

ABSTRACT
NOD-like receptor family pyrin domain containing three (NLRP3) inflammasome-mediated pyroptotic cell death and inflammation contribute to the pathogenesis of bronchial asthma, and it is reported that Substance P (SP) plays important role in the process, however, the detailed molecular mechanisms by which SP participates in the aggravation of bronchial asthma have not been fully studied. Here, our clinical data showed that SP and its receptor Neurokinin-1 receptor (NK1R) were significantly elevated in the plasma and peripheral blood mononuclear cell (PBMC) collected from patients with bronchial asthma, and further pre-clinical experiments evidenced that SP suppressed cell viability, accelerated lactate dehydrogenase (LDH) release, and upregulated ASC, Caspase-1, NLRP3, IL-1β and IL-18 to promote pyroptotic cell death and cellular inflammation in the human bronchial epithelial cells and asthmatic mice models in vitro and in vivo. Interestingly, SP-induced pyroptotic cell death was reversed by NK1R inhibitor L732138. Then, we uncovered the underlying mechanisms, and found that SP activated the downstream PI3K/AKT/NF-κB signal pathway in a NK1R-dependent manner, and blockage of this pathway by both PI3K inhibitor (LY294002) and NF-κB inhibitor (MG132) reversed SP-induced pyroptotic cell death and recovered cell viability in bronchial epithelial cells. Collectively, we concluded that SP interacted with its receptor NK1R to activate the PI3K/AKT/NF-κB pathway, which further triggered NLRP3-mediated pyroptotic cell death in the bronchial epithelial cells, resulting in the aggravation of bronchial asthma.
KEYWORDS: Substance P, bronchial asthma, pyroptotic cell death, inflammatory factors
Introduction
Bronchial asthma is a heterogeneous disease characterized by chronic inflammation involving multiple cells and cellular components [1]. Airway epithelial cells dysfunction is a hallmark of the pathogenesis of bronchial asthma [2,3]. Due to its complex pathogenesis, the efficacy of current therapeutic strategies for bronchial asthma is severely limited [2]. SP is an 11 amino acid neuropeptide widely distributed in mammalian tissues and body fluids [4,5], which exerts its biological functions through three different neurokinin (NK-1, NK-2, NK-3) receptors, among which NK1R is preferentially activated by SP [6,7]. In addition, SP is involved in regulating many physiological and pathophysiological processes, including fibrotic diseases [8], cardiomyopathies [9] and asthma [10,11]. In our previous studies, we uncovered that SP accelerates neurogenic airway inflammation through NK1R [12], and the NK1R antagonist WIN62577 hampered airway smooth muscle cells proliferation and migration [13]. Therefore, we speculated that SP/NK1R aggravates the progression of bronchial asthma by regulating cellular inflammatory response. However, the molecular mechanisms by which SP regulates bronchial asthma have not been fully delineated.
Cell pyroptosis is a kind of inflammation-associated programmed cell death, which is typically characterized with cell swelling until the cell membrane ruptures, formation of inflammasomes, the activation of the caspase family and Gasdermin proteins, the release of LDH and the secretion of a large number of pro-inflammatory factors, leading to a strong inflammatory response [14–17]. According to recent publications, pyroptotic cell death is involved in regulating the development of many diseases, including inflammatory bowel disease [18], Parkinson’s disease [19], cancers [20] and bronchial asthma [4]. Especially, blockage of NLRP3 inflammasome-mediated pyroptotic cell death ameliorates asthma in mice models [4]. As previously reported, various types of cell death can be regulated by SP [18], for example, researchers noticed that SP causes neuronal cell death at the cellular level [19]. Also, in acute brain injury, elevated SP was harmful to neuron survival and motor function, and upregulated SP accelerates neurotoxin 6-hydroxydopamine (6-OHDA)-induced cell death [20], and conversely, SP deletion inhibits oxidative stress and cell death induced by scratch injury after traumatic brain injury (TBI) [21]. However, it is still unclear whether SP regulates the NLRP3-mediated pyroptotic cell death to participate in the regulation of bronchial asthma.
Recent studies have shown that the PI3K/Akt/NF-κB signal transduction pathway is closely associated with the development of various diseases, including cancer [22], asthma [23], spinal cord injury [24], and chronic colitis [25]. Interestingly, nuclear factor (NF)-κB is a transcriptional regulator, which plays an important role in the inflammatory pathway of bronchial asthma [23], and the expressions of NF-κB and its downstream target genes can be controlled by phosphoinositide 3-kinase (PI3K)/The protein kinase B (AKT) signaling pathway, which also plays a vital role in the inflammatory process of cells [26,27]. Additionally, previous findings disclose that SP/NK-1 R activated two convergent pro-inflammatory signaling pathways, PI3K-Akt and NF-κB [28]. Strikingly, the PI3K/Akt/NF-κB signal pathway is reported to regulate both cell pyroptosis and cellular inflammation [24,25,29], specifically, activation of this signal pathway contribute to NLRP3 inflammasome-mediated pyroptotic cell death in spinal cord injury (SCI) [24], HUVECs [29] and chronic colitis [25].
Thus, based on the existed information, this study investigated the involvement of SP in regulating bronchial asthma development, and we found that SP NK1R-dependently activated the downstream PI3K/AKT/NF-κB signaling pathway, resulting in the activation of the NLRP3 inflammasome-mediated pyroptotic cell death and aggravation of bronchial asthma. Our data, for the first time, elucidated the detailed molecular mechanisms by which SP aggravate the development of bronchial asthma, which provided potential treatment strategies for this disease.
Materials and methods
Patients and samples
The bronchial asthma and normal controls plasma and peripheral blood mononuclear cell (PBMC) were collected from Shengjing Hospital of China Medical University. Normal controls had no history of lung disease, allergy, respiratory infection, or exposure to tobacco smoke. This study was approved by the Ethics Committee of Shengjing Hospital of China Medical University, and each participant signed an informed consent form. All procedures were carried out in accordance with the 1975 Declaration of Helsinki.
