Fig. 4e). Finally, we investigated cardiomyocyte biomarkers of which immunolabelling decreases with ischaemia22. Left ventricle immunohistochemistry staining for the biomarker troponin I revealed decreased immunolabelling with prolonged ischaemia, and significantly lower immunolabelling intensity in the ECMO versus OrganEx group (Extended Data Fig. 6l,m).
Liver cellular recovery was assessed using immunostaining for albumin and factor V, which are non-structural liver-synthesized proteins with a high abundance and short half-life, respectively. Compared with the OrganEx group, the immunolabelling intensities of both proteins were comparable in the 0 h WIT group and significantly diminished in the 7 h WIT and ECMO groups (Fig. 4f,g and Extended Data Fig. 6n,o).
In the kidneys, although many cellular features were preserved similar to the 0 h WIT group in the OrganEx groups, including tissue integrity (Fig. 3h and Extended Data Fig. 3d,e), cell death (Extended Data Figs. 4 and 5), molecular and proliferative injury responses (Extended Data Fig. 3f–j), and cellular metabolic indicators (Fig. 4c), the primary kidney functional metric, urine output, was minimal. Yet hypothermic perfusion is known to slow kidney function in patients with healthy organs23,24, and extracorporeal perfusion circuits can perturb endocrine, humoral and neural factors regulating glomerular filtration even when renal perfusion and cellular health is adequate25. Longer recovery time also may be required as low urine output often follows shock resuscitation26.
Next, we conducted continuous electroencephalography (EEG) analysis of the brain in the OrganEx and ECMO groups, and no signs of global network activity were detected (Extended Data Fig. 6a–e). In the OrganEx group, we hypothesize that this could be due to inadequate brain recovery overall, requirement for longer duration for recovery to unfold, neuronal activity antagonists within the perfusate, anaesthesia, hypothermic perfusion effects3,23 or any combination thereof. Interestingly, while receiving carotid injection of contrast for cerebrovascular fluoroscopic imaging, OrganEx-perfused animals exhibited complex, non-purposeful, non-stereotyped movements of the head, neck and torso from coordinated agonist/antagonist actions across multiple joints and muscle units. This was not observed during imaging of sedated alive or ECMO-treated animals (Extended Data Fig. 6f). EEG patterns during these movements were not interpretable owing to movement-induced artefacts, but were flat immediately before and after the movements (Extended Data Fig. 6g). Whether these movements were initiated from preferential interruption of cerebral descending inhibition of motor patterns or from positive action at the subcortical, spinal, peripheral nerve or neuromotor unit levels is difficult to determine. However, the ability for them to be executed does indicate the preservation of efferent motor output function at least at the level of the spinal cervical cord or its roots.
We next sought to investigate the longer-term actions of OrganEx perfusion on cellular viability. However, owing to regulatory constraints and the inability to extend the perfusion protocol beyond 6 h, we used organotypic hippocampal brain slice cultures (BSCs) to monitor features of tissues that were previously exposed to different perfusion interventions. BSCs were prepared at the beginning of the experiments (0 h WIT), at the end of ECMO and OrganEx perfusions, and from time-matched 7 h WIT controls, and were cultured for 14 days while assessing tissue integrity and protein synthesis using Click-iT assay screening27. BSCs from the 7 h WIT group failed these measures owing to severe tissue degradation. On the basis of visual inspection and DAPI staining, BSCs from the ECMO group were more fragile than OrganEx samples, and most disintegrated by day 14 (Fig. 4h,i). BSC tissue integrity was preserved until day 14 in the OrganEx group equal to the 0 h WIT group despite having had one additional ischaemic exposure (once during the initial 1 h warm ischaemia, and once during brain extraction, 5–10 min). Similarly, protein synthesis was comparable until day 14 in the OrganEx and 0 h WIT groups and decreased in the ECMO group across different hippocampal regions (Fig. 4j,k and Extended Data Fig. 6h–k).
Cell-specific transcriptomic changes
To investigate transcriptomic responses to distinct ischaemic exposures and the effects of the OrganEx intervention, we performed single-nucleus RNA-sequencing (snRNA-seq; Methods). The computational analysis of snRNA-seq data revealed major transcriptomically defined cell types (t-types) that were comparable to publicly available human and mouse single-cell datasets (Extended Data Fig. 7). However, we also identified prominent transcriptomic distinctions between the same t-types across all of the experimental groups (Extended Data Figs. 9–12c). This extensive cellular taxonomic resource expands on previous studies28–33 and enables the systematic investigation of transcriptomic changes in multiple porcine organs and cell types exposed to distinct WITs and reperfusion conditions (Extended Data Figs. 8, 9–12a,b; http://resources.sestanlab.org/OrganEx).
To compare cell type responsiveness to ischaemia based on transcriptomic changes across experimental groups, we performed Augur prioritization34 and highlighted t-types with the greatest transcriptomic divergence. This identified prominent changes in neurons, cardiomyocytes, hepatocytes and proximal convoluted tubule (PCT) cells, consistent with t-types validated in earlier studies and prompting detailed subanalysis (Fig. 5a–d). First, we evaluated whether patterns of transcriptomic changes within the OrganEx group versus the other groups reflect molecular and cellular changes observed in previous studies by assessing for transcriptomic enrichment of corresponding gene sets (Methods). Comparisons between the OrganEx group and the other groups revealed significant enrichment of gene sets facilitating cytoskeletal assembly, DNA repair, ATP metabolism, and suppression of apoptosis and other major cell death pathways across all major cell types in the organs investigated (Fig. 5a–d and Extended Data Figs. 9–12f–g). These data corroborate our earlier findings by demonstrating that OrganEx both inhibited progression of cellular injury (for example, cytoarchitecture, cell death, DNA fragmentation) and promoted repair by modulating cellular pathways at the transcriptomic level. Similarly, genes encoding proteins involved in cell death, oxidative injury and inflammatory signalling (Supplementary Table 4) are broadly regulated in favour of cell survival in the OrganEx group compared with ECMO in all of the organs studied. This correlates with choices of pharmacological compounds in the OrganEx perfusate, which had been included using a hypothesis-based, rational-polytherapy approach to modulate these pathways (Extended Data Figs. 9–12d).
Further investigation of the effects of OrganEx on glial inflammatory responses underlying brain injury progression after ischaemia showed that hippocampal microglial pro-inflammatory transcriptional enhancement was absent in the OrganEx group. By contrast, microglial inflammatory and astrocytic pan-reactive transcriptomic signatures were upregulated in the ECMO and 1 h WIT groups, respectively35,36 (Fig. 5a). Combined with immunofluorescence analyses of microglial IBA1 and astrocytic GFAP staining (Fig. 3a–d and Extended Data Fig. 2f–l), these findings demonstrate that OrganEx intervention modulates the glial inflammatory response.
We next evaluated transcriptomic signatures of tissue and cellular functioning in the heart, liver and kidney samples. Cardiomyocytes in the OrganEx group exhibited enrichment of genes orchestrating action potential formation and the well-described shift37 towards glycolytic metabolism after ischaemia signifying cardiomyocyte viability (Fig. 5b). In the liver and kidneys, hepatocytes and PCT cells were enriched for cytochrome P450 and PCT transporter genes, respectively, in OrganEx versus other groups—although not quite to the levels of the 0 h WIT controls, suggesting preservation of organ-specific functions (Fig. 5c,d). Liver acute-phase reactant and kidney injury marker genes also were notably lower after OrganEx reperfusion compared with after ECMO or 7 h WIT (Fig. 5c,d). These data corroborate earlier findings on tissue integrity and cellular activity (Figs. 3e–h and 4b–g and Extended Data Figs. 3d–j and 6l–o).
To identify transcriptomic patterns across experimental groups more systematically, and to determine functional gene modules, we performed co-expression analysis of differentially expressed genes across groups38. Eigengenes of each module designated key gene expression trends, with subsequent Gene Ontology (GO) analyses highlighting relevant biological pathways (Extended Data Figs. 9–12e and Supplementary Table 5). Here OrganEx and ECMO had divergent trends across different modules. Gene modules with eigengene increases in the OrganEx group featured GO terms related to cellular upkeep and organ-specific functions. By contrast, modules with increased eigengenes in the ECMO group had GO terms related to cell death (liver and kidneys).
Finally, analysis of ligand–receptor pairings39 of t-types showed reduced interactions related to inflammatory pathways (for example, IL-1, IL-6, ICAM, VCAM) in OrganEx versus ECMO groups (Extended Data Figs. 9–12h,i). This mirrors earlier findings that OrganEx reperfusion after ischaemia diminishes markers of inflammation associated with overall decreased cellular injury and augmented repair/protection processes compared with ECMO (Extended Data Figs. 5a–e and 9–12d).
Taken together, the snRNA-seq analysis supports our cellular- and tissue-level findings of decreased cellular injury and the initiation of certain molecular and cellular repair processes after OrganEx intervention.
Discussion
Here we described the OrganEx technology and its potential to support the recovery of key molecular and cellular processes in multiple porcine organs after prolonged global warm ischaemia. This also demonstrates the underappreciated capacity of the large mammalian body for restoration of haemodynamic and metabolic parameters after circulatory arrest or other severe ischaemic stress. Furthermore, these results bolster previous findings from our isolated post mortem porcine brain study3 that indicate that cellular deterioration is a more protracted process, not scripted within narrowly defined timeframes. This application of the OrganEx technology demonstrates that cellular demise can be halted, and their state can be shifted towards recovery at molecular and cellular levels, even after prolonged warm ischaemia. Furthermore, we conducted a comprehensive single-cell transcriptomic analysis of multiple porcine organs providing a unique resource for future studies on cell types, ischaemia and reperfusion.
Although some cellular viability can be restored after prolonged ischaemia in tissue cultures7 or isolated organs3,8–11, clinical scenarios typically involve shorter-duration ischaemia in the setting of cardiac arrest40,41 or regional perfusions in the setting of organ transplant42,43. Using a rational polytherapy approach built on optimized perfusion dynamics and augmentations to an acellular synthetic perfusate, the OrganEx technology was able to bridge previous clinical–translational gaps by restoring circulation and metabolic homeostasis across the whole body. This quells deleterious processes caused by disturbed cellular environments and a lack of oxygen, representing a distinct feature of this technology and an essential control for multiple non-specific injury mechanisms that affect end-organ recovery and overall prognosis after global ischaemia. Potential applications of this technology are manifold and could provide pathways in ischaemia research and advance related clinical disciplines. OrganEx has the potential to extend the limits of allowable warm ischaemia times through regional abdominal/thoracic reperfusion44, thereby increasing organ availability for transplantation. This approach would require obligatory antecedent clamping of the aorta/carotid arteries to prevent brain recirculation in the organ donor. Conversely, if any future refinements of the technology could be aimed at the recovery of the brain function after injury, then the brain circulation would remain patent. In this regard, OrganEx technology may improve outcomes in extracorporeal cardiopulmonary resuscitation45 with the needed circulatory support, or the OrganEx perfusate could aid in recovery wherein cardiac function is preserved but the brain is damaged, as seen in stroke. Thus, a clear distinction should be made before possible use of OrganEx technology regarding the inclusion of the brain circulation.
Although our studies demonstrate important cellular protection and repair processes in vital organs across meaningful time points, questions remain concerning organ recovery over an extended timeframe. As repeating all of the experiments over additional, extended durations to comprise a full longitudinal study were not feasible under current regulatory constraints, we used long-term organotypic slice culture preparations of the hippocampus to study post-perfusion cell survivability in the most ischaemia-sensitive tissue possible. This demonstrated that OrganEx intervention provides enduring effects on cellular recovery after transfer to the extended survival conditions. However, we do not know whether the subsequent in vitro cellular recovery and viability recapitulates conducting equally long OrganEx perfusions of the whole body.
Overall, further optimization and expansion of our technology will be needed to fully understand its broader effects on ischaemic tissues and recovery. Although the in vivo results here show promise to bridge gaps between basic research on ischaemia and transplantation medicine or clinical resuscitation science, understanding the recovery of brain function requires careful consideration. In particular, thoughtful evaluation is needed to elucidate why head and neck movements occurred after contrast injection only in the OrganEx group. With rigorous ethical review from researchers, bioethicists and institutional boards, future characterization of these movements may provide better understanding of neuromotor biology and ischaemic pathophysiology after cardiac arrest or stroke.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-022-05016-1.
Methods
Overview of the OrganEx perfusion system and perfusate
Overview of the OrganEx perfusion system.
The perfusion system consists of the main closed-loop circuit directly connected to an animal, and it includes a centrifugal pump (Medtronic Bio-Console 560, Medtronic) that drives the mixture of autologous blood and OrganEx perfusate through the oxygenator (Affinity Fusion, Medtronic) and custom-made pulsatility generator into the animal arterial system. The oxygenator is connected to a refrigerated bath (Polystat, Cole-Parmer) for temperature control and the gas blender (Sechrist Industries), for control of dissolved gases and anaesthesia infusion. The perfusion system has a fluid reservoir, which is used to prime the system and hold the supplement fluid. In parallel, an automated haemodiafiltration system and a reservoir are connected to the main circuit (Fig. 1b). The automated haemodiafiltration system is used to exchange the plasma fraction against a custom-made dialysis exchange solution. The haemodiafiltration system consists of a roller-pump (Cobe Shiley, Stockert), dialyser (Diacap Pro 13H, Braun) and two peristaltic pumps (Masterflex L/S, Cole-Parmer) integrated with level sensor (eTape, Milone Technologies) and custom-made logical controller. Two infusion pumps (Sigma Spectrum, Baxter Healthcare) are connected to the arterial side of the main circuit supplementing heparin and pharmacological compounds of the perfusate. The CDI blood parameter module and the haematocrit/oxygen saturation probe (Terumo Cardiovascular Systems) are connected on the arterial and venous side, respectively, along with pressure (PendoTECH) and flow sensors (Bio-Probe TX50, Medtronic). OrganEx perfusion system components, logical controllers and sensors are connected to a computer for automated control and data gathering. Detailed schematics are available on request.
Preparation and application of the OrganEx perfusate.
The OrganEx perfusate is a final mixture of a custom-made priming solution (Supplementary Table 2), Hemopure (HbO2 Therapeutics), custom-made dialysis exchange solution (Supplementary Table 3) and the solution of pharmacological compounds (Supplementary Table 1). In detail, before connecting an animal, the OrganEx perfusion system is flooded and primed with 2,200 ml of custom-made priming solution (5,000 ml), followed by infusion of 1,000 ml of Hemopure into the system. These solutions are left to mix and equilibrate throughout the perfusion system, after which 600 ml is dialysed out of the perfusion system to achieve the desired concentrations of electrolytes and oncotic agents in the perfusion system and prepare it for addition of the autologous blood. Before initiation of the perfusion protocol, animal femoral vessels are cannulated and connected to the main circuit. At 30 min of WIT, 5,000 USP units of heparin (Sigma-Aldrich) is administered into the system, followed by approximately 1,000 ml of venous blood from the dead animal, which is drained into the perfusion system. At this point, the circulatory volume in the OrganEx perfusion system is approximately 3,600 ml, of which 2,600 ml is the perfusate and 1,000 ml is autologous blood. Next, the mixture of the perfusate and autologous blood is left to equilibrate, and is counter dialysed against the residual priming solution over 30 min to allow for correction of metabolic derangements in the drained venous blood. In parallel, approximately 1,600 ml of fluid is filtered out of the perfusion system over 30 min while the residual fluid is cooled to 28 °C, yielding a final volume of 2,000 ml in the OrganEx perfusion system. Next, at 1 h WIT, 1,000 ml of the perfusate and autologous blood mixture is infused back into the animal, ensuring circulatory system filling after venous drainage, and the perfusion protocol is initiated. The remaining 1,000 ml of the mixture is stored in the reservoir and used for fluid supplementation, if required. After infusion of the perfusate and autologous blood mixture, pharmacological compounds and dialysis exchange solution, containing amino acids, vitamins and inorganic salts, are continuously infused into the main perfusion circuit by the infusion pump and haemodiafiltration system, respectively. The OrganEx perfusion system uses an automated haemodiafiltration circuit that corrects and maintains certain metabolic and electrolyte parameters by performing 1:1 (v:v) exchange of solutes and particles smaller than 40 kDa against a custom dialysis exchange solution (20,000 ml), while maintaining euvolaemia. Haemodiafiltration flux was kept at 30–35 ml kg−1 h−1 throughout 6 h perfusion.
Overview of the ECMO perfusion system.
The ECMO perfusion system was assembled according to the clinical standard14. The ECMO perfusion system has the main closed-loop circuit directly connected to an animal and consists of a centrifugal pump (Bio-Console 560, Medtronic) that drives autologous blood through the oxygenator (Affinity Fusion, Medtronic) into the animal arterial system. The oxygenator is connected to a refrigerated bath (Polystat, Cole-Parmer) for temperature control and the gas blender (Sechrist Industries), for control of dissolved gases and anaesthesia infusion. The perfusion system has a fluid reservoir, which is used to prime the system and hold the supplement fluid. Furthermore, ECMO perfusion system contained the CDI blood parameter module, and the haematocrit/oxygen saturation probe (Terumo Cardiovascular Systems) are connected on the arterial and venous side, respectively, along with the pressure (PendoTECH) and flow sensors (Bio-Probe TX50, Medtronic). All of the probes and sensors of the ECMO perfusion system are connected to a computer to enable data gathering. Detailed schematics are available on request.
The ECMO perfusion system is primed with 1,000 ml 0.9% sodium chloride (Baxter Healthcare) and 5,000 USP units of heparin (Sigma-Aldrich). After initiation of the perfusion protocol, the reservoir is disconnected from the main circuit.
Preordained essential principles in animal use
This research project was approved and overseen by Yale’s Institutional Animal Care and Use Committee and guided by an external advisory and ethics committee. Before starting the experiments, careful consideration of animals’ autonomy, previous research and the goals of the current study were used to guide the design of the study and its experiments. This yielded three preordained essential principles which we followed during the research performed in this Article.
First, we sought to minimize the animal number and any potential discomfort and suffering. Second, because the initial experimental outcomes and efficacy of OrganEx technology were uncertain, we focused on an internally controlled, proof-of-concept study that might demonstrate molecular and cellular viability first, before proceeding to possible survival studies. Finally, the perfusion time was selected on the basis of our previous study of the isolated porcine brain showing that 6 h of perfusion with BrainEx technology was enough to initiate processes associated with molecular and cellular recovery as well as expected clinical timeframes for the onset and propagation of end-organ hypoperfusion injury.
