

ABSTRACT
The mammalian gut contains a large, complex community of microorganisms collectively termed the microbiota. It is increasingly appreciated that gut microbes are closely integrated into mammalian physiology, participating in metabolic symbiosis, promoting immune function and signaling to a wide variety of distant cells, including the brain, via circulating metabolites. Recent advances indicate that microglia, the brain’s resident immune cells, are influenced by microbial metabolites at all stages of life, under both physiological and pathological conditions. The pathways by which microbiota regulate microglial function are therefore of interest for investigating links between neurological disorders and gut microbiome changes. In this review, we discuss the effects and mechanisms of microbiota-microglia signaling in steady state, as well as evidence for the involvement of this signaling axis in CNS pathologies.
KEYWORDS: Microglia, development, microbiota, neurological disorders, gut-derived metabolites, gut-brain axis
Introduction
It is increasingly appreciated that mammalian physiology depends not only on the host genome, but also by intimate interactions with commensal microbes which have been shaped by millions of years of co-evolution. Some such interactions are mutualistic, such as co-operative food digestion and nutrient synthesis, though some aspects of physiology are influenced by the mere presence of commensals or by infection by pathogenic species. Thus, the physiology of most animals can be best understood as a meta-organism comprising the host and a huge diversity of commensal microorganisms collectively referred to as the microbiota. Host-microbiota interactions are increasingly understood to be fundamental in shaping diverse aspects of mammalian biology, including development, metabolism, immunity and neurological function. The link between intestinal microbiota and brain function has been extensively described, and is commonly referred to as the microbiota-gut-brain axis.1
Microglia are one of the four primary cell types comprising the mammalian central nervous system (CNS). As the brain’s primary immune population, they contribute not only to the development and maintenance of the CNS, but also critically regulate CNS disease states such as neurodegeneration, autoimmunity and neurodevelopmental disorders.2 Although separated from commensal microbes by both the gut barrier and blood–brain barrier, the presence of intestinal microbes has been demonstrated to regulate microglial phenotype and functions, both at steady state and under disease conditions. Given the increasing interest in the gut microbiome as a regulator of neurological disorders, direct communication between intestinal microbiota and microglia is an intriguing concept which may prove to be a central mechanism linking the microbiome to various brain diseases. In this review, we explore recent advances in understanding the mechanistic links between gut flora and microglia, as well as their consequences for neurological disorders.
Microglia and brain development
Microglia are the brain’s tissue-resident macrophages, comprising between 5% and 15% of all cells in the healthy brain. Microglia originate as primitive c-kit+ macrophages in the embryonic yolk sac, which proceed to colonize the embryonic brain at around E9.5, quickly spreading to all developing brain structures.3–6 Due to the establishment of the blood–brain barrier at E13.5, the brain is shielded from subsequent waves of hematopoiesis originating in the fetal liver and bone marrow. Thus, there is no replacement of yolk-sac-derived microglia with monocyte-derived cells under homeostatic conditions,7 as is observed in other tissues.8 Instead, microglia self-renew throughout life via tightly regulated local proliferation and apoptosis.9,10 After first colonizing the brain at E9.5, microglia undergo several stepwise maturation stages (Figure 1).11,12 The first major transition involves a switch from early to pre-microglia at around E14.5.11,12 Pre-microglia then gradually acquire adult-like properties after birth, maturing fully into adult microglia beginning around the second or third postnatal week.11,12 Adult microglia exhibit a stereotypical, relatively homogenous signature enriched for genes involved in immunosurveillance, including the purinergic receptors P2ry12 and P2ry13, as well as Trem2 and Cd14.11,13 By contrast, earlier developmental ages are marked by far greater microglial diversity, with distinct microglial subsets exhibiting spatiotemporally regulated signatures and functions.13–15
Figure 1.
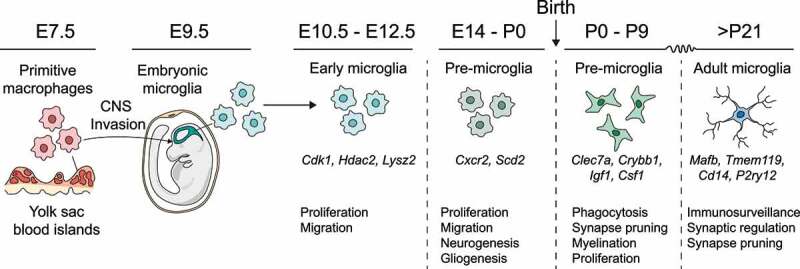
Microglial development. Microglia emerge from yolk-sac precursors and colonize the developing brain at around E9.5. Subsequently, they undergo a series of stepwise transformations, acquiring diverse developmental phenotypes as pre-microglia before maturing into adult microglia during the second or third postnatal week.
The timing of microglial entry into the brain means they are well positioned to influence CNS development. For instance, in the early postnatal brain microglia provide critical trophic support to oligodendrocyte precursor cells in the corpus callosum and cerebellar white matter, and loss of microglia during this time leads to later defects in myelination.16,17 Microglia also influence neuronal development, with some neuron subsets reliant on microglia for trophic signaling and correct positioning.18 Additionally, microglia are known to engulf apoptotic cells throughout development and adulthood, which is critical to tissue homeostasis. Interestingly, microglia are also capable of inducing developmental death of otherwise viable cells via direct engulfment, in a process termed “phagoptosis”.19 Through this process, microglia can directly fine-tune cell numbers during CNS development, including the extent of neurogenesis.20,21
Perhaps the best-studied function of microglia in neurodevelopment is complement-dependent synapse elimination, whereby superfluous synapses are tagged sequentially by C1q and C3, leading to their engulfment by CR3-expressing microglia.22–24 Loss of complement components or depletion of microglia therefore inhibits developmental circuit refinement, resulting in functional connectivity deficits in adults.25,26 Interestingly, aberrant synaptic refinement is postulated to be a central mechanism governing some psychiatric disorders, in particular schizophrenia, which has been linked to numerous polymorphisms affecting immune receptors including the complement system.27 Moreover, various neurodegenerative pathologies including Alzheimer’s disease have been suggested to encompass some degree of aberrant synaptic elimination by microglia.28 Thus, microglia are capable of directly shaping CNS architecture through a variety of mechanisms. Tight regulation of these processes is important for optimal CNS function, and disturbances to microglia during critical developmental windows can affect the course of brain maturation and susceptibility to neuropsychiatric and neurodevelopmental disorders.26,29 Similarly, microglial dysfunction in aging may play a key role in neurodegeneration and cognitive impairment
Microglia in CNS homeostasis and disease
Adult microglia exhibit several marked adaptations to their role as the primary immunocompetent cells of the brain. Firstly, their unique transcriptomic signature encompasses a large repertoire of genes involved in immunosurveillance.30 These include pattern recognition receptors such as toll-like receptors (TLRs), scavenger receptors and triggering receptor expressed on myeloid cells 2 (TREM2). Moreover, mature microglia express the receptors P2RY12 and P2RY13, conferring exquisite sensitivity to purinergic stimuli. Secondly, their long, elaborately branched processes are remarkably motile, undergoing extremely dynamic, rapid rearrangements and making contact with surrounding CNS cells on a constant basis.31,32 Thus, via their extensive sensome and unceasing motility, microglia scan the entire CNS space once every few hours, and are the primary responders to any disturbance in CNS homeostasis.
Following CNS insults such as injury, infection and neurodegeneration, microglia sense local cues and undergo transformation to reactive phenotypes. In general, these reactive microglia downregulate some homeostatic markers such as P2ry12, Tmem119 and Mafb, and induce genes involved in effector functions, many of which vary depending on the specific insult. Recent studies support the existence of multiple states of reactive microglia which can be induced by different pathologies such as neurodegeneration and autoimmunity and engage diverse reactive functions including phagocytosis, proliferation, antigen presentation and inflammatory cytokine secretion.13,14,33–35 Whilst microglial responses are undoubtedly critical for effective resolution of insults and preventing damage to the CNS, there is ample evidence to suggest that inappropriate microglial activity can be a causative factor in various CNS diseases.35 Microglia are therefore a key candidate for transducing environmental factors such as microbiome-related signaling to the exacerbation or amelioration of CNS pathology.
Microbiome-microglia interactions
The gut microbiota are known to exert extensive effects on host physiology via a variety of mechanisms. The most prominent mutualistic function of gut bacteria is metabolism – collectively the bacteria inhabiting the gastrointestinal tract contain many more metabolic genes than humans possess, thus allowing much greater metabolic flexibility than would otherwise be possible.36 In this sense, the gut microbiome is often described as an additional metabolic organ, assisting with fiber fermentation, tryptophan metabolism and detoxification of xenobiotics, amongst a wide range of other pathways. However, the effect of bacterial metabolism on mammalian physiology goes far beyond simple nutrient provision. Hundreds of bioactive metabolites are produced by the microbiota, circulating throughout every organ and affecting the activity of huge numbers of target cells.37–39
Of particular relevance to the CNS is the concept of the gut-brain axis, which describes numerous pathways linking gut physiology to brain function.1 The microbiota are critical players in the gut-brain axis, and can affect CNS physiology by directly modulating neurotransmission and neurodevelopment, by signaling through the vagus nerve, as well as by modifying endogenous metabolism, endocrine signaling and immune system activity (Figure 1).1,40,41 Studies in germ-free mice raised without any exposure to microbes, as well as in mice which have undergone microbiota depletion via broad-spectrum antibiotics, have revealed striking dependence of mammalian physiology on microbially derived cues. It is now increasingly appreciated that microglia are important target cells in the microbiota-gut-brain axis and may be critical in transducing changes in microbiome composition to altered brain function.
Microbiota modulate microglia in development and homeostasis
Microglial phenotypes are instructed by the microbiota throughout life, even prior to birth (Figure 1). Recent data indicate that microglia from embryos of germ-free mice exhibit large differences when compared to those of SPF embryos at E18.5.12 This effect was reported to be highly sex-dependent, with male embryos displaying far greater numbers of differentially expressed genes than females.12 Lack of a maternal microbiome resulted in greater microglial density in the cortex at E14.5 and E16.5, which persisted to E18.5 in male embryos. By contrast, the transcriptional effect of the microbiota on male mice was diminished postnatally, with females displaying much greater transcriptomic differences between germ-free and SPF microglia at P20.12 These findings add to a growing body of evidence that fetal neurodevelopment is influenced by cues derived from the maternal microbiota and place microglia as key players in mediating their effects.