Asthmatic mouse model
BALB/c mice (6–8 weeks) were obtained from Shanghai SLAC Experimental Animal Center (China). According to the experimental protocols provided by the previous publications, Ovalbumin (OVA) without adjuvant was used to sensitize mice with seven intraperitoneal injections of OVA every other day [10]. Then, the mice were challenged by OVA nebulization on the 33–40th day. OVA sensitized mice (day 50) received 0.5 mg SP/ml saline, and control mice received saline. L732138, a selective antagonist of NK1 receptor, purchased from Sigma, dissolved in 70% of DMSO in saline. L-732138 (500 μg in 50 μl DMSO 70%) or DMSO 70% alone (50 μl) was administered intraperitoneally in OVA-sensitized mice with experimental airway hypersensitivity 1 h before intrabronchial SP administration. Finally, all mice were killed by intraperitoneal injection of a lethal dose of pentobarbital for analysis. All animal experiments in this study are in compliance with the principles of management and use of experimental animals approved by the Ethics Committee of Shengjing Hospital of China Medical University.
Cell culture
16-HBE and BEAS-2B cells were obtained from the Procell Life Science & Technology Co., Ltd (Wuhan, China). All cells were maintained at 37°C in 5% CO2 using Endothelial Cell Medium (ECM, ScienCell Research Laboratories, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) and 1% (v/v) penicillin/streptomycin, and 1% endothelial cell growth factors (Invitrogen, Carlsbad, CA, USA).
Cell viability assay
16HBE (Procell Life Science & Technology Co., Ltd. CL-0249) and BEAS-2B cells (Procell Life Science & Technology Co., Ltd. CL-0496) were harvested after stimulation with different concentrations of SP and then cell viability tested by Cell Counting Kit-8 (CCK-8, Yisheng Biotechnology Co., Ltd., Shanghai, China. 40203ES60), following the manufacturer’s instructions.
LDH release assay
After different treatments, the lactate dehydrogenase (LDH) levels in the culture supernatants or BALF were detected using a LDH assay kit (Promega, Madison, Wisconsin, USA. J2380-10 mL).
ELISA
Bronchial epithelial cells were plated onto 96-well plates and cultured at 37°C overnight. Plasma from patients with bronchial asthma and healthy individuals was collected and stored at −80°C. Then, after different treatments, plasma or culture supernatant were collected for measurements using ELISA kits (Milliplex® Luminex premix 13-plex kit, Merck. HSTCMAG28SPMX13), according to the recommended protocols.
RT-qPCR
Total RNA was extracted from the PBMC using TRIzol reagent. RT-qPCR was performed on the Biosystems 7000 Sequence Detection System (Applied Biosystems, Foster City, CA) to evaluate the relative expression level of mRNA. The primer sequence design was as previously reported [30–34]. U6 snRNA (noncoding small nuclear RNA-001973) was used as an internal reference gene, and the relative expression level was checked using the 2−ΔΔCT method.
Western blot
Protein samples of the same amount were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene fluoride (PVDF) membrane, followed by blocking with 5% nonfat milk. Incubate the primary antibody against NKIR (Sigma-Aldrich (Shanghai) Trading Co. Ltd., Shanghai, China, 1:1000, SAB4502913), NLRP3 (Proteintech, Chicago, USA, 1:1000, 19771-1-AP), ASC (Santa Cruz, USA, 1:500, sc-22514-R), Caspase-1 (Proteintech, Chicago, USA, 1:1000, 22915-1-AP), PI3K (Abcam, Shanghai, China, 1:1000, ab32089), phosphorylated (p)-PI3K (Abcam, Shanghai, China, 1:1000, ab278545), mTOR (Abcam, Shanghai, China, 1:2000, ab32028), p-mTOR (Abcam, Shanghai, China, 1:5000, ab109268), or GAPDH (Proteintech, Chicago, USA, 1:2000, 60004-1-lg) and membrane overnight at 4°C, then washed and incubated with the anti-rabbit IgG secondary antibody (Abcam, Shanghai, China, 1:5000, ab190495) for 1 h at room temperature. Densitometry analysis was conducted using ImageJ software (National Institutes of Health, Bethesda, MD).
Immunofluorescence staining assay
Cells and tissues were fixed in 3.7% formaldehyde for 20 min and blocked with 5% skim milk in PBS containing 0.2% Triton X-100 for 5 min. Next, exposed to the primary antibodies of ASC and caspase-1 (Santa Cruz Biotechnology, USA) at 4°C overnight. Next, secondary antibody (Dako, Glostrup, Denmark) was incubated with the cells for 1 h. Afterward, the cells were mounted with Vectashield mounting medium containing DAPI. The results were observed under a Leica CTR 4000 fluorescence microscope.
Statistical analysis
Each experimental condition was performed independently in triplicate. Statistical analyses were conducted using GraphPad Prism 8 software. P values for comparisons between two groups were calculated using unpaired student’s t-test, and for three groups or more were calculated using one-way ANOVA (*P-value < 0.05).
Results
Higher substance P/NK1R levels and pro-inflammatory cytokines in patients with bronchial asthma
In order to analyze the correlations of SP/NK1R system with the development of bronchial asthma, we collected blood and PBMC samples from 20 patients with bronchial asthma and 12 healthy people, and we initially measured the plasma levels of SP in the above 32 plasma samples by ELISA. The results uncovered that the content of SP in the plasma of patients with bronchial asthma was significantly higher than that of healthy controls (Figure 1(a)). As previously reported, SP regulates many disease-related processes through interacting with its receptor NK1R [35,36]. Thus, RT-qPCR and western blot were performed to examine NK1R level in the patients’ PBMC. As expected, the results revealed that NK1R was also significantly upregulated in PBMC collected from bronchial asthma patients in contrast with their normal counterparts (Figure 1(b,c)), indicating that the SP/NK1R system was closely relevant to the development of bronchial asthma. According to recent studies, super-inflammation is considered as important contributor for the aggravation of bronchial asthma, and various pro-inflammatory cytokines are reported to participate in the regulation of this process [37–39]. To validate the above information, we performed the following experiments, and found that the mRNA and protein levels of the cytokines, including TNF-α, TGF-β1, IL-2, IL-5, IL-18 and IL-1β, were all significantly elevated in the bronchial asthma patients’ plasma (Figure S1A-F and Figure 1(d-i)) and PBMC (Figure 1(j-o) and Figure S2G-L) compared to the normal participants. Also, the correlation analysis results supported that SP levels were positively related with all of the above cytokines in patients’ serum (Figure 1(p-u)). Moreover, we analyzed the clinical characteristics of the subjects in Supplementary Table S1, and found that the higher the grade of bronchial asthma, the higher the concentration of SP. Those data suggested that SP/NK1R system and its associated super-inflammation were closely relevant to the progression of bronchial asthma.