Animal anaesthesia and surgical protocol
Experimental animals were procured from the local farm breeder, female domestic pigs (Sus scrofa domesticus; 10–12 weeks of age (~30–35 kg)). All of the animals were housed at Yale School of Medicine Division of Animal Care’s facilities at a minimum of 3 days before the experiment.
Before the experimental protocol, all of the animals received a fentanyl patch, 50 μg h−1 (Duragesic, Henry Schein) for sedation. To induce anaesthesia, 6 mg kg−1 of telazol (Henry Schein) and 2.2 mg kg−1 of xylazine (Henry Schein) were administered. Next, animals were intubated and connected to the ventilator using FiO2 of 40% and FiN2 of 60% with standard parameters of tidal volume 10–15 ml kg−1 and a frequency of 14–16 breaths per minute, along with 1–2% isoflurane (Henry Schein). After ventricular fibrillation and induction of cardiac arrest, ventilation was stopped for 1 h. After initiation of the perfusion protocol, in ECMO and OrganEx groups, ventilation was continued using a tidal volume of 3–4 ml kg−1, a frequency 5 breaths per minute, positive end-expiratory pressure (PEEP) of 10 cm H2O, low inflation pressure, FiO2 50% and FiN2 50%. During the 6 h perfusion protocol, 0.5% isoflurane was administered through the vapourizer connected to the gas blender.
Cardiac arrest and subsequent circulatory collapse were induced by ventricular fibrillation through the substernal window by applying a 9 V battery to the myocardial wall. Before ventricular fibrillation, the animals received 7,000 USP units of heparin (Sigma-Aldrich). To connect the circulatory system of the animal to the ECMO or OrganEx perfusion system, an incision was made in the right inguinal region exposing the femoral artery and vein (Fig. 1a). Both, arterial and venous cannulas were inserted into the femoral artery and vein, respectively. The artery was cannulated with 14 Fr and the vein with 19 Fr cannula (Edwards Lifesciences). The tip of the venous cannula was placed in the inferior vena cava opening of the right atrium, and arterial cannula was positioned inferior to renal arteries.
Perfusion protocol and monitoring of physiological and metabolic parameters
At the start of the perfusion protocol in the OrganEx group, a mixture of the perfusate and autologous blood was slowly infused over 5 min resulting in an average flow rate of 600 ml min−1 at the end of infusion. After this step, the flow rate was gradually increased over the next 20 min to a targeted flow rate of 80–100 ml kg−1 min−1 or the highest possible flow rate without introducing overspinning of the centrifugal pump. Throughout the flow-rate ramping-up process, pulsatility was set to oscillate around the mean flow rate at approximately ±10% of the given total flow rate. Residual mixture of the synthetic perfusate and autologous blood, which was stored in the reservoir, was used for fluid supplementation at 1–2 ml kg−1 h−1. Targeted arterial pressure was set to 40–70 mmHg, and it was controlled with phenylephrine, if required, but not exceeding 2 mg h−1. Similarly, in the ECMO group, the flow rate was gradually increased over 25 min, targeting flow rates and arterial pressures as in the OrganEx group. Ringer’s lactate (Baxter Healthcare) was used as a supplementation fluid at 3–4 ml kg−1 h−1. In both the ECMO and OrganEx group, the hypothermic perfusion protocol at 28 °C was used throughout the entire 6 h of the perfusion protocol.
The animals in both ECMO and OrganEx groups received 50 ml of 8.4% sodium bicarbonate (Henry Schein) during the first hour of perfusion. Glucose was supplemented according to the blood levels with the goal of maintaining euglycemia. Protamine (25 mg) was administered immediately after initiation of the perfusion protocol to control the activated clotting time, which was maintained between 180 and 220 s with titrated heparin administration. In both of the perfusion groups, partial pressure of arterial CO2 and O2 were targeted to 35 and 250 mmHg through a gas blender, respectively.
Electrocardiogram (ECG) assessment was performed with four leads placed at each corner of the trunk. Real-time arterial and central venous pressure monitoring was performed through cutdown of the brachial artery and jugular vein, respectively. Urine output was measured through a Foley catheter. Animal core temperature was continuously monitored using a rectal probe. Monitoring of ECG, pressure and temperature was performed using the Philips IntelliVue MP50. During the preoperative procedure, temperature was kept at 37 °C using a heating pad, which was turned off after ventricular fibrillation. EEG was monitored using the Natus long-term monitoring (LTM) system and EMU40 breakout box (Natus Medical). Six electrodes were placed subcutaneously along the scalp (Extended Data Fig. 6a) at the start of the sedation. EEG monitoring was conducted throughout the entire 6 h perfusion protocol.
Baseline and hourly arterial and venous samples were collected from the arterial and venous cannulas respectively. Sixty microlitres of each sample were immediately analysed using the GEM4000 clinical blood analyser system (Instrumentation Laboratory). Continuous monitoring of blood electrolytes and haemoglobin concentration and saturation were performed using CDI-500 system (Terumo Cardiovascular Systems).
Radiographic and ultrasound imaging of circulation
Fluoroscopy.
Imaging of the abdominal and head blood vessels was performed using the Philips Allura Xper FD20 system. In selected animals that underwent fluoroscopy, baseline physiological imaging was performed before the induction of ventricular fibrillation in both the ECMO and OrganEx experimental protocols. The contrast-injecting catheter was introduced through the femoral artery cutdown and positioned in the suprarenal aorta for renal imaging and in the common carotid artery for brain imaging. Omnipaque Contrast 350 mg ml−1 (General Electric), 24 ml and 45 ml were introduced using Medrad power injector (Bayer Vital) for brain and kidney imaging acquisition. After baseline imaging, all of the animals underwent additional fluoroscopy at hours 3 and 6 of perfusion. In the ECMO and OrganEx groups imaging of abdominal blood vessels was modified by placing the contrast-injecting catheter into the infrarenal aorta due to the reversal of arterial flow direction, a consequence of the femoral artery/vein perfusion approach in both ECMO and OrganEx groups. The reconstructed images were saved in DICOM format and further post-processed using RadiAnt DICOM Viewer software (Medixant).
Ultrasonography.
Perfusion dynamics was monitored using Triplex Ultrasonography (Spectral Doppler, Colour Doppler, and B-mode) using the LOGIQe portable ultrasound system (General Electric) and an 8L-RS linear array probe (General Electric). In all of the assessed animals, the left ophthalmic artery, common carotid artery and intrarenal arteries were used to profile perfusion dynamics. Power waveform analysis was performed using Frq 4.4 MHz, Gn 17, SV 2 and DR 40.
Cell nucleus isolation
After the appropriate experimental workflow, regions of interest were extracted from each organ and frozen at −80 °C. To ensure consistency between the samples, all dissections were performed by the same person. Cell nucleus isolation from each organ (brain, heart, liver, kidneys) were treated the same according to our already established protocol46,47 with some modifications to acknowledge each organ’s specific structural qualities and to have identical buffers to enable interorgan comparison within the same experimental animal. To avoid experimental bias, nucleus isolation was performed by the same person blinded for the replicates of experimental conditions. Furthermore, to randomly and fully represent the full tissue section, each tissue was pulverized to fine powder in liquid nitrogen with mortar and pestle (Coorstek). All of the reagents were molecular-biology grade and sourced from Sigma-Aldrich unless stated otherwise. Small amounts of pulverized tissue (5–10 mg) were then added into 1 ml of ice-cold lysis buffer (buffer A is 250 mM sucrose, 25 mM KCl, 5 mM MgCl2, 10 mM NaCl, 10 mM Tris-HCl (pH 7.4), protease inhibitors without EDTA (Roche), RNase inhibitor (80 U ml−1) (Roche), 1 mM DTT, 1% BSA (m/v) (Gemini Bio-Products), 0.1% NP-40 (v/v), 0.1% Tween-20 (v/v) (Bio-Rad), 0.01% Digitonin (m/v) (Thermo Fisher Scientific). For lysis of heart, 0.1% Triton X-100 (v/v) was additionally added. DTT, RNase Protector, protease inhibitors and all detergents were added immediately before use. The suspension was transferred to a 2 ml Dounce tissue homogenizer and lysed with constant pressure and without introduction of air with pestle A (30×) and pestle B (30×). The homogenate was strained through a prewetted 40 μm tube top cell strainer (Thermo Fisher Scientific). All subsequent centrifugation was performed in a refrigerated, bench-top centrifuge with swing-out rotor (Eppendorf). Heart lysate was centrifuged at 100g for 5 min at 4 °C, the pellet of myofibrils and non-dissociated connective tissue was discarded and the supernatant was retained. All lysates (brain, liver, kidneys) and heart supernatant (after centrifugation at 100g) were centrifuged at 1,000g for 10 min at 4 °C, pellets were saved and resuspended in 0.4 ml resuspension buffer (buffer B is buffer A without the detergents). The final 0.4 ml of solution was mixed with 0.4 ml (1:1) of Optiprep solution (buffer C is iodixanol 50% (v/v), 25 mM KCl, 5 mM MgCl2, 10 mM NaCl, 10 mM Tris-HCl (pH 7.4), protease inhibitors without EDTA, RNase inhibitor (80 U ml−1), 1 mM DTT, 1% BSA (m/v)). The suspension (25% iodixanol final) was mixed 10× head over head and overlayed on 0.6 ml of 29% iodixanol cushion (appropriate mix of buffer B and buffer C). The tubes were then centrifuged at 3,000g, for 30 min at 4 °C. After centrifugation, the supernatant was removed and a total of 1 ml of wash buffer (buffer D is 25 mM KCl, 5 mM MgCl2, 10 mM NaCl, 10 mM Tris-HCl (pH 7.4), RNase inhibitor (80 U ml−1), 1 mM DTT, 1% BSA (m/v), 0.1% Tween-20 (v/v), in DPBS (without Ca2+ and Mg2+) (Gibco)) was added in tubes and centrifuged at 1,000g for 10 min at 4 °C. The supernatants were then completely removed, pellets were gently dissolved by adding 100 μl of resuspension buffer (buffer E is buffer D without detergent) and pipetting 30 times with a 1 ml pipette tip, pooled and filtered through 20 μm cell strainer. Finally, nuclei were counted on haemocytometer and diluted to 1 million per ml.
Single-nucleus microfluidic capture and cDNA synthesis
Extracted nucleus samples were placed onto ice and processed in the laboratory within 15 min for snRNA-seq with targeted nucleus recovery of 10,000 nuclei on the microfluidic Chromium System (10x Genomics) by following the protocol (CG000315_ChromiumNextGEMSingleCell3’_GeneExpression_v3.1(DualIndex)_RevA) from the manufacturer, using the Chromium Single Cell 3ʹ GEM, Library & Gel Bead Kit v3.1, (PN-1000268, 10x Genomics) and Chromium Single Cell G Chip Kit (PN-1000120, 10x Genomics), Dual Index Kit TT Set A (PN-1000215, 10x Genomics) on the Chromium Controller (10x Genomics). As the input material were nuclei, cDNA was amplified for 14 cycles.
snRNA-seq library preparation and sequencing
After cDNA amplification, clean-up and construction of sample-indexed libraries and their amplification was performed according to the manufacturer’s instructions (CG000315_ChromiumNextGEMSingleCell3’_GeneExpression_v3.1(DualIndex)_RevA), with the amplification step directly dependent on the quantity of input cDNA.
To reach a sequencing depth of 20,000 raw reads per nucleus, single-nucleus libraries were run using paired-end sequencing with single indexing on the NovaSeq 6000 S4 (Illumina) system according to the manufacturer’s instructions (Illumina; 10x Genomics). To avoid lane bias, multiple uniquely indexed samples were mixed and distributed over several lanes.
Single-nucleus transcriptome analysis
Quality control and analysis of single-nucleus transcriptome data.
Sequencing reads were aligned to the reference pig genome (susScr11) with the combined exon–intron gene annotations from NCBI RefSeq using CellRanger v.5.0.1. pipeline48, which also performed UMI counting, barcode counting and distinguishing true cells from background. The filtered count matrices were then moralized by library size using the NormalizeData function in Seurat49. To compare cellular and transcriptomic changes across conditions, feature selection was first performed for each batch and the features from the same conditions were summarized using the SelectIntegrationFeatures function. The union of the highly variable genes across conditions were then passed to the data integration pipeline in Seurat to generate a batch-corrected expression matrix. To reveal the cellular diversity among the cells, we scaled the integrated data and then performed dimension reduction using principal component analysis (PCA), selecting principal components using elbow plot. We then identified cell clusters in the k-nearest neighbour graph and visualized clustering results by UMAP50. The initial clustering analysis revealed some low-quality clusters with a low number of unique molecular identifiers and normally high mitochondria percentage and no meaningful cluster markers, which were all removed for downstream analysis. To annotate the identity of the cell clusters, we then integrated the pig data with published human data from the same organ51–54 using the same data integration pipeline described above. We then calculated cluster markers using the FindMarkers function and manually removed the doublet clusters showing high expression of markers of two different type of cells. To gain more accurate cell annotations and clearer UMAP visualizations, the same pipelines of data integration, dimension reduction and cell clustering were reperformed on the filtered data.
Global transcriptomic comparisons with public datasets.
To validate the cluster annotation of our data, we performed global transcriptomic comparison with public reference datasets for the four organs51–54. For each organ, we calculated the log-transformed average expression for each cell cluster followed by performing pairwise Pearson correlation between the clusters in our dataset and the corresponding dataset using the highly variable genes. The resulted correlation coefficients were visualized on gradient heat maps (Extended Data Fig. 7c–f).
Cell type annotations.
The cell type classification for hippocampus data (Extended Data Fig. 7c) was based on the gene markers derived from recent data in adult human hippocampus and entorhinal cortex53. We initially classified the cells to several major groups based on marker gene expression: excitatory neurons (SLC17A7+), inhibitory neurons (GAD1+), oligodendrocyte progenitor cells (PDGFRA+), oligodendrocytes (PLP1+), astrocytes (AQP4+), microglia (PTPRC+) and vascular cells (COL1A1+). Owing to the high heterogeneity present in excitatory and inhibitory neuron populations, we further subclustered these two populations. For excitatory neurons, they were classified to mature granule cells (PROX1+), mossy cells (ADCYAP1+), CA2–4 excitatory neurons (FREM1+GALNT3+), CA1 and subiculum excitatory neurons (SATB 2+BCL11B+TLE−), and entorhinal cortex upper layer (CUX2+) and deep layer (TLE4+) excitatory neurons. For inhibitory neurons, they were classified on the basis of their developmental origins, either derived from medial ganglionic eminence (MGE, LHX6+) and caudal ganglionic eminence (CGE, ARADB2+). For a small group of cells connecting granule cells on the UMAP that are devoid of all these markers but marked by DCX and CALB2 expression, they were annotated as neuroblast cells, intermediate cell populations in pig adult neurogenesis.
The heart data (Extended Data Fig. 7d) were annotated based on the gene markers derived from recent data in adult human heart52. We classified the cells on the basis of marker expression: cardiomyocytes (MYH7+FHL2+), immune cells (PTPRC+), pericytes (RGS5+ABCC9+), smooth muscle cells (MYH11+ACTA2+), endothelial cells (CDH5+PECAM1+), fibroblast-like cells (DCN+GSN+) and neuronal cells (NRXN1+XKR4+). The endothelial cells have two subgroups that have differential expression of VWF and TBX1. Immune cells were further classified to myeloid cells (BANK1+C1QA+) and lymphoid cells (SKAP1+CD8A+).
The kidney data (Extended Data Fig. 7e) were annotated based on the gene markers derived from recent data in adult human kidney51. We classified the cells on the basis of marker expression: proximal tubule (CUBN+LRP2+), connecting tubule and principal cells (SAMD5+LINGO2+), loop of Henle (NHSL2+UMOD2+), intercalated cells (HEPACAM2+SLC26A7+), podocytes (PTPRQ+PTPRO+), immune cells (PTPRC+), endothelium (CHRM3+PTPRB+) and fibroblasts (PRKG1+FBLN5+). Immune cells were further classified as myeloid cells (BANK1+MARCH1+) and lymphoid cells (SKAP1+THSD7B+).
The liver data (Extended Data Fig. 7f) were annotated based on the gene markers derived from recent data in adult human liver54. We classified the cells on the basis of marker expression: hepatocytes (APOB+PCK1+), stellate cells (RELN+ACTA2+), cholangiocytes (CFTR+PKHD1+), immune cells (PTPRC+) and endothelial cells (FLT1+PECAM1+). We further classified immune cells into multiple subgroups: B cells (MS4A1+), plasma cells (JCHAIN+MZB1+), natural killer cell and T cells (SKAP1+), myeloid cells (CD163+EMR4+, which are predominantly Kupffer cells).
Heterogeneity of cells.
The FindMarkers function from Seurat55 was used to determine marker genes for high resolution clusters. P-value adjustment was performed using Bonferroni correction with the cut-off set at 0.05. The top 10 genes for each cluster were ranked by fold changes and were visualized on a heat map by using the DoHeatmap function in Seurat. We randomly sampled our datasets to have 1,000 cells per condition for each t-type before differential expression analysis.
Cell type prioritization using Augur.
To find the cell populations that exhibit high degree of transcriptomic changes, we used Augur34 to prioritize the cell types between each pair of conditions. As there are three samples per condition, we performed the Augur analysis on all of the nine sample pairs in each condition pair using the high-resolution cell clusters identified using Seurat. The median of the calculated AUC scores of each cluster were then visualized on the UMAP layout. Comparisons of the AUC scores for each specific cell type and a given condition pair were performed by comparing the AUC score of the specific cell type of interest and the AUC scores of all of the other cell types in that given condition pair using one-tailed Wilcoxon rank-sum tests.
Differential expression analysis and GO analysis.
We first calculated differentially expressed genes (DEGs) between OrganEx and other conditions (0 h WIT, 1 h WIT, 7 h WIT, ECMO) in hippocampal neurons, cardiomyocytes in the heart, hepatocytes in the liver and proximal convoluted tubule cells in the kidneys (major t-types for each organ). Furthermore, we determined DEGs between 0 h WIT and 1 h WIT, as well as 0 h WIT and 7 h WIT. DEGs were calculated using the Seurat FindMarkers function55 and defined at the cut-off criteria of being expressed by at least 10% of cells in either population and have an expression ratio greater than 0.1, average log2[FC] of greater than or less than 0.1. Multiple-testing adjustment for false-discovery rate (FDR) cut-off was set at 0.05. The top 15 DEGs ranked by absolute values of log2[FC], were visualized using the EnhancedVolcano package56. Furthermore, we performed GO enrichment analysis in ischaemic conditions (0 h WIT versus 1 h WIT and 0 h versus 7 h WIT) in major t-types of all organs. To maintain the number of DEGs comparable, we retained the top 800 DEGs in both ways ranked by fold changes. Enrichment analysis of biological pathways was performed using the enrichGO function from clusterProfiler57, which uses one-tailed Fisher’s exact tests to calculate the significance. In the case of multiple testing, P values were adjusted for FDR with the cut-off set at 0.05 and qvalueCutoff = 0.2. The top 10 biological processes ranked by P value were visualized using bar plot by a custom ggplot2 script.
