

ABSTRACT
A major challenge in natural killer (NK) cell immunotherapy is the limited persistence of NK cells in vivo. However, the proliferation of NK cells is dependent on cytokines such as interleukin-2 (IL-2). Although IL-2 is a critical cytokine for NK cell activation and survival, IL-2 administration in adoptive NK cell therapy can induce adverse toxicities. To improve the persistence of NK cells and attenuate the systemic toxicity of IL-2, we constructed a cell-restricted artificial IL-2, named membrane-bound IL-2 (mbIL-2), comprising human IL-2 and human IL-2Rα joined by a classic linker. We found that mbIL-2-activated NK-92 cells can survive and proliferate in vitro and in vivo, independent of exogenous IL-2, while mbIL-2-expressing NK-92 cells do not support bystander cell survival or proliferation. Additionally, mbIL-2 enhanced NK-92 cell-mediated antitumor activity by tuning the IL-2 receptor downstream signals and NK cell receptor repertoire expression. To conclude, our novel mbIL-2 improves NK-92 cell persistence and enhances NK-92 cell-mediated antitumor activity. NK-92 cells genetically modified to express the novel mbIL-2 with potential significance for clinical development.
KEYWORDS: IL-2, IL-2Rα, natural killer cells, NK cells persistence, cancer, immunotherapy, NKG2D
1. Introduction
Natural killer (NK) cells are cytotoxic lymphocytes of the innate immune system, which play a vital role in anti-viral and antitumor responses, therefore, have promising clinical use.1,2 To recognize tumor cells, NK cells, unlike T cells, rely on a set of activating and inhibitory receptors, without prior sensitization or major histocompatibility complex (MHC) class I restriction.3–5 In humans, primary NK cell immunodeficiency is associated with increased risk of malignancy and exacerbated outcomes.6,7 In cancer patients, the degree of NK cell infiltration in tumor tissues is related to the prognosis of certain patient cohorts.8,9 Despite their strong antitumor activity, NK cells face significant challenges, including limited persistence in vivo and obstacles in reaching the tumor sites,10 which hamper their effectiveness. Therefore, different strategies have been developed to enhance NK cell activity and proliferation in cancer patients, such as modification of cytokines or their receptors to specifically activate NK cells.11–13
Cytokines are pivotal for the development, maturation, activation, proliferation, and survival of NK cells.14 Some cytokines, including IL-2, IL-15, IL-18, IL-21, and IFN-γ, have been employed to restore attenuated NK cell function in tumor microenvironment.1,15 Among them, IL-2 is one of the most studied cytokine activators of NK cells and has several positive antitumor effects in NK cell immunotherapy. Studies have shown that NK cells in IL-2-deficient mice exhibit impaired cytotoxicity and IFN-γ production.16 IL-2 also improves NK cell proliferation and promotes the production of perforin and granzyme B.17,18 Numerous attempts have been made to use exogenous IL-2 to activate ex vivo NK cell proliferation or to administer IL-2 systemically to cancer patients in vivo to augment the proliferation and cytotoxic function of NK cells.19–21 However, IL-2 could induce regulatory T (Treg) cell activation and lead to severe toxicity, including severe vascular leak syndrome and neurotoxicity.14,22,23 Studies have shown that IL-2 activated Treg cell inhibit NK cell antitumor or antiviral responses. Mechanistically, Treg cell suppress NK cell antitumor activity through cell–cell contact and TGF-β. Depletion of Treg cell combination with IL-2 therapy promoted NK cell mediated antitumor activity.24–26
To limit the systemic toxicity of IL-2 and to avoid bystander cell activation, Konstantinidis et al. constructed an endoplasmic reticulum (ER)-retained IL-2.12 They demonstrated that ER-retained IL-2 provided NK-92 cell autocrine growth stimulation without support for bystander cell growth. Moreover, the cytotoxicity of ER-retained IL-2 expressed NK-92 cells was not changed compared with parental NK-92 cells. Youssef et al. engineered a chimeric IL-2-IL-2Rβ fusion protein. Compared to the secreting-IL-2, chimeric IL-2-IL-2Rβ fusion protein-expressing NK-92 cells were more activated and cytotoxic, and they displayed superior antitumor effects in vivo.27 Although the mechanism behind chimeric IL-2-IL-2Rβ fusion protein for NK activation is unclear, this study offers a potential strategy to activate NK cells and avoid the side effects caused by IL-2.27
IL-2 displays its activity through three classes of IL-2 receptor complexes: a low-affinity receptor IL-2Rα, an intermediate-affinity dimeric receptor complex containing IL-2Rβ and IL-2Rγc, and a high-affinity trimeric receptor complex formed by IL-2Rα, IL-2Rβ, and IL-2Rγc. The high-affinity trimeric IL-2Rαβγc is highly expressed by Treg cells, whereas activated NK cells mostly express the intermediate-affinity dimeric receptor complex IL-2Rβγc.14,28,29 However, the low-affinity receptor IL-2Rα is essential for the sensitivity of human NK cells.30 To simulate the biological process of high-affinity trimeric receptor complex in NK cells and avoid the side effects caused by IL-2, we constructed a cell-restricted artificial IL-2, named membrane-bound IL-2 (mbIL-2), by fusion of human IL-2 and human IL-2Rα with a (G4S)3 linker. Here, we demonstrated that mbIL-2 can vigorously improve NK-92 cell survival and proliferation, as well as enhance NK-92 cell-mediated antitumor activity by tuning IL-2 receptor downstream signals and NK cell receptor repertoire expression. To conclude, the novel mbIL-2 was indicated as a useful strategy for improving adoptive NK cell immunotherapy.
2. Material and methods
2.1. Cell lines and cell culture conditions
K562, 293 T, PC3, HO-8910, A375, SK-mel-28, A549, PC9, SW620, HCT116, U87, LN18, LN229, and NK-92 cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). K562, PC3, and PC9 cells were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS). 293 T, HO-8910, A375, SK-mel-28, A549, SW620, HCT116, U87, LN18, and LN229 cells were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% FBS. NK-92 cells were maintained in α-minimum essential medium (α-MEM) supplemented with 0.2 mM inositol, 0.1 mM 2-mercaptoethanol, 0.02 mM folic acid, 200 U/ml recombinant human IL-2 (PeproTech, Cranbury, NJ, USA), 12.5% horse serum, and 12.5% FBS.
2.2. Plasmid construction
To construct the membrane-bound IL-2 (mbIL-2), human IL-2 (GenBank NM_000586.3) was fused to a classic linker (G4S)331 that was directly linked to human IL-2Rα (GenBank NM_000417.3). After validating the sequence, mbIL-2 was cloned into the pCDH-MSCV-MCS-EF1α+Puro plasmid (SBI, Palo Alto, CA, USA) for lentiviral production (Figure 1a).
Figure 1.
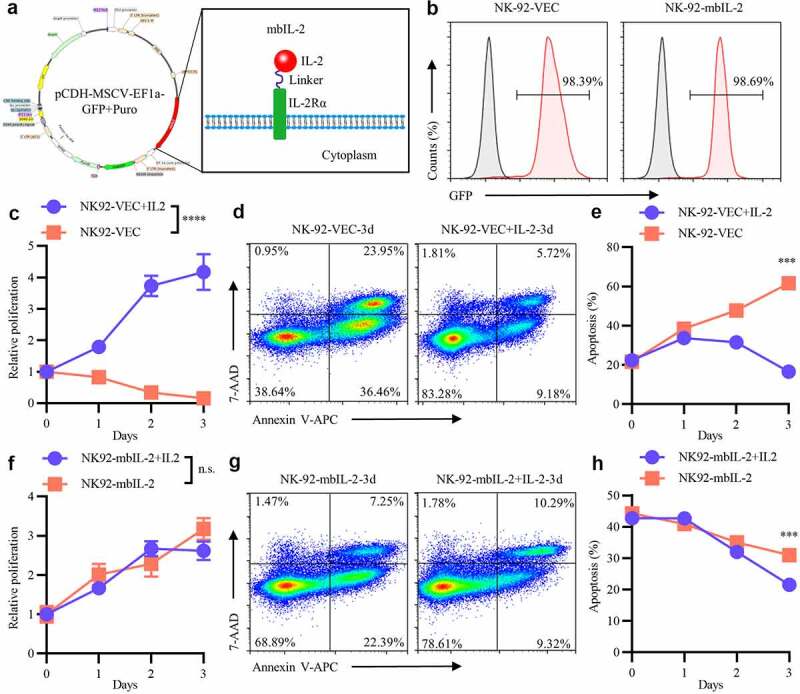
MbIL-2 maintains NK-92 cells survival and proliferation in vitro.
a: Schematic representation of mbIL-2 expression plasmids. b: Representative flow cytometry analysis showing the expression of mbIL-2 in NK-92 cells. NK-92 cells transduced with lentiviral for flow cytometry analysis. c: CCK8 assay of NK-92-VEC cell proliferation (n = 6). NK-92-VEC cells maintained with or without 200 U/ml IL-2 for assays. d–e: Analysis of the proportion of NK-92-VEC apoptotic cells under IL-2 culture conditions. NK-92-VEC cells maintained with or without 200 U/ml IL-2 for assays. d: Representative flow cytometry analysis showing the proportion of NK-92-VEC apoptotic cells under IL-2 condition culture after three days. e: Quantification and statistical analysis of the data (n = 3). f: CCK8 assay of NK-92-mbIL-2 cell proliferation (n = 6). NK-92-mbIL-2 cells maintained with or without 200 U/ml IL-2 for assays. g–h: Analysis of the proportion of NK-92-mbIL-2 apoptotic cells under IL-2 culture conditions. NK-92-mbIL-2 cells maintained with or without 200 U/ml IL-2 for assays. g: Representative flow cytometry analysis showing the proportion of NK-92-mbIL-2 apoptotic cells under IL-2 condition culture after three days. h: Quantification and statistical analysis of the data (n = 3).
