

Abstract
Cardiotoxicity, or the development of unwarranted cardiovascular side-effects of oncologic therapies, can involve all aspects of cardiovascular disease. The development of cardiac fibrosis is a dreaded complication that leads to cardiac mechanical dysfunction, tachyarrhythmias, and an increase in cardiovascular mortality. This review details established and putative mechanisms leading to fibroblast activation, myofibroblast transdifferentiation, and the development of replacement or interstitial cardiac fibrosis as a consequence of cancer treatments. Clinical and imaging strategies for cardiac fibrosis assessment as well as emerging antifibrotic therapeutics will also be addressed.
Introduction
The adult mammalian heart has a negligeable ability to regenerate – estimated at no more than ~ 1% replacement of cardiomyocytes per year [1–4] – and thus rather heals through cardiac repair mechanisms leading to scar tissue formation. In adult hearts, fibroblasts account for ~25% of the cell population [5], provide support to cardiomyocytes, and contribute to multiple signaling processes [6]. The majority of the cardiac extracellular matrix (ECM) is constituted by type I and III collagen [7,8]. Cardiac fibrosis requires the activation of cardiac fibroblasts – abundant in the interstitial and perivascular space – and transdifferentiation to myofibroblasts, resulting in the excess deposition of ECM proteins, which may play a reparative role but also negatively impact cardiac function [9]. In addition to activation of the renin-angiotensin-aldosterone system [10,11], β-adrenergic stimulation [12,13], and the Wnt pathway [14,15], key mediators of fibrogenic activation include growth factors, e.g. transforming growth factor (TGF)-β and platelet-derived growth factors (PDGF), and cytokines, e.g. tumor necrosis factor (TNF)-α, and interleukin (IL)-1, -4, -6, and -10 [16].
Cardiac fibrosis may occur as a consequence of epicardial and microvascular ischemic heart disease, inherited cardiomyopathies, pressure overload such as aortic valve stenosis or systemic hypertension, and certain oncologic treatments, amongst others. There are three main types of cardiac fibrosis based on histopathology, namely replacement fibrosis, interstitial fibrosis, and perivascular fibrosis [9]. Whether interstitial and perivascular fibrosis have distinct pathobiological mechanisms remains controversial and is addressed elsewhere [9]. Others also distinguish infiltrative interstitial fibrosis as observed in Fabry disease with diffuse cellular glycolipid accumulation [17].
Replacement or reparative cardiac fibrosis occurs as a result of sudden cardiomyocyte necrosis as observed following acute myocardial infarction and also reported in the setting of radiation therapy. Given the negligeable ability of cardiomyocytes to regenerate, the collagen-rich scar in this setting serves a critical role, i.e. maintaining the structural integrity of the heart, albeit at the price of a deterioration in left ventricular systolic function. However, in other pathologies such as systemic hypertension, diabetes mellitus, and obesity, interstitial cardiac fibrosis occurs in the absence of significant cardiomyocyte death, likely as a result of prolonged activation of fibrogenic stimuli, and predominantly involves the interstitium and perivascular space. Interstitial fibrosis can develop insidiously [9], and initially manifests as heart failure with preserved ejection fraction (HFpEF) due to increased interstitial stiffness leading to a reduction in ventricular compliance and diastolic filling. Finally, perivascular fibrosis is most prominent in hypertensive heart disease, and is associated with impaired microvascular function leading to perturbation of myocardial blood flow [18].
The end-result of replacement, interstitial, and perivascular fibrosis is often cardiac dysfunction, either diastolic, systolic, or their combination, leading to heart failure with reduced and/or preserved ejection fraction, and an increased propensity for both atrial and ventricular tachyarrhythmias most notably atrial fibrillation [19], ventricular tachycardia [20], and sudden cardiac death [21]. Indeed, excess ECM proteins, most prominently collagen, act as ‘scars’ from a functional standpoint, and are centrally implicated in the occurrence of focal electrical re-entry circuits, which may deteriorate to potentially life-threatening ventricular tachy-arrhythmias [22], and further contribute to the degradation of ventricular function.
Expansion of the ECM leading to cardiac fibrosis is associated with increased mortality [23]. Cardiac remodeling, defined as a change in geometry with ensuing worsening function following injury, occurs as a result of crosstalk between cardiac cells and the ECM: on the one hand, cells secrete ECM molecules with regulatory properties, on the other, changes in the ECM lead to cellular responses [9,24].
This review details known and putative associations between oncologic therapies and the development of cardiomyocyte injury, fibroblast activation, and subsequent deposition of excess ECM proteins leading to cardiac fibrosis.
Anthracyclines
Severe anthracycline-induced cardiotoxicity resulting in depressed systolic function and heart failure can afflict up to 25% of patients [25,26]. Ample evidence supports that anthracycline-induced cardiac injury is multi-factorial and -genic, with no single mechanism fully explaining all aspects of the injury process. Prior studies indicate anthracycline-induced cardiotoxicity results from a combination of deoxyribonucleic acid (DNA) damage, oxidative stress, and metabolic perturbations, leading to the activation of all forms of cell death [27,28].
Anthracycline chemotherapy intercalates in the DNA and induces single- and double-strand DNA breaks in target cells in a topoisomerase (Top)-2-dependent manner [29]. By producing temporary single- or double-stranded DNA breaks, Top regulates topological changes during DNA replication, transcription, or recombination [30]. Top-2α is overexpressed in tumors and is the molecular basis of anthracycline anticancer activity [31,32]. Adult cardiomyocytes express Top-2β but not Top-2α [31], and Top-2β is also an anthracycline target, forming a Top-2β–anthracycline–DNA ternary cleavage complex that induces DNA strand breaks and ensuing cell death [33,34]. Furthermore, anthracycline/Top-2β bind to selective promoters, significantly affecting the cardiomyocyte transcriptome [34,35]. Ensuingly, key antioxidative enzymes are reduced, providing a mechanism linking anthracycline-induced reactive oxygen species (ROS) production (O2•− superoxide anion, H2O2 hydrogen peroxide, and OH• hydroxyl radical) [36,37] in a Top-2β-dependent manner. An additional major pathway of anthracycline-induced cardiotoxicity is through mitochondrial complex I NADH dehydrogenase mediated redox cycling with the quinone moiety of anthracyclines, leading to the generation of excess reactive oxygen species (ROS) [36,38] with ensuing DNA damage [36,38–40]. Furthermore, anthracyclines can interact directly with iron to form complexes, catalyzing a Fenton reaction, i.e. the Fe2+ mediated conversion of H2O2 to OH•, supported by experimental studies in which excess iron accumulation worsens anthracycline-induced cardiotoxicity [41,42].
Anthracyclines such as doxorubicin are known to lead to cardiac fibrosis (Fig. 1) [40,43,44]. In particular, anthracycline-induced cardiomyocyte injury and death leads to an inflammatory response that induces fibroblast activation [45]. Indeed, anthracyclines damage mitochondria, causing the release of mitochondrial DNA, peptides, and lipids, that become damage-associated molecular patterns (DAMPs) resulting in innate immune system activation [46]. Given the abundance of mitochondria required to maintain cardiac energy consumption and function, these mitochondrial DAMPs can lead to a significant response amplification, mediated by pattern recognition receptors, primarily Toll-like receptor (TLR) -9 [47]. In turn, this activates the pro-inflammatory transcription factor nuclear factor (NF)-κB, induces the expression of inflammatory cytokines such as TNF-α, and the recruitment of inflammatory cells [48,49]. The ensuing chronic inflammation triggers TGF-β1 activation which promotes the conversion of fibroblasts to myofibroblasts with ensuing collagen synthesis and deposition [9,50,51]. Both TGF-β1 and its signal transducer SMAD (similar to mothers against decapentaplegic) -3 are consistently induced by anthracyclines in the heart, and during all phases of the injury process [52].
Figure 1. Progression of anthracycline cardiotoxicity from cardiomyocyte injury and death to fibroblast activation and interstitial fibrosis.