Figure 1.
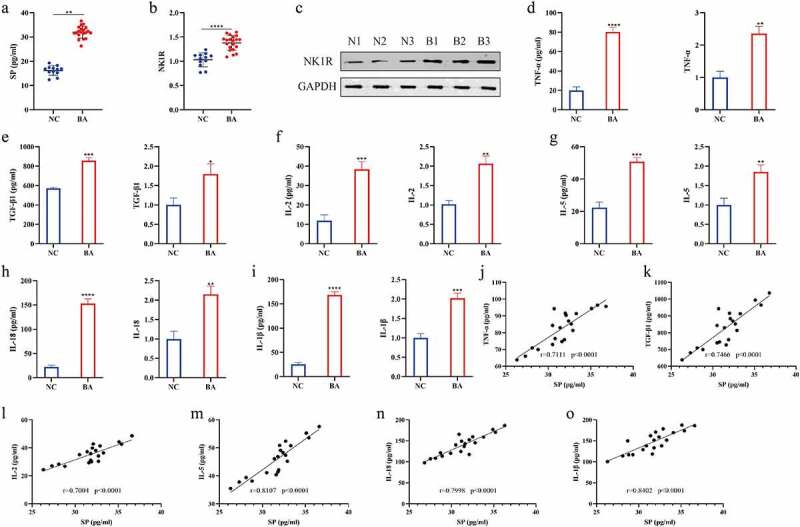
Higher substance P/NK1R levels and pro-inflammatory cytokines in patients with bronchial asthma. (a): ELISA was used to measure the concentrations of substance P in the plasma. (b): The expression of NK1R mRNA of bronchial asthma patients and normal control subjects. (c): The protein expression of NK1R of bronchial asthma patients and normal control subjects was detected by Western blot. “B1”, “B2”, “B3” represented “bronchial asthma patients 1, 2, or 3”, “N1”, “N2”, “N3” represented “normal control 1, 2, or 3”. (d-i): The concentration of pro-inflammatory cytokines, including TNF-α, TGF-β1, IL-2, IL-5, IL-18 and IL-1β, in plasma of patients with bronchial asthma was measured by ELISA (Left), The mRNA expression of pro-inflammatory cytokines (TNF-α, TGF-β1, IL-2, IL-5, IL-18 and IL-1β) in PBMC of bronchial asthma patients and normal control subjects was measured by RT-qPCR (Right). (j-o): Correlation analysis between SP and pro-inflammatory cytokines (TNF-α, TGF-β1, IL-2, IL-5, IL-18 and IL-1β) in plasma of patients of bronchial asthma was performed using Pearson correlation test. “BA” represented “bronchial asthma”, “NC” represented “normal control”. Each experiment was conducted in triplicate. *P < 0.05, ** P < 0.01, ***P < 0.001, ****P < 0.0001.
SP triggered pyroptotic cell death in human bronchial epithelial cell lines
According to the existed information, IL-1β and IL-18 are important effector cytokines of pyroptotic cell death, which has been proved to be closely associated with cellular inflammation and bronchial asthma [40,41], suggesting that cell pyroptosis may play important role in bronchial asthma. To ask whether SP participated in the regulation of bronchial asthma through regulating cell pyroptosis, the human bronchial epithelial cell lines (16-HBE and BEAS-2B) were subjected to different concentrations of SP treatment (0, 0.1, 0.5 and 1.0 μM) for in vitro experiments. The CCK-8 assay (Figure 2(a,b)) and colony formation assay (Figure 2(c,d)) showed that SP dose-dependently suppressed cell viability in the bronchial epithelial cells. Since 0.5 μM of SP significantly suppressed cell viability in the two human bronchial epithelial cell lines, we selected this concentration of SP for further experiments, and we found that SP triggered pyroptotic cell death in both 16-HBE and BEAS-2B cells (Figure 2(e-k)). Specifically, the immunofluorescence staining assay results showed that SP significantly upregulated Caspase-1/ASC levels in both 16-HBE and BEAS-2B cells (Figure 2(e)). Also, SP treatment promoted LDH release (figure 2(f)), increased the expression levels of NLPR3, ASC and Caspase-1 proteins (Figure 2(g-i)), and promoted IL-1β and IL-18 secretion in cells’ supernatants of 16-HBE and BEAS-2B cells (Figure 2(j,k)).
Figure 2.
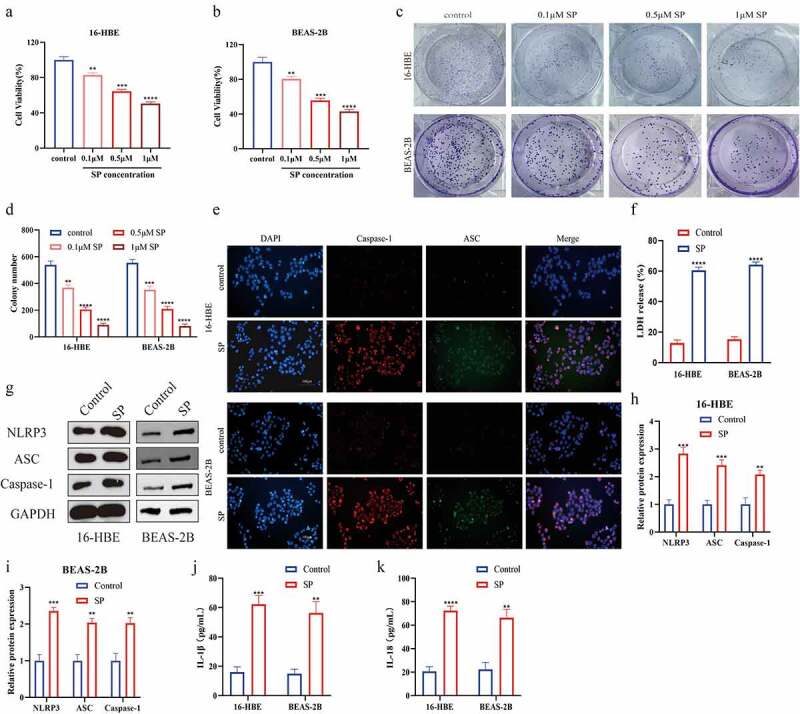
Substance P triggered pyroptotic cell death in human bronchial epithelial cell lines. (a, b): 16-HBE and BEAS-2B cells were treated with different doses (0, 0.1, 0.5 and 1 μM) of SP for 8 h. Cell viability was determined using the CCK8 assay. (c, d): The cell viability of 16-HBE and BEAS-2B cells were treated by SP was detected by colony formation. (e): ASC and Caspase-1 immunofluorescence of 16-HBE and BEAS-2B cells treated with 0.5 μM SP. (f): The release of LDH from SP-treated 16-HBE and BEAS-2B cells was measured by LDH assay kit. (g-i): Protein levels of NLRP3, ASC and Caspase-1 were detected by Western blot. (j): The levels of IL-1β were determined by ELISA. (k): The le ansvels of IL-18 were determined by ELISA. Each experiment was conducted in triplicate. *P < 0.05, ** P < 0.01, ***P < 0.001, ****P < 0.0001.
