

Abstract
Melanoma is a highly aggressive cancer originating from melanocytes. Its etiopathogenesis is strongly related to genetic, epigenetic, and environmental factors. Melanomas encountered in clinical practice are predominantly sporadic, whereas hereditary melanomas account for approximately 10% of the cases. Hereditary melanomas mainly develop due to mutations in the cyclin-dependent kinase 2A (CDKN2A) gene, which encodes two tumor suppressor proteins involved in the cell cycle regulation. CDKN2A, along with CDK4, TERT, and POT1 genes, are high-risk genes for melanoma. Among the genes that carry a moderate risk are MC1R and MITF, whose protein products are involved in melanin synthesis. The environment also contributes to the development of melanoma. Patients at risk of melanoma should be offered genetic counseling to discuss genetic testing options and the importance of skin UV protection, avoidance of sun exposure, and regular preventive dermatological examinations. Although cancer screening cannot prevent the development of the disease, it allows for early diagnosis when the survival rate is the highest.
Keywords: Melanoma, genetics, hereditary syndromes, genetic counseling
INTRODUCTION
Cutaneous melanoma is a malignant skin tumor that develops from melanocytes that produce melanin. Hippocrates first described melanoma in the 5th century B.C. as a black tumor (Greek, melas = black, oma = tumor); preserved medical texts from the late 16th century also mention incurable black tumors [1].
There are four main histological subtypes of melanomas: Superficial spreading melanoma (70%), nodular melanoma (15-30%), lentigo maligna melanoma (4-10%), and acral lentiginous melanoma (<5%) [2]. In addition to the skin, melanomas may also develop in the eye, upper respiratory, gastrointestinal, and genitourinary systems. Although it accounts for only 5% of all skin cancers, it has the highest mortality rate if not diagnosed early. Its incidence increases annually by 3-7%, and the number of newly diagnosed patients doubles every 10 years, making melanoma the most rapidly increasing cancer diagnosis in the white population [3].
The occurrence of melanoma highly depends on the geographic area, that is, its incidence is the highest in countries with the greatest number of sunny days, such as New Zealand and Australia [4,5]. Therefore, these countries have intensified the primary prevention measures, including education about melanoma and raising awareness about the risk of overexposure to the sun, which has helped reduce the incidence rate [6].
Melanoma risk factors may be classified into three groups: genetic, epigenetic, and environmental [7]. Genetic factors include family history, Fitzpatrick skin Types 1 or 2 (pale skin that easily burns and never tans, and red hair), and defects in DNA repair mechanisms [8]. These risk factors are the main topic of this article, especially genes associated with high or moderate risk of melanoma, hereditary syndromes, and the current genetic counseling approach in at-risk populations.
GENES THAT INCREASE THE RISK OF MELANOMA
Most malignant tumors in the human body have multifactorial causes, that is, they result from the complex interactions between genes and environment, or in other words, the interplay between genetics and epigenetics [9]. Such tumors are sporadic [10]. They arise from the cells that have accumulated mutations throughout life, eventually leading to their malignant transformation. A small fraction (approximately 10%) of all malignant tumors is hereditary. Unlike sporadic tumors, hereditary tumors occur in persons born with a mutated gene [11]. This phenomenon is called germline mutation or malignant variation. It is either inherited from one parent or occurs during gametogenesis, and consequently, a mutated gene is present in every cell of the body [12,13]. However, not everyone who inherits such a mutation will develop melanoma because this also depends on gene penetrance, expressed as a proportion of mutation carriers who develop a disease. For example, if gene penetrance is 100%, all carriers of gene mutation develop the disease; if gene penetrance is 50%, then 50% of mutation carriers develop the disease [14]. Whether or not a gene will have a phenotypical expression depends on other factors that increase or decrease the risk. In the case of melanoma, the other factors include the number of moles and sun exposure [15].
Cyclin-dependent kinase 2A (CDKN2A) gene
It is estimated that ~10% of all melanoma cases diagnosed in 2002 were hereditary, with 40-60% of them occurring due to the mutation of the gene coding for CDKN2A [16,17]. William Norris first observed the potential heredity of melanoma in 1820. However, his observation went unnoticed until 1968, when Lynch and Krush first reported on the relationship between pancreatic cancer, multiple moles, and melanoma. Ten years later, Clark described dysplastic nevi in several members of one family and called it the “B-K mole syndrome” [18].
Henry T. Lynch suggested “familial atypical multiple mole melanoma (FAMMM)” instead of “B-K mole syndrome.” The first mutation of the CDKN2A gene in FAMMM was reported in 1992 [19,20]. FAMMM is inherited as an autosomal dominant trait and is characterized by multiple melanocytic moles (>50 nevi) and positive family history. It is associated with germline mutations of CDKN2A. Some mutation carriers may be prone to pancreatic cancer or other malignancies [17].
The CDKN2A gene is located at the short arm of chromosome 9 at the 9p21.3 locus. The locus encodes two proteins interacting with two tumor suppressors: Retinoblastoma protein (Rb) and p53 protein (cellular tumor antigen p53 or tumor suppressor p53) [21].
The gene contains two promoters. When activated, each promoter leads to a different primary transcript, either alpha (α) or beta (β). Each transcript contains a specific exon 1, 1α, and 1β, respectively, whereas they share the exons 2 and 3. The promoter leading to the β transcript is located upstream of the promoter leading to the α transcript. Exon 1 variants being spliced to the shared exon 2 cause the formation of an open reading frame. Thus, exon 2 is read differently due to different starting points in the two transcripts, and the process of translation results in two utterly different proteins. The protein encoded by the α transcript consists of 156 amino acids and is called p16INK4a, while the translation of the β transcript results in the protein called p14ARF, which contains 132 amino acids (Figure 1, Table 1) [22].
FIGURE 1.
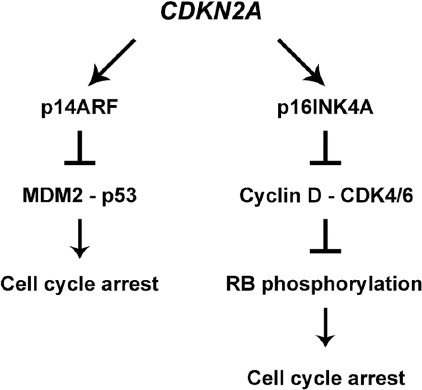
CDKN2A gene encodes several different isoforms, of which isoform 4 encodes p14ARF protein while isoform 1 is responsible for p16INK4A protein. Both proteins arrest the cell cycle: p14ARF acts through p53 protein while p16INK4A blocks the Cyclin D/CDK4/6 complex, affecting the pRB phosphorylation.
TABLE 1.
An overview of the genes that are involved in hereditary melanoma susceptibility.

Although different, they both influence the progression of the cell cycle. The most critical control point in the mammalian cell cycle is the G1 phase because it precedes the DNA replication in the S-phase. Thus, the replication of damaged DNA has to be prevented to avoid mutations [23].
The proteins involved in cell cycle regulation belong to two main groups: those that stimulate the cell cycle and those that stop it. The progression of the cell cycle is helped significantly by a group of kinases called CDK, which exert their function by binding to another cyclin protein. After the CDK-cyclin heterodimer is formed, kinase may phosphorylate the target proteins and stimulate the cell cycle [24].
