

CONSPECTUS
Biologically active peptides are a major growing class of drugs, but their therapeutic potential is constrained by several limitations including bioavailability and poor pharmacokinetics. The attachment of functional groups like lipids has proven to be a robust and effective strategy for improving their therapeutic potential. Biochemical and bioactivity-guided screening efforts have identified the cyanobactins as a large class of ribosomally synthesized and post-translationally modified peptides (RiPPs) that are modified with lipids. These lipids are attached by the F superfamily of peptide prenyltransferase enzymes that utilize 5-carbon (prenylation) or 10-carbon (geranylation) donors. The chemical structures of various cyanobactins initially showed isoprenoid attachments on Ser, Thr, or Tyr. Biochemical characterization of the F prenyltransferases from the corresponding clusters show that the different enzymes have different acceptor residue specificities but are otherwise remarkably sequence tolerant. Hence, these enzymes are well suited for biotechnological applications. The crystal structure of the Tyr O-prenyltransferase PagF reveals that the F enzyme share a domain architecture reminiscent of a canonical ABBA prenyltransferase fold but lacks secondary structural elements necessary to form an enclosed active site. Binding of only either cyclic or linear peptides is sufficient to close the active site to allow for productive catalysis, explaining why these enzymes cannot use isolated amino acids as substrates.
Almost all characterized isoprenylated cyanobactins are modified with 5-carbon isoprenoids. However, chemical characterization demonstrates that the piricyclamides are modified with a 10-carbon geranyl moiety, and in vitro reconstitution of the corresponding PirF shows that the enzyme is a geranyltransferase. Structural analysis of PirF shows an active site nearly identical to that of the PagF prenyltransferase but with a single amino acid substitution. Of note, mutation at this residue in PagF or PirF can completely switch the isoprenoid donor specificity of these enzymes. Recent efforts have resulted in significant expansion of the F family with enzymes identified that can carry out C-prenylations of Trp, N-prenylations of Trp and bis N-prenylations of Arg. Additional genome-guided efforts based on the sequence of F enzymes identify linear cyanobactins that are α-N-prenylated and α-C-methylated by a bifunctional prenyltransferase/methyltransferase fusion, and a bis-α-N- and α-C-prenylated linear peptide. The discovery of these different classes of prenyltransferases with diverse acceptor residue specificities expands the biosynthetic toolkit for enzymatic prenylation of peptide substrates.
In this Account, we review the current knowledge scope of the F family of peptide prenyltransferases, focusing on the biochemical, structure-function, and chemical characterization studies that have been carried out in our laboratories. These enzymes are easily amenable for diversity-oriented synthetic efforts as they can accommodate substrate peptides of diverse sequences and are thus attractive catalysts for use in synthetic biology approaches to generate high-value peptidic therapeutics.
Graphical Abstract
INTRODUCTION
Biologically active peptides represent a major growing class of therapeutics with more than 80 peptides drug that have reached the market and nearly 400–600 drug-candidates that are currently in preclinical studies.5–7 The remarkable potency, selectivity, and low immunogenicity of peptides, along with the ease of production had drawn significant pharmaceutical interest, starting with the commercially successful production of insulin, leuprolide and goserelin in the 1980s.8,9 Given the technological ease of peptide synthesis, the success of biologics and increased investment in research efforts, the market for peptide-based therapeutics will continue to expand.10 However, the therapeutic potential of peptides is constrained by several limitations including low oral bioavailability, poor pharmacokinetics behavior, and low plasma stability of medium and short length peptides. Numerous strategies have been adopted to improve the pharmacological profiles of peptides, such as cyclization, modification of the amide backbone, use of non-natural amino acids, and the attachment of functional groups such as phosphates, sugars and/or lipids.11,12
Lipidation has proven to be a robust and effective strategy for improving the therapeutic potential of peptide therapeutics.13,14 Successful examples of lipidated peptide therapeutics include the cyclic lipopeptide antibiotics daptomycin (Cubicin RF)15 and polymyxin B (Poly-Rx),16 the anti-diabetic glucagon-like peptide-1 analogs liraglutide (Victoza)17 and semaglutide (Ozempic),18 and the insulin detemir (Levemir) (Figure 1).19 The covalent incorporation of lipids onto peptide can dramatically enhances receptor selectivity, and enzymatic stability.20 Lipidation can also enhance bioavailability by promoting the interactions of peptides with the cell membrane, improving uptake,21 allowing for non-specific interactions with serum albumin to reduce renal filtration,22 and affecting the oligomeric state of the peptide to limit proteolytic degradation. Lastly, in contrast to other modifications, such as covalent attachment of polyethylene glycol (PEGylation), lipidation of peptides does not significantly affect in vitro activity.23
Figure 1.
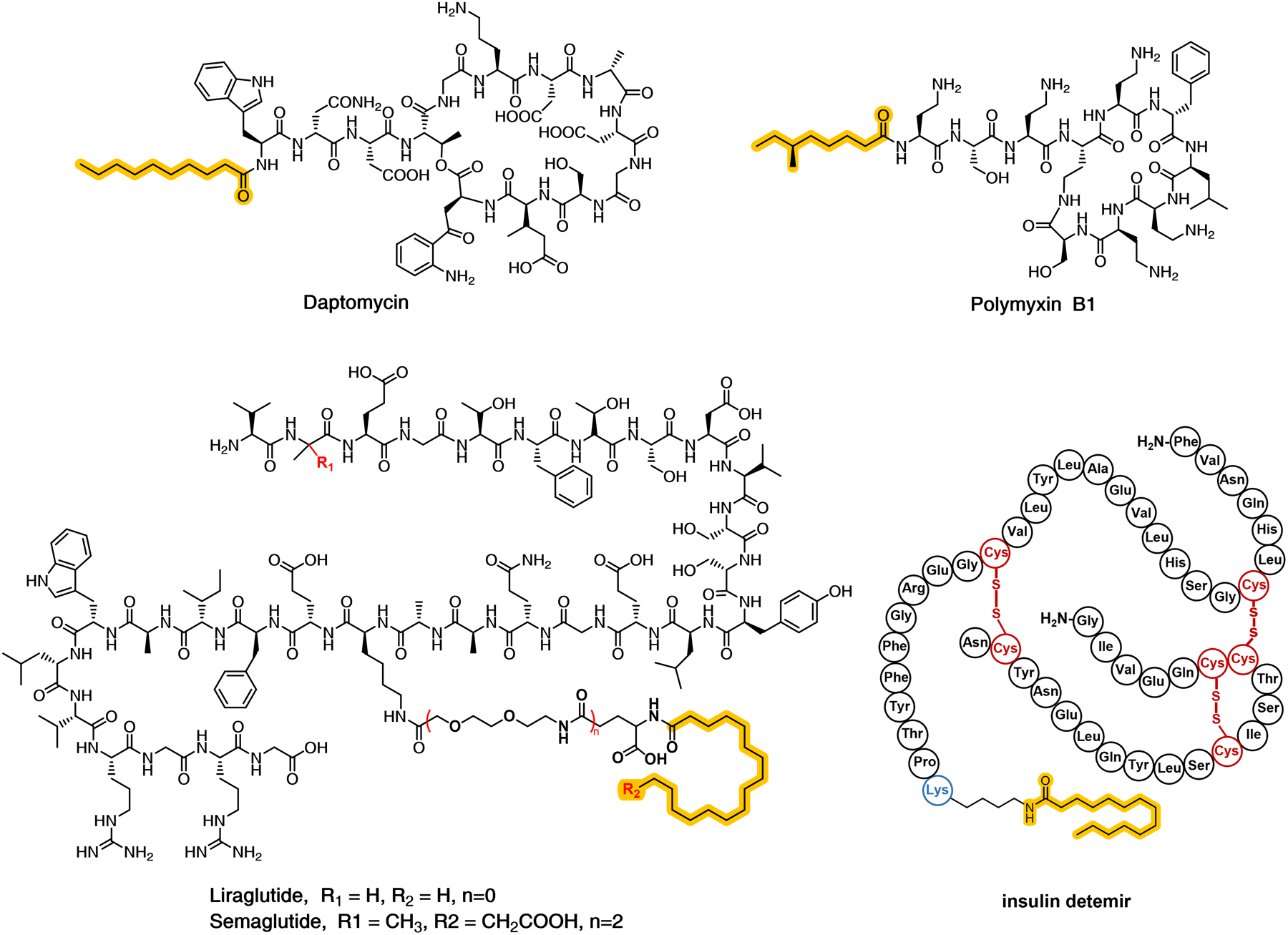
Chemical structures of representative lipid-attached peptides that have been approved for therapeutic use. Each of these compounds is either a natural product or derived from a biological compound. The lipopeptide natural products are commonly produced by a combination of a non-ribosomal peptide synthetase (peptide portion) and a polyketide or fatty acid synthase (lipid component).
Lipidation of peptides and proteins is an established post-translational modification.24 Protein lipidation, including prenylation, myristoylation, and palmitoylation, are known to modulate biological function; for example, the oncogenic activity of members of the RAS superfamily is modulated by prenylation.25 In addition, naturally occurring lipopeptides are widespread among bacteria and fungi, and several characterized samples have known therapeutic potential.26,27 Many such lipopeptides are produced by non-ribosomal peptide synthetases (NRPSs), and demonstrated bioactivities include antifungal (iturins and eichnocandins),28 antimicrobial (surfactins, daptomycin and polymyxin),29 and antineoplastic (kailuins), among others. The lipid is synthesized independently by either the fatty acid synthase (FAS) or by dedicated polyketide synthetases (PKSs).30 Transacylation is catalyzed by a condensation domain that transfers the thioester-activated lipid to a peptide substrate loaded on a peptidyl carrier protein (PCP).31 The substrate scope of the NRPS/PKS or NRPS/FAS systems has not been studied, but the condensation enzymes likely only function on their native substrates.
Biochemical and bioactivity-guided screening efforts also identify several classes of ribosomally derived lipidated peptides that fall into the category of ribosomally synthesized and post-translationally modified peptides (RiPPs) (Figure 2).32,33 The earliest characterized of these ribosomal lipopeptides was the ComX pheromone that mediates intracellular signaling in the Gram-positive bacterium Bacillus subtilis.34 Structural studies of ComX purified from a heterologous expression system demonstrate that the signal is a six-residue peptide that is C3-prenylated on Trp-4, followed by a spontaneous rearrangement that yields a tricyclic structure (Figure 2).35 The absolute stereochemistry was determined using nuclear magnetic resonance and confirmed using a synthetic standard. The membrane-associated prenyltransferase ComQ carries out post-translational modification on the peptide.36 A second example of ribosomally derived lipopeptides are the lipolanthines, such as microvionin, that consist of a lanthipeptide with an N-terminal guanidino fatty acid (Figure 2).37 Reconstitution studies demonstrate that the acyl chain is assembled by a dedicated PKS, and a GNAT-superfamily member catalyzes transfer of an ACP-linked acyl chain onto the modified peptide, analogous to the hybrid NRPS/PKS systems.38 More recently, the selidamides, such as phaeornamide, are identified as RiPPs containing a single lanthionine ring and an acyl chain attached to a lysine residue (Figure 2). Heterologous expression studies suggest that acyl transfer utilizes fatty acid from cellular pools, but this has not been confirmed by in vitro reconstitution studies.39
Figure 2.
