

Abstract
Background:
PDGFRA D842V mutations occur in 5–10% of gastrointestinal stromal tumours (GISTs), and previously approved tyrosine kinase inhibitors (TKIs) are inactive against this mutation. Consequently, patients have a poor prognosis. We present an updated analysis of avapritinib efficacy and long-term safety in this patient population.
Methods:
NAVIGATOR (NCT02508532), a two-part, open-label, dose-escalation/dose-expansion phase I study, enrolled adult patients with unresectable GISTs. Patients with PDGFRA D842V-mutant GIST were a prespecified subgroup within the overall safety population, which included patients who received ≥1 avapritinib dose. Primary end-points were overall response rate (ORR) and avapritinib safety profile. Secondary end-points were clinical benefit rate (CBR), duration of response (DOR) and progression-free survival (PFS). Overall survival (OS) was an exploratory end-point.
Results:
Between 7 October 2015 and 9 March 2020, 250 patients enrolled in the safety population; 56 patients were included in the PDGFRA D842V population, 11 were TKI-naïve. At data cut-off, median follow-up was 27.5 months. Safety profile was comparable between the overall safety and PDGFRA D842V populations. In the PDGFRA D842V population, the most frequent adverse events were nausea (38 [68%] patients) and diarrhoea (37 [66%]), and cognitive effects occurred in 32 (57%) patients. The ORR was 91% (51/56 patients). The CBR was 98% (55/56 patients). The median DOR was 27.6 months (95% confidence interval [CI]: 17.6–not reached [NR]); median PFS was 34.0 months (95% CI: 22.9–NR). Median OS was not reached.
Conclusion:
Targeting PDGFRA D842V-mutant GIST with avapritinib resulted in an unprecedented, durable clinical benefit, with a manageable safety profile. Avapritinib should be considered as first-line therapy for these patients.
Keywords: Avapritinib, Gastrointestinal stromal tumours, Phase 1, PDGFRA
1. Introduction
Over 85% of gastrointestinal stromal tumours (GISTs) are driven by oncogenic mutations in genes encoding KIT or platelet-derived growth factor receptor A (PDGFRA) receptor tyrosine kinases [1,2]. Between 5% and 6% of GISTs harbour PDGFRA D842V mutations. Patients with unresectable or metastatic PDGFRA D842V-mutant GIST have a poor prognosis, as imatinib and other approved kinase inhibitors have no proven efficacy for this mutation [3–6]. Studies of these agents have shown median progression-free survival (PFS) of 3–6 months and median overall survival (OS) of approximately 15 months [5,7,8]. These factors highlight the unmet need for patients with advanced PDGFRA D842V-mutant GIST.
Avapritinib (BLU-285, Blueprint Medicines Corporation, Cambridge, Massachusetts, USA) is a selective, potent inhibitor of mutated KIT and PDGFRA. Avapritinib is approved in the USA for treatment of adults with unresectable or metastatic GIST harbouring a PDGFRA exon 18 mutation, including PDGFRA D842V mutations, and in the European Union for adults with unresectable or metastatic GIST harbouring a PDGFRA D842V mutation [9,10]. Approvals were based on previously published findings from the NAVIGATOR study (NCT02508532). The maximum tolerated dose (MTD) determined in NAVIGATOR part 1 was avapritinib 400 mg once daily (QD); the recommended phase II dose (RP2D) was 300 mg QD [11].
As responses to avapritinib have proved durable in this population compared with other approved tyrosine kinase inhibitors (TKIs), long-term toxicities have become more clinically relevant. As such, we present updated, longer-term analyses of avapritinib safety from NAVIGATOR parts 1 and 2 and efficacy in patients with PDGFRA D842V-mutant GIST.
2. Materials and methods
2.1. Study design and patient population
NAVIGATOR (NCT02508532), a two-part, open-label, non-randomised, dose-escalation/dose-expansion phase I study conducted at 17 sites across nine countries [11], evaluated avapritinib safety and efficacy in patients with unresectable or metastatic GIST. Part 1 (dose escalation) enrolled patients with unresectable or metastatic GIST, with a starting dose of 30 mg QD, increasing until the MTD was reached. TKI-naïve patients with PDGFRA-mutant GIST were permitted in part 1, as no treatments were approved for this patient population. Part 2 (dose expansion) enrolled patients with unresectable or metastatic GIST in three prespecified groups (Fig. 1). Patients enrolled in part 2 received a 400 mg QD starting dose (MTD), subsequently reduced to 300 mg QD, identified as the RP2D.
Fig. 1.
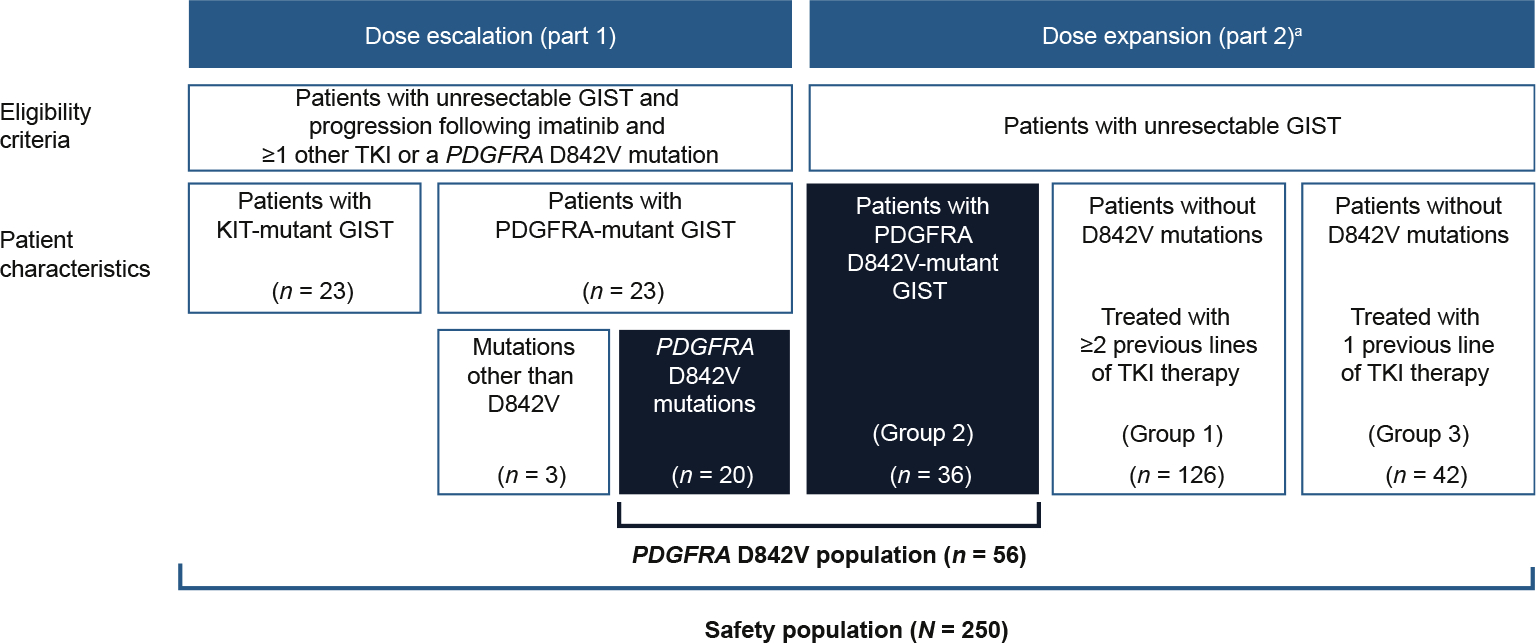
Patient disposition, aEnrolment at data cut-off 9 March 2020. GIST, gastrointestinal stromal tumour; PDGFRA, platelet-derived growth factor receptor A; TKI, tyrosine kinase inhibitor.