The proteins that stop the cell cycle are called antiproliferative proteins; they are the products of tumor suppressor gene activity. Two well-known tumor suppressor genes are RB1 and TP53 [25]. The Rb protein’s function is to halt the cell cycle in the G1 phase, which is accomplished by binding the Rb protein to the E2F transcription factor that stimulates the transcription of many genes responsible for the DNA replication process. In its active state, Rb is unphosphorylated or hypophosphorylated, binds to E2F and stops the cell progression at the restriction point (R-point) in the G1 phase. However, when it is phosphorylated by CDK bound to cyclin, the Rb protein conformation is altered, leading to a release of bound E2F, which triggers the transcription of many genes whose protein products stimulate DNA replication [26].
The p53 protein is also known as “the guardian angel” of the human genome because its expression is increased in cells that suffer DNA damage. It acts as a transcription factor that stimulates p21 gene transcription [27]. The gene encodes the protein that binds to the CDK-cyclin complex, thus preventing it from phosphorylating the Rb protein and halting the cell cycle. Thus, the damaged DNA should not replicate and create a mutation. At that moment, the cell should wait for repair before it continues the cycle. Moreover, if the repair does not occur, p21 may stimulate the apoptosis of the cell and prevent the occurrence of mutation [28]. p53 in the cell is bound to another protein called mouse double minute 2 homolog (MDM2), which protects it from degradation and becomes active only after being released from the complex.
The protein products of the CDKN2A gene exert their activity at the checkpoint of the cell progression from the G1 to S phase. p14ARF inhibits the MDM2 protein and its ubiquitin ligase activity, releasing p53 and making it free to stop the cell cycle through p21 (Figure 1) [29,30].
On the other hand, p16INK4a inhibits the cyclin D-CDK4/6 complex, preventing it from phosphorylating the Rb protein, which then remains active and does not allow E2F protein to transcribe the genes needed for the cell to enter the S-phase, consequently keeping the cell in the G1 phase and not allowing the progression of the cell cycle toward DNA replication (Figure 1) [29,31]. Thus, the two protein products of the CDKN2A gene bring the cell cycle to a halt in the same G1 phase by acting through two different mechanisms.
The CDKN2A gene mutations have different effects on the synthesis of p16INK4a and p14ARF proteins, as these are formed by transcription resulting from two different reading frames. There are four types of mutations: deletions, insertions, duplications, and substitutions [32]. According to their effects on protein synthesis, they may also be divided into missense, nonsense, and frameshift mutations. The mutation affecting p16INK4a is most frequently located at exon 1a, which corresponds to intron 1, which also harbors ~1/3 of the mutations regarding p14ARF. These mutations can result in an incompletely synthesized protein because the intron mutations may cause the incorrect processing of the primary transcript. Sometimes, the protein is not synthesized because the primary transcript cannot reach the cytoplasm through nuclear pores [33]. Notably, these mutations may increase the efficacy of immune checkpoint inhibitors, such as ipilimumab (anti-CTLA-4 monoclonal antibody) and anti-PD-1 (programmed cell death-1) antibodies pembrolizumab and nivolumab, possibly due to increased mutation load in CDKN2A mutated tumors [34].
CDK4
CDK4 is a serine/threonine kinase responsible for the progression of the cell cycle from the G1 to S phase [35]. It exerts its intracellular function only after binding to cyclin D and phosphorylating the retinoblastoma protein at a single point [36]. The result of such monophosphorylation is the release of transcription factor E2F, which triggers the transcription of the cyclin E gene CCNE1 and its binding to CDK2. The new cyclin E-CDK2 complex additionally hyperphosphorylates the Rb protein at other serine and threonine phosphorylation sites and facilitates the progression of the cell cycle (Table 1) [37,38].
Based on the GenoMEL centers’ study that involved 2137 cutaneous melanoma patients originating from 466 families with at least 3 cutaneous melanoma cases per family, the frequency of the CDK4 mutations is 2-3% [16].
The CDK4 gene is located at the short arm of chromosome 12 (12q14). It consists of 8 exons and is mutated in about 4% of melanoma cases [39]. The missense mutation at codon 24 of the second exon triggers the change in the activity of the protein product of this gene from a protooncogene to a dominant oncogene. This change results from histidine (R24H) or cysteine (R24C) being incorporated instead of arginine in codon 24, thus preventing p16 from binding to CDK4 protein and regulating its activity [40]. The median age of melanoma diagnosis in families with this mutation is 39 years, with an estimated lifetime penetrance of 74% [41].
Sporadic missense and silent mutations of this gene have also been reported in other cancers, such as endometrial cancer. Its expression is altered in ~2% of all cancers, including lung adenocarcinoma, liposarcoma, and glioblastoma. It is also the reason why this mutated form of the CDK4 gene is a well-chosen target for innovative drugs, such as palbociclib, ribociclib, and abemaciclib (CDK4/6 inhibitors) [42]. Notably, palbociclib has been approved to treat estrogen-positive breast cancer with a high proliferation index (measured by Ki-67), and clinical trials investigating its effectiveness in CDK4 mutated melanomas are underway [43,44].
Telomerase reverse transcriptase (TERT)
The TERT gene encoding the protein part of telomerase reverse transcriptase is located at the short arm of chromosome 5, locus 5p15.33. Telomerase is a ribonucleoprotein that acts as a reverse transcriptase – a function performed by TERT (Table 1). The other ribonucleic part comprises a long non-coding RNA – telomerase RNA (TR or TER) [45,46]. If the cells did not contain telomerase, the chromatids would become ever shorter with every DNA replication because DNA polymerase catalyzes the addition of a new deoxyribonucleoside triphosphate only in a 5’-3’ direction. In other words, it would be beneficial only for one newly synthesized DNA chain. In contrast, the other one would be shorter and shorter with each replication, and this is where telomerase comes into play and prevents such shortening of the chromatids [46]. However, in most cells in the body, telomeres actually do shorten, and the activity of telomerases is needed only in cells such as germ cells, lymphocytes, keratinocytes, endometrial cells, hematopoietic stem cells, and epithelial cells of the intestines, esophagus, and cervix [47]. Maintaining the same length of telomeres is the characteristic of many cancers, including melanoma. Mutations in the TERT gene are characteristic of both sporadic and hereditary melanomas. Specific mutations in the promoter of this gene generate the binding site for the family of ETS (E-twenty-six-specific sequence or E26 transforming sequence) transformation factors, leading to the increased TERT gene transcription [48,49]. The two most common mutations in the gene promoter result from the transition of cytosine to thymine. They are located within 100 bp from the transcription starting site and are called C228T and C250T (chromosome 5, 1,295,228 C>T and 1,295,250 C>T, respectively) [50].
Since these mutations were detected in 77% of intermediate melanocytic tumors and melanoma in situ cases, they mark the beginning of malignant transformation [51]. The described mutations in the TERT promoter region indicate poorer prognosis and can be used as a marker of shorter survival of these patients [52,53]. Inhibition of the activity of this gene and related protein can be a potential therapeutic target for melanoma patients [54].
Protection of telomeres protein 1 (POT1)
The POT1 gene is located at the long arm of chromosome 7 (7q31.33). Its protein product, POT1, is part of the protective protein complex, shelterin or telosome, included in the regulation of telomere length, maintenance of chromosomal stability, prevention of aberrant chromosome separation, and protection from unnecessary recombination repair (Table 1) [55]. It is a heterohexamer built of telomeric repeat factor (TRF) 1, TRF2, repressor activator protein 1, TERF1-interacting nuclear factor 2, tripeptidyl-peptidase 1 (TPP1), and POT1 subunits [56]. The POT1 protein consists of 634 amino acids; it is the only part of the complex that can bind directly to a DNA sequence using the oligonucleotide/oligosaccharide-binding (OB) fold domains OB1 and OB2 at the N-terminal. POT1 blocks the function of ataxia telangiectasia and Rad3-related (ART) protein responsible for initiating the DNA break repair through forming a heterodimer with POT1 protein. The heterodimer recruits telomerase to elongate the ends of the chromosome. The same protein may also have the opposite action, that is, prevent the elongation of telomeres by competitive inhibition with telomerase at the 3’ end of a single-stranded DNA molecule. The mutations leading to POT1 inhibition, or mutations resulting in the loss of a binding site for TPP1, increase telomerase activity and are related to various malignancies, including melanoma [57].