SP triggered pyroptotic cell death in asthma mice and human bronchial epithelial cells in a NK1R-dependent manner
We next validated that SP also triggered pyroptotic cell death in asthmatic mice in vivo. Specifically, establishment and SP treatment of BALB/c mice asthma model (Figure 3(a)), and the Real-Time qPCR and ELISA analysis verified that SP increased the expression levels of TNF-α, TGF-β1, IL-2, IL-5, IL-18 and IL-1β in both mice airway epithelium (Figure 3(b-g)) and serum (Figure 3(h-m)) to trigger super-inflammation in vivo. Then, we validated that SP upregulated Caspase-1 expressions (Figure 3(o)) and facilitate LDH release (Figure 3(n)) to promote cell pyroptosis in mice bronchial tissues. In addition, as it had been indicated in the previous literatures [42], SP often exerts its biological functions through interacting with its receptor NK1R, the mice were then co-administered with NK1R inhibitor L732138 for the rescuing experiments. As expected, blockage of NK1R suppressed pro-inflammatory cytokines secretion (Figure 3(b-m)), and restrain cell pyroptosis (Figure 3(n,o)) in SP-treated asthmatic mice models. Consistently, the above in vivo results were verified by the following in vitro experiments, which showed that SP NK1R-dependenlty upregulated IL-1β and IL-18 (Figure S2A, B), and promoted LDH release in the 16-HBE and BEAS-2B cell (Figure S2C), supporting the notion that SP activated the NLRP3 inflammasome-mediated pyroptotic cell death in bronchial asthma through interacting with its receptor NK1R.
Figure 3.
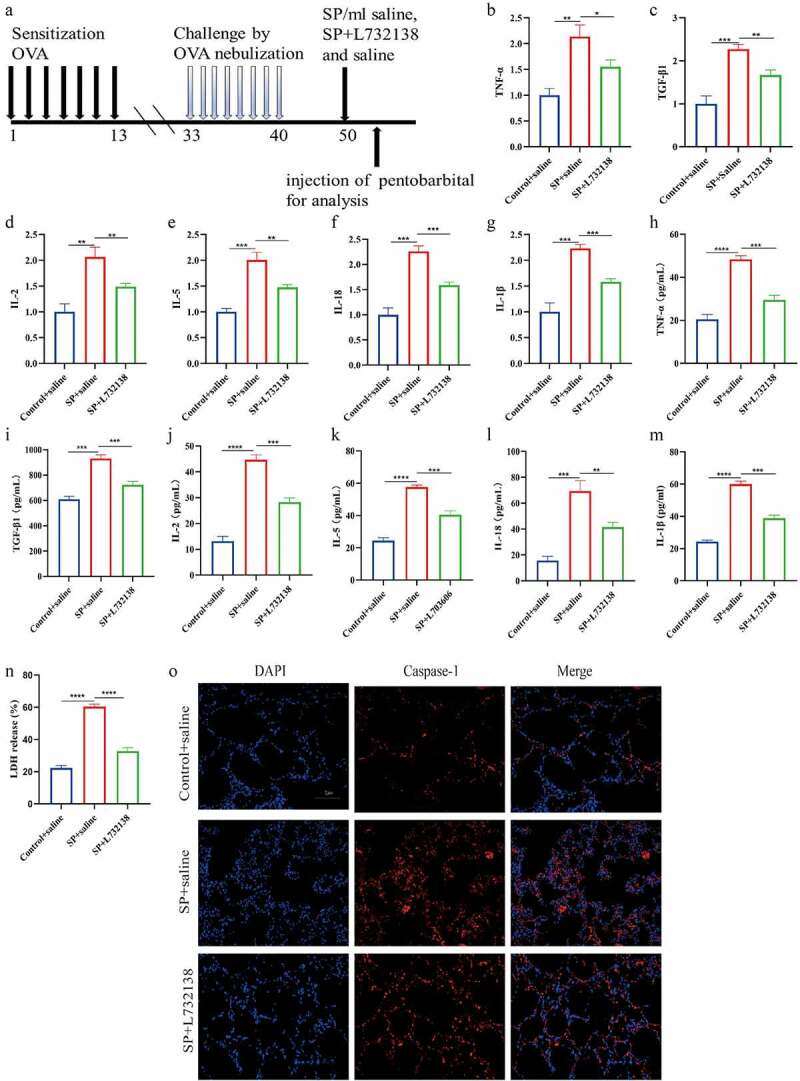
SP triggered pyroptotic cell death in asthma mice in a NK1R-dependent manner. (a) Overview of sensitization experiments in experimental asthma mice. (b-g): The mRNA expression of pro-inflammatory cytokines (TNF-α, TGF-β1, IL-2, IL-5, IL-18 and IL-1β) of asthma mice. (h-m): The expression levels of pro-inflammatory cytokines (TNF-α, TGF-β1, IL-2, IL-5, IL-18 and IL-1β) in serum of asthma mice. (n): The release of LDH was measured by LDH assay kit. (o): Immunofluorescence staining for Caspase-1 in airway epithelial were assessed. Each experiment was conducted in triplicate. *P < 0.05, ** P < 0.01, ***P < 0.001, ****P < 0.0001.