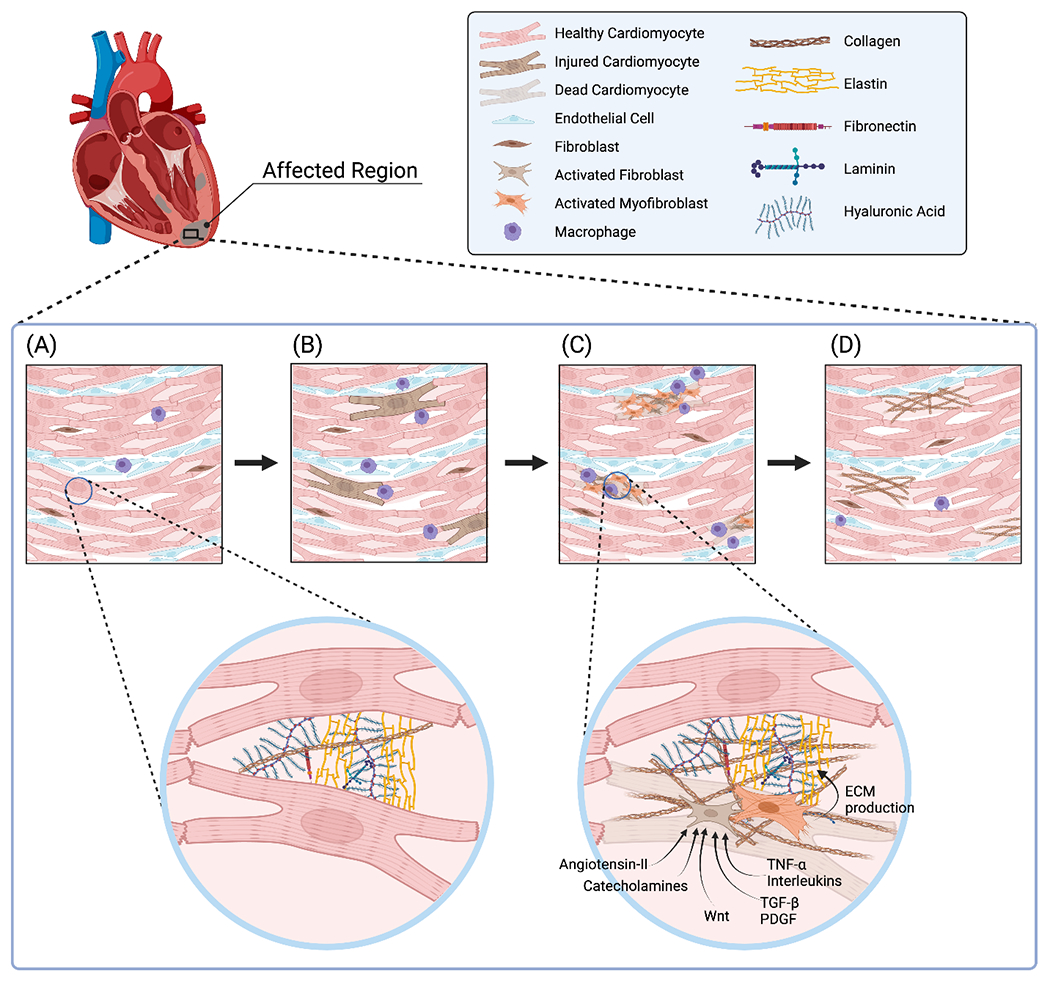
Anthracycline chemotherapy affects the heart in a ‘patchy’ manner. (A) Segment of normal left ventricular myocardium prior to chemotherapy. (B) In the setting of anthracycline treatment, cardiomyocyte injury develops with ensuing inflammatory infiltration, primarily macrophages. (C) With progression of anthracycline toxicity, cardiomyocyte death occurs, leading to activation of fibroblasts and myofibroblasts that produce excess extracellular matrix proteins, primarily collagen. (D) In the chronic phase of anthracycline cardiotoxicity, scattered dead cardiomyocytes have been replaced with collagen leading to interstitial cardiac fibrosis, setting the stage for ensuing cardiac mechanical complications.
ECM: extracellular matrix. PDGF: platelet-derived growth factor. TGF-β: transforming growth factor-β. TNF-α: tumor necrosis factor-α. Wnt: wingless and int-1.
In addition to cardiomyocyte injury and death, anthracyclines also directly promote the release of pro-fibrotic factors from the myocardium. Experimental evidence suggests that low-dose doxorubicin exposure can lead to fibroblast activation and perivascular fibrosis in the absence of cardiomyocyte injury [53]. Indeed, anthracyclines induce TGF-β release from cardiac endothelial cells [54] and fibroblasts [55]. Furthermore, PDGF-A and -B are induced 4-5-fold in the ventricles of doxorubicin treated mice [56]. Doxorubicin also increases plasma angiotensin-II levels 3-fold [57], mediated in part by enhanced renin [58] and angiotensin-converting enzyme [59,60] activities. Importantly, anthracyclines promote the trans-differentiation of cardiac fibroblasts to myofibroblasts [55]. Further research established the expression of additional fibroblast activation markers in response to anthracyclines. Indeed, daunorubicin treatment is associated with increased cardiac fibroblast expression of vimentin [61], a protein previously implicated in fibroblast proliferation and differentiation [62]. Tenascin-C, another marker of fibroblast activation, is induced in doxorubicin-treated pigs [63]. Similarly, connective tissue growth factor (CTGF) is a matricellular protein secreted by activated fibroblasts to mediate TGF-β activity during the fibrotic response to anthracycline therapy (64), and further has an autocrine function by amplifying fibroblast activation, thus creating a positive feedback loop [65].
Following fibroblast/myofibroblast activation, there is increased deposition of collagen type I and III in the ECM, leading to cardiac interstitial fibrosis [52]. In addition, doxorubicin also results in fibronectin deposition in the ECM [66]. Importantly, tissue inhibitor of metalloproteinase (TIMP) -1 that hinders matrix metalloproteinase (MMP) activity, is induced by anthracycline treatment, thus impeding ECM degradation and indirectly promoting collagen accumulation [53]. Similarly, the matricellular protein thrombospondin (TSP) -2 – released mainly by fibroblasts – is induced by doxorubicin, and also inhibits MMP proteolytic activity [67]. The net effect of these molecular actions caused by anthracyclines is ECM accumulation, leading to interstitial fibrosis. It is important to note that the cardiac injury process induced by anthracyclines and ensuing interstitial fibrosis is spatially heterogeneous (Fig. 1), with unaffected regions of the myocardium compensating through augmentation of function, thus ‘masking’ early cardiotoxicity from a functional standpoint [68].
Trastuzumab
Trastuzumab cardiotoxicity is mediated by inhibition of cardiomyocyte human epidermal growth factor receptor-2 (also known as ErbB2) which interferes with mitochondrial integrity through Bcl-x proteins, leading to adenosine triphosphate (ATP) depletion, interference with the repair and survival of cardiomyocytes, and mechanical failure [69,70]. In mice, treatment with an anti-ErbB2 antibody further leads to ventricular myofibril disarray [71]. Additional animal studies demonstrate trastuzumab-mediated activation of cardiomyocyte apoptosis [72] which – similar to anthracyclines – leads to cardiac interstitial fibrosis [73,74] that can be characterized by cardiac magnetic resonance imaging (MRI) [75,76].
VEGF Signaling Inhibitors
Angiogenesis inhibitors, or vascular endothelial growth factor (VEGF) signaling pathway inhibitors (VSPi), block VEGF signaling through various mechanisms, i.e. (i) anti-VEGF antibodies (e.g., bevacizumab), (ii) anti-VEGF receptor antibodies (e.g., ramucirumab), and (iii) VEGFR intracellular domain tyrosine kinase inhibitors (TKIs, e.g., sorafenib, sunitinib) [77]. VSPi are implicated in the onset of hypertension in up to 80% of patients [78,79]. Dissecting the mechanisms thereof is of paramount importance given treatment of hypertension associated with VSPi is challenging as demonstrated by its abrupt onset and requirement for multiple anti-hypertensive medications [78]. Whereas the exact pathobiology leading to severe hypertension remains to be determined, putative pathways include endothelial dysfunction with decreased nitric oxide production [80,81], and glomerular injury with podocyte apoptosis [82,83]. Refractory hypertension is a well-known risk factor for the development of cardiac interstitial fibrosis [9] and has been reported in certain animal studies of VSPi-induced cardiotoxicity [84,85]. In addition, VSPi have been associated with accelerated atherosclerosis [86] as well as coronary microvascular dysfunction (sunitinib) [87], however the contributions thereof to the development of ischemic heart disease and subsequent cardiac fibrosis have not been investigated to date. Importantly, cardiotoxicity by VSPi is reversible in up to 80% of patients upon withdrawal [88].
Bruton’s Tyrosine Kinase Inhibitors
Bruton’s tyrosine kinase inhibitors (BTKi) are increasingly used in B lymphocyte malignancies [89]. A first-generation BTKi, ibrutinib, has been implicated in cardiac fibrosis, a consequence of its lack of selectivity leading to frequent off-target actions [90]. Indeed, mice treated with ibrutinib develop left atrial fibrosis with increased deposition of fibronectin, collagen-I and -III, left atrial enlargement, and atrial fibrillation [90,91]. Using chemo-proteomic profiling and genetically modified mice, the cardiotoxic effect of ibrutinib was demonstrated to be due to the inhibition of CSK (C-terminal Src kinase), itself a non-receptor tyrosine kinase that functions as a master negative regulator of Src family tyrosine kinases (SFKs) [92]. Second-generation BTKi (e.g. acalabrutinib, zanubrutinib) are more selective, leading to fewer cardiovascular off-target effects. A recent meta-analysis indicated an 87% decrease in symptomatic or life-threatening atrial fibrillation, and a 38% decrease in severe hypertension (defined as ≥ 160/110 mmHg or malignant hypertension) with second- vs. first-generation BTKi [93]. To date, second-generation BTKi have not been implicated in cardiac fibrosis [94].
Immune Checkpoint Inhibitors
Cardiac complications of immune checkpoint inhibitors (ICI) that act on different co-stimulatory molecules expressed by T lymphocytes and antigen-presenting cells are rare – with the caveat of likely being under-recognized [95–97] – but potentially fatal [98]. The pathobiology of cardiac fibrosis in the setting of ICI treatment has not been explored [99,100]. Putative mechanisms leading to fibroblast activation, transdifferentiation to myofibroblasts, and replacement fibrosis – not demonstrated to date in experimental models – may involve ICI-induced (i) direct cytotoxic T-cell mediated cardiomyocyte death [101], (ii) chronic inflammation with ensuing oxidative stress [102], and (iii) accelerated coronary atherosclerosis [103] leading to ischemic heart disease and cardiomyocyte death. Whether ICI also has a direct effect on fibroblasts should also be scrutinized. Cardiac fibrosis is overall a very rare complication in ICI [98,104].