According to one study, POT1 seems to be one of the most commonly mutated genes in hereditary melanoma, along with CDKN2A [58]. Moreover, other studies indicated that these mutations were more common than TERT mutations [59,60].
Melanocortin 1 receptor (MC1R)
MC1R is located at the long arm of chromosome 16 (16q24.3). It encodes the MC1R, which belongs to the family of the G protein-coupled receptors (GPCR). The extracellular GPCR domain binds ligands, whereas the intracellular GPCR domain activates adenylyl cyclase and cAMP synthesis through G protein [61]. One of the most critical roles of the MC1R is melanin biosynthesis, which occurs in the melanocyte organelles called melanosomes and results from the binding of α-melanocyte-stimulating hormone (αMSH) and agouti signaling protein (Table 1). The binding of αMSH to the MSH receptor (MSH-R) activates adenylyl cyclase, catalyzing cAMP production. The result is the synthesis of eumelanin from tyrosine. ASIP competes for the same receptor, that is, it acts antagonistically by blocking the expression of microphthalmia-associated transcription factor (MITF). MITF is the main factor in melanin synthesis because it regulates the activity of tyrosine-related protein 1 (TRP1) and tyrosinase [62]. The binding of ASIP to MSH-R inhibits eumelanin synthesis and stimulates the production of pheomelanin [63].
Interestingly, MC1R variants may substantially increase the penetrance of CDKN2A mutations and the risk of melanoma in affected families, particularly multiple MC1R variants and red hair color variants [64]. Identifying polymorphisms and mutations of the MC1R gene would enable a better understanding of melanoma susceptibility and potential treatments [65-67].
Melanocyte-inducing transcription factor or microphthalmia-associated transcription factor (MITF)
Melanocyte-inducing transcription factor (MITF) gene, or microphthalmia-associated transcription factor gene, is located at the short arm of chromosome 3 (3p13). It encodes the transcription factor called basic-helix-loop-helix-leucine zipper, which it uses to bind to DNA [68]. This domain recognizes specific sequences in the target gene promoters, such as tyrosinase (TYR). It is essential for regulating the expression of TYR and TYR-related proteins, such as TYR-related protein 1 and, therefore, plays a central role in regulating melanin synthesis in melanocytes (Table 1) [69]. Among several isoforms of the MITF gene, only MITF-M is specific for melanocytes [70]. Wnt, TGF-beta, and RTK are only some of the signaling pathways related to the expression of this gene [71].
MELANOMA-ASSOCIATED SYNDROMES
Melanomas are part of several hereditary syndromes. Two of them, FAMMM and BRCA1-associated protein-1 (BAP1) tumor predisposition syndrome, a malignant tumor syndrome (including melanoma) associated with the mutation of the BAP1 gene, are described in the following paragraphs.
FAMMM syndrome
The first record of FAMMM syndrome dated from 1820, when Norris described the development of a tumor from a brownish mole that recurred after removal in a patient with about 40 more similar skin lesions and enlarged lymph nodes [72]. The disease was so extensive that the only option was palliative care. The autopsy found that the tumor spread throughout the body, including the heart and lungs. The family history revealed that the patient’s father died of the same disease, and the siblings also had numerous nevi. Norris concluded that it was a hereditary disorder [73].
In 1968, Lynch and Krush described four families with multiple melanomas, including a family where the proband (the first affected family member who seeks medical attention and whose findings raise the suspicion of a hereditary disease) developed the disease at age 26. In 1980, the hereditary nature of this syndrome was confirmed: it showed an autosomal dominant pattern of inheritance. In the 1990s, Lynch reported that the syndrome was associated with other cancers, especially pancreatic carcinoma [74]. The association between this syndrome and pancreatic cancer was explicitly observed in patients carrying the p16-Leiden mutation in the CDKN2A gene (deletion of 19 base pairs in exon 2 of the gene CDKN2A; NM_000077.4: c.225_243del19 (p.p75fs)) [75,76]. The mutation carriers were also prone to esophageal cancer [77,78]. Many other tumors are related to this syndrome, including lung, breast, liver, and brain tumors [79]. The loss of CDKN2A heterozygosity is considered the first step in developing melanoma in patients with FAMMM syndrome [80].
Diagnostic criteria for FAMMM syndrome are as follows:
Melanoma in one or more first- or second-degree relatives
Total body nevi count >50, including atypical nevi (asymmetric, raised above the skin, varying in color, and size)
Nevi showing specific histological features, including asymmetry, subepidermal fibroplasia, and lentiginous melanocytic hyperplasia (spindle or epithelioid melanocytes forming nests of different sizes and merging with adjacent rete ridges, and creating bridges), and dermal lymphocyte infiltrates [72].
These patients are referred to genetic counseling, genetic testing, and follow-up. Examination intervals depend on the number of close relatives with the disease and the nevi count. The usual follow-up interval is 6 months. Dermoscopy is the method of choice, but the importance of self-examination should not be underestimated. The use of smartphone applications for examination, which may become available soon, also holds potential [73].
BAP1 tumor predisposition syndrome
This syndrome, caused by mutations in the BAP1 gene, is characterized by uveal melanoma, mesothelioma, and (less often) skin melanoma. Other malignancies may also develop, including kidney, bladder, brain, and soft-tissue tumors [81]. A tumor suppressor gene called BAP1 codes for ubiquitin carboxy-terminal hydroxylase BAP1. It removes ubiquitin from other proteins, making them more resistant to degradation. It also interferes with their interaction with other proteins. BAP1 is involved in various cellular processes, regulating the cell cycle, transcription, chromatin organization, DNA repair, and apoptosis (Table 1) [82]. This protein, made of 729 amino acids, consists of three main domains: the N-terminal domain that removes ubiquitin, the middle domain that binds the nuclear transcription co-factor called host cell factor 1, and the C-terminal domain that interacts with other proteins [83].
The disease is inherited in an autosomal dominant pattern, and mutations that affect the nuclear localization signal (the sequence of amino acids that directs the protein into the nucleus) or catalytic domain for ubiquitin removal are believed to cause the most severe clinical presentations [84]. BAP1 gene is often mutated in cases of uveal melanoma, which accounts for 3–5% of all diagnosed melanomas [85]. The carriers of BAP1 gene mutations are also prone to developing clear cell renal cell carcinoma or mesothelioma [86,87]. More recent studies suggest that BAP1 mutations may indicate a poorer prognosis for these patients [88].
Mutation of the BAP1 gene usually manifests as the growth of melanocytic BAP1-associated intradermal tumors (MBAITs). These tumors are raised above the skin surface, are about 5 mm in diameter, and are pigmented or skin-colored. They were previously called atypical Spitz tumors; however, later, it was shown that they differ histologically and morphologically from typical and atypical Spitz tumors. They usually occur in the second decade of life [81]. The number of lesions increases with time but varies from patient to patient [72]. If this gene mutation is suspected, the patient should be referred to genetic counseling, with testing and follow-up measures arranged for the patient and the entire family [89].