Interactions between microglia and colonizing commensals are also important for early postnatal neurodevelopment.42 Germ-free pups exhibit defective synaptic pruning in Purkinje cells, which is linked to decreased numbers of amoeboid microglia and aberrantly low expression of CD45, CD68 and scavenger receptors.42 Intriguingly, both microglial phenotype and synaptic pruning can be rescued by colonization of GF pups with either conventional microbiota or a consortium of Bifidobacteria, which dominate the early postnatal microbiome in humans.42 However, the mechanism regulating communication between Bifidobacteria and cerebellar microglia is still unclear, and might be direct (eg. via circulating metabolites) or could involve other cell types, or indeed could be secondary to effects on the Purkinje neurons themselves. Nonetheless, this study suggests microglia could be critical in linking early-life neuroimmune interactions and neurodevelopment to microbiome composition. Further studies should seek to determine how early gut microbiota influence microglia, and whether this communication axis could affect the genesis of neurodevelopmental disorders.
In adult animals, gut bacteria exert a constitutive influence on microglial physiology via release of short-chain fatty acids (SCFA) (Figure 1). Microglia from adult germ-free animals display a distinctive phenotype characterized by increased density and aberrant morphology, with longer processes and overlapping territories.43,44 Intriguingly, the transcriptomic phenotype of germ-free microglia appears to be immature compared to SPF controls, with increased expression of proliferative markers and decreased expression of genes relating to immune function (eg Cd86, Ly86, Hif1a) and other markers of mature adult microglia (eg. Cst7, Neurl3).11,43 It is therefore increasingly appreciated that colonization with gut microbes may provide an environmental cue driving microglia to assume their fully mature phenotype, which occurs naturally around the same time. Moreover, since ablation of the microbiome in SPF animals via broad-spectrum antibiotics largely phenocopies the effect of germ-free housing on microglial phenotype, this instruction clearly occurs constitutively, rendering microglia sensitive to real-time perturbations in gut microbe composition.43 Interestingly, colonization of germ-free animals with a defined repertoire of 15 species known as altered Schaedler flora (ASF) was insufficient to reproduce the effect of SPF housing.43 This suggests that complex microbiota may be better able to support microglial maturity than other, defined communities. However, it is unclear whether the inability of ASF flora to replicate the effects of SPF housing is truly related to complexity, or rather to insufficient production of particular metabolites such as SCFAs, which might be adequately provided by different species in a similarly defined consortium. Remarkably, it was recently reported that, of the 3 major SCFAs produced by the gut biota (acetate, propionate and butyrate), only acetate is in fact necessary and sufficient for microglial maturity, with neither propionate nor butyrate able to influence the phenotype of germ-free microglia.44 Thus, production of acetate alone may be one of the most critical parameters when considering the effects of microbial communities on microglial state.
The immature phenotype of germ-free microglia is not limited to effects on morphology and gene expression in homeostasis. Instead, germ-free microglia also exhibit defective responses to immune stimuli. In SPF mice, intracerebroventricular (i.c) injection of the bacterial endotoxin lipopolysaccharide (LPS) induces a robust TLR4- and NF-κB-dependent response resulting in upregulation of inflammatory mediators including TNF-α, IL-1ß and CCL2. In germ-free animals, this response is markedly diminished, indicating an immunologically immature state.43 Similarly, infection with lymphocytic choriomeningitis virus (LCMV) in germ-free animals produces lower induction of Il1b and Tnfa, as well as the immediate-early genes Fosb, c-fos and c-jun compared to SPF controls.43 Interestingly however, germ-free microglia displayed normal induction of the antiviral signaling molecules Cxcl10, Irf7 and Isg15 following LCMV infection.43 This could indicate that lack of microbiota induces a specific signaling defect in microglia which impairs certain response pathways but leaves typical antiviral signaling mediated by interferons and RNA sensing pathways intact. The exact nature and mechanism of the signaling alterations induced by lack of microbiota are still unclarified, but could prove critical when drawing mechanistic links between microbiota perturbations and microglial disease responses.
Mechanisms of microbiota-microglia communication
Given the separation between microglia and commensal microbes, interactions are generally only possible via circulatory factors or via altered activity in an intermediate cellfor example, neuronal signaling via the vagus nerve. On the simplest possible level, molecules produced by gut microbes can enter the blood through the intestinal wall, subsequently entering the brain and acting on microglia directly to alter their physiology (Figure 2). However, microglial phenotypes are also sensitive to changes in other CNS cells such as astrocytes, as well as to alterations in peripheral immunity and metabolism. Indeed, relatively few studies have so far demonstrated clear-cut examples of direct microbiota-microglia communication, and in most cases the involvement of additional cell types or signal relays cannot be excluded. Moreover, direct or indirect modulation of microglia by the microbiota can likely occur both in the form of constitutive signaling and epigenetic effects imprinted developmentally. The latter possibility remains somewhat underexplored compared to constitutive signaling pathways, but two studies have so far confirmed differences in chromatin accessibility and histone modification patterns between SPF and germ-free microglia.12,44
Figure 2.
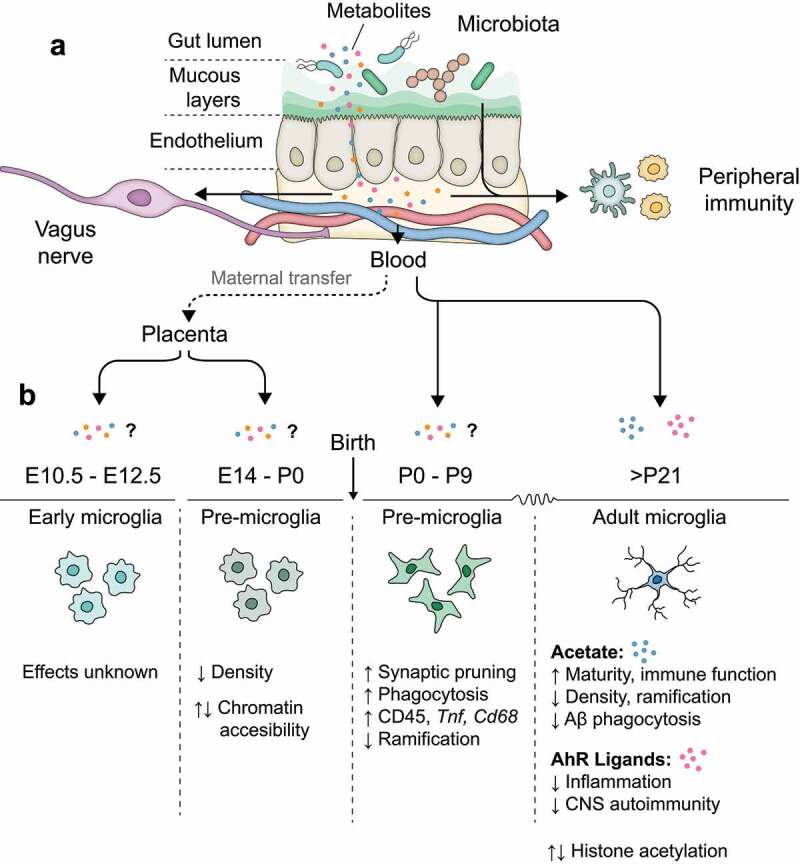
Microbiota-microglia interactions. (a) Microbiota are confined to the gut lumen, separated from the lamina propria by gut endothelial cells. Microbiota-derived metabolites can penetrate the endothelium and enter the bloodstream, as well as influencing local immune cells and vagal afferents. (b) Effects of microbial metabolites on microglial function under steady-state across developmental stages. Evidence suggests that microglial density, epigenetics and transcriptomics are affected by microbiota at all stages of life.
Currently the best-studied microglia-microbe interactions occur via circulating SCFAs, which are produced by gut bacteria via fiber fermentation. The three most abundant SCFAs produced by the microbiota are acetate, propionate and butyrate, consisting of 2-, 3- and 4-carbon chains, respectively. It was recently discovered that acetate alone is able to reverse many features of the germ-free microglial phenotype, while propionate and butyrate are entirely ineffective.44 Thus, while SCFAs had generally been regarded as a single class of metabolite, it appears that their effects on microglia are divergent, with acetate being the primary mediator of microglia-microbiota signaling.44 The mechanism by which acetate affects microglial phenotype remains largely unclarified, though several potential pathways have been identified.
Recent data using radiolabelled carbon tracing indicate that gut-derived acetate in the blood is capable of entering the brain and is incorporated into the microglial Krebs cycle.44 This same report also linked acetate availability to metabolic alterations in germ-free microglia. Specifically, germ-free microglia exhibit increased mitochondrial mass compared to SPF controls, which is coupled to reduced activity of the mitochondrial electron transport chain complex II.44 This defect is rescuable by acetate supplementation and may relate to lack of acetyl Co-A, resulting in increased generation of oxaloacetic acid, a known complex II inhibitor.44 Whilst it is unclear whether this metabolic defect is related to the other functional alterations in germ-free microglia, it is feasible given the now widely recognized role of metabolism as a central regulator of macrophage function.45,46 These data therefore support a model whereby microbiota modulate microglia by providing a carbon source required for normal metabolism, thus forming a direct link between diet, gut microbiota composition and microglial state. This idea is particularly compelling given that the primary receptor for acetate, FFAR2,47 is not expressed in the brain, thus making direct effects of acetate on microglia via GPCR signaling unlikely.43
It is especially interesting to consider the effects of acetate on microglial metabolism in the 5xFAD AD model, where germ-free animals exhibit greater microglial phagocytosis and reduced plaque pathology, which is reversible by acetate supplementation.44,48 Exposure to Aβ is well described to induce a shift in microglial metabolism toward glycolysis, which over time supports inflammatory polarization and subsequent inhibition of phagocytosis, thus promoting accumulation of amyloid plaques.49–51 Indeed, GF microglia exhibit lower induction of glycolytic and proinflammatory genes in the 5xFAD model than their SPF counterparts, which is largely dependent on acetate.44 Thus, acetate availability appears to constitutively modulate both dependence on glycolysis and inflammation in amyloid-exposed microglia, which may result in phagocytic suppression and increased plaque accumulation. However, exactly why this occurs remains unclarified.