Inclusion criteria were as specified in Heinrich et al. [11]. Part 1 primary end-points were determination of MTD/RP2D of avapritinib and safety profile. Part 2 primary end-points were evaluation of overall response rate (ORR) in prespecified cohorts and avapritinib safety profile. Secondary end-points included clinical benefit rate (CBR), duration of response (DOR) and PFS. OS was a prespecified exploratory end-point. All end-points were previously defined in Heinrich et al. [11].
Tumour mutation status was determined by local testing and centrally confirmed using DNA from archival or new tumour biopsy samples. NAVIGATOR was performed in accordance with the ethical principles of the Declaration of Helsinki and was consistent with the International Conference on Harmonisation of Good Clinical Practice. The institutional review board or independent ethics committee of each site approved the study. All patients provided written informed consent.
2.2. Statistical analysis
Data cut-off was 9 March 2020, more than 15 months after the original data cut-off [11]. The safety population included all patients from parts 1 and 2 who received ≥1 dose of avapritinib. The PDGFRA D842V population (prespecified subgroup) included patients in the safety population with PDGFRA D842V-mutant GIST. A sample size of 31 patients with PDGFRA D842V-mutant GIST allowed testing the null hypothesis of ORR ≤10% versus the alternative hypothesis of ORR ≥35% with 90% power, assuming two-sided type I error rate of 0.05.
Efficacy analyses in the PDGFRA D842V population included patients with ≥1 target lesion and ≥1 postbaseline disease assessment by central radiologic review. The primary end-point was assessed by central radiology per modified Response Evaluation Criteria in Solid Tumors v1.1; ORR was estimated using frequency, percentage and two-sided 95% confidence interval (CI) based on the Cloppere–Pearson exact binomial distribution. Kaplane–Meier methods were used to estimate DOR, PFS and OS at specific time points. Efficacy results were summarised by starting dose. A time-varying Cox regression model was used to estimate the effects of age, sex, baseline Eastern Cooperative Oncology Group (ECOG) performance status, prior TKIs (all fixed covariates) and avapritinib dose (50 mg QD increments; time-varying covariate, assessed continually) on PFS. All statistical analyses were performed with SAS (version 9.3 or higher).
3. Results
3.1. Patients
Between 7 October 2015 and 9 March 2020, 250 patients were enrolled in parts 1 and 2, received ≥1 dose of study drug, and were included in the safety population (Fig. 1), of whom 67% received a 300 mg starting dose. At data cut-off, 205 (82%) patients had discontinued treatment (Fig. 2), most frequently due to disease progression (128 [51%] patients) and adverse events (AEs) (54 [22%] patients), deemed treatment-related in 37 (15%) patients.
Fig. 2.
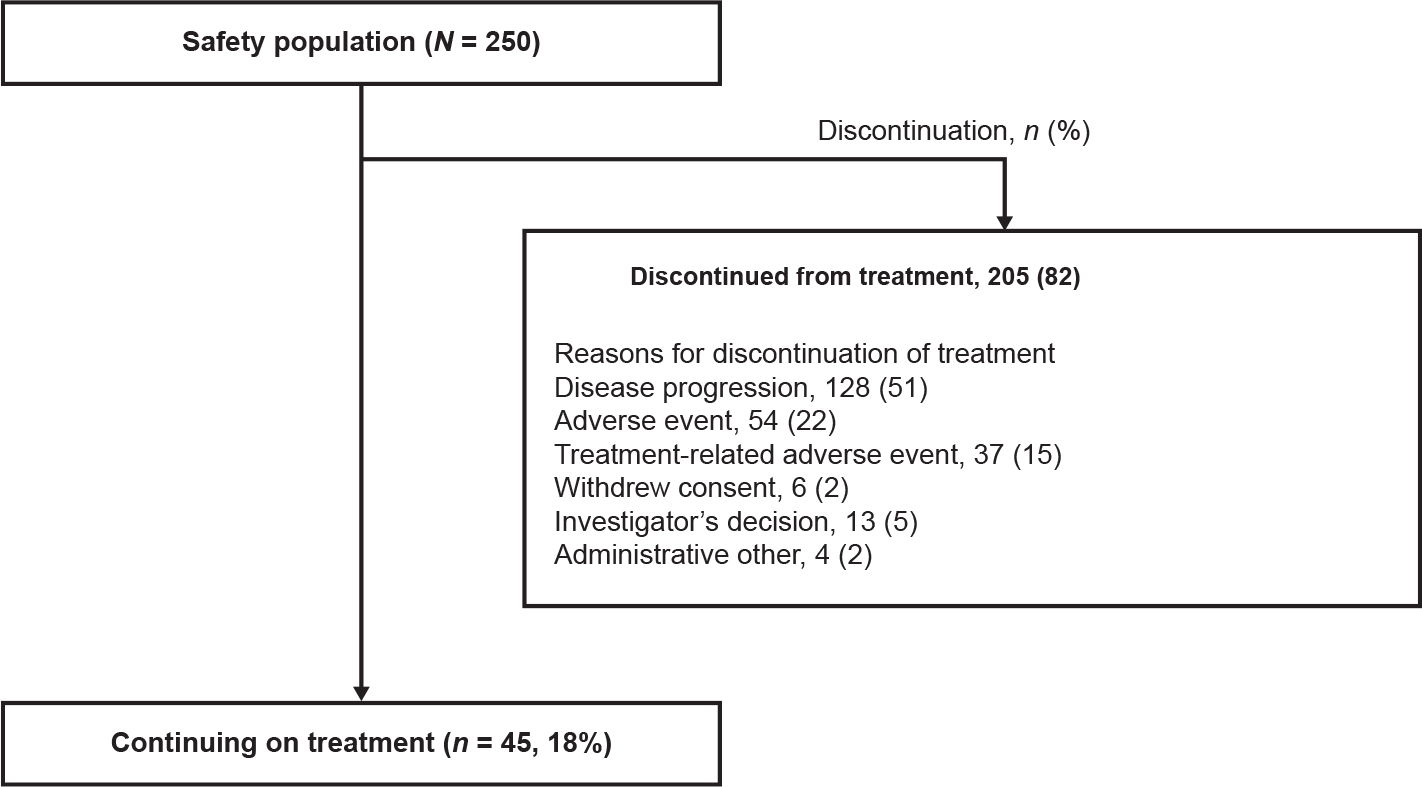
Treatment discontinuations in the safety population.