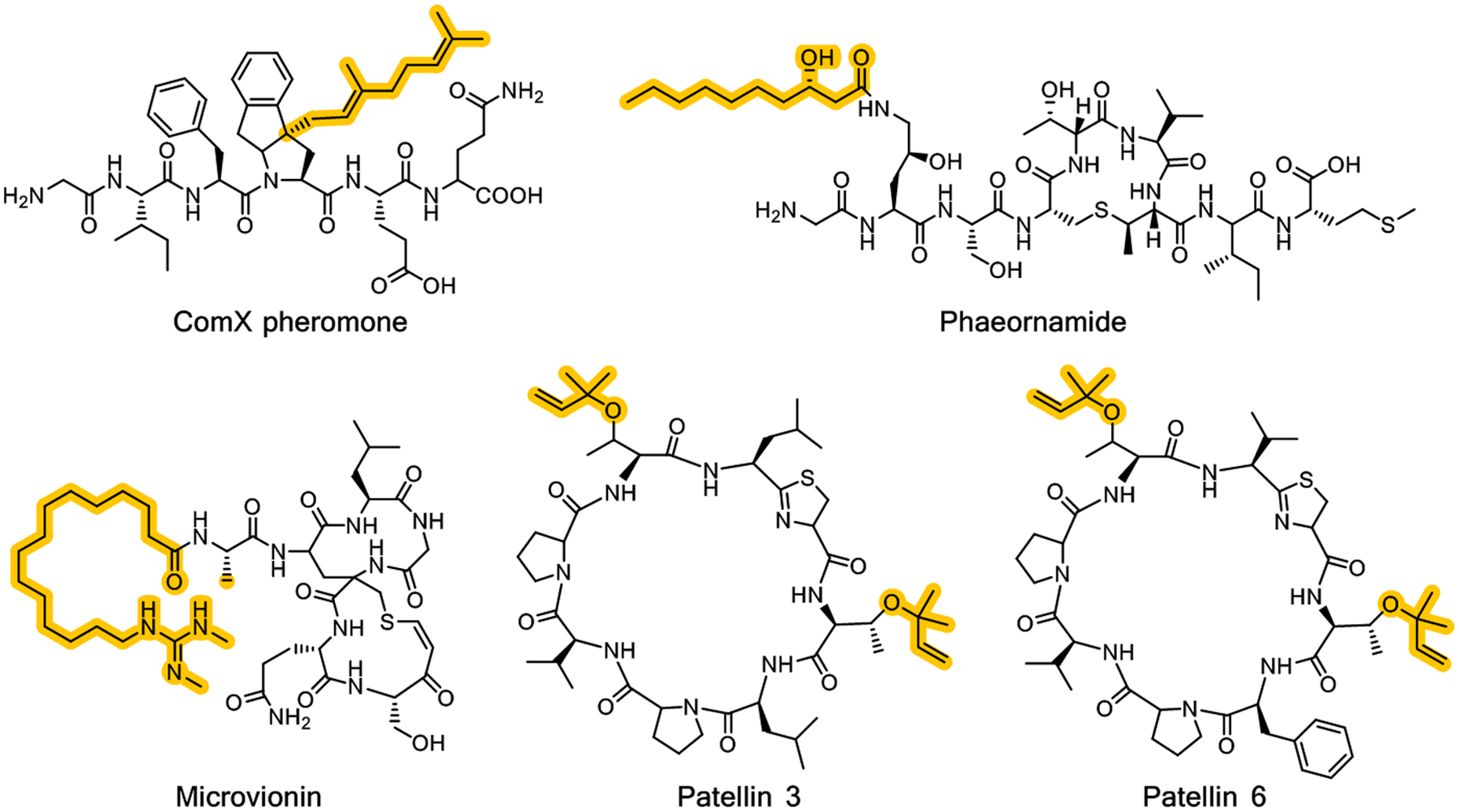
Chemical structures of bioactive lipopeptides that are products of ribosomally synthesized and post-translationally modified peptide (RiPP) pathway. The lipids are derived from isoprenoids or fatty acids and conjugated to the peptide by prenyltransferases or acyltransferases, respectively.
The most widely studied of the ribosomally derived lipopeptides are the cyanobactin class of RiPP natural products (Figure 3).40,41 The first isolated prenylated cyanobactins were the patellins, isolated from the marine invertebrates Lissoclinum patella,42 where they are biosynthesized by obligate symbiont cyanobacteria of the Prochloron genus.43 The patellin structures reveal a cyclic peptide containing a thiazole residue and reverse O-prenylation at Ser or Thr residues. Reconstitution of the patellin biosynthetic cluster in E. coli confirmed that the constituent open reading frames encode all enzymes necessary for the post-translational modification including prenylation.43 A second class of modification observed in cyanobactins is the forward O-prenylation on Tyr, as observed in the cyclic peptides prenylagaramides A and B, isolated from Planktothrix agardhii NIES-596.44 Sequencing and genomic analysis identified a plausible biosynthetic cluster for prenylagaramides production. Chemical analysis of cyclic peptides isolated from the strain confirmed the presence of several products with O-prenylation on Tyr.45
Figure 3.
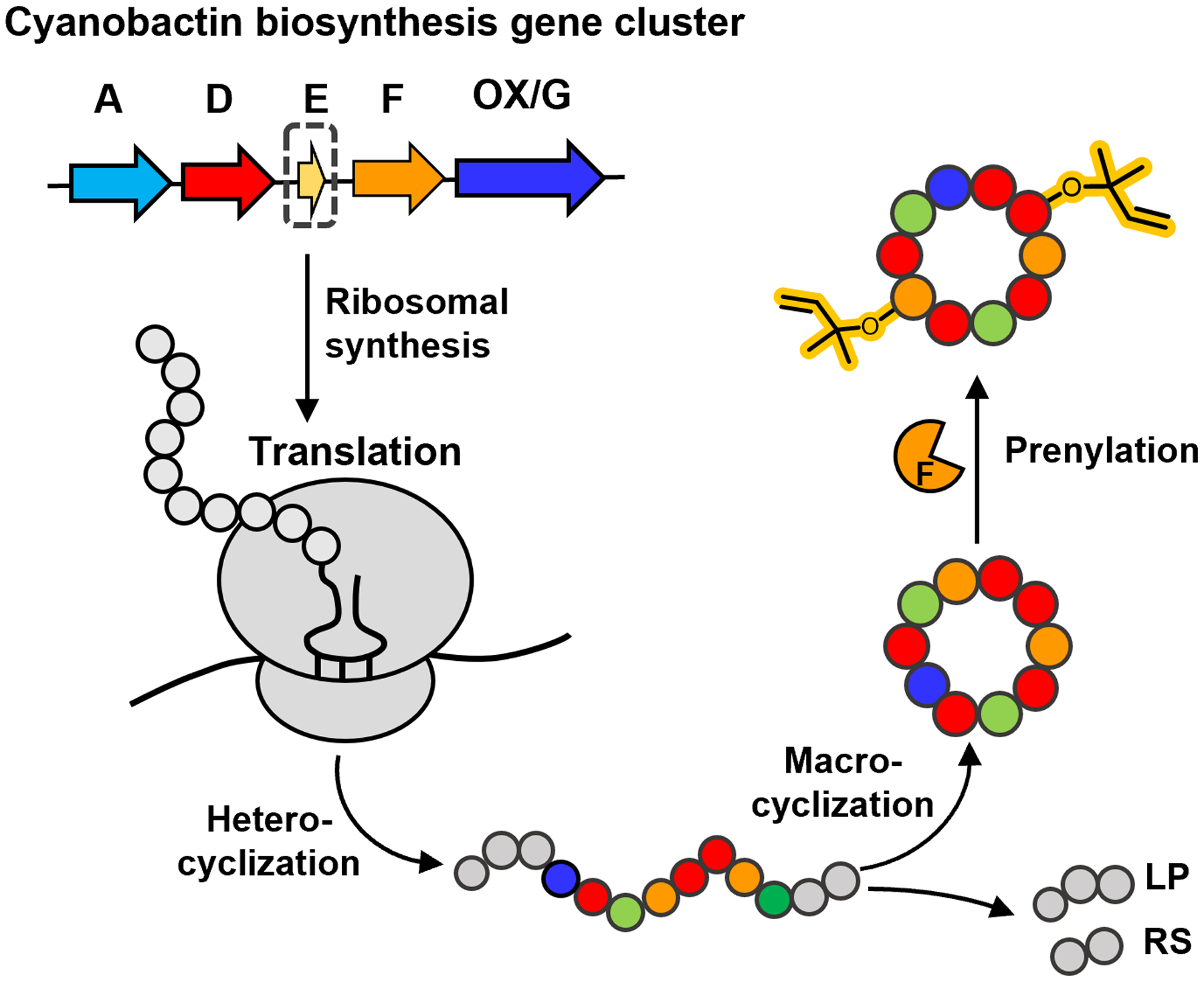
General scheme for the biosynthesis of cyanobactins. The precursor is ribosomally synthesized and consists of a leader sequence (LP), core peptide (CP) and recognition sequence (RS). General post-translational modifications include heterocyclization by enzymes D and OX, proteolysis by enzymes A and G, macrocyclization by enzyme G and prenylation of the modified peptide by enzyme F to elaborate the final product.
THE CYANOBACTIN F CLASS OF RiPP PRENYLTRANSFERASES
Initial efforts to identify the requisite catalyst for isoprene addition in cyanobactin biosynthetic clusters were hindered by the lack of any genes with identifiable sequence signatures to known prenyltransferase classes. Detailed bioinformatics analysis of several putative cyanobactin pathways discovered by metagenomic sequencing identified homologs of truF1 present in all pathways with prenylated products.43 However, neither the TruF1 gene product nor any homologs show sequence similarities to known classes of prenyltransferases that transfer allylic prenyl moieties to small molecule acceptors.1 Recombinantly expressed and purified homolog LynF from Lyngbya aestuarii unequivocally establishes that the enzyme catalyzes prenyl transfer a series of plausible substrates (Figure 4).1 As no known natural products had been identified from this pathway during the time of the study, synthetic substrates corresponding to the core sequence and variants were tested. The LynE precursor peptide contains repeat sequence of ACMPCYP and was devoid of any Ser or Thr residues, which had been previously identified as the sites of prenylation in the patellamides.42 Synthetic substrates utilized Pro residues as mimics for the Cys-derived thiazol(in)e residues.