GENETIC COUNSELING
The National Society of Genetic Counselors probably gave the best definition of genetic counseling in 2006: “Genetic counseling is the process of helping people understand and adapt to the medical, psychological, and familial implications of genetic contributions to disease. This process integrates the interpretation to assess the chance of disease, education, and counseling” [90]. The advantage of genetic counseling is to have a better understanding of basic concepts related to genetics, such as mutation, mutation of germinative or somatic cells, early tumor markers, targeted therapy, and molecular analysis [91], and reduction of anxiety related to a possible positive test result and its long-term effect on the patient’s quality of life [92].
The counseling process includes the assessment of disease probability in patients and other members of their families. For that purpose, the consensus on the testing protocol is essential. For example, to test a person for a pathogenic variant of the CDKN2A gene, there should be at least three first- or second-degree relatives on the same side of the family with the disease plus a positive prediction test, such as GenoMELPREDICT [93] or evidence of pathogenetic variant of this gene in a family member [94]. When making recommendations, the following factors should be considered: number of family members with a confirmed diagnosis of the skin or ocular melanoma; melanoma before the age of 40; and presence of pancreatic cancer or some other malignancy [95]. These evaluations are best supported by research in families with known mutations such as the mutation CDKN2A c.256G > A (Ala86Thr), that is, replacement of guanine by adenine at position 256, resulting in the incorporation of threonine instead of alanine [96].
Genetic counseling is essential not only for the possibility of testing but also for the risk calculation and modification of risk behavior, which play an equally important role in the etiology of the disease since the information received during counseling may positively change the behavior of the person [92]. Specifically, in hereditary melanoma, avoiding prolonged exposure to UV light is of utmost importance [97]. Usually, it is imperative in children not to get tested for adult hereditary tumors. However, in the case of hereditary melanoma, there are indications that genetic testing in children could be justified in the case of CDKN2A gene mutations [98]. It was reported that high-quality genetic counseling contributed to a decreased number of hours of UV light exposure in hereditary mutation carriers and non-carriers alike [99].
The person who comes for genetic counseling should be explained the advantages and disadvantages of genetic testing to help them decide whether to accept it. The advantage of early identification of mutation carriers is implementing thorough lifelong monitoring of the carrier and their family members by digital dermoscopy and photography, thus detecting the melanoma in its earliest stage. In the case of CDKN2A gene mutation, other malignancies should also be considered, especially pancreatic cancer [17].
The disadvantage of genetic testing is possible anxiety due to the increased risk of melanoma if the test results are positive. It is advisable to refer the mutation carrier to psychological counseling in such cases. A negative test result (absence of mutation), on the other hand, may give a false sense of security, which is also a disadvantage. Therefore, it is essential to highlight that familial malignant melanoma accounts for only 10% of all melanoma cases, whereas the remaining 90% are sporadic. Psychoeducation can help these patients understand the importance of sunscreen use, self-examination, and regular preventive dermatological check-ups.
CONCLUSIONS
Melanoma is an aggressive malignancy with high metastatic potential. Sporadic melanoma is prevalent in clinical practice, whereas familial malignant melanoma accounts for approximately 10% of the cases. The highest proportion of familial malignant melanoma cases arises from the mutations in the CDKN2A gene, which codes for two tumor-suppressor proteins, p14ARF and p16INK4a. Mutations of the CDKN2A carry a high risk of melanoma, together with CDK4, TERT, and POT1 gene mutations. They encode proteins responsible for cell cycle regulation (CDKN2A and CDK4) or telomeres length control (TERT and POT1). The genes that carry a moderate melanoma risk include MC1R and MITF, whose protein products are involved in melanin synthesis. Since environmental influence plays a role in melanoma development, at-risk patient groups should be offered genetic counseling. During the counseling, along with the option of genetic testing, the patients should be advised to protect their skin from UV light, avoid sun exposure, and keep regular preventive check-ups with their dermatologist. Regular examinations may not prevent the development of the disease, but they increase the probability of early diagnosis when the survival rate is the highest. As none of the above-mentioned genes can be individually held responsible for causing melanoma, the most significant advantage of genetic counseling is psychoeducation.
Footnotes
Conflict of interest: The authors declare no conflicts of interest.
Funding: Authors received no specific funding for this work.
REFERENCES
- 1.Abdo JF, Sharma A, Sharma R. Role of heredity in melanoma susceptibility:A primer for the practicing surgeon. Surg Clin North Am. 2020;100(1):13–28. doi: 10.1016/j.suc.2019.09.006. https://doi.org/10.1016/j.suc.2019.09.006. [DOI] [PubMed] [Google Scholar]
- 2.Argenziano G. Cutaneous Melanoma:A Pocket Guide for Diagnosis and Management. London, United Kingdom: Elsevier, Academic Press; 2017. [Google Scholar]
- 3.Garbe C, Melanoma BJ, Bolognia JL, Schaffer JV, Cerroni L. Dermatology. 4th ed. Vol. 2. Netherlands: Elsevier; 2018. pp. 1989–2019. [Google Scholar]
- 4.Vanderwalde A, Spetzler D, Xiao N, Gatalica Z, Marshall J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 aand tumor mutational burden in 11,348 patients. Cancer Med. 2018;7(3):746–56. doi: 10.1002/cam4.1372. https://doi.org/10.1002/cam4.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sneyd MJ, Cox B. A comparison of trends in melanoma mortality in New Zealand and Australia:The two countries with the highest melanoma incidence and mortality in the world. BMC Cancer. 2013;13:372. doi: 10.1186/1471-2407-13-372. https://doi.org/10.1186/1471-2407-13-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee GC, Kunitake H, Stafford C, Bordeianou LG, Francone TD, Ricciardi R. High risk of proximal and local neoplasms in 2206 apatients with anogenital extramammary paget's disease. Dis Colon Rectum. 2019;62(11):1283–93. doi: 10.1097/DCR.0000000000001487. https://doi.org/10.1097/dcr.0000000000001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D'Orazio AJ, Jarrett S, Marsch A, Lagrew J, Cleary L. Melanoma-Epidemiology, Genetics and Risk Factors. India: InTech; 2013. https://doi.org/10.5772/55172. [Google Scholar]