Gene set enrichment analysis.
To assess whether gene sets of interest are upregulated in a specific condition (for example, 0 h WIT, 1 h WIT, 7 h WIT, ECMO and OrganEx), we performed gene set enrichment analysis in hippocampal neurons, astrocytes and microglial cells, cardiomyocytes in the heart, hepatocytes in the liver and proximal convoluted tubule cells in the kidney. This method is commonly used in GO enrichment analysis and has been widely applied in multiple published studies46,47,53. Specifically, we set all of the expressed genes (expressed in at least one cell) as the gene universe and considered each set of condition-enriched genes as a sampling from the gene universe. The gene set enrichment, performed using hypergeometric tests (also named one-tailed Fisher’s exact test), is an assessment of whether genes from a given gene set are over-represented in condition-enriched genes compared with drawing from the gene universe by chance. To identify condition-enriched genes in the above-mentioned t-types, we performed differential expression analysis using the Seurat FindMarkers function55. In brief, we took one condition group, compared its expression profiles with the rest of the conditions using Wilcoxon rank-sum test. For any given comparison, genes with a FDR of less than 0.01 were considered to be statistically significant and were retained. As the Wilcoxon rank-sum test can be biased by the differences in cell numbers, that is, more cells lead to more DEGs, we randomly sampled our datasets to have 1,000 cells per condition in each t-type before differential expression. With the condition-enriched genes and certain selected gene sets downloaded from Gene Ontology (http://geneontology.org/), we were able to assess the significance of gene set enrichment in a given condition. P values of less than 0.05 were considered to be significant. Significance of the enrichment was visualized in a dot plot in which the size and colour of the dot shows significance as −log10[P]. As shown in Fig. 5a–d, we tested enrichment in all conditions in all organs for positive regulation of DNA repair (GO:0045739), negative regulation of apoptotic process (GO:0043066), positive regulation of cytoskeleton organization (GO:0051495) and ATP metabolic process (GO:0046034). The same approach was used in the assessment of functional enrichment analysis for each organ. Thus, in the hippocampus, we tested in microglial cell enrichment for pro-inflammatory markers35, and in astrocytes for pan-reactive markers36 as shown in Fig. 5a. Cardiomyocytes in the heart were tested for cardiac muscle cell action potential (GO:0086001), fatty acid beta-oxidation (GO:0006635) and glycolysis (GO:0006096) as can be seen in Fig. 5b. In the liver, we tested in hepatocyte for enrichment of acute phase reactants58 and all expressed CYP isoforms (Fig. 5c) and, finally, proximal tubule cells in the kidney were tested for injury molecule genes33 and PCT transporter genes59 (Fig. 5d).
Gene set expression enrichment analysis.
We also evaluated the expression dynamics of selected cell death-related gene set across conditions60–63. Dot plots were first generated using Seurat DotPlot function55 (Extended Data Figs. 9–12f). Next, for each gene of the given gene set, we tested its expression enrichment in each condition by comparing its expression in the given condition to that of other conditions. Similarly, we used Wilcoxon Rank Sum test to measure the significance. The resulting log-transformed P values (−log10[P]) across conditions were visualized in a bar plot (Extended Data Figs. 9–12g).
Hierarchical clustering for top DEGs.
To acquire the dynamic changes in the transcriptome among different conditions and time points for each tissue, we identified the top DEGs for all of the paired conditions: 0 h versus 1 h, 0 h versus 7 h, 0 h versus ECMO, 0 h versus OrganEx, 1 h versus 7 h, 1 h versus ECMO, 1 h versus OrganEx, 7 h versus ECMO, 7 h versus OrganEx, and ECMO versus OrganEx using the FindMarkers() function in Seurat49. For the hippocampus, we selected significant DEGs with an average log2[FC] of greater than 0.5 or less than −0.5. We merged the top 50 upregulated genes and 50 downregulated DEGs (in total 100 genes) from each paired condition for the following analysis. To identify gene expression patterns, we calculated the average expression of the merged DEGs across t-types and experimental groups and defined the correlation coefficients subtracted from 1 as gene–gene distances, which was passed to hierarchical clustering using the hclust function in R with ward.D2 algorithm. To define gene modules (clusters), we parcellated the genes using the cutreeDynamic function from the R WGCNA package38 with the settings minClusterSize = 45, sensitivity = 2. We then plotted the scaled eigengenes, the first principal component of the expression matrix of each module, to show the expression trend of the genes in each module. Using TopGO64, we performed the GO analysis for the genes in each module, and used Fisher’s exact test to calculate the P values. For the heart, kidneys and liver, we used the same methods as for the hippocampal data to select the genes. To keep the numbers of selected genes comparable with the hippocampus samples, we retained significant DEGs with average log2[FC] of greater than 0.75 or less than −0.75 in heart data, significant DEGs with an average log2[FC] greater than 1.00 or less than −1.00 in kidney data, and selected significant DEGs with an average log2[FC] greater than 1.75 or less than −1.75 in the liver data. Then we used the same settings to build the gene expression network using hierarchical clustering38. We plotted the scaled eigengenes of each module and used the same method (TopGO) for GO analysis.
Evaluation of the perfusate components effects.
To evaluate the effects of the perfusate components on OrganEx, we compared the expression enrichment of the related pathways (cell death, inflammation, and oxidative response) between the OrganEx and ECMO conditions (Supplementary Table 4). Specifically, we calculated the AUC scores using the AUCell package65 and performed a one-sided Wilcoxon rank-sum test to evaluate the significance of the pathway enrichment.
Trajectory analysis.
We used monocle2 (ref. 66) and custom R scripts to conduct pseudotime analysis for the hippocampus, heart, liver and kidneys. We followed the recommended analysis protocol, except using the FindMarkers function from Seurat package to perform pairwise comparisons across different conditions to find the statistically significant up- and downregulated genes. To reduce false-positive results, some parameters were customized on the basis of computational permutation. For example, it was required that a minimum percentage of expressed cells for each gene in either condition was higher than 0.1, and fold change greater than 1.25. The maximum number of cells in either condition was downsampled to 1,000 cells to balance the comparison. Consequently, the DEGs identified by Seurat were used as the informative genes to order cells using the setOrderingFilter function from monocle2, and the advanced nonlinear reconstruction algorithm called DDRTree was chosen to execute data dimensional reduction.
Cell–cell communication analysis.
Cell–cell interactions based on the expression of known ligand–receptor pairs in different t-types were inferred using CellChat (v.1.1.3)39. We followed the official CellChat workflow for analysing multiple datasets (0 h WIT, 1 h WIT, 7 h WIT, ECMO and OrganEx). We first randomly downsampled each dataset to 1,000 cells per t-type to balance the comparison. Next, we loaded normalized counts into CellChat. Next, we selected the CellChatDB. human database for cell–cell communication analysis. We then applied the preprocessing functions identifyOverExpressedGenes and identifyOverExpressedInteractions with the standard parameter set. We next computed communication probability between interacting cell groups with a truncated mean set at 0.1. Subsequently, we applied filterCommunication, computeCommunProbPathway and aggregateNet using the standard parameters. To identify conserved and context-specific signalling pathways, we applied the rankNet function on the netP data slot, which showed in a stacked bar plot the overall information flow of each signalling pathway. To determine the strength of the reactions and the t-types involved in each signalling pathway, we performed netAnalysis_signalingRole_heatmap and visualized the overall signalling by aggregating outgoing and incoming signalling together.
Tissue processing and histology
Tissue preparation.
After the completion of each experimental protocol, the brain, heart, lungs, kidneys, liver and pancreas were sampled and immersion-fixed in a solution containing 10% (w/v) neutral buffered formalin with gentle shaking. Sampling was performed by the same person at each experimental time point, randomly from the same region of each organ. After fixation, each tissue piece was processed and embedded into a paraffin block using the Excelsior tissue processor (Thermo Fisher Scientific). These paraffin blocks were trimmed on the Shandon Finesse 325 microtome (Thermo Fisher Scientific) to 5 μm sections. Sections were mounted on TruBond 380 adhesive slides and allowed to dry overnight at room temperature. All of the slides for the following tissue analysis were processed with deparaffinization and rehydration as previously described3. In brief, the sections were deparaffinized in 2 changes of Histo-Clear II (64111–04, Electron Microscopy Sciences) for 10 min each. The slides were then transferred to 100% alcohol, for two changes, 10 min each, and then transferred once through 95%, 70% and 50% alcohol for 5 min each. The slides were then rinsed in water and washed in wash buffer (0.05% Tween-20 in 1× PBS) for 10 min.
Tissue staining with H&E, cresyl violet (Nissl), the TUNEL assay and PAS.
All heart, lung, liver, kidney and pancreas slides were stained simultaneously for H&E using the automatic Shandon Linistain slide stainer (Thermo Fisher Scientific).
For Nissl staining, brain sections (hippocampus and PFC) were stained with 0.1% cresyl violet solution (Abcam, ab246816) for 5 min and were rinsed quickly in 1 change of distilled water. These sections were then dehydrated quickly in absolute alcohol and later cleared in Histo-Clear II and finally cover-slipped with Prolong Gold Antifade Mountant (Thermo Fisher Scientific, P36934).
All of the slides for the TUNEL assay (Millipore, S7101, 3542625) were processed simultaneously. They were fist deparaffinized, rehydrated and incubated with proteinase K (20 μg ml−1 in PBS) for 30 min at 37 °C. Slides were rinsed with PBS and incubated with 3% H2O2 in PBS for 10 min at room temperature to block endogenous peroxidase activity, followed by PBS washing and incubation in 0.1% Triton X-100 in 0.1% sodium citrate for 2 min on ice (4 °C). The sections were incubated with a mixture of TdT solution and fluorescein isothiocyanate dUTP solution in a humidified chamber at 37 °C for 60 min. This was followed by washings with PBS and incubation with anti-fluorescein antibody Fab fragments conjugated with horseradish peroxidase in a humidified chamber at 37 °C for 30 min. After washing with PBS, methyl green counterstain was applied to stain for nuclei.
All slides for PAS staining were processed simultaneously. They were first deparaffinized and rehydrated followed by incubation for 10 min in 0.5% periodic acid solution. The slides were then washed in three changes of distilled water after being incubated for 15 min in Schiff reagent. After incubation, the slides were washed in distilled water for 10 min and counterstained for 1 min in haematoxylin with acetic acid. After counterstaining, the sections were again washed in distilled water, differentiated in acid alcohol (one dip) and then rinsed before adding bluing solution (two dips). Finally, the slides were dehydrated with 95% and absolute alcohol and cleared with xylene before mounting in resin.
Immunohistochemistry.
All slides used for a specific immunohistochemistry staining were processed simultaneously. Slides containing formalin-fixed paraffin-embedded histological sections were first deparaffinized in 2 changes of Histo-Clear II (64111–04, Electron Microscopy Sciences) for 10 min each. The slides were then transferred to 100% alcohol, for two changes, 10 min each, and then transferred once through 95%, 70% and 50% alcohol for 5 min each. The slides were then rinsed in water and washed in wash buffer (0.05% Tween-20 in 1× PBS) for 10 min. The slides were then placed into a chamber filled with antigen retrieval buffer (10× R-Buffer A diluted to 1× by water, pH 6, 62706–10, Electron Microscopy Sciences). The slides then underwent heat-mediated antigen retrieval in the Unique Retriever system (Electron Microscopy Sciences). After antigen retrieval, the slides were washed in wash buffer and blocked for 1 h in 10% normal goat serum diluted in wash buffer at room temperature. The slides were then incubated in primary antibodies at 4 °C overnight at the following dilutions: rabbit anti-NeuN (1/1,000, Abcam, ab177487, GR3275112–10), mouse anti-GFAP (1/1,000, Sigma-Aldrich, G3893–100UL, 0000082460), rabbit anti-Iba1 (1/500, Wako, 019–19741, PTR2404), mouse anti-albumin (1/500, Abcam, 4A1C11, GR3215248–15), mouse anti-beta actin (1/500, Invitrogen, AC-15, 01003256), rabbit cleaved caspase-3 (1/50, R&D Systems, MAB835, KHK0821021), rabbit anti-RIPK3 (1:200, Aviva Systems Biology, ARP31513_T100), rabbit anti-IL-1B (1:200, Thermo Fisher Scientific, ASC0912), rabbit anti-GPX4 (1:200, Abcam, ab125066), rabbit anti-cardiac troponin I (1:100, Abcam, ab47003), rabbit anti-factor V (1:100, Abcam, ab234849), mouse anti-cytokeratin 8+18+19 (1:500, Abcam, ab41825), rabbit anti-ki-67 (1:200, Abcam, ab15580) and rabbit anti-TIM1 (1:200, Abcam, ab78494). The next day, the slides were washed and then incubated with fluorescently tagged secondary anti-rabbit (Cell Signaling Technology, 8889S, 12) and anti-mouse (Abcam, ab150113, GR3370569–1) antibodies at a dilution of 1/500 for 1 h at room temperature. After staining with secondary antibodies, slides were washed and incubated in DAPI (1/1,000 for 5 min) at room temperature. Finally, slides were washed and mounted with coverslips using Prolong Gold Antifade Mountant (P36934, Thermo Fisher Scientific). TSA kit (Superboost, Thermo Fisher Scientific, B40915) was used to amplify the IHC signals.
Microscopy and image processing
Tissue sections were imaged using an LSM880 confocal microscope (Zeiss) equipped with a motorized stage using 10× (0.3 NA) or 20× (0.8 NA) objective lenses with identical settings across all experimental conditions. The lasers used were as follows: argon 458, 488 and 514; diode 405; and DPSS 561–10. The DPSS 561–10 laser intensity was increased during imaging of the control perfusate samples for the intravascular haemoglobin fluorescence study to obtain a background signal that was comparable to the other groups. Images were acquired at either 1,024 × 1,024 or 2,048 × 2,048 pixel resolution. The images are either representative confocal tile scans, high-magnification maximum intensity z-stack projections (approximately 7–9 μm stacks; ~1 μm per z-step) or high-magnification confocal images. Alternatively, histological images were acquired using the Aperio CS2 Pathology Slide Scanner (Leica) as described above. Image adjustments were uniformly applied to all of the experimental conditions in Zeiss Zen. Digitized images were assembled in Zeiss Zen, ImageScope and Adobe Illustrator.
Histological data analysis and quantification
H&E staining pathology injury score.
All of the H&E slides were scanned. Four images were randomly selected from each slide and from the corresponding areas. Blinded observers scored each image accordingly. The criteria for heart were nuclear damage, myocyte vacuolization, widened spaces between myofibers and oedema. The criteria for the lungs were nuclear damage, pneumocyte vacuolization and haemorrhage. The criteria for the kidneys were nuclear damage, tubule vacuolization, haemorrhage and tubule damage. The criteria for the liver were nuclear damage, tissue vacuolization, hepatocyte vacuolization and congestion. The criteria for the pancreas were nuclear damage, cell vacuolization, haemorrhage and oedema.
Cresyl violet (Nissl) staining injury score.
Two images were taken per region of interest (hippocampal CA1 region or PFC) and were evaluated by a observer in a blinded manner using the cell counter function in ImageJ according to previously established criteria3.
Kidney PAS staining pathology injury score.
All kidney PAS-stained slides were examined by kidney pathologist in a blinded manner. Score (0–3) was assigned depending on the severity of the damage which included: Bowman space dilation, tubular dilation, tubular vacuolization, brush border disruption and the presence of the casts.
Brain immunofluorescence cell analysis and quantification.
All of the images were normalized to the regions of interest (hippocampal CA1, CA3 and DG) using ImageJ and randomized. The numbers of labelled astrocytes (GFAP+ cells) and microglia (IBA1+ cells) were quantified manually by a blinded observer using the cell counter plugin and averaged on the basis of the acquired area/cells. The particle analysis of astrocytes was performed using a custom pipeline in the open-source software CellProfiler67, in which a uniform threshold was set on GFAP to identify GFAP+ skeleton objects. The number of GFAP+ fragments in each area was then auto-counted and averaged according to the number of the astrocytes. The mean intensity of NeuN per neuron was quantified by a custom pipeline in which neurons were identified on the basis of the DAPI segmented objects expressing NeuN. Data were analysed and plotted in GraphPad Prism9. All data are shown as mean ± s.e.m. n values refer to the number of biological replicates.
Kidney, liver and heart immunofluorescence analysis.
In the kidneys, three images were taken randomly of the kidney cortex. All of the images were randomized and analysed by a blinded observer. Glomerulus and the proximal convoluted tubule were manually selected as the regions of interest in the four same-size circles in corresponding regions across images. The mean intensity of ACTB and TIM1 was calculated using ImageJ. The number of Ki-67+ cells was manually counted.
In the liver, immunolabelling quantification was performed the same for albumin and factor V. Three images were randomly taken from the same region of the liver. The mean intensity of albumin and factor V was automatically quantified using ImageJ across the whole slide.
In the heart, three images were taken randomly from the left ventricle just slightly below the epicardium. All of the images were randomized, after which a blinded observer selected cardiac muscle tissue as a region of interest. The amount of cytoplasmic troponin I was then quantified using mean intensity function in ImageJ.
Cell death pathway quantification and analysis (actCASP3, IL-1B, RIPK3 and GPX4).
For the kidneys, liver, heart, and pancreas, three images were taken randomly from each slide and from the corresponding areas of each organ. actCASP3+ cell intensity was quantified manually by a blinded observer using ImageJ. Mean immunolabelling intensity of IL-1B, RIPK3 and GPX4 in the kidneys, liver and heart was quantified using ImageJ with the background subtraction and measure functions. In the brain, the number of actCASP3+ and RIPK3+ cells were counted manually in the hippocampal CA1 region and prefrontal cortex. Expression of IL-1B and GPX4 in hippocampal CA1 was measured in cells that were manually selected in the granule cell layer using ImageJ measure function.
TUNEL quantification and analysis.
All of the images were taken using a bright- field microscope followed by a random same-size snapshots of the slides. A blinded observer analysed randomized images using Fiji to separate channels of nuclei staining (haematoxylin) and DNA fragments (DAB). Total DNA fragments were quantified on the basis of the total DAB intensity using CellProfiler67.