Additional explanations for the maturation-inducing effect of acetate on microglia include protein acetylation and activation of GPCR signaling pathways. Indeed, microglia in mice lacking the SCFA receptor FFAR2 exhibit similar defects to those from germ-free animals despite a lack of FFAR2 expression in the brain, indicating that microglia could be affected by SCFA signaling in other organs.43 However, supplementation of germ-free animals with propionate, another FFAR2 agonist, does not affect microglial phenotype, with acetate being the only SCFA capable of reproducing microbiota-induced microglial maturation. As such, it is possible that FFAR2 is dispensable for the effect of acetate on microglia, but its deletion phenocopies acetate deprivation via a separate, unclarified mechanism.
Protein acetylation constitutes another route by which acetate availability might directly influence microglial physiology. Acetylation is an extremely common post-translational modification with profound consequences for the function of various immunological proteins.52 For example, the transcription factor NF-κB, which is critical for the generation of inflammatory responses, relies on acetylation of the p50 and p65 subunits for DNA binding and full transcriptional activity, respectively.53,54 LPS elicits classical NFκB-dependent responses which are blunted severely in germ-free microglia. This raises the possibility that the dimished LPS responsivity observed in the absence of microbiota could be due to lower acetate availability leading to insufficient acetylation of NF-κB or other immunologically important proteins. However, to date no studies have examined the effect of microbial acetate on acetylation of the general microglial proteome.
Another facet to protein acetylation involves epigenetic modification of histone proteins by histone acetyltransferases (HATs). Acetylated histones generally result in more accessible chromatin and increased gene transcription,55 and are a key mechanism regulating immune cell effector functions following stimulation.56,57 Acetate supplementation produces increased histone acetylation in various organs,58 including the brain,59 and microbiota have been reported to regulate patterns of acetylation across a variety of histone sites via release of SCFAs.58 Gut microbiota-derived acetate may affect histone acetylation by increasing the availability of acetyl-CoA, a necessary substrate for HAT activity, or via inhibition of histone deacetylases (HDACs).59 In microglia, gut microbiota have been shown to regulate acetylation at the 9th lysine of histone 3 (H3K9),44 a modification associated with increased promoter activity. Whilst levels of H3K9 acetylation appear to decrease overall under GF conditions, many genes also display increased H3K9 acetylation compared to SPF microglia.44 This is in agreement with a previous report which identified differentially accessible chromatin regions between GF and SPF microglia via ATAC-Seq beginning in early development.12 Thus, the microbiota clearly regulate microglial histone acetylation, which could underly some aspects of their effect on microglial function. However, whether or not this effect can be attributed to acetate is unclear, since propionate and butyrate are also known HDAC inhibitors capable of influencing epigenetic state60–62 and the effect of individual SCFAs on microglial histone modification has not been assessed. Moreover, it is unknown whether microglial histone modification patterns are controlled dynamically by the microbiota or are imprinted during development and remain static thereafter, as was recently suggested for the liver.63 Finally, it would be helpful to dissect precisely which aspects of the germ-free microglial phenotype are dependent on epigenetic modifications, and whether this could account for the differing effects of antibiotic treatment in SPF animals versus acetate supplementation in GF conditions.43,44,48
Taken together, the evidence for a direct microbiota-microglia communication axis mediated by acetate is compelling, but key questions remain around the precise nature of the effect, which may involve multiple parallel functions of acetate as both a signaling molecule and metabolic substrate. A major question moving forward is what role other microbial metabolites might play in instructing microglial phenotype. Acetate supplementation reverses most of the metabolic and phenotypic differences between GF and SPF microglia, but does not completely normalize the transcriptome under either homeostatic or pathological conditions.44 This could be explained by lingering epigenetic differences between GF and SPF animals, and/or could imply the presence of extra signaling factors which regulate microglial phenotype more subtly. Aryl hydrocarbon receptor (AhR) ligands have been demonstrated to influence microglial state during CNS autoimmunity,64 but their effects under homeostatic conditions have not been reported. Microglia are also affected by microbial products during embryonic and postnatal development, but the molecular players are entirely unknown. Acetate has been shown to be differentially abundant in SPF versus germ-free embryos, along with a variety of compounds including the AhR ligand indoxyl-3-sulfate, trimethylamine-N-oxide (TMAO), hippurate, N, N, N-trimethyl-5-aminovalerate (TMAV) and imidazole propionate.65–67 One recent study identified defective thalamocortical axon development in germ-free embryos, which is rescued by maternal supplementation with the latter 4 compounds listed above, but not by SCFAs including acetate. Whether embryonic and early postnatal microglia could also be regulated by these compounds, acetate, or AhR ligands would provide an interesting basis for future studies.
Microglia-microbiome communication in disease
Microglia are active regulators of a variety of CNS pathological states, including neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease, as well as neuroinflammatory and neuropsychiatric conditions.2 Signaling from the microbiota to microglia has therefore been suggested as an environmental factor capable of promoting or ameliorating disease. In support of this idea, microbiota-dependent influences on microglial states have been reported to exhibit a high degree of sex-dependence,12 which could help account for the marked differences in susceptibility to certain CNS diseases observed between men and women. A vast quantity of literature has in recent years been generated suggesting roles for microbiome dysbiosis in the pathology of numerous neurological conditions. Since this is too extensive to review in full, we focus here on critically reviewing the most compelling evidence linking communication between microbiota and microglia to neurological disorders.
Alzheimer’s disease
Alzheimer’s disease (AD) is a chronic neurodegenerative disease characterized by progressive brain atrophy and profound memory loss, which is accompanied by deposition of amyloid plaques and overabundance of hyperphosphorylated tau protein.68 Amyloid plaques are composed of high-order aggregates of amyloid-beta (Aβ), a peptide produced by cleavage of amyloid precursor protein (APP).69 While the pathology of AD is complex and incompletely understood, excessive accumulation of Aβ is thought to be a major trigger of subsequent disease processes such as tau hyperphosphorylation, synaptic dysfunction and neuronal loss.69–71
The microbiota are proposed to play a major role in AD, which could occur at least partially via microglia-mediated mechanisms. Several studies have reported decreased richness and diversity of fecal microbiota in AD patients compared with healthy controls.72,73 More specific differences on the level of individual phyla and genera have been reported, but in some cases are inconsistent between different studies72–77 (reviewed by78). For instance, Vogt et al.72 noted decreased abundance of Actinobacteria in AD patients, whereas Ling et al.73 observed an increase. Similarly, increased abundance of Bacteroides is noted by some, but not other studies.72,73,77 One relatively consistent point, however, is that the abundance of Firmicutes appears to be generally decreased in AD patients.72–77 Additionally, some functional relationships have been proposed between AD pathology and the balance of inflammatory versus anti-inflammatory species such as Eschieria/Shigella, E. rectalis and F. prauznitii.74,75 While it is entirely feasible that changes in gut microbiota aggravate or promote AD, and evidence exists for altered microbiome composition in AD patients, human clinical studies have thus far been entirely correlative. Moreover, since AD-associated pathological changes begin decades prior to the onset of clinical symptoms,70 the most relevant changes in gut biota may be missed by studies focusing on patients with diagnosed AD, and future studies tracking temporal changes in microbiome composition prior to AD onset may be more informative.
Whilst mechanistic associations between gut microbiota variation and AD pathology are difficult to obtain in humans, there is now a wealth of corroborating data confirming that mouse models of AD are extremely sensitive to changes in gut bacteria. The first study to address this issue demonstrated that antibiotic administration to APP/PS1 mice (high-dose for one week followed by chronic low-dose) reduced amyloid plaque burden.79 Subsequent studies have validated that germ-free housing or antibiotic treatment reduce plaque pathology in the APPSwe/PS1ΔE9,80,81 APPPS1-2181–83 and 5xFAD44,48 models. Mechanistically, this effect seems to be independent of amyloid processing pathways,48 and instead reflects altered microglial activity.44,48 Microglia from germ-free animals exhibit greater induction of phagocytic genes such as Axl, Apoe and Clec7a, as well as higher expression of reactivity markers such as Cst7 compared to SPF controls.44,48,84 This is coupled to increased levels of amyloid phagocytosis in germ-free microglia, indicating that microbiota interactions direct critical microglial functions in AD models.44,48 Interestingly, recent studies report that, similarly to the steady-state phenotypic differences observed in germ-free mice, the effect of microbiota on amyloid pathology appears to be mediated primarily SCFAs, specifically acetate.44,84 Thus, clear evidence exists for an acetate-mediated microbiota-microglia signaling axis which promotes amyloid accumulation in AD mouse models. Most intriguingly, a recent study in human AD patients reported that amyloid burden correlates positively with serum acetate levels, but negatively with serum butyrate and propionate.85 This supports a model whereby acetate may act via microglia to promote amyloid pathology in AD, whereas butyrate and propionate are unable to modulate microglial physiology and instead are protective due to effects on other cell types. While causative associations between individual microbes or metabolites and human AD remain unproven, these results suggest that AD interventions targeting gut-derived SCFAs should pay close attention to the role of acetate versus propionate and butyrate in modulating amyloid burden.
Parkinson’s disease
Parkinson’s disease (PD) is a complex neuropathology involving degeneration of dopaminergic neurons in various brain regions such as the substantia nigra, along with extensive intracellular accumulation of aggregated α-synuclein protein.86 Symptoms include progressive motor dysfunction along with cognitive impairment, depression, and sleep disruption.