- 8.Gupta V, Sharma VK. Skin typing:Fitzpatrick grading and others. Clin Dermatol. 2019;37(5):430–6. doi: 10.1016/j.clindermatol.2019.07.010. https://doi.org/10.1016/j.clindermatol.2019.07.010. [DOI] [PubMed] [Google Scholar]
- 9.Morani AC, Hanafy AK, Ramani NS, Katabathina VS, Yedururi S, Dasyam AK, et al. Hereditary and sporadic pancreatic ductal adenocarcinoma:Current update on genetics and imaging. Radiol Imaging Cancer. 2020;2(2):190020. doi: 10.1148/rycan.2020190020. https://doi.org/10.1148/rycan.2020190020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang GA, Wiggins JM, Corless BC, Syeda MM, Tadepalli JS, Blake S, et al. TERT, BRAF, and NRAS mutational heterogeneity between paired primary and metastatic melanoma tumors. J Investig Dermatol. 2020;140(8):1609–18e7. doi: 10.1016/j.jid.2020.01.027. https://doi.org/10.1016/j.jid.2020.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bateman AC. DNA mismatch repair proteins:Scientific update and practical guide. J Clin Pathol. 2021;74(4):264–8. doi: 10.1136/jclinpath-2020-207281. https://doi.org/10.1136/jclinpath-2020-207281. [DOI] [PubMed] [Google Scholar]
- 12.Evans DG, van Veen EM, Byers HJ, Evans SJ, Burghel GJ, Woodward ER, et al. High likelihood of actionable pathogenic variant detection in breast cancer genes in women with very early onset breast cancer. J Med Genet. 2021;59(2):115–21. doi: 10.1136/jmedgenet-2020-107347. https://doi.org/10.1136/jmedgenet-2020-107347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petridis C, Arora I, Shah V, Megalios A, Moss C, Mera A, et al. Frequency of pathogenic germline variants in BRCA1, BRCA2, PALB2, CHEK2 aand TP53 in ductal carcinoma in situ diagnosed in women under the age of 50?years. Breast Cancer Res. 2019;21(1):58. doi: 10.1186/s13058-019-1143-y. https://doi.org/10.1186/s13058-019-1143-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hallquist ML, Tricou EP, Ormond KE, Savatt JM, Coughlin CR, Faucett WA, et al. Application of a framework to guide genetic testing communication across clinical indications. Genome Med. 2021;13(1):71. doi: 10.1186/s13073-021-00887-x. https://doi.org/10.1186/s13073-021-00887-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bishop JN, Harland M, Randerson-Moor J, Bishop DT. Management of familial melanoma. Lancet Oncol. 2007;8(1):46–54. doi: 10.1016/S1470-2045(06)71010-5. https://doi.org/10.1016/s1470-2045(06)71010-5. [DOI] [PubMed] [Google Scholar]
- 16.Goldstein AM, Chan M, Harland M, Gillanders EM, Hayward NK, Avril MF, et al. High-risk melanoma susceptibility genes and pancreatic cancer, neural system tumors, and uveal melanoma across GenoMEL. Cancer Res. 2006;66(20):9818–28. doi: 10.1158/0008-5472.CAN-06-0494. https://doi.org/10.1158/0008-5472.iacan-06-0494. [DOI] [PubMed] [Google Scholar]
- 17.Soura E, Eliades PJ, Shannon K, Stratigos AJ, Tsao H. Hereditary melanoma:Update on syndromes and management:Genetics of familial atypical multiple mole melanoma syndrome. J Am Acad Dermatol. 2016;74(3):395–407. doi: 10.1016/j.jaad.2015.08.038. https://doi.org/10.1016/j.jaad.2015.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clark WH, Jr, Reimer RR, Greene M, Ainsworth AM, Mastrangelo MJ. Origin of familial malignant melanomas from heritable melanocytic lesions. The B-K mole syndrome. Arch Dermatol. 1978;114(5):732–8. https://doi.org/10.1001/archderm.114.5.732. [PubMed] [Google Scholar]
- 19.Hussussian CJ, Struewing JP, Goldstein AM, Higgins PA, Ally DS, Sheahan MD, et al. Germline p16 amutations in familial melanoma. Nat Genet. 1994;8(1):15–21. doi: 10.1038/ng0994-15. https://doi.org/10.1038/ng0994-15. [DOI] [PubMed] [Google Scholar]
- 20.Cannon-Albright LA, Goldgar DE, Meyer LJ, Lewis CM, Anderson DE, Fountain JW, et al. Assignment of a locus for familial melanoma, MLM, to chromosome 9p13-p22. Science. 1992;258(5085):1148–52. doi: 10.1126/science.1439824. https://doi.org/10.1126/science.1439824. [DOI] [PubMed] [Google Scholar]
- 21.González-Gil C, Ribera J, Ribera JM, Genescà E. The yin and yang-like clinical implications of the CDKN2A/ARF/CDKN2B gene cluster in acute lymphoblastic Leukemia. Genes (Basel) 2021;12(1):79. doi: 10.3390/genes12010079. https://doi.org/10.3390/genes12010079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fontana R, Ranieri M, La Mantia G, Vivo M. Dual role of the alternative reading frame ARF protein in cancer. Biomolecules. 2019;9(3):87. doi: 10.3390/biom9030087. https://doi.org/10.3390/biom9030087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tyson JJ, Csikasz-Nagy A, Novak B. The dynamics of cell cycle regulation. BioEssays. 2002;24(12):1095–109. doi: 10.1002/bies.10191. https://doi.org/10.1002/bies.10191. [DOI] [PubMed] [Google Scholar]
- 24.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer:A changing paradigm. Nat Rev Cancer. 2009;9(3):153–66. doi: 10.1038/nrc2602. https://doi.org/10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 25.Jiang Z, Jones R, Liu JC, Deng T, Robinson T, Chung PE, et al. RB1 aand p53 at the crossroad of EMT and triple-negative breast cancer. Cell Cycle (Georgetown, Tex) 2011;10(10):1563–70. doi: 10.4161/cc.10.10.15703. https://doi.org/10.4161/cc.10.10.15703. [DOI] [PubMed] [Google Scholar]
- 26.Dick FA, Rubin SM. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol. 2013;14(5):297–306. doi: 10.1038/nrm3567. https://doi.org/10.1038/nrm3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zilfou JT, Lowe SW. Tumor suppressive functions of p53. Cold Spring Harbor Perspect Biol. 2009;1(5):a001883. doi: 10.1101/cshperspect.a001883. https://doi.org/10.1101/cshperspect.a001883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karimian A, Ahmadi Y, Yousefi B. Multiple functions of p21 ain cell cycle apoptosis and transcriptional regulation after DNA damage. DNA Repair. 2016;42:63–71. doi: 10.1016/j.dnarep.2016.04.008. https://doi.org/10.1016/j.dnarep.2016.04.008. [DOI] [PubMed] [Google Scholar]
- 29.Brown VL, Harwood CA, Crook T, Cronin JG, Kelsell DP, Proby CM. p16INK4a and p14ARF tumor suppressor genes are commonly inactivated in cutaneous squamous cell carcinoma. J Investig Dermatol. 2004;122(5):1284–92. doi: 10.1111/j.0022-202X.2004.22501.x. https://doi.org/10.1111/j.0022-202x.2004.22501.iax. [DOI] [PubMed] [Google Scholar]
- 30.Weber HO, Samuel T, Rauch P, Funk JO. Human p14(ARF)-mediated cell cycle arrest strictly depends on intact p53 asignaling pathways. Oncogene. 2002;21(20):3207–12. doi: 10.1038/sj.onc.1205429. https://doi.org/10.1038/sj.onc.1205429. [DOI] [PubMed] [Google Scholar]