The PDGFRA D842V population included 56 patients; part 1 included 20 patients, part 2 included 36 patients. At data cut-off, median follow-up was 27.5 months for the PDGFRA D842V population, and 30 (54%) patients had discontinued treatment (Fig. A1). The most common reasons for treatment discontinuation were AEs (19 [34%] patients), deemed treatment-related in 13 (23%) patients and disease progression in 5 (9%) patients. Treatment-related AEs (TRAEs) leading to discontinuation were most commonly nervous system disorders (8 [14%]), psychiatric disorders (4 [7%]) and gastrointestinal disorders (1 [2%]).
In total, 38 patients with PDGFRA D842V-mutant GIST received an avapritinib 300/400 mg starting dose, 28 of whom received a 300 mg starting dose. In these patients, median follow-up was 25.5 months at the time of data cut-off; 19 (50%) discontinued treatment by data cut-off. Discontinuation was due to AEs in 13 (34%) patients, deemed treatment-related in 8 (21%) patients, and disease progression in 2 (5%) patients. TRAEs leading to discontinuation were most commonly nervous system disorders (4 [11%]), psychiatric disorders (3 [8%]) and gastrointestinal disorders (n = 1 [3%]).
Baseline demographics and clinical characteristics were similar between the PDGFRA D842V and safety populations (Table 1). Most patients in the PDGFRA D842V and safety populations had ≥1 prior TKI therapy at baseline (80% and 96%, respectively; Table A1). In the PDGFRA D842V population, 11 patients were TKI-naïve.
Table 1.
Baseline patient demographics and clinical characteristics by avapritinib starting dose in the PDGFRA D842V population.
| Demographic/characteristic | Avapritinib starting dose |
|||
|---|---|---|---|---|
| 300 mg (n = 28) | 400 mg (n = 10) | 300/400 mg (n = 38) | All dosesa (n = 56) | |
|
| ||||
| Age, years, median (range) | 63 (29–90) | 66 (35–70) | 64 (29–90) | 64 (25–90) |
| Age group, years, n (%) | ||||
| <65 | 18 (64) | 4 (40) | 22 (58) | 31 (55) |
| ≥65 | 10 (36) | 6 (60) | 16 (42) | 25 (45) |
| Sex, n (%) | ||||
| Female | 10 (36) | 3 (30) | 13 (34) | 17 (30) |
| Male | 18 (64) | 7 (70) | 25 (66) | 39 (70) |
| Race, n (%) | ||||
| Asian | 6 (21) | 0 | 6 (16) | 6 (11) |
| Black or African American | 3 (11) | 0 | 3 (8) | 4 (7) |
| White | 17 (61) | 8 (80) | 25 (66) | 39 (70) |
| Unknown | 1 (4) | 2 (20) | 3 (8) | 6 (11) |
| Other | 1 (4) | 0 | 1 (3) | 1 (2) |
| ECOG PS, n (%) | ||||
| 0 | 9 (32) | 4 (40) | 13 (34) | 21 (38) |
| 1 | 17 (61) | 6 (60) | 23 (61) | 32 (57) |
| 2 | 2 (7) | 0 | 2 (5) | 3 (5) |
| Primary tumour site of GIST, n (%) | ||||
| Stomach | 21 (75) | 8 (80) | 29 (76) | 46 (82) |
| Peritoneum | 3 (11) | 0 | 3 (8) | 3 (5) |
| Jejunum or ileum | 1 (4) | 0 | 1 (3) | 2 (4) |
| Omentum | 1 (4) | 1 (10) | 2 (5) | 2 (4) |
| Duodenum | 0 | 1 (10) | 1 (3) | 1 (2) |
| Rectum | 1 (4) | 0 | 1 (3) | 1 (2) |
| Colon | 1 (4) | 0 | 1 (3) | 1 (2) |
| Metastatic disease, n (%) | 27 (96) | 10 (100) | 37 (97) | 54 (96) |
| Largest target lesion size,b n (%) | ||||
| ≤5 cm | 12 (43) | 4 (40) | 16 (42) | 21 (38) |
| >5 to ≤10 cm | 9 (32) | 4 (40) | 13 (34) | 16 (29) |
| >10 cm | 7 (25) | 2 (20) | 9 (24) | 19 (34) |
| Stage at screening visit (TNM), n (%) | ||||
| Stage III | 1 (4) | 0 | 1 (3) | 2 (4) |
| Stage IV | 13 (46) | 6 (60) | 19 (50) | 29 (52) |
| Unknown | 14 (50) | 4 (40) | 18 (47) | 25 (45) |
ECOG PS, Eastern Cooperative Oncology Group performance status; GIST, gastrointestinal stromal tumour; PDGFRA, platelet-derived growth factor receptor A; TNM, tumour, node, metastasis.
Includes 17 patients with <300 mg starting doses and 1 patient with a 600 mg starting dose.
Lesion size by central radiographic assessment.
3.2. Safety
The avapritinib safety profiles in the PDGFRA D842V and safety populations were similar, despite longer median treatment duration in the PDGFRA D842V population (23.2 months [range = 1.2–52.0]) compared with the safety population (6.6 months [range = 0.2–52.0]; Table 2). TRAEs were reported by all patients in the PDGFRA D842V population and 98% of patients in the safety population (Table A2). In the PDGFRA D842V population, the most commonly reported any-cause AEs were nausea (38 [68%] of 56 patients), diarrhoea (37 [66%]) and anaemia (37 [66%]). In total, 54 (96%) patients had dose modifications (50 [89%] had dose interruptions, 41 [73%] had dose reductions). The safety profile in patients with PDGFRA D842V-mutant GIST receiving starting doses of 300/400 mg was reflective of the PDGFRA D842V population at all doses (Table A3).