2.3. Lentivirus production and transfection
Lentivirus were produced in 293 T cells as previously described,32 then NK-92 cells were transfected using the spinoculation method. Briefly, 2 × 105 NK-92 cells were seeded in 24-well plates (Corning, Corning, NY, USA) with 5 μg/mL polybrene and the lentiviral supernatants. The plates were centrifuged at 1000 × g for 60 min at room temperature and cultured at 37°C, as described above. NK-92 cells were infected with lentivirus carrying mbIL-2 (NK-92-mbIL-2) or empty vector (NK-92-VEC).
2.4. Cell proliferation and apoptosis assay
To detect cell proliferation, 1 × 104 NK-92-VEC or NK-92-mbIL-2 cells were seeded in 96-well plates per well. After 24, 48, and 72 h of incubation, cell activity was tested using the Cell Counting Kit-8 (Dojindo, Mashiki, Japan) following the manufacturer instructions.
To detect apoptosis levels, the cells were harvested and stained with Annexin V-APC and 7-AAD (BioLegend, San Diego, CA, USA) following the manufacturer instructions, and then analyzed using a NovoCyte flow cytometer.
2.5. Immunoblot
Cells were lysed in RIPA lysis buffer with fresh protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA) and SuperNuclease (SinoBiological, Beijing, China) on ice for 30 min. We measured protein samples using a standard bicinchoninic acid assay (Thermo Fisher, Waltham, MA, USA). Proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and proteins were transferred to polyvinylidene fluoride membranes. The membranes were blocked with tris-buffered saline with 0.1% Tween® 20 Detergent plus 5% nonfat dry milk for 2 h, followed by incubation with primary antibodies overnight at 2–8°C. All primary antibodies were purchased from Abcam (Cambridge, UK) or Cell Signaling Technology (Danvers, MA, USA) unless otherwise indicated. Species-specific horseradish peroxidase-conjugated secondary antibodies (1:10,000) were incubated with the membranes for 1 h at room temperature. Immunoreactive signals were detected using an enhanced chemiluminescence (ECL) system using an ECL kit (Millipore, Burlington, MA, USA). All loading samples were normalized to the internal reference protein, glyceraldehyde 3-phosphate dehydrogenase.
2.6. Cytotoxicity assay
To evaluate the in vitro lytic activity of NK-92-VEC and NK-92-mbIL-2 cells, we performed a cytotoxicity assay as previously described.33 Briefly, the target cells were co-cultured with effector cells in a U-bottom 96-well plate for 4 h at 37°C at different ratios. Each experiment was performed in triplicates. After co-culture, cells were collected and stained with 7-AAD (BioLegend, USA). The cell mixture was analyzed using a NovoCyte flow cytometer. Dead target cells were determined as GFP-negative and 7-AAD-postive. Spontaneous target cell lysis in the absence of effector cells was subtracted to calculate specific cytotoxicity.
2.7. ELISA and cytokine releasing assay
IL-2 was detected using an ELISA kit (MultiSciences Biotech Co., Hangzhou, Zhejiang Province, China) according to the manufacturer instructions. To detect NK cell cytokine release, NK-92-VEC or NK-92-mbIL-2 cells were co-cultured with K562 target cells at effector: target ratios of 1:1 for 12 h. The supernatant of the co-culture system was harvested. The expression levels of IFN-γ were detected using an ELISA kit (MultiSciences, China), according to the manufacturer instructions.
2.8. Degranulation assay
We co-cultured 1 × 105 NK-92-VEC or NK-92-mbIL-2 cells with 1 × 105 K562 cells in 200 μl of K562 culture medium with allophycocyanin-conjugated anti-CD107a antibody (BioLegend, USA). After 1 h, 100 ug/ml monensin (BD Biosciences, San Jose, CA, USA) was added to the co-culture system and incubated for 4 h and the cells were analyzed by flow cytometry. All GFP+-CD107a+ cells were degranulated NK cells. Spontaneous NK cell degranulation in the absence of target cells was subtracted to calculate specific proportion of NK cell degranulation.
2.9. RNA sequencing analysis
NK-92-VEC cells were cultured with 200 U/ml recombinant human IL-2 as per ATCC recommendation, and NK-92-mbIL-2 cells were cultured without exogenous IL-2 condition medium. The cells were collected and washed thrice with phosphate buffered saline, and total RNAs were extracted using TRIzol reagent (Invitrogen, Waltham, MA, USA). Subsequently, they were treated with DNase I to remove genomic DNA. The Majorbio Company (Shanghai, China) performed library construction and RNA sequencing using Illumina NovaSeq 6000 (Illumina, San Diego, CA, USA). The raw data were processed by Fastp and SeqPrep with the default parameters and aligned to the reference genome (http://asia.ensembl.org/Homo_sapiens/Info/Index) in orientation mode using Bowtie2. For bioinformatics analysis, fragments per transcript per million reads were used to calculate the expression levels, which were quantified using RSEM tools. The differential gene expression analysis of NK-92-VEC and NK-92-mbIL-2 cells was performed using DESeq2 software, with a fold-change≥2 and adjusted p-value < 0.05.
The gene set enrichment analysis (GSEA) was enriched in gene lists extracted from MSigDB to determine enrichment in gene sets from the immunologic signatures. The enrichment analysis of Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways was conducted and visualized using the clusterProfiler package.
For quantitative real-time polymerase-chain reaction (qRT-PCR), total RNA was extracted using TRIzol reagent (Invitrogen, USA) and cDNA was synthesized using the PrimeScript™ RT reagent Kit (TAKARA, Beijing, China). qRT-PCR was conducted using TB Green Premix Ex Taq II (TAKARA, China) according to the manufacturer instructions. Each reaction was performed in triplicates. The primer sequences used for the qRT-PCR are listed in Supplementary Table S1.
2.10. Xenograft model
Six- to eight-week-old female NCG mice (NOD/ShiLtJGpt-Prkdcem26Cd52Il2rgem26Cd22/Gpt) were purchased from ChengDu GemPharmatech Co. Ltd. The experimental protocol was approved by the Ethics Committee of West China Hospital, Sichuan University, China. To detect cell proliferation in vivo, NK-92-VEC and NK-92-mbIL-2 cells were collected and 5 × 106 NK-92-VEC or NK-92-mbIL-2 cells were injected intravenously (n = 5). After 21 days of inoculation, tissue-resident NK-92-VEC or NK-92-mbIL-2 cells were examined by flow cytometry. To compare the cytotoxicity of the cells in vivo, K562-luc cells were collected, and 1 × 106 cells suspended in RPMI-1640 with Matrigel Matrix (Corning, USA) were subcutaneously injected to establish a xenograft model. After the volume of the tumors reached 100–200 mm3, 5 × 106 NK-92-VEC or NK-92-mbIL-2 cells were injected intratumorally (n = 5). Subsequently, the volume of the tumors was measured periodically. To detect the cytotoxicity of the cells in the in-situ model in vivo, K562-luc cells were collected, and 1 × 106 cells suspended in RPMI-1640 were injected intravenously to establish an in-situ model of leukemia. After four days, 5 × 106 NK-92-VEC or NK-92-mbIL-2 cells were injected intravenously (n = 5). Tumor burden was measured by in vivo bioluminescence using the Xenogen-IVIS Imaging System. All experiments were performed following the guidelines of the Animal Care and Use Committee of the West China Hospital, Sichuan University, China.
2.11. Statistical analysis
Data were processed using GraphPad Prism (version 8.0) and expressed as the mean ± standard deviation (n ≥ 3). Statistical analyses were performed using Student’s t-test or two-way analysis of variance (ANOVA). Differences were considered statistically significant at P < 0.05.
3. Results
3.1. Construction of mbIL-2 and preparation of NK-92-mbIL-2 cells
Mechanistically, IL-2 initially binds to IL-2Rα and then recruits IL-2Rβ and IL-2Rγc to form high-affinity trimeric receptor complexes.34 To simulate this biological process, mbIL-2 was constructed by the fusion of human IL-2 and human IL-2Rα with a (G4S)3 linker (Figure 1a). NK-92 cells, whose survival and proliferation depend on exogenous IL-2,35 were infected with lentiviruses carrying mbIL-2 (NK-92-mbIL-2) or empty vector (NK-92-VEC) DNAs. For the lentiviruses had GFP reporter gene, after transduction, the GFP+-NK-92 cells were enriched and expanded for subsequent experiments (Figure 1b).
3.2. Role of mbIL-2 in NK-92 cell survival and proliferation
Consistent with previous studies, exogenous IL-2 is required for the proliferation of NK-92 cells.36 When exogenous IL-2 was applied to the cell culture, NK-92-VEC cells proliferated normally and avoided apoptosis (Figures 1c–e). However, without exogenous IL-2, NK-92-VEC cell proliferation was significantly inhibited and apoptosis gradually increased with culture time (Figures 1c–e). In contrast, NK-92-mbIL-2 cells proliferated normally and avoided apoptosis irrespective of exogenous IL-2 conditional culture (Figures 1f-h). These data suggested that mbIL-2 supported NK-92 cell survival and proliferation, which were exogenous IL-2 independent. Moreover, NK-92-mbIL-2 cells were maintained without exogenous IL-2 for more than one year in our lab. Unless otherwise specified, the NK-92-VEC cells were maintained with 200 U/ml recombinant human IL-2, and the NK-92-mbIL-2 cells were maintained without exogenous IL-2 for subsequent experiments.