The PI3K/AKT/ NF-κB signaling pathway could be activated by SP
Next, we investigated the underlying mechanisms by which SP activated cell pyroptosis in bronchial asthma. As previously reported, the PI3K/AKT/NF-κB signal transduction pathway is closely related with pyroptotic cell death [24,25], and this pathway can be activated by SP [22]. Also, this signal pathway is often aberrantly activated in bronchial asthma [43]. Thus, we asked whether the SP/NK1R system activated the PI3K/AKT/NF-κB signal pathway in bronchial asthma. To achieve this, the 16-HBE and BEAS-2B cells were administered with SP and NK1R inhibitor L732138, and the expression levels of the PI3K/AKT/NF-κB signaling pathway-related proteins in 16-HBE and BEAS-2B cells were determined by Western blot. As shown in Figure 4(a-h), SP increased the expression levels of phosphorylated PI3K (p-PI3K), Akt (p-Akt) and NF-κB (p-NF-κB) to activate the PI3K/AKT/NF-κB signaling pathway in 16-HBE and BEAS-2B cells, but did not alter the expression status of total PI3K, Akt and NF-κB. Interestingly, SP-induced activation of the PI3K/AKT/NF-κB signaling pathway was reversed by co-treating cells with L732138, suggesting that SP also activated the PI3K/AKT/NF-κB signaling pathway through interacting with its receptor NK1R.
Figure 4.
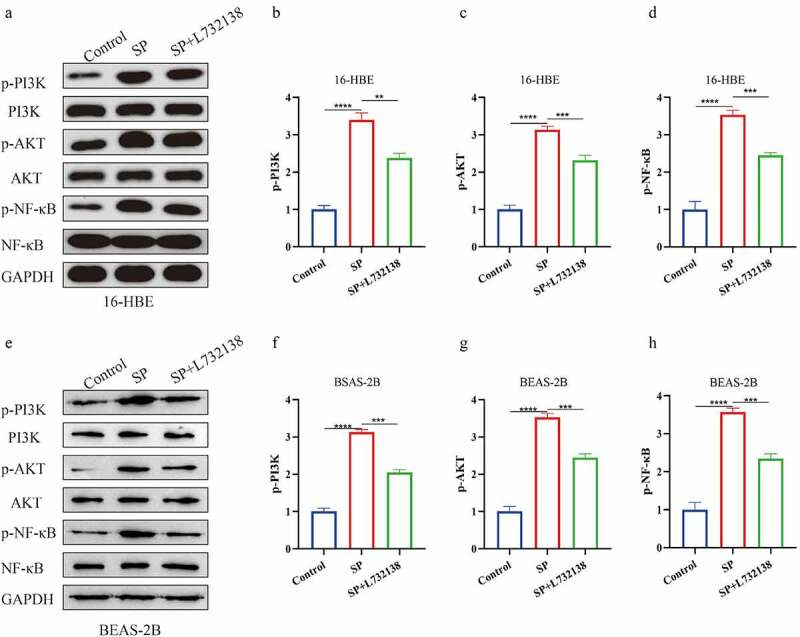
The PI3K/AKT/ NF-κB signaling pathway could be activated by SP. (a): The expression of proteins related to PI3K/AKT/NF-κB signaling pathway in 16-HBE cells was measured by Western blot. (b-d): Gray statistical value of PI3K/AKT/NF-κB signaling pathway related proteins in Western Blot. (e): The expression of proteins related to PI3K/AKT/NF-κB signaling pathway in BEAS-2B cells was measured by Western blot. (f-h): Gray statistical value of PI3K/AKT/NF-κB signaling pathway related proteins in Western Blot. Each experiment was conducted in triplicate. *P < 0.05, ** P < 0.01, ***P < 0.001, ****P < 0.0001.
SP activated the PI3K/AKT/NF-κB signaling pathway to induce pyroptotic cell death and cellular inflammation in the bronchial epithelial cells
Given that the PI3K/AKT/NF-κB signaling pathway can be activated by SP [22], and this pathway is reported to regulate NLRP3-mediated pyroptotic cell death [44–46], we finally explored whether SP triggered pyroptotic cell death in the human bronchial epithelial cells through modulating this pathway. Thus, the 16-HBE and BEAS-2B cells were, respectively, administered with SP, PI3K inhibitor LY294002, and NF-κB inhibitor MG132, which were grouped as follows: Control, SP alone group, SP plus MG132 group (SP+MG132), and SP plus LY294002 group (SP+LY294002). As shown in Figure 5(a-c), the suppressing effects of SP on cell viability were reversed by co-treating cells with both MG132 and LY294002. Also, the immunofluorescence staining assay (Figure 5(d,e)) uncovered that the levels of Caspase-1 and ASC were curbed in SP-treated cells by MG132 and LY294002. Similarly, the promoting effects of SP on LDH release in the cells were restrained by blocking the PI3K/AKT/NF-κB signaling pathway (figure 5(f,g)), and Western Blot analysis (Figure 5(h,i)) verified that both MG132 and LY294002 decreased the expression levels of Caspase-1, ASC and NLRP3 to inactivate SP-induced pyroptotic cell death in the bronchial epithelial cells. Finally, the cells’ supernatants were collected, and our ELISA assay results supported that SP promoted secretion of IL-1β and IL-18 to facilitate cellular inflammation, which were also suppressed by MG132 and LY294002 (Figure 5(j,k)). Those data suggested that SP promoted cell pyroptosis and cellular inflammation in bronchial asthma by activating the PI3K/AKT/NF-κB signaling pathway.
Figure 5.
