

Abstract
The neurodegenerative diseases Alzheimer’s disease (AD) and Parkinson’s disease (PD) both have a myriad of risk factors including genetics, environmental exposures, and lifestyle. However, aging is the strongest risk factor for both diseases. Aging also profoundly influences the immune system, with immunosenescence perhaps the most prominent outcome. Through genetics, mouse models, and pathology, there is a growing appreciation of the role the immune system plays in neurodegenerative diseases. In this review, we explore the intersection of aging and the immune system in AD and PD.
Keywords: Alzheimer’s disease, Parkinson’s disease, Microglia, T cells, Immunosenescence
Introduction
Over the past decade, it has become clear that the immune system plays a central role in neurodegenerative diseases [1, 2]. Parkinson’s disease (PD) and Alzheimer’s disease (AD) are both neurodegenerative diseases with immune and genetic components, but they each have distinct pathological and clinical phenotypes. The study of the innate immune system in these diseases has become a major focus for the field. Despite these significant research efforts, most of the immune processes that have been implicated in AD and PD remain poorly understood. The resident central nervous system (CNS) innate immune cells, microglia, have been the focus of these efforts thus far. However, there is also a vital need to understand the role of infiltrating adaptive immune cells, specifically T cells, that may either maintain healthy neurons in the affected regions of the brain or lead to neuronal loss in disease contexts.
Aging is the largest risk factor for both AD and PD, and importantly also severely impacts the immune system’s fitness [3, 4]. By definition, the predominant form of AD, late-onset AD (LOAD), is a disease of aging as one must be over 65 years of age to be diagnosed with LOAD. In contrast, early-onset Alzheimer’s disease (EOAD) presents before age 65 and occurs in only 5% of cases [5]. The global burden of AD is only expected to grow as the world’s aging population continues to increase. Aging is also a primary risk factor for Parkinson’s disease [6]. The incidence of Parkinsonism (an umbrella term that refers to a group of disorders that cause movement disturbances as classically seen in Parkinson’s disease such as tremors, bradykinesia, and rigidity) increases in the elderly and becomes very common in populations over age 65 [7]. In addition, age of onset for PD affects disease progression, with those developing late-onset disease exhibiting more severe and rapid disease progression [8, 9]. Patients who were older at the time of disease onset exhibited more severe bradykinesia and rigidity and were more likely to have a balance disorder [8]. Hence, understanding the contribution of aging to these diseases is critical for segregating subsets of patients for correct clinical treatments.
Aging and the immune system
Reduced efficacy of vaccinations and increased susceptibility to viruses in older adults are classical examples of the effects of aging on the immune system [3, 10, 11]. These effects are usually attributed to immunosenescence, a process that leads to changes in all immune cells and an inability to mount productive responses against pathogens and vaccinations. Cellular senescence, now termed replicative senescence, was originally defined as the loss of proliferative ability in replication-competent cells. Replicative senescence is thought to be a protective mechanism, designed to prevent stressed cells from undergoing malignant transformation [12]. The signature proteins that are upregulated that maintain senescence are the cyclin-dependent kinase inhibitors p21WAF1/Cip1 and p16INK4a [13]. Senescent cells are increased in many different tissues with age, including the CNS [14–17]. The presence of senescent cells in the CNS may alter neuropathology in neurodegenerative disease, as clearance of senescent cells from the CNS was found to be beneficial in a mouse model of tauopathy [14].
Senescent cells also share a senescence-associated secretory phenotype (SASP). This includes pro-inflammatory cytokines, growth modulators, and chemotactic proteins. Interleukin-6 (IL-6), a pro-inflammatory cytokine, is perhaps the cytokine most associated with SASP. IL-8/C-X-C motif chemokine ligand 8 (CXCL8), a chemokine that recruits cells expressing C-X-C motif chemokine receptor 1 (CXCR1) as well as CXCR2 (often neutrophils), is also a key component of SASP. Immune cells produce many of the same molecules as seen in SASP in response to infections. These responses are designed to target damaged cells and pathogens and are normally robust, acute, and often self-limiting. This protective response is very much in contrast with chronic inflammation which tends to be long-lasting, and the host inflammatory response is responsible for tissue damage [18]. This low level, chronic inflammation which often occurs with aging and senescence has been termed inflammaging [19]. Because of the production of pro-inflammatory cytokines and chemokines that occurs with inflammation, the theory that anti-inflammatory treatments will be beneficial for diseases of aging has arisen [20]. But the pro-inflammatory cytokines may indicate a senescent immune response that has reduced functionality in other activities, such as phagocytosis [21]. In this case, either removing the senescent cells or pushing the cell back towards a robust acute response might be a better approach.