To determine the persistence of NK-92-mbIL-2 in vivo, we examined their distribution in vivo. DiR-labeled NK-92-mbIL-2 cells were injected into immunocompromised NCG mice tail vein. After three days of infusion, NK-92-mbIL-2 cells were distributed mainly in the liver and spleen (Figure 2a). Subsequently, 5 × 106 NK-92-VEC or NK-92-mbIL-2 cells were systemically engrafted into immunocompromised NCG mice by intravenous inoculation. Mice were sacrificed 21 days after transplantation, and tissue-resident NK-92-VEC or NK-92-mbIL-2 cells were examined by flow cytometry (Figure 2b). As expected, NK-92-mbIL-2 cells preferentially resided in the liver and spleen. In contrast, NK-92-VEC cells were not detected in the liver or spleen (Figures 2c–f). Compared to those of NK-92-VEC controls, the spleens of NK-92-mbIL-2 groups showed an increase in size (Figure 2b). However, NK-92-VEC or NK-92-mbIL-2 cells were not detected in the blood of the mice (Fig. S1). These data suggest that NK-92-mbIL-2 cells reside primarily in the liver and spleen, and exhibit long-term persistence in vivo.
Figure 2.
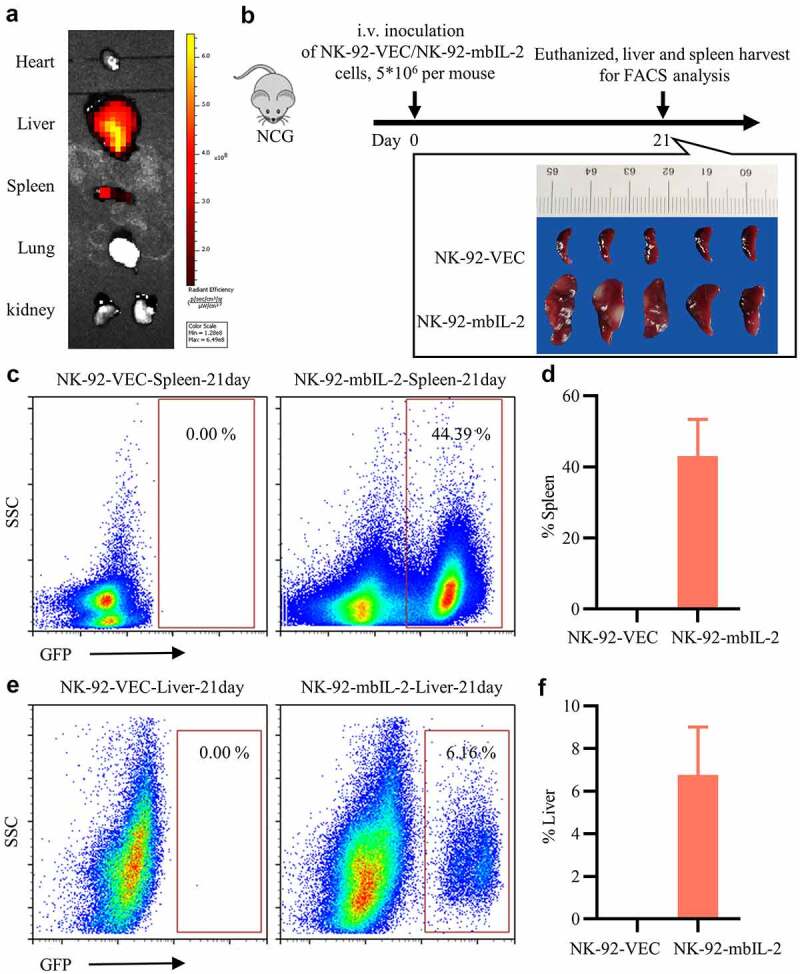
MbIL-2 supports NK-92 cells long-term persistence in vivo.
a: Representative fluorescence images of mice after intravenous injection of DiR-labeled NK-92-mbIL-2 cells after three days. b: NCG mice implanted intravenously with NK-92-VEC or NK-92-mbIL-2 cells. The mice were sacrificed 21 days after transplantation for analysis. NK-92-VEC maintained with 200 U/ml IL-2 and NK-92-mbIL-2 maintained without IL-2 in vitro for experiments. c: Representative flow cytometry analysis showing the proportion of NK-92-VEC or NK-92-mbIL-2 cells residing in the spleen. d: Quantification and statistical analysis of the data in C (n = 5). e: Representative flow cytometry analysis showing the proportion of NK-92-VEC or NK-92-mbIL-2 cells residing in living cells. f: Quantification and statistical analysis of the data in E (n = 5).
To summarize, these data indicate that mbIL-2 alone can support NK-92 cell proliferation and persistence both in vitro and in vivo.
3.3. Role of mbIL-2 in bystander cell survival and proliferation
As mbIL-2 was predicted to localize to the cell membrane, similar to IL-2Rα, we further determined the subcellular localization of mbIL-2. During NK-92-mbIL-2 cell culture, the supernatants were evaluated for IL-2 by ELISA at different times. As expected, the IL-2 concentrations in the supernatant did not change significantly over time, and the values were below the detection limits of the ELISA kit (Figure 3a). These data suggest that mbIL-2 is a non-secretory fusion protein. And flow cytometry analysis showed that mbIL-2 can be detected on the cell surface (Fig. S5). For better observation, we constructed an mbIL-2-GFP fusion protein, in which the mbIL-2 C-terminus was linked to the GFP protein N-terminus with a linker. After lentiviral transduction of 293 T cells, the mbIL-2-GFP fusion protein localized to the cell membrane and formed aggregates (Figure 3b). As a control, 293 T cells transduced with GFP lentiviral vector showed green fluorescence distributed throughout the cell (Figure 3b). These data suggest that mbIL-2 is a non-secretory fusion protein that is to some degree localized on the cell membrane.
Figure 3.
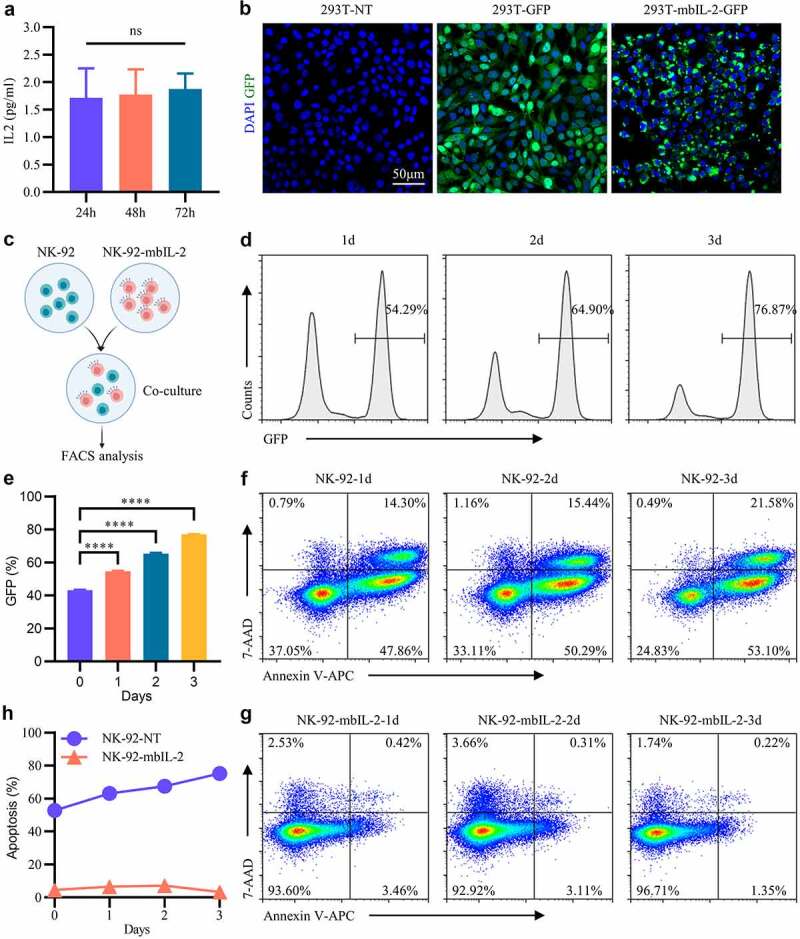
MbIL-2 does not support bystander cell survival and proliferation.
a: ELISA detects IL-2 in NK-92-mbIL-2 cell culture supernatants at different times (n = 3). NK-92-mbIL-2 maintained without IL-2 and collected culture supernatants for assays. b: Representative confocal laser images of 293 T-NT, 293 T-GFP, and 293 T-mbIL-2-GFP. 293 T-NT represent 293 T cells not-transduced lentiviral. 293 T-GFP represent 293 T cells transduced empty lentiviral carrying GFP reporter gene. 293 T-mbIL-2-GFP represent 293 T cells transduced lentiviral carrying mbIL-2-GFP fusion protein. c: Schematic diagram of NK-92-mbIL-2 cell co-culture with NK-92 cells. At the co-culture time, IL-2 was not added. d: Representative flow cytometry analysis showing the proportion of NK-92-mbIL-2 in co-culture assays shown by C. GFP positive cells represent NK-92-mbIL-2 cells which carrying GFP reporter gene. GFP negative cells represent non-transduced NK-92 cells. e: Quantification and statistical analysis of the data in D (n = 3). f: Representative flow cytometry analysis showing the proportion of apoptotic non-transduced NK-92 cells in co-culture assays shown in C. g: Representative flow cytometry analysis showing the proportion of apoptotic NK-92-mbIL-2 cells in co-culture assays shown in C. h: Quantification and statistical analysis of the data in F and G (n = 3).