Gut-brain signaling has long been suggested to be of particular relevance for PD, especially considering that α-synuclein pathology is often observed in the gut,87 and that gut inflammation and motility problems are common in PD patients. It has even been postulated that α-synuclein aggregates may in some cases originate in the gut and propagate to the brain in a prion-like manner via the vagus nerve, though this idea remains controversial.87–89 A role for the microbiota in regulating PD pathology could therefore be multifaceted, influencing the local generation of toxic α-synuclein species or distantly affecting microglial phenotypes in affected regions of the CNS, as occurs in AD models. Some studies have demonstrated altered gut microbiota composition in PD patients, though the results are likely affected by drug treatment and, as with AD, it is unclear whether gut flora changes precede brain pathology.90
To date, the only direct evidence for involvement of the gut microbiota in PD comes from rodent studies. In an α-synuclein overexpression (ASO) model, GF mice were protected against gut motility defects, microglial reactivity, brain inflammation and ultimately motor deficits.91 Similarly to in AD models, the detrimental effects of complex gut biota on neuropathology in ASO mice are reproducible by supplementation with SCFAs.91 Interestingly, colonization of GF ASO mice with microbiota samples from PD patients generally (but not invariably) produced greater motor impairment than with samples from healthy controls.91 However, it is unknown whether this effect is due to differences in SCFA production between PD-derived and control microbiota or whether other mechanisms are responsible.
Most studies assessing SCFA levels in PD patients have found decreased concentrations in fecal samples, suggesting dampened SCFA production.92–95 However, it has also been suggested that, due to the frequent alterations in gut function accompanying PD, stool SCFA measurements may not accurately represent systemic exposure compared to healthy controls.95 Measurements of circulating SCFAs in serum are less common, but have been variously reported as decreased,96 increased,95 or unchanged97 in PD patients. Moreover, correlations between individual SCFAs and clinical symptoms are most often reported for propionate and butyrate,95,97 which in mice do not directly affect microglia.44 Thus, there is currently little evidence to support acetate-mediated microbiota-microglia signaling as a causative mechanism in PD patients. While this does not rule out a role for the microbiota in modulating microglial phenotype in human PD, it may be that other metabolites are responsible,98 or indeed a more complex mechanism such as via systemic immunity, gut barrier integrity or neuronal signaling. Further mechanistic studies in mouse models may prove useful to elucidate exactly why PD patient-derived microbiota elicit greater neurodegeneration in mouse models, and whether microglia are critically involved.
Multiple Sclerosis
Multiple Sclerosis (MS) is an autoimmune condition generally caused by autoreactive immune responses directed against myelin components, resulting in the formation of brain lesions exhibiting prominent inflammation, demyelination and blood–brain barrier disruption.99 While the disease course of MS is highly variable, most patients eventually experience irreversible neurological damage, impairment and disability which continues to worsen over time. The causes of MS are unknown, though previous infection with Epstein-Barr virus (EBV) appears near-universal amongst MS patients, and may promote the generation of cross-reactive T cells via molecular mimicry.100 As a primarily immune-mediated condition, immunomodulatory therapies in MS have shown some clinical success in reducing relapse frequency and slowing disease course.99
Multiple states of reactive microglia can be observed in MS, and likely play multifaceted roles in regulating the progression of pathology.34,101 One the one hand, microglia can contribute to the intense inflammation typical of MS lesions, inducing neuronal damage and demyelination.101 On the other hand, microglia are also critical for phagocytosis of myelin debris and stimulating oligodendrocyte differentiation and remyelination.101 Thus, microglia constitute a key target cell for any interventions aimed at modifying the course of MS. Unlike typical neurodegenerative conditions, the peripheral immune system is heavily involved in MS, and overall pathology is influenced by the properties of both CNS-resident cells and infiltrating peripheral cells. Thus, whilst there is evidence that gut bacteria are capable of modifying MS pathology, it must be considered that their effects can be mediated not only through changes in microglia but also in peripheral immunity. Indeed, a variety of studies have demonstrated that gut flora are required for the generation of autoreactive T-cell responses in experimental autoimmune encephalomyelitis (EAE), a commonly used model of MS.102,103 Thus, germ-free animals are generally resistant to EAE pathology due to defective T-cell differentiation. In humans, differences in the ability of gut bacteria to stimulate inflammatory versus regulatory T cell responses may constitute a key environmental factor determining MS risk.104 It is also for this reason that GF animals are of limited use for studying microbiota-microglia signaling in EAE, and therefore only few studies have successfully demonstrated mechanistic links between gut biota and microglial phenotypes in EAE models.
One key study examining microglia-microbiota crosstalk in EAE did so by focusing on the aryl hydrocarbon receptor (AhR), a major mediator of gut-immune signaling.64 Mice harboring AhR deletion in microglia following transient CX3CR1-CreERT2-mediated recombination exhibit exacerbated clinical scores after immunization with MOG peptide.64 This was found to be due to reduced AhR-dependent secretion of TGF-α and increased production of VEGF-B from microglia, causing increased pro-inflammatory astrocyte polarization. AhR ligands are produced by the gut microbiota via tryptophan metabolism, and restriction of dietary tryptophan in WT mice phenocopied the effect of microglial AhR deletion.64 Conversely, supplementing a tryptophan-deficient diet with AhR ligands such as indole-3-carbinole (I3C) rescued the worsened EAE pathology in WT, but not microglial AhR-deficient mice.64 Thus, gut flora are capable of influencing microglial phenotypes in EAE independently of changes in T cell-mediated immunity, and may be critical in transducing diet-related environmental variables to EAE risk. These results build on a previous study indicating that AhR ligands can also signal via astrocytes, and that AhR signaling may form part of the protective influence of interferon-beta in MS.105 Thus, microbial tryptophan metabolism and AhR ligands are a promising lead for harnessing endogenous microglia-microbial signaling pathways in MS, and efforts to validate their role in human subjects are ongoing.106
Autism spectrum disorder
Autism Spectrum Disorder (ASD) comprises a group of related neurodevelopmental disorders whose manifestations vary widely in both type and severity but can include learning and social impairment as well as repetitive behaviors.107 The etiology of ASD is poorly understood, but is generally thought to be a multi-hit process involving genetic predisposition in combination with environmental triggers during critical developmental periods.108 Gastrointestinal symptoms are common in individuals with ASD, suggesting a link between gut function and ASD symptoms.109 The gut microbiota is a prime example of an environmental factor which is established during early development and proceeds to regulate both brain and gut physiology, and is often dysregulated in ASD.1 There is currently intense interest in microbiota-based therapies for ASD, some of which have already shown promising results in initial trials.110
Microglia are presumed to be involved in ASD pathology due to their intimate contribution to neurodevelopment and ability to regulate brain function through release of inflammatory mediators and neurotrophic molecules.111 Indeed, some degree of microglial abnormalities and brain pathology have been noted in ASD patients.112–116 However, altered microglial states could occur downstream of neuronal abnormalities in ASD and definitive causal associations are still lacking.
Currently some of the best evidence for causative roles of microglia and microbiota in ASD come from models of maternal immune activation (MIA). Viral and bacterial infections during pregnancy are a known risk factor for the development of ASD in humans,117,118 and offspring of mice injected with the viral mimetic poly(I:C) during pregnancy exhibit a phenotype which shares some features with ASD, including social interaction deficits and increased repetitive-like behaviors.119 This effect depends on induction of an acute inflammatory response in the maternal serum, including IL-6 and IL-17a.120–122 Maternal cytokine elevation results in subsequent production of inflammatory mediators in the fetal brain,123 and increased concentrations of some cytokines can be detected during postnatal development,124 though precisely which mediators are altered at which time points vary between studies.125 Microglia from MIA offspring display an altered phenotype characterized by premature induction of the mature gene signature in neonatal pups, as well as decreased phagocytosis and higher basal expression of inflammatory genes in adulthood.11,126 While these results in combination with human data suggest that microglial dysfunction can promote ASD pathogenesis, the exact contribution of microglia to cognitive and behavioral outcomes is still unclear, and many studies report conflicting results with regard to microglial reactivity following MIA.127
MIA pathogenesis is also strongly linked to gut microbiota. Segmented filamentous bacteria (SFB) in the maternal gut are required for induction of the IL-17a response following poly(I:C) exposure, and mice lacking SFB do not show the expected behavioral deficits in MIA offspring.122 Thus, the ability of viral infection to promote ASD may depend on the presence of commensals which direct maternal immune responses. Gut bacteria may also have constitutive effects on ASD pathology, since colonization of GF mice with gut biota derived from individuals with ASD promoted behavioral symptoms typically observed in ASD mouse models to a greater extent than that from neurotypical controls.128 This effect has been ascribed to differential abundance of microbial metabolites, which is commonly observed in ASD,129 leading to altered expression of ASD-linked genes in the brain.128 Conversely, treatment of ASD model mice with normal human commensals such as B. fragilis has been shown to reduce the severity of behavioral symptoms.130 These studies argue in favor of a causative role of microbiota alterations in ASD pathology, both in regulating predisposition via maternal immune activation and in promoting behavioral symptoms in later development, acting as an environmental “hit” in concert with other environmental and genetic factors. Given the abundant microbial metabolite alterations in ASD patients,129 it is feasible that their effects are mediated at least partially via alterations in microglial activity, though to date no studies have confirmed this.
In contrast to this idea, one recent study has questioned the causality between gut microbiota composition and ASD, suggesting instead that the behavioral symptoms of ASD drive microbiota alterations via less varied food intake.131 Moreover, most studies to date investigating treatment of ASD patients with pre- or probiotic formulations have been described as either inconclusive or unreliable by a recent meta-analysis.110 Despite this, the first trial of fecal microbiota transfer in ASD has reported promising outcomes.132,133 Thus, the role of both the microbiota and microglia as targets for ASD are still somewhat controversial, but remain an active and promising area of research.
Outlook and future challenges
To date, research into microglial modulation by microbiota has uncovered several promising phenomena of potential relevance to neurological disorders. However, the field is still in its infancy and significant challenges in the interpretation and translation of current findings into disease settings remain to be addressed. The first major caveat to microbiome research in general is that studies in humans largely produce correlative data with little mechanistic insight, and establishing causality between human diseases and microbiome alterations is extremely difficult. This is particularly true of diseases such as AD which exhibit long periods of clinical latency before symptoms manifest. It is for this reason that mouse models are generally relied upon for mechanistic studies seeking to dissect the contribution of microbiota perturbations to disease. However, translating mouse findings back into humans has a notoriously low success rate, which has been attributed not only to issues in preclinical study design, but also to more fundamental flaws in the accuracy with which human diseases are modeled in mice.