Functional organ assessments
Heart contractility measurements.
A small piece of the apical portion of the hearts’ left ventricle was excised and placed in the carbonated Tyrode’s solution (140 mM NaCl, 6 mM KCl, 10 mM glucose, 10 mM HEPES, 1 mM MgCl2, 1.8 mM CaCl2, pH 7.4). The tissue was then dissected into 1–2 cm3 cubes and placed onto a holder with the epicardium facing down so the cutting align with the cardiac myofibril orientation. Using a vibratome, the heart was sliced at 150 μm thickness at 4 °C. After slicing, spontaneous rate and rhythm of heart contraction were characterized and recorded using a slice microscope with temperature control set to 37 °C (Scientifica SliceScope). Beating frequency was quantified within the time course of 30 s.
Glucose assays.
At the end of each perfusion, two identical tissue samples with 5 mm length were acquired from each organ (kidney, heart and cerebral cortex) using a 3 mm biopsy punch (Miltex, Integra) and were placed in cold PBS (fresh) and PFA (fix overnight) solutions, respectively. The fresh tissues and the fixed controls were incubated in 2-NBDG working solution (Cayman, 11046, 100 μM) for 30 min at 37 °C followed by 15 min of wash in PBS. The images were acquired immediately using a widefield microscope (Zeiss) under a ×2.5 objective (Ex: 465–495 nm, Em: 520–560 nm) with exposure time of 295 ms (kidney and heart) and 50 ms (cerebral cortex). Images were analysed using ImageJ to quantify the mean intensity of the region of interest with normalization based on the fixed controls.
Organotypic hippocampus culture and nascent protein synthesis assay.
Hippocampus was isolated at the end of appropriate experimental protocol and sectioned using a vibratome at 250 μm thickness in carbonated NMDG-HEPES aCSF solution (92 mM NMDG, 2.5 mM KCl, 1.25 mM NaH2PO4, 30 mM NaHCO3, 20 mM HEPES, 25 mM glucose, 2 mM thiourea, 5 mM sodium ascorbate, 3 mM sodium pyruvate, 0.5 mM CaCl2·2H2O and 10 mM MgSO4·7H2O, pH 7.3). Hippocampal slices were then collected and transferred onto culture membranes (Falcon culture insert, 0.4 μm) and cultured in a six-well culture dish with 1.5 ml medium (48% DMEM/F-12 (Gibco), 48% Neurobasal (Gibco), 1× N-2 (Gibco), 1× B-27 (Gibco), 1× Glutamax (Gibco), 1× NEAA (Gibco), 1× penicillin–streptomycin (Gibco)). The plates were maintained in an incubator at 37 °C with 5% CO2 for up to 2 weeks. The amino acid methionine analogue azidohomoalanine (Thermo Fisher Scientific) was added to the hippocampal slice at a final concentration of 50 μM in HEPES-buffered solution (HBS) followed by the incubation of 6 h at 37 °C. Slices were washed with PBS and immediately fixed in 4% paraformaldehyde at 4 °C overnight. Detection of nascent protein synthesis was performed using the modified Click-iT l-azidohomoalanine Alexa Fluor 488 Protein Synthesis HCS Assay kit (Thermo Fisher Scientific). To visualize neurons simultaneously, the slices were then blocked with 5% NDS for 1 h at room temperature, and incubated with rabbit anti-NeuN antibodies (abcam, 1:1,000) overnight at 4 °C followed by incubation with anti-rabbit AlexaFluor647 (1:500) for 1 h at room temperature. The slices were then co-stained with DAPI to visualize nuclei. Images were acquired using the Zeiss LSM800 confocal microscope with ×20 objective. The combined z-stack images of DAPI, AlexaFluor 488 and AlexaFluor 647 channels were acquired with optical slice thickness of around 20 μm and images of maximum projection are shown. Proteins detected around the nucleus (perinuclear) are the most abundant newly synthesized proteins in cells27, and were quantified using ImageJ intensity measurement function.
Image visualization
Depiction of the pig in Fig. 1a and pig’s head in Extended Data Fig. 6a were adapted from BioRender with postprocessing using Adobe Illustrator and Adobe Photoshop.
Statistical analysis and reproducibility
All data are reported as mean ± s.e.m. with data analysis being conducted using one-way ANOVA with Dunnett’s post hoc multiple-comparisons adjustment in reference to the OrganEx perfusion group, or unpaired t-tests for comparisons between two groups. Fisher’s exact tests were used to compare the occurrence of QRS complexes in the OrganEx and ECMO groups during the perfusion protocol. Details of the number of replicates in each experimental group together with appropriate statistical analyses are shown below. Significance was set at P < 0.05. All statistical analysis and plotting were performed in GraphPad 9 (GraphPad) or in Python. All of the figures were created using Adobe Illustrator (Adobe Systems).
Further information.
Owing to space limitations in the figure legends, further information regarding statistical values and reproducibility of the results is given below.
In Fig. 2c–e, the results are taken at the baseline (where applicable) and every hour throughout the perfusion experiment from the ECMO and OrganEx perfusion groups. Each group consisted of n = 6 biological replicates. In Fig. 2c, total flow rate: P values for hours 1–6, 1 h: <0.0001, 2 h: <0.0001, 3 h: <0.0001, 4 h: <0.0001, 5 h: <0.0001, 6 h: <0.0001; t values for hours 1–6, 1 h: 12.34, 2 h: 8.120, 3 h: 8.869, 4 h: 9.683, 5 h: 17.64, 6 h: 22.96. Brachial arterial pressure: P values for hours 0–6, 0 h: 0.9704, 1 h: <0.0001, 2 h: 0.0002, 3 h: 0.0003, 4 h: 0.0006, 5 h: 0.0027, 6 h: <0.0001; t values for hours 0–6, 0 h: 0.03807, 1 h: 12.24, 2 h: 5.645, 3 h: 5.480, 4 h: 4.890, 5 h: 3.953, 6 h: 7.590. In Fig. 2d, mixed venous O2 saturation: P values for hours 0–6, 0 h: 0.8996, 1 h: 0.0837, 2 h: 0.0002, 3 h: 0.0003, 4 h: 0.0015, 5 h: <0.0001, 6 h: 0.0036; t values for hours 1–6, 0 h: 0.1295, 1 h: 1.920, 2 h: 5.582, 3 h: 5.453, 4 h: 4.338, 5 h: 7.691, 6 h: 3.781. In Fig. 2e, serum K+: P values for hours 0–6, 0 h: 0.8365, 1 h: 0.0001, 2 h: 0.0008, 3 h: <0.0001, 4 h: <0.0001, 5 h: <0.0001, 6 h: <0.0001; t values for hours 0–6, 0 h: 0.2118, 1 h: 6.156, 2 h: 4.707, 3 h: 7.224, 4 h: 12.24, 5 h: 7.401, 6 h: 9.358. Serum pH: P values for hours 0–6, 0 h: 0.3489, 1 h: <0.0001, 2 h: <0.0001, 3 h: <0.0001, 4 h: <0.0001, 5 h: <0.0001, 6 h: <0.0001; t values for hours 0–6, 0 h: 0.9827, 1 h: 6.285, 2 h: 9.256, 3 h: 9.668, 4 h: 8.427, 5 h: 7.056, 6 h: 7.034.
In Fig. 3b–d, data points are from a representative brain per condition; the experiment was repeated in n = 3 independent brains per condition. In Fig. 3b, one-way ANOVA (P = 0.0001, F4,10 = 19.12) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 7 h WIT: P = 0.009; OrganEx versus ECMO: P = 0.003. In Fig. 3c, one-way ANOVA (P = 0.0098, F4,10 = 6.035) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 7 h WIT: P = 0.0059; OrganEx versus ECMO: P = 0.0356. In Fig. 3d, one-way ANOVA (P = 0.0076, F4,10 = 6.495) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO: P = 0.0109.
In Fig. 3f–h, data points are from a representative organ (heart, liver and kidney) per condition, the experiment was repeated in n = 5 independent organs per condition. In Fig. 3f, one-way ANOVA (P < 0.0001, F4,20 = 52.16) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 1 h WIT: P = 0.0429; OrganEx versus 7 h WIT: P < 0.0001; OrganEx versus ECMO: P < 0.0001. In Fig. 3g, one-way ANOVA (P < 0.0001, F4,20 = 50.79) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 1 h WIT: P = 0.006; OrganEx versus 7 h WIT: P < 0.0001; OrganEx versus ECMO: P = 0.0009. In Fig. 3g, one-way ANOVA (P < 0.0001, F4,20 = 50.79) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 1 h WIT: P = 0.006; OrganEx versus 7 h WIT: P < 0.0001. OrganEx versus ECMO: P = 0.0009; in Fig. 3h, one-way ANOVA (P < 0.0001, F4,20 = 17.79) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 1 h WIT: P = 0.0001; OrganEx versus 7 h WIT: P < 0.0001; OrganEx versus ECMO: P = 0.0022.
In Fig. 3j,k, data points are from a representative kidney per condition, the experiment was repeated in n = 3 independent kidneys per condition. In Fig. 3j, one-way ANOVA (P = 0.0262, F4,10 = 4.398) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO: P = 0.0103. In Fig. 3k, one-way ANOVA (P = 0.0057, F4,10 = 7.082) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO: P = 0.0023.
In Fig. 4a–c, data points are from a representative organ (brain, heart and kidney) per condition, the experiment was repeated in n = 3 independent organs per condition. In Fig. 4a, one-way ANOVA (P = 0.003, F2,6 = 17,73) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO: P = 0.0024. In Fig. 4b, one-way ANOVA (P = 0.0033, F2,6 = 17,07) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO: P = 0.0033. In Fig. 4c, one-way ANOVA (P = 0.0033, F2,6 = 6.453) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO: P = 0.0320.
In Fig. 4d, each data points are from representative perfusion experiment per condition, the experiment was repeated n = 5 times. A two-sided Fisher’s exact t-test was used: P = 0.0476.
In Fig. 4g, each data point is from a representative liver per condition, the condition was repeated n = 3 times. One-way ANOVA (P < 0.0001, F4,10 = 15.52) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 1 h WIT: P = 0.0002; OrganEx versus 7 h WIT: P = 0.0005; OrganEx versus ECMO: P = 0.0007.
In Fig. 4i,k, each data point is from the representative hippocampal slice per condition, the experiment was repeated n = 3–5 times per condition. In Fig. 4i, for day 1: one-way ANOVA (P = 0.0352, F2,10 = 4.766) with post hoc Dunnett’s adjustment was performed; for day 14: one-way ANOVA (P = 0.0027, F2,10 = 11.35) with post hoc Dunnett’s adjustment was performed, Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO: P = 0.0026. In Fig. 4k, for day 1: one-way ANOVA (P = 0.0077, F2,10 = 8.246) with post hoc Dunnett’s adjustment was performed, Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO: P = 0.0216; for day 14: one-way ANOVA (P = 0.0270, F2,9 = 5.544) with post hoc Dunnett’s adjustment was performed, Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO: P = 0.027.
In Extended Data Fig. 1c,d, each data point is from the representative perfusion experiment per condition, the experiment was repeated n = 6 times. In Extended Data Fig. 1c, arterial cannula pressure: P values for hours 1–6, 1 h: <0.0001, 2 h: <0.0001, 3 h: <0.0001, 4 h: <0.0001, 5 h: <0.0001, 6 h: <0.0001; t values for hours 1–6, 1 h: 12.34, 2 h: 8.120, 3 h: 8.869, 4 h: 9.683, 5 h: 17.64, 6 h: 22.96. In Extended Data Fig. 1c, venous cannula pressure: P values for hours 1–6, 1 h: <0.0001, 2 h: 0.0306, 3 h: 0.0237, 4 h: 0.0003, 5 h: 0.0069, 6 h: 0.0027; t values for hours 1–6, 1 h: 9.058, 2 h: 2.516, 3 h: 2.665, 4 h: 5.516, 5 h: 3.386, 6 h: 3.946. In Extended Data Fig. 1d, O2 delivery: P values for hours 1–6, 1 h: <0.0001, 2 h: <0.0001, 3 h: <0.0001, 4 h: <0.0001, 5 h: <0.0001, 6 h: <0.0001; t values for hours 1–6, 1 h: 10.44, 2 h: 6.957, 3 h: 8.578, 4 h: 9.462, 5 h: 14.32, 6 h: 18.56. In Extended Data Fig. 1d, O2 consumption: P values for hours 1–6, 1 h: 0.8432, 2 h: 0.1815, 3 h: 0.3195, 4 h: 0.3667, 5 h: 0.2152, 6 h: 0.0394; t values for hours 1–6, 1 h: 0.2030, 2 h: 1.436, 3 h: 1.048, 4 h: 0.9455, 5 h: 1.323, 6 h: 2.368.
In Extended Data Fig. 2b–e, data points are from a representative brain per condition, the experiment was repeated in n = 3 independent brains per condition. In Extended Data Fig. 2b, one-way ANOVA (P = 0.0008, F4,10 = 11.95) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO: P = 0.0097; in Extended Data Fig. 2c, one-way ANOVA (P = 0.0003, F4,10 = 14.64) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO: P = 0.0006. In Extended Data Fig. 2d, one-way ANOVA (P < 0.001, F4,10 = 44.47) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 7 h WIT: P < 0.001; OrganEx versus ECMO: P < 0.001. In Extended Data Fig. 2e, one-way ANOVA (P < 0.001, F4,10 = 60.70) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 0 h WIT: P = 0.0024; OrganEx versus 1 h WIT: P < 0.001; OrganEx versus 7 h WIT: P < 0.001; OrganEx versus ECMO: P = 0.0159.
In Extended Data Fig. 2g–l, data points are from a representative brain per condition, the experiment was repeated in n = 3 independent brains per condition. In Extended Data Fig. 2g, one-way ANOVA (P = 0.011, F4,10 = 5.819) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 1 h WIT: P = 0.0287; OrganEx versus 7 h WIT: P = 0.0045; OrganEx versus ECMO: P = 0.0149. In Extended Data Fig. 2i, one-way ANOVA (P < 0.0003, F4,10 = 32.20) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 0 h WIT: P = 0.0243; OrganEx versus 7 h WIT: P = 0.0157; OrganEx versus ECMO: P = 0.0001. In Extended Data Fig. 2j, one-way ANOVA (P = 0.0231, F4,10 = 4.590) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 7 h WIT: P = 0.0137. In Extended Data Fig. 2k, one-way ANOVA (P = 0.0002, F4,10 = 17.45) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 1 h WIT: P = 0.09338; OrganEx versus 7 h WIT: P < 0.0001; OrganEx versus ECMO: P = 0.0157. In Extended Data Fig. 2l, one-way ANOVA (P = 0.0032, F4,10 = 8.285) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO: P = 0.0198.
In Extended Data Fig. 3b,c,e, data points are from a representative organ (lung, pancreas, kidney) per condition, the experiment was repeated in n = 5 independent organs per condition. In Extended Data Fig. 3b, one-way ANOVA (P < 0.0001, F4,20 = 45.78) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 7 h WIT: P < 0.0001; OrganEx versus ECMO: P < 0.0001. In Extended Data Fig. 3c, one-way ANOVA (P < 0.0001, F4,20 = 19.80) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 1 h WIT: P = 0.0009; OrganEx versus 7 h WIT: P < 0.0001; OrganEx versus ECMO: P = 0.0009. In Extended Data Fig. 3e, one-way ANOVA (P < 0.001, F4,20 = 1.915) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO: P = 0.0410.
In Extended Data Fig. 3g–j, data points are from a representative kidney per condition, the experiment was repeated in n = 3 independent kidneys per condition. In Extended Data Fig. 3g, one-way ANOVA (P < 0.01, F4,10 = 7.983) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 7 h WIT: P = 0.0159; OrganEx versus ECMO: P = 0.0118. In Extended Data Fig. 3i, one-way ANOVA (P < 0.01, F4,10 = 8.286) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 7 h WIT: P = 0.0092; OrganEx versus ECMO: P = 0.0084. In Extended Data Fig. 3j, one-way ANOVA (P < 0.01, F4,10 = 11.12) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 7 h WIT: P = 0.002; OrganEx versus ECMO: P = 0.0022.
In Extended Data Fig. 4b–e, data points are from a representative organ (heart, liver, kidney and pancreas) per condition, the experiment was repeated in n = 3 independent organs per condition. In Extended Data Fig. 4b, one-way ANOVA (P < 0.0001, F4,10 = 22.48) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 7 h WIT: P = 0.0018; OrganEx versus ECMO: P < 0.0001. In Extended Data Fig. 4c, one-way ANOVA (P = 0.0020, F4,10 = 9.392) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 1 h WIT: P = 0.004; OrganEx versus ECMO: P = 0.001. In Extended Data Fig. 4d, one-way ANOVA (P < 0.0001, F4,10 = 23.50) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 1 h WIT: P = 0.0022; OrganEx versus 7 h WIT: P = 0.0002; OrganEx versus ECMO: P < 0.0001. In Extended Data Fig. 4e, one-way ANOVA (P = 0.0008, F4,10 = 12.10) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO WIT: P = 0.0008.
In Extended Data Fig. 4g–j, data points are from a representative organ (heart, liver, kidney and pancreas) per condition, the experiment was repeated in n = 5 independent organs per condition. In Extended Data Fig. 4g, one-way ANOVA (P < 0.0001, F4,20 = 14.35) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO: P < 0.0001. In Extended Data Fig. 4h, one-way ANOVA (P < 0.0001, F4,20 = 13.26) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO: P < 0.0001. In Extended Data Fig. 4i, one-way ANOVA (P = 0.0002, F4,20 = 9.110) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO: P = 0.0012. In Extended Data Fig. 4j, one-way ANOVA (P = 0.0387, F4,20 = 3.102) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO: P = 0.0216.
In Extended Data Fig. 4l,m, each data points are from a representative brain per condition, the experiment was repeated in n = 3 independent brains per condition. In Extended Data Fig. 4l, one-way ANOVA (P < 0.0001, F4,10 = 37.55) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 0 h WIT: P = 0.0083; OrganEx versus 1 h WIT: P = 0.0002; OrganEx versus 7 h WIT: P = 0.016. In Extended Data Fig. 4m, one-way ANOVA (P < 0.0001, F4,10 = 66.81) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 0 h WIT: P = 0.0001; OrganEx versus 1 h WIT: P < 0.0001.
In Extended Data Fig. 4o,p, each data points are from a representative brain per condition, the experiment was repeated in n = 5 independent brains per condition. In Extended Data Fig. 4o, one-way ANOVA (P = 0.0547, F4,20 = 2.784) with post hoc Dunnett’s adjustment was performed. In Extended Data Fig. 4p, one-way ANOVA (P = 0.1005, F4,20 = 2.244) with post hoc Dunnett’s adjustment was performed.
