

ABSTRACT
Epidemiological projections point to acquisition of ever-expanding multidrug resistance (MDR) by Escherichia coli, a commensal of the digestive tract and a source of urinary tract pathogens. Bioinformatics analyses of a large collection of E. coli genomes from EnteroBase, enriched in clinical isolates of worldwide origins, suggest the Cytotoxic Necrotizing Factor 1 (CNF1)-toxin encoding gene, cnf1, is preferentially distributed in four common sequence types (ST) encompassing the pandemic E. coli MDR lineage ST131. This lineage is responsible for a majority of extraintestinal infections that escape first-line antibiotic treatment, with known enhanced capacities to colonize the gastrointestinal tract. Statistical projections based on this dataset point to a global expansion of cnf1-positive multidrug-resistant ST131 strains from subclade H30Rx/C2, accounting for a rising prevalence of cnf1-positive strains in ST131. Despite the absence of phylogeographical signals, cnf1-positive isolates segregated into clusters in the ST131-H30Rx/C2 phylogeny, sharing a similar profile of virulence factors and the same cnf1 allele. The suggested dominant expansion of cnf1-positive strains in ST131-H30Rx/C2 led us to uncover the competitive advantage conferred by cnf1 for gut colonization to the clinical strain EC131GY ST131-H30Rx/C2 versus cnf1-deleted isogenic strain. Complementation experiments showed that colon tissue invasion was compromised in the absence of deamidase activity on Rho GTPases by CNF1. Hence, gut colonization factor function of cnf1 was confirmed for another clinical strain ST131-H30Rx/C2. In addition, functional analysis of the cnf1-positive clinical strain EC131GY ST131-H30Rx/C2 and a cnf1-deleted isogenic strain showed no detectable impact of the CNF1 gene on bacterial fitness and inflammation during the acute phase of bladder monoinfection. Together these data argue for an absence of role of CNF1 in virulence during UTI, while enhancing gut colonization capacities of ST131-H30Rx/C2 and suggested expansion of cnf1-positive MDR isolates in subclade ST131-H30Rx/C2.
KEYWORDS: Escherichia coli, ExPEC, ST131, CNF1, rho GTPases, gastrointestinal tract, colonization, UTI
Introduction
Extraintestinal pathogenic Escherichia coli (ExPEC) form a heterogenic phylogenetic group characterized by the presence of specific virulence factors (VFs) conferring elevated risks of contracting severe forms of extra-intestinal infections, such as urinary tract infections (UTI). UTI are common infections that affect more than 150 million individuals, annually, and are the second cause of antibiotic prescription.1 Clinical studies document a high prevalence of the cytotoxic necrotizing factor 1 (cnf1)-encoding gene in uropathogenic strains of E. coli (UPEC), which belong to the larger group of ExPEC, and its presence in the microbiota of healthy patients.2–4 CNF1 is a paradigm of bacterial deamidase AB toxins activating Rho GTPases.5–8 The cnf1 gene belongs to the prototypic pathogenicity island (PAI) IIJ96 from the O4:K6 E. coli strain J96, that also contains an alpha-hemolysin (HlyA) encoding operon, a UclD adhesin tipped F17-like chaperone-usher (CU) fimbriae, and the PapGII adhesin tipped pyelonephritis-associated pili (pap) operon.9,10 Despite hypotheses that CNF1 plays a role in urovirulence,3 attempts to define fitness advantages conferred by this toxin in mouse models of UTI have led to opposing conclusions.11–14 UTI are inflammatory diseases, although whether CNF1 modulates inflammation, including neutrophil infiltration, into the bladder warrants clarification.11–13 This is particularly of interest as, in an animal model of bacteremia, CNF1 exerts a paradoxical host-protective effect antagonized by the action of the genetically associated alpha-hemolysin, further blurring the role of CNF1 in pathogenesis.15–17 Cell biology studies established that CNF1 confers high invasive capacities of epithelial cells to E. coli, similar to other Rho GTPase activating factors found in Enterobacteriaceae.18,19 Three types of CNF-like toxins have been described in E. coli strains, sharing high amino acid sequence identities.20–23 However, isolates expressing the CNF2 and CNF3 toxins are rarely detected in extraintestinal infections in humans. In the clinic, CNF1 is not linked to specific pathophysiological outcomes, in contrast to other known bacterial AB toxins from E. coli, such as Shiga-like toxins or the heat-labile toxin.
E. coli represents the predominant facultative aerobic bacteria of the gut microbiota, as well as an extraintestinal opportunistic pathogen.24,25 The gut is a known reservoir for uropathogenic bacteria, including, notably extended-spectrum beta-latamase (ESBL)-producing E. coli.26–29 Only a few sequence types (STs) within the E. coli population account for more than half of all E. coli strains responsible for extraintestinal infections not causally related to antibiotic resistance.24,30 The globally disseminated E. coli ST131 has emerged as the predominant lineage responsible for worldwide dissemination of the ESBL encoding gene blaCTX-M-15 and the rise of multidrug resistant MDR extraintestinal infections.31,32 This well-defined sequence type is structured into three different clades, with the fluoroquinolone-resistant clade C strains subdivided into two subclades comprised of H30R/C1 and the dominant expanding H30Rx/C2, frequently carrying blaCTX-M-15.33–35
One reason for the unprecedented success of E. coli ST131-H30 clade C may be its intrinsic capacity to persist in the gastrointestinal tract (GIT) in competition with other strains of E. coli.27,36–38 Enhanced colonization capacities of the GIT by E. coli ST131 may promote inter-individual transmission, favoring its dissemination in the human population and other hosts, as compared to other lineages.,27,39–41 as well as account for a lack of a phylogeographical signal among these strains.42 The remarkable fitness of this lineage strongly supports the idea of a step-wise acquisition of factors promoting GIT colonization, potentially scattered among UPEC populations, as well as promoting bacterial virulence or pathogenicity in the context of extraintestinal infections.43
To better appreciate cnf1 dynamics, we performed a large-scale screening of the toxin gene distribution in a large dataset of E. coli genomes deposited in EnteroBase.44 The observed increase of cnf1-positive strains in the ST131-H30Rx/C2 lineage led us to hypothesize that cnf1 may confer a competitive advantage to colonize the GIT. Indeed, the wildtype strain EC131GY from lineage ST131-H30Rx/C2 outcompeted a cnf1-deleted variant when concurrently inoculated into the GIT, arguing for a role of CNF1 in EC131GY selection within the gut that might be linked to CNF1 deamidase activity on Rho GTPases to promote tissue invasion. Surprisingly, we observed no differences in fitness or inflammation in monoinfections of the urinary tract linked to the presence or absence of cnf1. Collectively, these data support although CNF1 does not impact host response to UTI, it acts as an intestinal colonization factor during competition in the GIT.
Results
Analysis of the distribution of cnf genes in a large collection of E. coli genomes
At the start of this study, we mined large genomic datasets from EnteroBase to gain more insight into the distribution of the cnf1 gene and its close homologs in the E. coli population.44 EnteroBase represents an integrated software environment widely used to define the population structure of several bacterial genera, including pathogens. Quantitative information on the collection of 141,234 E. coli genomes deposited in EnteroBase are reported in supplementary Figure S1. This collection, starting in 1900, aggregates genomes from strains collected worldwide, but mainly from Europe and North America, and from a wide range of sources but primarily human clinical isolates (Sup. Figure S1a, S1b, S1c). Using a Hidden Markov Model (HMM) approach coupled to amino acid pairwise distance calculation, we retrieved cnf-like positive strains and characterized each type of cnf sequence. In total, we identified 6,411 cnf-positive strains (4.5% of all E. coli isolates) with a remarkable dominance of cnf1 (87.8%, n = 5,634), as compared to cnf2 (8.6%, n = 554) and cnf3 (3.5%, n = 223). These strains displayed only one type of cnf-like encoding gene. The prevalent cnf1 gene in this genomic dataset was widely distributed among isolates of all origins but most notably in the groups denoted humans (5.4% of n = 48,518 human isolates) and companion animals (24.1% of n = 2,652 companion animal isolates) (Sup. Figure S1c).
We next studied the distribution of cnf1 among E. coli phylogenetic groups and sequence types (STs). The cnf1 gene was preferentially associated with isolates from the phylogroup B2, representing 24.3% of n = 22,305 retrieved genome sequences (Sup. Figure S1d). We observed a tight association of cnf1 with the most frequently encountered ExPEC STs (Table 1) (P < 2.2 10−16, Chi-square association test). Notably, a majority of the 5,634 cnf1-positive strains segregated among four STs: ST131 (24.5% of cnf1-positive strains, n = 1,382), ST73 (23.2%, n = 1,308), ST12 (12.4%, n = 699), and ST127 (10.7%, n = 601). The remaining 29.2% of cnf1-positive strains were widely distributed among 266 other STs. Interestingly, we noticed a steady increase of the percentage of cnf1-positive strains in the E. coli ST131 lineage from 13% in 2009 to 23% in 2019 (Figure 1), while this percentage fluctuated around high values in ST73, ST12, and ST127. This analysis revealed a close association of cnf1 with common ExPEC lineages and a surprising convergent distribution of cnf1 in the four lineages ST131, ST73, ST12, and ST127.
Table 1.
Distribution of phylogroups and sequence types among E. coli cnf-positive strains from EnteroBase. The total number and the percentage of each phylogroup and most dominant sequence types (STs) among cnf-positive strains are indicated.