Radiation Therapy
Radiation therapy is an integral part of the armamentarium against multiple chest cancers, including lymphomas, lung cancer, and breast cancer [105]. However, radiotherapy may lead to a myriad of cardiovascular complications, including accelerated coronary artery disease, conduction system abnormalities, valvular disease, pericardial disease, and cardiac fibrosis. Whereas the underlying pathobiological mechanisms leading to cardiac fibrosis following radiotherapy are not entirely understood, certain features of the disease process have been explored (Fig. 2).
Figure 2. Radiation therapy-induced endothelial and cardiomyocyte injury leading to fibroblast activation and cardiac fibrosis.
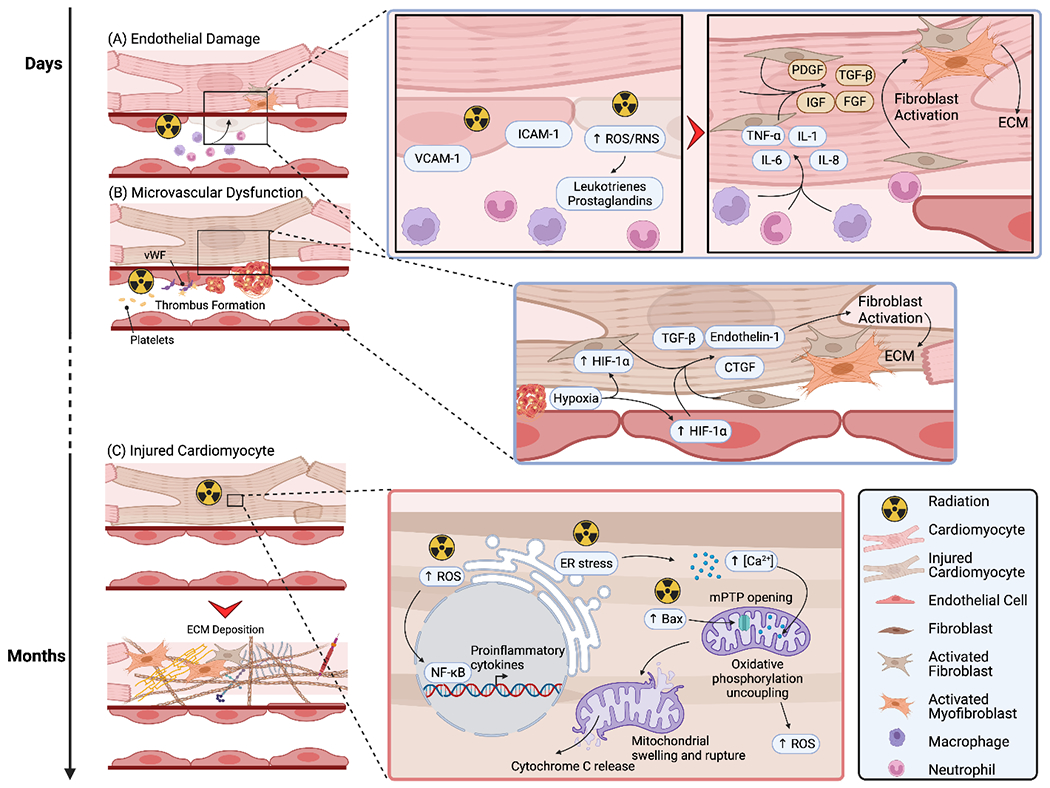
Mechanisms leading to cardiac fibrosis following radiation therapy. (A) Following radiation, endothelial cells rapidly express adhesion molecules, leading to the recruitment of leukocytes. ROS-mediated release of leukotrienes and prostaglandins further induce capillary permeability and leukocyte extravasation. Leukocytes, mainly monocytes and neutrophils, subsequently release pro-inflammatory cytokines, stimulating cardiomyocytes and fibroblasts to secrete pro-fibrotic mediators with ensuing fibroblast activation, ECM production, and cardiac fibrosis. (B) Radiation therapy is complicated by collagen exposure in the sub-endothelial matrix, with ensuing binding of von Willebrand Factor, platelet adhesion, and thrombus formation. Downstream, hypoxia occurs, leading to endothelial cell and fibroblast release of HIF-1α, in turn promoting the secretion of pro-fibrotic factors, and fibroblast activation. (C) Radiation can also directly injure cardiomyocytes, albeit at a later time-point. Three mechanisms have been proposed, namely an increase in ROS production, ER stress leading to excess calcium accumulation in the mitochondria, and increased Bax expression which translocates to the mitochondrial membrane and opens the mitochondrial membrane transition pore. Combined, these lead to mitochondrial swelling and rupture, cytochrome c release, and activation of cell death pathways. Following cardiomyocyte death, replacement cardiac fibrosis occurs.
Bax: Bcl-2 associated X-protein. CTGF: connective tissue growth factor. ECM: extracellular matrix. ER: endoplasmic reticulum. HIF-1α: hypoxia-inducible factor-1α. ICAM-1: intercellular adhesion molecule-1. IGF: insulin-like growth factor. IL: interleukin. mPTP: mitochondrial membrane transition pore. NF-κB: nuclear factor-κB. PDGF: platelet-derived growth factor. RNS: reactive nitrogen species. ROS: reactive oxygen species. TGF-β: transforming growth factor-β. TNF-α: tumor necrosis factor-α. VCAM-1: vascular cell adhesion molecule-1. vWF: von Willebrand Factor.
Endothelial injury of the dense myocardial capillary network and ensuing microvascular dysfunction plays a central role. Following radiation, there is a rapid rise in endothelial ROS production, mediated in part by the up-regulation of the NADPH oxidases NOX-2 and -4 [106], with ensuing NO scavenging and peroxynitrite (ONOO−) formation, implicated in protein nitrosylation [107]. This endothelial dysfunction leads to the release of eicosanoids such as leukotrienes and prostaglandins that increase capillary permeability and leukocyte extravasation [108]. In addition, irradiated endothelial cells express adhesion molecules such as vascular cell adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1), promoting the recruitment of neutrophils and monocytes [109,110]. These leukocytes secrete inflammatory mediators, including TNF-α, IL-1, -6, and -8, and signal to neighboring cardiomyocytes and fibroblasts, rapidly promoting the release of pro-fibrotic cytokines such as TGF-β, PDGF, insulin-like growth factor (IGF), and basic fibroblast growth factor (FGF), as well as myofibroblast transdifferentiation [105,111,112].
Furthermore, endothelial injury results in exposure of the sub-endothelial matrix, permitting binding of von Willebrand factor via its collagen interaction sites, followed by a conformational change that enables platelet glycoprotein binding and ensuing thrombus formation and vascular occlusion [113,114]. This decrease in endothelial and capillary density results in microvascular disease, ischemia, cardiomyocyte death, and cardiac fibrosis [105]. In addition, chronic hypoxia leads to the expression of hypoxia-inducible factor (HIF) -1α, implicated in the stimulation of pro-fibrotic mediators such as TGF-β, endothelin-1, and CTGF, thus further contributing to replacement cardiac fibrosis [115,116].
Cardiomyocytes are relatively resistant to radiation injury due to their very low rate of proliferation [1–4], however direct radiation-induced cardiomyocyte injury has been reported. First, radiation induces ROS [117] which interact with the transcription factor NF-κB, resulting in its nuclear translocation and proinflammatory cytokine production, leading to chronic inflammation [118,119]. Second, radiation induces the cardiomyocyte endoplasmic reticulum to release excess calcium ions, leading to mitochondrial swelling, mitochondrial membrane permeabilization, and oxidative phosphorylation uncoupling [120,121]. Third, experimental studies have described an increase in Bax levels following radiation [120,122]. Bax then translocates from the cytoplasm to the mitochondrial membrane where it opens the mitochondrial membrane transition pore (mPTP), leading to membrane depolarization and rupture, release of cytochrome c into the cytoplasm, and cardiomyocyte death [123]. Although less well characterized, these direct radiation-induced cardiomyocyte death mechanisms also implicate replacement fibrosis, as above [119].
Strategies for Cardiac Fibrosis Assessment
Animal studies present the distinct advantage of direct tissue collection and histological staining at predefined timepoints [124], in addition to targeted genetic manipulation and controlled environmental factors. Following histology staining, for example with picrosirius red, and digital scanning, the fraction of cardiac fibrosis may be quantified using pixel-based segmentation and clustering [125].
Whereas endomyocardial biopsies in humans are the gold standard for visualization of fibrosis, this approach is limited by its invasive nature [126]. Circulating biomarkers [127] including galectin-3, secreted by inflammatory cells and fibroblasts, and soluble ST-2 (suppression of tumorigenicity-2), a member of the IL-1 receptor family, correlate with the degree of cardiac fibrosis [128–130]. The carboxy-terminal pro-peptide of pro-collagen type I and the amino-terminal pro-peptide of pro-collagen type III are additional circulating biomarkers that correlate with the collagen volume fraction in cardiac fibrosis [131]. However, this strategy is not used in routine clinical practice, in part due to a lack of validation.