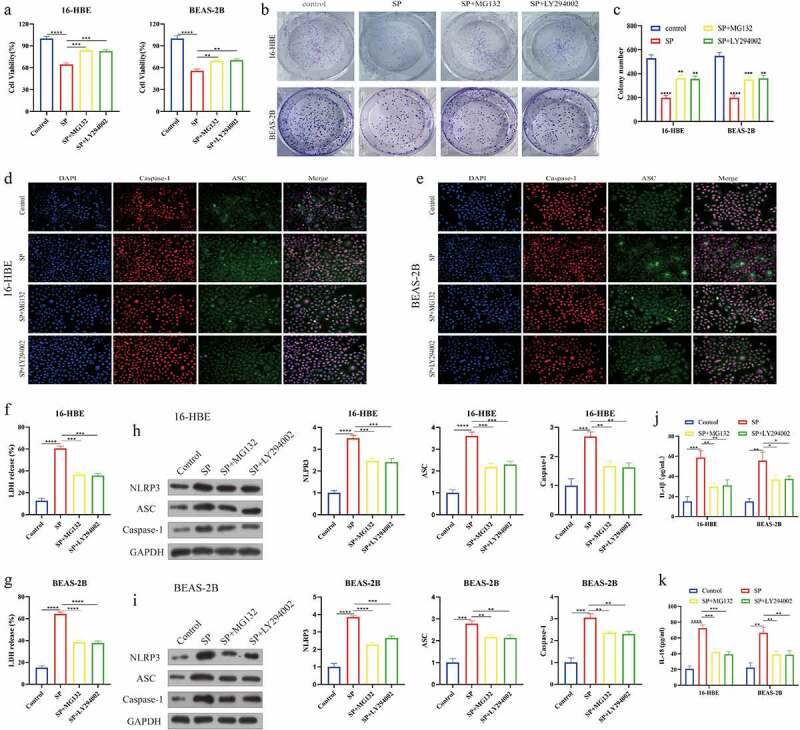
SP activated the PI3K/AKT/NF-κB signaling pathway to induce pyroptotic cell death and cellular inflammation in the bronchial epithelial cells. (a): CCK8 analysis of cell viability treated with LY294002 or MG132. (b, c) Colony formation assays with control, SP, SP+MG132 and SP+LY294002 in 16-HBE and BEAS-2B cells. (d): Caspase-1 and ASC immunofluorescence of 16-HBE cells treated with LY294002 or MG132. (e): Caspase-1 and ASC immunofluorescence of BEAS-2B cells treated with LY294002 or MG132. (f, g): The release of LDH from LY294002 or MG132 treated 16-HBE and BEAS-2B cells was measured by LDH assay kit. (h, i): Protein levels of NLRP3, ASC and Caspase-1 were detected by Western blot. (j, k): The levels of IL-1β and IL-18 after LY294002 or MG132 treatment were determined by ELISA. Each experiment was conducted in triplicate. *P < 0.05, ** P < 0.01, ***P < 0.001, ****P < 0.0001.
Discussion
Currently, the pathogenesis of bronchial asthma is very complicated, and various pathogenic factors that contribute to the development of this disease, which seriously limits the development of novel treatment strategies for this disease [47,48]. SP is a pleiotropic neuropeptide belonging to the tachykinin family, and SP is highly expressed in inflamed airways. According to recent literatures, SP plays a key role in regulating the development of a variety of diseases, such as leukemia [48], epilepsy [7], fibrotic [6,8], various cancers [22,35] and asthma [10,49]. Also, it is reported that SP exerts its biological functions through interacting with its receptor NK1R [42], and NK1R itself is associated with the development of inflammation-associated diseases [50]. However, the involvement of the SP/NK1R system in modulating the progression of bronchial asthma has not been studied. Thus, in our study, we verified that both SP and NK1R were significantly upregulated in the plasma and PBMC collected from the patients with bronchial asthma, and the expression levels of the SP was positively related with the pro-inflammatory cytokines (TNF-α, TGF-β1, IL-2, IL-5, IL-18 and IL-1β), suggesting that SP/NK1R system was involved in regulating bronchial asthma, and this process was associated with super-inflammation, which were supported by the existed publications [50].
Cell pyroptosis is a type of programmed inflammation-associated cell death, which is reported to be related with the occurrence of many diseases, such as atherosclerosis [51], multiple sclerosis [52], melanoma [53], spinal cord injury [44] and asthma [3]. In particular, Panganiban et al. demonstrate that GSDMB-mediated epithelial cell pyroptosis is involved in the pathogenesis of asthma [54], and NLRP3 inflammasome-mediated pyroptosis is also closely related to abnormal inflammation-related colitis [25], which were supported by our results that NLRP3 inflammasome-mediated pyroptotic cell death and cellular inflammation contributed to the aggravation of bronchial asthma. In addition, SP is capable of promoting various types of cell death [19–21], unfortunately, it is still unclear whether SP participated in the regulation of cell pyroptosis. In our study, we firstly verified that SP upregulated NLRP3, ASC, Caspase-1, IL-1β and IL-18 to activate cell pyroptosis in the in vitro and in vivo bronchial asthma models. Moreover, since SP is known to exert its biological functions through interacting with its receptor NK1R [42], and we validated that SP NK1R-dependently triggered pyroptotic cell death in the bronchial asthma models.
Aberrant activation or inactivation of the classical PI3K/Akt/NF-κB signaling pathway is identified as critical contributor for the development of atherosclerosis [27], cancer [22,55,56], asthma [43,57], and lung diseases [57]. Especially, this signal pathway also affects the progression of asthma, and data from the existed literatures suggest that the PI3K/AKT/NF-κB signaling pathway participates in the regulation of asthma through influencing super-inflammation [43,57]. Interestingly, previous data illustrate that the PI3K/AKT/NF-κB pathway can be activated by SP [22,55,56], which were verified by our results that SP activated this pathway in human bronchial epithelial cells through interacting with its receptor NK1R. Moreover, as previously described, the PI3K/AKT/NF-κB signaling pathway is critical signal that regulates cell pyroptosis and cellular inflammation in diabetic kidney disease [45], abdominal aortic aneurysm [58] and atherosclerosis [46], and based on the fact that the SP/NK1R system is able to activate both this pathway and cell pyroptosis, we validated that blockage of the PI3K/Akt/NF-κB signaling pathway abrogated the promoting effects of SP on cell pyroptosis and inflammation in the bronchial epithelial cells.
Conclusions
In summary, we concluded that SP and its receptor NK1R promoted bronchial asthma progression by inducing pyroptotic cell death and cellular inflammation through activating the PI3K/Akt/NF-κB signaling pathway. However, more clinical samples and further in-depth studies are needed. These findings revealed the regulatory mechanism of SP in the inflammatory response of bronchial asthma and provided a theoretical basis for the treatment of bronchial asthma.