Figure 4.
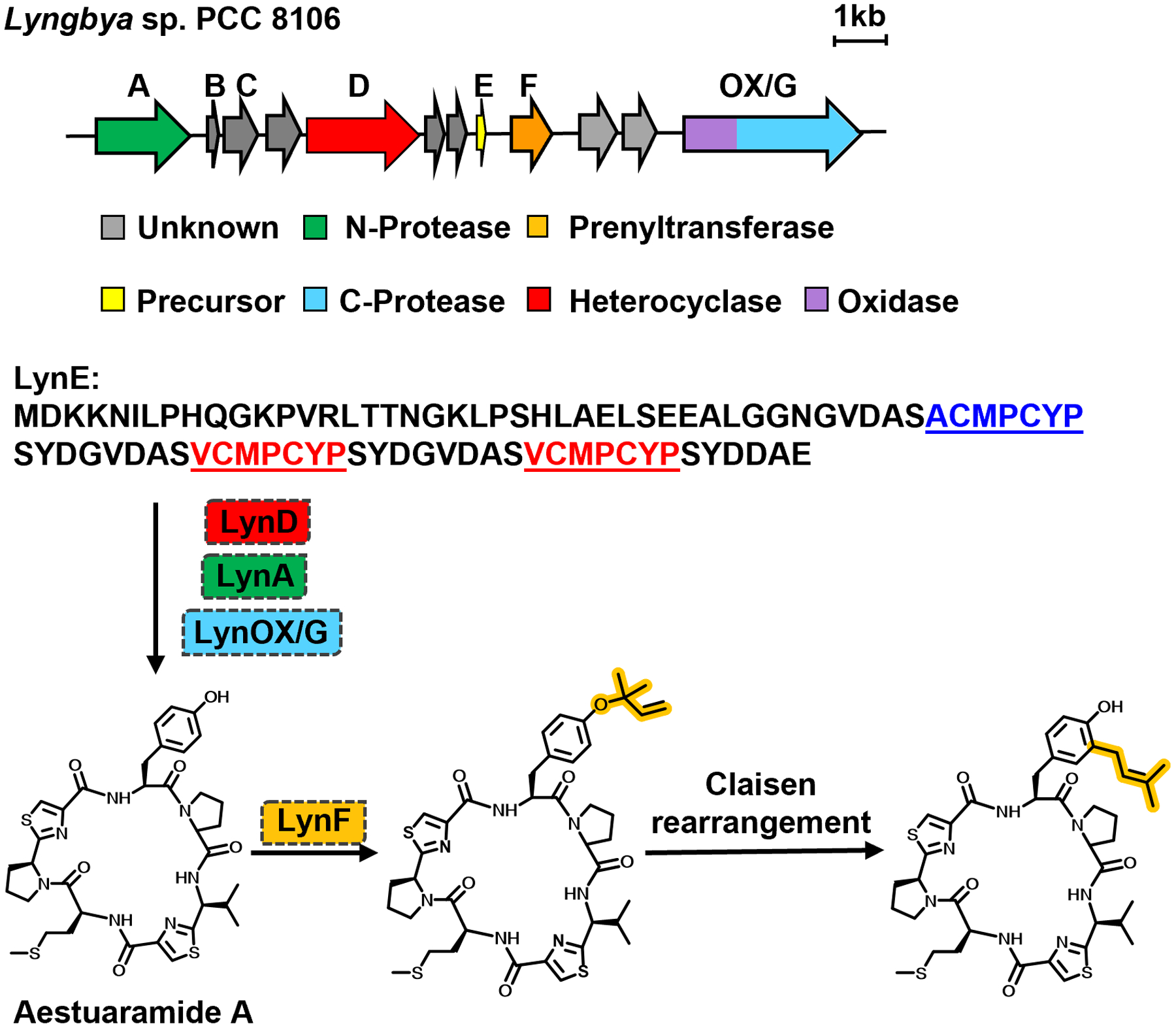
The biosynthetic pathway to aestuaramide A from Lyngbya sp. The precursor peptide contains multiple core sequences that are arranged as cassettes. Heterocyclization and macrocyclization of each core is carried out by the canonical biosynthetic enzymes. Incubation of purified aestuaramide A with recombinant LynF results in O-prenylation on Tyr, which subsequently undergoes a Claisen rearrangement to produce a Tyr forward C-prenylated product.
When incubated with the isoprene donor dimethylallyl pyrophosphate (DMAPP) and magnesium chloride, LynF catalyzed the prenylation on Tyr residues on synthetic peptide substrates (Figure 4).1 Notably, LynF demonstrates tolerance for a broad range of peptide substrates, but prenyl transfer is specific for Tyr and other amino acids are not modified. While LynF was able to effectively prenylate the Tyr residue on cyclic[APMPPYP] and large analogs, it could also process the linear peptide, albeit much less effectively. While L-Tyr itself was not a substrate of LynF, small-molecule phenolics such as N-ter-butyloxycarbonyl (Boc)-L-Tyr and N-acetyl-L-Tyr are prenylated. Structural analysis of LynF modified cyclic[APMPPYP] and N-(Boc)-L-Tyr shows that the final product was forward prenylated on the ortho-carbon of Tyr. More detailed time-course analysis shows that LynF carries out the reverse O-prenylation on phenolics, followed by a non-enzymatic Claisen-type sigmatropic rearrangement to produce the final C-prenylated product (Figure 4).
As noted, the initial biochemical reconstitution studies of LynF were carried out on synthetic peptides that are structural analogs of the presumed natural products. To confirm the role of the F enzymes on bona fide substrates, the thiazol(in)e-containing cyclic peptides aestuaramides were identified by screening cultures of Lyngbya aestuarii PCC 8106.46 Congeners of the aestuaramides include peptides with reverse O-prenylation on Tyr, as well as those without the isoprene moiety. Incubation of the latter peptide with recombinant LynF and the allylic donor DMAPP results in O-prenylation on Tyr. Furthermore, the isolated prenylated peptide underwent a spontaneously rearrangement to yield a stable Tyr C-prenylated species, providing direct evidence for LynF as a peptide prenyltransferase.46 The presence of modified cyclic peptides lacking the isoprene in cell extracts is consistent with isoprenylation as a late-stage post-translational modification that is independent of the leader peptide or recognition sequences.
Subsequently, optimization of heterologous expression conditions facilitated purification of TruF1.47 Reconstitution studies with either defined cyclic peptide substrates or with the precursor peptide and other enzymes from the biosynthetic pathway shows that this enzyme catalyzes the expected reverse O-prenylation on Ser and Thr residues as observed in the isolated natural product pattelins. These studies establish LynF and TruF1 as founding members of the cyanobactin F family peptide prenyltransferases.47 As studies with natural and synthetic substrates conclusively demonstrate that prenylation is a late-stage enzymatic modification, it is likely that the F family enzymes may be used as broadly substrate tolerant biotechnology tools for peptide lipidation. Indeed, in vivo library generation experiments defined the selectivity rules for TruF1 Ser/Thr prenyltransferase,48,49 while saturation mutagenesis using PagF Tyr prenyltransferase defined an exceptionally broad scope for modification of linear and cyclic peptides.50
STRUCTURAL BASIS FOR F ENZYME SUBSTRATE SPECIFICITIES
The first structure of a F family prenyltransferase is that of PagF from Planktothrix agardhii that carries out forward Tyr O-prenylation in the biosynthesis of the prenylagaramides.2 The 1.8 Å resolution structure of PagF consists of 10 antiparallel β-strands surround by 10 α-helices that form a barrel structure (Figure 5). The architecture is similar to those of aromatic prenyltransferases of the ABBA and indole PTase family,51,52 despite the lack of detectable similarities in sequence. Additionally, PagF is roughly two-thirds the size of these ABBA prenyltransferases, and the crystal structure shows that the base of the canonical PTase fold, necessary to form the hydrophobic active site barrel, is absent in the structure of PagF.2 As a result, the active site of PagF is solvent exposed, and this barrel must be enclosed to prevent non-productive quenching of the DMAPP-derived allylic carbocation.
Figure 5.
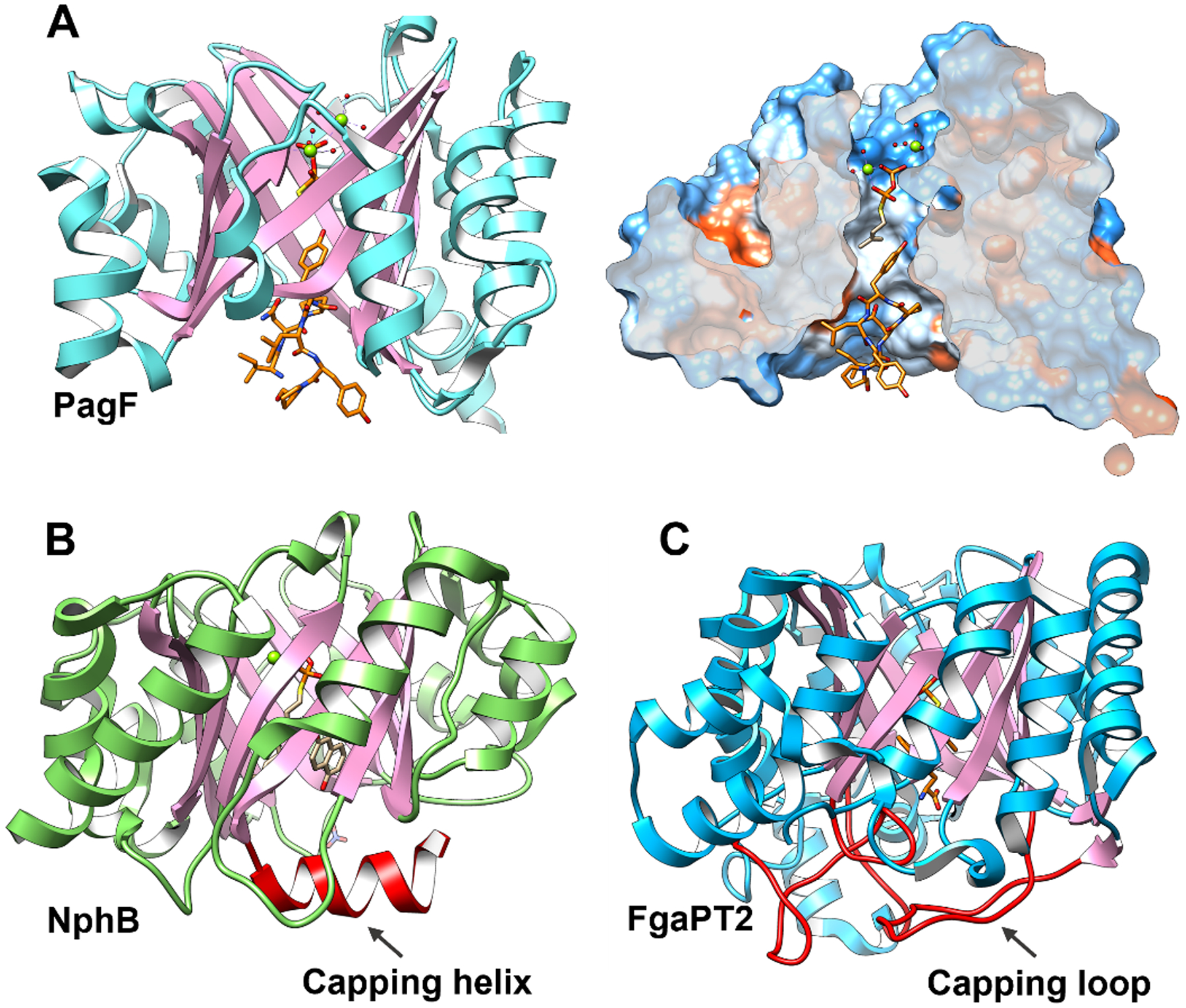
(A) Cocrystal structure of PagF (PDB ID 5TU6) and substrate DMSPP with cyclic[INPYLYP]. A surface cutaway diagram shows that the large cyclic substrate is bound at the base of the active site barrel to plug the solvent-exposed channel. The crystal structure of small molecular prenyltransferases, NphB (B) (PDB ID 1ZCW) and FgaPT2 (C) (PDB ID 3I4X), show solvent-occluded active sites in which the base of hydrophobic barrels is encapsulated by secondary structural elements (colored in red). Helices are colored in cyan (PagF), green (NphB) or blue (FgaPT2) and strands are colored in pink.