Table 2.
Any-cause adverse events occurring in ≥20% of patients in the safety population and the PDGFRA D842V population.
| Preferred term, n (%) | PDGFRA D842V population (n = 56) | Safety population (N = 250) |
|---|---|---|
|
| ||
| Nausea | 38 (68) | 161 (64) |
| Fatigue | 35 (63) | 157 (63) |
| Anaemia | 37 (66) | 136 (54) |
| Diarrhoea | 37 (66) | 112 (45) |
| Periorbital oedema | 27 (48) | 110 (44) |
| Vomiting | 21 (38) | 106 (42) |
| Decreased appetite | 23 (41) | 101 (40) |
| Increased lacrimation | 21 (38) | 88 (35) |
| Memory impairment | 23 (41) | 81 (32) |
| Peripheral oedema | 21 (38) | 80 (32) |
| Abdominal pain | 19 (34) | 64 (26) |
| Constipation | 12 (21) | 64 (26) |
| Hair colour changes | 16 (29) | 62 (25) |
| Dizziness | 16 (29) | 59 (24) |
| Face oedema | 13 (23) | 57 (23) |
| Increased blood bilirubin | 16 (29) | 54 (22) |
| Hypokalaemia | 14 (25) | 48 (19) |
| Headache | 13 (23) | 48 (19) |
| Dysgeusia | 13 (23) | 47 (19) |
| Decreased weight | 15 (27) | 46 (18) |
| Dyspepsia | 13 (23) | 44 (18) |
| Cough | 15 (27) | 39 (16) |
| Neutropenia | 14 (25) | 29 (12) |
| Upper respiratory tract infection | 12 (21) | 27 (11) |
PDGFRA, platelet-derived growth factor receptor A.
Cognitive effects (combined term including memory impairment, confusional state, cognitive disorder, encephalopathy; any cause) and intracranial bleeding (combined term including intracranial haemorrhage, cerebral haemorrhage, subdural haematoma; any cause) were previously determined AEs of special interest [11]. In the safety population, any-grade cognitive effects occurred in 115 (46%) patients, while any-grade intracranial bleeding occurred in 7 (3%) patients. In the PDGFRA D842V population, any-grade cognitive effects occurred in 32 (57%) patients, while any-grade intracranial bleeding occurred in 3 (5%) patients (Table A4).
In the PDGFRA D842V population, deaths due to AEs were reported in 9 (16%) patients. One death was considered treatment-related (Table A5).
3.3. Overall response rate
In the PDGFRA D842V population ORR was 91% (51/56 patients; Table 3), irrespective of prior therapy. For 300/400 mg starting doses, ORR was 95% (36/38 patients; 5 [13%] patients achieved a complete response [CR], and 31 [82%] patients achieved a partial response [PR]; Fig. 3). Among the 27 patients receiving a 300 mg starting dose, ORR was 96% (95% CI: 82–100). Median time-to-first response across all doses was 61 days (range = 52–757) and was consistent at 300 mg and 400 mg starting doses (Table 3), but was numerically longer with starting doses <300 mg (91 days [range = 53–508]). At all doses, disease control rate was 100%. For TKI-naïve patients, ORR was 91% (10/11 patients); 3 (27%) patients achieved a CR and 7 (64%) patients achieved a PR. For TKI-naïve patients with a 300/400 mg starting dose, ORR was 100% (5/5 patients); 2 (40%) patients achieved a CR and 3 (60%) patients achieved a PR.
Table 3.
ORR and time-to-first response by starting dose in the PDGFRA D842V population.
| Avapritinib starting dose |
||||
|---|---|---|---|---|
| 300 mg (n = 28) | 400 mg (n = 10) | 300/400 mg (n = 38) | All dosesa (n = 56) | |
|
| ||||
| ORR,b n (%) | 27 (96) | 9 (90) | 36 (95) | 51 (91) |
| 95% CI | 82–100 | 56–100 | 82–99 | 80–97 |
| CR | 3 (11) | 2 (20) | 5 (13) | 7 (13) |
| PR | 24 (86) | 7 (70) | 31 (82) | 44 (79) |
| SD, n (%) | 1 (4) | 1 (10) | 2 (5) | 5 (9) |
| Progressive disease, n (%) | 0 | 0 | 0 | 0 |
| Clinical benefit rate,c n (%) | 28 (100) | 9 (90) | 37 (97) | 55 (98) |
| 95% CI | 88–100 | 56–100 | 86–100 | 90–100 |
| Disease control rate,d n (%) | 28 (100) | 10 (100) | 38 (100) | 56 (100) |
| 95% CI | 88–100 | 69–100 | 91–100 | 94–100 |
| Median time-to-first response,e days (range) | 61 (53–224) | 57 (52–757) | 60 (52–757) | 61 (52–757) |
CI, confidence interval; CR, complete response; mRECIST, modified Response Evaluation Criteria in Solid Tumors; ORR, overall response rate; PDGFRA, platelet-derived growth factor receptor A; PR, partial response; SD, stable disease.
Per mRECIST v1.1.
Includes 17 patients with <300 mg starting doses and 1 patient with 600 mg starting dose.
CR or PR.
CR or PR of any duration or SD for ≥16 weeks from start of treatment.
CR, PR or SD.
Fig. 3.

Maximal percentage change from baseline in sum of target lesion diameters in the PDGFRA D842V population. CR, complete response; PD, progressive disease; PDGFRA, platelet-derived growth factor receptor A; PR, partial response; SD, stable disease.
3.4. Secondary end-points
In the PDGFRA D842V population, the CBR was 98% (55/56 patients) at all doses (Table 3), and 97% in patients receiving the 300/400 mg starting dose. The median DOR was 27.6 months (95% CI: 17.6–not reached [NR]; Fig. 4) for all starting doses. For the 300/400 mg starting doses, the DOR was 22.1 months (95% CI: 14.3–NR) and the 12-month DOR was 73% (95% CI: 59–88). These data were immature at cut-off. For TKI-naïve patients, the CBR was 100%; the median DOR was 22.1 months (95% CI: 9.2–NR) at all starting doses and 22.1 months (95% CI: 3.7–NR) for TKI-naïve patients receiving the 300/400 mg starting doses.
Fig. 4.