| Phylogroups | ST | Number of strains |
Percentage of Phylogroup or Sequence type in CNF-positive strains |
||||||
|---|---|---|---|---|---|---|---|---|---|
| All | CNF+ | CNF1+ | CNF2+ | CNF3+ | CNF1 | CNF2 | CNF3 | ||
| A | Total A | 34,982 | 51 | 0 | 28 | 23 | 0 | 5.05 | 10.31 |
| ST10 | 8,748 | 24 | 0 | 17 | 7 | 0.0 | 3.1 | 3.1 | |
| ST342 | 325 | 16 | 0 | 0 | 16 | 0.0 | 0.0 | 7.2 | |
| B1 | Total B1 | 37,262 | 527 | 96 | 373 | 58 | 1.7 | 67.3 | 26.0 |
| ST101 | 938 | 93 | 24 | 69 | 0 | 0.4 | 12.5 | 0.0 | |
| ST392 | 79 | 66 | 0 | 66 | 0 | 0.0 | 11.9 | 0.0 | |
| ST58 | 1,487 | 44 | 9 | 35 | 0 | 0.2 | 6.3 | 0.0 | |
| ST29 | 496 | 35 | 0 | 0 | 35 | 0.0 | 0.0 | 15.7 | |
| ST2217 | 46 | 31 | 0 | 31 | 0 | 0.0 | 5.6 | 0.0 | |
| ST5738 | 24 | 23 | 0 | 23 | 0 | 0.0 | 4.2 | 0.0 | |
| ST21 | 5,082 | 10 | 0 | 0 | 10 | 0.0 | 0.0 | 4.5 | |
| ST343 | 134 | 2 | 0 | 0 | 2 | 0.0 | 0.0 | 0.9 | |
| ST2836 | 63 | 2 | 0 | 0 | 2 | 0.0 | 0.0 | 0.9 | |
| ST4063 | 3 | 2 | 0 | 0 | 2 | 0.0 | 0.0 | 0.9 | |
| B2 | Total B2 | 22,305 | 5,478 | 5,414 | 63 | 1 | 96.1 | 11.4 | 0.4 |
| ST131 | 9,242 | 1,383 | 1,382 | 0 | 1 | 24.5 | 0.0 | 0.4 | |
| ST73 | 2,071 | 1,308 | 1,308 | 0 | 0 | 23.2 | 0.0 | 0.0 | |
| ST12 | 809 | 699 | 699 | 0 | 0 | 12.4 | 0.0 | 0.0 | |
| ST127 | 709 | 601 | 601 | 0 | 0 | 10.7 | 0.0 | 0.0 | |
| ST372 | 366 | 206 | 206 | 0 | 0 | 3.7 | 0.0 | 0.0 | |
| ST95 | 1,882 | 173 | 147 | 26 | 0 | 2.6 | 4.7 | 0.0 | |
| ST141 | 360 | 164 | 164 | 0 | 0 | 2.9 | 0.0 | 0.0 | |
| ST998 | 175 | 149 | 149 | 0 | 0 | 2.6 | 0.0 | 0.0 | |
| ST80 | 152 | 109 | 105 | 4 | 0 | 1.9 | 0.7 | 0.0 | |
| ST537 | 50 | 35 | 35 | 0 | 0 | 0.6 | 0.0 | 0.0 | |
| ST647 | 28 | 26 | 0 | 26 | 0 | 0.0 | 4.7 | 0.0 | |
| C | Total C | 3,465 | 56 | 45 | 10 | 1 | 0.8 | 1.8 | 0.4 |
| D | Total D | 9,905 | 37 | 20 | 13 | 4 | 0.4 | 2.3 | 1.8 |
| E | Total E | 16,391 | 155 | 7 | 14 | 134 | 0.1 | 2.5 | 60.1 |
| ST11 | 13,639 | 113 | 0 | 0 | 113 | 0.0 | 0.0 | 50.7 | |
| ST5592 | 5 | 5 | 0 | 0 | 5 | 0.0 | 0.0 | 2.2 | |
| ST11457 | 4 | 4 | 0 | 0 | 4 | 0.0 | 0.0 | 1.8 | |
| F | Total F | 2,957 | 38 | 37 | 0 | 1 | 0.7 | 0.0 | 0.4 |
| G | Total G | 1,862 | 34 | 0 | 34 | 0 | 0.0 | 6.1 | 0.0 |
| ST117 | 1,383 | 31 | 0 | 31 | 0 | 0.0 | 5.6 | 0.0 | |
| Clade I | Total CI | 406 | 18 | 0 | 18 | 0 | 0.0 | 3.2 | 0.0 |
| ST3057 | 41 | 11 | 0 | 11 | 0 | 0.0 | 2.0 | 0.0 | |
| Clade II | Total CII | 6 | 0 | 0 | 0 | 0 | 0.0 | 0.0 | 0.0 |
| Clade III | Total CIII | 39 | 0 | 0 | 0 | 0 | 0.0 | 0.0 | 0.0 |
| Clade IV | Total CIV | 39 | 0 | 0 | 0 | 0 | 0.0 | 0.0 | 0.0 |
| Clade V | Total CV | 166 | 0 | 0 | 0 | 0 | 0.0 | 0.0 | 0.0 |
| Other 358 STs | 34,599 | 1,044 | 803 | 215 | 26 | 14.3 | 38.8 | 11.7 | |
Figure 1.
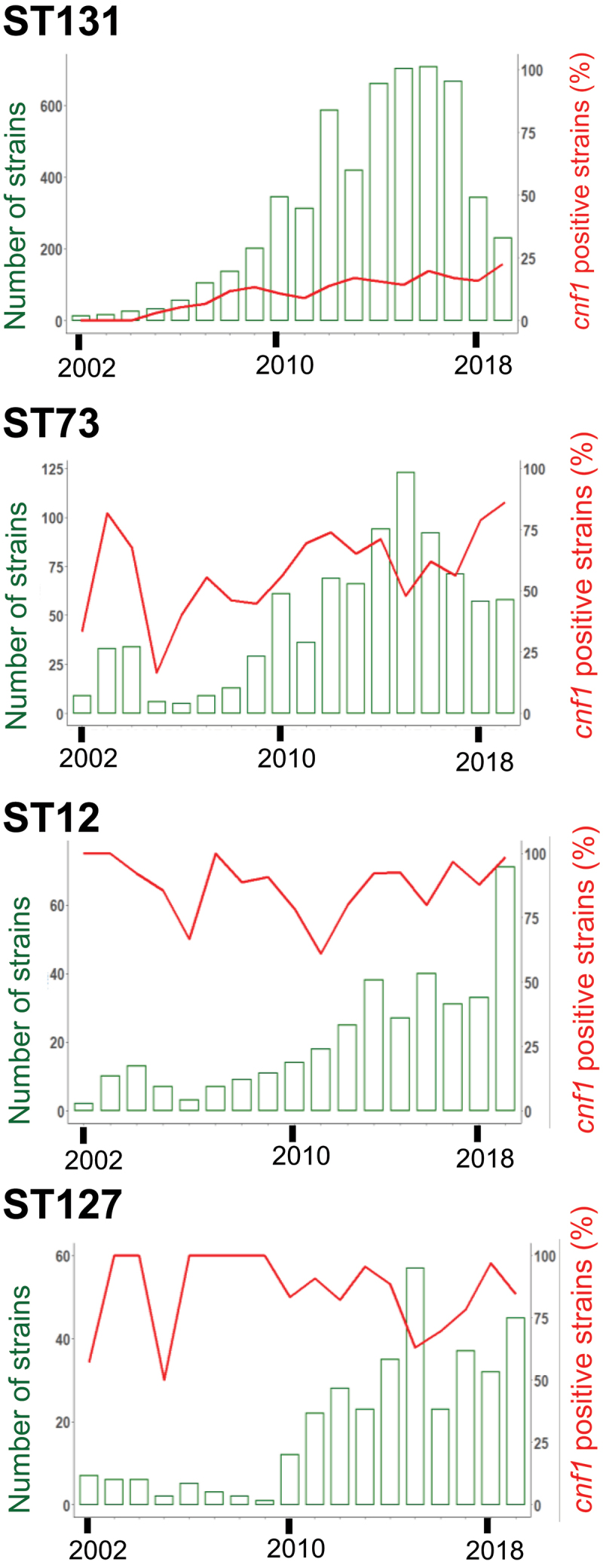
Prevalence overtime in representative E. coli sequence types bearing cnf1. Bar chart show number of E. coli strains from ST131, ST73, ST12 and ST127 isolated each year during the period 2002–2019, left y-axis. Percentages of cnf1-positive strains per year, right y-axis.
Cnf1-positive strains segregate into monophyletic groups in ST131 phylogeny
The rising prevalence of cnf1 in E. coli ST131 over time motivated us to study cnf1 distribution in this ST. EnteroBase contained 9,242 genomes of E. coli ST131 at the time of analysis (November 2020). To facilitate genomic analysis, we retained 5,231 genomes isolated from 1967 to 2018. We built a Maximum Likelihood phylogenetic tree based on a total of 37,304 non-recombinant single nucleotide polymorphism (SNPs). Phylogenetic distribution of strains showed an expected dominant population of clade C (76%, n = 3,981; 99% fimH30), as compared to clade A (11%, n = 569; 92% fimH41) and B (13%, n = 68; 62% fimH22) (Figure 2a, Sup. Figure S2a). We also found an expected co-distribution of parC (S80I/E84V) and gyrA (S83L/D87N) alleles, which confer resistance to fluoroquinolones in most strains from clade C (99.84%, n = 3,975 strains), and a tight association of the blaCTX-M-15 ESBL gene (85%, n = 2,194 isolates) with strains from subclade H30Rx/C2 (P< 2.2e−16, Chi-square association test). The high number of strains gave enough resolution to distinguish two sublineages, C2_1 and C2_2, originating from C2_0 (Figure 2a). From available metadata, we verified the absence of overall geographical and temporal links in the phylogenetic distribution of E. coli ST131 strains (Sup. Figure S2b).
Figure 2.
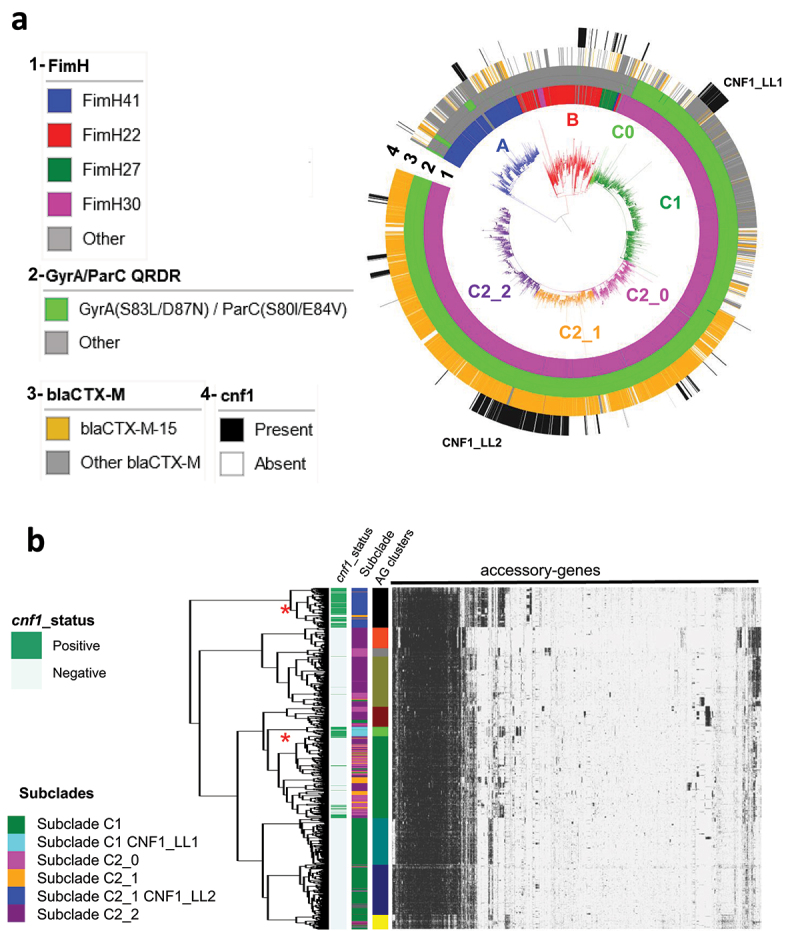
Dynamic of CNF1-encoding gene in E. coli ST131 from EnteroBase. A) Maximum likelihood phylogeny of E. coli ST131 from EnteroBase (Sup. Figure S2 for extended information). The phylogeny was constructed with 5,231 genomes for a total of 37,304 non-recombinant core-genome SNPs. The different clades and subclades A, B, C0, C1, C2_0, C2_1, C2_2 are highlighted in blue, red, light green, green, pink, Orange and purple respectively. From inside to outside circles are indicated (1) fimH alleles, (2) gyrA and parC alleles conferring resistance to fluoroquinolones (shown in green), (3) positive strains for blaCTX-M-15 (shown in Orange) and (4) strains bearing cnf1 gene (shown in black).) Hierarchical clustering of strains from clade C (n = 3981 strains) based on their accessory gene content. The pan-genome is composed of 51,742 genes including 2,672 genes that are present in 98% of the strains. The graph displays the 7,678 genes identified as present in at least 50 and less than 3,930 genomes. The colored annotation indicates (from left to right) the presence of cnf1 (CNF1_status), subclades (C1, C1 CNF1_LL1, C2_0, C2_1, C2_1 CNF1_LL2, C2_2) and accessory genes cluster (AG_clusters). Large lineages of cnf1-positive strains in clades C1 and C2_1 are denoted CNF1_LL1 and CNF1_LL2, respectively. Red stars indicate the two large lineages of cnf1-positive strains.