Ternary complex structures of PagF bound to the inert prenyl donor analog dimethylallyl S-thiolodiphosphate (DMSPP)/Mg2+ and either N-(Boc)-L-Tyr (2.1 Å resolution), a Tyr-Tyr-Tyr tripeptide (1.9 Å resolution), or cyclic[INPYLYP] (2.2 Å resolution), the latter of which is the physiological substrate of this enzyme50 show substrate binding to a hydrophobic barrel at the base of the protein. Each of these structures show that the peptide substrate binding provides an enclosed hydrophobic cavity that would shield the allylic carbocation from solvent. The amino acid L-Tyr would be too small to encapsulate the solvent-exposed channel, explaining why it is not modified by PagF. By contrast, in structures of canonical ABBA small molecule prenyltransferase such as NphB or FgaPT2, secondary structural elements from the protein, rather than substrate, occlude the base of the respective hydrophobic barrel (Figure 5).53,54
Michaelis-Menten kinetics analysis on PagF using a range of synthetic substrates and the allylic donor yielded turnover numbers consistent with those observed for small molecule prenyltransferases.2 However, the catalytic efficiencies (i.e. kcat/KM) correlate directly with the volume of the substrate with an increase in catalytic efficiency of 4,500-fold for cyclic[INPYLYP] (792 Å3 volume), as compared to that for N-(Boc)-L-Tyr (259 Å3 volume). In particular, the Km values for the substrates tested decrease with the size of the substrate, consistent with the role of the cyclic peptide substrate as a more effective plug of the PagF hydrophobic active site. This is consistent with the observation that F enzymes can function only on cyclic or large peptide substrates and not free amino acids.
A superposition of the cocrystal structures resulted in near identical placement of the isoprene acceptor Tyr, with geometry and distance to the allylic donor favoring forward, rather than reverse O-prenylation. Moreover, the residue following the substrate Tyr is anchored through hydrogen bonding and van der Waals contact in an adjacent hydrophobic patch. These observations suggest that PagF could modify Tyr residues in the context of nearly any substrate provided that the following residue was either aliphatic or aromatic. Two random peptides with a Tyr-Leu-Tyr motif attached at the amino terminus were effectively modified by PagF showing that F enzymes may be used for prenylation on non-native peptide of interest.2
ISOPRENE DONOR SPECIFICITY OF F ENZYMES
Chemical characterization of prenylated cyanobactins, along with subsequent in vitro reconstitution of the corresponding F prenyltransferases, demonstrate that the enzymes exhibit different chemoselectivities for the modified residue.45 Notably, almost all these natural products exclusively contain residues that are modified with C5 isoprenoids and their corresponding F enzyme use DMAPP as the donor. An LC-MS based screening effort of freshwater cyanobacteria to identify peptides with an isoprene unit linked to a heteroatom resulted in the discovery of several new cyanobactins.55 Notably, analysis of extracts of Microcystis aeruginosa PCC 7005 produced a fragmentation pattern consistent with the loss of 136 Da consistent with a C10 geranyl group, rather than the expected 68 Da loss that would occur upon fragmentation of a heteroatom linked to a C5 isoprene. These compounds were termed piricyclamides, and sequencing of the putative biosynthetic cluster identified F family homolog PirF. However, the final natural product could not be obtained in quantities necessary for either identification of the residue that was modified or for complete structure determination.55
In vitro testing of recombinantly purified PirF against a series of N-(Boc) substrates using geranyl pyrophosphate (GPP) as the isoprene donor showed that only N-(Boc)-L-Tyr is modified by the enzyme (Figure 6).3 No activity is observed using DMAPP (C5), farnesyl pyrophosphate (C15) or geranylgeranyl pyrophosphate (C20), indicating that PirF selectively uses the C10-carbon donor GPP. As with PagF, the substrate scope of PirF is broad, and the enzyme could carry out GPP-dependent geranylation on Tyr-Tyr-Tyr tripeptide as well as presumptive substrate cyclic[MSGVDYYNP]. Kinetic characterization using the cyclic and linear substrates shows that both were equally competent substrates. NMR characterization of Tyr-Tyr-Tyr that is in vitro geranylated demonstrates that PirF carries out O-geranylation on Tyr. As the natural substrate contains two Tyr residues, additional characterization shows that the major product (60%) consisted of O-geranylation on the first Tyr. Using the synthetic substrates as standards, more detailed analysis of cyanobacterial extracts shows that the major product (87%) contained a geranyl moiety on the first Tyr, similar to the trend observed with purified enzyme functioning on the synthetic cyclic peptide substrate. PirF represents the first characterized enzyme that can catalyze the forward O-geranylation on a Tyr residue in a peptide substrate.3
Figure 6.
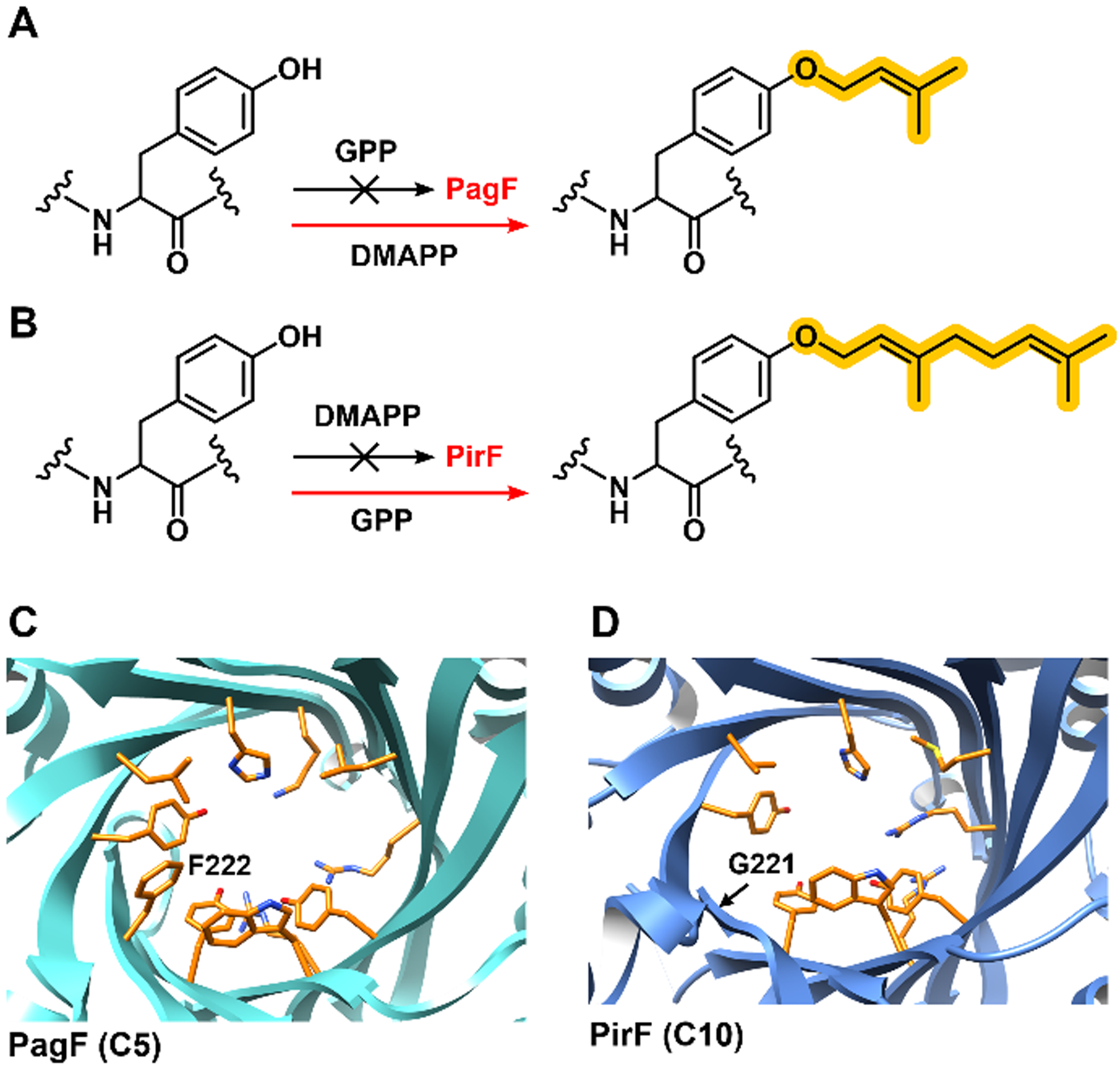
(A) PagF catalyzes transfer of C5 dimethylallyl donor DMAPP on Tyr and cannot transfer C10 isoprenes using GPP. (B) In constrast, PirF (PDB ID 6PGM) carries out C10 transfer from GPP but show no activity using DMAPP. A comparison of the crystal structures of (C) PagF (PDB ID 5TU6) and (D) PirF (PDB ID 6PGM) shows that the distinct difference between the two enzymes is that F222 in the PagF active site is replaced with a G221 in PirF.