Germ-free mice, for instance, have provided invaluable insights into the essential role of gut microbes in mammalian biology, but are clearly highly unphysiological and unsuited to modeling the effects of subtle changes to a complex microbiota such as those observed in humans. Many additional gnotobiotic mouse models exist, harboring varying numbers of known bacterial strains, though other than the ASF model their effects on microglial function remain uncharacterized. These models could provide a useful reductionist approach to identify particular bacterial strains which might promote microglial function, either via acetate release or other mechanisms. However, these suffer the same drawback of questionable translational relevance.
Theoretically, this limitation can be overcome by use of fecal microbiota derived from human subjects. However, these models should also be interpreted with caution due to various issues including incomplete colonization of the recipient animal and the absence of additional relevant host and environmental factors.134,135 It is also increasingly appreciated that even SPF mice have unnaturally sparse microbiota lacking many natural symbionts and pathogens, which results in relatively immature immune system highly dissimilar to that of humans.136–139 Interestingly, colonization of laboratory mouse strains with complex “wild” biota or infection with pathogens results in a much closer approximation of human immunity than can be achieved in SPF animals.136–139 However, it is currently unclear whether microglial function is affected by colonization with wild biota, or whether its effects relate purely to microbial diversity or rather to the presence of specific immunostimulatory microbes absent from SPF mice. Nonetheless, conventional SPF mice clearly suffer from a paucity of microbial stimulation at baseline, which could render investigation of microbiota-immune interactions prone to artifacts.
Thus, to maximize the impact of microbiota-based therapies on human medicine it is important to establish robust causal relationships in human-relevant models. It would be intriguing for future efforts to establish whether the presence of wild-like microbiota affects microglial function, since this could provide a more robust platform for investigating human-relevant pathways of microbiota-microglia communication and increase the translational success of any resulting strategies. It is equally critical for future studies in human patients to gather high-quality data combining microbiota profiling with metabolomic analyses, ideally including longitudinal study designs and diverse patient cohorts to maximize mechanistic interpretability and identify high-confidence targets for animal model validation. It should also be appreciated that, although direct signaling between microbiota and microglia is a compelling concept, optimal clinical success will likely only come by targeting multiple systems including gut and metabolic function, as well as peripheral immunity.
Currently, the only robustly defined routes of direct communication between microbiota and microglia occur via acetate and AhR ligands, the latter of which has only been demonstrated in the context of EAE.64 Given the great diversity of microbially derived molecules permeating the body, it seems likely that at least some are capable of modulating microglia in ways which remain to be discovered, especially considering that the bulk of studies to date have been performed in a single strain of laboratory mouse under conditions of artificially low microbial diversity. It would therefore be interesting for future studies to dissect the effect of both wild mouse microbiota and transplanted human microbiota on microglial phenotype, with particular focus on the underlying metabolites and mechanisms. Extra care should also be given to examining the relationship between microbiota and microglia at different developmental stages. Embryonic, early postnatal and adult microglia are not only intrinsically different, but also very likely experience divergent sets of microbial cues. Thus, the existence of developmentally specific signaling axes should be considered. Another currently overlooked facet to microbiota research is the role of non-bacterial species such as fungi. Fungi have already been demonstrated to promote social behavior via IL-17 signaling in neurons,140 and to modify Alzheimer’s pathology in APP/PS1 mice via an unclarified pathway.141 Thus, fungi may provide additional routes of microglial modulation by microbiota and are deserving of further study in both humans and mice.
Funding Statement
. M.P. was supported by the Sobek Foundation, the Ernst-Jung Foundation, the Novo Nordisk Prize, the DFG (SFB 992, SFB1160, CRC/TRR 167 “NeuroMac”, Reinhart-Koselleck-Grant, Gottfried Wilhelm Leibniz Prize), Alzheimer Forschung Initiative e.V. (AFI) and the Ministry of Science, Research and Arts, Baden-Wuerttemberg (Sonderlinie “Neuroinflammation”). This study was supported by the DFG under Germany’s Excellence Strategy (CIBSS – EXC-2189 – Project ID390939984).
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.Cryan JF, O’Riordan KJ, Cowan CSM, Sandhu KV, Bastiaanssen TFS, Boehme M, Codagnone MG, Cussotto S, Fulling C, Golubeva AV, et al. The microbiota-gut-brain axis. Physiol Rev. 2019;99(4):1877–19. doi: 10.1152/physrev.00018.2018. [DOI] [PubMed] [Google Scholar]
- 2.Prinz M, Masuda T, Wheeler MA, Quintana FJ.. Microglia and central nervous system-associated macrophages-from origin to disease modulation. Annu Rev Immunol. 2021;39(1):251–277. doi: 10.1146/annurev-immunol-093019-110159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, Wieghofer P, Heinrich A, Riemke P, Hölscher C, et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci. 2013;16(3):273–280. doi: 10.1038/nn.3318. [DOI] [PubMed] [Google Scholar]
- 5.Schulz C, Perdiguero EG, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SEW, Pollard JW, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336(6077):86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 6.Masuda T, Amann L, Monaco G, Sankowski R, Staszewski O, Krueger M, Del Gaudio F, He L, Paterson N, Nent E, et al. Specification of CNS macrophage subsets occurs postnatally in defined niches. Nature. 2022;604(7907):740–748. doi: 10.1038/s41586-022-04596-2. [DOI] [PubMed] [Google Scholar]
- 7.Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch U-K, Mack M, Heikenwalder M, Brück W, Priller J, Prinz M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10(12):1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- 8.Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. 2014;41(1):21–35. doi: 10.1016/j.immuni.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Askew K, Li K, Olmos-Alonso A, Garcia-Moreno F, Liang Y, Richardson P, Tipton T, Chapman MA, Riecken K, Beccari S, et al. Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell Rep. 2017;18(2):391–405. doi: 10.1016/j.celrep.2016.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tay TL, Mai D, Dautzenberg J, Fernández-Klett F, Lin G, Datta M, Drougard A, Stempfl T, Ardura-Fabregat A, Staszewski O, et al. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat Neurosci. 2017;20(6):793–803. doi: 10.1038/nn.4547. [DOI] [PubMed] [Google Scholar]
- 11.Matcovitch-Natan O, Winter DR, Giladi A, Vargas Aguilar S, Spinrad A, Sarrazin S, Ben-Yehuda H, David E, Zelada González F, Perrin P, et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science. 2016;353(6301):aad8670. doi: 10.1126/science.aad8670. [DOI] [PubMed] [Google Scholar]
- 12.Thion MS, Low D, Silvin A, Chen J, Grisel P, Schulte-Schrepping J, Blecher R, Ulas T, Squarzoni P, Hoeffel G, et al. Microbiome influences prenatal and adult microglia in a sex-specific manner. Cell. 2018;172(3):500–516.e16. doi: 10.1016/j.cell.2017.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Masuda T, Sankowski R, Staszewski O, Böttcher C, Amann L, Scheiwe C, Nessler S, Kunz P, van Loo G, Coenen VA, et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature. 2019;566(7744):388–392. doi: 10.1038/s41586-019-0924-x. [DOI] [PubMed] [Google Scholar]
- 14.Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, Walker AJ, Gergits F, Segel M, Nemesh J, et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity. 2019;50(1):253–271.e6. doi: 10.1016/j.immuni.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Q, Cheng Z, Zhou L, Darmanis S, Neff NF, Okamoto J, Gulati G, Bennett ML, Sun LO, Clarke LE . Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron. 2019;101(2):207–223.e10. doi: 10.1016/j.neuron.2018.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hagemeyer N, Hanft K-M, Akriditou M-A, Unger N, Park ES, Stanley ER, Staszewski O, Dimou L, Prinz M. Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathol. 2017;134(3):441–458. doi: 10.1007/s00401-017-1747-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wlodarczyk A, Holtman IR, Krueger M, Yogev N, Bruttger J, Khorooshi R, Benmamar‐Badel A, Boer‐Bergsma JJ, Martin NA, Karram K, et al. A novel microglial subset plays a key role in myelinogenesis in developing brain. EMBO J. 2017;36(22):3292–3308. doi: 10.15252/embj.201696056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ueno M, Fujita Y, Tanaka T, Nakamura Y, Kikuta J, Ishii M, Yamashita T. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci. 2013;16(5):543–551. doi: 10.1038/nn.3358. [DOI] [PubMed] [Google Scholar]
- 19.Brown GC, Neher JJ. Microglial phagocytosis of live neurons. Nat Rev Neurosci. 2014;15(4):209–216. doi: 10.1038/nrn3710. [DOI] [PubMed] [Google Scholar]