- 31.Azazmeh N, Assouline B, Winter E, Ruppo S, Nevo Y, Maly A, et al. Chronic expression of p16(INK4a) in the epidermis induces Wnt-mediated hyperplasia and promotes tumor initiation. Nat Commun. 2020;11(1):2711. doi: 10.1038/s41467-020-16475-3. https://doi.org/10.1038/s41467-020-16475-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pellegrini C, Maturo MG, Martorelli C, Suppa M, Antonini A, Kostaki D, et al. Characterization of melanoma susceptibility genes in high-risk patients from Central Italy. Melanoma Res. 2017;27(3):258–67. doi: 10.1097/CMR.0000000000000323. https://doi.org/10.1097/cmr.0000000000000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan SH, Chiang J, Ngeow J. CDKN2A germline alterations and the relevance of genotype-phenotype associations in cancer predisposition. Hereditary Cancer Clin Pract. 2021;19(1):21. doi: 10.1186/s13053-021-00178-x. https://doi.org/10.1186/s13053-021-00178-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Helgadottir H, Ghiorzo P, van Doorn R, Puig S, Levin M, Kefford R, et al. Efficacy of novel immunotherapy regimens in patients with metastatic melanoma with germline CDKN2A mutations. J Med Genet. 2020;57(5):316–21. doi: 10.1136/jmedgenet-2018-105610. https://doi.org/10.1136/jmedgenet-2018-105610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deshpande A, Sicinski P, Hinds PW. Cyclins and cdks in development and cancer:A perspective. Oncogene. 2005;24(17):2909–15. doi: 10.1038/sj.onc.1208618. https://doi.org/10.1038/sj.onc.120↪. [DOI] [PubMed] [Google Scholar]
- 36.Narasimha AM, Kaulich M, Shapiro GS, Choi YJ, Sicinski P, Dowdy SF. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. eLife. 2014;3:02872. doi: 10.7554/eLife.02872. https://doi.org/10.7554/elife.02872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knudsen ES, Wang JY. Dual mechanisms for the inhibition of E2F binding to RB by cyclin-dependent kinase-mediated RB phosphorylation. Mol Cell Biol. 1997;17(10):5771–83. doi: 10.1128/mcb.17.10.5771. https://doi.org/10.1128/mcb.17.10.5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kitagawa M, Higashi H, Jung HK, Suzuki-Takahashi I, Ikeda M, Tamai K, et al. The consensus motif for phosphorylation by cyclin D1-Cdk4 ais different from that for phosphorylation by cyclin A/E-Cdk2. EMBO J. 1996;15(24):7060–9. https://doi.org/10.1002/j.1460-2075.1996.iatb01097.x. [PMC free article] [PubMed] [Google Scholar]
- 39.AACR Project GENIE. Powering precision medicine through an international consortium Cancer Discov. 2017;7(8):818–31. doi: 10.1158/2159-8290.CD-17-0151. https://doi.org/10.1158/2159-8290.iacd-17-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Read J, Wadt KA, Hayward NK. Melanoma genetics. J Med Genet. 2016;53(1):1. doi: 10.1136/jmedgenet-2015-103150. [DOI] [PubMed] [Google Scholar]
- 41.Puntervoll HE, Yang XR, Vetti HH, Bachmann IM, Avril MF, Benfodda M, et al. Melanoma prone families with CDK4 agermline mutation:Phenotypic profile and associations with MC1R variants. J Med Genet. 2013;50(4):264. doi: 10.1136/jmedgenet-2012-101455. https://doi.org/10.3989/alqantara.2020.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wimberly H, Brown JR, Schalper K, Haack H, Silver MR, Nixon C, et al. PD-L1 aexpression correlates with tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy in breast cancer. Cancer Immunol Res. 2015;3(4):326–32. doi: 10.1158/2326-6066.CIR-14-0133. https://doi.org/10.1158/2326-6066.iacir-14-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.AbuHammad S, Cullinane C, Martin C, Bacolas Z, Ward T, Chen H, et al. Regulation of PRMT5-MDM4 aaxis is critical in the response to CDK4/6 inhibitors in melanoma. Proc Natl Acad Sci U S Am. 2019;116(36):17990–8000. doi: 10.1073/pnas.1901323116. https://doi.org/10.1073/pnas.2005945117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Young RJ, Waldeck K, Martin C, Foo JH, Cameron DP, Kirby L, et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 ainhibitor PD0332991 in melanoma cell lines. Pigment Cell Melanoma Res. 2014;27(4):590–600. doi: 10.1111/pcmr.12228. https://doi.org/10.1111/pcmr.12228. [DOI] [PubMed] [Google Scholar]
- 45.McNally EJ, Luncsford PJ, Armanios M. Long telomeres and cancer risk:The price of cellular immortality. J Clin Investig. 2019;129(9):3474–81. doi: 10.1172/JCI120851. https://doi.org/10.1172/jci120851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stone MD. Detailed view of human telomerase enzyme invites rethink of its structure. Nature. 2018;557(7704):174–5. doi: 10.1038/d41586-018-04756-3. https://doi.org/10.1038/d41586-018-04756-3. [DOI] [PubMed] [Google Scholar]
- 47.Meyerson M. Role of telomerase in normal and cancer cells. J Clin Oncol. 2000;18(13):2626–34. doi: 10.1200/JCO.2000.18.13.2626. [DOI] [PubMed] [Google Scholar]
- 48.Sizemore GM, Pitarresi JR, Balakrishnan S, Ostrowski MC. The ETS family of oncogenic transcription factors in solid tumours. Nat Rev Cancer. 2017;17(6):337–51. doi: 10.1038/nrc.2017.20. https://doi.org/10.1038/nrc.2017.20. [DOI] [PubMed] [Google Scholar]
- 49.Urso C. Melanocytic skin neoplasms:What lesson from genomic aberrations? Am J Dermatopathol. 2019;41(9):623–9. doi: 10.1097/DAD.0000000000001341. https://doi.org/10.1097/dad.0000000000001341. [DOI] [PubMed] [Google Scholar]
- 50.Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339(6122):957–9. doi: 10.1126/science.1229259. https://doi.org/10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shain AH, Yeh I, Kovalyshyn I, Sriharan A, Talevich E, Gagnon A, et al. The genetic evolution of melanoma from precursor lesions. N Engl J Med. 2015;373(20):1926–36. doi: 10.1056/NEJMoa1502583. https://doi.org/10.1056/nejmoa1502583. [DOI] [PubMed] [Google Scholar]
- 52.van Poppelen NM, de Bruyn DP, Bicer T, Verdijk R, Naus N, Mensink H, et al. Genetics of ocular melanoma:Insights into genetics, inheritance and testing. Int J Mol Sci. 2020;22(1):336. doi: 10.3390/ijms22010336. https://doi.org/10.3390/ijms22010336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Andrés-Lencina JJ, Rachakonda S, García-Casado Z, Srinivas N, Skorokhod A, Requena C, et al. TERT promoter mutation subtypes and survival in stage I and II melanoma patients. Int J Cancer. 2019;144(5):1027–36. doi: 10.1002/ijc.31780. https://doi.org/10.1002/ijc.31780. [DOI] [PubMed] [Google Scholar]