We next analyzed the distribution of cnf1-positive strains in E. coli ST131 phylogeny (n = 725, cnf1-positive E. coli) (Figure 2a, black stripes). The cnf1-positive strains were preferentially associated with subclade C2 (n = 520) (p < 2.2 10−16, Chi-square association test), as compared to subclade C1 (n = 101), clade B (n = 72), or clade A (n = 32) (Figure 2a). Strikingly, most cnf1-positive strains segregated into lineages in all clades and subclades with a noticeable distribution of cnf1-positive ST131 strains in two large lineages (LL) in H30R/C1 (n = 101 cnf1-positive strains/107 strains in CNF1_LL1) and in H30Rx/C2_1 (n = 396 cnf1-positive strains/425 strains in the CNF1_LL2) (Figure 2a). We then analyzed the diversity of cnf1 alleles to define their distribution in the phylogeny of ST131 (Sup. Table S1). A similar analysis was performed with the alpha-hemolysin encoding gene, hlyA. We found a wide co-distribution of one combination of alleles of cnf1 (allele P1cnf1, 85,1%) and hlyA (allele P1hlyA, 77,2%) in E. coli ST131 clade A and C, whereas strains from clade B displayed a large range of combinations of various alleles (Sup. Figure S2a). Together, our data point to a clonal expansion of worldwide disseminated ST131-H30 strains with the same allele of cnf1. This prompted us to perform a clustering analysis of ST131-H30 strains according to their accessory gene contents. We generated a pan-genome matrix of 51,742 coding sequences from the n = 3,981 strains of clade C. The dataset of accessory genes was built from n = 7,678 sequences that were present in at least 50 and no more than 3,931 strains. We conducted a hierarchical clustering of strains and retained 10 distinct accessory gene clusters. Strikingly, this revealed a conservation between phylogenetically defined groups CNF1_LL1 and CNF1_LL2 and groups defined by their accessory gene contents (Figure 2b). Indeed, the hierarchical clustering was most evident for CNF1_LL2, showing a differential enrichment determined with Scoary of n = 1,434 genes as compared to other strains from clade C (P < 0.05, Bonferroni-adjusted correction). Together, these data point toward intensive group-specific diversification of accessory gene content in cnf1-positive clusters in ST131-H30.
E. coli ST131 cnf1-positive strains segregate between two clade-specific virulence profiles
We then defined strain contents in virulence factors (VF) and acquired antibiotic-resistance genes (RG) to perform an unbiased analysis of their distribution into clusters, using a latent block model approach, as described in the materials and methods. The unsupervised clustering procedure identified a total of 10 RG-clusters and 7 VF-clusters (Figure 3a). Differences in number of VFs and RGs among clusters were all significant (Figure 3b). We found that cnf1-positive strains were scattered among several RG clusters (Figure 3a, left panel). By contrast, most cnf1-positive strains segregated into the VF4 cluster (84% of cnf1-positive strains, n = 609) with the remaining 16% strains distributed between VF1 (15%) and other VF clusters (1%) (Figure 3a, right panel). In contrast to the scattered distribution of RG-clusters into the phylogeny, we observed a distribution of well-defined groups of VF-clusters (Figure 3c). A majority of cnf1-positive strains from clade A and B were part of cluster VF1, whereas cnf1-positive strains from clade C were part of cluster VF4. With a median of 33 virulence factors, VF4-positive strains displayed the largest number of virulence factors. The VF1 profile was more specifically defined by the presence of genes encoding the IbeA invasin and IroN Salmochelin siderophore receptor (Sup. Figure S3a). By contrast, the VF4 profile was more specifically defined by cnf1 and hlyA (respectively 54% and 61% in VF4) and also encompassed genes encoding the UclD adhesin that caps the F17-like chaperone-usher (CU) fimbriae cluster and the PapG II adhesin from the pyelonephritis-associated pili (pap) operon (Sup. Figure S3a). 9,45 Analysis of several complete sequences of cnf1-bearing PAI from ST131-H30 showed a conservation of a module containing genes defining VF4 (Sup. Figure S3b). Indeed, elements best defining VF4 were genetically associated and displayed high synteny with cnf1-bearing pathogenicity islands (PAI) IIJ96 from the O4:K6 E. coli strain J96.
Figure 3.
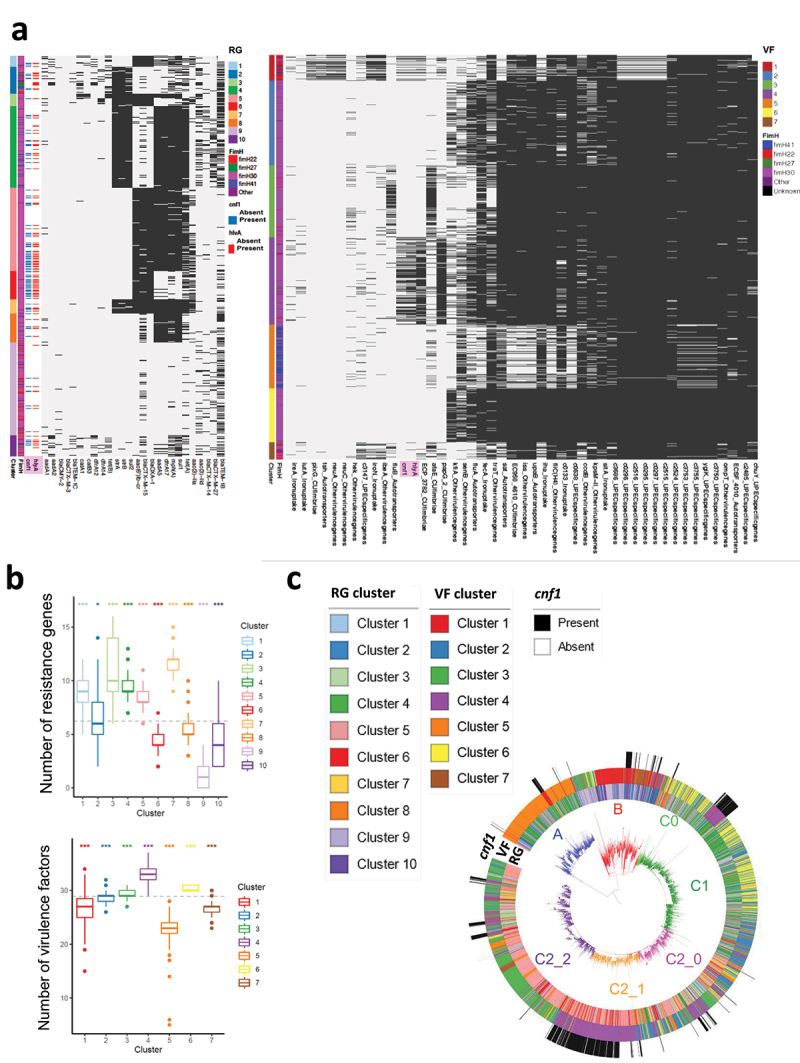
Co-clustering of acquired antibiotic-resistance gene and virulence factors in E. coli ST131. A) Heatmaps show clusters of antibiotic acquired-resistance gene (RG) (left panel) or virulence gene (VF) (right panel) profiles (Sup. table S2) constructed using a binary latent block model between strains by row and RGs or VFs by column. Black lines indicate the presence of RG or VF in each strain. Annotations are displayed on the right of each heatmap: information about strain clusters and fimH alleles together with hlyA and cnf1 carriage. B) Box-and-whisker plot showing the distribution of strains according to their content of acquired antibiotic-resistance genes (upper panel) or content of virulence factors (lower panel). The dotted line shows the mean number of RG or VF. All one-versus-all comparisons of VF and RG contents between clusters (*P < 0.05, ***P < 0.001). C) RG, VF clusters and cnf1 carriage are displayed on the E. coli ST131 phylogenetic tree. The different clades and subclades A, B, C0, C1, C2_0, C2_1, C2_2 are highlighted in blue, red, light green, green, pink, Orange and purple respectively.
Cnf1-positive strains display dominant expansion in ST131-H30Rx/C2
We next analyzed the temporal distribution of cnf1-positive strains within clades and subclades. Using a Generalized Linear Models (GLM) approach, we first verified within our dataset the increase of fimH30-positive isolates over time (clade C) in E. coli ST131 that was maximal in H30Rx/C2 (P < 2.2 10−16, Chi-square association test) (Figure 4a). We also noted a significant increase in the proportion of cnf1-positive strains over time in E. coli ST131 (Figure 4b, top panel). The GLM was then fitted on years, clades, and subclades. We tested the significance of the year effect and P-values were corrected for multiple comparisons using Tukey’s method. The year effect was not significant for clade A, B, or subclade H30R/C1 (Figure 4b). Instead, we observed a significant increase of the proportion of cnf1-positive strains within H30Rx/C2 over time (P = 1.25 10−11). In addition, the GLM fitted curves predicted that the prevalence of cnf1-positive strains within H30Rx/C2 subclade would be approximately 50% (confidence interval of 95% [43% to 58%] in 2018; [47% to 64%] in 2019). Predictive values were compared to the prevalence of cnf1 in ST131 strains isolated in 2018 or 2019 in a second independent dataset up-loaded from EnteroBase in September 2020. The prevalence of cnf1-positive strains within the subclade H30Rx/C2 was 45% in 2018 and 48% in 2019, confirming the prediction of a dominant expansion of cnf1-positive strains within ST131-H30Rx/C2.