In Extended Data Fig. 5, each data point is from a representative brain specimen per condition, the experiment was repeated n = 3 times per condition. In Extended Data Fig. 5b, one-way ANOVA (P = 0.0013, F4,10 = 10.52) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 7 h WIT: P = 0.002; OrganEx versus ECMO: P = 0.0418. In Extended Data Fig. 5c, one-way ANOVA (P = 0.0002, F4,10 = 17.06) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 1 h WIT: P = 0.0386; OrganEx versus 7 h WIT: P = 0.0302. In Extended Data Fig. 5d, one-way ANOVA (P < 0.0001, F4,10 = 23.67) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO: P = 0.0001. In Extended Data Fig. 5e, one-way ANOVA (P = 0.0003, F4,10 = 14.74) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO: P = 0.0006. In Extended Data Fig. 5g, one-way ANOVA (P = 0.0264, F4,10 = 4.389) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 7 h WIT: P = 0.0301; OrganEx versus ECMO: P = 0.0270. In Extended Data Fig. 5h, one-way ANOVA (P = 0.0082, F4,10 = 6.365) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 1 h WIT: P = 0.0082; OrganEx versus 7 h WIT: P = 0.005; OrganEx versus ECMO: P = 0.0124. In Extended Data Fig. 5i, one-way ANOVA (P = 0.0012, F4,10 = 10.81) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 7 h WIT: P = 0.001; OrganEx versus ECMO: P = 0.0012. In Extended Data Fig. 5j, one-way ANOVA (P = 0.0001, F4,10 = 18.96) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 7 h WIT: P = 0.0048; OrganEx versus ECMO: P = 0.0049. In Extended Data Fig. 5l, one-way ANOVA (P = 0.0264, F4,10 = 7.430) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 1 h WIT: P = 0.0437; OrganEx versus 7 h WIT: P = 0.0437; OrganEx versus ECMO: P = 0.0437. In Extended Data Fig. 5m, one-way ANOVA (P = 0.0082, F4,10 = 6.365) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 1 h WIT: P = 0.0082; OrganEx versus 7 h WIT: P = 0.005; OrganEx versus ECMO: P = 0.0124. In Extended Data Fig. 5n, one-way ANOVA (P = 0.0002, F4,10 = 16.23) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 7 h WIT: P = 0.0013; OrganEx versus ECMO: P = 0.044. In Extended Data Fig. 5o, one-way ANOVA (P = 0.0024, F4,10 = 9.035) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 1 h WIT: P = 0.0019; OrganEx versus 7 h WIT: P = 0.0062; OrganEx versus ECMO: P = 0.0034.
In Extended Data Fig. 6j,k, each data point is from a representative hippocampal slice per condition, the experiment was repeated n = 3–5 times per condition. In Extended Data Fig. 6j, for day 1: one-way ANOVA (P = 0.0078, F2,9 = 8.749) with post hoc Dunnett’s adjustment was performed, Dunnett’s multiple-comparisons test resulted in OrganEx versus ECMO: P = 0.0245; for day 7: one-way ANOVA (P = 0.0126, F2,10 = 6.995) with post hoc Dunnett’s adjustment was performed, Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO: P = 0.0086; for day 14: one-way ANOVA (P = 0.005, F2,9 = 10.12) with post hoc Dunnett’s adjustment was performed, Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO: P = 0.0037. In Extended Data Fig. 6k, for day 1: one-way ANOVA (P = 0.0406, F2,9 = 4.670) with post hoc Dunnett’s adjustment was performed, Dunnett’s multiple-comparisons test resulted in OrganEx versus ECMO: P = 0.0375; for day 7: one-way ANOVA (P = 0.0278, F2,9 = 5.476) with post hoc Dunnett’s adjustment was performed, Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO: P = 0. 0406; for day 14: an unpaired two-tailed t-test was performed: P = 0.8691, t = 0.1735.
In Extended Data Fig. 6m,o, data points are from a representative organs (heart, liver) per condition, the experiment was repeated in n = 3 independent organs per condition. In Extended Data Fig. 6m, one-way ANOVA (P = 0.0207, F4,10 = 4.760) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus ECMO: P = 0.00434. In Extended Data Fig. 6o, one-way ANOVA (P = 0.0063, F4,10 = 6.863) with post hoc Dunnett’s adjustment was performed; Dunnett’s multiple-comparisons test resulted in: OrganEx versus 7 h WIT: P = 0.0064; OrganEx versus ECMO: P = 0.0163.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The snRNA-seq dataset was deposited at the NCBI’s Gene Expression Omnibus68 and is accessible through GEO Series accession number GSE183448.
Code availability
The source code used to analyse the data presented in this paper is deposited and publicly available at GitHub (https://github.com/sestanlab/OrganEx).
Extended Data
Extended Data Fig. 1 |. Analysis of circulation and blood/perfusate properties after 1h of warm ischaemia and perfusion interventions.
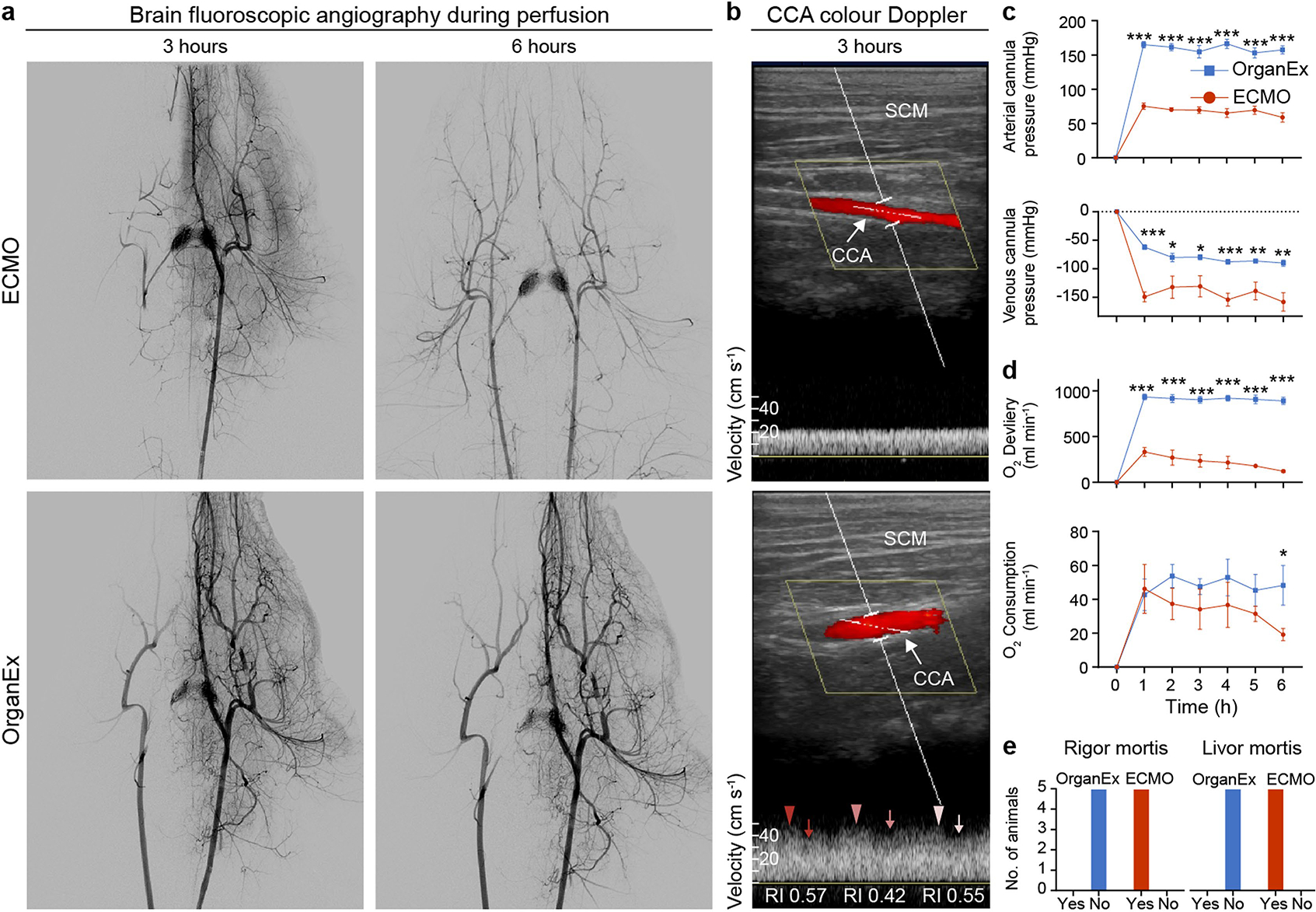
a, Representative fluoroscopy images of autologous blood flow (ECMO intervention, up) or a mixture of autologous blood and the perfusate (OrganEx intervention, below) in the head captured after 3 and 6 h respectively of perfusion, showing robust restoration of the circulation in the OrganEx group. A contrast catheter was placed in the left common carotid artery (CCA), except in the ECMO group at 6 h timepoint where contrast catheter could not be advanced beyond aortic arch in to the left CCA due to pronounced vasoconstriction, thus resulting in bilateral CCA filling. n = 6. b, Representative colour Doppler images of the CCA demonstrating robust flow in OrganEx group. Ultrasound waveform analysis demonstrated that OrganEx produced pulsatile, biphasic flow pattern (lower panel). SCM, sternocleidomastoid muscle; RI, resistive index. n = 6. c, Longitudinal change in arterial and venous cannula pressures throughout the perfusion demonstrating robust perfusion in OrganEx group. d, Time-dependent changes in oxygen delivery and consumption demonstrating increased oxygen delivery and stable oxygen consumption over the perfusion period in OrganEx group. n = 6. e, Presence of classical signs of death (rigor and livor mortis) in ECMO as compared to OrganEx group at the experimental endpoint. Data presented are mean ± s.e.m. Two-tailed unpaired t-test was performed. For more detailed information on statistics and reproducibility, see methods. *P < 0.05, **P < 0.01, ***P < 0.001.
Extended Data Fig. 2 |. Nissl staining and immunohistochemical analysis of the hippocampal CA1 region and the prefrontal cortex (PFC).
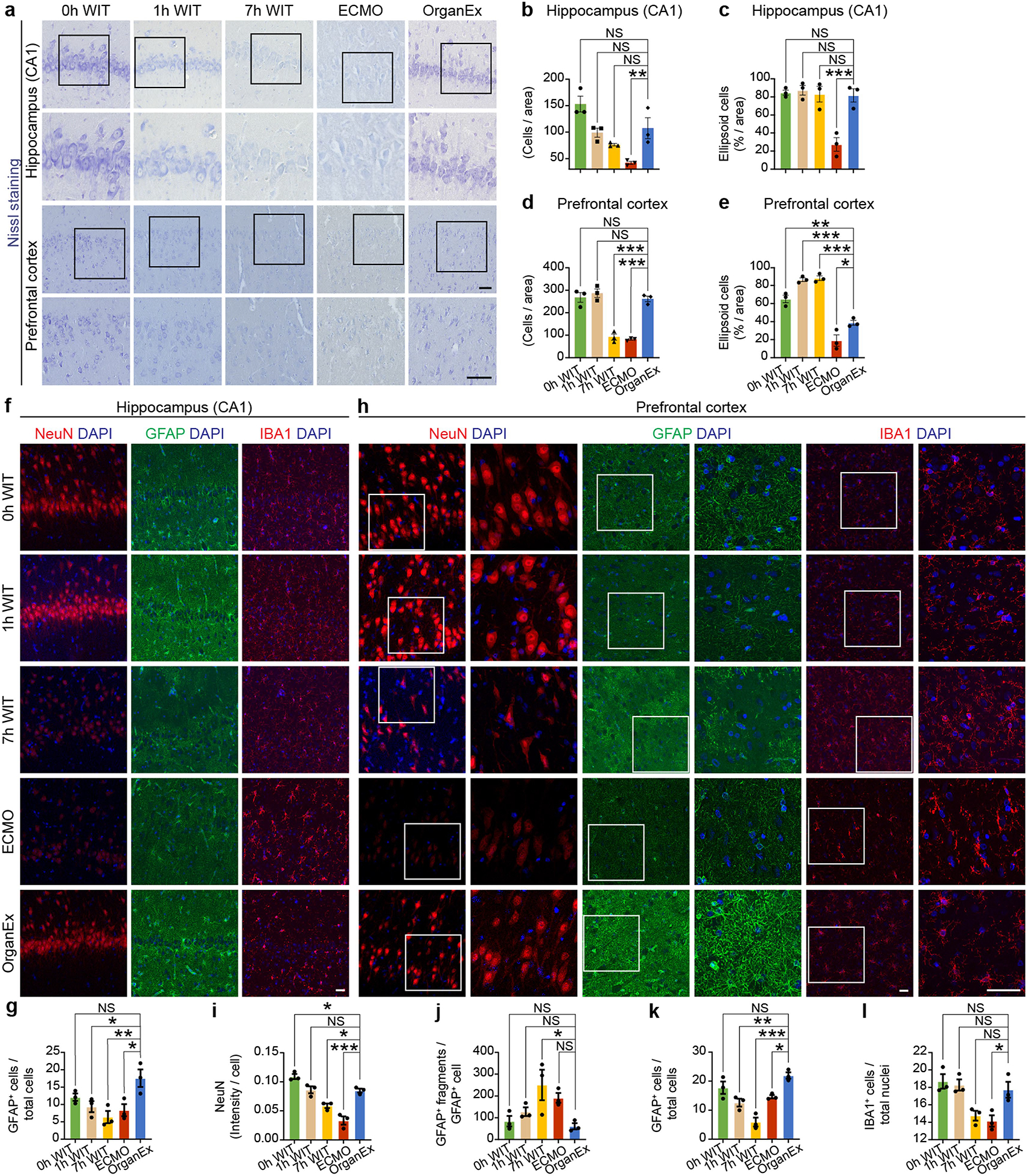
a, Representative images of Nissl staining of the CA1 (up) and PFC (below). b, c, Quantification of the number of cells per standardized area (b) and percentage of ellipsoid cells per area (c) in the CA1 between the experimental groups. d, e, Quantification of the number of cells per standardized area (d) and percentage of ellipsoid cells per area (e) in the PFC between the experimental groups. n = 3. f, h, Representative confocal images of immunofluorescent staining for neurons (NeuN), astrocytes (GFAP), and microglia (IBA1) counterstained with DAPI nuclear stain in CA1 (f) and PFC (h). g, Quantification of GFAP immunoreactivity in hippocampal CA1 region depicting comparable immunoreactivity between OrganEx and 0h WIT group, with a significant increase compared to the other groups. i, j, k, l, Quantification of NeuN immunolabeling intensity (i), number of GFAP+ fragments (j), and number of GFAP+ cells (k) depict similar trends between the groups as seen in the CA1. Microglia number (l) shows comparable results between OrganEx and 0h WIT with different dynamics seen in the ECMO group. n = 3. Scale bars, 50 μm. Data presented are mean ± s.e.m. One-way ANOVA with post-hoc Dunnett’s adjustments was performed. For more detailed information on statistics and reproducibility, see methods. *P < 0.05, **P < 0.01, ***P < 0.001.
Extended Data Fig. 3 |. Representative images of H&E staining across assessed peripheral organs and kidney periodic acid-Schiff (PAS) staining and immunolabeling for HACVR1 and Ki-67.
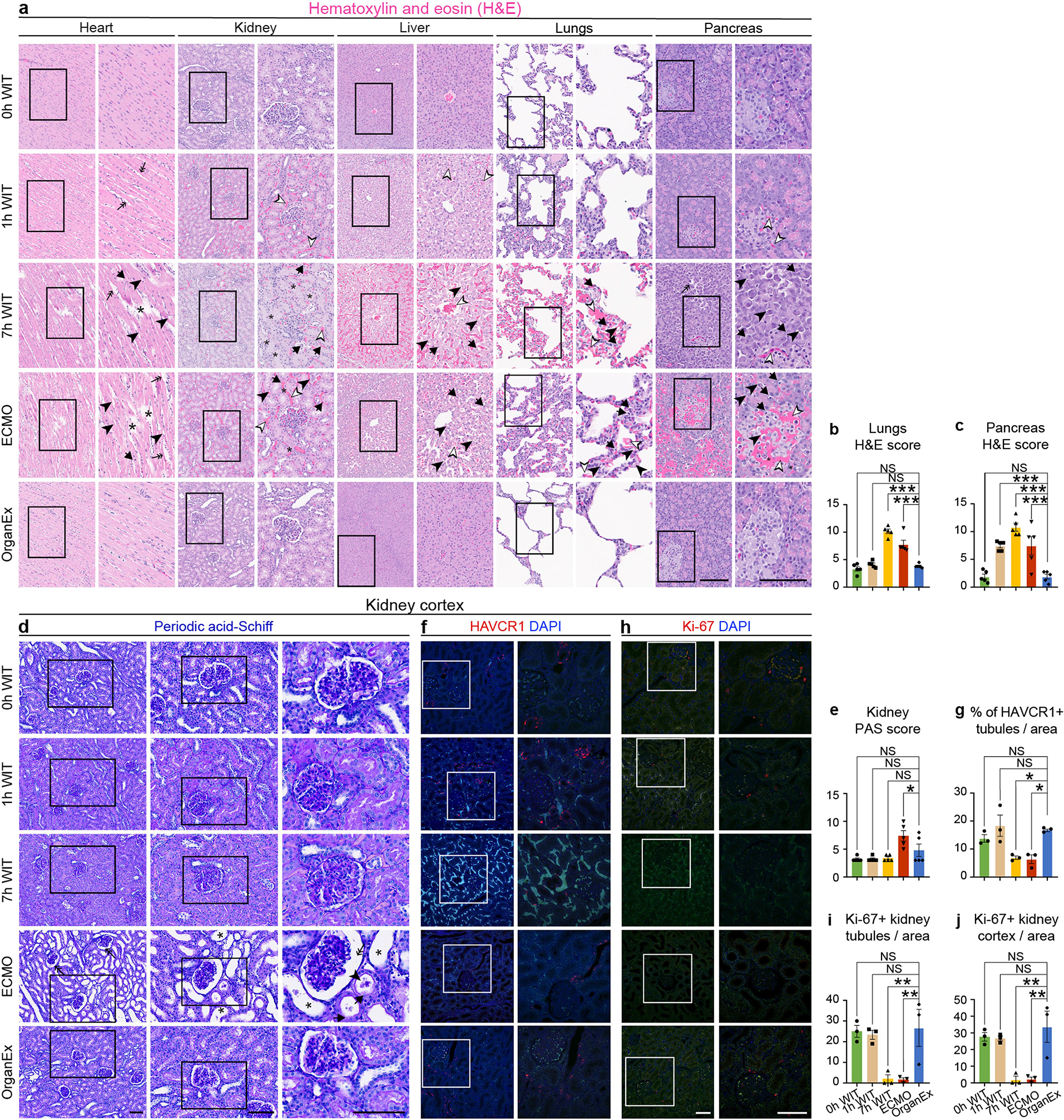
a, Representative images of the H&E staining in heart, kidney, liver, lungs, and pancreas. Arrows point to nuclear damage, asterisks point to disrupted tissue integrity, empty arrowheads point to haemorrhage, full arrowheads point to cell vacuolization, double arrows point to tissue oedema. b, c, H&E histopathological scores in lungs (b) and pancreas (c). d, Representative images of PAS staining of the kidney. Arrows point to disrupted brush border, full arrowheads point to the presence of casts, asterisks point to tubular dilation, double arrows point to the Bowman space dilation. e, Kidney PAS histopathological damage score. n = 5. f, h, Representative confocal images of immunofluorescent staining for HAVCR1 and Ki-67 in kidney, respectively. g, Quantification of HAVCR1 immunolabeling signal intensity. i, j, Quantification of the kidney Ki-67 positive staining. HACVR1 and Ki-67 immunolabeling quantification results follow a similar pattern seen with other organs with comparable results between 0h WIT and OrganEx group and significant decrease in the 7h WIT and ECMO groups. n = 3. Scale bars,100 μm. Data presented are mean ± s.e.m. One-way ANOVA with post-hoc Dunnett’s adjustments was performed. For more detailed information on statistics and reproducibility, see methods. *P < 0.05, **P < 0.01.