Considering the systemic toxicity of IL-2 limiting its application,22,23 we evaluated whether NK-92-mbIL-2 cells would support the survival and proliferation of bystander non-transduced NK-92 cells. NK-92-mbIL-2 and non-transduced NK-92 cells were co-cultured at a 1:1 ratio and analyzed at different points of time (Figure 3c). As the co-culture time increased, the proportion of NK-92-mbIL-2 cells increased, and the proportion of non-transduced NK-92 cells decreased gradually (Figures 3d–e). Additionally, the proportion of apoptotic non-transduced NK-92 cells increased gradually over time (Figures 3f-h) and the proportion of apoptotic NK-92-mbIL-2 cells was maintained at a lower ratio (Figures 3g–h). In the co-culture system, the non-transduced NK-92 cells have similar apoptosis proportion compared with non-transduced NK-92 cells cultured alone (Fig. S4A). After 15 days co-culturing, non-transduced NK-92 cells almost undetectable by flow cytometry (Fig. S4B). These data suggest that in the co-culture system, non-transduced NK-92 cells could not maintain normal growth and proliferation.
To summarize, these data indicate that mbIL-2 can be detected on the cell membrane and mbIL-2-transduced NK-92 cells did not maintain normal growth and proliferation of bystander non-transduced NK-92 cells.
3.4. Role of mbIL-2 in NK-92 cell-mediated cytotoxicity
Since IL-2 is critical for NK cell activation, we tested whether mbIL-2-expressed NK cells enhanced their cytotoxicity against tumor cells and the difference between mbIL-2-expressed NK cells and exogenous IL-2 activated NK cells. After NK-92-VEC or NK-92-mbIL-2 cells were co-cultured with K562 cells at different ratios, K562 cell lysis was detected. And IL-2 was added to NK-92-VEC cells, but not NK-92-mbIL-2 cells at the time of co-culturing. Compared with exogenous IL-2-activated NK-92-VEC cells, NK-92-mbIL-2 cells exerted superior neutralization of K562 cells (Figure 4a). To confirm the enhanced cytotoxic activity of NK-92-mbIL-2 cells, we measured CD107α degranulation and IFN-γ secretion by NK cells against K562 cells. After cell culture, NK-92-mbIL-2 cells had a higher proportion of CD107α-positive cells and higher levels of IFN-γ was released compared to the corresponding results of NK-92-VEC cells activated by exogenous IL-2 (Figures 4b-c). These data indicate that mbIL-2 enhanced NK-92 cell-mediated cytotoxicity in vitro and was superior to exogenous IL-2.
Figure 4.
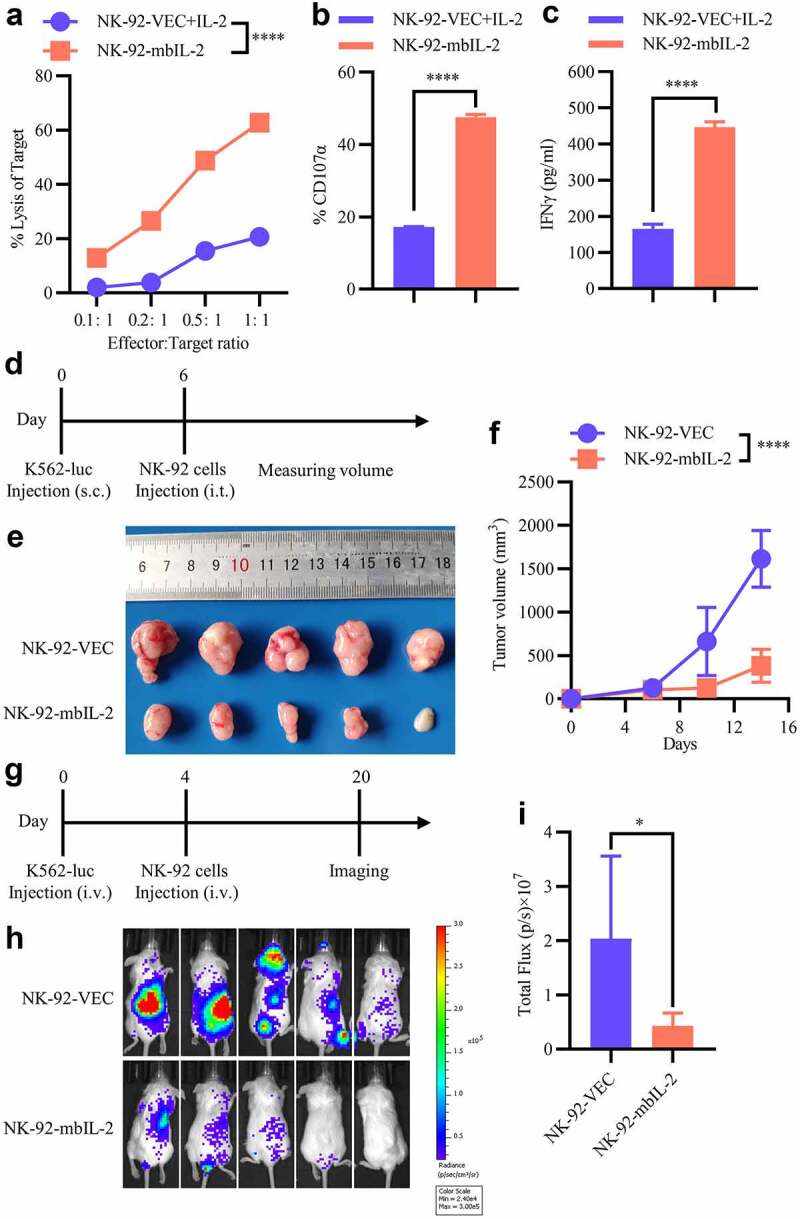
MbIL-2 enhances the antitumor activity of NK-92 cells.
a: Direct lysis of NK cells against target cells K562. Effector and target cells were co-incubated for 4 h at the indicated effector: target ratio. Flow cytometric analysis of the proportion of GFP−7-AAD+ cells (n = 3). b: Flow cytometry analysis of the proportion of GFP+-CD107α+ cells after NK cells were co-incubated with target cells at 1:1 for 5 h (n = 3). c: ELISA data showing the release of IFN-γ by NK cells after co-incubation with target cells at 1:1 for 12 h (n = 3). d–f: In vivo studies using K562-luc cells in a mouse subcutaneous xenograft model treated with NK-92-VEC and NK-92-mbIL-2 cells. d: Schematic diagram of the study. 1 × 106 K562-luc cells with Matrigel Matrix were subcutaneously injected to establish a xenograft model. After the volume of the tumors reached 100–200 mm3, 5 × 106 NK-92-VEC or NK-92-mbIL-2 cells were injected intratumorally (n = 5). Subsequently, the volume of the tumors was measured periodically. e: Representative image of tumor burden after the mice were sacrificed (n = 5). f: Tumor burden was periodically determined (n = 5). g–i: In vivo study using K562-luc cells in a mouse orthotopic xenograft model treated with NK-92-VEC and NK-92-mbIL-2 cells. g: Schematic diagram of the study. 1 × 106 K562-luc cells were injected intravenously to establish an in-situ model of leukemia. After four days, 5 × 106 NK-92-VEC or NK-92-mbIL-2 cells were injected intravenously (n = 5). Tumor burden was measured by in vivo bioluminescence using the Xenogen-IVIS Imaging System. h: Representative bioluminescent image of the tumor burden after mice were treated (n = 5). i: Quantification and statistical analysis of the data in H (n = 5).
To investigate the in vivo antitumor activity of NK-92-mbIL-2 cells, we constructed a K562-luc xenograft model. Mice were grouped into two types (five mice per group), one group type received one intratumoral infusion (5 × 106/mouse) of NK-92-VEC and the other group type received NK-92-mbIL-2 cells. Then, the NK-92-VEC group mice were intraperitoneally administrated with IL-2 (1000 U/mouse) for every 2 days until the end of experiment. And the volume of the tumors was measured periodically (Figure 4d). Compared to the mice treated with NK-92-VEC, mice treated with an infusion of NK-92-mbIL-2 cells showed significantly suppressed tumor burden (Figures 4e–f). As the K562 cell line is a human erythroleukemia cell line, we constructed a K562-luc orthotopic leukemia model by intravenous injection. With similar grouping of mice as mentioned above, one group type received one intravenous infusion (5 × 106/mouse) of NK-92-VEC and the other group type received NK-92-mbIL-2 cells. Then, the NK-92-VEC group mice were intraperitoneally administrated with IL-2 (1000 U/mouse) for every 2 days until the end of the experiment. And tumor growth was monitored by measuring changes in tumor bioluminescence (Figure 4g). Sixteen days after NK cell infusion, the tumor bioluminescence of NK-92-mbIL-2-treated groups was significantly lower than that of NK-92-VEC-treated groups (Figures 4h–i). These data suggest that mbIL-2 enhances NK-92 cell-mediated cytotoxicity in vivo.
To summarize, these data indicate that mbIL-2 enhances NK-92 cell-mediated cytotoxicity in vitro and in vivo and superior to exogenous IL-2-activated NK-92 cells.
3.5. Role of mbIL-2 in NK-92 cell-mediated pan-cancer antitumor activity
After establishing that mbIL-2 enhances NK-92 cell-mediated cytotoxicity, we tested whether mbIL-2 enhances NK-92 cell-mediated pan-cancer antitumor activity. We detected NK-92-mbIL-2 and NK-92-VEC cell-mediated cytotoxicity in prostate cancer (PRAD), ovarian cancer (OV), melanoma (SKCM), lung cancer (LUAD), colorectal cancer (COADREAD), and glioblastoma (GBM). As shown in Figs. S2A–F, NK-92-mbIL-2 cells had better antitumor activity than NK-92-VEC cells against these cancer cells. In this study, LN18 cell line was found to be an NK-insensitive GBM cell line. However, LN18 cells became sensitive to mbIL-2-expressed NK-92 cells (Fig. S2F). These data indicate that mbIL-2 enhances NK-92 cell-mediated pan-cancer antitumor activity.
