

ABSTRACT
In a recent study in Nature Immunology, Musella et al. demonstrate that suboptimal type I interferon (IFN-I) signaling in tumors undergoing immunogenic cell death (ICD) facilitates the accumulation of cancer stem cells (CSCs) by triggering the epigenetic regulator lysine demethylase 1B (KDM1B). KDM1B stands out as a promising target for the development of novel strategies to improve anti-cancer responses driven by ICD.
KEYWORDS: Chemotherapy, immunogenic cell death, epigenetic immune escape, cancer stem cells, interferon type I, KDM1B
Type I interferons (IFN-I) are a broad class of inflammatory cytokines that are secreted when host defenses are compromised. In cancer, the role of IFN-I has been considered essential for immunogenic responses of anti-cancer treatments including radiation therapy and chemotherapy.1–3 However, evidence also indicate that IFN-I can promote negative feedback and immunosuppression. For instance, low IFN-I responsiveness epigenetically imprinted predicts survival increase of anti-programmed cell death protein 1 (PD-1) while high responsiveness to IFN-I was associated with poor survival outcome.4 Therefore, like chronic infection, ongoing IFN-I response may be a central driver of immune dysfunction and resistance to treatments.5–7
The mechanisms responsible for immune evasion subsequent to a suboptimal IFN-I response remain elusive. Recently, Musella et al. demonstrate that the expansion of cancer stem cells (CSC; a rare population of cancer cells with great treatment resistance, immunoevasive and metastatic properties) is associated with IFN-I response.8
Musella et al. first investigated in vitro the induction of putative CSCs markers; namely prominin 1 (Prom 1, also known as CD133), CD24 and CD44; in multiple epithelial and mesenchymal cancer cells in response to IFN-I. They found that IFN-I promotes the enrichment of CD133+ CD24+ CD44+ CSCs in most of the cell lines tested except for the non-tumorigenic MCF10A and the highly CSC-enriched HMLER cells (presumably due to their high expression of CSC markers at baseline). In line with the heterogeneity attributed to CSCs, mouse fibrosarcoma MCA205 cells presented two subpopulations of IFN-I- induced CSCs: CD133+ CD24+ CD44+ low (CD44L) and the CD133+ CD24+ CD44+ high (CD44H). Moreover, MCA205 cells exposed to IFN-I led to an increase of CSC markers in both CD133+ (CSC) and CD133- (non-CSC) cells, indicating that IFN-I drives stemness in both subsets; findings that were further substantiated by morphological, phenotypical and functional evidence.8
Interestingly, local treatment with a single dose of 105 U IFN-I promoted the expansion of CSC within the MCA205 tumors growing in syngeneic animals as compared to repeated administration of IFN-I (7 injections of 2 × 104 U each administered over the entire course of the experiment);8 therefore highlighting distinct responses of the tumor microenvironment in respect to the magnitude of induction and timing of IFN-I signaling.
Next, the authors exposed pre-validated MCA205-derived clones deficient for the major nucleic acid sensors and signal transducers of the IFN-I pathway to oxaliplatin, an ICD inducer capable to stimulate IFN-I response. Confirming their previous findings, MCA205 parental cells exhibited an expansion of CD44H and CD44L CSC subsets post oxaliplatin treatment; an effect that was partially reduced in most of IFN-I deficient clones. This observation was further supported at the transcriptional level with decreased expression of CSC-related genes such as Sox2, Nanog, Myc and Oct3/4 in IFN-I deficient MCA205 cells. To ask if similar effect can be obtained in vivo, Musella and colleagues have treated MCA205 tumors growing in syngeneic animals with cisplatin, a chemotherapeutic agent unable to induce ICD, and confirmed the absence of CSC expansion into the tumors. Importantly, doxorubicin-mediated ICD induction combined with repeated injections of 2 × 104 U IFN-I prevented the induction of CSC-like cells thus resulting in an improved tumor control and mice survival.8 Therefore, these data demonstrate that accumulation of CSC from suboptimal IFN-I response can be reverted with repeated administration of IFN-I.