Extended Data Fig. 4 |. Analysis of cell death across experimental conditions and organs.
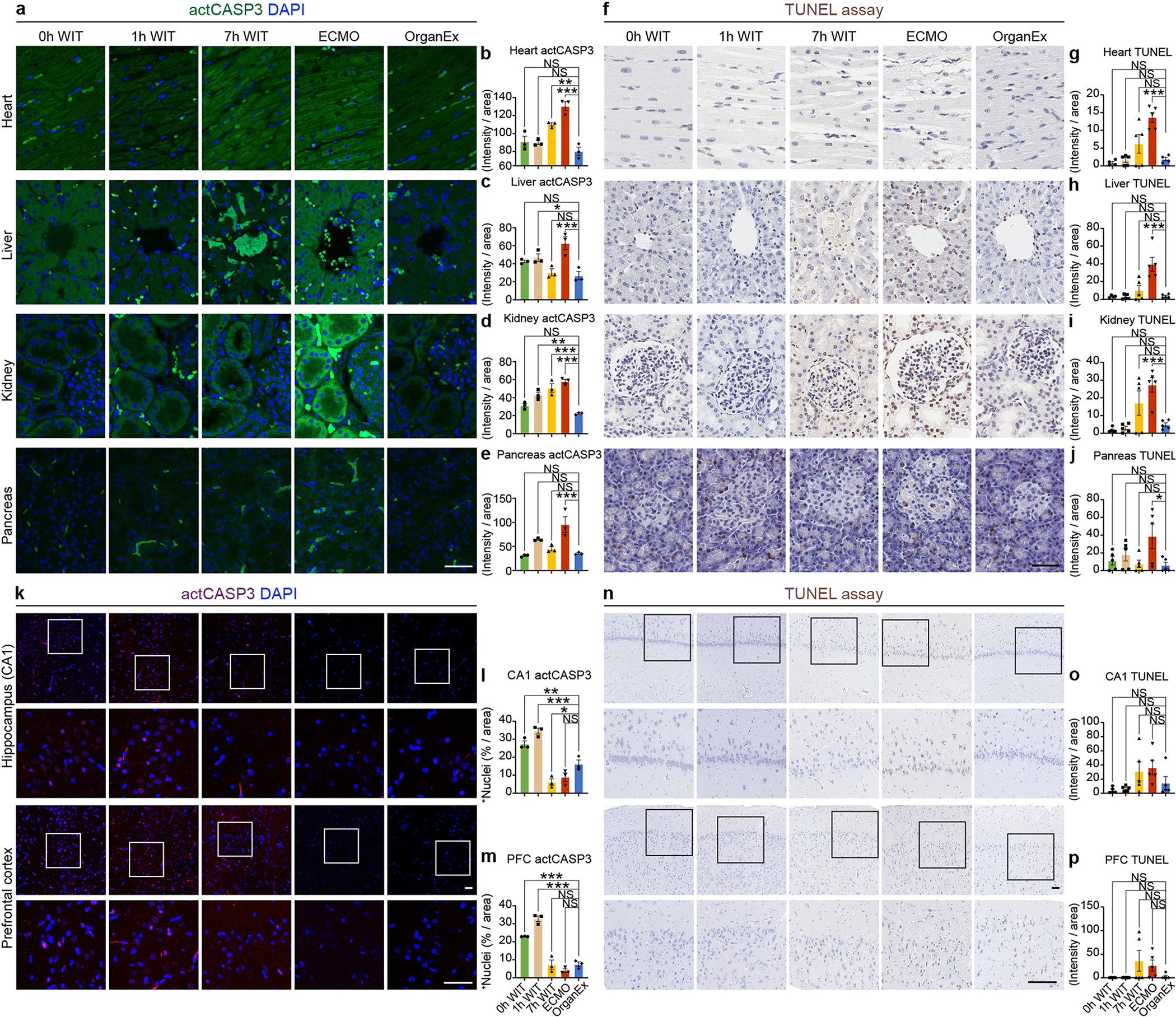
a, f, k, n, Representative confocal images of immunofluorescent staining for activated caspase 3 (actCASP3) and TUNEL assay in heart, liver, kidney, pancreas and brain. b-e, Quantification of actCASP3 immunolabeling signal intensity in heart (b), liver (c), kidney (d), and pancreas (e). n = 3. g-j, Normalized total intensity of TUNEL signal in heart (g), liver (h), kidney (i), and pancreas (j). n = 3. l, m, Percentage of actCASP3 positively stained nuclei in the CA1 (l) and PFC (m). n = 5. o, p, Normalized total intensity of TUNEL signal in CA1 (o) and PFC (p). n = 5. Scale bars, 50 μm. Data presented are mean ± s.e.m. One-way ANOVA with post-hoc Dunnett’s adjustments was performed. For more detailed information on statistics and reproducibility, see methods. *P < 0.05, **P < 0.01, ***P < 0.001.
Extended Data Fig. 5 |. Evaluation of different cell death pathways by immunohistochemical staining for important molecules in pyroptosis (IL-1B), necroptosis (RIPK3) and ferroptosis (GPX4) across the experimental conditions.
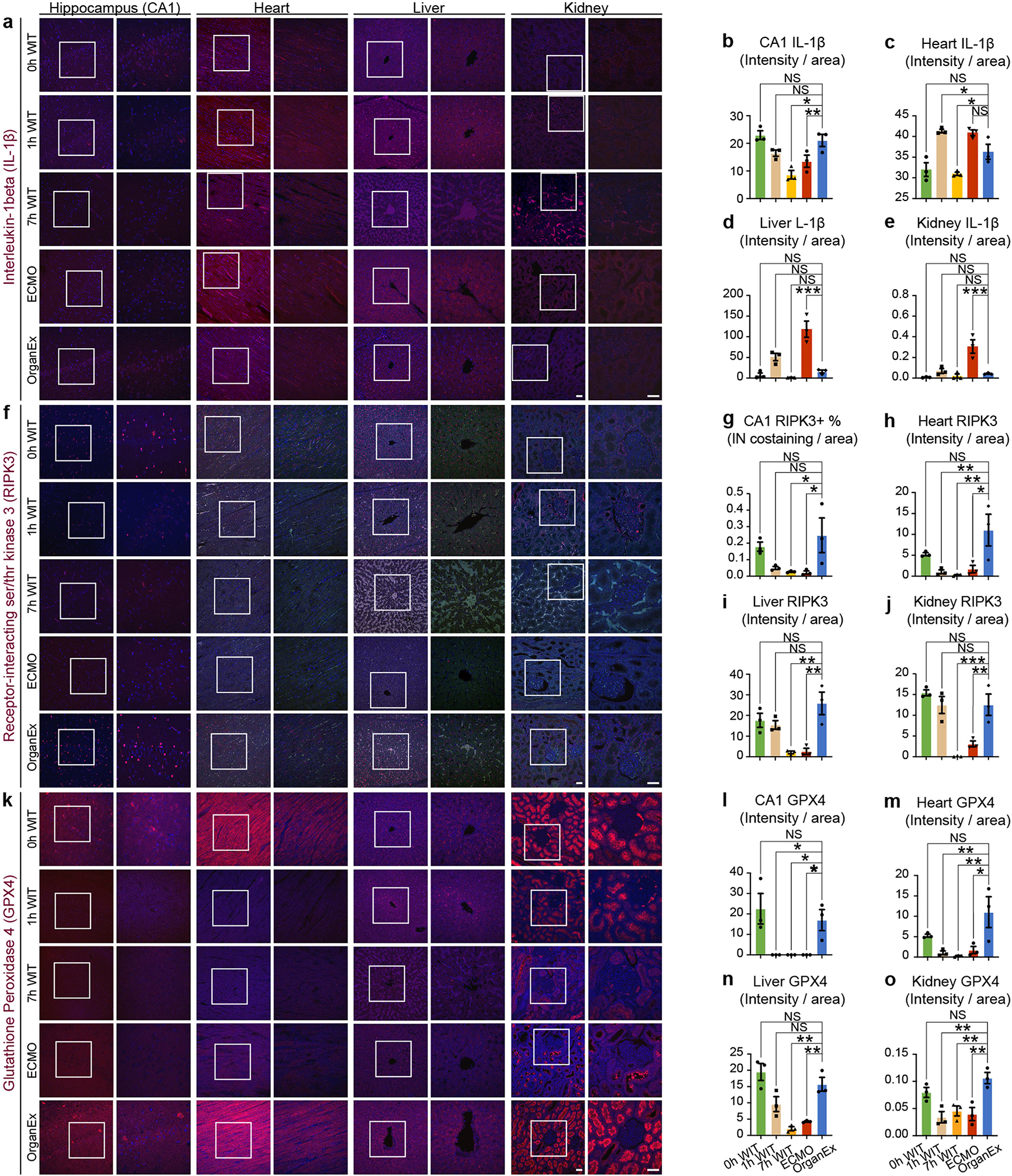
a, f, k, Representative confocal images of immunofluorescent staining for pyroptosis marker IL-1B, necroptosis marker RIPK3, and ferroptosis marker GPX4, each co-stained with DAPI nuclear stain in CA1, heart, liver, and kidney. b-e, Quantification of IL-1B immunolabeling signal intensity in CA1 (b), heart (c), liver (d), and kidney (e). n = 3. g-j, Quantification of RIPK3 positive intranuclear co-staining in CA1 (g), and immunolabeling signal intensity heart (h), liver (i), kidney (j). n = 3. l-o, Quantification of GPX4 immunolabeling signal intensity in CA1 (l), heart (m), liver (n), and kidney (o). n = 3. Scale bars, 50 μm left and right panels. Data presented are mean ± s.e.m. One-way ANOVA with post-hoc Dunnett’s adjustments was performed. For more detailed information on statistics and reproducibility, see methods. *P < 0.05, **P < 0.01, ***P < 0.001. IN, intranuclear.
Extended Data Fig. 6 |. EEG setup and recordings, click-iT chemistry and immunohistochemical analysis of factor V and troponin I.
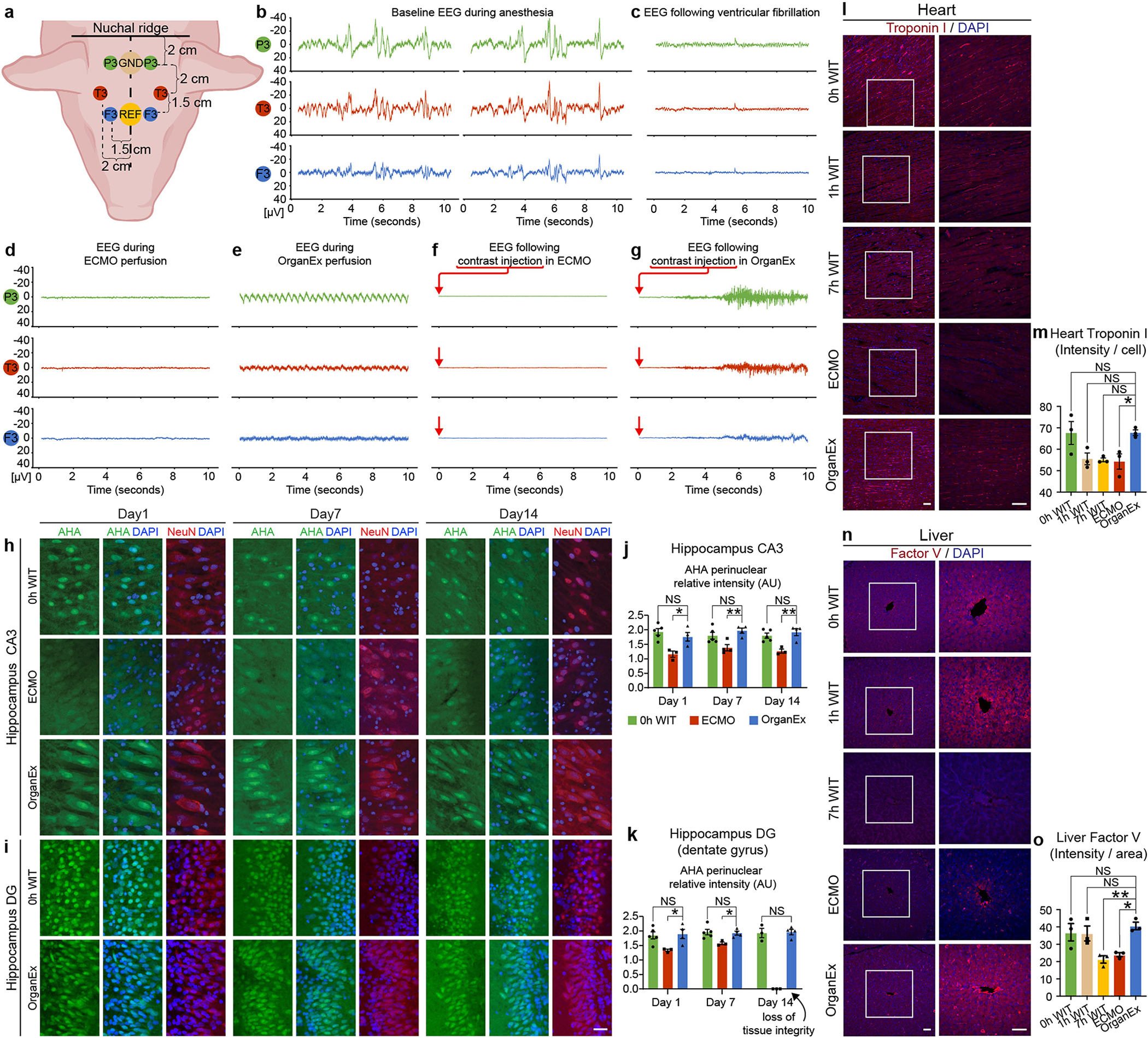
a, Placement of EEG electrodes on the porcine scalp. b, Representative snapshot of the EEG recordings after administration of anaesthesia and before the induction of cardiac arrest by ventricular fibrillation. c, Representative snapshot of the EEG recordings immediately following the ventricular fibrillation. d, Representative snapshot of the EEG during ECMO intervention at around 2h of perfusion protocol. e, Representative snapshot of the EEG during OrganEx intervention at around 2h of perfusion protocol, showing a light pulsatile artefact. f, g, Representative snapshot of the EEG recordings following contrast injection at 3h in ECMO and OrganEx animals, respectively. OrganEx EEG snapshot is consistent with a possible muscle-movement artefact. GND, ground electrode; REF, reference electrode. h, i, Representative confocal images of AHA through Click-iT chemistry in newly synthesized proteins with DAPI nuclear stain in the long-term organotypic hippocampal slice culture in CA3 (h) and DG (i) subregions. j, k, Relative intensity of nascent protein around nuclei in hippocampal CA3 (j) and DG (k) region showing comparable protein synthesis between OrganEx and 0h WIT up to 14 days in culture. n = 3–5. l, Representative confocal images of immunofluorescent staining for troponin I in the heart. m, Quantification of troponin I immunolabeling signal intensity in heart. A decreased trend in immunolabeling intensity was observed with ischaemia time and a significant decrease in immunolabeling intensity in ECMO compared to the OrganEx group. n = 3. n, Representative confocal images of immunofluorescent staining for factor V in liver. o, Quantification of factor V immunolabeling signal intensity in liver follows a similar pattern seen with other organs with comparable results between 0h WIT, 1h WIT, and OrganEx group and a significant decrease in 7h WIT and ECMO groups. n = 3. Scale bars, 50 μm. Data presented are mean ± s.e.m. For more detailed information on statistics and reproducibility, see methods. *P < 0.05, **P < 0.01. AU, arbitrary units.
Extended Data Fig. 7 |. Quality control of snRNA-seq data in healthy and varying ischaemic conditions in the hippocampus, heart, liver, and kidney.

Through transcriptomic integration and iterative clustering, we generated a taxonomy of t-types in healthy organs and brain, heart, liver, and kidney that experienced ischaemia (1h WIT, 7h WIT, ECMO and OrganEx), representing presumptive major cell types across organs of interest. These major t-types were further subdivided into high-resolution subclusters that were transcriptomically comparable to publicly available human single-cell datasets and that were marked by distinct expression profiles (c-f)51–54. a, Bar plot showing the number of cells/nuclei across organs and biological replicates. b, Violin plot showing the distribution of the number of unique molecular identifiers – UMIs (upper panel) and genes (lower panel) detected across all biological replicates. c-f, respective analyses of snRNA-seq in the hippocampus (c), heart (d), liver (e), and kidney (f). The left upper corner depicts detailed UMAP layout showing all t-types in the respective organs. The right side depicts the expression of top t-type markers. The left lower corner depicts transcriptomic correlation between the t-type taxonomy defined in this study and that of previous human and mouse datasets51–54. c., cells; LSECs, liver sinusoidal endothelial cells; end., endothelium; prog., progenitor; perit., peritubular; TAL, thick ascending limb; dend., dendritic; CNT, connecting tubule.