Echocardiography may be used for the rapid assessment of cardiac structure, dimensions, function, and remodeling. 2-D [132] or 3-D speckle [133] tracking echocardiography with strain measurement further permits the assessment of segmental ventricular function, reported to correlate with cardiac fibrosis [134]. Whereas echocardiography does not have the spatial resolution to directly image interstitial fibrosis, ensuing changes in diastolic function can be readily measured and help unmask the disease, albeit with limited specificity [135].
For in vivo characterization, cardiac MRI is the reference modality for detection of cardiac fibrosis in both human and animal studies [136]. Two cardiac magnetic resonance imaging (MRI) methods are routinely used for fibrosis detection; (i) late gadolinium enhancement, and (ii) T1 mapping, alone or in combination with an extracellular contrast agent to determine the extracellular volume fraction calculated by pre- and post-contrast T1 measurements [137–139]. Whereas late gadolinium enhancement is ideally suited for the assessment of replacement (or reparative) cardiac fibrosis, T1 mapping is favored for the detection of diffuse interstitial fibrosis, for example in the setting of anthracycline-induced cardiotoxicity, and quantification of the ECM expansion by measurement of the extracellular volume fraction (Fig.1) [140–142]. Using serial cardiac MRI, Farhad et al. demonstrated in a chronic doxorubicin mouse injury model that 10 weeks after the first dose of chemotherapy, left ventricular ejection fraction (LVEF) deteriorated and cardiac fibrosis developed, as measured by T1 mapping and validated histologically using Masson’s trichrome staining [143]. Importantly, cardiac fibrosis also predicted late mortality in mice, whereas the deterioration in LVEF did not [143]. Similar experiments and findings in mice [144], rats [145,146], rabbits [125,147], and pigs [148] have been reported by other groups.
These experimental observations indicating a strong correlation between cardiac fibrosis and left ventricular dysfunction [23,141,149] have translational implications. Whereas on the one hand abnormal T1 mapping and extracellular volume expansion in anthracycline treated patients may warrant closer follow-up and/or changes in chemotherapy, on the other hand normal T1 mapping may allow for longer surveillance intervals [142].
Nuclear imaging permits molecular targeting of cellular and molecular contributors to cardiac fibrosis. Mostly studied in the setting of replacement cardiac fibrosis following myocardial infarction, nuclear imaging molecular strategies should be explored further in experimental models of oncologic treatments. Such radiotracers include 99mTc-labeled cyanine-5.5 RGD imaging peptide (99mTc-CRIP) [150,151] and 18F-labeled RGD [152,153]. The RGD peptide (containing the arginine-glycine-aspartate motif) binds to integrins such as αvβ3, expressed on the cell membrane of myofibroblasts [154]. Caution must be exerted however given these integrins are also expressed by endothelial cells, particularly during neo-angiogenesis [155], decreasing their specificity for cardiac fibrosis assessment.
Recently, fibroblast activation protein (FAP) has gained significant traction in nuclear imaging, with expanding applications in oncologic diseases. 68Ga-fibroblast activation protein inhibitor (FAPI) positron emission tomography (PET) was initially developed to detect FAP-expressing, cancer-associated fibroblasts. Retrospective studies have indicated potential applicability in cardiac diseases, with myocardial FAPI signals correlating with underlying metabolic disease [156], coronary artery disease, and LVEF [157] in patients with cancer. Interestingly, a small prospective study observed an association between right ventricular 68Ga-FAPI signals and the severity of right ventricular dysfunction and pulmonary hypertension, suggesting detection of right ventricular fibrosis [158]. Furthermore, 68Ga-FAPI predicts the degradation of ventricular dysfunction following acute myocardial infarction, and may provide a novel biomarker of left ventricular remodeling [159]. In this setting, experimental and clinical studies evaluating 68Ga-FAPI PET in oncologic therapies implicated in the development of cardiac fibrosis warrant evaluation.
Limitations
The scientific literature retraced here is heterogeneous in nature, with limited studies conducted using pertinent genetically modified models. Thus, definitive results are currently lacking in this field. This review may provide a framework for further evaluation of the molecular mechanisms culminating in fibroblast activation and cardiac fibrosis in the context of established and emerging oncologic therapies. As such, additional work is needed in fibroblast-specific gene editing models to determine whether well-established pro-fibrotic signaling pathways, e.g. the mitogen-activated protein kinase p38α [160] or the TGF-β downstream mediator SMAD-3 [161] can protect from oncologic treatment mediated cardiac fibrosis and dysfunction.
Conclusions and Future Directions
Improved long-term cancer survival has led to an increase in the incidence of adverse cardiac side-effects of cancer treatments [25,26]. The exponential growth of cancer therapies warrants (i) monitoring of adverse cardiovascular side-effects such as cardiac fibrosis, (ii) developing pertinent experimental strategies to dissect underlying pathobiological mechanisms leading to cardiac fibrosis, and (iii) exploring novel therapeutic strategies targeting cardiac fibrosis [162].
Once established, the direct targeting of cardiac fibrosis has proven difficult in the clinical setting [163–165]. Beyond previously explored antifibrotic therapeutics, a promising new development involves targeting of the epigenetic machinery, particularly inhibition of histone acetyltransferases (HATs) and histone deacetylases (HDACs) [162]. This breakthrough advance centered on chromatin-level modification permits gene expression changes – independent of gene editing – downstream of cellular activation pathways and upstream of transcriptional changes leading to ECM protein production and deposition. HAT p300 is targeted by curcumin [166,167], leading to decreased cardiac fibrosis, however with the caveat of a lack of curcumin target specificity [162]. Recently, a novel compound A-485 was identified with high p300 specificity [168], setting the stage for its evaluation in cardiac fibrosis models. Furthermore, HDAC inhibition with compound ITF2357/givinostat mitigates cardiac fibrosis in experimental models [169,170].
Another shift in therapeutic strategy involves engineered CD8+ T-cells [171,172]. By directing T-cell specificity using a chimeric antigen receptor (CAR-T cells) towards cardiac fibroblasts expressing FAP – a cell surface glycoprotein minimally expressed in normal heart tissue but significantly induced in activated fibroblasts – immunotherapy may successfully target pathological cardiac fibrosis [171]. These results bear great promise given CAR-T cells are already approved by the FDA in patients with leukemia or lymphoma [173], thus setting the stage for clinical trial evaluation in patients with cardiac fibrosis. Given the dynamic nature of FAP expression by fibroblasts, i.e. during ‘active’ ECM deposition, patient selection will be critical to increase the likelihood of trial success [174]. Specifically, patients undergoing active cancer therapy, particularly those with limited therapeutic options in whom the chemotherapeutic and/or radiotherapeutic strategies necessitate continuation despite the development of off-target cardiac fibrosis, as opposed to patients with fully established excess cardiac ECM deposition where cardiac fibroblasts have returned to a more quiescent state, may stand to benefit the most from a CAR-T cell strategy. This will almost certainly require an imaging-based patient selection process, either by cardiac MRI or FAPI PET imaging, for the early and accurate identification of cardiac fibrosis prior to randomization to CAR-T cell immunotherapy.
Acknowledgements
The author thanks Jeremy Jong for assistance with the figure which was created with BioRender.com.
Funding
Supported by VA Merit BX004558, UCLA Cardiovascular Discovery Fund/Lauren B. Leichtman and Arthur E. Levine Investigator Award, and NIH NCATS UCLA CTSI UL1TR00188.
Footnotes
Disclosures
None.
References and recommended reading
• of special interest
• • of outstanding interest
- 1.Bergmann O, Bhardwaj RD, Bernard S et al. Evidence for cardiomyocyte renewal in humans. Science 2009;324:98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Senyo SE, Steinhauser ML, Pizzimenti CL et al. Mammalian heart renewal by pre-existing cardiomyocytes. Nature 2013;493:433–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kimura W, Xiao F, Canseco DC et al. Hypoxia fate mapping identifies cycling cardiomyocytes in the adult heart. Nature 2015;523:226–30. [DOI] [PubMed] [Google Scholar]
- 4.Karra R, Poss KD. Redirecting cardiac growth mechanisms for therapeutic regeneration. J Clin Invest 2017;127:427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banerjee I, Fuseler JW, Price RL, et al. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Physiol Heart Circ Physiol 2007;293:H1883–91. [DOI] [PubMed] [Google Scholar]
- 6.Hall C, Gehmlich K, Denning C, Pavlovic D. Complex Relationship Between Cardiac Fibroblasts and Cardiomyocytes in Health and Disease. J Am Heart Assoc 2021;10:e019338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weber KT. Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol 1989;13:1637–52. [DOI] [PubMed] [Google Scholar]
- 8.Silva AC, Pereira C, Fonseca A, et al. Bearing My Heart: The Role of Extracellular Matrix on Cardiac Development, Homeostasis, and Injury Response. Front Cell Dev Biol 2020;8:621644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frangogiannis NG. Cardiac fibrosis. Cardiovasc Res 2021;117:1450–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]; • • Authoritative review detailing the cellular and molecular basis of cardiac fibrosis in various diseases and the development of fibrosis-targeting therapies.