- 20.Wakselman S, Béchade C, Roumier A, Bernard D, Triller A, Bessis A. Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J Neurosci. 2008;28(32):8138–8143. doi: 10.1523/JNEUROSCI.1006-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cunningham CL, Martínez-Cerdeño V, Noctor SC. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J Neurosci. 2013;33(10):4216–4233. doi: 10.1523/JNEUROSCI.3441-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 23.Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 24.Schafer DP, Lehrman E, Kautzman A, Koyama R, Mardinly A, Yamasaki R, Ransohoff R, Greenberg M, Barres B, Stevens B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frost JL, Schafer DP. Microglia: architects of the developing nervous system. Trends Cell Biol. 2016;26(8):587–597. doi: 10.1016/j.tcb.2016.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paolicelli RC, Ferretti MT. Function and dysfunction of microglia during brain development: consequences for synapses and neural circuits. Front Synaptic Neurosci. 2017;9(9). doi: 10.3389/fnsyn.2017.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woo JJ, Pouget JG, Zai CC, Kennedy JL. The complement system in schizophrenia: where are we now and what’s next? Mol Psychiatry. 2020;25(1):114–130. doi: 10.1038/s41380-019-0479-0. [DOI] [PubMed] [Google Scholar]
- 28.Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352(6286):712–716. doi: 10.1126/science.aad8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014;15(5):300–312. doi: 10.1038/nrn3722. [DOI] [PubMed] [Google Scholar]
- 30.Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang L-C, Means TK, El Khoury J. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 2013;16(12):1896–1905. doi: 10.1038/nn.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davalos D, Grutzendler J, Yang G, Kim J, Zuo Y, Jung S, Littman D, Dustin ML, Gan W. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8(6):752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 32.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 33.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. A unique microglia type associated with restricting development of alzheimer’s disease. Cell. 2017;169(7):1276–1290.e17. doi: 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 34.Jordão MJC, Sankowski R, Brendecke SM, Locatelli G, Tai Y-H, Tay TL, Schramm E, Armbruster S, Hagemeyer N, Groß O, et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science. 2019;363(6425). doi: 10.1126/science.aat7554. [DOI] [PubMed] [Google Scholar]
- 35.Prinz M, Jung S, Priller J. Microglia biology: one century of evolving concepts. Cell. 2019;179(2):292–311. doi: 10.1016/j.cell.2019.08.053. [DOI] [PubMed] [Google Scholar]
- 36.Tierney BT, Yang Z, Luber JM, Beaudin M, Wibowo MC, Baek C, Mehlenbacher E, Patel CJ, Kostic AD. The landscape of genetic content in the gut and oral human microbiome. Cell Host Microbe. 2019;26(2):283–295.e8. doi: 10.1016/j.chom.2019.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dodd D, Spitzer MH, Van Treuren W, Merrill BD, Hryckowian AJ, Higginbottom SK, Le A, Cowan TM, Nolan GP, Fischbach MA, et al. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature. 2017;551(7682):648–652. doi: 10.1038/nature24661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uchimura Y, Fuhrer T, Li H, Lawson MA, Zimmermann M, Yilmaz B, Zindel J, Ronchi F, Sorribas M, Hapfelmeier S, et al. Antibodies set boundaries limiting microbial metabolite penetration and the resultant mammalian host response. Immunity. 2018;49(3):545–559.e5. doi: 10.1016/j.immuni.2018.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lai Y, Liu C-W, Yang Y, Hsiao Y-C, Ru H, Lu K. High-coverage metabolomics uncovers microbiota-driven biochemical landscape of interorgan transport and gut-brain communication in mice. Nat Commun. 2021;12(1):6000. doi: 10.1038/s41467-021-26209-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fung TC, Olson CA, Hsiao EY. Interactions between the microbiota, immune and nervous systems in health and disease. Nat Neurosci. 2017;20(2):145–155. doi: 10.1038/nn.4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Needham BD, Kaddurah-Daouk R, Mazmanian SK. Gut microbial molecules in behavioural and neurodegenerative conditions. Nat Rev Neurosci. 2020;21(12):717–731. doi: 10.1038/s41583-020-00381-0. [DOI] [PubMed] [Google Scholar]
- 42.Luck B, Engevik MA, Ganesh BP, Lackey EP, Lin T, Balderas M, Major A, Runge J, Luna RA, Sillitoe RV, et al. Bifidobacteria shape host neural circuits during postnatal development by promoting synapse formation and microglial function. Sci Rep. 2020;10(1):7737. doi: 10.1038/s41598-020-64173-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Erny D, Hrabě de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, Keren-Shaul H, Mahlakoiv T, Jakobshagen K, Buch T, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. 2015;18(7):965–977. doi: 10.1038/nn.4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Erny D, Dokalis N, Mezö C, Castoldi A, Mossad O, Staszewski O, Frosch M, Villa M, Fuchs V, Mayer A, et al. Microbiota-derived acetate enables the metabolic fitness of the brain innate immune system during health and disease. Cell Metab. 2021;33(11):2260–2276.e7. doi: 10.1016/j.cmet.2021.10.010. [DOI] [PubMed] [Google Scholar]
- 45.Borst K, Schwabenland M, Prinz M. Microglia metabolism in health and disease. Neurochem Int. 2019;130:104331. doi: 10.1016/j.neuint.2018.11.006. [DOI] [PubMed] [Google Scholar]
- 46.Caputa G, Castoldi A, Pearce EJ. Metabolic adaptations of tissue-resident immune cells. Nature Immunology. 2019;20(7):793–801. doi: 10.1038/s41590-019-0407-0. [DOI] [PubMed] [Google Scholar]
- 47.Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, Muir AI, Wigglesworth MJ, Kinghorn I, Fraser NJ, et al. The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids *. J Biol Chem. 2003;278(13):11312–11319. doi: 10.1074/jbc.M211609200. [DOI] [PubMed] [Google Scholar]
- 48.Mezö C, Dokalis N, Mossad O, Staszewski O, Neuber J, Yilmaz B, Schnepf D, de Agüero MG, Ganal-Vonarburg SC, Macpherson AJ, et al. Different effects of constitutive and induced microbiota modulation on microglia in a mouse model of Alzheimer’s disease. Acta Neuropathol Commun. 2020;8(1):119. doi: 10.1186/s40478-020-00988-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng T-C, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493(7434):674–678. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baik SH, Kang S, Lee W, Choi H, Chung S, Kim J-I, Mook-Jung I. A breakdown in metabolic reprogramming causes microglia dysfunction in alzheimer’s disease. Cell Metab. 2019;30(3):493–507.e6. doi: 10.1016/j.cmet.2019.06.005. [DOI] [PubMed] [Google Scholar]
- 51.Shippy DC, Ulland TK. Microglial immunometabolism in alzheimer’s disease. Front Cell Neurosci. 2020;14:563446. doi: 10.3389/fncel.2020.563446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cameron AM, Lawless SJ, Pearce EJ. Metabolism and acetylation in innate immune cell function and fate. Semin Immunol. 2016;28(5):408–416. doi: 10.1016/j.smim.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen L-F, Greene WC. Regulation of distinct biological activities of the NF-kappaB transcription factor complex by acetylation. J Mol Med. 2003;81(9):549–557. doi: 10.1007/s00109-003-0469-0. [DOI] [PubMed] [Google Scholar]
- 54.Chen L, Mu Y, Greene WC. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 2002;21(23):6539–6548. doi: 10.1093/emboj/cdf660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447(7143):407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 56.Lauterbach MA, Hanke JE, Serefidou M, Mangan MSJ, Kolbe -C-C, Hess T, Rothe M, Kaiser R, Hoss F, Gehlen J, et al. Toll-like receptor signaling rewires macrophage metabolism and promotes histone acetylation via ATP-citrate lyase. Immunity. 2019;51(6):997–1011.e7. doi: 10.1016/j.immuni.2019.11.009. [DOI] [PubMed] [Google Scholar]
- 57.Qiu J, Villa M, Sanin DE, Buck MD, O’Sullivan D, Ching R, Matsushita M, Grzes KM, Winkler F, Chang C-H, et al. Acetate promotes T cell effector function during glucose restriction. Cell Rep. 2019;27(7):2063–2074.e5. doi: 10.1016/j.celrep.2019.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Krautkramer KA, Kreznar JH, Romano KA, Vivas EI, Barrett-Wilt GA, Rabaglia ME, Keller MP, Attie AD, Rey FE, Denu JM. Diet-microbiota interactions mediate global epigenetic programming in multiple host tissues. Mol Cell. 2016;64(5):982–992. doi: 10.1016/j.molcel.2016.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Soliman ML, Rosenberger TA. Acetate supplementation increases brain histone acetylation and inhibits histone deacetylase activity and expression. Mol Cell Biochem. 2011;352(1–2):173–180. doi: 10.1007/s11010-011-0751-3. [DOI] [PubMed] [Google Scholar]
- 60.Candido EP, Reeves R, Davie JR. Sodium butyrate inhibits histone deacetylation in cultured cells. Cell. 1978;14(1):105–113. doi: 10.1016/0092-8674(78)90305-7. [DOI] [PubMed] [Google Scholar]
- 61.Vidali G, Boffa LC, Bradbury EM, Allfrey VG. Butyrate suppression of histone deacetylation leads to accumulation of multiacetylated forms of histones H3 and H4 and increased DNase I sensitivity of the associated DNA sequences. Proc Natl Acad Sci U S A. 1978;75(5):2239–2243. doi: 10.1073/pnas.75.5.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Waldecker M, Kautenburger T, Daumann H, Busch C, Schrenk D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J Nutr Biochem. 2008;19(9):587–593. doi: 10.1016/j.jnutbio.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 63.Saiman Y, Shen TCD, Lund PJ, Gershuni VM, Jang C, Patel S, Jung S, Furth EE, Friedman ES, Chau L. Global microbiota-dependent histone acetylation patterns are irreversible and independent of short chain fatty acids. Hepatology. 2021;74(6):3427–3440. doi: 10.1002/hep.32043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rothhammer V, Borucki DM, Tjon EC, Takenaka MC, Chao -C-C, Ardura-Fabregat A, de Lima KA, Gutiérrez-Vázquez C, Hewson P, Staszewski O, et al. Microglial control of astrocytes in response to microbial metabolites. Nature. 2018;557(7707):724–728. doi: 10.1038/s41586-018-0119-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vuong HE, Pronovost GN, Williams DW, Coley EJL, Siegler EL, Qiu A, Kazantsev M, Wilson CJ, Rendon T, Hsiao EY. The maternal microbiome modulates fetal neurodevelopment in mice. Nature. 2020;586(7828):281–286. doi: 10.1038/s41586-020-2745-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kimura I, Miyamoto J, Ohue-Kitano R, Watanabe K, Yamada T, Onuki M, Aoki R, Isobe Y, Kashihara D, Inoue D, et al. Maternal gut microbiota in pregnancy influences offspring metabolic phenotype in mice. Science. 2020;367(6481):eaaw8429. doi: 10.1126/science.aaw8429. [DOI] [PubMed] [Google Scholar]