The observation that PirF carries out forward O-geranylation on Tyr, while PagF carries out forward O-prenylation on Tyr is notable given that the two enzymes share more than 70% sequence identity. To provide a quantitative context for the respective post-translational modifications, Michael-Menten kinetics were carried out on PagF and PirF using a Tyr-Tyr-Tyr model peptide as substrate.4 As expected PagF carries out DMAPP-dependent prenylation on the substrate with a kcat/KM of 2.7 × 104 M−1 min−1 but shows no activity using GPP. In contrast, PirF carries out GPP-dependent geranylation with a kcat/KM of 5.1 × 104 M−1 min−1 but is inactive using DMAPP as the isoprene donor.
The 2.3 Å resolution crystal structure of PirF shows the expected F family fold and a remarkable near conservation of all residues in the vicinity of the active site with a single exception: Phe222 in the edge of the active site in PagF is replaced with a Gly221 in the structure of PirF creating the necessary cavity to accommodate the longer C10 isoprene donor (Figure 6).4 Remarkably, mutation at this single residue completely switches the specificity for the allylic donor. The PagF F222A and F222G variants are effective C10-geranyltransferases (kcat/KM of 3.9 × 104 M−1 min−1 and 7.4 × 104 M−1 min−1) but with a 10-fold and 30-fold loss in C5-prenyltransferase activity, respectively. Hence, mutations at this residue do not simply expand the scope but rather completely switch tolerance for the isoprene donor. The 1.85 Å resolution cocrystal structure of F222A PagF in complex with GPP and Mg2+ shows that the C10 moiety of GPP extends out of the active site and occupies the cavity created by substitution of the bulky Phe222 with an Ala.4 Presumably, the F222G variant would provide lesser steric hindrance, and may explain the greater geranyltransferase activity of this variant.
CHEMOSELECTIVITY OF F ENZYMES FOR ISOPRENYLATION
In the past few years, studies further characterize F family enzymes with previously unanticipated chemoselectivities, guided by fragmentation-based screening methods and/or genome-mining based on the knowledge of the cyanobactin biosynthetic pathways (Figure 7). While TruF1 was demonstrated to catalyze reverse O-prenylation on Ser and Thr residues in the pattelins, forward Ser/Thr O-prenylation had not been previously identified in any isolated natural product. Bioinformatics analysis identifies the tolF gene in the freshwater cyanobacterium Tolypothrix sp. PCC 7601 and phylogenetic analysis suggests that TolF shares evolutionary relationship with TruF1, despite limited sequence identity (~48%). Large scale isolation of prenylated cyanobactins from this organism yielded tolypamide, a cyclic azolic cyanobactin that is forward O-prenylated on Thr. Heterologously expressed TolF demonstrates Thr forward O-prenyltransferase activity on the native azolic peptide and linear and cyclic peptide analogs.56
Figure 7.
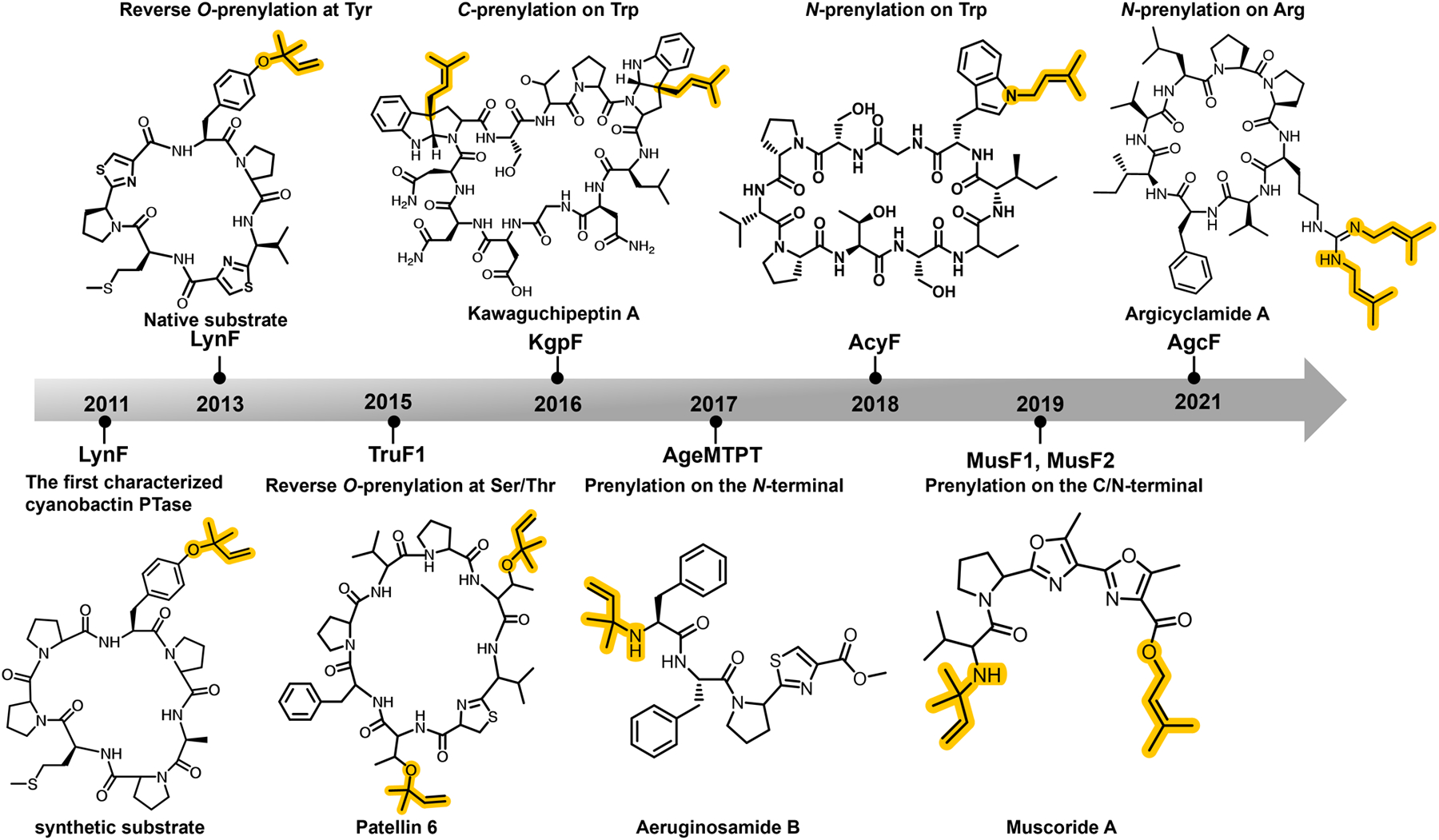
The timeline of the identification and characterization of new cyanobactin prenyltransferases (PTases) and chemical structures of the modified final products.
Nearly three decades ago, the cyclic peptide kawaguchipeptin A from the cyanobacterium Microcystis aeruginosa NIES-88 was isolated in a screening effort, and structural elucidation showed the presence of two isoprene groups attached at C3 on two different Trp residues.57 Decades later, the kawaguchipeptins are shown to be the product of a cyanobactin pathway by genome sequencing and heterologous production in E. coli.58 The corresponding F family prenyltransferase from this cluster, KgpF carries out bis-prenylation on heterologougly produced cyclic[WLNGDNNWSTP] but only modifies a single Trp residue in the linear peptide. Although the aromatic C-prenylated aestuaramides have been characterized, this modification is the result of a spontaneous Claisen condensation following O-prenylation on Tyr. In contrast, aromatic C3-prenylation is the product of a direct post-translational modification by KgpF (Figure 7).58 Tryptophan prenylation at C3 has been reported for oscillatorin as determined by NMR analysis but the enzyme responsible for the modification has not been characterized.59
The C3-prenylation on a peptide Trp substrate is reminiscent of the modification observed in the ComX pheromone produced by B. subtilis and related bacilli.35 To formally assign the configuration of the isoprene attached at C3, fluorenylmethyloxycarbonyl (Fmoc)-L-Tyr was used as substrate surrogate and prenylated using recombinant KgpF.60 Comparison of the enzymatically produced product against synthetic C3 stereoisomeric standards conclusively shows the stereoconfiguration of the modified Trp residue in kawaguchipeptin. Notably, the observed stereochemistry was opposite to that observed for the C3 modification on ComX.60 As the ComX prenyltransferase (ComQ) is not an F family or canonical ABBA type prenyltransferase, the basis for the different stereochemical outcomes has not been determined. The cyclic peptides trikoramides, from Symploca hydnoides, also contain two Trp residues that are C3-prenylated but with a stereochemistry opposite to that observed in the structure of kawaguchipeptin; the biosynthetic gene cluster for this natural product has not been identified.61
Cyanobactins with isoprene modifications of Trp heteroatoms have also been reported. The chemical structure of the croissamide, isolated from the free-living cyanobacterium Symploca sp., reveals a cyclic peptide with a reverse N-prenylation on Trp.62 Although the biosynthetic origin of this natural product has not yet been confirmed, it is very likely a cyanobactin RiPP. A LC-MS-based screening effort of extracts from filamentous cyanobacteria of the genus Anabaena identified cyclic peptides termed anacyclamides in 27 strains, some of which were prenylated.63 However, insufficient production levels precluded structural determination of any of the anacyclamides. Subsequent heterologous production of the biosynthetic cluster in E. coli and in vitro studies of the AcyF prenyltransferase conclusively demonstrates that the enzyme catalyzes forward N-prenylation on Trp (Figure 7).64 No activity was observed for AcyF when GPP was used as the isoprenoid donor. The rationale for the difference in chemo- and regio-specificities between AcyF (N-prenylation on Trp) and KgpF (aromatic C3-prenylation on Trp) is not yet been determined.
Screening efforts using LC-MS profiling of extracts of Microcystis aeruginosa NIES-88 (the producing strain of kawaguchipeptins) identifies ion peaks corresponding to masses that differ by one and two multiples of 68 atomic mass units, which is consistent with singly and doubly prenylated products.65 Large-scale isolation and chemical analysis of the product is consistent with a cyclic peptide with the attachment of either one or two isoprene moieties on Arg. Precise sequencing of M. aeruginosa NIES-88 identified a precursor peptide (AgcE1) with core sequencing corresponding to the final product but absent of any syntenic biosynthetic genes. A second putative precursor (AgcE2) was identified near a previously uncharacterized F family enzyme and named AgcF but without the biosynthetic proteases that are necessary for macrocyclication.66–68 Heterologous expression studies reveal that the precursor peptide is macrocyclized by the kawaguchipeptin biosynthetic machinery and reconstitution studies show that AgcF catalyzes the bis-N-prenylation on the cyclic product (Figure 7). Moreover, AgcF could also utilize GPP as a isoprenoid donor, in contrast to other characterized F enzymes that show strict donor specificity.65 While guanidine prenylation has been previously identified in alkaloids69 and Arg prenylation has been observed in the natural products microguanidine and aeruginoguanidines,70 these studies are the first to establish a biosynthetic origin for this modification.