Figure 4.
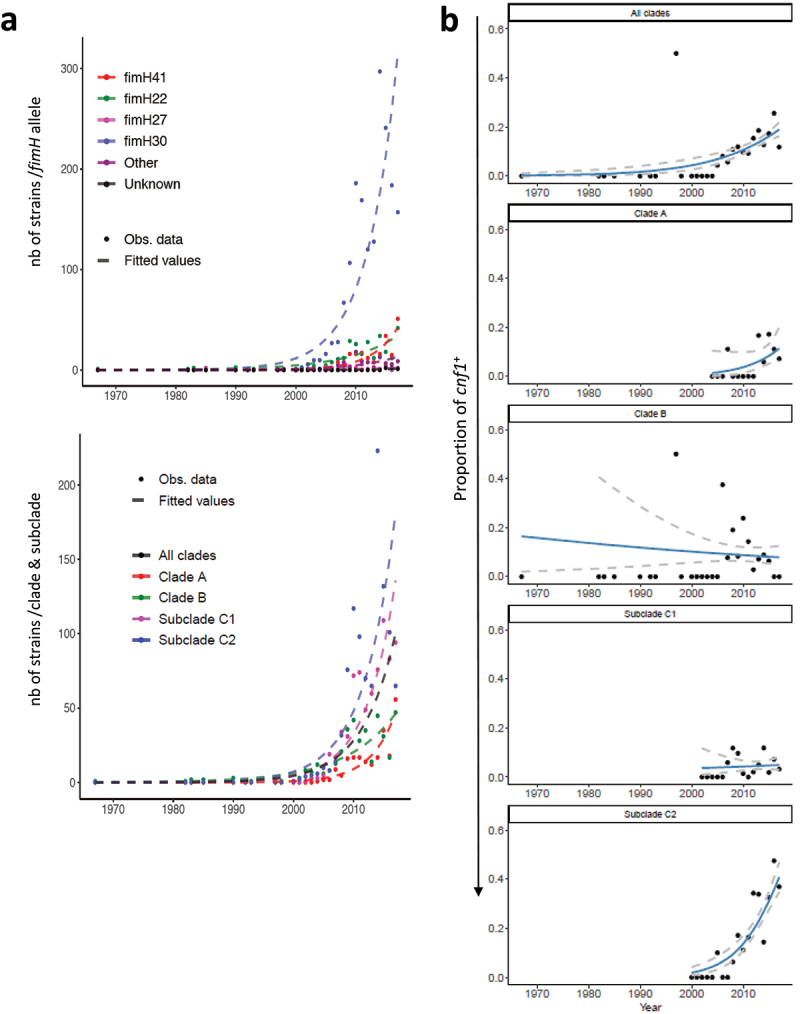
Increase over the years in the proportion of cnf1-positive strains in E. coli ST131 H30Rx/C2. A) Distribution of fimH alleles (upper panel) or clades/subclades (lower panel) within the study population of E. coli ST131. Both figures show observed counts per year (dots) and data fitted lines (dashed lines) with a generalized linear model (Poisson regression). B) Increase of the proportion of cnf1-positive strains in the whole E. coli ST131 population along time (top panel, P = 7.41 10–7) and by clades and subclades. The black dots represent the observed proportion of cnf1-positive strains by year with fitted line of a logistic regression model (blue curves). Dashed gray lines display the 95% confidence intervals. The P-values are not significant for clade A (P = 0.287), B (P = 0.952), H30R/C1 (P = 0.992) and significant for H30Rx/C2 (P = 1.25 10−11).
Cnf1 confers a competitive advantage for gut colonization in two ST131-H30Rx/C2 strains
The dominant expansion of cnf1-positive strains in ST131 H30Rx/C2 prompted us to explore whether CNF1 might function as a virulence factor in UTI and a colonization factor in the gastrointestinal tract, a natural environment for E. coli. We selected a VF4/cnf1-positive strain of E. coli ST131 H30Rx/C2, here referred to as EC131GY (Sup. Figure S4). This strain displays a prototypic cnf1-bearing PAI IIJ96 (Sup. Figure S3b). We generated an EC131GY strain in which cnf1 was replaced with a kanamycin resistance cassette (EC131GY Δcnf1::kanr) and verified the absence of CNF1 expression (Sup. Figure S5a). We then verified, in vitro, the absence of fitness cost due to the kanamycin resistance cassette as shown by equal growth of the parental and Δcnf1::kanr EC131GY strains, and the absence of competition between the strains when grown together (Sup. Figure S5b and S5c). Next, in mono-microbial bladder infections, we observed no difference in the number of colony-forming units (CFU) between wildtype EC131GY and the Δcnf1::kanr strain at 1, 3, and 7 d post-infection (Figure 5a). In addition, we observed indistinguishable responses in 20 variables of the innate immune response between the two infections at 24 hours post-infection (Sup Figure S6A-S6E). This included no observed difference after infection with either of the two strains in inflammatory cytokine expression, or in proportions of resident macrophage subsets, dendritic cells, monocytes, neutrophils, NK, or lymphoid cell populations (Sup Figure S6B-S6E).
Figure 5.
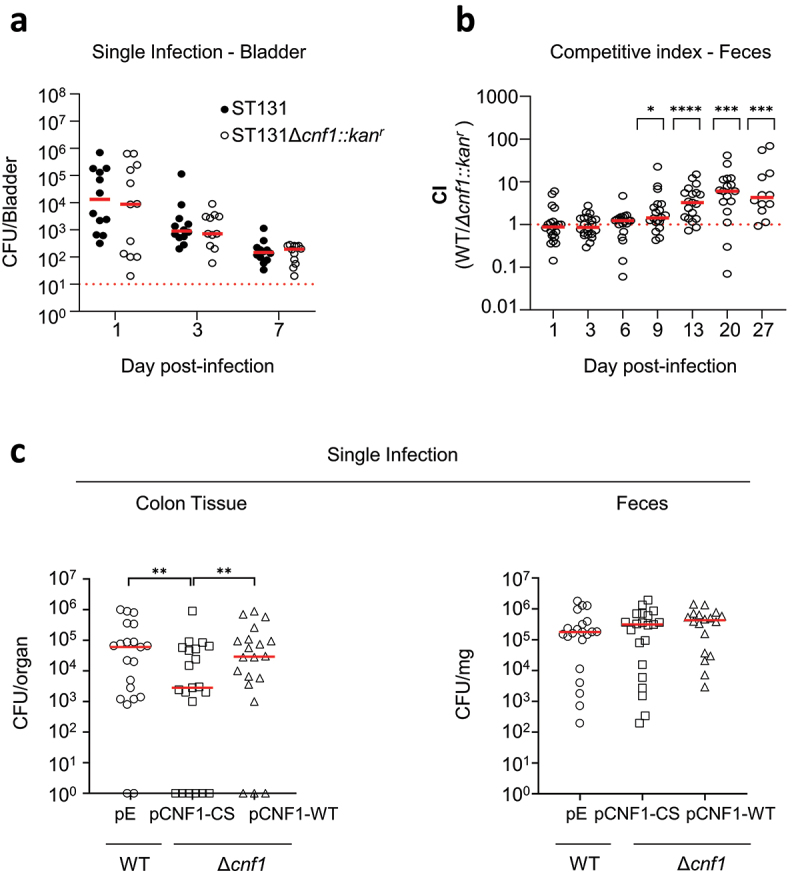
CNF1 promotes ST131-H30Rx/C2 intestinal colonization. A) For urinary tract infection, mice were infected separately with wildtype EC131GY (WT) and EC131GY Δcnf1::kanr (Δcnf1::kanr) via intravesical instillation of the bladder. B-C) For GIT colonization, mice were pretreated with streptomycin and subsequently infected concurrently via oral gavage with EC131GY WT and Δcnf1::kanr (B), or with the following stains alone: EC131GY wildtype with an empty vector (WT + pE) as control, EC131GY Δcnf1 with a vector encoding catalytically inactive cnf1 (Δcnf1 + pCNF1-CS), or EC131GY Δcnf1 with a vector encoding cnf1 (Δcnf1 + pCNF1-WT) (C). Levels of viable bacteria in bladder homogenates, feces, or colonic tissue after ex vivo gentamicin treatment were assessed at indicated times by measuring colony forming units (CFU). Data show CFU per bladder at 1, 3, and 7 d post-infection (A), competitive index (CI) in feces at indicated days post-infection (B), CFU in colon tissues and feces at 72 hours post-infection (C) for each animal and medians (red bar). (A and C) bladders are n = 9–10, two replicates at day 1, 3, 7 and feces CI, n = 22–24, three replicates. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 and ns: non-significant by Wilcoxon signed-rank test. (C) colon tissues and feces n = 20–21, four replicates. A mixed effect model adjusted on the conditions and dates. **P < 0.01 (Δcnf1 + pCNF1-CS vs WT + pE), **P < 0.01 (Δcnf1 + pCNF1-CS vs Δcnf1 + pCNF1-WT), and P = 0.98 (WT + pE vs Δcnf1 + pCNF1-WT).
We then explored the impact of cnf1 on GIT colonization by competitive infection with wildtype EC131GY and EC131GY Δcnf1::kanr, using intra-gastric gavage, to model the natural environment in which several strains of E. coli are present. Longitudinal measurements of CFU in the feces showed that cnf1 conferred an advantage to wildtype EC131GY over the EC131GY Δcnf1::kanr isogenic strain for gut colonization from 9 d after oral gavage, which persisted over 27 d (Figure 5b). We confirmed the competitive advantage for gut colonization conferred by cnf1 in another clinical isolate, BLSE2018-86 from ST131 H30Rx/C2, which had an advantage over the cnf1 mutant from 6 d post-inoculation (Sup. Figure S4 and S7). We next performed a quantitative approach to assess the impact of CNF1 deamidase activity on the efficiency of colon tissue invasion by EC131GY. The strain EC131GY Δcnf1 was transformed with plasmids expressing wildtype CNF1 (Δcnf1 + pCNF1-WT) or the catalytically-inactive mutant C866S (Δcnf1 + pCNF1-CS), while the wildtype CNF1-producing EC131GY was transformed with an empty vector as a control (WT + pE). These strains showed equal growth kinetics in vitro (Sup. Figure S5b). Equivalent quantities of each strain were delivered by oral gavage. At an early time point of 72 hours post-infection, we enumerated CFU in feces and colon tissue after ex vivo gentamicin treatment of tissues, a membrane non-permeant aminoglycoside. While we observed no difference in CFU in gentamicin-treated colonic tissues between the two strains expressing wildtype CNF1, we observed a significant lower number of CFU for EC131GY Δcnf1 expressing the catalytically inactive C866S form of the CNF1 toxin (Figure 5c). We interpret these data to support that invasion of colon tissue by EC131GY is mediated, at least in part, by CNF1 deamidase activity. Together, these data uncover an advantage conferred by CNF1 for GIT colonization by two clinical strains of ST131-H30Rx/C2 subclade.