- 10.Kagami S, Border WA, Miller DE, Noble NA. Angiotensin-Ii Stimulates Extracellular-Matrix Protein-Synthesis through Induction of Transforming Growth-Factor-Beta Expression in Rat Glomerular Mesangial Cells. J Clin Invest 1994;93:2431–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weber KT, Sun Y, Bhattacharya SK, et al. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat Rev Cardiol 2013;10:15–26. [DOI] [PubMed] [Google Scholar]
- 12.Benjamin IJ, Jalil JE, Tan LB, et al. Isoproterenol-induced myocardial fibrosis in relation to myocyte necrosis. Circ Res 1989;65:657–70. [DOI] [PubMed] [Google Scholar]
- 13.Nguyen MN, Kiriazis H, Ruggiero D et al. Spontaneous ventricular tachyarrhythmias in beta2-adrenoceptor transgenic mice in relation to cardiac interstitial fibrosis. Am J Physiol Heart Circ Physiol 2015;309:H946–57. [DOI] [PubMed] [Google Scholar]
- 14.Duan J, Gherghe C, Liu D et al. Wnt1/betacatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J 2012;31:429–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiang FL, Fang M, Yutzey KE. Loss of beta-catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice. Nat Commun 2017;8:712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li R, Frangogiannis NG. Chemokines in cardiac fibrosis. Curr Opin Physiol 2021;19:80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hinderer S, Schenke-Layland K. Cardiac fibrosis - A short review of causes and therapeutic strategies. Adv Drug Deliv Rev 2019;146:77–82. [DOI] [PubMed] [Google Scholar]
- 18.Dai Z, Aoki T, Fukumoto Y, Shimokawa H. Coronary perivascular fibrosis is associated with impairment of coronary blood flow in patients with non-ischemic heart failure. J Cardiol 2012;60:416–21. [DOI] [PubMed] [Google Scholar]
- 19.Sohns C, Marrouche NF. Atrial fibrillation and cardiac fibrosis. Eur Heart J 2020;41:1123–1131. [DOI] [PubMed] [Google Scholar]
- 20.Disertori M, Mase M, Ravelli F. Myocardial fibrosis predicts ventricular tachyarrhythmias. Trends Cardiovasc Med 2017;27:363–372. [DOI] [PubMed] [Google Scholar]
- 21.Gulati A, Jabbour A, Ismail TF et al. Association of fibrosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. JAMA 2013;309:896–908. [DOI] [PubMed] [Google Scholar]
- 22.Martin R, Maury P, Bisceglia C et al. Characteristics of Scar-Related Ventricular Tachycardia Circuits Using Ultra-High-Density Mapping: A Multi-Center Study. Circ Arrhythm Electrophysiol 2018;11:e006569. [DOI] [PubMed] [Google Scholar]
- 23.Wong TC, Piehler K, Meier CG et al. Association between extracellular matrix expansion quantified by cardiovascular magnetic resonance and short-term mortality. Circulation 2012;126:1206–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frangogiannis NG. Matricellular proteins in cardiac adaptation and disease. Physiol Rev 2012;92:635–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang HM, Moudgil R, Scarabelli T, et al. Cardiovascular Complications of Cancer Therapy: Best Practices in Diagnosis, Prevention, and Management: Part 1. J Am Coll Cardiol 2017;70:2536–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang HM, Okwuosa TM, Scarabelli T, et al. Cardiovascular Complications of Cancer Therapy: Best Practices in Diagnosis, Prevention, and Management: Part 2. J Am Coll Cardiol 2017;70:2552–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carvalho FS, Burgeiro A, Garcia R, et al. Doxorubicin-induced cardiotoxicity: from bioenergetic failure and cell death to cardiomyopathy. Med Res Rev 2014;34:106–35. [DOI] [PubMed] [Google Scholar]
- 28.Ewer MS, Ewer SM. Cardiotoxicity of anticancer treatments. Nat Rev Cardiol 2015;12:547–58. [DOI] [PubMed] [Google Scholar]
- 29.Tewey KM, Rowe TC, Yang L, et al. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science 1984;226:466–8. [DOI] [PubMed] [Google Scholar]
- 30.Stingele J, Bellelli R, Boulton SJ. Mechanisms of DNA-protein crosslink repair. Nat Rev Mol Cell Biol 2017;18:563–573. [DOI] [PubMed] [Google Scholar]
- 31.Capranico G, Tinelli S, Austin CA, et al. Different patterns of gene expression of topoisomerase II isoforms in differentiated tissues during murine development. Biochim Biophys Acta 1992;1132:43–8. [DOI] [PubMed] [Google Scholar]
- 32.Damiani RM, Moura DJ, Viau CM, et al. Pathways of cardiac toxicity: comparison between chemotherapeutic drugs doxorubicin and mitoxantrone. Arch Toxicol 2016;90:2063–2076. [DOI] [PubMed] [Google Scholar]
- 33.Lyu YL, Kerrigan JE, Lin CP et al. Topoisomerase IIbeta mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res 2007;67:8839–46. [DOI] [PubMed] [Google Scholar]
- 34.Zhang S, Liu X, Bawa-Khalfe T et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med 2012;18:1639–42. [DOI] [PubMed] [Google Scholar]; • Article establishing a major pathway of anthracycline-induced cardiotoxicity, namely that Top-2β is also an anthracycline target, forming a Top-2β–anthracycline–DNA ternary cleavage complex that induces DNA strand breaks and ensuing cardiomyocyte death.
- 35.Yeh ET, Chang HM. Oncocardiology-Past, Present, and Future: A Review. JAMA Cardiol 2016;1:1066–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davies KJ, Doroshow JH. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J Biol Chem 1986;261:3060–7. [PubMed] [Google Scholar]
- 37.Cappetta D, De Angelis A, Sapio L et al. Oxidative Stress and Cellular Response to Doxorubicin: A Common Factor in the Complex Milieu of Anthracycline Cardiotoxicity. Oxid Med Cell Longev 2017;2017:1521020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doroshow JH, Davies KJ. Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J Biol Chem 1986;261:3068–74. [PubMed] [Google Scholar]
- 39.Berthiaume JM, Wallace KB. Adriamycin-induced oxidative mitochondrial cardiotoxicity. Cell Biol Toxicol 2007;23:15–25. [DOI] [PubMed] [Google Scholar]
- 40.Renu K, V GA, P BT, Arunachalam S. Molecular mechanism of doxorubicin-induced cardiomyopathy - An update. Eur J Pharmacol 2018;818:241–253. [DOI] [PubMed] [Google Scholar]
- 41.Panjrath GS, Patel V, Valdiviezo CI, et al. Potentiation of Doxorubicin cardiotoxicity by iron loading in a rodent model. J Am Coll Cardiol 2007;49:2457–64. [DOI] [PubMed] [Google Scholar]
- 42.Ichikawa Y, Ghanefar M, Bayeva M et al. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Invest 2014;124:617–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nebigil CG, Desaubry L. Updates in Anthracycline-Mediated Cardiotoxicity. Front Pharmacol 2018;9:1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poulin F, Thavendiranathan P. Cardiotoxicity Due to Chemotherapy: Role of Cardiac Imaging. Curr Cardiol Rep 2015;17. [DOI] [PubMed] [Google Scholar]
- 45.Davis J, Molkentin JD. Myofibroblasts: trust your heart and let fate decide. J Mol Cell Cardiol 2014;70:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakayama H, Otsu K. Mitochondrial DNA as an inflammatory mediator in cardiovascular diseases. Biochem J 2018;475:839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hamel Y, Mauvais FX, Madrange M et al. Compromised mitochondrial quality control triggers lipin1-related rhabdomyolysis. Cell Rep Med 2021;2:100370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-kappaB in the heart: to be or not to NF-kappaB. Circ Res 2011;108:1122–32. [DOI] [PubMed] [Google Scholar]
- 49.Martin SJ. Cell death and inflammation: the case for IL-1 family cytokines as the canonical DAMPs of the immune system. Febs J 2016;283:2599–2615. [DOI] [PubMed] [Google Scholar]
- 50.Frangogiannis NG. The immune system and cardiac repair. Pharmacol Res 2008;58:88–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hartupee J, Mann DL. Role of inflammatory cells in fibroblast activation. J Mol Cell Cardiol 2016;93:143–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leerink JM, van de Ruit M, Feijen EAM et al. Extracellular matrix remodeling in animal models of anthracycline-induced cardiomyopathy: a meta-analysis. J Mol Med (Berl) 2021;99:1195–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tanaka R, Umemura M, Narikawa M et al. Reactive fibrosis precedes doxorubicin-induced heart failure through sterile inflammation. ESC Heart Fail 2020;7:588–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun Z, Schriewer J, Tang M et al. The TGF-beta pathway mediates doxorubicin effects on cardiac endothelial cells. J Mol Cell Cardiol 2016;90:129–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Narikawa M, Umemura M, Tanaka R et al. Doxorubicin induces trans-differentiation and MMP1 expression in cardiac fibroblasts via cell death-independent pathways. PLoS One 2019;14:e0221940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baguma-Nibasheka M, Feridooni T, Zhang F, Pasumarthi KBS. Regulation of Transplanted Cell Homing by FGF1 and PDGFB after Doxorubicin Myocardial Injury. Cells 2021;10:2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng M, Kang YM, Liu W, et al. Inhibition of cyclooxygenase-2 reduces hypothalamic excitation in rats with adriamycin-induced heart failure. PLoS One 2012;7:e48771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rashikh A, Pillai KK, Najmi AK. Protective effect of a direct renin inhibitor in acute murine model of cardiotoxicity and nephrotoxicity. Fundam Clin Pharmacol 2014;28:489–500. [DOI] [PubMed] [Google Scholar]