Genome mining using the sequences of the two biosynthetic proteases common amongst all cyanobactin biosynthetic clusters identified putative biosynthetic clusters in Microcystis and Oscillatoria that contained precursor peptide predicted to produce final produces of chains lengths between three to five amino acids each.71 Chemical screening of extracts from representative cyanobacterial species identified prenylated peptides with fragmentation patterns that corresponded to the bioinformatics prediction. Detailed structural analysis demonstrated that the linear cyanobactins are short peptides that contain a thioazole, methyl esterification of the carboxy terminus and reverse α-N-prenylation at the amino terminus.71 These peptides were named aeruginosamide B and C as they are structurally related to a linear tetrapeptide that had previously been isolated and characterized but of unknown biosynthetic origins.72 The linear RiPP peptides expand the chemical diversity of cyanobactins and point to the likelihood of ribosomal biosynthetic origin for other linear peptides that had previously been characterized.
Sequence analysis of the aeruginosamide biosynthetic gene cluster identified a single open reading frame that showed sequence similarities to the F family enzymes but with an amino terminal fusion of a S-adenosylmethionine (SAM) dependent methyltransferase domain. Reconstitution studies demonstrated that this AgeMTPT fusion catalyzed both the α-N-prenylation at the amino terminus and methylation of the carboxy terminus (Figure 7).73 The order of the biosynthetic steps was firmly established using purified enzymes and synthetic substrate and showed that the prenyltransferase domain could modify several biosynthetic intermediates, but the methyltransferase domain could only function on the thioazoline containing core peptide. Lastly, unlike macrocylase enzymes from canonical cyanobactin biosynthetic pathways, the homologous AgeG produced linear products.73
The peptide alkaloid muscoride A is produced by the freshwater cyanobacterium Nostoc muscorum IAM M-14, and consists of a bis-methyloxazole residues and prenylation at both the amino and carboxy terminus.74 The amino terminus is modified by a reverse α-N-prenylation while the carboxy terminus is modified by forward α-O-prenylation. The presence of the bis-oxazole has drawn interest from various total synthesis efforts,75–77 but the biosynthetic origins had only recently been elucidated. Chemical screening of extracts of N. muscorum PCC 7906 identified a mass consistent with that of muscoride A.78 Mining of the sequenced genome using the F family sequences as a probe identified a plausible cyanobactin-like biosynthetic pathway. The validity of the biosynthetic cluster was established by heterologous production in E. coli and comparison against the isolated natural product as a standard. Notably, the biosynthetic pathway contains two F enzymes that show 66% sequence identity. Heterologous production of the cluster with deletion of either or both prenyltransferases showed that MusF1 catalyzed the forward α-O-prenylation at the carboxy terminus, and MusF2 catalyzed only the reverse α-N-prenylation at the amino terminus (Figure 7).78 The enzymes seemingly functioned independently in the in vivo production assays, but this has not been biochemically confirmed. Remarkably, despite sharing 66% sequence identity, the two homologs demonstrate different and strict regio- and chemo-specificities. The basis for these preferences has yet to be determined.
IMPLICATIONS FOR PEPTIDE DIVERSIFICATION AND THERAPEUTIC DESIGN
The broad tolerance of F enzymes for isoprenylation of peptide substrates regardless of sequence suggests that they may have utility in biotechnology, and any such use would benefit from detailed knowledge of the acceptor residue chemoselectivities of each F enzymes. To this end, we use the Enzyme Similarity Tool from the Enzyme Function Initiative (EFI-EST)79 to generate a sequence similarity network for the F homologs of known sequences from UniProtKB database (IPR031037). At an alignment score threshold of 86 (~53% sequence identity), the sequences cluster together with respect to their expected chemoselectivities, based on the functions of known enzymes (Figure 8). While the stereochemistry of the final product cannot be discerned by bioinformatics, the utility of the tools such as the EFI-EST should facilitate rapid identification of novel catalysts in newly sequence cyanobacterial genomes for further in vitro efforts.
Figure 8.
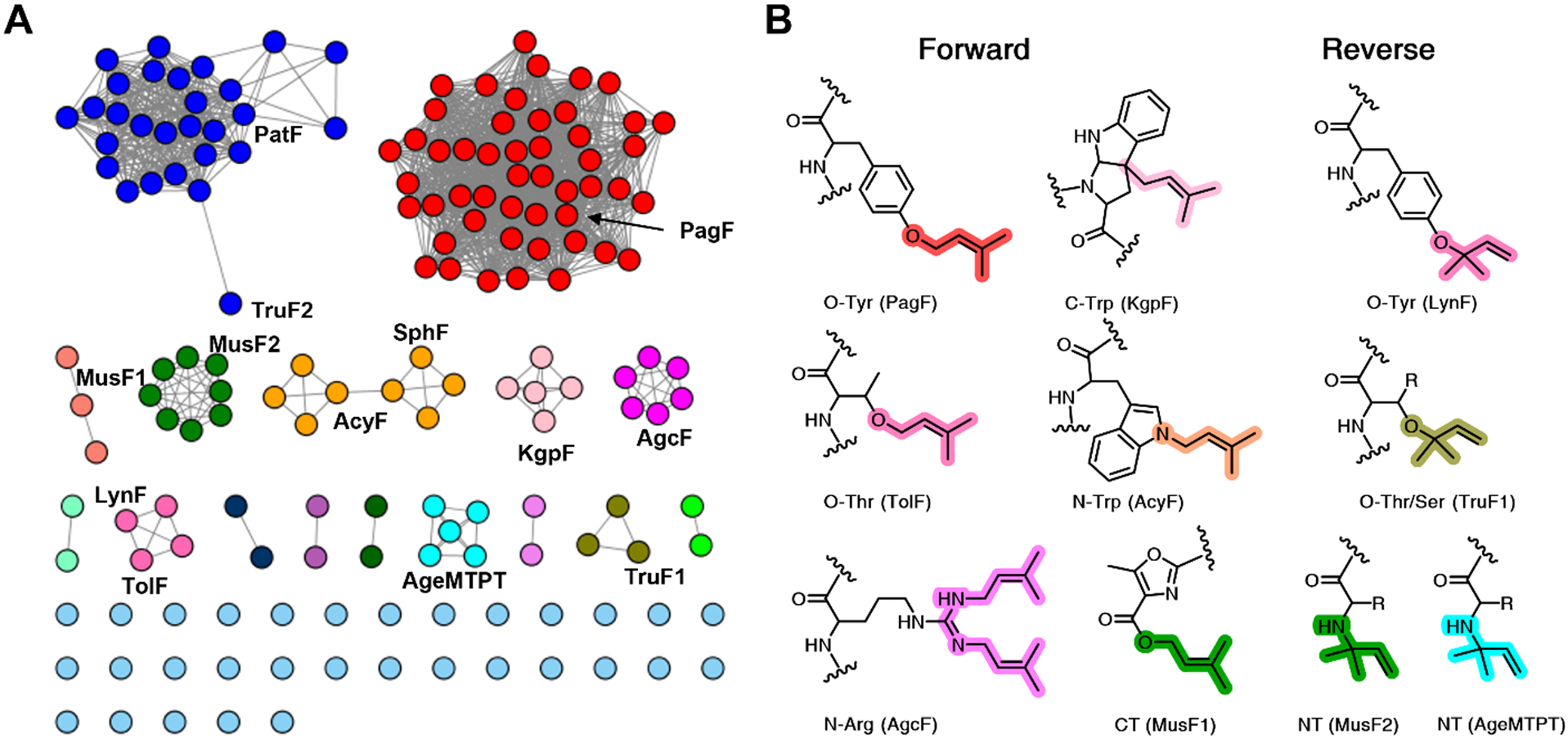
(A) Sequence similarity network of F family enzymes generated using InterPro IPR031037 to query the EFI-EST tools and visualized in Cytoscape. Nodes are clustered with an alignment score threshold of 86 (~53% sequence identity). At this threshold, the sequences cluster together according to their expected chemoselectivities. Sequences which do not cluster with any other are shown as cyan circles. (B) Known types of post-translational prenylation shown by the residue chemospecificity, along with the names of the enzymes that install these modifications.
These enzymes are easily amenable for diversity-oriented synthetic efforts and are thus attractive catalysts for use in synthetic biology approaches to generate high-value peptidic therapeutics.48,50 Moreover, simple mutational changes can alter isoprene donor specificity and further expand the scope of biocatalysts available for synthetic efforts.4 We anticipate that knowledge of structure-function relationships among F enzymes will benefit future studies in which these catalysts are deployed in the production of large libraries of lipidated peptide products or in directing the synthesis of bioactive compounds with desired modifications.
ACKNOWLEDGEMENTS
This work was supported by NIH NIGMS grants GM131347 to S.K.N. and GM122521 to E.W.S.
Biographies
Yiwu Zheng received his Ph.D. in Chemistry from Xiamen University in the laboratory of Prof. Chuanliu Wu. He joined the Department of Biochemistry at the University of Illinois as a post-doctoral researcher in the Prof. Satish K. Nair’s laboratory. His research interest is in understanding the mechanism of natural product biosynthesis, particularly those that fall into the class of ribosomally synthesized and post-translationally modified peptides.
Ying Cong received her bachelor’s degree from Tianjin University. She joined the Department of Medicinal Chemistry at the University of Utah as a Ph.D. student in Prof. Eric W. Schmidt’s laboratory. Her research mainly focuses on enzyme modification and protein engineering of natural products, and particularly on ribosomally synthesized and post-translationally modified peptide biosynthesis.
Eric W. Schmidt holds the William Droschkey Endowed Chair in Pharmacy at the University of Utah and is Distinguished Professor of Medicinal Chemistry. After receiving a B.S. in Chemistry from the University of California, San Diego, he trained in marine natural products chemistry and biology with the late D. John Faulkner at the Scripps Institution of Oceanography (UCSD). He went on to a postdoctoral fellowship focused on fungal natural products biosynthesis in the laboratory of Craig A. Townsend in the Department of Chemistry at The Johns Hopkins University. Schmidt has been on the faculty at the University of Utah since 2001.
Satish K. Nair holds the Gregorio Weber Chair and is the Head of the Department of Biochemistry at the University of Illinois Urbana-Champaign. He obtained his ScB (with Honors) from Brown University in Chemistry and his Ph.D. in the Department of Chemistry at the University of Pennsylvania where he trained under David W. Christianson. Following a Leukemia Society of America post-doctoral fellowship at Rockefeller University (with Stephen K. Burley), he began his independent academic career at the University of Illinois in 2001. At Illinois, Nair also serves as the Director for the Center for Biophysics and Quantitative Biology and is the Co-Director for the Center for Macromolecular CryoEM.