Extended Data Fig. 8 |. Single-nucleus transcriptome analysis in healthy and varying ischaemic conditions in the hippocampus, heart, liver, and kidney.
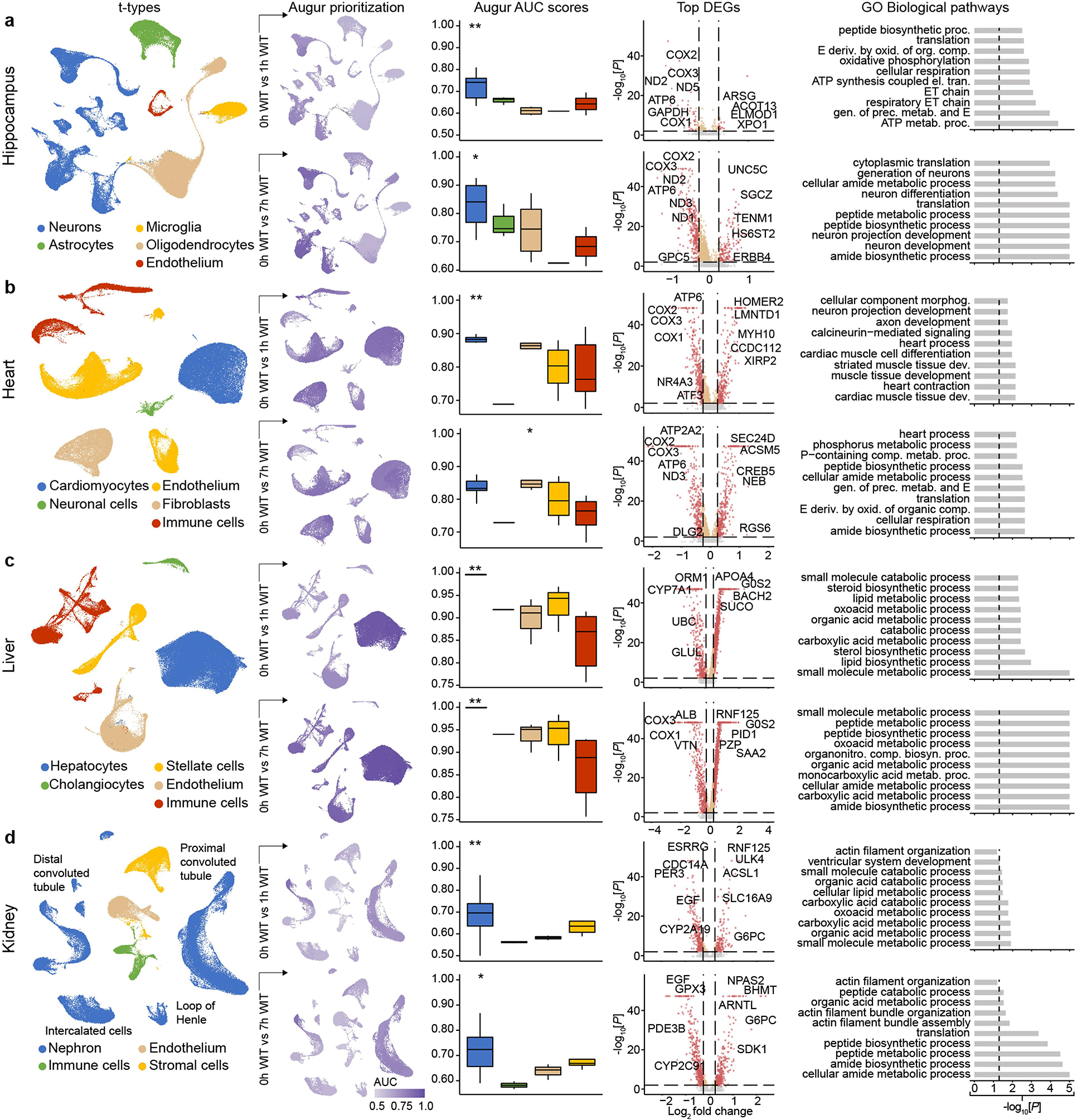
a-d, From left to right: UMAP layout showing major t-types; UMAP layout, coloured by Augur cell type prioritization (AUC) between 0h WIT compared to 1h (up) and 7h WIT (down); statistical comparison of Augur AUC scores between 0h WIT and 1h (up) and 7h (down) of WIT; Volcano plot showing top DEGs in major annotated t-types between 0h and 1h WIT (up), or 0h and 7h WIT (down); GO terms associated with the genes up and downregulated in detected nuclei between 0h and 1h WIT (up), or 0h and 7h WIT (down) with their nominal P-value in respective major annotated t-types.
Extended Data Fig. 9 |. Hippocampal single-nucleus transcriptome analysis comparing OrganEx to other experimental conditions.
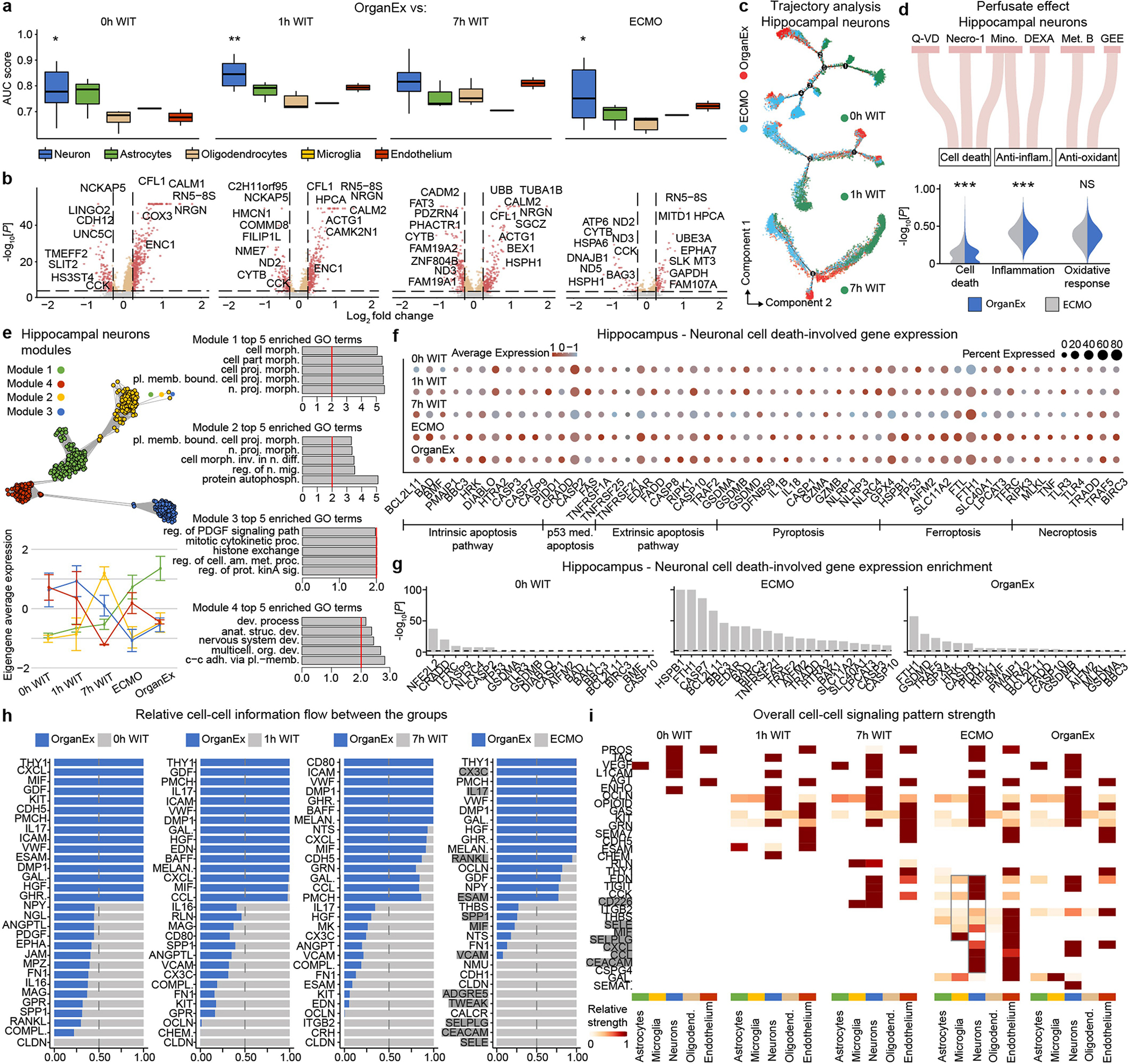
a, AUC scores of the Augur cell type prioritization between OrganEx and other groups. b, Volcano plot showing DEGs in hippocampal neurons between OrganEx and 0h WIT, 1h WIT, 7h WIT, and ECMO. c, Trajectories of hippocampal neurons. Colour indicates different experimental groups. d, Sankey plot showing perfusate components and violin plots showing their effects on hippocampal neurons between the OrganEx and ECMO groups. e, Hierarchical clustering of the top DEGs across experimental groups and derived functional gene modules (upper left). Eigengene average expression trends exhibit distinct trends between ECMO and OrganEx groups (lower left) of modules whose enriched GO terms are predominantly related to cellular function (right) (Supplementary Table 5). f, Expression of the genes involved in cell-death pathways in neurons. g, Gene expression enrichment of the genes involved in cell-death pathways in neurons. h, Stacked bar plot showing relative information flow for each signalling pa pathway across experimental group pairs. Significant signalling pathways were ranked based on differences in the overall information flow within the inferred networks between OrganEx and 0h WIT, 1h WIT, 7h WIT, and ECMO. Genes important in inflammation are highlighted grey. i, Overall signalling patterns across all experimental conditions. Genes important in inflammation are highlighted grey. Necro-1, necrostatin-1; Mino, minocycline; DEXA, dexamethasone; Met. B, methylene blue; GEE, Glutathione Ethyl Ester. *P < 0.05, **P < 0.01, ***P < 0.001, NS: not significant.
Extended Data Fig. 10 |. Heart single-nucleus transcriptome analysis comparing OrganEx to other experimental conditions.
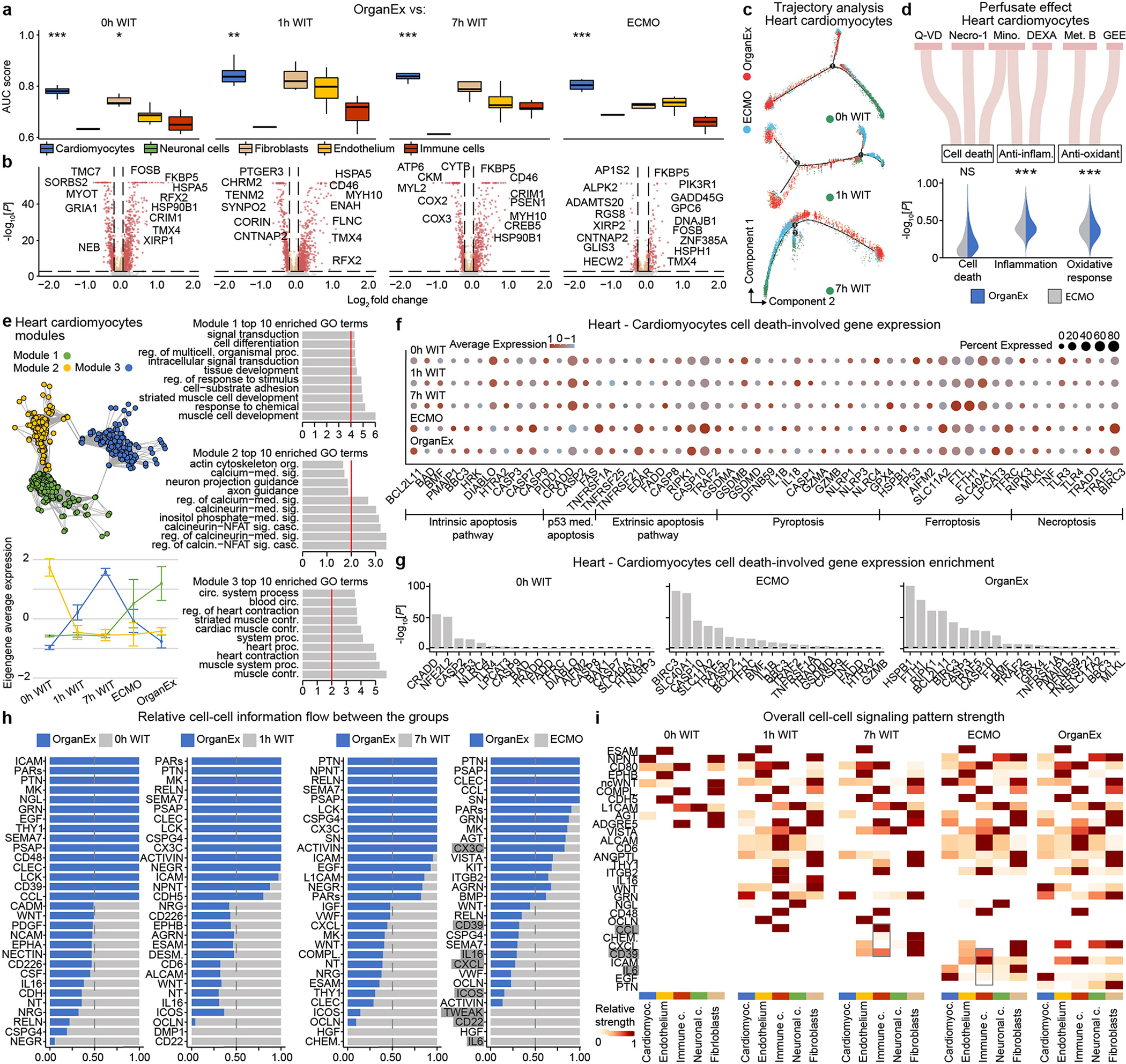
a, AUC scores of the Augur cell type prioritization between OrganEx and other groups. b, Volcano plot showing the DEGs in cardiomyocytes between OrganEx and 0h WIT, 1h WIT, 7h WIT, and ECMO. c, Trajectories of heart cardiomyocytes. Colour indicates different experimental groups. d, Sankey plot showing perfusate components and violin plots showing their effects on cardiomyocytes between the OrganEx and ECMO groups. e, Hierarchical clustering of the top DEGs across experimental groups and derived functional gene modules (upper left). Eigengene average expression trends exhibit distinct trends between ECMO and OrganEx groups (lower left) of modules whose enriched GO terms are predominantly related to cellular function (right) (Supplementary Table 5). f, Expression of the genes involved in cell-death pathways in cardiomyocytes. g, Gene expression enrichment of the genes involved in cell-death pathways in cardiomyocytes. h, Stacked bar plot showing relative information flow for each signalling pathway across experimental group pairs. Significant signalling pathways were ranked based on differences in the overall information flow within the inferred networks between OrganEx and 0h WIT, 1h WIT, 7h WIT, and ECMO. Genes important in inflammation are highlighted grey. i, Overall signalling patterns across all experimental conditions. Genes important in inflammation are highlighted grey. Necro-1, necrostatin-1; Mino, minocycline; DEXA, dexamethasone; Met. B, methylene blue; GEE, Glutathione Ethyl Ester. *P < 0.05, **P < 0.01, ***P < 0.001, NS: not significant.
Extended Data Fig. 11 |. Liver single-nucleus transcriptome analysis comparing OrganEx to other experimental conditions.
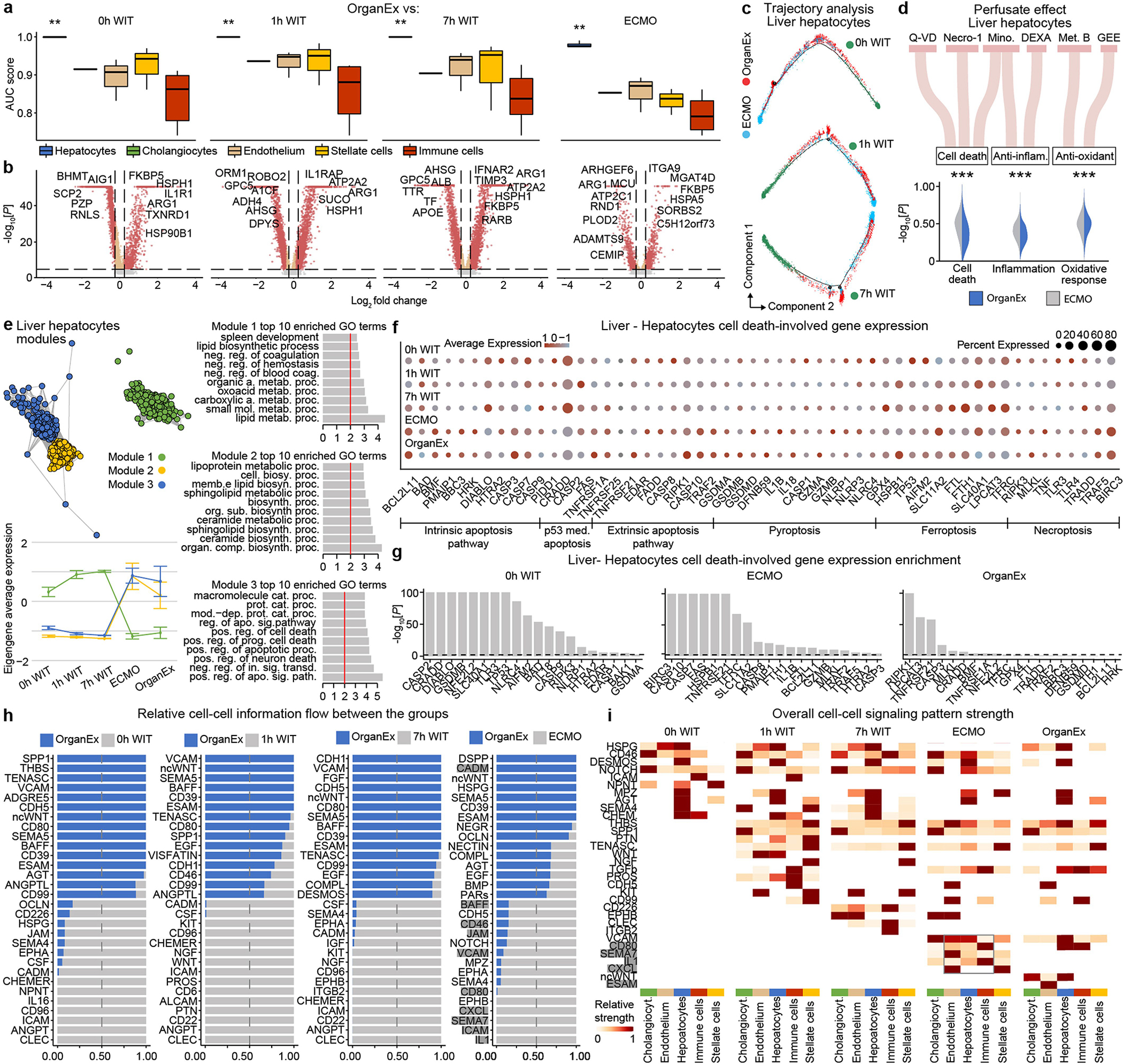
a, AUC scores of the Augur cell type prioritization between OrganEx and other groups. b, Volcano plot showing DEGs in hepatocytes between OrganEx and 0h WIT, 1h WIT, 7h WIT, and ECMO. c, Trajectories of liver hepatocytes. Colour indicates different experimental groups. d, Sankey plot showing perfusate components and violin plots showing their effects on hepatocytes between the OrganEx and ECMO. e, Hierarchical clustering of the top DEGs across experimental groups and derived functional gene modules (upper left). Eigengene average expression trends exhibit distinct trends between ECMO and OrganEx groups (lower left) of modules whose enriched GO terms are predominantly related to cellular function or cell death (right) (Supplementary Table 5). f, Expression of the genes involved in cell-death pathways in hepatocytes. g, Gene expression enrichment of the genes involved in cell-death pathways in hepatocytes. h, Stacked bar plot showing relative information flow for each signalling pathway across experimental group pairs. Significant signalling pathways were ranked based on differences in the overall information flow within the inferred networks between OrganEx and 0h WIT, 1h WIT, 7h WIT, and ECMO. Genes important in inflammation are highlighted grey. i, Overall signalling patterns across all experimental conditions. Genes important in inflammation are highlighted grey. Necro-1, necrostatin-1; Mino, minocycline; DEXA, dexamethasone; Met. B, methylene blue; GEE, Glutathione Ethyl Ester. DEXA, dexamethasone; Met. B, methylene blue; GEE, Glutathione Ethyl Ester. *P < 0.05, **P < 0.01, ***P < 0.001, NS: not significant.