- 59.Zhang J, Herman EH, Ferrans VJ. Effects of ICRF-186 [(L)1,2-bis(3,5-dioxopiperazinyl-1-yl)propane] on the toxicity of doxorubicin in spontaneously hypertensive rats. Toxicology 1994;92:179–92. [DOI] [PubMed] [Google Scholar]
- 60.Okumura K, Jin D, Takai S, Miyazaki M. Beneficial effects of angiotensin-converting enzyme inhibition in adriamycin-induced cardiomyopathy in hamsters. Jpn J Pharmacol 2002;88:183–8. [DOI] [PubMed] [Google Scholar]
- 61.Lencova-Popelova O, Jirkovsky E, Mazurova Y et al. Molecular remodeling of left and right ventricular myocardium in chronic anthracycline cardiotoxicity and post-treatment follow up. PLoS One 2014;9:e96055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cheng F, Shen Y, Mohanasundaram P et al. Vimentin coordinates fibroblast proliferation and keratinocyte differentiation in wound healing via TGF-beta-Slug signaling. Proc Natl Acad Sci U S A 2016;113:E4320–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gyongyosi M, Lukovic D, Zlabinger K et al. Liposomal doxorubicin attenuates cardiotoxicity via induction of interferon-related DNA damage resistance. Cardiovasc Res 2020;116:970–982. [DOI] [PubMed] [Google Scholar]
- 64.Chuva de Sousa Lopes SM, Feijen A, Korving J et al. Connective tissue growth factor expression and Smad signaling during mouse heart development and myocardial infarction. Dev Dyn 2004;231:542–50. [DOI] [PubMed] [Google Scholar]
- 65.Dorn LE, Petrosino JM, Wright P, Accornero F. CTGF/CCN2 is an autocrine regulator of cardiac fibrosis. J Mol Cell Cardiol 2018;121:205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cappetta D, Esposito G, Piegari E et al. SIRT1 activation attenuates diastolic dysfunction by reducing cardiac fibrosis in a model of anthracycline cardiomyopathy. Int J Cardiol 2016;205:99–110. [DOI] [PubMed] [Google Scholar]
- 67.van Almen GC, Swinnen M, Carai P et al. Absence of thrombospondin-2 increases cardiomyocyte damage and matrix disruption in doxorubicin-induced cardiomyopathy. J Mol Cell Cardiol 2011;51:318–28. [DOI] [PubMed] [Google Scholar]
- 68.Moslehi J, Amgalan D, Kitsis RN. Grounding Cardio-Oncology in Basic and Clinical Science. Circulation 2017;136:3–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crone SA, Zhao YY, Fan L et al. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat Med 2002;8:459–465. [DOI] [PubMed] [Google Scholar]
- 70.Ewer MS, O’Shaughnessy JA. Cardiac toxicity of trastuzumab-related regimens in HER2-overexpressing breast cancer. Clin Breast Cancer 2007;7:600–7. [PubMed] [Google Scholar]
- 71.Sawyer DB, Zuppinger C, Miller TA, et al. Modulation of anthracycline-induced myofibrillar disarray in rat ventricular myocytes by neuregulin-1beta and anti-erbB2: potential mechanism for trastuzumab-induced cardiotoxicity. Circulation 2002;105:1551–4. [DOI] [PubMed] [Google Scholar]
- 72.ElZarrad MK, Mukhopadhyay P, Mohan N et al. Trastuzumab alters the expression of genes essential for cardiac function and induces ultrastructural changes of cardiomyocytes in mice. PLoS One 2013;8:e79543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Riccio G, Esposito G, Leoncini E et al. Cardiotoxic effects, or lack thereof, of anti-ErbB2 immunoagents. FASEB J 2009;23:3171–8. [DOI] [PubMed] [Google Scholar]
- 74.Riccio G, Coppola C, Piscopo G et al. Trastuzumab and target-therapy side effects. Hum Vacc Immunother 2016;12:1124–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Florido R, Smith KL, Cuomo KK, Russell SD. Cardiotoxicity From Human Epidermal Growth Factor Receptor-2 (HER2) Targeted Therapies. J Am Heart Assoc 2017;6:e006915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bouwer NI, Jager A, Liesting C et al. Cardiac monitoring in HER2-positive patients on trastuzumab treatment: A review and implications for clinical practice. Breast 2020;52:33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Force T, Krause DS, Van Etten RA. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat Rev Cancer 2007;7:332–44. [DOI] [PubMed] [Google Scholar]
- 78.Patel S, Dushenkov A, Jungsuwadee P, et al. Team-Based Approach to Management of Hypertension Associated with Angiogenesis Inhibitors. J Cardiovasc Transl Res 2020;13:463–477. [DOI] [PubMed] [Google Scholar]
- 79.Qi WX, He AN, Shen Z, Yao Y. Incidence and risk of hypertension with a novel multi-targeted kinase inhibitor axitinib in cancer patients: a systematic review and meta-analysis. Br J Clin Pharmacol 2013;76:348–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.He H, Venema VJ, Guo XL, et al. Vascular endothelial growth factor signals endothelial cell production of nitric oxide and prostacyclin through Flk-1/KDR activation of c-Src. J Biol Chem 1999;274:25130–25135. [DOI] [PubMed] [Google Scholar]
- 81.Boe AE, Eren M, Murphy SB et al. Plasminogen activator inhibitor-1 antagonist TM5441 attenuates Nomega-nitro-L-arginine methyl ester-induced hypertension and vascular senescence. Circulation 2013;128:2318–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Muller-Deile J, Worthmann K, Saleem M, et al. The balance of autocrine VEGF-A and VEGF-C determines podocyte survival. Am J Physiol Renal Physiol 2009;297:F1656–67. [DOI] [PubMed] [Google Scholar]
- 83.Lankhorst S, Baelde HJ, Kappers MHW et al. Greater Sensitivity of Blood Pressure Than Renal Toxicity to Tyrosine Kinase Receptor Inhibition With Sunitinib. Hypertension 2015;66:543–549. [DOI] [PubMed] [Google Scholar]
- 84.Yoon YS, Uchida S, Masuo O et al. Progressive attenuation of myocardial vascular endothelial growth factor expression is a seminal event in diabetic cardiomyopathy: restoration of microvascular homeostasis and recovery of cardiac function in diabetic cardiomyopathy after replenishment of local vascular endothelial growth factor. Circulation 2005;111:2073–2085. [DOI] [PubMed] [Google Scholar]
- 85.Park CW, Kim HW, Lim JH et al. Vascular endothelial growth factor inhibition by dRK6 causes endothelial apoptosis, fibrosis, and inflammation in the heart via the Akt/eNOS axis in db/db mice. Diabetes 2009;58:2666–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Winnik S, Lohmann C, Siciliani G et al. Systemic VEGF inhibition accelerates experimental atherosclerosis and disrupts endothelial homeostasis--implications for cardiovascular safety. Int J Cardiol 2013;168:2453–61. [DOI] [PubMed] [Google Scholar]
- 87.Chintalgattu V, Rees ML, Culver JC et al. Coronary microvascular pericytes are the cellular target of sunitinib malate-induced cardiotoxicity. Sci Transl Med 2013;5:187ra69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Touyz RM, Herrmann J. Cardiotoxicity with vascular endothelial growth factor inhibitor therapy. NPJ Precis Oncol 2018;2:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fleming MR, Xiao L, Jackson KD, Beckman JA, Barac A, Moslehi JJ. Vascular Impact of Cancer Therapies: The Case of BTK (Bruton Tyrosine Kinase) Inhibitors. Circ Res 2021;128:1973–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xiao L, Salem JE, Clauss S et al. Ibrutinib-Mediated Atrial Fibrillation Attributable to Inhibition of C-Terminal Src Kinase. Circulation 2020;142:2443–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jiang L, Li L, Ruan Y et al. Ibrutinib promotes atrial fibrillation by inducing structural remodeling and calcium dysregulation in the atrium. Heart Rhythm 2019;16:1374–1382. [DOI] [PubMed] [Google Scholar]
- 92.Okada M. Regulation of the SRC family kinases by Csk. Int J Biol Sci 2012;8:1385–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Abushukair H, Syaj S, Ababneh O et al. First- versus second-generation Bruton tyrosine kinase inhibitors in Waldenstrom’s Macroglobulinemia: A systematic review and meta-analysis. Am J Hematol 2022;97:942–950. [DOI] [PubMed] [Google Scholar]
- 94.Estupinan HY, Berglof A, Zain R, Smith CIE. Comparative Analysis of BTK Inhibitors and Mechanisms Underlying Adverse Effects. Front Cell Dev Biol 2021;9:630942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Salem JE, Manouchehri A, Moey M et al. Cardiovascular toxicities associated with immune checkpoint inhibitors: an observational, retrospective, pharmacovigilance study. Lancet Oncol 2018;19:1579–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang L, Awadalla M, Mahmood SS et al. Cardiovascular magnetic resonance in immune checkpoint inhibitor-associated myocarditis. Eur Heart J 2020;41:1733–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Palaskas N, Lopez-Mattei J, Durand JB, et al. Immune Checkpoint Inhibitor Myocarditis: Pathophysiological Characteristics, Diagnosis, and Treatment. J Am Heart Assoc 2020;9:e013757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ball S, Ghosh RK, Wongsaengsak S et al. Cardiovascular Toxicities of Immune Checkpoint Inhibitors. J Am Coll Cardiol 2019;74:1714–1727. [DOI] [PubMed] [Google Scholar]