- 54.Liang WS, Hendricks W, Kiefer J, Schmidt J, Sekar S, Carpten J, et al. Integrated genomic analyses reveal frequent TERT aberrations in acral melanoma. Genome Res. 2017;27(4):524–32. doi: 10.1101/gr.213348.116. https://doi.org/10.1101/gr.213348.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Robles-Espinoza CD, Harland M, Ramsay AJ, Aoude LG, Quesada V, Ding Z, et al. POT1 aloss-of-function variants predispose to familial melanoma. Nat Genet. 2014;46(5):478–81. doi: 10.1038/ng.2947. https://doi.org/10.1038/ng.2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ye JZ, Donigian JR, van Overbeek M, Loayza D, Luo Y, Krutchinsky AN, et al. TIN2 abinds TRF1 and TRF2 simultaneously and stabilizes the TRF2 complex on telomeres. J Biol Chem. 2004;279(45):47264–71. doi: 10.1074/jbc.M409047200. https://doi.org/10.1074/jbc.m409047200. [DOI] [PubMed] [Google Scholar]
- 57.Calvete O, Garcia-Pavia P, Domínguez F, Bougeard G, Kunze K, Braeuninger A, et al. The wide spectrum of POT1 agene variants correlates with multiple cancer types . Eur J Hum Genet. 2017;25(11):1278–81. doi: 10.1038/ejhg.2017.134. https://doi.org/10.1038/ejhg.2017.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trigueros-Motos L. Mutations in POT1 apredispose to familial cutaneous malignant Melanoma. 2014;86(3):217–8. doi: 10.1111/cge.12416. https://doi.org/10.1111/cge.12416. [DOI] [PubMed] [Google Scholar]
- 59.Potrony M, Puig-Butille JA, Ribera-Sola M, Iyer V, Robles-Espinoza CD, Aguilera P, et al. POT1 agermline mutations but not TERT promoter mutations are implicated in melanoma susceptibility in a large cohort of Spanish melanoma families . Br J Dermatol. 2019;181(1):105–13. doi: 10.1111/bjd.17443. https://doi.org/10.1111/bjd.17443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Toland AE. POT1 apathogenic variants:Not all telomere pathway genes are equal in risk of hereditary cutaneous melanoma. Br J Dermatol. 2019;181(1):14–5. doi: 10.1111/bjd.17728. https://doi.org/10.1111/bjd.17728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Horrell E, Boulanger M, D'Orazio J. Melanocortin 1 areceptor:Structure function,nd regulation. Front Genet. 2016;7:95. doi: 10.3389/fgene.2016.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang W, Wang S, Gu J, Gao Y, Wang Z, Zhang K, et al. Synergistic tumoricidal effect of combined hPD-L1 avaccine and HER2 gene vaccine . Biochem Biophys Res Commun. 2018;497(1):394–400. doi: 10.1016/j.bbrc.2018.02.092. https://doi.org/10.1016/j.bbrc.2018.02.092. [DOI] [PubMed] [Google Scholar]
- 63.Saleha S, Khan TA, Zafar S. MC1R gene variants involvement in human OCA phenotype. J Open Life Sci. 2016;11(1):142–50. https://doi.org/10.1515/biol-2016-0020. [Google Scholar]
- 64.Fargnoli MC, Gandini S, Peris K, Maisonneuve P, Raimondi S. MC1R variants increase melanoma risk in families with CDKN2A mutations:A meta-analysis. Eur J Cancer. 2010;46(8):1413–20. doi: 10.1016/j.ejca.2010.01.027. https://doi.org/10.1016/j.ejca.2010.01.027. [DOI] [PubMed] [Google Scholar]
- 65.Herraiz C, Martínez-Vicente I, Maresca V. The α-melanocyte-stimulating hormone/melanocortin-1 areceptor interaction:A driver of pleiotropic effects beyond pigmentation. Pigment Cell Melanoma Res. 2021;34(4):748–61. doi: 10.1111/pcmr.12980. https://doi.org/10.1111/pcmr.12980. [DOI] [PubMed] [Google Scholar]
- 66.Fuiten AM, Fankhauser RG, Smit DJ, Stark MS, Enright TF, Wood MA, et al. Genetic analysis of multiple primary melanomas arising within the boundaries of congenital nevi depigmentosa. Pigment Cell Melanoma Res. 2021;34(6):1123–30. doi: 10.1111/pcmr.12979. https://doi.org/10.1111/pcmr.12979. [DOI] [PubMed] [Google Scholar]
- 67.Zanna I, Caini S, Raimondi S, Saieva C, Masala G, Massi D, et al. Germline MC1R variants and frequency of somatic BRAF, NRAS, and TERT mutations in melanoma:Literature review and meta-analysis. Mol Carcinogen. 2021;60(3):167–71. doi: 10.1002/mc.23280. https://doi.org/10.1002/mc.23280. [DOI] [PubMed] [Google Scholar]
- 68.Zarei M, Giannikou K, Du H, Liu HJ, Duarte M, Johnson S, et al. MITF is a driver oncogene and potential therapeutic target in kidney angiomyolipoma tumors through transcriptional regulation of CYR61. Oncogene. 2021;40(1):112–26. doi: 10.1038/s41388-020-01504-8. https://doi.org/10.1038/s41388-020-01504-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bristot IJ, Kehl Dias C, Chapola H, Parsons RB, Klamt F. Metabolic rewiring in melanoma drug-resistant cells. Crit Rev Oncol Hematol. 2020;153:102995. doi: 10.1016/j.critrevonc.2020.102995. https://doi.org/10.1016/j.critrevonc.2020.102995. [DOI] [PubMed] [Google Scholar]
- 70.Yajima I, Kumasaka MY, Thang ND, Goto Y, Takeda K, Iida M, et al. Molecular network associated with MITF in skin melanoma development and progression. J skin Cancer. 2011;2011:730170. doi: 10.1155/2011/730170. https://doi.org/10.1155/2011/730170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Koludrovic D, Davidson I. MITF, the Janus transcription factor of melanoma. Future Oncol. 2013;9(2):235–44. doi: 10.2217/fon.12.177. https://doi.org/10.2217/fon.12.177. [DOI] [PubMed] [Google Scholar]
- 72.Schierbeck J, Vestergaard T, Bygum A. Skin cancer associated genodermatoses:A literature review. Acta Dermatovenereol. 2019;99(4):360–9. doi: 10.2340/00015555-3123. https://doi.org/10.2340/00015555-3123. [DOI] [PubMed] [Google Scholar]
- 73.Lynch HT, Shaw TG. Familial atypical multiple mole melanoma (FAMMM) syndrome:History, genetics, and heterogeneity. Familial Cancer. 2016;15(3):487–91. doi: 10.1007/s10689-016-9888-2. https://doi.org/10.1007/s10689-016-9888-2. [DOI] [PubMed] [Google Scholar]
- 74.Ibrahim I, Sibinga Mulder BG, Bonsing B, Morreau H, Farina Sarasqueta A, Inderson A, et al. Risk of multiple pancreatic cancers in CDKN2A-p16-Leiden mutation carriers. Eur J Hum Genet. 2018;26(8):1227–9. doi: 10.1038/s41431-018-0170-y. https://doi.org/10.1038/s41431-018-0170-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Potjer TP, van der, Stoep N, Houwing-Duistermaat JJ, Konings IC, Aalfs CM, van den, Akker PC, et al. Pancreatic cancer-associated gene polymorphisms in a nation-wide cohort of p16-Leiden germline mutation carriers;a case-control study. BMC Res Notes. 2015;8(1):264. doi: 10.1186/s13104-015-1235-4. https://doi.org/10.1186/s13104-015-1235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Oldenburg RA, de Vos tot Nederveen Cappel WH, van Puijenbroek M, van den, Ouweland A, Bakker E, Griffioen G, et al. Extending the p16-leiden tumour spectrum by respiratory tract tumours. J Med Genet. 2004;41(3):31. doi: 10.1136/jmg.2003.012336. https://doi.org/10.1136/jmg.2003.012336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.van der, Wilk BJ, Noordman BJ, Atmodimedjo PN, Dinjens WN, Laheij RJ, Wagner A, et al. Development of esophageal squamous cell cancer in patients with FAMMM syndrome:Two clinical reports. Eur J Med Genet. 2020;63(3):103840. doi: 10.1016/j.ejmg.2020.103840. https://doi.org/10.1016/j.ejmg.2020.103840. [DOI] [PubMed] [Google Scholar]