- 67.Pessa-Morikawa T, Husso A, Kärkkäinen O, Koistinen V, Hanhineva K, Iivanainen A, Niku M. Maternal microbiota-derived metabolic profile in fetal murine intestine, brain and placenta. BMC Microbiol. 2022;22(1):46. doi: 10.1186/s12866-022-02457-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Knopman DS, Amieva H, Petersen RC, Chételat G, Holtzman D, Hyman B, Nixon R, Jones D. Alzheimer disease. Nat Rev Dis Primer. 2021;7:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8(6):595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jack CR, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9(1):119. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 72.Vogt NM, Kerby RL, Dill-McFarland KA, Harding SJ, Merluzzi AP, Johnson SC, Carlsson CM, Asthana S, Zetterberg H, Blennow K, et al. Gut microbiome alterations in Alzheimer’s disease. Sci Rep. 2017;7(1):13537. doi: 10.1038/s41598-017-13601-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ling Z, Zhu M, Yan X, Cheng Y, Shao L, Liu X, Jiang R, Wu S. Structural and Functional dysbiosis of fecal microbiota in chinese patients with alzheimer’s disease. Front Cell Dev Biol. 2021;8, 634069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cattaneo A, Cattane N, Galluzzi S, Provasi S, Lopizzo N, Festari C, Ferrari C, Guerra UP, Paghera B, Muscio C, et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol Aging. 2017;49:60–68. doi: 10.1016/j.neurobiolaging.2016.08.019. [DOI] [PubMed] [Google Scholar]
- 75.Haran JP, Bhattarai SK, Foley SE, Dutta P, Ward DV, Bucci V, McCormick BA. Alzheimer’s Disease microbiome is associated with dysregulation of the anti-inflammatory P-glycoprotein pathway. mBio. 2019;10(3):e00632–19. doi: 10.1128/mBio.00632-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li B, He Y, Ma J, Huang P, Du J, Cao L, Wang Y, Xiao Q, Tang H, Chen S. Mild cognitive impairment has similar alterations as Alzheimer’s disease in gut microbiota. Alzheimers Dement. 2019;15(10):1357–1366. doi: 10.1016/j.jalz.2019.07.002. [DOI] [PubMed] [Google Scholar]
- 77.Liu P, Wu L, Peng G, Han Y, Tang R, Ge J, Zhang L, Jia L, Yue S, Zhou K, et al. Altered microbiomes distinguish Alzheimer’s disease from amnestic mild cognitive impairment and health in a Chinese cohort. Brain Behav Immun. 2019;80:633–643. [DOI] [PubMed] [Google Scholar]
- 78.Wu S, Liu X, Jiang R, Yan X, Ling Z. Roles and mechanisms of gut microbiota in patients with Alzheimer’s Disease. Front Aging Neurosci. 2021;13. doi: 10.3389/fnagi.2021.650047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Minter MR, Zhang C, Leone V, Ringus DL, Zhang X, Oyler-Castrillo P, Musch MW, Liao F, Ward JF, Holtzman DM, et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci Rep. 2016;6(1):30028. doi: 10.1038/srep30028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Minter MR, Hinterleitner R, Meisel M, Zhang C, Leone V, Zhang X, Oyler-Castrillo P, Zhang X, Musch MW, Shen X, et al. Antibiotic-induced perturbations in microbial diversity during post-natal development alters amyloid pathology in an aged APPSWE/PS1ΔE9 murine model of Alzheimer’s disease. Sci Rep. 2017;7(1):10411. doi: 10.1038/s41598-017-11047-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dodiya HB, Frith M, Sidebottom A, Cao Y, Koval J, Chang E, Sisodia SS. Synergistic depletion of gut microbial consortia, but not individual antibiotics, reduces amyloidosis in APPPS1-21 Alzheimer’s transgenic mice. Sci Rep. 2020;10(1):8183. doi: 10.1038/s41598-020-64797-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Harach T, Marungruang N, Duthilleul N, Cheatham V, Mc Coy KD, Frisoni G, Neher JJ, Fåk F, Jucker M, Lasser T, et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci Rep. 2017;7(1):41802. doi: 10.1038/srep41802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dodiya HB, Kuntz T, Shaik SM, Baufeld C, Leibowitz J, Zhang X, Gottel N, Zhang X, Butovsky O, Gilbert JA, et al. Sex-specific effects of microbiome perturbations on cerebral Aβ amyloidosis and microglia phenotypes. J Exp Med. 2019;216(7):1542–1560. doi: 10.1084/jem.20182386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Colombo AV, Sadler, RK, Llovera, G, Singh, V, Roth, S, Heindl, S, Sebastian Monasor, L, Verhoeven, A, Peters, Finn, Parhizkar, Samira, et al. Microbiota-derived short chain fatty acids modulate microglia and promote Aβ plaque deposition. eLife. 2021;10:e59826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Marizzoni M, Cattaneo A, Mirabelli P, Festari C, Lopizzo N, Nicolosi V, Mombelli E, Mazzelli M, Luongo D, Naviglio D, et al. Short-chain fatty acids and lipopolysaccharide as mediators between gut dysbiosis and amyloid pathology in alzheimer’s disease. J Alzheimers Dis. 2020;78(2):683–697. doi: 10.3233/JAD-200306. [DOI] [PubMed] [Google Scholar]
- 86.Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, Schrag A-E, Lang AE. Parkinson disease. Nat Rev Dis Primer. 2017;3(1):1–21. doi: 10.1038/nrdp.2017.13. [DOI] [PubMed] [Google Scholar]
- 87.Braak H, Tredici KD, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197–211. doi: 10.1016/S0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 88.Svensson E, Horváth-Puhó E, Thomsen RW, Djurhuus JC, Pedersen L, Borghammer P, Sørensen HT. Vagotomy and subsequent risk of Parkinson’s disease. Ann Neurol. 2015;78(4):522–529. doi: 10.1002/ana.24448. [DOI] [PubMed] [Google Scholar]
- 89.Lionnet A, Leclair-Visonneau L, Neunlist M, Murayama S, Takao M, Adler CH, Derkinderen P, Beach TG. Does Parkinson’s disease start in the gut? Acta Neuropathol. 2018;135(1):1–12. doi: 10.1007/s00401-017-1777-8. [DOI] [PubMed] [Google Scholar]
- 90.Klann EM, Dissanayake U, Gurrala A, Farrer M, Shukla AW, Ramirez-Zamora A, Mai V, Vedam-Mai V. The gut–brain axis and its relation to parkinson’s disease: a review. Front Aging Neurosci. 2022;13:782082. doi: 10.3389/fnagi.2021.782082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, Challis C, Schretter CE, Rocha S, Gradinaru V, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of parkinson’s disease. Cell. 2016;167(6):1469–1480.e12. doi: 10.1016/j.cell.2016.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Unger MM, Spiegel J, Dillmann K-U, Grundmann D, Philippeit H, Bürmann J, Faßbender K, Schwiertz A, Schäfer K-H. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat Disord. 2016;32:66–72. doi: 10.1016/j.parkreldis.2016.08.019. [DOI] [PubMed] [Google Scholar]
- 93.Aho VTE, Houser MC, Pereira PAB, Chang J, Rudi K, Paulin L, Hertzberg V, Auvinen P, Tansey MG, Scheperjans F. Relationships of gut microbiota, short-chain fatty acids, inflammation, and the gut barrier in Parkinson’s disease. Mol Neurodegener. 2021;16(1):6. doi: 10.1186/s13024-021-00427-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tan AH, Chong CW, Lim S-Y, Yap IKS, Teh CSJ, Loke MF, Song S-L, Tan JY, Ang BH, Tan YQ, et al. Gut microbial ecosystem in Parkinson disease: new clinicobiological insights from multi-omics. Ann Neurol. 2021;89(3):546–559. doi: 10.1002/ana.25982. [DOI] [PubMed] [Google Scholar]
- 95.Chen S-J, Chen -C-C, Liao H-Y, Lin Y-T, Wu Y-W, Liou J-M, Wu M-S, Kuo C-H, Lin C-H. Association of Fecal and plasma levels of short-chain fatty acids with gut microbiota and clinical severity in patients with parkinson disease. Neurology. 2022;98(8):e848–e858. doi: 10.1212/WNL.0000000000013225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wu G, Jiang Z, Pu Y, Chen S, Wang T, Wang Y, Xu X, Wang S, Jin M, Yao Y. Serum short-chain fatty acids and its correlation with motor and non-motor symptoms in Parkinson’s disease patients. BMC Neurol. 2022;22(1):13. doi: 10.1186/s12883-021-02544-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shin C, Lim Y, Lim H, Ahn T-B. Plasma short-chain fatty acids in patients with parkinson’s disease. Mov Disord. 2020;35(6):1021–1027. doi: 10.1002/mds.28016. [DOI] [PubMed] [Google Scholar]
- 98.van Kessel SP, El Aidy S. Bacterial metabolites mirror altered gut microbiota composition in patients with parkinson’s disease. J Park Dis. 2019;9:S359–S370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Filippi M, Bar-Or, A, Piehl, F, Preziosa, P, Solari, A, Vukusic, S, Rocca, M. Multiple sclerosis. Nat Rev Dis Primer. 2018;4:1–27. [DOI] [PubMed] [Google Scholar]
- 100.Olsson T, Barcellos LF, Alfredsson L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol. 2017;13(1):25–36. doi: 10.1038/nrneurol.2016.187. [DOI] [PubMed] [Google Scholar]
- 101.Voet S, Prinz M, van Loo G. Microglia in central nervous system inflammation and multiple sclerosis pathology. Trends Mol Med. 2019;25(2):112–123. doi: 10.1016/j.molmed.2018.11.005. [DOI] [PubMed] [Google Scholar]
- 102.Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, Wekerle H, Krishnamoorthy G. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature. 2011;479(7374):538–541. doi: 10.1038/nature10554. [DOI] [PubMed] [Google Scholar]