REFERENCES
- (1).McIntosh JA; Donia MS; Nair SK; Schmidt EW Enzymatic basis of ribosomal peptide prenylation in cyanobacteria. J Am Chem Soc 2011, 133, 13698–13705. [DOI] [PMC free article] [PubMed] [Google Scholar]; This work describes the first biochemical reconstitution studies demonstrating that the F family enzymes catalyze prenylation on peptide substrates. LynF carries out reverse O-prenylation on Tyr, which then undergoes a spontaneous Claisen rearrangement to yield an aromatic C-prenylated product.
- (2).Hao Y; Pierce E; Roe D; Morita M; McIntosh JA; Agarwal V; Cheatham TE 3rd; Schmidt EW; Nair SK Molecular basis for the broad substrate selectivity of a peptide prenyltransferase. Proc Natl Acad Sci U S A 2016, 113, 14037–14042. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes the first crystal structure of an F family enzyme, PagF that catalyzes forward O-prenylation on Tyr. The structural and biochemical data provide a rationale for why these enzymes work only on peptide substrates and not on isolated amino acids.
- (3).Morita M; Hao Y; Jokela JK; Sardar D; Lin Z; Sivonen K; Nair SK; Schmidt EW Post-translational tyrosine geranylation in cyanobactin biosynthesis. J Am Chem Soc 2018, 140, 6044–6048. [DOI] [PMC free article] [PubMed] [Google Scholar]; In this paper, PirF is shown to catalyze GPP-dependent O-geranylation on a peptide Tyr, which is a new enzymatic post-translational modification. Chemical characterization of piricyclamides 7005E1, along with comparisons of enzymatically synthesized standards establish the regioselectivity of the enzyme.
- (4).Estrada P; Morita M; Hao Y; Schmidt EW; Nair SK A single amino acid switch alters the isoprene donor specificity in ribosomally synthesized and post-Translationally modified peptide prenyltransferases. J Am Chem Soc 2018, 140, 8124–8127. [DOI] [PMC free article] [PubMed] [Google Scholar]; Replacement of a single residue near the active site is shown to switch the isoprenoid donor specificity of PirF (a geranyltransferase) and PagF (a prenytransferase). Structural analysis of the PagF variant shows how the mutation facilitates GPP binding.
- (5).Fosgerau K; Hoffmann T Peptide therapeutics: current status and future directions. Drug Discov Today 2015, 20, 122–128. [DOI] [PubMed] [Google Scholar]
- (6).Kaspar AA; Reichert JM Future directions for peptide therapeutics development. Drug Discov Today 2013, 18, 807–817. [DOI] [PubMed] [Google Scholar]
- (7).Kowalczyk R; Harris PWR; Williams GM; Yang SH; Brimble MA Peptide Lipidation - A synthetic strategy to afford peptide based therapeutics. Adv Exp Med Biol 2017, 1030, 185–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lau JL; Dunn MK Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg Med Chem 2018, 26, 2700–2707. [DOI] [PubMed] [Google Scholar]
- (9).Muttenthaler M; King GF; Adams DJ; Alewood PF Trends in peptide drug discovery. Nat Rev Drug Discov 2021, 20, 309–325. [DOI] [PubMed] [Google Scholar]
- (10).Nestor JJ Jr. The medicinal chemistry of peptides. Curr Med Chem 2009, 16, 4399–4418. [DOI] [PubMed] [Google Scholar]
- (11).Adessi C; Soto C Converting a peptide into a drug: strategies to improve stability and bioavailability. Curr Med Chem 2002, 9, 963–978. [DOI] [PubMed] [Google Scholar]
- (12).Gentilucci L; De Marco R; Cerisoli L Chemical modifications designed to improve peptide stability: incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization. Curr Pharm Des 2010, 16, 3185–3203. [DOI] [PubMed] [Google Scholar]
- (13).Rounds T; Straus SK Lipidation of antimicrobial peptides as a design strategy for future alternatives to antibiotics. Int J Mol Sci 2020, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zhang L; Bulaj G Converting peptides into drug leads by lipidation. Curr Med Chem 2012, 19, 1602–1618. [DOI] [PubMed] [Google Scholar]
- (15).Baltz RH Daptomycin: mechanisms of action and resistance, and biosynthetic engineering. Curr Opin Chem Biol 2009, 13, 144–151. [DOI] [PubMed] [Google Scholar]
- (16).Kadar B; Kocsis B; Nagy K; Szabo D The renaissance of polymyxins. Curr Med Chem 2013, 20, 3759–3773. [DOI] [PubMed] [Google Scholar]
- (17).Jackson SH; Martin TS; Jones JD; Seal D; Emanuel F Liraglutide (victoza): the first once-daily incretin mimetic injection for type-2 diabetes. P T 2010, 35, 498–529. [PMC free article] [PubMed] [Google Scholar]
- (18).Knudsen LB; Lau J The discovery and development of liraglutide and semaglutide. Front Endocrinol (Lausanne) 2019, 10, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Tibaldi JM Evolution of insulin development: focus on key parameters. Adv Ther 2012, 29, 590–619. [DOI] [PubMed] [Google Scholar]
- (20).Menacho-Melgar R; Decker JS; Hennigan JN; Lynch MD A review of lipidation in the development of advanced protein and peptide therapeutics. J Control Release 2019, 295, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Epand RM Biophysical studies of lipopeptide-membrane interactions. Biopolymers 1997, 43, 15–24. [DOI] [PubMed] [Google Scholar]
- (22).van Witteloostuijn SB; Pedersen SL; Jensen KJ Half-Life extension of biopharmaceuticals using chemical methods: alternatives to PEGylation. ChemMedChem 2016, 11, 2474–2495. [DOI] [PubMed] [Google Scholar]
- (23).Knudsen LB; Nielsen PF; Huusfeldt PO; Johansen NL; Madsen K; Pedersen FZ; Thogersen H; Wilken M; Agerso H Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J Med Chem 2000, 43, 1664–1669. [DOI] [PubMed] [Google Scholar]
- (24).Resh MD Covalent lipid modifications of proteins. Curr Biol 2013, 23, R431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wang M; Casey PJ Protein prenylation: unique fats make their mark on biology. Nat Rev Mol Cell Biol 2016, 17, 110–122. [DOI] [PubMed] [Google Scholar]
- (26).Cochrane SA; Vederas JC Lipopeptides from Bacillus and Paenibacillus spp.: A Gold Mine of Antibiotic Candidates. Med Res Rev 2016, 36, 4–31. [DOI] [PubMed] [Google Scholar]
- (27).Raaijmakers JM; De Bruijn I; Nybroe O; Ongena M Natural functions of lipopeptides from Bacillus and Pseudomonas: more than surfactants and antibiotics. FEMS Microbiol Rev 2010, 34, 1037–1062. [DOI] [PubMed] [Google Scholar]
- (28).Ramachandran R; Shrivastava M; Narayanan NN; Thakur RL; Chakrabarti A; Roy U Evaluation of antifungal efficacy of three new cyclic lipopeptides of the class bacillomycin from Bacillus subtilis RLID 12.1. Antimicrob Agents Chemother 2018, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Jujjavarapu SE; Dhagat S; Kurrey V Identification of Novel Ligands for Therapeutic Lipopeptides: Daptomycin, Surfactin and Polymyxin. Curr Drug Targets 2018, 19, 1589–1598. [DOI] [PubMed] [Google Scholar]
- (30).Robbins T; Liu YC; Cane DE; Khosla C Structure and mechanism of assembly line polyketide synthases. Curr Opin Struct Biol 2016, 41, 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Gotze S; Stallforth P Structure elucidation of bacterial nonribosomal lipopeptides. Org Biomol Chem 2020, 18, 1710–1727. [DOI] [PubMed] [Google Scholar]
- (32).Arnison PG; Bibb MJ; Bierbaum G; Bowers AA; Bugni TS; Bulaj G; Camarero JA; Campopiano DJ; Challis GL; Clardy J; Cotter PD; Craik DJ; Dawson M; Dittmann E; Donadio S; Dorrestein PC; Entian KD; Fischbach MA; Garavelli JS; Goransson U; Gruber CW; Haft DH; Hemscheidt TK; Hertweck C; Hill C; Horswill AR; Jaspars M; Kelly WL; Klinman JP; Kuipers OP; Link AJ; Liu W; Marahiel MA; Mitchell DA; Moll GN; Moore BS; Muller R; Nair SK; Nes IF; Norris GE; Olivera BM; Onaka H; Patchett ML; Piel J; Reaney MJ; Rebuffat S; Ross RP; Sahl HG; Schmidt EW; Selsted ME; Severinov K; Shen B; Sivonen K; Smith L; Stein T; Sussmuth RD; Tagg JR; Tang GL; Truman AW; Vederas JC; Walsh CT; Walton JD; Wenzel SC; Willey JM; van der Donk WA Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat Prod Rep 2013, 30, 108–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Montalban-Lopez M; Scott TA; Ramesh S; Rahman IR; van Heel AJ; Viel JH; Bandarian V; Dittmann E; Genilloud O; Goto Y; Grande Burgos MJ; Hill C; Kim S; Koehnke J; Latham JA; Link AJ; Martinez B; Nair SK; Nicolet Y; Rebuffat S; Sahl HG; Sareen D; Schmidt EW; Schmitt L; Severinov K; Sussmuth RD; Truman AW; Wang H; Weng JK; van Wezel GP; Zhang Q; Zhong J; Piel J; Mitchell DA; Kuipers OP; van der Donk WA New developments in RiPP discovery, enzymology and engineering. Nat Prod Rep 2021, 38, 130–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Shank EA; Kolter R Extracellular signaling and multicellularity in Bacillus subtilis. Curr Opin Microbiol 2011, 14, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Okada M; Sato I; Cho SJ; Iwata H; Nishio T; Dubnau D; Sakagami Y Structure of the Bacillus subtilis quorum-sensing peptide pheromone ComX. Nat Chem Biol 2005, 1, 23–24. [DOI] [PubMed] [Google Scholar]
- (36).Magnuson R; Solomon J; Grossman AD Biochemical and genetic characterization of a competence pheromone from B. subtilis. Cell 1994, 77, 207–216. [DOI] [PubMed] [Google Scholar]
- (37).Wiebach V; Mainz A; Siegert MJ; Jungmann NA; Lesquame G; Tirat S; Dreux-Zigha A; Aszodi J; Le Beller D; Sussmuth RD The anti-staphylococcal lipolanthines are ribosomally synthesized lipopeptides. Nat Chem Biol 2018, 14, 652–654. [DOI] [PubMed] [Google Scholar]
- (38).Kozakai R; Ono T; Hoshino S; Takahashi H; Katsuyama Y; Sugai Y; Ozaki T; Teramoto K; Teramoto K; Tanaka K; Abe I; Asamizu S; Onaka H Acyltransferase that catalyses the condensation of polyketide and peptide moieties of goadvionin hybrid lipopeptides. Nat Chem 2020, 12, 869–877. [DOI] [PubMed] [Google Scholar]