Supplementary Material
Funding Statement
The author(s) reported there is no funding associated with the work featured in this article.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Availability of data and materials
All data generated or analyzed during this study are included in this published article (and its supplementary information files).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15384101.2022.2092166
References
- [1].Izuhara K, Matsumoto H, Ohta S, et al. Recent developments regarding periostin in bronchial asthma. Allergol Int. 2015;18:S3–S10. [DOI] [PubMed] [Google Scholar]
- [2].Yuan L, Du X, Tang S, et al. ITGB4 deficiency induces senescence of airway epithelial cells through p53 activation. FEBS J. 2019;286:1191–1203. [DOI] [PubMed] [Google Scholar]
- [3].Zhuang J, Cui H, Zhuang L, et al. Bronchial epithelial pyroptosis promotes airway inflammation in a murine model of toluene diisocyanate-induced asthma. Biomed Pharmacother. 2020;125:109925. [DOI] [PubMed] [Google Scholar]
- [4].Zieglgänsberger W. Substance P and pain chronicity. Cell Tissue Res. 2019;375(1):227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Fxz L, Xu F, Lin X, et al. The role of substance P in the regulation of bone and cartilage metabolic activity. Front Endocrinol (Lausanne). 2020;11:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sloniecka M, Danielson P. Substance P induces fibrotic changes through activation of the RhoA/ROCK pathway in an in vitro human corneal fibrosis model. J Mol Med (Berl). 2019;97(10):1477–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chi G, Huang Z, Li X, et al. Substance P regulation in epilepsy. Curr Neuropharmacol. 2018;16(1):43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Peng L, Agogo GO, Guo J, et al. Substance P and fibrotic diseases. Neuropeptides. 2019;76:101941. [DOI] [PubMed] [Google Scholar]
- [9].Dvorakova MC, Kruzliak P, Rabkin SW. Role of neuropeptides in cardiomyopathies. Peptides. 2014;61:1–6. [DOI] [PubMed] [Google Scholar]
- [10].Hens G, Raap U, Vanoirbeek J, et al. selective nasal allergen provocation induces substance p- mediated bronchial hyperresponsiveness. Am J Respir Cell Mol Biol. 2011;44(4):517–523. [DOI] [PubMed] [Google Scholar]
- [11].Xu CW, Guo SC, Zheng ZW, et al. Effect of gamma-aminobutyric acid treatment on plasma substance P and calcitonin gene-related peptide levels in children with asthma. Chin J Contemp Pediatr. 2013;15(2):102–104. [PubMed] [Google Scholar]
- [12].Miao L, Shang YX. Inhaled corticosteroids inhibit substance P receptor expression in asthmatic rat airway smooth muscle cells. BMC Pulm Med. 2012;12:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Miao L, Shang YX, Wei B, et al. The effect of substance P on asthmatic rat airway smooth muscle cell proliferation, migration, and cytoplasmic calcium concentration in vitro. J Inflam. 2011;8(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu X, Zhang Z, Ruan J, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535(7610):153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Si MM, Karki R, Kanneganti T. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev. 2017;277(1):61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wu X, Zhang H, Qi W, et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018;9(2):171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fang Y, Tian S, Pan Y, et al. Pyroptosis: a new frontier in cancer. Biomedecine Pharmacotherapie. 2019;121:109595. [DOI] [PubMed] [Google Scholar]
- [18].Hartman ML. Non-apoptotic cell death signaling pathways in melanoma. Int J Mol Sci. 2020;21(8):2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Vink R, Heuvel C. Substance P antagonists as a therapeutic approach to improving outcome following traumatic brain injury. Neurotherapeutics. 2010;7(1):74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Thornton E, Tran T, Vink R. A substance P mediated pathway contributes to 6-hydroxydopamine induced cell death. Neurosci Lett. 2010;481(1):64–67. [DOI] [PubMed] [Google Scholar]
- [21].Li Q, Wu X, Yang Y, et al. Tachykinin NK1 receptor antagonist L-733,060 and substance P deletion exert neuroprotection through inhibiting oxidative stress and cell death after traumatic brain injury in mice. Int J Biochem Cell Biol. 2019;107:154–165. [DOI] [PubMed] [Google Scholar]
- [22].Javid H, Asadi J, Avval FZ, et al. The role of substance P/neurokinin 1 receptor in the pathogenesis of esophageal squamous cell carcinoma through constitutively active PI3K/Akt/NF-κB signal transduction pathways. Mol Biol Rep. 2020;47(3):2253–2263. [DOI] [PubMed] [Google Scholar]
- [23].Shi H, Liu J, Lu A. Expression profiles of PI3K, NF-κB, and STAT1 in peripheral blood mononuclear cells in children with bronchial asthma. Zhongguo Dang Dai Er Ke Za Zhi. 2016;18(7):614–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Xu S, Wang J, Jiang J, et al. TLR4 promotes microglial pyroptosis via lncRNA-F630028O10Rik by activating PI3K/AKT pathway after spinal cord injury. Cell Death Dis. 2020;11(8):693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chen X, Liu G, Yuan Y, et al. NEK7 interacts with NLRP3 to modulate the pyroptosis in inflammatory bowel disease via NF-κB signaling. Cell Death Dis. 2019;10(12):906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kang Q, Liu W, Liu H, et al. Effect of compound Chuanxiong capsule on inflammatory reaction and PI3K/Akt/NF-κB signaling pathway in atherosclerosis. Evidence Based Complementray Altern Med. 2015;2015:584596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tao Y, Songping Y, Chao M, et al. SIRT4 suppresses the PI3K/Akt/NFκB signaling pathway and attenuates HUVEC injury induced by oxLDL. Mol Med Rep. 2019;19(6):4973–4979. [DOI] [PubMed] [Google Scholar]
- [28].Sun J, Ramnath RD, Tamizhselvi R, et al. Role of protein kinase C and phosphoinositide 3-kinase-Akt in substance P-induced proinflammatory pathways in mouse macrophages. FASEB J. 2009;81(4):997–1010. [DOI] [PubMed] [Google Scholar]
- [29].Yang L, Li T. Apolipoprotein M and sphingosine-1-phosphate complex alleviates TNF-α-induced endothelial cell injury and inflammation through PI3K/AKT signaling pathway. BMC Cardiovasc Disord. 2019;19(1):279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wu H, Zhao Y, Huang Q, et al. NK 1R/5-HT1AR interaction is related to the regulation of melanogenesis. FASEB J. 2018;32(6):3193–3214. [DOI] [PubMed] [Google Scholar]