Extended Data Fig. 12 |. Kidney single-nucleus transcriptome analysis comparing OrganEx to other experimental conditions.
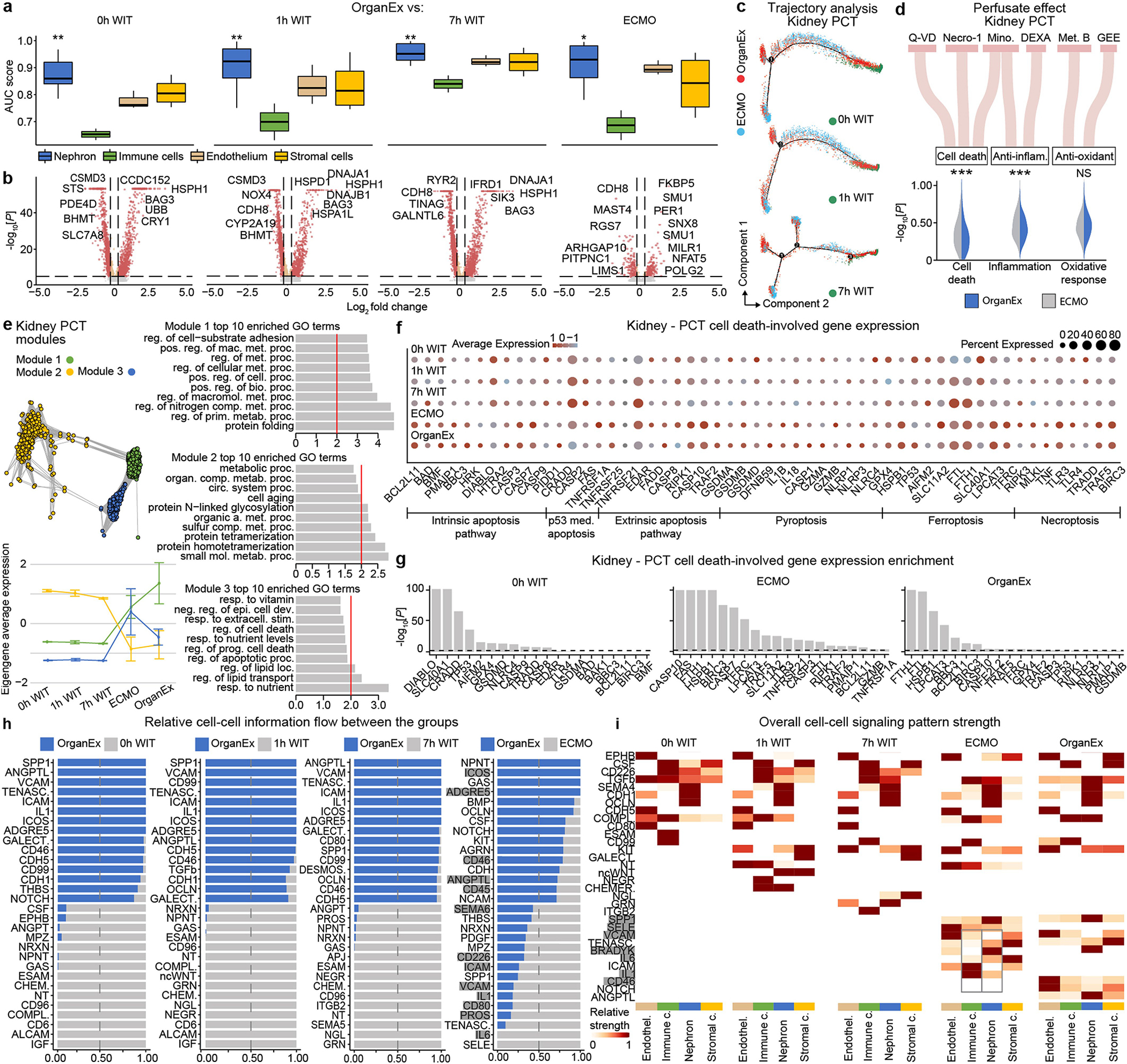
a, AUC scores of the Augur cell type prioritization between OrganEx and other groups. b, Volcano plot showing DEGs in PCT between OrganEx and 0h WIT, 1h WIT, 7h WIT, and ECMO. c, Trajectories of kidney PCTs. Colour indicates pseudotime progression and different cell states, respectively. d, Sankey plot showing perfusate components and violin plots showing their effects on PCT between the OrganEx and ECMO groups. e, Hierarchical clustering of the top DEGs across experimental groups and derived functional gene modules (upper left). Eigengene average expression trends exhibit distinct trends between ECMO and OrganEx groups (lower left) of modules whose enriched GO terms are predominantly related to cellular function or cell death (right) (Supplementary Table 5). f, Expression of the genes involved in cell-death pathways in PCT. g, Gene expression enrichment of the genes involved in cell-death pathways in PCT. h, Stacked bar plot showing relative information flow for each signalling pathway across experimental group pairs. Significant signalling pathways were ranked based on differences in the overall information flow within the inferred networks between OrganEx and 0h WIT, 1h WIT, 7h WIT, and ECMO. Genes important in inflammation are highlighted grey. i, Overall signalling patterns across all experimental conditions. Genes important in inflammation are highlighted grey. PCT, proximal convoluted tubules; DCT, distal convoluted tubules; Necro-1, necrostatin-1; Mino, minocycline; DEXA, dexamethasone; Met. B, methylene blue; GEE, Glutathione Ethyl Ester. *P < 0.05, **P < 0.01, ***P < 0.001, NS: not significant.
Supplementary Material
Acknowledgements
We thank the staff at HbO2 Therapeutics for providing the Hemopure product; S. G. Waxman for providing us with insights into central nervous system assessments; N. Guerrera, C. Hawley, M. Mamarian and C. Romero for their help in the operating room; T. Wing for assistance with the EEG; C. Booth, A. Brooks, A. Nugent, G. Terwilliger and M. Schadt for help with histopathology and staining; T. Rajabipour for help with the perfusion circuit; P. Heerdt for help with animal perfusions; K. Henderson for assistance with slide imaging; R. Khozein for providing us with EEG equipment; the members of the external advisory and ethics committee for assistance and guidance throughout this research; various members of our laboratory community for their comments on the manuscript; and the staff at the Yale Macaque Brain Resource (grant to A. Duque, NIMH R01MH113257) for the use of the Aperio CS2 scanner. This work was supported by the NIH BRAIN Initiative grants MH117064, MH117064–01S1, R21DK128662, T32GM136651, F30HD106694 and Schmidt Futures.
Footnotes
Competing interests D.A., Z.V. and N.S. have disclosed these findings to the Yale Office of Cooperative Research, which has filed a patent to ensure broad use of the technology. All protocols, methods, perfusate formulations and components of the OrganEx technology remain freely available for academic and non-profit research. Although the Hemopure product was provided in accordance with a material transfer agreement between HbO2 Therapeutics and Yale University through N.S., the Company had no influence on the study design or interpretation of the results. No author has a financial stake in, or receives compensation from, HbO2 Therapeutics.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-022-05016-1.
Peer review information Nature thanks Amir Bashan, Rafael Kramann and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Reprints and permissions information is available at http://www.nature.com/reprints.
References
- 1.Lee P, Chandel NS & Simon MC Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 21, 268–283 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daniele SG et al. Brain vulnerability and viability after ischaemia. Nat. Rev. Neurosci. 22, 553–572 (2021). [DOI] [PubMed] [Google Scholar]
- 3.Vrselja Z et al. Restoration of brain circulation and cellular functions hours post-mortem. Nature 568, 336–343 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsia CC, Schmitz A, Lambertz M, Perry SF & Maina JN Evolution of air breathing: oxygen homeostasis and the transitions from water to land and sky. Compr. Physiol. 3, 849–915 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eltzschig HK & Eckle T Ischemia and reperfusion-from mechanism to translation. Nat. Med. 17, 1391–1401 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iadecola C, Buckwalter MS & Anrather J Immune responses to stroke: mechanisms, modulation, and therapeutic potential. J. Clin. Invest. 130, 2777–2788 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trump BF & Harris CC Human tissues in biomedical research. Hum. Pathol. 10, 245–248 (1979). [DOI] [PubMed] [Google Scholar]
- 8.Brasile L et al. Overcoming severe renal ischemia: the role of ex vivo warm perfusion. Transplantation 73, 897–901 (2002). [DOI] [PubMed] [Google Scholar]
- 9.García Sáez D et al. Ex vivo heart perfusion after cardiocirculatory death; a porcine model. J. Surg. Res. 195, 311–314 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Schön MR et al. Liver transplantation after organ preservation with normothermic extracorporeal perfusion. Ann. Surg. 233, 114–123 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Charles EJ et al. Ex vivo assessment of porcine donation after circulatory death lungs that undergo increasing warm ischemia times. Transplant Direct 4, e405 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taunyane IC et al. Preserved brain morphology after controlled automated reperfusion of the whole body following normothermic circulatory arrest time of up to 20 minutes. Eur. J. Cardiothorac. Surg. 50, 1025–1034 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Grunau B et al. Comparing the prognosis of those with initial shockable and non-shockable rhythms with increasing durations of CPR: informing minimum durations of resuscitation. Resuscitation 101, 50–56 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Lequier L, Horton SB, McMullan DM & Bartlett RH Extracorporeal membrane oxygenation circuitry. Pediatr. Crit. Care Med. 14, S7–S12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirino T Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 239, 57–69 (1982). [DOI] [PubMed] [Google Scholar]
- 16.Pulsinelli WA, Brierley JB & Plum F Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann. Neurol. 11, 491–498 (1982). [DOI] [PubMed] [Google Scholar]
- 17.Unal-Cevik I, Kilinç M, Gürsoy-Ozdemir Y, Gurer G & Dalkara T Loss of NeuN immunoreactivity after cerebral ischemia does not indicate neuronal cell loss: a cautionary note. Brain Res. 1015, 169–174 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Kroemer G et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 16, 3–11 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang PL et al. Kidney injury molecule-1 expression in transplant biopsies is a sensitive measure of cell injury. Kidney Int. 73, 608–614 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nadasdy T, Laszik Z, Blick KE, Johnson LD & Silva FG Proliferative activity of intrinsic cell populations in the normal human kidney. J. Am. Soc. Nephrol. 4, 2032–2039 (1994). [DOI] [PubMed] [Google Scholar]
- 21.Dunn AF, Catterton MA, Dixon DD & Pompano RR Spatially resolved measurement of dynamic glucose uptake in live ex vivo tissues. Anal. Chim. Acta 1141, 47–56 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fishbein MC, Wang T, Matijasevic M, Hong L & Apple FS Myocardial tissue troponins T and I. An immunohistochemical study in experimental models of myocardial ischemia. Cardiovasc. Pathol. 12, 65–71 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Brown DJ, Brugger H, Boyd J & Paal P Accidental hypothermia. N. Engl. J. Med. 367, 1930–1938 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Guluma KZ et al. Therapeutic hypothermia is associated with a decrease in urine output in acute stroke patients. Resuscitation 81, 1642–1647 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Villa G, Katz N & Ronco C Extracorporeal membrane oxygenation and the kidney. Cardiorenal Med. 6, 50–60 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tujjar O et al. Acute kidney injury after cardiac arrest. Crit. Care 19, 169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dieterich DC et al. In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat. Neurosci. 13, 897–905 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Movahed M, Brockie S, Hong J & Fehlings MG Transcriptomic hallmarks of ischemia-reperfusion injury. Cells 10, 1838 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang J et al. Effects of ischemia on gene expression. J. Surg. Res. 99, 222–227 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Molenaar B et al. Single-cell transcriptomics following ischemic injury identifies a role for B2M in cardiac repair. Commun. Biol. 4, 146 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Androvic P et al. Decoding the transcriptional response to ischemic stroke in young and aged mouse brain. Cell Rep. 31, 107777 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Ferreira PG et al. The effects of death and post-mortem cold ischemia on human tissue transcriptomes. Nat. Commun. 9, 490 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kirita Y, Wu H, Uchimura K, Wilson PC & Humphreys BD Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proc. Natl Acad. Sci. USA 117, 15874–15883 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skinnider MA et al. Cell type prioritization in single-cell data. Nat. Biotechnol. 39, 30–34 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jurga AM, Paleczna M & Kuter KZ Overview of general and discriminating markers of differential microglia phenotypes. Front. Cell Neurosci. 14, 198 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liddelow SA et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lopaschuk GD & Stanley WC Glucose metabolism in the ischemic heart. Circulation 95, 313–315 (1997). [DOI] [PubMed] [Google Scholar]
- 38.Langfelder P & Horvath S WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jin S et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Markmann JF et al. Impact of portable normothermic blood-based machine perfusion on outcomes of liver transplant: the OCS Liver PROTECT randomized clinical trial. JAMA Surg. 157, 189–198 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Carlis R et al. How to preserve liver grafts from circulatory death with long warm ischemia? A retrospective Italian cohort study with normothermic regional perfusion and hypothermic oxygenated perfusion. Transplantation 105, 2385–2396 (2021). [DOI] [PubMed] [Google Scholar]
- 42.Smith DE et al. Early experience with donation after circulatory death heart transplantation using normothermic regional perfusion in the United States. J. Thorac. Cardiovasc. Surg. 164, 557–568.e1 (2022). [DOI] [PubMed] [Google Scholar]
- 43.Sellers MT et al. Early United States experience with liver donation after circulatory determination of death using thoraco-abdominal normothermic regional perfusion: a multi-institutional observational study. Clin. Transplant. 36, e14659 (2022). [DOI] [PubMed] [Google Scholar]
- 44.De Beule J et al. A systematic review and meta-analyses of regional perfusion in donation after circulatory death solid organ transplantation. Transpl. Int. 34, 2046–2060 (2021). [DOI] [PubMed] [Google Scholar]
- 45.De Charrière A et al. ECMO in cardiac arrest: a narrative review of the literature. J. Clin. Med. 10, 534 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu Y et al. Spatiotemporal transcriptomic divergence across human and macaque brain development. Science 362, eaat8077 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li M et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362, eaat7615 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng GX et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stuart T et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kobak D & Linderman GC Initialization is critical for preserving global data structure in both t-SNE and UMAP. Nat. Biotechnol. 39, 156–157 (2021). [DOI] [PubMed] [Google Scholar]
- 51.Stewart BJ et al. Spatiotemporal immune zonation of the human kidney. Science 365, 1461–1466 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Litviňuková M et al. Cells of the adult human heart. Nature 588, 466–472 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Franjic D et al. Transcriptomic taxonomy and neurogenic trajectories of adult human, macaque, and pig hippocampal and entorhinal cells. Neuron 110, 452–469 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.MacParland SA et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 9, 4383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hao Y et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blighe K, Rana S, Lewis M EnhancedVolcano: publication-ready volcano plots with enhanced colouring and labeling (2018); https://github.com/kevinblighe/EnhancedVolcano
- 57.Yu G, Wang LG, Han Y & He QY clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Laposata M Laboratory Medicine: The Diagnosis of Disease in the Clinical Laboratory 364 (McGraw-Hill Education, 2012). [Google Scholar]
- 59.Lee JW, Chou C-L & Knepper MA Deep sequencing in microdissected renal tubules identifies nephron segment–specific transcriptomes. J. Am. Soc. Nephrol. 26, 2669–2677 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cavalcante GC et al. A cell’s fate: an overview of the molecular biology and genetics of apoptosis. Int. J. Mol. Sci. 20, 4133 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu P et al. Pyroptosis: mechanisms and diseases. Signal Transduct. Target. Ther. 6, 128 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li J et al. Ferroptosis: past present and future. Cell Death Dis. 11, 88 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dhuriya YK & Sharma D Necroptosis: a regulated inflammatory mode of cell death. J. Neuroinflammation 15, 199 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alexa A & Rahnenfuhrer J topGO: enrichment analysis for Gene Ontology. R package version 2.48.0 (2022). [Google Scholar]
- 65.Aibar S et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Trapnell C et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McQuin C et al. CellProfiler 3.0: next-generation image processing for biology. PLoS Biol. 16, e2005970 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Edgar R, Domrachev M & Lash AE Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The snRNA-seq dataset was deposited at the NCBI’s Gene Expression Omnibus68 and is accessible through GEO Series accession number GSE183448.