- 99.Zhu H, Ivanovic M, Nguyen A, et al. Immune checkpoint inhibitor cardiotoxicity: Breaking barriers in the cardiovascular immune landscape. J Mol Cell Cardiol 2021;160:121–127. [DOI] [PubMed] [Google Scholar]
- 100.Moslehi J, Lichtman AH, Sharpe AH, et al. Immune checkpoint inhibitor-associated myocarditis: manifestations and mechanisms. J Clin Invest 2021;131:e145186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Grabie N, Lichtman AH, Padera R. T cell checkpoint regulators in the heart. Cardiovasc Res 2019;115:869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhazykbayeva S, Pabel S, Mugge A, et al. The molecular mechanisms associated with the physiological responses to inflammation and oxidative stress in cardiovascular diseases. Biophys Rev 2020;12:947–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Drobni ZD, Alvi RM, Taron J et al. Association Between Immune Checkpoint Inhibitors With Cardiovascular Events and Atherosclerotic Plaque. Circulation 2020;142:2299–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Voskens CJ, Goldinger SM, Loquai C et al. The price of tumor control: an analysis of rare side effects of anti-CTLA-4 therapy in metastatic melanoma from the ipilimumab network. PLoS One 2013;8:e53745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang B, Wang H, Zhang M et al. Radiation-induced myocardial fibrosis: Mechanisms underlying its pathogenesis and therapeutic strategies. J Cell Mol Med 2020;24:7717–7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Farhood B, Goradel NH, Mortezaee K et al. Intercellular communications-redox interactions in radiation toxicity; potential targets for radiation mitigation. J Cell Commun Signal 2019;13:3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Venkatesulu BP, Mahadevan LS, Aliru ML et al. Radiation-Induced Endothelial Vascular Injury: A Review of Possible Mechanisms. JACC Basic Transl Sci 2018;3:563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Seemann I, Gabriels K, Visser NL et al. Irradiation induced modest changes in murine cardiac function despite progressive structural damage to the myocardium and microvasculature. Radiother Oncol 2012;103:143–50. [DOI] [PubMed] [Google Scholar]
- 109.Heckmann M, Douwes K, Peter R, Degitz K. Vascular activation of adhesion molecule mRNA and cell surface expression by ionizing radiation. Exp Cell Res 1998;238:148–54. [DOI] [PubMed] [Google Scholar]
- 110.Quarmby S, Hunter RD, Kumar S. Irradiation induced expression of CD31, ICAM-1 and VCAM-1 in human microvascular endothelial cells. Anticancer Res 2000;20:3375–81. [PubMed] [Google Scholar]
- 111.Lindner D, Zietsch C, Tank J et al. Cardiac fibroblasts support cardiac inflammation in heart failure. Basic Res Cardiol 2014;109:428. [DOI] [PubMed] [Google Scholar]
- 112.Thomas TP, Grisanti LA. The Dynamic Interplay Between Cardiac Inflammation and Fibrosis. Front Physiol 2020;11:529075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Boerma M, Kruse JJ, van Loenen M et al. Increased deposition of von Willebrand factor in the rat heart after local ionizing irradiation. Strahlenther Onkol 2004;180:109–16. [DOI] [PubMed] [Google Scholar]
- 114.Lof A, Muller JP, Brehm MA. A biophysical view on von Willebrand factor activation. J Cell Physiol 2018;233:799–810. [DOI] [PubMed] [Google Scholar]
- 115.Distler JH, Jungel A, Pileckyte M et al. Hypoxia-induced increase in the production of extracellular matrix proteins in systemic sclerosis. Arthritis Rheum 2007;56:4203–15. [DOI] [PubMed] [Google Scholar]
- 116.Yarnold J, Brotons MC. Pathogenetic mechanisms in radiation fibrosis. Radiother Oncol 2010;97:149–61. [DOI] [PubMed] [Google Scholar]
- 117.Vorotnikova E, Rosenthal RA, Tries M, et al. Novel Synthetic SOD/Catalase Mimetics Can Mitigate Capillary Endothelial Cell Apoptosis Caused by Ionizing Radiation. Radiat Res 2010;173:748–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Halle M, Gabrielsen A, Paulsson-Berne G et al. Sustained Inflammation Due to Nuclear Factor-Kappa B Activation in Irradiated Human Arteries. Journal of the American College of Cardiology 2010;55:1227–1236. [DOI] [PubMed] [Google Scholar]
- 119.Cuomo JR, Sharma GK, Conger PD, Weintraub NL. Novel concepts in radiation-induced cardiovascular disease. World J Cardiol 2016;8:504–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sridharan V, Aykin-Burns N, Tripathi P et al. Radiation-induced alterations in mitochondria of the rat heart. Radiat Res 2014;181:324–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Livingston K, Schlaak RA, Puckett LL, Bergom C. The Role of Mitochondrial Dysfunction in Radiation-Induced Heart Disease: From Bench to Bedside. Front Cardiovasc Med 2020;7:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Salata C, Ferreira-Machado SC, De Andrade CB, et al. Apoptosis induction of cardiomyocytes and subsequent fibrosis after irradiation and neoadjuvant chemotherapy. Int J Radiat Biol 2014;90:284–90. [DOI] [PubMed] [Google Scholar]
- 123.Kinnally KW, Peixoto PM, Ryu SY, Dejean LM. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim Biophys Acta 2011;1813:616–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rai V, Sharma P, Agrawal S, Agrawal DK. Relevance of mouse models of cardiac fibrosis and hypertrophy in cardiac research. Mol Cell Biochem 2017;424:123–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hong YJ, Park HS, Park JK et al. Early Detection and Serial Monitoring of Anthracycline-Induced Cardiotoxicity Using T1-mapping Cardiac Magnetic Resonance Imaging: An Animal Study. Sci Rep 2017;7:2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Seferovic PM, Tsutsui H, McNamara DM et al. Heart Failure Association of the ESC, Heart Failure Society of America and Japanese Heart Failure Society Position statement on endomyocardial biopsy. Eur J Heart Fail 2021;23:854–871. [DOI] [PubMed] [Google Scholar]
- 127.Liu T, Song D, Dong J et al. Current Understanding of the Pathophysiology of Myocardial Fibrosis and Its Quantitative Assessment in Heart Failure. Front Physiol 2017;8:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ho JE, Liu C, Lyass A et al. Galectin-3, a marker of cardiac fibrosis, predicts incident heart failure in the community. J Am Coll Cardiol 2012;60:1249–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Passino C, Barison A, Vergaro G et al. Markers of fibrosis, inflammation, and remodeling pathways in heart failure. Clin Chim Acta 2015;443:29–38. [DOI] [PubMed] [Google Scholar]
- 130.Bayes-Genis A, de Antonio M, Vila J et al. Head-to-head comparison of 2 myocardial fibrosis biomarkers for long-term heart failure risk stratification: ST2 versus galectin-3. J Am Coll Cardiol 2014;63:158–66. [DOI] [PubMed] [Google Scholar]
- 131.Lopez B, Gonzalez A, Ravassa S et al. Circulating Biomarkers of Myocardial Fibrosis: The Need for a Reappraisal. J Am Coll Cardiol 2015;65:2449–56. [DOI] [PubMed] [Google Scholar]
- 132.Popovic ZB, Kwon DH, Mishra M et al. Association between regional ventricular function and myocardial fibrosis in hypertrophic cardiomyopathy assessed by speckle tracking echocardiography and delayed hyperenhancement magnetic resonance imaging. J Am Soc Echocardiogr 2008;21:1299–305. [DOI] [PubMed] [Google Scholar]
- 133.Wang J, Zhang Y, Zhang L et al. Assessment of Myocardial Fibrosis Using Two-Dimensional and Three-Dimensional Speckle Tracking Echocardiography in Dilated Cardiomyopathy With Advanced Heart Failure. J Card Fail 2021;27:651–661. [DOI] [PubMed] [Google Scholar]
- 134.Plana JC, Thavendiranathan P, Bucciarelli-Ducci C, Lancellotti P. Multi-Modality Imaging in the Assessment of Cardiovascular Toxicity in the Cancer Patient. JACC Cardiovasc Imaging 2018;11:1173–1186. [DOI] [PubMed] [Google Scholar]
- 135.Celutkiene J, Pudil R, Lopez-Fernandez T et al. Role of cardiovascular imaging in cancer patients receiving cardiotoxic therapies: a position statement on behalf of the Heart Failure Association (HFA), the European Association of Cardiovascular Imaging (EACVI) and the Cardio-Oncology Council of the European Society of Cardiology (ESC). Eur J Heart Fail 2020;22:1504–1524. [DOI] [PubMed] [Google Scholar]; • Societal outline of the evidence supporting various imaging modalities for the assessment of cardiotoxicity.