- [31].GE M, D M, BM J, et al. The usefulness of competitive PCR: airway gene expression of IL-5, IL-4, IL-4, IL-4δ2, IL-2, and IFNγ in asthma. Thorax. 2001;56(7):541–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chen LX, Xu CM, Gao F, et al. Associations of IL-18 and IL-9 expressions and gene polymorphisms with asthma. Eur Rev Med Pharmacol Sci. 2020;24(12):6931–6938. [DOI] [PubMed] [Google Scholar]
- [33].Persson IM, Menzel M, Ramu S, et al. IL-1β mediates lung neutrophilia and IL-33 expression in a mouse model of viral-induced asthma exacerbation. Respir Res. 2018;19(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Huang H, Lu H, Liang L, et al. MicroRNA-744 inhibits proliferation of bronchial epithelial cells by regulating smad3 pathway via targeting transforming growth factor-β1 (TGF-β1) in severe asthma. Med Sci Monit. 2019;25:2159–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Darban RA, Korfi F, Hashemy I, et al. The effect of SP/NK1R on the expression and activity of catalase and superoxide dismutase in glioblastoma cancer cells. Biochem Res Int. 2021;2021:6620708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Han C, Du D, Wen Y, et al. Chaiqin chengqi decoction ameliorates acute pancreatitis in mice via inhibition of neuron activation-mediated acinar cell SP/NK1R signaling pathways. J Ethnopharmacol. 2021;274(6):114029. [DOI] [PubMed] [Google Scholar]
- [37].Kianian F, Kadkhodaee M, Sadeghipour HR, et al. An overview of high-mobility group box 1, a potent pro-inflammatory cytokine in asthma. J Basic Clin Physiol Pharmacol. 2020;31(6): 20190363. [DOI] [PubMed] [Google Scholar]
- [38].Yin H, Liu MH, Gao F, et al. Pro-inflammatory and pro-fibrotic role of long non-coding RNA RMRP in pediatric asthma through targeting microRNA-206/CCL2 axis. J Biol Regulators Homeostatic Agents. 2021;35(1):71–83. [DOI] [PubMed] [Google Scholar]
- [39].Szollosi DE, Ghoneim O, Manzoor MK, et al. Novel piperazino-enaminones suppress pro-inflammatory cytokines and inhibit chemokine receptor CCR2. Inflammation. 2016;39(6):2053–2061. [DOI] [PubMed] [Google Scholar]
- [40].Fang Y, Tian S, Pan Y, et al. Pyroptosis: a new frontier in cancer. Biomed Pharmacother. 2020;121:109595. [DOI] [PubMed] [Google Scholar]
- [41].Yuan H, Hideki H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41(12):1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mehrabani N, Vaezi Kakhki MR, Javid H, et al. The SP/NK1R system-mediated ROS generation in GBM cells through inhibiting glutaredoxin protein. Neurol Res Int. 2021;2021:9966000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wu D, Li S, Liu X, et al. Alpinetin prevents inflammatory responses in OVA-induced allergic asthma through modulating PI3K/AKT/NF-κB and HO-1 signaling pathways in mice. Int Immunopharmacol. 2020;89:107073. [DOI] [PubMed] [Google Scholar]
- [44].Xu S, Shao M, Ma X, et al. CD73 alleviates GSDMD-mediated pyroptosis in spinal cord injury through PI3K/AKT/Foxo1 signaling. Clin Transl Med. 2021;11(1):e269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Liu BH, Tu Y, Ni GX, et al. Total flavones of abelmoschus manihot ameliorates podocyte pyroptosis and injury in high glucose conditions by targeting METTL3-dependent m(6)A modification-mediated NLRP3-inflammasome activation and PTEN/PI3K/Akt signaling. Front Pharmacol. 2021;12:667644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Xu S, Chen H, Ni H, et al. Targeting HDAC6 attenuates nicotine-induced macrophage pyroptosis via NF-κB/NLRP3 pathway. Atherosclerosis. 2020;317(2021):1–9. [DOI] [PubMed] [Google Scholar]
- [47].Li X, Wang B, Huang M, et al. miR-30a-3p participates in the development of asthma by targeting CCR3. Open Med. 2020;15(1):483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Toutenhoofd P, Zee J. Severe bronchial asthma. Ned Tijdschr Geneeskd. 2013;157(33):A6117. [PubMed] [Google Scholar]
- [49].Chen Y, Gao Y, Lu W, et al. Influence of acupuncture on the expression of VIP, SP, NKA and NKB, cAMP/cGMP and HE content and treatment of bronchial asthma in rats. Cell Mol Biol. 2020;66(5):29–35. [PubMed] [Google Scholar]
- [50].Martinez AN, Philipp MT. Substance P and antagonists of the neurokinin-1 receptor in neuroinflammation associated with infectious and neurodegenerative diseases of the central nervous system. J Neurolo Neuromed. 2016;1(2):29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Liu X, Zhang Y, Yang BF. Melatonin prevents endothelial cell pyroptosis via regulation of long noncoding RNA MEG3/miR-223/NLRP3 axis. J Pineal Res. 2018;64(2):269–270. [DOI] [PubMed] [Google Scholar]
- [52].Mckenzie BA, Mamik MK, Saito LB, et al. Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Pro Natl Acad Sci U S A. 2018;115(26):E6065–E6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Erkes DA, Cai W, Sanchez IM, et al. Mutant BRAF and MEK inhibitors regulate the tumor immune microenvironment via pyroptosis. Cancer Discov. 2020;10(2):254–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Panganiban RA, Sun M, Dahlin A, et al. A functional splice variant associated with decreased asthma risk abolishes the ability of gasdermin B to induce epithelial cell pyroptosis. J Allergy Clin Immunol. 2018;142(5):1469–1478.e1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ghahremanloo A, Javid H, Afshari AR, et al. Investigation of the role of neurokinin-1 receptor inhibition using aprepitant in the apoptotic cell death through PI3K/Akt/NF-kappaB signal transduction pathways in colon cancer cells. Biomed Res Int. 2021;2021:1383878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Javid H, Afshari AR, Zahedi Avval F, et al. Aprepitant promotes caspase-dependent apoptotic cell death and G2/M arrest through PI3K/Akt/NF-kappaB axis in cancer stem-like esophageal squamous cell carcinoma spheres. Biomed Res Int. 2021;2021:8808214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Chen X, Yang J, Shen H, et al. Muc5ac production inhibited by decreased lncRNA H19 via PI3K/Akt/NF-kB in asthma. J Asthma Allergy. 2021;14:1033–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Xiong JM, Liu H, Chen J, et al. Curcumin nicotinate suppresses abdominal aortic aneurysm pyroptosis via lncRNA PVT1/miR-26a/KLF4 axis through regulating the PI3K/AKT signaling pathway. Toxicol Res (Camb). 2021;10(3):651–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its supplementary information files).