Discussion
E. coli ST131 has rapidly become a globally dominant lineage of ExPEC responsible for UTI that are resistant to antibiotic treatments, globally. Independently of the acquisition of multidrug-resistance genes, advantages ascribed to the ST131 lineage encompass an increased capacity to colonize the GIT, although molecular determinants enhancing gut colonization remain to be defined.41,46 Here, we report that cnf1 enhances the capacity of ST131 H30/Rx/C2 to compete with the cnf1-negative isogenic strains for gut colonization. Moreover, CNF1 deamidase activity enhances EC131GY capacity to invade colon tissues. These findings represent a change of paradigm for the CNF1 toxin by providing evidence that CNF1 has no detectable impact on inflammation during the first 7 d of UTI and enhances enhances ST131 H30/Rx/C2 competitive advantage of gut colonization. Although, more work is needed to clarify the role of cnf1 in other aspects of UTI virulence, such as the formation of bladder reservoirs or in recurrent UTI, our data clearly assigned to CNF1 a function of gut colonization factor.
In parallel, statistical analysis of more than five thousand isolates of E. coli ST131 from EnteroBase, suggests an intercontinental expansion of cnf1-positive H30Rx/C2 strains among human clinical isolates from subclade C2 and ST131. This hypothesis is supported by the distribution of a large cluster of VF4/cnf1-positive H30Rx/C2 strains onto the phylogeny sharing the same alleles of cnf1 and hlyA toxin genes. Indeed, the expansion of a phylogenetic subcluster of ST131-H30Rx/C2 strains bearing cnf1-positive PAI within the subclade C2 has recently been highlighted for companion dog isolates.47,48 Thus, although the EnteroBase E. coli dataset likely includes some bias in the sampling of strains with a high abundance of strains responsible for acute human infections and from specific continents, this database is well-adapted to study toxin gene distribution in human clinical isolates of E. coli. Further in support of a possible advantage conferred by the acquisition of cnf1, we found a high coverage of strains positive for cnf1 in the common clinical STs, ST73, ST12, and ST127, but not in ST95.24 Collectively, these findings suggest that cnf1 may enhance the capacity of GIT colonization by some ExPEC, in turn contributing to the dissemination of a few E. coli lineages and sublineages, such as ST131-H30Rx/C2.
The intestinal tract is a key reservoir for ExPEC strains.49 A previous study suggested that E. coli ST131 has a high capacity to invade human intestinal epithelial cells and persistently colonize mouse GIT in a type I pili-dependent manner.46 Our findings, that cnf1 gives a competitive advantage for GIT colonization and that CNF1 toxin deamidase activity enhances invasion of colonic tissue, raises the interest of defining the relationships between cnf1 and type I pili for tissue colonization. This includes both bladder and gut tissue colonization. Indeed, our data showing an absence of difference in inflammation during UTI does not rule out a potential effect of CNF1 in bladder tissue colonization and chronic carriage, leading to recurrent UTI. Indeed, cell biology studies showed that CNF1 promotes epithelial cell invasion, including bladder epithelial cells, by E. coli through its capacity to activate host Rho GTPase signaling.19,23,50,51 In line with this, CNF1 deamidase exacerbates Rho GTPase signaling, and notably Rac1, to promote type I pili-mediated host cell invasion.52,53 Enhanced capacities to invade and colonize intestinal tissues may also involve factors encoded within PAI IIEC131GY-like from ST131 H30Rx/C2. Indeed, cnf1-bearing PAIs contain a core set of genes encoding the F17-like pili, the P-fimbriae tipped with PapG class II adhesin, and the HlyA toxin, as well as a gene encoding hemagglutinin in E. coli K1 (Hek).54–56 The cnf1-bearing PAIs also include elements of oxidative stress adaptation, namely the methionine sulfoxide reductase complex MsrPQ encoding genes yedYZ, which may work against CNF1-generated oxidative stress.57,58
Large-scale phylogenetic reconstruction of ST131 genomes from EnteroBase showed an expected phylogenetic distribution within clades and subclades of genetic traits defining this lineage. We report a stable population of cnf1-positive strains in H30R/C1 in EnteroBase, contrasting with the expansion of cnf1-positive strains in H30Rx/C2. Moreover, we observed a high prevalence of cnf1-positive strains in a few STs commonly responsible for extraintestinal infections. It will be of interest to decipher the interplay of cnf1 in gut colonization by H30R/C1, as well as ST73, ST12, and ST127 that display lower acquired resistance gene content as compared to E. coli strains from ST131 subclade C2.24,59 This should help draw the relationship between strain-specific profiles of antibiotic resistance and the function of cnf1 in gut colonization linked to bacterial dissemination. This may also help define epistatic relationships between cnf1 function as a gut colonization factor and strain-specific genetic backgrounds, including regulatory factors of cnf1-gene expression, toxin secretion, and strain-dependent adaptation to the gut environment including invasion of specific niches in the intestine.49,60,61
Material and methods
E. coli genomes dataset
The dataset corresponds to 141,234 E. coli genome sequences retrieved from EnteroBase (November 2020) (http://enterobase.warwick.ac.uk) 44 Strains’ metadata (collection year, continent, source niche of isolation and sequence type) were also retrieved (Sup. Table S3). Assemblies were downloaded in GenBank format and proteomes generated using annotations provided in GenBank files.
In silico detection and typing of cnf-like toxin encoding genes
The search for cnf genes in E. coli genomes was carried out with a domain specific Hidden Markov Models (HMM) profile built with 16 representative sequences of CNF1 catalytic domain (Sup. Table S4) using HMMER (http://hmmer.org/) 62 Protein sequences from positive hits were extracted from EnteroBase annotated E. coli proteomes and submitted to Clustal Omega for the computation of pairwise distances of the sequences, along with representative sequences of CNF-like toxin (CNF1 (AAA85196.1), CNF2 (WP_012775889.1) and CNF3 (WP_02231387.1)). Distances were used to determine the type of toxin with a threshold value of 0.1. In total 2.7% of HMM-positive sequences with a threshold value above 0.1 against all type of CNF-like toxin or below 0.1 against at least two type of CNF-like toxin were excluded from the analysis.
ST131 dataset structure and phylogenomic analysis
The database used for phylogenetic and statistical analyses consists of whole-genome sequences of E. coli ST131 isolates collected by mining EnteroBase from 1967 to 2018.44 Leaning on Find ST(s) tool from EnteroBase, we retained a total of 5,231 genome assemblies and associated metadata, including information of the isolation date, country and source of isolates (Sup. table S5). Phylogeny of ST131 isolates was resolved using core non-recombinant SNPs defined with Parsnp (in total 37,304 SNPs)63 and Gubbins v2.3.4.64 A maximum-likelihood tree was then estimated with RAxML v8.2.8 applying a general time-reversible substitution-model with a gamma distribution rate across sites and with an ascertainment bias correction65 and the resulting tree was edited with the interactive Tree of Life (iTol) v4 program.66 Chi-square association test was used to evaluate the significant association of cnf1 and blaCTX-M-15 with subclade C2.
Pan-genome analysis
The pangenome of E. coli ST131 was estimated using Roary, a high-speed pan genome pipeline analysis tool.67 Roary returns as output, the gene presence/absence matrix. The matrix was curated to retain genes present in at least 50 genomes and less than 3931 genomes (7678 sequences), that constituted our accessory genes pool dataset. Hierarchical clustering analysis was then conducted according to the Ward’s minimum variance-derived method. The Ward’s method is a clustering criterion that aggregates observations into clusters to minimize the within-cluster variance. The method was implemented using the pheatmap package in R (cran.r-project.org/web/packages/pheatmap/index.html). The gene presence/absence file generated by Roary was further analyzed using Scoary with a significant Bonferroni-adjusted P-value < 0.05 for genes associated to cnf1-positive lineages (Sup. Table S8).68
In silico antimicrobial resistance and virulence-associated markers
GyrA and ParC protein sequences were retrieved from the EnteroBase annotated genomes, and aligned with the mafft L-INS-I approach.69 After a visual inspection of the alignment, in-house customized perl scripts (https://github.com/rpatinonavarrete/QRDR) were used to identify the amino acids at the quinolone resistance-determining region (QRDR) (positions 83 and 87, and 80 and 84 in GyrA and ParC, respectively). Search for cnf1 and hlyA alleles in ST131 genomes dataset was carried out by Blastn analysis. Sequences were next aligned with Muscle70 and curated to remove incomplete sequences. SNPs were then extracted using SNP-sites.71 To determine strain specific VF profiles, annotated VFs from UPEC described in34 were translated and pBLASTed against ST131 genomes dataset considering only hits with e-value < 10–5 and identical matches > 95% (sup. Table S2).72 Acquired antibiotic-resistance genes (RGs) in ST131 genomes were defined with ResFinder.73
Co-clustering method
Statistical analyses were performed using R software version 3.6.0. A total of 20 strains from the collection of 5,231 strains of E. coli ST131 were removed from the analysis due to incomplete associated metadata. The clustering of strains with specific virulence or acquired antibiotic-resistance gene profiles was performed with binary latent block model, implemented in the R package blockcluster.74 The co-clustering of both virulence or resistance genes and strains was performed with a binary latent block model, implemented in the R package blockcluster.74 This package implements an Expectation Maximization algorithm to compute the maximum likelihood estimator of the parameters of the mixture of Bernoulli distributions used for co-clustering. As proposed by the authors,74 the number of clusters was estimated by maximizing the integrated complete-data likelihood criterion (ICL) on a bidimensional grid of parameters making this unsupervised classification procedure automatic.
Generalized linear model
Proportion of cnf1 along time was modeled using a generalized linear model fitted with binomial distribution and logit link. The model was adjusted on the effect of years and clades with an interaction between these two factors. We used the Tukey’s HSD test which adjusts the P-values for multiple comparisons (5 comparisons, one by clade and one for gathered clades). First, to test if the evolution of cnf1 proportion was either specific to each clade or global, the significance of the interaction term was tested with a likelihood ratio test, which compares the above-mentioned model against the null model, with no interaction. Then, we investigated the possible increase of the proportion of cnf1 within each clade. The significance of the slope coefficient for each clade was tested by computing contrasts of the above model. P-values were adjusted for multiplicity using single-step correction method. The distribution of fimH alleles and clades/subclades within the study population of E. coli ST131 was analyzed with a similar approach, except that a Poisson regression model was used to model counting data. The hypothesis testing strategy to investigate the significance of the increase of fimH alleles and clades/subclades along time is discussed above.