3.6. Role of mbIL-2 in NK-92 cell IL-2 receptors downstream signals
To understand the mechanism by which mbIL-2 activates NK cells, we suspected that mbIL-2 activates NK cells dependent on IL-2-receptors downstream signals. Indeed, the bioactivity of IL-2 relies on IL-2 receptor-related JAK-STAT kinases and initially for the Janus family tyrosine.37 Some studies have shown that tofacitinib, a JAK3 kinase inhibitor,37 can inhibit the IL-2 signaling.38 NK-92-VEC+IL-2 or NK-92-mbIL-2 cells cultured with 1 μM tofacitinib and detected at different points of time (Figure 5a). As the culture time increased, the proportion of apoptotic NK-92-VEC or NK-92-mbIL-2 cells increased gradually (Figures 5a–b) while the proliferation of NK-92-VEC and NK-92-mbIL-2 cells was inhibited (Figure 5c). These data suggest that similar to exogenous IL-2, mbIL-2 activates NK-92 cells through the classical IL-2-receptor JAK-STAT signaling pathway.
Figure 5.
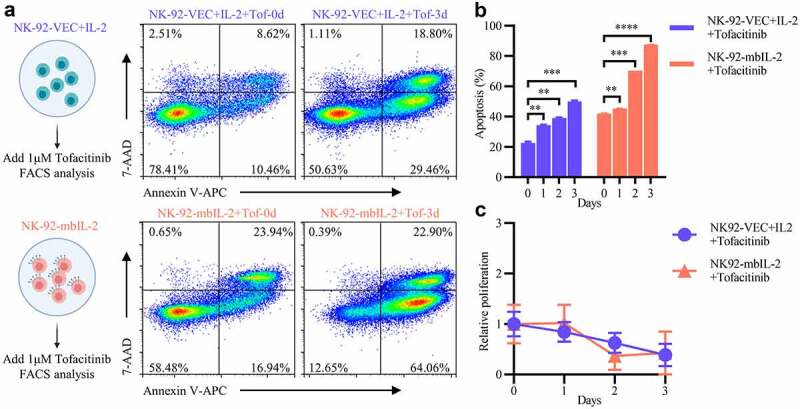
MbIL-2 activation of NK cells relies on classic IL-2 receptor signal transduction.
a: Representative flow cytometric analysis of NK-92-VEC and NK-92-mbIL-2 cells treated with tofacitinib. NK-92-VEC maintained with 200 U/ml IL-2 and add 1 μM tofacitinib for assays. NK-92-mbIL-2 maintained without IL-2 and add 1 μM tofacitinib for assays. b: Quantification and statistical analysis of data from A (n = 3). c: CCK8 assay of cell proliferation in NK-92-VEC and NK-92-mbIL-2 cells treated with tofacitinib (n = 6). NK-92-VEC maintained with 200 U/ml IL-2 and add 1 μM tofacitinib for assays. NK-92-mbIL-2 maintained without IL-2 and add 1 μM tofacitinib for assays.
To further determine the differences between mbIL-2 and exogenous IL-2 activated downstream signals, we compared the global gene expression profiles of NK-92-VEC (n = 3) and NK-92-mbIL-2 cells (n = 3) (Figures 6a–b). A total of 1625 differential genes were identified (fold-change ≥ 2 and p-value < 0.05), which included 932 upregulated genes and 693 downregulated genes (Figure 6c). For the GSEA analysis, the “Unstim vs IL-2 stim NK cell UP” gene set represent up-regulated genes in NK cells activated by exogenous IL-2. And the “Unstim vs IL-2 stim NK cell DN” gene set represent down-regulated genes in NK cells activated by exogenous IL-2.39 Compared with exogenous IL-2 activated NK-92-VEC cells, the “Unstim vs IL-2 stim NK cell UP” gene set negatively correlated with NK-92-mbIL-2 cells. However, “Unstim vs IL-2 stim NK cell DN” gene set were exactly opposite (Figure 6d). These data suggest that, in contrast to those of exogenous IL-2, the downstream signals of mbIL-2-activated IL-2 receptors might be weaker.
Figure 6.
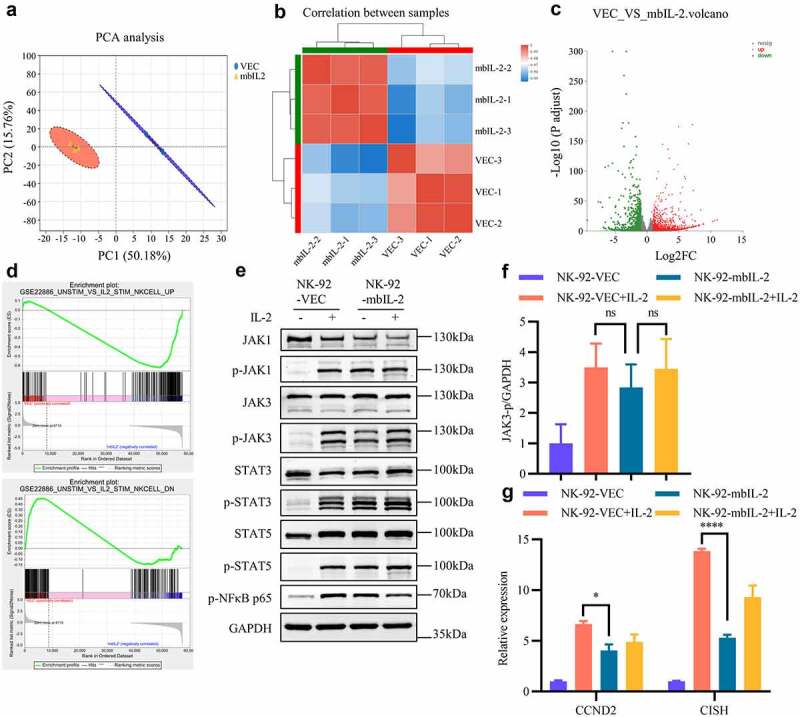
MbIL-2 tunes IL-2-receptor downstream signals of NK cells.
a: PCA of NK-92-VEC and NK-92-mbIL-2 cell transcriptome profiles (n = 3). NK-92-VEC cells maintained with 200 U/ml IL-2 and NK-92-mbIL-2 cells maintained without IL-2 for RNA-sequencing. b: Correlation analysis of the NK-92-VEC and NK-92-mbIL-2 cell transcriptome profiles (n = 3). c: Differential gene expression analysis of NK-92-VEC and NK-92-mbIL-2 cell transcriptome profiles (n = 3). d: The GSEA of NK-92-VEC and NK-92-mbIL-2 cell transcriptome profiles. The top gene set represent NK cells up-regulated genes activated by exogenous IL-2. The bottom gene set represent NK cells down-regulated genes activated by exogenous IL-2. e: Immunoblot analysis of JAK-STAT kinase phosphorylation in NK-92-VEC and NK-92-mbIL-2 cells. NK-92-VEC cells maintained with 200 U/ml IL-2 or without IL-2 condition culture 24 h for assays. NK-92-mbIL-2 cells maintained without IL-2 or with 200 U/ml IL-2 condition culture 24 h for assays. f: Quantification and statistical analysis of the data in E (n = 3). g: qPCR analysis of CCND2 and CISH gene expression in NK-92-VEC and NK-92-mbIL-2 cells. NK-92-VEC cells maintained with 200 U/ml IL-2 or without IL-2 condition culture 24 h for assays. NK-92-mbIL-2 cells maintained without IL-2 or with 200 U/ml IL-2 condition culture 24 h for assays.
To validate whether the differences were related to JAK-STAT activation, we examined the levels of the phosphorylated JAK-STAT signaling pathway in NK-92-VEC and NK-92-mbIL-2 cells in response to exogenous IL-2 stimulation and non-stimulation. From Figure 6e, the JAK-STAT pathway (including JAK1, JAK3, STAT3, STAT5, and NF-κB) had similar phosphorylation levels in NK-92-VEC and NK-92-mbIL-2 cells. Although not significantly different, NK-92-mbIL-2 had a lower phosphorylation ratio in JAK3 than in exogenous IL-2 stimulated NK-92-VEC cells (Figure 6f). Consistently, the expression of IL-2 downstream genes CCND2 and CISH was downregulated in NK-92-mbIL-2 cells (Figure 6g). Of these, CISH is an important cytokine-induced, including IL-2 and IL-15,40 negative feedback inhibitor in NK cells.41 These data suggest that IL-2 receptors are activated by mbIL-2 with lower negative feedback than exogenous IL-2.
To summarize, these data suggest that mbIL-2, compared to exogenous IL-2, might tune NK cell IL-2 receptor downstream signals, resulting a change in corresponding genes expression.
3.7. Role of mbIL-2 in activating receptors for NK-92 cell-mediated cytotoxicity
Upon establishing that mbIL-2 enhanced NK cell-mediated cytotoxicity better than exogenous IL-2, we determined that mbIL-2 and exogenous IL-2 activated the NK cell receptor repertoire, which is essential for NK cells to recognize tumor cells.3 Unsupervised cluster analysis revealed that most of the activating receptors were upregulated, and inhibitory receptors were downregulated in the NK-92-mbIL-2 groups (Figure 7a). The relative expression of these genes, detected by qPCR, was usually consistent with the cluster analysis results (Figures 7b–c and S3D). As the receptors are expressed on the cell membrane, we determined receptor expression by flow cytometry, which also revealed that activating receptors NKG2D and DNAM1 were upregulated in the NK-92-mbIL-2 groups (Figures 7d). These data suggest that mbIL-2 upregulates the activating receptors and downregulates the inhibitory receptors of NK cells, which might explain why NK-92-mbIL-2 cells have greater cytotoxicity than NK-92-VEC control cells.
Figure 7.