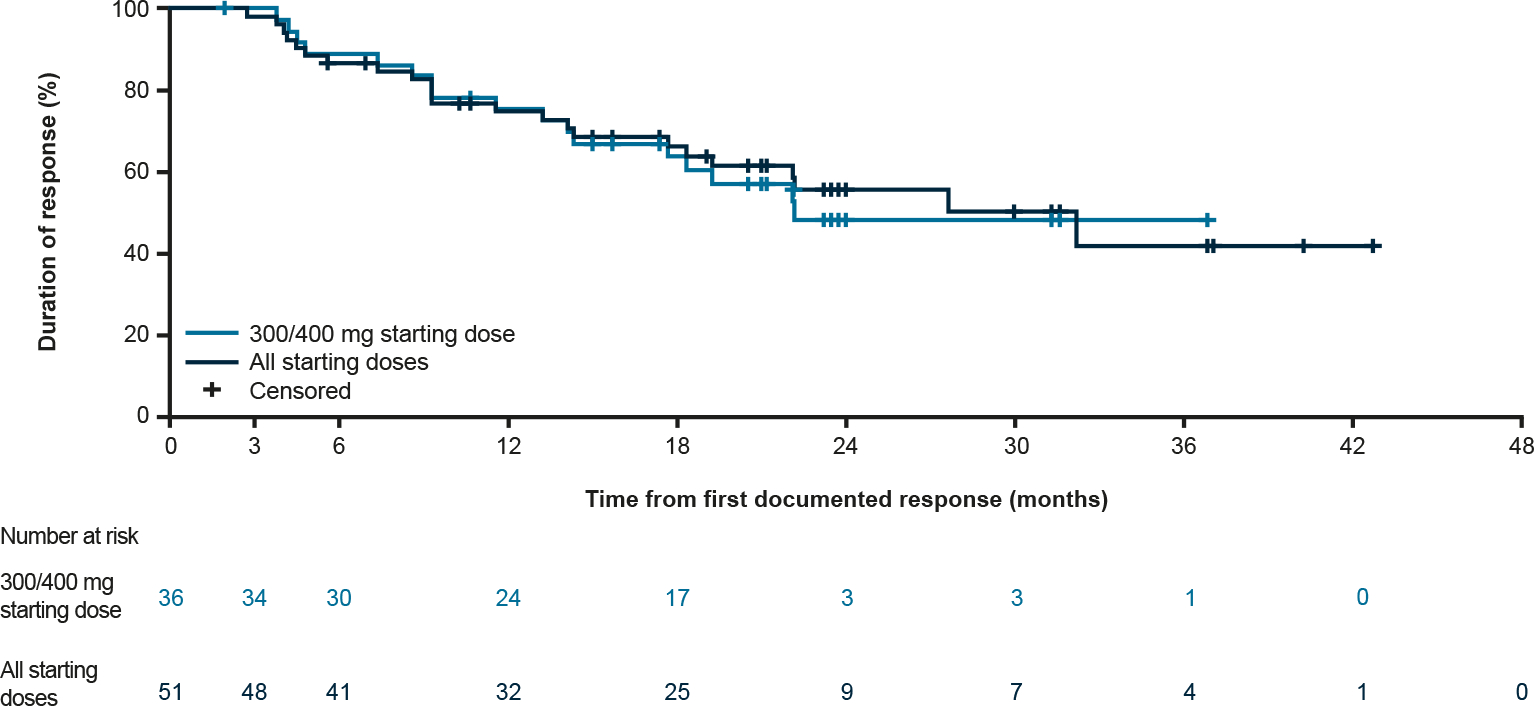
Duration of response in the PDGFRA D842V population, PDGFRA, platelet-derived growth factor receptor A.
The median PFS for the PDGFRA D842V population at all starting doses was 34.0 months (95% CI: 22.9–NR; Fig. 5), and 27.6 months (95% CI: 19.5–NR) for the 300/400 mg starting doses. For all starting doses, PFS at 3, 6 and 12 months was 100% (95% CI: 100–100), 92% (95% CI: 85–100) and 83% (95% CI: 72–93), respectively. For TKI-naïve patients, median PFS was 27.6 months (95% CI: 13.1–NR) and 27.6 months (95% CI: 5.5–27.6) for those receiving the 300/400 mg starting doses.
Fig. 5.
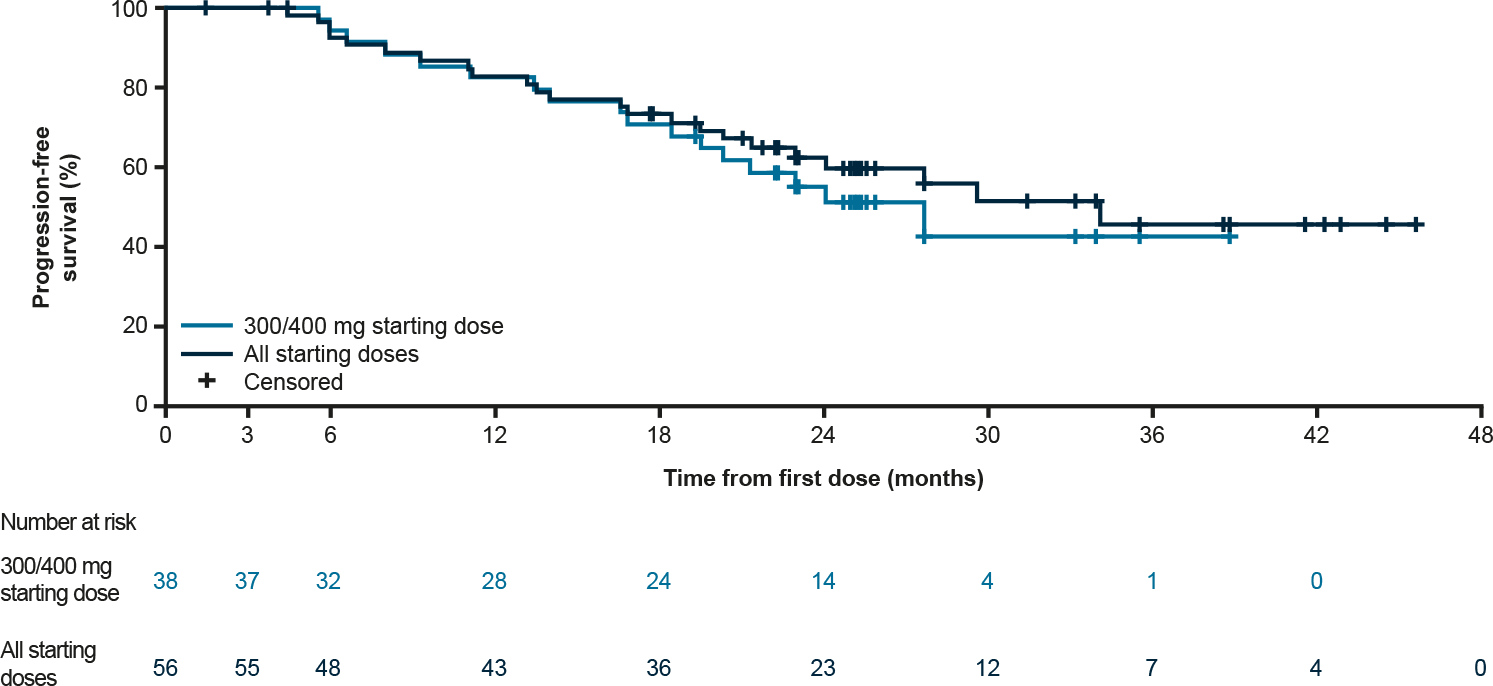
Progression-free survival in the PDGFRA D842V population, PDGFRA, platelet-derived growth factor receptor A.