Construction of bacterial strains
The ST131 strain H1-001-0141-G-Y, here referred to as EC131GY, was originally isolated from a patient suffering from bacteremia (Sup. table S6).75 The strain is naturally resistant to ampicillin, to cefotaxime (CMI >256 mg/L) and is susceptible to gentamicin (CMI 0.5 mg/L). A streptomycin-resistant isolate was selected and used to engineer the cnf1 mutant strain. Deletion of cnf1 gene from the chromosome of EC131GY was achieved by gene replacement with kanamycin resistance (EC131GY Δcnf1::kanr) using the Lambda Red recombination system for gene replacement as previously described.76 Briefly, primers for amplification of the kanamycin cassette and the flanking FRT regions in pKD4 have been designed to target the first and the last 81 nucleotides of the cnf1 gene (Sup. table S7). The resulting PCR product was purified using commercial kits (Macherey Nagel). The strain carrying the temperature-sensitive helper plasmid pKOBEG coding for the Lambda red recombinase system was processed as previously described.76 The resulting mutants were tested for the gene replacement by PCR with primers listed in the supplementary table S7, and pKOBEG plasmid loss was verified on LB agar plates with chloramphenicol. The kanamycin cassette in EC131GY Δcnf1::kanr was removed with pCP20 expressing flippase, as reported in,77 to generate EC131GY Δcnf1. Resulting colonies were verified by PCR with primers listed in the supplementary table S7. The cnf1 gene including its promoter region was cloned BamHI and KpnI in pISN1 bearing chloramphenicol resistance,78 a gift from Petra Dersch, here referred to as pCNF1-WT (Sup. table S6). The plasmid encoding the catalytically-inactive mutant C866S (pCNF1-CS) was generated by site-directed mutagenesis using oligonucleotides listed in supplementary table S7. The stain BLSE2018-86 was isolated from a patient suffering from UTI.47 All mutants were verified for growth in LB by performing growth curves in a FLUOstar Omega microplate reader. Briefly, starting from a fresh overnight culture, bacteria were diluted 1/100 in 5 mL LB supplemented with streptomycin 200 µg/mL. 200 μL of each culture were placed as 5 replicates in a 96 flat bottom plate (Greiner) and incubated for 12 hours at 37°C with 120 rpm orbital shaking. Absorbance at 600 nm was measured every 10 minutes.
Western blot
Bacterial pellets were collected in RIPA buffer. The lysates were boiled in 1x Laemmli buffer for 5 minutes at 100°C and resolved on 8% SDS-PAGE, transferred to nitrocellulose membrane (GE Healthcare). The proteins were colored with ponceau S (Biorad) and the membrane was blocked with 5% milk in TBS-T (Euromedex). Membranes were incubated with the primary antibody: CNF1 (Santa Cruz sc52655 clone NG8 1/1000), RNA Polymerase (Biolegend 699907 clone NT73 1/1000) and rabbit serum (1/1000) against the conserved amino acids 914–936 of HlyA, as previously described.79 Membranes were washed with TBS-T and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h. Signals were observed using Immobion Western Chemiluminescent HRP Substrate (Merck).
Mouse colonization model
Local Animal Studies Committee and National Research Council approved all procedures used for the mouse experiments described in the present study (APAFIS#26133-202006221228936 v1, 2016–0010). For intravesical infection: urinary tract infection was induced in female C57BL/6 mice aged 6–7 weeks (Charles River), as previously described.80,81 Briefly, a single colony of EC131GY or the cnf1 mutant was inoculated in 10 mL LB medium with antibiotics and incubated at 37°C under static conditions for 18 h. Mice were infected with a total of 107 CFU of bacteria in 50 μL PBS via a urinary catheter under anesthesia. To calculate CFU, bladders were aseptically removed and homogenized in 1 mL of PBS. Serial dilutions were plated on LB agar plates with antibiotics, as required. For gut colonization, groups of female C57BL/6 mice aged 6–7 weeks (Charles River) were pretreated with a single dose of streptomycin (1 g/kg in 200 µL water) per os 1 d prior to gavage, as described in82 and infected with the strains derived from EC131GY or BLSE2018-86. Mice were infected per os with 2 × 109 CFU of each strain either alone or in 1:1 mix (WT: mutant strains) for the competitive index (CI) in 200 μL PBS. Fecal pellets were collected from every individual mouse at indicated times, weighed and homogenized in 500 μL phosphate-buffed saline (PBS) pH 7.2 by vigorous vortexing. CFUs were determined by plating serial dilutions on selective LB agar plates. Strains were prepared for infection as follows: a single colony of EC131GY or BLSE2018-86 or their derivative was inoculated in 10 mL selective LB medium and incubated at 37°C under static conditions for 24 h. Bacteria were then inoculated in 25 mL fresh selective LB medium at 1:1000 dilution and incubated at 37°C under static conditions for 18–24 h. Bacteria were then washed twice in cold PBS, and concentrated in PBS at approximately 2 × 109 CFU per 200 μL. Inocula titers were verified in parallel for each infection. The value of CI was calculated as: CFU WT output strain/CFU mutant output strain, with the verification in each experiment that CFU WT input strain/CFU mutant input strain was very close to 1. A Wilcoxon signed-rank test was performed to assess the statistical significance of differences in CI over time. Statistical analyses were performed with GraphPad Prism 9. CFU in colon tissues were assessed upon treatment ex vivo in gentamicin 100 µg/mL for 2 hours. Washed tissues were homogenized in PBS using IKA T25D Ultra Turrax homogenizer and CFU were determined by plating serial dilutions on selective agar. To assess the statistical significance of colonic tissue invasion, a linear mixed model was applied to the Log10 values of CFU. This model was adjusted on conditions EC131GY Δcnf1 + pCNF1-CS or pCNF1-WT and EC131GY + empty vector (pE) as fixed effect and on the date of experiment as random effect. Comparison performed using contrasts within this model and P-values adjusted using Tukey correction in R software.
Enzyme-linked immunosorbent assay (ELISA)
IL-6, TNF-α, and CXCL1 were measured in bladder tissue homogenates (also used for CFU measurements) using the R&D Systems DuoSet ELISA kits according to manufacturer’s protocols with no changes, except, due to limited sample volumes, 45 μL of experimental samples were used instead of 100 μL.
Flow cytometry
Mice were sacrificed at 24 hours post-infection (PI) and the bladders removed. Single-cell homogenates were prepared by incubating minced bladders in 0.34 Units/mL Liberase TM (Roche) diluted in PBS at 37°C for 1 hour, with manual agitation every 15 minutes.80 Digested tissue was filtered using a 100 μm filter (Miltenyi), washed, blocked with Fc Block (Rat anti-mouse CD16/CD32, BD Biosciences), and immunostained (Supplementary Table S9). Samples were acquired on a BD Fortessa (BD Biosciences) and analyzed using FlowJo Version 10.7.1 software.
Supplementary Material
Acknowledgments
We thank François-Xavier WEILL for fruitful discussions. The plasmids pKOBEG and p3xFlag-CmR CNF1 wildtype were kindly provided by Jean-Marc GHIGO and Petra DERSCH, respectively.
Funding Statement
This work was supported by Inserm Transversal Programme on Microbiota, the French National Research Agency (ANR-10-LABX-62-IBEID, INCEPTION), ANR-17-CE17-0014 and ANR-21-CE15-0006, the “Fondation ARC” PJA 20191209650, the “Ligue Nationale contre le Cancer Subvention de Recherche Scientifique” RS20/75-63, the “Fondation pour la Recherche Médicale” (Equipe FRM 2016, DEQ20161136698).
Disclosure statement
The authors report there are no competing interests to declare.
Author contributions
Bioinformatics analyses were performed by L. T-M., S. D-D., R. P-N. and analysed by E. L., L. L., A. M., P. G. and E. D. Statistical analyses were performed by M-A. N. and E. P. In vivo experiments were coordinated by A. M., M. A. I., O. D. and performed by M-A. N., A. M. and L. R-F. with strains engineered by S.P. and A.M. The research was coordinated by E. L. and manuscript drafted with L. T-M. and L. L. together with A. M., M. A. I, O. D., E. D., R. P-N. and P. G. Manuscript was reviewed and approved by all authors.