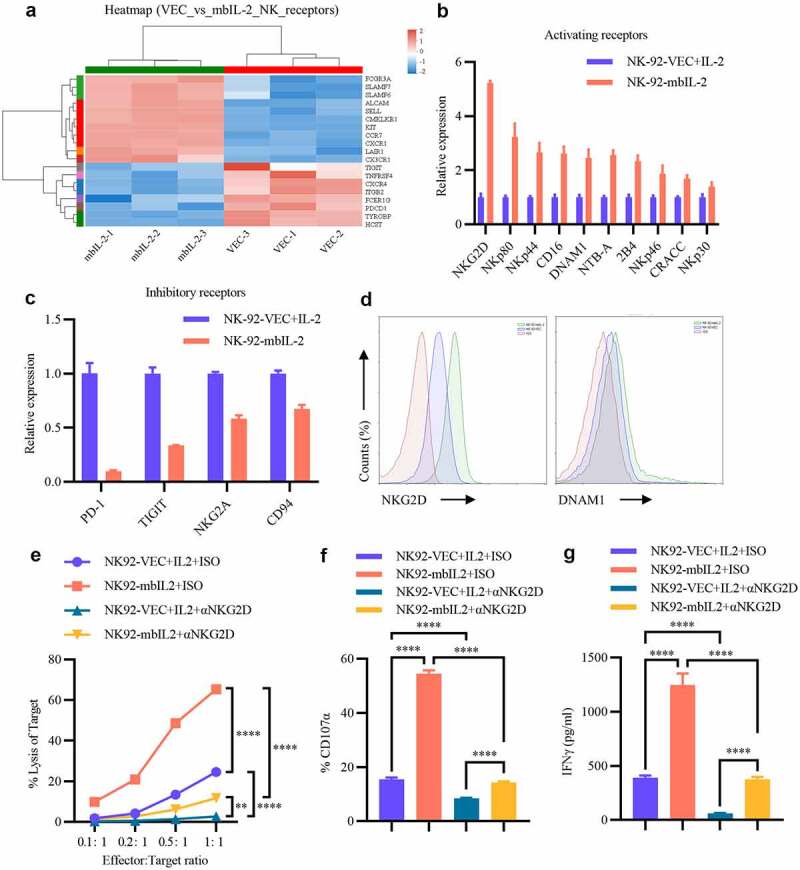
MbIL-2 regulates receptor repertoire to enhance NK cell cytotoxicity.
a: Unsupervised cluster analysis of NK cell receptors in NK-92-VEC cells and NK-92-mbIL-2 cell transcriptome profiles (n = 3). NK-92-VEC maintained with 200 U/ml IL-2 and NK-92-mbIL-2 maintained without IL-2 for RNA-sequencing. b: qPCR analysis of NK cell activating receptor gene expression in NK-92-VEC and NK-92-mbIL-2 cells (n = 3). NK-92-VEC maintained with 200 U/ml IL-2 and NK-92-mbIL-2 maintained without IL-2 for assays. c: qPCR analysis of NK cell inhibitory receptor gene expression in NK-92-VEC and NK-92-mbIL-2 cells (n = 3). NK-92-VEC maintained with 200 U/ml IL-2 and NK-92-mbIL-2 maintained without IL-2 for assays. d: Flow cytometric analysis of NKG2D and DNAM1 expression in NK-92-VEC and NK-92-mbIL-2 cells. NK-92-VEC maintained with 200 U/ml IL-2 and NK-92-mbIL-2 maintained without IL-2 for assays. e: Direct lysis of NK cells against target cell K562. Effector cells were blocked with NKG2D antibody or isotype antibody for 1 h and then co-incubated with the target cells for 4 h at the indicated effector: target ratio. Flow cytometric analysis of the proportion of GFP−7-AAD+ cells (n = 3). At the co-culture time, exogenous IL-2 was added to the NK-92-VEC cells and not added to the NK-92-mbIL-2 cells for assays. f: Flow cytometric analysis of the proportion of GFP+-CD107α+ cells after NK cells were blocked by NKG2D antibody or isotype antibody for 1 h co-incubation with target cells at 1:1 for 12 h (n = 3). At the co-culture time, exogenous IL-2 was added to the NK-92-VEC cells and not added to the NK-92-mbIL-2 cells for assays. g: ELISA data showing the release of IFN-γ by NK cells blocked by the NKG2D antibody or isotype antibody for 1 h after co-incubation with target cells at 1:1 for 5 h (n = 3). At the co-culture time, exogenous IL-2 was added to the NK-92-VEC cells and not added to the NK-92-mbIL-2 cells for assays.
To validate whether mbIL-2 enhances NK-92 cell-mediated cytotoxicity depending on these receptors, receptor block experiments were conducted. After blocking the NKG2D receptor, which had the highest upregulated fold-change of these receptors (Figures 7b-d), the cytotoxicity of NK-92-VEC and NK-92-mbIL-2 cells was reduced (Figure 7e). Nevertheless, NK-92-mbIL-2 cells still showed higher cytotoxicity than NK-92-VEC cells (Figure 7e). The degranulation assay showed that the proportion of CD107α-positive cells decreased when the NKG2D receptor was blocked, and NK-92-mbIL-2 cells, compared to NK-92-VEC cells, had a higher proportion of CD107α-positive cells (Figure 7f). Furthermore, IFN-γ cytokines were detected to evaluate the NK cell activation status, revealing that the cytokines released by NK-92-mbIL-2 cells were significantly higher than those released by NK-92-GFP cells, even though the NKG2D receptor was blocked (Figure 7g). However, cytokine secretion decreased under blocked NKG2D receptor compared to that in the groups where NKG2D receptor was not blocked (Figure 7g). These data suggest that blocking the NKG2D receptor would reduce mbIL-2-enhanced NK cell-mediated cytotoxicity. Other upregulated activating receptors might explain why NK-92-mbIL-2 has higher cytotoxicity than NK-92-VEC cells under blocked NKG2D receptor conditions.
To summarize, these data suggest that mbIL-2 could upregulate the expression of many activating receptors in NK-92 cells, which are essential for mbIL-2 enhanced NK-92 cell-mediated cytotoxicity.
3.8. Role of mbIL-2 in chemotaxis of NK-92 cells
Reaching tumor sites is another challenge for NK cells.42 KEGG and GO enrichment analyses of differentially expressed genes (p < .05) of NK-92-mbIL-2 cells compared with those of NK-92-VEC cells revealed that cell chemotaxis, cell–cell adhesion, and cell migration terms were significantly enriched (Figs. S3A–B). These data suggest that mbIL-2 might enhances the chemotaxis and migration effects of NK-92 cells. To detect the relative expression of NK cell chemokine receptor genes, we found that ChemR23, CX3CR1, CXCR1, CCR7, and SELL were upregulated in NK-92-mbIL-2 cells compared to those in NK-92-VEC cells (Fig. S3C). Evidently, the adhesion molecule, ALCAM, was upregulated in NK-92-mbIL-2 cells. Among them, CXCR1, a chemokine receptor of the pro-inflammatory cytokine IL-8, improves the NK cell migration to tumor sites and enhances NK cell antitumor responses.43 For functional assays, our results shown that mbIL-2 improved the chemotaxis of NK-92 cells induced by IL-8 (Figs. S3E and S3F).
To summarize, these data suggest that mbIL-2 upregulates chemokine receptor and adhesion molecule expression in NK-92 cells, which might improve the chemotaxis and migration of NK-92 cells.
4. Discussion
In adoptive NK cell immunotherapy, infused NK cells are present in vivo for less than two weeks after infusion.20,44 Although the short lifespan of NK cells confers safety, it also limits the therapeutic efficiency of NK cells. IL-2 is a critical cytokine that activates NK cells and has been frequently used for NK cell proliferation.19 However, clinical trials on NK cell infusion by IL-2 have not yet been established.3 Although, IL-2 can activate NK cells to improve the in vivo persistence and enhance the antitumor effects of NK cell immunotherapy, the administration of IL-2 can also induce Treg cell activation and severe toxicity.14,22,23 In despite of the high-affinity IL-2 receptor complex formed by IL-2Rα, IL-2Rβ, and IL-2Rγc, activated NK cells mostly express IL-2Rβ and IL-2Rγc receptors.14,28,29 Studies indicated IL-2Rα essential for the sensitivity of human NK cells.30 Therefore, we speculated that simulating high-affinity IL-2Rαβγc trimeric receptor complexes would benefit NK cell activation and persistence. In parallel, to limit the systemic toxicity of IL-2, we constructed a cell-restricted mbIL-2 by fusion of human IL-2 and human IL-2Rα with a (G4S)3 linker.
To validate the function of mbIL-2, we utilized a lentivirus carrying mbIL-2 to transfect NK-92 cells that otherwise depend on exogenous IL-2 for survival and proliferation.35 Similar to exogenous IL-2, mbIL-2 can support NK-92 cell survival and proliferation independent of exogenous IL-2. In vivo, NK-92 cells expressing mbIL-2 primarily reside in the liver and spleen and show long-term persistence. We also observed spleen enlargement following the resistance of NK-92-mbIL-2 cells. Regarding NK-92 cell is a tumor-derived NK cell line, long-term persistence of NK-92-mbIL-2 might have the tumorigenic potential and raise safety concerns. To overcome this issue, we proposed four solutions: First, pre-irradiation NK-92-mbIL-2 cells before application in vivo, which inhibit NK-92-mbIL-2 cells proliferation and retain its anti-tumor cytotoxicity. Second, add safety switches, such as inducible suicide gene, to the NK-92-mbIL-2 cells. Third, use small-molecule, such as tofacitinib, to inhibit IL-2 receptors activity of NK-92-mbIL-2 cells when the cell completed it bio-function or safety events occurred. Finally, a safer primary NK cell to replace NK-92 cell expression mbIL-2.