- 136.Melendez GC, Hundley WG. Is Myocardial Fibrosis a New Frontier for Discovery in Cardiotoxicity Related to the Administration of Anthracyclines? Circ Cardiovasc Imaging 2016;9:e005797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Flett AS, Hayward MP, Ashworth MT et al. Equilibrium contrast cardiovascular magnetic resonance for the measurement of diffuse myocardial fibrosis: preliminary validation in humans. Circulation 2010;122:138–44. [DOI] [PubMed] [Google Scholar]
- 138.Jordan JH, Vasu S, Morgan TM et al. Anthracycline-Associated T1 Mapping Characteristics Are Elevated Independent of the Presence of Cardiovascular Comorbidities in Cancer Survivors. Circ Cardiovasc Imaging 2016;9:e004325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Pezel T, Viallon M, Croisille P et al. Imaging Interstitial Fibrosis, Left Ventricular Remodeling, and Function in Stage A and B Heart Failure. JACC Cardiovasc Imaging 2021;14:1038–1052. [DOI] [PubMed] [Google Scholar]
- 140.Neilan TG, Coelho-Filho OR, Pena-Herrera D et al. Left ventricular mass in patients with a cardiomyopathy after treatment with anthracyclines. Am J Cardiol 2012;110:1679–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Neilan TG, Coelho-Filho OR, Shah RV et al. Myocardial extracellular volume by cardiac magnetic resonance imaging in patients treated with anthracycline-based chemotherapy. Am J Cardiol 2013;111:717–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ambale-Venkatesh B, Lima JA. Cardiac MRI: a central prognostic tool in myocardial fibrosis. Nat Rev Cardiol 2015;12:18–29. [DOI] [PubMed] [Google Scholar]
- 143.Farhad H, Staziaki PV, Addison D et al. Characterization of the Changes in Cardiac Structure and Function in Mice Treated With Anthracyclines Using Serial Cardiac Magnetic Resonance Imaging. Circ Cardiovasc Imaging 2016;9:e003584. [DOI] [PMC free article] [PubMed] [Google Scholar]; • • Mouse study with translational implications characterizing anthracycline-induced cardiotoxicity with the development of cardiac fibrosis, association with left ventricular dysfunction, and mortality prediction.
- 144.Noel CV, Rainusso N, Robertson M et al. Early detection of myocardial changes with and without dexrazoxane using serial magnetic resonance imaging in a pre-clinical mouse model. Cardiooncology. 2021;7:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cottin Y, Ribuot C, Maupoil V et al. Early incidence of adriamycin treatment on cardiac parameters in the rat. Can J Physiol Pharmacol 1994;72:140–5. [DOI] [PubMed] [Google Scholar]
- 146.Park HS, Hong YJ, Han K et al. Ultrahigh-field cardiovascular magnetic resonance T1 and T2 mapping for the assessment of anthracycline-induced cardiotoxicity in rat models: validation against histopathologic changes. J Cardiovasc Magn Reson 2021;23:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hong YJ, Kim TK, Hong D et al. Myocardial Characterization Using Dual-Energy CT in Doxorubicin-Induced DCM Comparison With CMR T1-Mapping and Histology in a Rabbit Model. JACC Cardiovasc Imaging 2016;9:836–845. [DOI] [PubMed] [Google Scholar]
- 148.Galan-Arriola C, Lobo M, Vilchez-Tschischke JP et al. Serial Magnetic Resonance Imaging to Identify Early Stages of Anthracycline-Induced Cardiotoxicity. J Am Coll Cardiol 2019;73:779–791. [DOI] [PubMed] [Google Scholar]
- 149.Schelbert EB, Piehler KM, Zareba KM et al. Myocardial Fibrosis Quantified by Extracellular Volume Is Associated With Subsequent Hospitalization for Heart Failure, Death, or Both Across the Spectrum of Ejection Fraction and Heart Failure Stage. J Am Heart Assoc 2015;4:e002613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.van den Borne SW, Isobe S, Verjans JW et al. Molecular imaging of interstitial alterations in remodeling myocardium after myocardial infarction. J Am Coll Cardiol 2008;52:2017–28. [DOI] [PubMed] [Google Scholar]
- 151.Verjans J, Wolters S, Laufer W et al. Early molecular imaging of interstitial changes in patients after myocardial infarction: comparison with delayed contrast-enhanced magnetic resonance imaging. J Nucl Cardiol 2010;17:1065–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Higuchi T, Bengel FM, Seidl S et al. Assessment of alphavbeta3 integrin expression after myocardial infarction by positron emission tomography. Cardiovasc Res 2008;78:395–403. [DOI] [PubMed] [Google Scholar]
- 153.Jenkins WS, Vesey AT, Stirrat C et al. Cardiac alphaVbeta3 integrin expression following acute myocardial infarction in humans. Heart 2017;103:607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hinz B. Masters and servants of the force: the role of matrix adhesions in myofibroblast force perception and transmission. Eur J Cell Biol 2006;85:175–81. [DOI] [PubMed] [Google Scholar]
- 155.Meoli DF, Sadeghi MM, Krassilnikova S et al. Noninvasive imaging of myocardial angiogenesis following experimental myocardial infarction. J Clin Invest 2004;113:1684–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Heckmann MB, Reinhardt F, Finke D et al. Relationship Between Cardiac Fibroblast Activation Protein Activity by Positron Emission Tomography and Cardiovascular Disease. Circ Cardiovasc Imaging 2020;13:e010628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Siebermair J, Kohler MI, Kupusovic J et al. Cardiac fibroblast activation detected by Ga-68 FAPI PET imaging as a potential novel biomarker of cardiac injury/remodeling. J Nucl Cardiol 2021;28:812–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gu Y, Han K, Zhang Z et al. (68)Ga-FAPI PET/CT for molecular assessment of fibroblast activation in right heart in pulmonary arterial hypertension: a single-center, pilot study. J Nucl Cardiol Epub 2022 Mar 23. [DOI] [PubMed] [Google Scholar]
- 159.Diekmann J, Koenig T, Thackeray JT et al. Cardiac Fibroblast Activation in Patients Early After Acute Myocardial Infarction: Integration with Magnetic Resonance Tissue Characterization and Subsequent Functional Outcome. J Nucl Med Epub 2022. Feb 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Molkentin JD, Bugg D, Ghearing N et al. Fibroblast-Specific Genetic Manipulation of p38 Mitogen-Activated Protein Kinase In Vivo Reveals Its Central Regulatory Role in Fibrosis. Circulation 2017;136:549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]; • • Article estbalishing that signals from diverse modes of injury converge on p38α MAP kinase within the fibroblast to program the fibrotic response and myofibroblast formation in vivo using conditional gene deletion and transgenic overexpression strategies.
- 161.Khalil H, Kanisicak O, Prasad V et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest 2017;127:3770–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]; • • Article dissecting the in vivo importance of TGF-β-Smad2/3 signaling in pressure overload-induced and fibroblast-mediated cardiac fibrosis using pertinent genetically modified mice.
- 162.Travers JG, Tharp CA, Rubino M, McKinsey TA. Therapeutic targets for cardiac fibrosis: from old school to next-gen. J Clin Invest 2022;132:e148554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Stratton MS, Koch KA, McKinsey TA. p38alpha: A Profibrotic Signaling Nexus. Circulation 2017;136:562–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Park S, Nguyen NB, Pezhouman A, Ardehali R. Cardiac fibrosis: potential therapeutic targets. Transl Res 2019;209:121–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sweeney M, Corden B, Cook SA. Targeting cardiac fibrosis in heart failure with preserved ejection fraction: mirage or miracle? EMBO Mol Med 2020;12:e10865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Morimoto T, Sunagawa Y, Kawamura T et al. The dietary compound curcumin inhibits p300 histone acetyltransferase activity and prevents heart failure in rats. J Clin Invest 2008;118:868–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bugyei-Twum A, Advani A, Advani SL et al. High glucose induces Smad activation via the transcriptional coregulator p300 and contributes to cardiac fibrosis and hypertrophy. Cardiovasc Diabetol 2014;13:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Weinert BT, Narita T, Satpathy S et al. Time-Resolved Analysis Reveals Rapid Dynamics and Broad Scope of the CBP/p300 Acetylome. Cell 2018;174:231–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jeong MY, Lin YH, Wennersten SA et al. Histone deacetylase activity governs diastolic dysfunction through a nongenomic mechanism. Sci Transl Med 2018;10:eaao0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Travers JG, Wennersten SA, Pena B et al. HDAC Inhibition Reverses Preexisting Diastolic Dysfunction and Blocks Covert Extracellular Matrix Remodeling. Circulation 2021;143:1874–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Aghajanian H, Kimura T, Rurik JG et al. Targeting cardiac fibrosis with engineered T cells. Nature 2019;573:430–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rurik JG, Tombacz I, Yadegari A et al. CAR T cells produced in vivo to treat cardiac injury. Science 2022;375:91–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.June CH, Sadelain M. Chimeric Antigen Receptor Therapy. N Engl J Med 2018;379:64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Friedman SL. Fighting Cardiac Fibrosis with CAR T Cells. N Engl J Med 2022;386:1576–1578. [DOI] [PubMed] [Google Scholar]