3.5. Exploratory end-points
At data cut-off, the median OS was NR. OS at 12, 24 and 36 months was 93% (95% CI: 85–100), 75% (95% CI: 63–87) and 61% (95% CI: 45–77), respectively. For patients receiving the 300/400 mg starting doses, OS was similar to the total PDGFRA D842V population (Fig. 6): at 12, 24 and 36 months, OS was 91% (95% CI: 82–100), 71% (95% CI: 55–86) and 71% (95% CI: 55–86), respectively. median OS was NR for TKI-naïve patients in the PDGFRA D842V population; OS at 24 months was 82% (95% CI: 59–100) for TKI-naïve patients and 80% (95% CI: 45–100) for TKI-naïve patients receiving a 300/400 mg starting dose. OS at 36 months was 72% (95% CI: 44–99) for all starting doses in TKI-naïve patients and was not estimable for TKI-naïve patients receiving 300/400 mg starting doses.
Fig. 6.

Overall survival in the PDGFRA D842V population, PDGFRA, platelet-derived growth factor receptor A.
In the PDGFRA D842V population, the time-varying Cox regression model indicated that high ECOG performance status was the most significant factor negatively impacting PFS (hazard ratio [HR]: 6.09; 95% CI: 2.36–15.74). However, each incremental 50 mg QD avapritinib dose increase over the study duration was associated with a 31% increase in PFS (HR: 0.69; 95% CI: 0.53–0.89; Table 4). Prior TKIs had no impact on PFS (HR: 0.34; 95% CI: 0.10–1.13).
Table 4.
Factors impacting hazard ratio for PFS in the PDGFRA D842V population.
| Parameter | HR | 95% CI | P value |
|---|---|---|---|
|
| |||
| Age | 1.05 | 0.99–1.11 | 0.086 |
| Sex | 1.42 | 0.54–3.73 | 0.479 |
| Avapritinib dosea | 0.69 | 0.53–0.89 | 0.005 |
| Prior TKI | 0.34 | 0.10–1.13 | 0.077 |
| ECOG PS | 6.09 | 2.36–15.74 | < 0.001 |
CI, confidence interval; ECOG PS, Eastern Cooperative Oncology Group performance status; HR, hazard ratio; QD, once daily; TKI, tyrosine kinase inhibitor. Bold values are statistically significant P values.
50 mg QD dose increments assessed continually for each patient throughout the treatment period.
4. Discussion
Patients with unresectable or metastatic PDGFRA D842V-mutant GIST have poor prognoses, as previously approved TKIs for GIST do not target PDGFRA D842V [3–8,11–13]. These updated analyses demonstrate the long-term, durable activity of avapritinib in patients with PDGFRA D842V-mutant GIST. With long-term follow-up of 27.5 months, the DOR (27.6 months) observed in this study is remarkable for these patients, 46% of whom were continuing treatment at data cut-off. Median DOR could not be determined at the previous data cut-off for the PDGFRA D842V population (median follow-up 15.9 months), as data were not mature [11]. The clinical activity with avapritinib shown here is unprecedented; no other approved TKIs have shown reliable, durable responses in this patient population [5–7].
ORRs were 95% in patients receiving 300/400 mg starting doses, 96% in patients receiving the 300 mg starting dose, and 91% in the overall PDGFRA D842V population. The latter represent slight increases over the previous cut-off (93% with the 300 mg starting dose; 88% overall) [11]. Although most patients were TKI-pretreated, responses were similar between TKI-naïve and TKI-pretreated patients; the ORR was 91% in both groups. As other TKIs do not target PDGFRA D842V, tumours are unlikely to have developed cross-resistance, and many of these patients progressed on several prior lines of treatment [5,7,8,12–14]. Nonetheless, as response rates were similar between TKI-pretreated and TKI-naïve patients, avapritinib should be considered before other treatments. Utilising ineffective TKIs as first-line treatment may allow increases in tumour load and progression to widespread metastatic disease, both of which are negative prognosticators of long-term survival [15,16]. At baseline, 96% of patients harbouring PDGFRA D842V mutations had metastatic disease, indicating that avapritinib is an option to address the severe unmet need in this patient population.
The DOR and PFS at all avapritinib starting doses were unprecedented compared with other agents in this patient population [5,7,8]. The median PFS was similar across all starting doses. However, exploratory analyses showed that incrementally higher avapritinib doses, assessed continually for each patient throughout the treatment period, were associated with longer PFS. This suggests that although dose modifications were frequently pursued to manage AEs, maintaining the highest tolerable avapritinib dose may be important to achieving the best patient outcomes while rigorously monitoring for toxicity. In line with this, time-to-response was faster with higher avapritinib starting doses compared with lower doses. In addition, lower ECOG performance status was associated with longer PFS, suggesting that initiating avapritinib as early as possible in the disease course should be considered. Use of blinded, independent radiology review for tumour measurements eliminated potential bias of investigator assessments. OS data were not sufficiently mature at data cut-off to allow full interpretation, but initial results are favourable.