Considering that IL-2 receptors are expressed on multiple immune cells, including T and NK cells. Among of these cells, Treg cell are more sensitive to IL-2 because of their high-affinity IL-2-receptor complexes. Administration of IL-2 can regulate the function of these cells and induce adverse toxicity.14,22,23 We evaluated the bystander effect by mbIL-2. Our results suggest that mbIL-2 does not aid in the activation nor supports the survival and proliferation of bystander cells. The subcellular localization of mbIL-2, a non-secretory fusion protein located on the cell membrane, may explain how it prevents the activation of bystander cells. Unlike engineered soluble IL-2 mutation,45,46 the mbIL-2 exhibits cell-expression specificity. Based on this, our results highlight that this cell-restricted IL-2 strategy might favorable for CAR-NK cell and CAR-T cell immunotherapies, whose preparation need to be modified through genetic engineering. Therefore, CAR-NK cell or CAR-T cell modified with mbIL-2 no additional preparation processing steps are required. Moreover, the mbIL-2 improves long-term persistence and activation of these cells, which benefit for CAR-NK cell and CAR-T cell immunotherapies.
To validate the extensiveness of mbIL-2 for NK cell mediated antitumor activity, we tested several cancer cell lines, including PRAD, OV, SKCM, LUAD, COADREAD, and GBM. Our results indicate that mbIL-2 expressed NK-92 cells showed superior cytotoxicity in all the tested cancer cells. In vivo, mbIL-2 can also enhance NK-92 cell-mediated cytotoxicity and superior to exogenous IL-2 activated NK-92 cells. In previous studies, Konstantinidis et al. constructed an endoplasmic reticulum (ER) retained IL-2 (ER-IL-2) for NK cells through linking IL-2 with ER retention signal ERRS.12 Youssef et al. constructed a CIRB for NK cells through fusing IL-2 with IL-2Rβ.27 Both ER-IL-2 and CIRB supported NK cells persistence independent of exogenous IL-2 and do not support bystander cells survival. Moreover, CIRB-NK-92 cells have been shown to displayed superior antitumor effects than parental cells. Besides of these studies, NK-92 cells also have some available formulations including NK-92MI (RRID: CVCL_3755), NK-92-41BB (RRID: CVCL_B5ZF), NK-92-CD16 (RRID: CVCL_B5ZE), NK-92.39 (RRID: CVCL_A0AP) and NoGFP-CD16.NK-92 (RRID: CVCL_V429). Among them, NK-92MI cells was engineered to express IL-2, which survival and persistence independent of exogenous IL-2. Studies have shown that NK-92MI cells are identical to the parental cells with similar cytotoxic activity, however, secreted IL-2 might cause systemic toxicity in vivo.47 Compared with NK-92MI cells, mbIL-2 is a novel cell-restricted IL-2 and do not support bystander cells survival, which imply the fewer side effects for application. Compared with ER-IL-2 and CIRB, mbIL-2 has been designed to simulate the high-affinity IL-2 trimeric receptor complexes for NK cell through fusing IL-2 and IL-2Rα. Similarly, mbIL-2 supported NK-92 cells persistence independent of exogenous IL-2. Our studies provide an alternative strategy to design immunotherapeutic NK cells.
As the bioactivity of natural IL-2 relies on IL-2-receptor complexes and receptor-related JAK-STAT kinases.37 When JAK3 phosphorylation was inhibited, mbIL-2 and exogenous IL-2 did not support the survival and proliferation of NK-92 cells, suggesting that mbIL-2 and exogenous IL-2 have similar initial signals. However, mbIL-2 and exogenous IL-2-activated NK-92 cells showed significant differences at the whole-transcriptome level. This is manifested by mbIL-2 negatively correlating IL-2 with the activated NK cell gene set and weaker negative feedback regulation of mbIL-2 activated NK-92 cells. We speculate that mbIL-2 might tune the NK-92 cell IL-2-receptor downstream signals, although the mechanisms by which it is achieved remain unknown. Given the complexity of the mechanisms and such work digressed for this study, we did not select it for further study.
The recognition and neutralization of target (tumor) cells by NK cells relies on the activating and inhibitory receptor repertoire expressed on the cell membrane.3,48 Through analysis, we found that mbIL-2 activated NK-92 cells were upregulated in most of the activating receptors, such as NKG2D, NKp80, and DNAM1, and inhibited the expression of most of the inhibitory receptors, such as PDCD1 and TIGIT. Among the activating receptors, NKG2D, which is critical for NK cell activation and immune surveillance,49 showed the highest fold-change in the upregulated activating receptors. To validate this receptor attribute to mbIL-2 enhanced NK-92 cell cytotoxicity, we used an NKG2D antibody to block NKG2D receptors, which resulted in reduced cytotoxicity for both mbIL-2 and exogenous IL-2 activated NK-92 cells. However, mbIL-2 activated NK-92 cells still exhibit higher cytotoxicity than exogenously activated NK cells. As stated above, the function of NK cells depends on the receptor repertoire expressed by these cells. We speculate that the upregulated activating receptors attributed to mbIL-2 enhanced NK-92 cell cytotoxicity, of which NKG2D is one of them, and downregulated inhibitory receptors may be attributed to reduced immune escape of the tumor cells from mbIL-2 activated NK-92 cells, for which further investigation is required.
In adoptive cell immunotherapy, migrating through obstacles to reach sites of solid tumors is another challenge for NK cells.42 Our results suggest that mbIL-2 activated NK-92 cells upregulated the chemokine receptor and adhesion molecule gene expression. Among these, the adhesion molecule ALCAM has been validated as favorable for engineered T cells across the blood–brain barrier in GBM.50 Moreover, the chemokine receptor CXCR1 improves NK cell migration to tumor sites and enhances NK cell antitumor responses.43 Our study suggests that mbIL-2 can possibly improve the chemotaxis and migration of NK-92 cells, which is beneficial for solid tumor therapy using NK-92 cells.
To summarize our study, we constructed a membrane-bound IL-2 to simulate high-affinity IL-2 trimeric receptor complexes for NK cells (Figure 8). MbIL-2 was found to maintain NK-92 cell survival and persistence, and enhance NK-92 cell-mediated antitumor activity by tuning IL-2-receptor downstream signals and NK cell receptor repertoire expression. Additionally, we found that mbIL-2 can possibly improve the chemotaxis and migration of NK-92 cells by upregulating genes. To conclude, mbIL-2 is a novel cell-restricted artificial IL-2 that improves NK-92 cell persistence and enhances NK-92 cell-mediated antitumor activity. NK-92 cells genetically modified to express the novel mbIL-2 with potential significance for clinical development.
Figure 8.
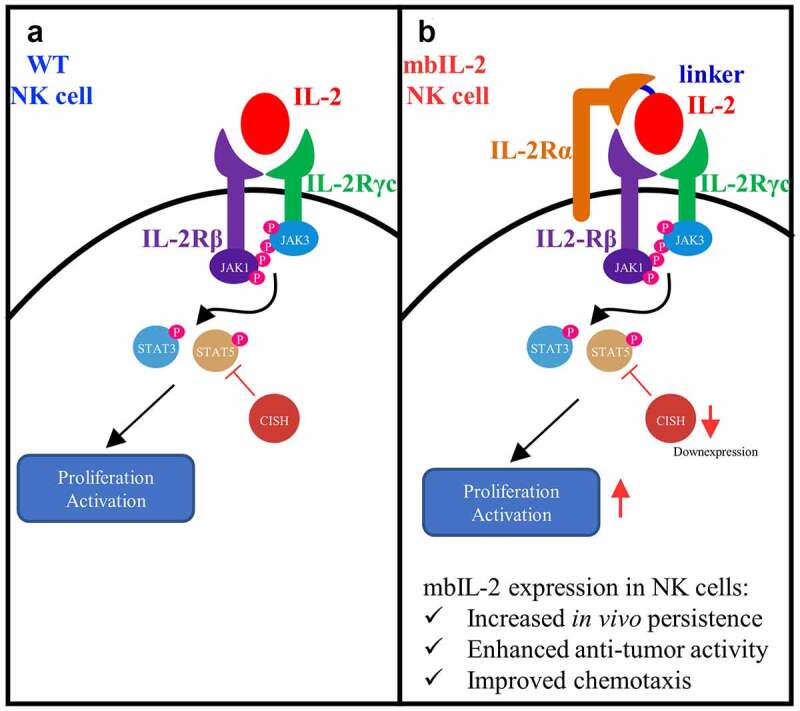
Schematic diagram of mbIL-2 activation NK cells.
a: IL-2 activation NK cells through an intermediate-affinity IL2 receptors, composed with IL-2Rβ and IL-2Rγc. b: MbIL-2 simulate the biological process of high-affinity IL-2 receptor and increased NK cells persistence and enhanced NK cells mediated anti-tumor activity and improved NK cell chemotaxis.
Supplementary Material
Acknowledgment
The authors would like to thank all the reviewers who participated in the review and Editage for its linguistic assistance during the preparation of this manuscript and Ms. Feifei Song for her very helpful advice and suggestions. This work was supported by grants from 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYGD20003) and the National Natural Science Foundation of China Program grant (81902918).
Funding Statement
This work was supported by the West China Hospital, Sichuan University [ZYGD20003] and National Natural Science Foundation of China [81902918].
Disclosure statement
The author reports no conflicts of interest in this work.
Data availability statement
All sequencing data are available through the NCBI Gene Expression Omnibus under the accession number GSE199391 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE199391]. All data included in this study are available upon request by contact with the corresponding author.