- (39).Hubrich F; Bosch NM; Chepkirui C; Morinaka BI; Rust M; Gugger M; Robinson SL; Vagstad AL; Piel J Ribosomally derived lipopeptides containing distinct fatty acyl moieties. Proc Natl Acad Sci U S A 2022, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Gu W; Dong SH; Sarkar S; Nair SK; Schmidt EW The biochemistry and structural biology of cyanobactin pathways: enabling combinatorial biosynthesis. Methods Enzymol 2018, 604, 113–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Sivonen K; Leikoski N; Fewer DP; Jokela J Cyanobactins-ribosomal cyclic peptides produced by cyanobacteria. Appl Microbiol Biotechnol 2010, 86, 1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Zabriskie TM; Foster MP; Stout TJ; Clardy J; Ireland CM Studies on the solution- and solid-state structure of patellin 2. J Am Chem Soc 1990, 112, 8080–8084. [Google Scholar]
- (43).Donia MS; Ravel J; Schmidt EW A global assembly line for cyanobactins. Nat Chem Biol 2008, 4, 341–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Murakami M; Itou Y; Ishida K; Shin HJ Prenylagaramides A and B, new cyclic peptides from two strains of Oscillatoria agardhii. J Nat Prod 1999, 62, 752–755. [DOI] [PubMed] [Google Scholar]
- (45).Donia MS; Schmidt EW Linking chemistry and genetics in the growing cyanobactin natural products family. Chem Biol 2011, 18, 508–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).McIntosh JA; Lin Z; Tianero MD; Schmidt EW Aestuaramides, a natural library of cyanobactin cyclic peptides resulting from isoprene-derived Claisen rearrangements. ACS Chem Biol 2013, 8, 877–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Sardar D; Lin Z; Schmidt EW Modularity of RiPP enzymes enables designed synthesis of decorated peptides. Chem Biol 2015, 22, 907–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Ruffner DE; Schmidt EW; Heemstra JR Assessing the combinatorial potential of the RiPP cyanobactin tru pathway. ACS Synth Biol 2015, 4, 482–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Tianero MD; Donia MS; Young TS; Schultz PG; Schmidt EW Ribosomal route to small-molecule diversity. J Am Chem Soc 2012, 134, 418–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Sarkar S; Gu W; Schmidt EW Expanding the chemical space of synthetic cyclic peptides using a promiscuous macrocyclase from prenylagaramide biosynthesis. ACS Catal 2020, 10, 7146–7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Heide L Prenyl transfer to aromatic substrates: genetics and enzymology. Curr Opin Chem Biol 2009, 13, 171–179. [DOI] [PubMed] [Google Scholar]
- (52).Tello M; Kuzuyama T; Heide L; Noel JP; Richard SB The ABBA family of aromatic prenyltransferases: broadening natural product diversity. Cell Mol Life Sci 2008, 65, 1459–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Kuzuyama T; Noel JP; Richard SB Structural basis for the promiscuous biosynthetic prenylation of aromatic natural products. Nature 2005, 435, 983–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Metzger U; Schall C; Zocher G; Unsold I; Stec E; Li SM; Heide L; Stehle T The structure of dimethylallyl tryptophan synthase reveals a common architecture of aromatic prenyltransferases in fungi and bacteria. Proc Natl Acad Sci U S A 2009, 106, 14309–14314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Leikoski N; Fewer DP; Jokela J; Alakoski P; Wahlsten M; Sivonen K Analysis of an inactive cyanobactin biosynthetic gene cluster leads to discovery of new natural products from strains of the genus Microcystis. PLoS One 2012, 7, e43002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Purushothaman M; Sarkar S; Morita M; Gugger M; Schmidt EW; Morinaka BI Genome-mining-based discovery of the cyclic peptide tolypamide and TolF, a Ser/Thr forward O-prenyltransferase. Angew Chem Int Ed Engl 2021, 60, 8460–8465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Ishida K; Matsuda H; Murakami M; Yamaguchi K Kawaguchipeptin B, an antibacterial cyclic undecapeptide from the cyanobacterium Microcystis aeruginosa. J Nat Prod 1997, 60, 724–726. [DOI] [PubMed] [Google Scholar]
- (58).Parajuli A; Kwak DH; Dalponte L; Leikoski N; Galica T; Umeobika U; Trembleau L; Bent A; Sivonen K; Wahlsten M; Wang H; Rizzi E; De Bellis G; Naismith J; Jaspars M; Liu X; Houssen W; Fewer DP A unique tryptophan C-prenyltransferase from the kawaguchipeptin biosynthetic pathway. Angew Chem Int Ed Engl 2016, 55, 3596–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Sano T; Kaya K Oscillatorin, a chymotrypsin inhibitor from toxic Oscillatoria agardhii. Tetradehron Let 1996, 37, 6873–6876. [DOI] [PubMed] [Google Scholar]
- (60).Okada M; Sugita T; Akita K; Nakashima Y; Tian T; Li C; Mori T; Abe I Stereospecific prenylation of tryptophan by a cyanobacterial post-translational modification enzyme. Org Biomol Chem 2016, 14, 9639–9644. [DOI] [PubMed] [Google Scholar]
- (61).Phyo MY; Ding CYG; Goh HC; Goh JX; Ong JFM; Chan SH; Yung PYM; Candra H; Tan LT Trikoramide A, a prenylated cyanobactin from the marine cyanobacterium Symploca hydnoides. J Nat Prod 2019, 82, 3482–3488. [DOI] [PubMed] [Google Scholar]
- (62).Iwasaki K; Iwasaki A; Sumimoto S; Sano T; Hitomi Y; Ohno O; Suenaga K Croissamide, a proline-rich cyclic peptide with an N-prenylated tryptophan from a marine cyanobacterium Symploca sp. Tetradehron Let 2018, 59, 3806–3809. [Google Scholar]
- (63).Leikoski N; Fewer DP; Jokela J; Wahlsten M; Rouhiainen L; Sivonen K Highly diverse cyanobactins in strains of the genus Anabaena. Appl Environ Microbiol 2010, 76, 701–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Dalponte L; Parajuli A; Younger E; Mattila A; Jokela J; Wahlsten M; Leikoski N; Sivonen K; Jarmusch SA; Houssen WE; Fewer DP N-Prenylation of tryptophan by an aromatic prenyltransferase from the cyanobactin biosynthetic pathway. Biochemistry 2018, 57, 6860–6867. [DOI] [PubMed] [Google Scholar]
- (65).Phan CS; Matsuda K; Balloo N; Fujita K; Wakimoto T; Okino T Argicyclamides A-C unveil enzymatic basis for guanidine bis-prenylation. J Am Chem Soc 2021, 143, 10083–10087. [DOI] [PubMed] [Google Scholar]
- (66).Agarwal V; Pierce E; McIntosh J; Schmidt EW; Nair SK Structures of cyanobactin maturation enzymes define a family of transamidating proteases. Chem Biol 2012, 19, 1411–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Koehnke J; Bent A; Houssen WE; Zollman D; Morawitz F; Shirran S; Vendome J; Nneoyiegbe AF; Trembleau L; Botting CH; Smith MC; Jaspars M; Naismith JH The mechanism of patellamide macrocyclization revealed by the characterization of the PatG macrocyclase domain. Nat Struct Mol Biol 2012, 19, 767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Lee J; McIntosh J; Hathaway BJ; Schmidt EW Using marine natural products to discover a protease that catalyzes peptide macrocyclization of diverse substrates. J Am Chem Soc 2009, 131, 2122–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Coqueiro A; Regasini LO; Stapleton P; da Silva Bolzani V; Gibbons S In vitro antibacterial activity of prenylated guanidine alkaloids from Pterogyne nitens and synthetic analogues. J Nat Prod 2014, 77, 1972–1975. [DOI] [PubMed] [Google Scholar]
- (70).Pancrace C; Ishida K; Briand E; Pichi DG; Weiz AR; Guljamow A; Scalvenzi T; Sassoon N; Hertweck C; Dittmann E; Gugger M Unique biosynthetic pathway in bloom-forming cyanobacterial genus Microcystis jointly assembles cytotoxic aeruginoguanidines and microguanidines. ACS Chem Biol 2019, 14, 67–75. [DOI] [PubMed] [Google Scholar]
- (71).Leikoski N; Liu L; Jokela J; Wahlsten M; Gugger M; Calteau A; Permi P; Kerfeld CA; Sivonen K; Fewer DP Genome mining expands the chemical diversity of the cyanobactin family to include highly modified linear peptides. Chem Biol 2013, 20, 1033–1043. [DOI] [PubMed] [Google Scholar]
- (72).Lawton LA; Morris LA; Jaspars M A bioactive modified peptide, aeruginosamide, isolated from the cyanobacterium Microcystis aeruginosa. J Org Chem 1999, 64, 5329–5332. [DOI] [PubMed] [Google Scholar]
- (73).Sardar D; Hao Y; Lin Z; Morita M; Nair SK; Schmidt EW Enzymatic N- and C-protection in cyanobactin RiPP natural products. J Am Chem Soc 2017, 139, 2884–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Nagatsu A; Kajitani H; Sakakibara J Muscoride A: A new oxazole peptide alkaloid from freshwater cyanobacterium Nostoc muscorum. Tetradehron Let 1995, 36, 4097–4100. [Google Scholar]
- (75).Amaike K; Muto K; Yamaguchi J; Itami K Decarbonylative C-H coupling of azoles and aryl esters: unprecedented nickel catalysis and application to the synthesis of muscoride A. J Am Chem Soc 2012, 134, 13573–13576. [DOI] [PubMed] [Google Scholar]
- (76).Coqueron PY; Didier C; Ciufolini MA Iterative oxazole assembly via alpha-chloroglycinates: total synthesis of (−)-muscoride A. Angew Chem Int Ed Engl 2003, 42, 1411–1414. [DOI] [PubMed] [Google Scholar]
- (77).Correa A; Cornella J; Martin R Nickel-catalyzed decarbonylative C-H coupling reactions: a strategy for preparing bis(heteroaryl) backbones. Angew Chem Int Ed Engl 2013, 52, 1878–1880. [DOI] [PubMed] [Google Scholar]
- (78).Mattila A; Andsten RM; Jumppanen M; Assante M; Jokela J; Wahlsten M; Mikula KM; Sigindere C; Kwak DH; Gugger M; Koskela H; Sivonen K; Liu X; Yli-Kauhaluoma J; Iwai H; Fewer DP Biosynthesis of the bis-prenylated alkaloids muscoride A and B. ACS Chem Biol 2019, 14, 2683–2690. [DOI] [PubMed] [Google Scholar]
- (79).Zallot R; Oberg N; Gerlt JA Discovery of new enzymatic functions and metabolic pathways using genomic enzymology web tools. Curr Opin Biotechnol 2021, 69, 77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]