- 103.Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci. 2011;108(supplement_1):4615–4622. doi: 10.1073/pnas.1000082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Berer K, Gerdes LA, Cekanaviciute E, Jia X, Xiao L, Xia Z, Liu C, Klotz L, Stauffer U, Baranzini SE. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc Natl Acad Sci. 2017;114(40):10719–10724. doi: 10.1073/pnas.1711233114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rothhammer V, Mascanfroni ID, Bunse L, Takenaka MC, Kenison JE, Mayo L, Chao -C-C, Patel B, Yan R, Blain M, et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med. 2016;22(6):586–597. doi: 10.1038/nm.4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tsaktanis T, Beyer T, Nirschl L, Linnerbauer M, Grummel V, Bussas M, Tjon E, Mühlau M, Korn T, Hemmer B, et al. Aryl hydrocarbon receptor plasma agonist activity correlates with disease activity in progressive MS. Neurol - Neuroimmunol Neuroinflammation. 2021;8(2):e933. doi: 10.1212/NXI.0000000000000933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lenroot RK, Yeung PK. Heterogeneity within autism spectrum disorders: what have we learned from neuroimaging studies? Front Hum Neurosci. 2013;7:733. doi: 10.3389/fnhum.2013.00733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lord C, Brugha T S, Charman T, Cusack J, Dumas G, Frazier T, Jones E, Jones R, Pickles A, State M, et al. Autism spectrum disorder. Nat Rev Dis Primer. 2020;6:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McElhanon BO, McCracken C, Karpen S, Sharp WG. Gastrointestinal symptoms in autism spectrum disorder: a meta-analysis. Pediatrics. 2014;133(5):872–883. doi: 10.1542/peds.2013-3995. [DOI] [PubMed] [Google Scholar]
- 110.Tan Q, Orsso CE, Deehan EC, Kung JY, Tun HM, Wine E, Madsen KL, Zwaigenbaum L, Haqq AM. Probiotics, prebiotics, synbiotics, and fecal microbiota transplantation in the treatment of behavioral symptoms of autism spectrum disorder: a systematic review. Autism Res. 2021;14(9):1820–1836. doi: 10.1002/aur.2560. [DOI] [PubMed] [Google Scholar]
- 111.Koyama R, Ikegaya Y. Microglia in the pathogenesis of autism spectrum disorders. Neurosci Res. 2015;100:1–5. doi: 10.1016/j.neures.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 112.Morgan JT. Abnormal microglial–neuronal spatial organization in the dorsolateral prefrontal cortex in autism. Brain Res. 2012;1456:72–81. doi: 10.1016/j.brainres.2012.03.036. [DOI] [PubMed] [Google Scholar]
- 113.Tetreault NA, Hakeem AY, Jiang S, Williams BA, Allman E, Wold BJ, Allman JM. Microglia in the cerebral cortex in autism. J Autism Dev Disord. 2012;42(12):2569–2584. doi: 10.1007/s10803-012-1513-0. [DOI] [PubMed] [Google Scholar]
- 114.Suzuki K, Sugihara G, Ouchi Y, Nakamura K, Futatsubashi M, Takebayashi K, Yoshihara Y, Omata K, Matsumoto K, Tsuchiya KJ, et al. Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry. 2013;70(1):49–58. doi: 10.1001/jamapsychiatry.2013.272. [DOI] [PubMed] [Google Scholar]
- 115.Gupta S, Ellis SE, Ashar FN, Moes A, Bader JS, Zhan J, West AB, Arking DE. Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat Commun. 2014;5(1):5748. doi: 10.1038/ncomms6748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Velmeshev D, Schirmer L, Jung D, Haeussler M, Perez Y, Mayer S, Bhaduri A, Goyal N, Rowitch DH, Kriegstein AR. Single-cell genomics identifies cell type–specific molecular changes in autism. Science. 2019;364(6441):685–689. doi: 10.1126/science.aav8130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Atladóttir HO, Thorsen P, Østergaard L, Schendel DE, Lemcke S, Abdallah M, Parner ET. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J Autism Dev Disord. 2010;40(12):1423–1430. doi: 10.1007/s10803-010-1006-y. [DOI] [PubMed] [Google Scholar]
- 118.Brown AS. Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev Neurobiol. 2012;72(10):1272–1276. doi: 10.1002/dneu.22024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Malkova NV, Yu CZ, Hsiao EY, Moore MJ, Patterson PH. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav Immun. 2012;26(4):607–616. doi: 10.1016/j.bbi.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Smith SEP, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27(40):10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, Hoeffer CA, Littman DR, Huh JR. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science. 2016;351(6276):933–939. doi: 10.1126/science.aad0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kim S, Kim H, Yim YS, Ha S, Atarashi K, Tan TG, Longman RS, Honda K, Littman DR, Choi GB, et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature. 2017;549(7673):528–532. doi: 10.1038/nature23910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I, Yee BK, Feldon J. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J Neurosci. 2006;26(18):4752. doi: 10.1523/JNEUROSCI.0099-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Garay PA, Hsiao EY, Patterson PH, McAllister AK. Maternal immune activation causes age- and region-specific changes in brain cytokines in offspring throughout development. Brain Behav Immun. 2013;31:54–68. doi: 10.1016/j.bbi.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hameete BC, Fernández-Calleja JMS, de Groot MWGDM, Oppewal TR, Tiemessen MM, Hogenkamp A, de Vries RBM, Groenink L. The poly(I:C)-induced maternal immune activation model; a systematic review and meta-analysis of cytokine levels in the offspring. Brain Behav Immun Health. 2020;11:100192. doi: 10.1016/j.bbih.2020.100192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mattei D, Ivanov A, Ferrai C, Jordan P, Guneykaya D, Buonfiglioli A, Schaafsma W, Przanowski P, Deuther-Conrad W, Brust P, et al. Maternal immune activation results in complex microglial transcriptome signature in the adult offspring that is reversed by minocycline treatment. Transl Psychiatry. 2017;7(5):e1120. doi: 10.1038/tp.2017.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Smolders S, Notter T, Smolders SMT, Rigo J-M, Brône B. Controversies and prospects about microglia in maternal immune activation models for neurodevelopmental disorders. Brain Behav Immun. 2018;73:51–65. doi: 10.1016/j.bbi.2018.06.001. [DOI] [PubMed] [Google Scholar]
- 128.Sharon G, Cruz NJ, Kang D-W, Gandal MJ, Wang B, Kim Y-M, Zink EM, Casey CP, Taylor BC, Lane CJ, et al. Human gut microbiota from autism spectrum disorder promote behavioral symptoms in mice. Cell. 2019;177(6):1600–1618.e17. doi: 10.1016/j.cell.2019.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Needham BD, Adame MD, Serena G, Rose DR, Preston GM, Conrad MC, Campbell AS, Donabedian DH, Fasano A, Ashwood P, et al. Plasma and fecal metabolite profiles in autism spectrum disorder. Biol Psychiatry. 2021;89(5):451–462. doi: 10.1016/j.biopsych.2020.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hsiao EY, McBride S, Hsien S, Sharon G, Hyde E, McCue T, Codelli J, Chow J, Reisman S, Petrosino J, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 2013;155(7):1451–1463. doi: 10.1016/j.cell.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yap CX, Henders AK, Alvares GA, Wood DLA, Krause L, Tyson GW, Restuadi R, Wallace L, McLaren T, Hansell NK, et al. Autism-related dietary preferences mediate autism-gut microbiome associations. Cell. 2021;184(24):5916–5931.e17. doi: 10.1016/j.cell.2021.10.015. [DOI] [PubMed] [Google Scholar]
- 132.Kang D-W, Adams JB, Gregory AC, Borody T, Chittick L, Fasano A, Khoruts A, Geis E, Maldonado J, McDonough-Means S, et al. Microbiota transfer therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: an open-label study. Microbiome. 2017;5(1):10. doi: 10.1186/s40168-016-0225-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kang D-W, Adams JB, Coleman DM, Pollard EL, Maldonado J, McDonough-Means S, Caporaso JG, Krajmalnik-Brown R. Long-term benefit of Microbiota Transfer Therapy on autism symptoms and gut microbiota. Sci Rep. 2019;9(1):5821. doi: 10.1038/s41598-019-42183-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Arrieta M-C, Walter J, Finlay BB. Human microbiota-associated mice: a model with challenges. Cell Host Microbe. 2016;19(5):575–578. doi: 10.1016/j.chom.2016.04.014. [DOI] [PubMed] [Google Scholar]
- 135.Walter J, Armet AM, Finlay BB, Shanahan F. Establishing or exaggerating causality for the gut microbiome: lessons from human microbiota-associated rodents. Cell. 2020;180(2):221–232. doi: 10.1016/j.cell.2019.12.025. [DOI] [PubMed] [Google Scholar]
- 136.Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, Thompson EA, Fraser KA, Rosato PC, Filali-Mouhim A, et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature. 2016;532(7600):512–516. doi: 10.1038/nature17655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Reese TA, Bi K, Kambal A, Filali-Mouhim A, Beura L, Bürger M, Pulendran B, Sekaly R-P, Jameson S, Masopust D, et al. Sequential infection with common pathogens promotes human-like immune gene expression and altered vaccine response. Cell Host Microbe. 2016;19(5):713–719. doi: 10.1016/j.chom.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rosshart SP, Vassallo BG, Angeletti D, Hutchinson DS, Morgan AP, Takeda K, Hickman HD, McCulloch JA, Badger JH, Ajami NJ, et al. Wild mouse gut microbiota promotes host fitness and improves disease resistance. Cell. 2017;171(5):1015–1028.e13. doi: 10.1016/j.cell.2017.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rosshart SP, Herz J, Vassallo BG, Hunter A, Wall MK, Badger JH, McCulloch JA, Anastasakis DG, Sarshad AA, Leonardi I, et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science. 2019;365(6452):eaaw4361. doi: 10.1126/science.aaw4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Leonardi I, Gao IH, Lin W-Y, Allen M, Li XV, Fiers WD, De Celie MB, Putzel GG, Yantiss RK, Johncilla M, et al. Mucosal fungi promote gut barrier function and social behavior via Type 17 immunity. Cell. 2022;185(5):831–846.e14. doi: 10.1016/j.cell.2022.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ye T, Yuan S, Kong Y, Yang H, Wei H, Zhang Y, Jin H, Yu Q, Liu J, Chen S, et al. Effect of probiotic fungi against cognitive impairment in mice via regulation of the fungal microbiota–gut–brain axis. J Agric Food Chem. 2022;70(29):9026–9038. doi: 10.1021/acs.jafc.2c03142. [DOI] [PubMed] [Google Scholar]