Data availability statement
Raw data are available at http://enterobase.warwick.ac.uk and processed data in supplementary tables.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2022.2121577
References
- 1.Klein RD, Hultgren SJ.. Urinary tract infections: microbial pathogenesis, host-pathogen interactions and new treatment strategies. Nat Rev Microbiol. 2020;18(4):211–20. doi: 10.1038/s41579-020-0324-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Landraud L, Gauthier M, Fosse T, Boquet P. Frequency of Escherichia coli strains producing the cytotoxic necrotizing factor (CNF1) in nosocomial urinary tract infections. Lett Appl Microbiol. 2000;30(3):213–216. doi: 10.1046/j.1472-765x.2000.00698.x. [DOI] [PubMed] [Google Scholar]
- 3.Dubois D, Delmas J, Cady A, Robin F, Sivignon A, Oswald E, Bonnet R. Cyclomodulins in Urosepsis Strains of Escherichia coli. J Clin Microbiol. 2010;48(6):2122–2129. doi: 10.1128/JCM.02365-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Erjavec M S, Žgur-Bertok D. Virulence potential for extraintestinal infections among commensal Escherichia coli isolated from healthy humans--the Trojan horse within our gut. FEMS Microbiol Lett. 2015;362(5). doi: 10.1093/femsle/fnu061. [DOI] [PubMed] [Google Scholar]
- 5.Flatau G, Lemichez E, Gauthier M, Chardin P, Paris S, Fiorentini C, Boquet P. Toxin-induced activation of the G protein p21 Rho by deamidation of glutamine. Nature. 1997;387(6634):729–733. doi: 10.1038/42743. [DOI] [PubMed] [Google Scholar]
- 6.Schmidt G, Sehr P, Wilm M, Selzer J, Mann M, Aktories K. Gln 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nature. 1997;387(6634):725–729. doi: 10.1038/42735. [DOI] [PubMed] [Google Scholar]
- 7.Aktories K, Barbieri JT. Bacterial cytotoxins: targeting eukaryotic switches. Nat Rev Microbiol. 2005;3(5):397–410. doi: 10.1038/nrmicro1150. [DOI] [PubMed] [Google Scholar]
- 8.Patel JC, Galan JE. Manipulation of the host actin cytoskeleton by Salmonella–all in the name of entry. Curr Opin Microbiol. 2005;8(1):10–15. doi: 10.1016/j.mib.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 9.Blum G, Falbo V, Caprioli A, Hacker J. Gene clusters encoding the cytotoxic necrotizing factor type 1, Prs-fimbriae and alpha-hemolysin form the pathogenicity island II of the uropathogenic Escherichia coli strain J96. FEMS Microbiol Lett. 1995;126(2):189–195. doi: 10.1111/j.1574-6968.1995.tb07415.x. [DOI] [PubMed] [Google Scholar]
- 10.Schneider G, Dobrindt U, Middendorf B, Hochhut B, Szijártó V, Emody L, Hacker J. Mobilisation and remobilisation of a large archetypal pathogenicity island of uropathogenic Escherichia coli in vitrosupport the role of conjugation for horizontal transfer of genomic islands. BMC Microbiol. 2011;11(1):210. doi: 10.1186/1471-2180-11-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rippere-Lampe KE, O’Brien AD, Conran R, Lockman HA, Barbieri JT. Mutation of the gene encoding cytotoxic necrotizing factor type 1 (cnf 1) attenuates the virulence of uropathogenic Escherichia coli. Infect Immun. 2001;69(6):3954–3964. doi: 10.1128/IAI.69.6.3954-3964.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rippere-Lampe KE, Lang M, Ceri H, Olson M, Lockman HA, O’Brien AD, Barbieri JT. Cytotoxic necrotizing factor type 1-positive Escherichia coli causes increased inflammation and tissue damage to the prostate in a rat prostatitis model. Infect Immun. 2001;69(10):6515–6519. doi: 10.1128/IAI.69.10.6515-6519.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lacerda Mariano L, Ingersoll MA. The immune response to infection in the bladder. Nat Rev Urol. 2020;17(8):439–458. doi: 10.1038/s41585-020-0350-8. [DOI] [PubMed] [Google Scholar]
- 14.Michaud JE, Kim KS, Harty W, Kasprenski M, Wang MH. Cytotoxic Necrotizing Factor-1 (CNF1) does not promote E. coli infection in a murine model of ascending pyelonephritis. BMC Microbiol. 2017;17(1):127. doi: 10.1186/s12866-017-1036-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Landraud L, Gibert M, Popoff MR, Boquet P, Gauthier M. Expression of cnf1 by Escherichia coli J96 involves a large upstream DNA region including the hlyCABD operon, and is regulated by the RfaH protein. Mol Microbiol. 2003;47(6):1653–1667. doi: 10.1046/j.1365-2958.2003.03391.x. [DOI] [PubMed] [Google Scholar]
- 16.Diabate M, Munro P, Garcia E, Jacquel A, Michel G, Obba S, Goncalves D, Luci C, Marchetti S, Demon D, et al. Escherichia coli α-hemolysin counteracts the anti-virulence innate immune response triggered by the rho Gtpase activating toxin CNF1 during Bacteremia. PLoS Pathog. 2015;11(3):e1004732. doi: 10.1371/journal.ppat.1004732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dufies O, Doye A, Courjon J, Torre C, Michel G, Loubatier C, Jacquel A, Chaintreuil P, Majoor A, Guinamard RR, et al. Escherichia coli Rho GTPase-activating toxin CNF1 mediates NLRP3 inflammasome activation via p21-activated kinases-1/2 during bacteraemia in mice. Nat Microbiol. 2021;6(3):401–412. doi: 10.1038/s41564-020-00832-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boquet P, Lemichez E. Bacterial virulence factors targeting Rho GTPases: parasitism or symbiosis? Trends Cell Biol. 2003;13(5):238–246. doi: 10.1016/S0962-8924(03)00037-0. [DOI] [PubMed] [Google Scholar]
- 19.Doye A, Mettouchi A, Bossis G, Clement R, Buisson-Touati C, Flatau G, Gagnoux L, Piechaczyk M, Boquet P, Lemichez E. CNF1 exploits the ubiquitin-proteasome machinery to restrict Rho GTPase activation for bacterial host cell invasion. Cell. 2002;111(4):553–564. doi: 10.1016/S0092-8674(02)01132-7. [DOI] [PubMed] [Google Scholar]
- 20.Falbo V, Pace T, Picci L, Pizzi E, Caprioli A. Isolation and nucleotide sequence of the gene encoding cytotoxic necrotizing factor 1 of Escherichia coli. Infect Immun. 1993;61(11):4909–4914. doi: 10.1128/iai.61.11.4909-4914.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orden JA, Dominguez-Bernal G, Martinez-Pulgarin S, Blanco M, Blanco JE, Mora A, Blanco J, Blanco J, de la Fuente R. Necrotoxigenic Escherichia coli from sheep and goats produce a new type of cytotoxic necrotizing factor (CNF3) associated with the eae and ehxA genes. Int Microbiol. 2007;10:47–55. [PubMed] [Google Scholar]
- 22.Oswald E, Sugai M, Labigne A, Wu HC, Fiorentini C, Boquet P, O’Brien AD. Cytotoxic necrotizing factor type 2 produced by virulent Escherichia coli modifies the small GTP-binding proteins Rho involved in assembly of actin stress fibers. Proc Natl Acad Sci U S A. 1994;91(9):3814–3818. doi: 10.1073/pnas.91.9.3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ho M, Mettouchi A, Wilson BA, Lemichez E. CNF1-like deamidase domains: common Lego bricks among cancer-promoting immunomodulatory bacterial virulence factors. Pathog Dis. 2018;76(5). doi: 10.1093/femspd/fty045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Denamur E, Clermont O, Bonacorsi S, Gordon D. The population genetics of pathogenic Escherichia coli. Nat Rev Microbiol. 2021;19(1):37–54. doi: 10.1038/s41579-020-0416-x. [DOI] [PubMed] [Google Scholar]
- 25.Tenaillon O, Skurnik D, Picard B, Denamur E. The population genetics of commensal Escherichia coli. Nat Rev Microbiol. 2010;8(3):207–217. doi: 10.1038/nrmicro2298. [DOI] [PubMed] [Google Scholar]
- 26.Nielsen KL, Dynesen P, Larsen P, Frimodt-Møller N. Faecal Escherichia coli from patients with E. coli urinary tract infection and healthy controls who have never had a urinary tract infection. J Med Microbiol. 2014;63(4):582–589. doi: 10.1099/jmm.0.068783-0. [DOI] [PubMed] [Google Scholar]
- 27.Johnson JR, Davis G, Clabots C, Johnston BD, Porter S, DebRoy C, Pomputius W, Ender PT, Cooperstock M, Slater BS, et al. Household Clustering of Escherichia coli sequence type 131 clinical and fecal isolates according to whole genome sequence analysis. Open Forum Infect Dis. 2016;3(3):ofw129. doi: 10.1093/ofid/ofw129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamamoto S, Tsukamoto T, Terai A, Kurazono H, Takeda Y, Yoshida O. Genetic evidence supporting the fecal-perineal-urethral hypothesis in cystitis caused by Escherichia coli. J Urol. 1997;157(3):1127–1129. doi: 10.1016/S0022-5347(01)65154-1. [DOI] [PubMed] [Google Scholar]
- 29.Moreno E, Andreu A, Pigrau C, Kuskowski MA, Johnson JR, Prats G. Relationship between Escherichia coli strains causing acute cystitis in women and the fecal e. coli population of the host. J Clin Microbiol. 2008;46(8):2529–2534. doi: 10.1128/JCM.00813-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kallonen T, Brodrick HJ, Harris SR, Corander J, Brown NM, Martin V, Peacock SJ, Parkhill J. Systematic longitudinal survey of invasive Escherichia coli in England demonstrates a stable population structure only transiently disturbed by the emergence of ST131. Genome Res. 2017;27(8):1437–1449. doi: 10.1101/gr.216606.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson JR, Johnston B, Clabots C, Kuskowski MA, Castanheira M. Escherichia coli sequence type st131 as the major cause of serious multidrug-resistant E. coli infections in the United States. Clin Infect Dis. 2010;51(3):286–294. doi: 10.1086/653932. [DOI] [PubMed] [Google Scholar]
- 32.Peirano G, Pitout JD. Molecular epidemiology of Escherichia coli producing CTX-M β-lactamases: the worldwide emergence of clone ST131 O25:H4. Int J Antimicrob Agents. 2010;35(4):316–321. doi: 10.1016/j.ijantimicag.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 33.Price LB, Johnson JR, Aziz M, Clabots C, Johnston B, Tchesnokova V, Nordstrom L, Billig M, Chattopadhyay S, Stegger M, et al. The epidemic of extended-spectrum-β-lactamase-producing Escherichia coli ST131 Is driven by a single highly pathogenic subclone, H 30-Rx. MBio. 2013;4(6):e00377–13. doi: 10.1128/mBio.00377-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petty NK, Ben Zakour NL, Stanton-Cook M, Skippington E, Totsika M, Forde BM, Phan M-D, Gomes Moriel D, Peters KM, Davies M, et al. Global dissemination of a multidrug resistant Escherichia col clone. Proc Natl Acad Sci U S A. 2014;111(15):5694–5699. doi: 10.1073/pnas.1322678111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ben Zakour NL, Alsheikh-Hussain AS, Ashcroft MM, Khanh Nhu NT, Roberts LW, Stanton-Cook M, Schembri MA, Beatson SA. Sequential Acquisition of Virulence and Fluoroquinolone Resistance Has Shaped the Evolution of Escherichia coli ST131. MBio. 2016;7(2):e00347–16. doi: 10.1128/mBio.00347-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Madigan T, Johnson JR, Clabots C, Johnston BD, Porter SB, Slater BS, Banerjee R. Extensive household outbreak of urinary tract infection and intestinal Colonization due to extended-spectrum β-lactamase–producing Escherichia coli sequence type 131. Clin Infect Dis. 2015;61(1):e5–12. doi: 10.1093/cid/civ273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tchesnokova VL, Rechkina E, Chan D, Haile HG, Larson L, Ferrier K, Schroeder DW, Solyanik T, Shibuya S, Hansen K, et al. Pandemic Uropathogenic Fluoroquinolone-resistant Escherichia coli have enhanced ability to persist in the gut and cause bacteriuria in healthy women. Clin Infect Dis. 2020;70(5):937–939. doi: 10.1093/cid/ciz547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shevchenko SG, Radey M, Tchesnokova V, Kisiela D, Sokurenko EV, Elkins CA. Escherichia coli clonobiome: assessing the strain diversity in feces and urine by deep amplicon sequencing. Appl Environ Microbiol. 