- 78.Middlebrooks CD, Stacey ML, Li Q, Snyder C, Shaw TG, Richardson-Nelson T, et al. Analysis of the CDKN2A gene in FAMMM syndrome families reveals early age of onset for additional syndromic cancers. Cancer Res. 2019;79(11):2992–3000. doi: 10.1158/0008-5472.CAN-18-1580. https://doi.org/10.1158/0008-5472.iacan-18-1580. [DOI] [PubMed] [Google Scholar]
- 79.Vasen HF, Gruis NA, Frants RR, van der, Velden PA, Hille ET, Bergman W. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 adeletion of p16 (p16-Leiden) Int J Cancer. 2000;87(6):809–11. https://doi.org/10.1002/1097-0215(20000915)87:6<809:aid-ijc8>3.0.iaco;2-u. [PubMed] [Google Scholar]
- 80.Christodoulou E, Nell RJ, Verdijk RM, Gruis NA, van der, Velden PA, van D, oorn R. Loss of wild-type CDKN2A is an early event in the development of melanoma in FAMMM syndrome. J Investig Dermatol. 2020;140(11):2298–301e3. doi: 10.1016/j.jid.2020.03.938. https://doi.org/10.1016/j.jid.2020.03.938. [DOI] [PubMed] [Google Scholar]
- 81.Ransohoff KJ, Jaju PD, Tang JY, Carbone M, Leachman S, Sarin KY. Familial skin cancer syndromes:Increased melanoma risk. J Am Acad Dermatol. 2016;74(3):423–34. doi: 10.1016/j.jaad.2015.09.070. https://doi.org/10.1016/j.jaad.2015.09.070. [DOI] [PubMed] [Google Scholar]
- 82.Louie BH, Kurzrock R. BAP1:Not just a BRCA1-associated protein. Cancer Treat Rev. 2020;90:102091. doi: 10.1016/j.ctrv.2020.102091. https://doi.org/10.1016/j.ctrv.2020.102091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Masoomian B, Shields JA, Shields CL. Overview of BAP1 acancer predisposition syndrome and the relationship to uveal melanoma. J Curr Ophthalmol. 2018;30(2):102–9. doi: 10.1016/j.joco.2018.02.005. https://doi.org/10.1016/j.joco.2018.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Laitman Y, Newberg J, Molho RB, Jin DX, Friedman E. The spectrum of tumors harboring BAP1 agene alterations. Cancer Genet. 2021;256-257:31–5. doi: 10.1016/j.cancergen.2021.03.007. https://doi.org/10.1016/j.cancergen.2021.03.007. [DOI] [PubMed] [Google Scholar]
- 85.Fallico M, Raciti G, Longo A, Reibaldi M, Bonfiglio V, Russo A, et al. Current molecular and clinical insights into uveal melanoma (Review) Int J Oncol. 2021;58(4):10. doi: 10.3892/ijo.2021.5190. https://doi.org/10.3892/ijo.2021.5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Goldberg Y, Laitman Y, Ben David M, Bazak L, Lidzbarsky G, Salmon LB, et al. Re-evaluating the pathogenicity of the c.783+2T>C BAP1 agermline variant . Hum Mutation. 2021;42(5):592–9. doi: 10.1002/humu.24189. https://doi.org/10.1002/humu.24189. [DOI] [PubMed] [Google Scholar]
- 87.Uner OE, See TR, Szalai E, Grossniklaus HE, Stålhammar G. Estimation of the timing of BAP1 amutation in uveal melanoma progression. Sci Rep. 2021;11(1):8923. doi: 10.1038/s41598-021-88390-6. https://doi.org/10.1038/s41598-021-88390-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang F, Luo M, Qu H, Cheng Y. BAP1 apromotes viability and migration of ECA109 cells through KLF5/CyclinD1/FGF-BP1. FEBS Open Bio. 2021;11(5):1497–503. doi: 10.1002/2211-5463.13105. https://doi.org/10.1002/2211-5463.13105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cabaret O, Perron E, Bressac-de Paillerets B, Soufir N, de l. a Fouchardière A. Occurrence of BAP1 agermline mutations in cutaneous melanocytic tumors with loss of BAP1-expression:A pilot study. Genes Chromosomes Cancer. 2017;56(9):691–4. doi: 10.1002/gcc.22473. https://doi.org/10.1002/gcc.22473. [DOI] [PubMed] [Google Scholar]
- 90.Skirton H, Cordier C, Ingvoldstad C, Taris N, Benjamin C. The role of the genetic counsellor:A systematic review of research evidence. Eur J Hum Genet. 2015;23(4):452–8. doi: 10.1038/ejhg.2014.116. https://doi.org/10.1038/ejhg.2014.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.McDaniels BA, Hianik RS, Bellcross C, Shaib WL, Switchenko J, Dixon MD, et al. The impact of genetic counseling educational tools on patients'knowledge of molecular testing terminology. J Cancer Educ. 2020;35(5):864–70. doi: 10.1007/s13187-019-01535-0. https://doi.org/10.1007/s13187-019-01535-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhu X, Leof ER, Rabe KG, McCormick JB, Petersen GM, Radecki B, reitkopf C. Psychological impact of learning CDKN2A variant status as a genetic research result. Public Health Genom. 2018;21((3-4)):154–63. doi: 10.1159/000496556. https://doi.org/10.1159/000496556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Taylor NJ, Mitra N, Qian L, Avril MF, Bishop DT, Bressac-de Paillerets B, et al. Estimating CDKN2A mutation carrier probability among global familial melanoma cases using GenoMELPREDICT. J Am Acad Dermatol. 2019;81(2):386–94. doi: 10.1016/j.jaad.2019.01.079. https://doi.org/10.1016/j.jaad.2019.01.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mittendorf EA, Philips AV, Meric-Bernstam F, Qiao N, Wu Y, Harrington S, et al. PD-L1 aexpression in triple-negative breast cancer. Cancer Immunol Res. 2014;2(4):361–70. doi: 10.1158/2326-6066.CIR-13-0127. https://doi.org/10.1158/2326-6066.iacir-13-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Holland EA, Lo S, Kelly B, Schmid H, Cust AE, Palmer JM, et al. FRAMe:Familial risk assessment of melanoma-a risk prediction tool to guide CDKN2A germline mutation testing in Australian familial melanoma. Familial Cancer. 2020;20(3):231–9. doi: 10.1007/s10689-020-00209-x. https://doi.org/10.1007/s10689-020-00209-x. [DOI] [PubMed] [Google Scholar]
- 96.Hemminki K, Srivastava A, Rachakonda S, Bandapalli O, Nagore E, Hemminki A, et al. Informing patients about their mutation tests:CDKN2A c.256G>A in melanoma as an example. Hereditary Cancer Clin Pract. 2020;18:15. doi: 10.1186/s13053-020-00146-x. https://doi.org/10.1186/s13053-020-00146-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Taber JM, Aspinwall LG, Drummond DM, Stump TK, Kohlmann W, Champine M, et al. Priority of risk (but not perceived magnitude of risk) predicts improved sun-protection behavior following genetic counseling for familial melanoma. Ann Behav Med. 2021;55(1):24–40. doi: 10.1093/abm/kaaa028. https://doi.org/10.1093/abm/kaaa028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wu YP, Aspinwall LG, Parsons B, Stump TK, Nottingham K, Kohlmann W, et al. Parent and child perspectives on family interactions related to melanoma risk and prevention after CDKN2A/p16 atesting of minor children . J Community Genet. 2020;11(3):321–9. doi: 10.1007/s12687-020-00453-9. https://doi.org/10.1007/s12687-020-00453-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stump TK, Aspinwall LG, Drummond DM, Taber JM, Kohlmann W, Champine M, et al. CDKN2A testing and genetic counseling promote reductions in objectively measured sun exposure one year later. Genet Med. 2020;22(1):26–34. doi: 10.1038/s41436-019-0608-9. https://doi.org/10.1038/s41436-019-0608-9. [DOI] [PMC free article] [PubMed] [Google Scholar]