To decipher the mechanism responsible for CSC enrichment post ICD, authors performed co-cultures experiments of MCA205, AT3 or CT26 cells with benzonase, RNAse and DNAse. While the magnitude of response is different, authors observed that only the degradation of all nucleic acids (i.e. benzonase) impaired the oxaliplatin-induced CSC expansion, thus suggesting that CSC accumulation depends on an intact IFN-I pathway. Importantly, the oxaliplatin-induced accumulation of CSC amongst MCA205 cells was reduced by the actin inhibitor cytochalasin D, indicating the contribution of extracellular vesicles (EV) that contain stemness-related transcriptional factors, namely Sox2, Nanog, Myc and Oct3/4.8 Globally, these data indicate that CSC expansion occurs via free and EV-mediated transfer of nucleic acids and stem-related genes. These data confirm and expand on previous findings demonstrating the transfer of IFN-I stimulatory nucleic acid species via EV.9
To further characterize in vitro and in vivo the expansion of CSC cells in the context of ICD, authors have evaluated their heterogeneity, finding that CD44H but not CD44L CSC MCA205 cells displayed greater chemoresistance, metastatic potential and immunoevasive properties as compared to parental MCA205. Specifically, parental MCA205 were superior to CSC-like MCA205 at inducing T-cell cross priming, consistent with the increase in expression of immunosuppressive markers such as PD-L1 and TIM-3 as well as the pro-metastatic cytokine secretome of CSC-like MCA205 cells.8
To determine the mechanism responsible for CSC rewiring post IFN-I, Musella et al. performed ATACseq and first analyzed loci that were closed in parental but opened in CD44H CSC MCA205 cells. Their findings identified genes controlling cancer stemness (Myc and Sox), epithelial-to-mesenchymal transition (Gata6 and Tfcp2) and immune escape (Gpr17 and Serpin) in CD44H CSC MCA205 cells. Importantly, loci opened in parental MCA205 but closed in CD44H CSC MCA205 belonged to antigen presentation machinery (Tap1, Tap2 and Ctsl). Integration with RNA sequencing confirmed these observations. Overall, the IFN-I induction of cancer stemness in MCA205 cells is associated with a substantial chromatin remodeling of many transcription factors binding motifs that largely belong to cancer stemness, tissue remodeling and immunoevasion.8
Next, the authors investigated the epigenic regulator KDM1B (lysine demethylase 1B) because of its critical role in chromatin remodeling in cancer evolution, cellular plasticity and immune escape. ATACseq of KDM1B-deficient MCA205 cells revealed that the lack of KDM1B fosters chromatin closing at loci that were linked with IFN-I-induced stemness while overexpression of KDM1B permit the opening of these loci. The epigenetic role of KDM1B was further validated by ChIP-seq analysis of CD44H CSC MCA205 cells, with evidence of KDM1B interactions with various genes involved in stemness maintenance, embryonic development, and invasiveness.8 Blockade of KDM1B with tranylcypromine (TCP) impaired the enrichment of CSC amongst MCA205 cells exposed to oxaliplatin. Importantly, TCP limited the expansion of CSC in response to doxorubicin amongst MCA205 tumors growing in an immunocompetent host, therefore improving tumor control. Consistently, KDM1B-overexpressing MCA205 tumors did increase the representation of CSC at baseline and promoted treatment resistance, and metastatic properties.8 Altogether, these data confirm the major role of KDM1B as an epigenetic driver of ICD-induced cancer stemness in the context of suboptimal IFN-I response.
Bioinformatic analysis of public databases of breast cancer patients responsive to anthracyclines revealed a positive correlation of KDM1B levels with transcriptional signatures of cancer stemness. Finally, Musella et al. took advantage of the METABRIC breast cancer patient cohort to perform a multivariate survival analysis, finding that the group of patients with a significant decreased of disease-specific survival displayed high KDM1B expression together with genes signatures associated with IFN-I and stemness.8 These data are in line with previous studies highlighting a negative prognostic role of IFN-I signatures in breast cancer patients.10
In summary, studies conducted by Musella and colleagues revealed that suboptimal induction of IFN-I in the context of ICD activates the epigenetic regulator KDM1B in cancer cells to drive treatment resistance and tumor progression (Figure 1). These findings shed light on KDM1B as a novel actionable target to improve anti-cancer responses driven by ICD.
Figure 1.
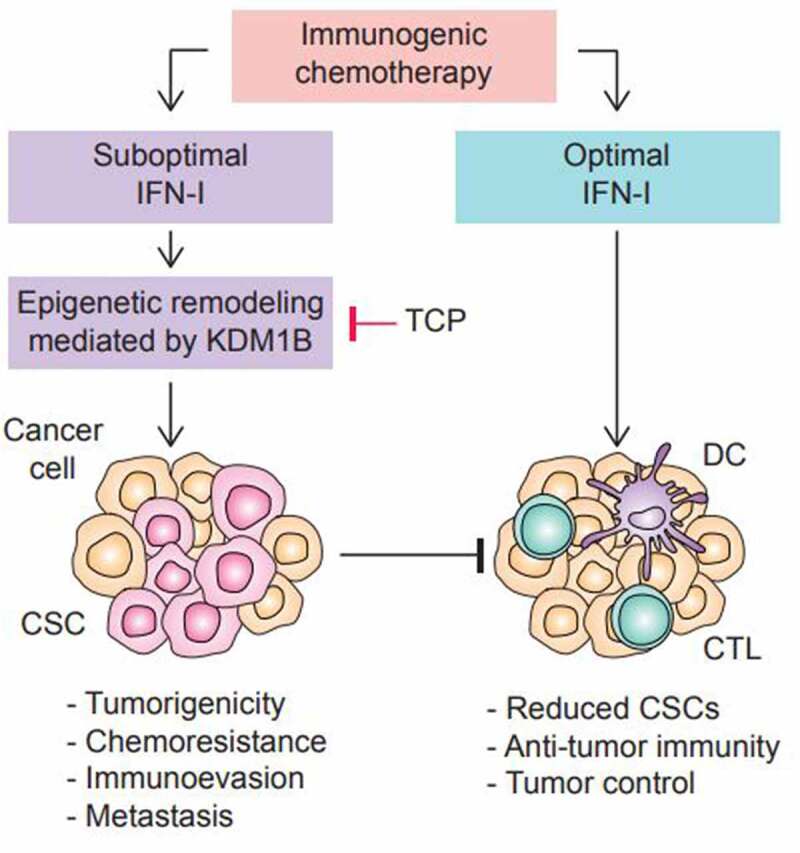
Chemotherapy-induced IFN-I promotes KDM1B-mediated CSCs accumulation and tumor resistance. Chemotherapeutics can induce immunogenic cell death (i.e., oxaliplatin or doxorubicin), resulting in the release of damage-associated molecular patterns (DAMPs), such as type I interferons (IFN-I), that stimulate anti-tumor immunity mediated by dendritic cells (DCs) and cytotoxic T lymphocytes (CTLs). Depending on the dose and timing of the IFN-I signaling, IFN-I induces an epigenetic remodeling mediated by the lysine demethylase 1B (KDM1B) that leads to the accumulation of cancer stem cells (CSCs). These CSCs promote tumorigenicity, chemoresistance, immunoevasion and metastasis. Inhibition of KDM1B with tranylcypromine (TCP) in combination with immunogenic chemotherapy, prevented the accumulation of CSCs and lead to tumor control.