2019;85(23). doi: 10.1128/AEM.01866-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gurnee EA, Ndao IM, Johnson JR, Johnston BD, Gonzalez MD, Burnham CAD, Hall-Moore CM, McGhee JE, Mellmann A, Warner BB, et al. Gut colonization of healthy children and their mothers with pathogenic ciprofloxacin-resistant Escherichia coli. J Infect Dis. 2015;212(12):1862–1868. doi: 10.1093/infdis/jiv278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laupland KB, Church DL, Vidakovich J, Mucenski M, Pitout JD. Community-onset extended-spectrum β-lactamase (ESBL) producing Escherichia coli: importance of international travel. J Infect. 2008;57(6):441–448. doi: 10.1016/j.jinf.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 41.Vimont S, Boyd A, Bleibtreu A, Bens M, Goujon JM, Garry L, Clermont O, Denamur E, Arlet G, Vandewalle A. The CTX-M-15-producing Escherichia coli clone O25b: H4-ST131 has high intestine colonization and urinary tract infection abilities. PLoS One. 2012;7(9):e46547. doi: 10.1371/journal.pone.0046547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McNally A, Oren Y, Kelly D, Pascoe B, Dunn S, Sreecharan T, Vehkala M, Välimäki N, Prentice MB, Ashour A, et al. Combined analysis of variation in core, accessory and regulatory genome regions provides a super-resolution view into the evolution of bacterial populations. PLoS Genet. 2016;12(9):e1006280. doi: 10.1371/journal.pgen.1006280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Le Gall T, Clermont O, Gouriou S, Picard B, Nassif X, Denamur E, Tenaillon O. Extraintestinal virulence is a coincidental by-product of commensalism in B2 phylogenetic group Escherichia coli strains. Mol Biol Evol. 2007;24(11):2373–2384. doi: 10.1093/molbev/msm172. [DOI] [PubMed] [Google Scholar]
- 44.Zhou Z, Alikhan NF, Mohamed K, Fan Y, Agama SG, Achtman M. The EnteroBase user’s guide, with case studies on Salmonella transmissions, Yersinia pestis phylogeny, and Escherichia core genomic diversity. Genome Res. 2020;30(1):138–152. doi: 10.1101/gr.251678.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bidet P, Bonacorsi S, Clermont O, De Montille C, Brahimi N, Bingen E. Multiple insertional events, restricted by the genetic background, have led to acquisition of pathogenicity island II J96 -Like domains among Escherichia coli strains of different clinical origins. Infect Immun. 2005;73(7):4081–4087. doi: 10.1128/IAI.73.7.4081-4087.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sarkar S, Hutton ML, Vagenas D, Ruter R, Schüller S, Lyras D, Schembri MA, Totsika M. Intestinal colonization traits of pandemic multidrug-resistant Escherichia coli ST131. J Infect Dis. 2018;218(6):979–990. doi: 10.1093/infdis/jiy031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bonnet R, Beyrouthy R, Haenni M, Nicolas-Chanoine MH, Dalmasso G, Madec JY, Bonomo RA. Host colonization as a major evolutionary force favoring the diversity and the emergence of the worldwide multidrug-resistant Escherichia coli ST131. mBio. 2021;12(4):e0145121. doi: 10.1128/mBio.01451-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Royer G, Darty MM, Clermont O, Condamine B, Laouenan C, Decousser J-W, Vallenet D, Lefort A, de Lastours V, Denamur E, et al. Phylogroup stability contrasts with high within sequence type complex dynamics of Escherichia coli bloodstream infection isolates over a 12-year period. Genome Med. 2021;13(1):77. doi: 10.1186/s13073-021-00892-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meador JP, Caldwell ME, Cohen PS, Conway T, McCormick BA. Escherichia coli pathotypes occupy distinct niches in the mouse intestine. Infect Immun. 2014;82(5):1931–1938. doi: 10.1128/IAI.01435-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Falzano L, Rivabene R, Fabbri A, Fiorentini C. Epithelial cells challenged with a Rac-activating E. coli cytotoxin acquire features of professional phagocytes. Toxicol In Vitro. 2002;16(4):421–425. doi: 10.1016/S0887-2333(02)00027-9. [DOI] [PubMed] [Google Scholar]
- 51.Visvikis O, Boyer L, Torrino S, Doye A, Lemonnier M, Lorès P, Rolando M, Flatau G, Mettouchi A, Bouvard D, et al. Escherichia coli producing CNF1 toxin hijacks tollip to trigger rac1-dependent cell invasion. Traffic. 2011;12(5):579–590. doi: 10.1111/j.1600-0854.2011.01174.x. [DOI] [PubMed] [Google Scholar]
- 52.Martinez JJ, Hultgren SJ. Requirement of Rho-family GTPases in the invasion of Type 1-piliated uropathogenic Escherichia coli. Cell Microbiol. 2002;4(1):19–28. doi: 10.1046/j.1462-5822.2002.00166.x. [DOI] [PubMed] [Google Scholar]
- 53.Petracchini SH, Doye D, Asnacios A, Fage A, Vitiello F, Balland E, Janel M, Lafont S, Gupta F, Ladoux M, et al. Optineurin links Hace1-dependent Rac ubiquitylation to integrin-mediated mechanotransduction to control bacterial invasion and cell division. BioRxiv. 2021;10(13):464032. doi: 10.1101/2021.10.13.464032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fagan RP, Smith SG. The Hek outer membrane protein of Escherichia coli is an auto-aggregating adhesin and invasin. FEMS Microbiol Lett. 2007;269(2):248–255. doi: 10.1111/j.1574-6968.2006.00628.x. [DOI] [PubMed] [Google Scholar]
- 55.Ristow LC, Welch RA. RTX toxins ambush immunity’s first cellular responders. Toxins (Basel). 2019;11(12):720. doi: 10.3390/toxins11120720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Geibel S, Waksman G. The molecular dissection of the chaperone-usher pathway. Biochim Biophys Acta. 2014;1843(8):1559–1567. doi: 10.1016/j.bbamcr.2013.09.023. [DOI] [PubMed] [Google Scholar]
- 57.Gennaris A, Ezraty B, Henry C, Agrebi R, Vergnes A, Oheix E, Bos J, Leverrier P, Espinosa L, Szewczyk J, et al. Repairing oxidized proteins in the bacterial envelope using respiratory chain electrons. Nature. 2015;528(7582):409–412. doi: 10.1038/nature15764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Falzano L, Rivabene R, Santini MT, Fabbri A, Fiorentini C. An Escherichia coli cytotoxin increases superoxide anion generation via rac in epithelial cells. Biochem Biophys Res Commun. 2001;283(5):1026–1030. doi: 10.1006/bbrc.2001.4894. [DOI] [PubMed] [Google Scholar]
- 59.Hertz FB, Nielsen JB, Schønning K, Littauer P, Knudsen JD, Løbner-Olesen A, Frimodt-Møller N. “Population structure of Drug-susceptible, -resistant and ESBL-producing Escherichia coli from community-acquired urinary tract infections”. BMC Microbiol. 2016;16(1):63. doi: 10.1186/s12866-016-0681-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Duprilot M, Baron A, Blanquart F, Dion S, Pouget C, Lettéron P, Flament-Simon SC, Clermont O, Denamur E, Nicolas-Chanoine MH. Success of Escherichia coli O25b:H4 sequence type 131 clade c associated with a decrease in virulence. Infect Immun. 2020;88(12). doi: 10.1128/IAI.00576-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thänert R, Choi J, Reske KA, Hink T, Thänert A, Wallace MA, Wang B, Seiler S, Cass C, Bost MH, et al. Persisting uropathogenic Escherichia coli lineages show signatures of niche-specific within-host adaptation mediated by mobile genetic elements. Cell Host Microbe. 2022;30(7):1034–1047.e6. doi: 10.1016/j.chom.2022.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mistry J, Finn RD, Eddy SR, Bateman A, Punta M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013;41(12):e121. doi: 10.1093/nar/gkt263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Treangen TJ, Ondov BD, Koren S, Phillippy AM. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014;15(11):524. doi: 10.1186/s13059-014-0524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Croucher NJ, Page AJ, Connor TR, Delaney AJ, Keane JA, Bentley SD, Parkhill J, Harris SR. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015;43(3):e15. doi: 10.1093/nar/gku1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Letunic I, Bork P. Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019;47(W1):W256–W259. doi: 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sitto F, Battistuzzi FU, Hall BG. Estimating Pangenomes with Roary. Mol Biol Evol. 2020;37(3):933–939. doi: 10.1093/molbev/msz284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brynildsrud O, Bohlin J, Scheffer L, Eldholm V. Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol. 2016;17(1):238. doi: 10.1186/s13059-016-1108-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Katoh K, Kuma K, Miyata T, Toh H. Improvement in the accuracy of multiple sequence alignment program MAFFT. Genome Inform. 2005;16:22–33. [PubMed] [Google Scholar]
- 70.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004;5(1):113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Page AJ, Taylor B, Delaney AJ, Soares J, Seemann T, Keane JA, Harris SR. SNP-sites: rapid efficient extraction of SNPs from multi-FASTA alignments. Microb Genom. 2016;2(4):e000056. doi: 10.1099/mgen.0.000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinform. 2009;10(1):421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, Aarestrup FM, Larsen MV. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67(11):2640–2644. doi: 10.1093/jac/dks261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bhatia PS, Lovleff S, Govaert G. blockcluster: an R package for model-based co-clustering. J Stat Softw. 2017;76(9):1–24. doi: 10.18637/jss.v076.i09. [DOI] [Google Scholar]
- 75.de Lastours V, Laouénan C, Royer G, Carbonnelle E, Lepeule R, Esposito-Farèse M, Clermont O, Duval X, Fantin B, Mentré F, et al. Mortality in Escherichia coli bloodstream infections: antibiotic resistance still does not make it. J Antimicrob Chemother. 2020;75(8):2334–2343. doi: 10.1093/jac/dkaa161. [DOI] [PubMed] [Google Scholar]
- 76.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97(12):6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cherepanov PP, Wackernagel W. Gene disruption in Escherichia coli: tcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995;158(1):9–14. doi: 10.1016/0378-1119(95)00193-A. [DOI] [PubMed] [Google Scholar]
- 78.Chaoprasid P, Lukat P, Mühlen S, Heidler T, Gazdag E-M, Dong S, Bi W, Rüter C, Kirchenwitz M, Steffen A, et al. CrYstal structure of bacterial cYtotoxic necrotizing factor CNF Y reveals molecular building blocks for intoxication. EMBO J. 2021;40(4):e105202. doi: 10.15252/embj.2020105202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cortajarena AL, Goni FM, Ostolaza H. A receptor-binding region in Escherichia coli α-Haemolysin. J Biol Chem. 2003;278(21):19159–19163. doi: 10.1074/jbc.M208552200. [DOI] [PubMed] [Google Scholar]
- 80.Mora-Bau G, Platt AM, van Rooijen N, Randolph GJ, Albert ML, Ingersoll MA, Gause WC. Macrophages subvert adaptive immunity to urinary tract infection. PLoS Pathog. 2015;11(7):e1005044. doi: 10.1371/journal.ppat.1005044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zychlinsky Scharff A, Albert ML, Ingersoll MA. Urinary tract infection in a small animal model: transurethral catheterization of male and female mice. J Vis Exp. 2017;130:54432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Spaulding CN, Klein RD, Ruer S, Kau AL, Schreiber HL, Cusumano ZT, Dodson KW, Pinkner JS, Fremont DH, Janetka JW, et al. Selective depletion of uropathogenic E. coli from the gut by a FimH antagonist. Nature. 2017;546(7659):528–532. doi: 10.1038/nature22972. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data are available at http://enterobase.warwick.ac.uk and processed data in supplementary tables.