Safety profiles in the PDGFRA D842V and safety populations were comparable with previously reported results, with no new safety signals identified [11]. This study of avapritinib provides the first documented long-term safety profile in patients with GIST, with a longer follow-up compared with previous studies of TKIs. Treatment discontinuations across the safety population were most often due to AEs (22%) (15% of which, 15% were deemed treatment-related). This is higher than reported with imatinib (7–16%; [17,18]), regorafenib (6%; [19]) or sunitinib (7%; [20]). The most frequent TRAEs leading to discontinuation were nervous system and psychiatric disorders, which includes cognitive effects and intracranial bleeding. In the PDGFRA D842V population, any-grade cognitive effects and intracranial bleeding were reported in 57% and 5% of patients, of which 2% and 5% were grade ≥III, respectively. Incidence of cognitive effects and intracranial bleeding were similar to that previously reported [11]. Cognitive effects were well managed by dose modification, with grade ≥II events improving in a median of 1.6 weeks [21]. Previous analyses have shown no association between incidence of cognitive effects and cumulative avapritinib dose [21]. Similar AEs of cognitive effects have been reported with other TKIs known to cross the blood–brain barrier (e.g., entrectinib, lorlatinib) despite not targeting PDGFRA [22–24]. Awareness, early recognition and management of these AEs is important. While the most commonly reported AEs (nausea, diarrhoea and anaemia) have been reported with imatinib, regorafenib and sunitinib [17–20], other AEs commonly reported with regorafenib (hand–foot skin reaction, hypertension) and sunitinib (skin discolouration) were uncommon with avapritinib and are likely reflective of differences in the overall kinase inhibitory spectrum of these TKIs [12].
NAVIGATOR is an open-label, single-arm, phase I study assessing avapritinib safety and efficacy in advanced GIST. As PDGFRA D842V-mutant GIST represent a small proportion (5–6%) of GIST cases, the study population was small despite pooling patients from parts 1 and 2. Nevertheless, this study was the largest prospective cohort of patients with PDGFRA D842V-mutant GIST.
4.1. Conclusion
The results presented here demonstrate targeting PDGFRA D842V with avapritinib provided an unprecedented, durable clinical benefit in this subgroup of patients with GIST, with a manageable safety profile. Avapritinib presents an effective first-line treatment for patients with advanced/metastatic PDGFRA D842V-mutant GIST, a previously untreatable GIST subtype.
Supplementary Material
Acknowledgements
The authors would like to thank the patients, their families and all investigators involved in this study. Medical writing support, including assisting authors with the development of the outline and initial draft and incorporation of comments, was provided by Natasha Tracey (PhD); editorial support, including figure preparation, formatting, proofreading and submission, was provided by Elke Sims (MLangTrans) both of Paragon, Knutsford, UK, supported by Blueprint Medicines Corporation according to Good Publication Practice guidelines (http://annals.org/aim/article/2424869/good-publication-practice-communicating-company-sponsored-medical-research-gpp3). Blueprint Medicines Corporation was involved in the study design and collection, analysis and interpretation of data, as well as data checking of information provided in the manuscript. However, ultimate responsibility for opinions, conclusions and data interpretation lies with the authors.
Funding
The NAVIGATOR study (NCT02508532) was supported by Blueprint Medicines Corporation, Cambridge, Massachusetts, USA.
Footnotes
Conflict of interest statement
Dr Jones reported receiving grants from MSD; and personal fees from Adaptimmune, Athenex, Blueprint Medicines Corporation, Clinigen, Eisai, Eli Lilly, Epizyme, Daichii Sankyo, Deciphera Pharmaceuticals, Helsinn, Immunedesign, Merck, PharmaMar, Tracon and UptoDate outside the submitted work.
Dr Serrano reported receiving other support from Bayer, Blueprint Medicines Corporation, Deciphera Pharmaceuticals, Eli Lilly, Novartis, Pfizer and PharmaMar; and grants from Bayer, Deciphera Pharmaceuticals and Pfizer outside the submitted work.
Dr von Mehren reported receiving other support from Blueprint Medicines Corporation during the conduct of the study; and other support from Arog Pharmaceuticals, Deciphera Pharmaceuticals, Exelixis and Novartis outside the submitted work.
Dr George reported receiving research support to her institution from Ariad, Bayer, Blueprint Medicines Corporation, Daiichi Sankyo, Deciphera Pharmaceuticals, Novartis and Pfizer; and advisory board/consulting fees from AstraZeneca, Blueprint Medicines Corporation, Daiichi Sankyo, Deciphera Pharmaceuticals and Eli Lilly.
Dr Heinrich reported receiving grants and personal fees from Blueprint Medicines Corporation; and personal fees and other support from Molecular MD during the conduct of the study; personal fees and other from Novartis; and grants and personal fees from Deciphera Pharmaceuticals outside the submitted work. Dr Heinrich also has a patent “Treatment of Gastrointestinal Stromal Tumors” licenced to Novartis, and a patent “Activating Mutations of PDGFRA” issued.
Dr Kang reported receiving personal fees from ALX Oncology, Amgen, Bristol Myers Squibb, Daehwa Pharmaceutical, MacroGenics, Novartis, Surface Oncology and Zymeworks outside the submitted work.
Dr Schöffski reported receiving personal fees from Deciphera Pharmaceuticals; other support from Adaptimmune, Blueprint Medicines Corporation, Deciphera Pharmaceuticals, Exelixis, Eisai, Eli Lilly, Ellipses Pharma, Genmab, Intellisphere, Loxo Oncology, Merck, Plexxikon, Servier and Transgene; and grants from Ipsen and MSD outside the submitted work.
Dr Cassier reported receiving personal fees from Blueprint Medicines Corporation during the conduct of the study; other support from AbbVie, Bayer, Bristol Myers Squibb, Eli Lilly, GlaxoSmithKline, Janssen, Merck Serono, MSD, Novartis and Roche/Genentech; personal fees from Amgen, Bristol Myers Squibb, MSD, Novartis and Roche/Genentech; non-financial support from MSD and Novartis; and grants from Novartis outside the submitted work.
Dr Mir reported receiving consulting fees from Eli Lilly, Janssen, Lundbeck, Pfizer, Roche, Servier and Vifor Pharma; and owns stock options from Amplitude Surgical, Transgene and Ipsen.
Dr Chawla reported receiving funding from ADI, Amgen, GlaxoSmithKline, Ignyta, Immix Bopharma, Inhibrx, Janssen, Karyopharm Therapeutics, Roche, SARC and Tracon outside the submitted work.
Dr Eskens has nothing to disclose. Dr Rutkowski reported receiving personal fees from Blueprint Medicines Corporation, Bristol Myers Squibb, Merck, MSD, Novartis, Pierre Fabre, Pfizer, Roche and Sanofi outside the submitted work.