Acknowledgments
MDM is supported by a 2022-SITC Nektar Therapeutics Equity and Inclusion in Cancer Immunotherapy fellowship. CVB is supported by grants from the Uncle Kory Foundation, the St-Baldrick’s Foundation and the StacheStrong Foundation.
Funding Statement
This work was supported by the Society for Immunotherapy of Cancer [2022-SITC Bektar postdoctoral fellowship];Uncle Kory Foundation;Stache Strong Foundation;St. Baldrick’s Foundation.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Disclosure statement
Authors have nothing to disclose pertaining to this work.
References
- 1.Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remédios C, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014;20(11):1301–3. doi: 10.1038/nm.3708. [DOI] [PubMed] [Google Scholar]
- 2.Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, Inghirami G, Coleman CN, Formenti SC, Demaria S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. 2017;8:15618. doi: 10.1038/ncomms15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamazaki T, Kirchmair A, Sato A, Buqué A, Rybstein M, Petroni G, Bloy N, Finotello F, Stafford L, Navarro Manzano E, et al. Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat Immunol. 2020;21(10):1160–1171. doi: 10.1038/s41590-020-0751-0. [DOI] [PubMed] [Google Scholar]
- 4.Boukhaled GM, Gadalla R, Elsaesser HJ, Abd-Rabbo D, Quevedo R, Yang SYC, Guo M, Wang BX, Noamani B, Gray D, et al. Pre-encoded responsiveness to type I interferon in the peripheral immune system defines outcome of PD1 blockade therapy. Nat Immunol. 2022;23(8):1273–1283. doi: 10.1038/s41590-022-01262-7. [DOI] [PubMed] [Google Scholar]
- 5.Zhang X, Wang S, Zhu Y, Zhang M, Zhao Y, Yan Z, Wang Q, Li X.. Double-edged effects of interferons on the regulation of cancer-immunity cycle. Oncoimmunology. 2021;10(1):1929005. doi: 10.1080/2162402X.2021.1929005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sprooten J, Garg AD. Type I interferons and endoplasmic reticulum stress in health and disease. Int Rev Cell Mol Biol. 2020;350:63–118. doi: 10.1016/bs.ircmb.2019.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gong W, Donnelly CR, Heath BR, Bellile E, Donnelly LA, Taner HF, Broses L, Brenner JC, Chinn SB, Ji -R-R, et al. Cancer-specific type-I interferon receptor signaling promotes cancer stemness and effector CD8+ T-cell exhaustion. Oncoimmunology. 2021;10(1):1997385. doi: 10.1080/2162402X.2021.1997385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Musella M, Guarracino A, Manduca N, Galassi C, Ruggiero E, Potenza A, Maccafeo E, Manic G, Mattiello L, Soliman Abdel Rehim S, et al. Type I IFNs promote cancer cell stemness by triggering the epigenetic regulator KDM1B. Nature Immunol. 2022. doi: 10.1038/s41590-022-01290-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greiner AC, Knebel E. Editors., in health professions education: a bridge to quality. 2003: Washington (DC). doi: 10.17226/10681. [DOI] [PubMed] [Google Scholar]
- 10.Rodriguez-Ruiz ME, Buqué A, Hensler M, Chen J, Bloy N, Petroni G, Sato A, Yamazaki T, Fucikova J, Galluzzi L, et al. Apoptotic caspases inhibit abscopal responses to radiation and identify a new prognostic biomarker for breast cancer patients. Oncoimmunology. 2019;8(11):e1655964. doi: 10.1080/2162402X.2019.1655964. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.