Authors’ contributions
Qi Xiong designed and conducted the experiments and drafted the manuscript. Hongxin Deng designed the experiments and supervised the research. All authors discussed the results and commented on the manuscript.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/2162402X.2022.2127282
References
- 1.Abel AM, Yang C, Thakar MS, Malarkannan S.. Natural Killer Cells: development, Maturation, and Clinical Utilization. Front Immunol. 2018;9:1869. doi: 10.3389/fimmu.2018.01869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gotthardt D, Trifinopoulos J, Sexl V, Putz EM. JAK/STAT cytokine signaling at the crossroad of NK cell development and maturation. Front Immunol. 2019;10:2590. doi: 10.3389/fimmu.2019.02590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shimasaki N, Jain A, Campana D. NK cells for cancer immunotherapy. Nat Rev Drug Discov. 2020;19:200–15. [DOI] [PubMed] [Google Scholar]
- 4.B HR, E NM, T HH, et al. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int J Cancer Res. 1975;16(2):230–239. [DOI] [PubMed] [Google Scholar]
- 5.Kiessling R, Klein E, Wigzell H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol. 1975;5:112–117. [DOI] [PubMed] [Google Scholar]
- 6.Lorenzi L, Tabellini G, Vermi W, et al. Occurrence of nodular lymphocyte-predominant hodgkin lymphoma in hermansky-pudlak type 2 syndrome is associated to natural killer and natural killer T cell defects. PloS one. 2013;8(11):e80131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orange JS. Natural killer cell deficiency. J Allergy Clin Immunol. 2013;132:515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.A AP, H BP, Hokland M, et al. NK cells and the tumour microenvironment: implications for NK-cell function and anti-tumour activity. Trends Immunol. 2003;24(11):603–609. [DOI] [PubMed] [Google Scholar]
- 9.Mandal R, Şenbabaoğlu Y, Desrichard A, et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight. 2016;1(17):e89829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daher M, Rezvani K. Outlook for new CAR-Based therapies with a focus on CAR NK cells: what lies beyond CAR-engineered T cells in the race against cancer. Cancer Discov. 2021;11(1):45–58. doi: 10.1158/2159-8290.CD-20-0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fujii R, Jochems C, R SR, Wong HC, Schlom J, Hodge JW. An IL-15 superagonist/IL-15Rα fusion complex protects and rescues NK cell-cytotoxic function from TGF-β1-mediated immunosuppression. Cancer Immunol Immunother? CII. 2018;67(4):675–689. doi: 10.1007/s00262-018-2121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Konstantinidis KV, Alici E, Aints A, Christensson B, Ljunggren H-G, Dilber MS. Targeting IL-2 to the endoplasmic reticulum confines autocrine growth stimulation to NK-92 cells. Exp Hematol. 2005;33(2):159–164. doi: 10.1016/j.exphem.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 13.Patil NK, Luan L, Bohannon JK, Guo Y, Hernandez A, Fensterheim B, Sherwood ER. IL-15 superagonist expands mCD8+ T, NK and NKT cells after burn injury but fails to improve outcome during burn wound infection. PloS one. 2016;11(2):e0148452. doi: 10.1371/journal.pone.0148452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Konjević GM, Vuletić AM, Mirjačić Martinović KM, Larsen AK, Jurišić VB. The role of cytokines in the regulation of NK cells in the tumor environment. Cytokine. 2019;117:30–40. doi: 10.1016/j.cyto.2019.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Wang D, Malarkannan S. Transcriptional regulation of natural killer cell development and functions. Cancers. 2020;12(6):1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kündig TM, Schorle H, Bachmann MF, Hengartner H, Zinkernagel RM, Horak I. Immune responses in interleukin-2-deficient mice. Science (New York, N Y). 1993;262(5136):1059–1061. doi: 10.1126/science.8235625. [DOI] [PubMed] [Google Scholar]
- 17.Gasteiger G, Hemmers S, Firth MA, Le Floc’h A, Huse M, Sun JC, Rudensky AY. IL-2–dependent tuning of NK cell sensitivity for target cells is controlled by regulatory T cells. J Exp Med. 2013;210(6):1167–1178. doi: 10.1084/jem.20122462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vuletić AM, Jovanić IP, Jurišić VB, Milovanović ZM, Nikolić SS, Tanić NT, Konjević GM. In-vitro activation of natural killer cells from regional lymph nodes of melanoma patients with interleukin-2 and interleukin-15. Melanoma Res. 2015;25(1):22–34. doi: 10.1097/CMR.0000000000000126. [DOI] [PubMed] [Google Scholar]
- 19.Cho D, Campana D. Expansion and activation of natural killer cells for cancer immunotherapy. Korean J Lab Med. 2009;29(2):89–96. doi: 10.3343/kjlm.2009.29.2.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, McKenna D, Le C, Defor TE, Burns LJ, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105(8):3051–3057. doi: 10.1182/blood-2004-07-2974. [DOI] [PubMed] [Google Scholar]
- 21.Myers JA, Miller JS. Exploring the NK cell platform for cancer immunotherapy. Nat Rev Clin Oncol. 2021;18(2):85–100. doi: 10.1038/s41571-020-0426-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Skrombolas D, Frelinger JG. Challenges and developing solutions for increasing the benefits of IL-2 treatment in tumor therapy. Expert Rev Clin Immunol. 2014;10(2):207–217. doi: 10.1586/1744666X.2014.875856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chavez ARDV, Buchser W, Basse PH, Liang X, Appleman LJ, Maranchie JK, Zeh H, De Vera ME, Lotze MT. Pharmacologic administration of interleukin-2. Ann N Y Acad Sci. 2009;1182(1):14–27. doi: 10.1111/j.1749-6632.2009.05160.x. [DOI] [PubMed] [Google Scholar]
- 24.Littwitz-Salomon E, Akhmetzyanova I, Vallet C, Francois S, Dittmer U, Gibbert K. Activated regulatory T cells suppress effector NK cell responses by an IL-2-mediated mechanism during an acute retroviral infection. Retrovirology. 2015;12(1):1–13. doi: 10.1186/s12977-015-0191-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.D HWH, Ames E, Álvarez M, et al. Combination therapy using IL-2 and anti-CD25 results in augmented natural killer cell–mediated antitumor responses. Biol Blood Marrow Transplant. 2008;14(10):1088–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.J SM, L TMW, Swann J, et al. CD4+CD25+ T regulatory cells suppress NK cell-mediated immunotherapy of cancer. J Immunol (Baltimore, Md ?: 1950). 2006;176(3):1582–1587. [DOI] [PubMed] [Google Scholar]
- 27.Jounaidi Y, F CJ, W MK, et al. Tethering IL2 to its receptor IL2Rβ enhances antitumor activity and expansion of natural killer NK92 cells. Cancer Res. 2017;77(21):5938–5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.K AA, Trotta E, Simeonov D R, et al. Revisiting IL-2: biology and therapeutic prospects. Sci Immunol. 2018;3(25):eaat1482. [DOI] [PubMed] [Google Scholar]
- 29.Spolski R, Li P, Leonard WJ. Biology and regulation of IL-2: from molecular mechanisms to human therapy. Nat Rev Immunol. 2018;18:648–659. [DOI] [PubMed] [Google Scholar]
- 30.Akman B, Hu X, Liu X, et al. PRDM1 decreases sensitivity of human NK cells to IL2‐induced cell expansion by directly repressing CD25 (IL2RA). J Leukoc Biol. 2021;109(5):901–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen X, Zaro JL, Shen W-C. Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev. 2013;65:1357–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown LY, Dong W, Kantor B. An Improved Protocol for the Production of Lentiviral Vectors. STAR protocols. 2020;1(3):100152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang Y, Zeng J, Liu T, et al. DNAM1 and 2B4 costimulatory domains enhance the cytotoxicity of anti-GPC3 chimeric antigen receptor-modified natural killer cells against hepatocellular cancer cells in vitro. Cancer Manag Res. 2020;12:3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, Lupardus P, L LS, et al. Structural biology of shared cytokine receptors. Annu Rev Immunol. 2009;27:29–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. 1994;8:652–658. [PubMed] [Google Scholar]
- 36.Nagashima S, Mailliard R, Kashii Y, et al. Stable transduction of the interleukin-2 gene into human natural killer cell lines and their phenotypic and functional characterization in vitro and in vivo. Blood. 1998;91(10):3850–3861. [PubMed] [Google Scholar]
- 37.Liao W, Lin J-X, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity. 2013;38:13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghoreschi K, I JM, Li X, et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J Immunol. 2011;186(7):4234–4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fujisaki H, Kakuda H, Shimasaki N, et al. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res. 2009;69(9):4010–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoshimura A, Nishinakamura H, Matsumura Y, et al. Negative regulation of cytokine signaling and immune responses by SOCS proteins. Arthritis Res Ther. 2005;7(3):100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.B DR, B KT, F DL, et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat Immunol. 2016;17(7):816–824. [DOI] [PubMed] [Google Scholar]
- 42.Biederstädt A, Rezvani K. Engineering the next generation of CAR-NK immunotherapies. Int J Hematol. 2021;114:554–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ng YY, Tay JCK, Wang S. CXCR1 expression to improve anti-cancer efficacy of intravenously injected CAR-NK cells in mice with peritoneal xenografts. Mol Ther Oncolytics. 2020;16:75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Romee R, Rosario M, M B-EM, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016;8(357):357ra123–357ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun Z, Ren Z, Yang K, et al. A next-generation tumor-targeting IL-2 preferentially promotes tumor-infiltrating CD8+ T-cell response and effective tumor control. Nat Commun. 2019;10(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mo F, Yu Z, Li P, et al. An engineered IL-2 partial agonist promotes CD8+ T cell stemness. Nature. 2021;597(7877):544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.K TY, Maki G, Miyagawa B, et al. Characterization of genetically altered, interleukin 2-independent natural killer cell lines suitable for adoptive cellular immunotherapy. Hum Gene Ther. 1999;10(8):1359–1373. [DOI] [PubMed] [Google Scholar]
- 48.Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer. 2016;16:7–19. [DOI] [PubMed] [Google Scholar]
- 49.Zingoni A, Molfetta R, Fionda C, et al. NKG2D and its ligands:“one for all, all for one. Front Immunol. 2018;9:476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Samaha H, Pignata A, Fousek K, et al. A homing system targets therapeutic T cells to brain cancer. Nature. 2018;561(7723):331–337. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data are available through the NCBI Gene Expression Omnibus under the accession number GSE199391 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE199391]. All data included in this study are available upon request by contact with the corresponding author.