Dr Tap reported receiving other support from Blueprint Medicines Corporation during the conduct of the study; receiving personal fees from Agios Pharmaceuticals, Blueprint Medicines Corporation, Daiichi Sankyo, Deciphera Pharmaceuticals, Eli Lilly, EMD Serono, Eisai, GlaxoSmithKline, Janssen, Immune Design, Loxo Oncology and NanoCarrier outside the submitted work; having a patent Companion Diagnostic for CDK4 inhibitors – 14/854,329 pending to MSKCC/SKI; attending scientific advisory boards for Atropos Therapeutics and Certis Oncology Solutions; being a consultant for Daiichi Sankyo; having stock ownership in Atropos Therapeutics and Daiichi Sankyo; and having involvement in an FDA ODAC meeting for pexidartinib.
Dr Zhou is a former employee of Blueprint Medicines Corporation.
Dr Roche reported receiving other support from Epizyme outside the submitted work; and being a current employee and shareholder of Blueprint Medicines Corporation.
Dr Bauer reported receiving grants from Blueprint Medicines Corporation, Incyte and Novartis; personal fees from Bayer, Blueprint Medicines Corporation, Deciphera Pharmaceuticals, Exelixis and Novartis during the conduct of the study; and personal fees from ADC Therapeutics, Daiichi Sankyo, Eli Lilly, Exelixis, Janssen-Cilag, Nanobiotix, PharmaMar and Plexxikon outside the submitted work.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejca.2020.12.008.
Data sharing
The anonymised derived data from this study that underlie the results reported in this article will be made available, beginning 12 months and ending 5 years following this article publication, to any investigators who sign a data access agreement and provide a methodologically sound proposal to medinfo@blueprintmedicines.com. The trial protocol will also be made available, as will a data fields dictionary.
References
- [1].Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol 2008;26:5352–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003;299:708–10. [DOI] [PubMed] [Google Scholar]
- [3].Corless CL, Schroeder A, Griffith D, et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol 2005;23:5357–64. [DOI] [PubMed] [Google Scholar]
- [4].Hirota S, Ohashi A, Nishida T, et al. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology 2003;125:660–7. [DOI] [PubMed] [Google Scholar]
- [5].Roubaud G, Kind M, Coindre JM, et al. Clinical activity of sorafenib in patients with advanced gastrointestinal stromal tumor bearing PDGFRA exon 18 mutation: a case series. Ann Oncol 2012;23:804–5. [DOI] [PubMed] [Google Scholar]
- [6].Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 2003;21:4342–9. [DOI] [PubMed] [Google Scholar]
- [7].Cassier PA, Fumagalli E, Rutkowski P, et al. Outcome of patients with platelet-derived growth factor receptor alpha-mutated gastrointestinal stromal tumors in the tyrosine kinase inhibitor era. Clin Canc Res 2012;18:4458–64. [DOI] [PubMed] [Google Scholar]
- [8].Yoo C, Ryu MH, Jo J, et al. Efficacy of imatinib in patients with platelet-derived growth factor receptor alpha-mutated gastrointestinal stromal tumors. Cancer Res Treat 2016;48:546–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Blueprint Medicines (Netherlands) B.V. AYVAKIT® (avapritinib). Summary of product characteristics. https://www.ema.europa.eu/en/documents/product-information/ayvakyt-epar-product-information_en.pdf. [Accessed 8 October 2020].
- [10].Blueprint Medicines Corporation. AYVAKIT® (avapritinib). Prescribing Information. https://www.blueprintmedicines.com/uspi/AYVAKIT.pdf. [Accessed 6 March 2020].
- [11].Heinrich MC, Jones RL, von Mehren M, et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): a multicentre, open-label, phase 1 trial. Lancet Oncol 2020;21:935–46. [DOI] [PubMed] [Google Scholar]
- [12].Evans EK, Gardino AK, Kim JL, et al. A precision therapy against cancers driven by KIT/PDGFRA mutations. Sci Transl Med 2017;9:eaao1690. [DOI] [PubMed] [Google Scholar]
- [13].Farag S, Somaiah N, Choi H, et al. Clinical characteristics and treatment outcome in a large multicentre observational cohort of PDGFRA exon 18 mutated gastrointestinal stromal tumour patients. Eur J Canc 2017;76:76–83. [DOI] [PubMed] [Google Scholar]
- [14].Park SH, Ryu MH, Ryoo BY, et al. Sorafenib in patients with metastatic gastrointestinal stromal tumors who failed two or more prior tyrosine kinase inhibitors: a phase II study of Korean gastrointestinal stromal tumors study group. Invest N Drugs 2012;30:2377–83. [DOI] [PubMed] [Google Scholar]
- [15].Hompland I, Bruland ØS, Hølmebakk T, et al. Prediction of long-term survival in patients with metastatic gastrointestinal stromal tumor: analysis of a large, single-institution cohort. Acta Oncol 2017;56:1317–23. [DOI] [PubMed] [Google Scholar]
- [16].Mehren Mv, Heinrich MC, Joensuu H, et al. Follow-up results after 9 years (yrs) of the ongoing, phase II B2222 trial of imatinib mesylate (IM) in patients (pts) with metastatic or unresectable KIT+ gastrointestinal stromal tumors (GIST). J Clin Oncol 2011;29. 10016–10016. [Google Scholar]
- [17].Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol 2008;26:626–32. [DOI] [PubMed] [Google Scholar]
- [18].Dematteo RP, Ballman KV, Antonescu CR, et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet 2009;373:1097–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Demetri GD, Reichardt P, Kang Y-K, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381:295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 2006;368:1329–38. [DOI] [PubMed] [Google Scholar]
- [21].Joseph CP, Abaricia SN, Angelis MA, George S, Jones RL. Avapritinib for the treatment of GIST: analysis of efficacy, safety, and patient management strategies at the recommended phase 2 dose. In: Connective Tissue Oncology Society annual meeting;2019. [Tokyo, Japan]. [Google Scholar]
- [22].Pfizer Inc. LORBRENA (lorlatinib) tablets. Prescribing information. http://labeling.pfizer.com/ShowLabeling.aspx?format=PDF&id=11140. [Accessed 6 March 2020]. [Google Scholar]
- [23].Genetech USA Inc. ROZLYTREK (entrectinib) capsules. Prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212725s000lbl.pdf. [Accessed 14 April 2020].
- [24].US Food and Drug Administration. NDA/BLA Multidisciplinary review and evaluation NDA 212608 AYVAKIT (avapritinib). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/212608Orig1s000MultidisciplineR.pdf. [Accessed 6 March 2020].
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The anonymised derived data from this study that underlie the results reported in this article will be made available, beginning 12 months and ending 5 years following this article publication, to any investigators who sign a data access agreement and provide a methodologically sound proposal to medinfo@blueprintmedicines.com. The trial protocol will also be made available, as will a data fields dictionary.