Alzheimer’s disease
AD is a progressive neurodegenerative disease and the most common cause of dementia. The condition can develop undetected for years until the clinical manifestation of cognitive impairment and memory issues appear, which then progressively worsen over time [22]. Physiological changes in the brain begin to occur years before the onset of symptoms [23, 24]. The symptoms of dementia cause deterioration of an individual’s independence, which dramatically impacts the daily lives of patients and their families. There are currently no viable therapeutic options to treat the cognitive symptoms of AD. Clinical trials for new drugs have not seen significant results in alleviating the disease [25]. Although recently approved drugs have been found to clear pathology, they struggle to improve cognitive outcomes [26]. This disconnect between the pathology and cognitive impairment indicates an unknown factor we have not accounted for thus far. A possible reason for the lack of treatment options in AD may be the divergence in AD pathology and cognitive impairment that has been described in the literature. Studies such as Boyle et al. have uncovered evidence that the known neuropathology of AD cannot fully account for dementia seen in these elderly patients. The majority of variation in cognitive decline in these patients remains unexplained [27].
Clinical presentations suggestive of Alzheimer’s dementia are classified by assessments of cognitive status. However, AD continues to be biologically defined by neuropathological hallmarks [28]. This pathology is characterized by aberrant extracellular amyloid-beta (Aβ) aggregates, which form diffuse and neuritic plaques, and hyperphosphorylated tau aggregates, which form intraneuronal neurofibrillary tangles [29]. These pathologies are progressively accompanied by loss of synapses, neuronal death, and gross brain atrophy [29]. In addition to examining the known hallmarks of AD, there has been a resurgence in examining the non-neuronal cells of the CNS in the AD brain. When Alois Alzheimer first characterized brains with AD, he described glial cells with abnormal morphology, which we now know to be microglia and astrocytes [30–33].
Parkinson’s disease
PD is the second most common neurodegenerative disorder affecting the elderly after AD [34, 35]. It is the most prevalent movement disorder, affecting over one million Americans and over four million individuals worldwide, and its incidence is expected to double by 2030 [36]. PD is a neurologic disease characterized by motor symptoms including tremors, rigidity, and postural instability. The clinical motor symptoms, such as shaking, rigidity, bradykinesia, and difficulty with walking and gait, presumably result from the accumulation of pathological processes that overwhelm the brain’s capacity to tolerate or compensate for their adverse effects. It is becoming clearer that the PD motor symptoms may only develop after years of ongoing neurodegenerative cell loss in the substantia nigra [37, 38].
The defining neuropathological features of PD are the loss of dopaminergic neurons in the substantia nigra and aggregation of alpha-synuclein protein, encoded by the SNCA gene, within neurons. Missense mutations in the SNCA gene as well as overproduction of wild-type alpha-synuclein can cause PD [39, 40]. Further reports found alpha-synuclein neuronal proteins present within Lewy bodies [41]. Lewy bodies are intracellular protein aggregates comprised mostly of alpha-synuclein, ubiquitin, and neurofilament, and their presence in neurons is a hallmark of PD pathology [42]. They are associated with activated microglia and dopaminergic neuron death [43, 44]. Indeed, alpha-synuclein induces microglial activation and morphological changes [45].
Parkinson’s research pioneer Arvid Carlsson first discovered that reduced dopamine levels caused PD-like symptoms [46], and subsequently proposed increasing dopamine levels through therapeutic intervention [47]. The dopamine precursor levodopa has since been successfully used to treat motor symptoms of PD. It may also be beneficial for cognitive decline in individuals with PD [48]. However, levodopa is not without its side effects, and there is a lack of effective strategies for the treatment of PD motor, cognitive, and behavioral symptoms beyond levodopa.
Microglia
In both AD and PD, there are indications from pathology, genetics, and murine studies that CNS-resident immune cells play an important role in disease pathogenesis. Several tissues have their own specialized, resident macrophage with generic innate immune functions as well as tissue-specific roles [49]. Microglia serve as the resident immune cell of the CNS. While once highly debated, it is now accepted that microglia originate from hematopoietic progenitors in the yolk sac and emigrate to the CNS before the development of the brain, in mice that are embryonic day 8.5–10.5 [50–53]. Microglia detect and react to any nearby pathological agents. They constantly survey the environment, respond to injury and pathogens, and perform tissue repair [54–56]. Microglia closely interact with neurons and impact neuronal function, as they partake in neurogenesis and synaptic pruning [54, 56, 57].
Dystrophic microglia are a morphologically described subset of microglia that appear to have fragmentation of their branches and beading in their processes [58, 59]. Interestingly, the number of microglia with a dystrophic morphology was found to be greater in cases with either Alzheimer’s disease, dementia with Lewy bodies, or limbic predominant age-related TDP-43 encephalopathy compared to age-matched controls [60]. It has been postulated that the dystrophic morphology of microglia represents senescent microglia [58]. Senescent microglia exhibit reduced phagocytic and migration capabilities in comparison to activated microglia [61]. Markers of senescence were found to be increased in microglia in patients with AD [62]. These deviations in microglia homeostasis may contribute to the pathology observed in AD and PD. The murine tauopathy model, MAPT P301S PS19 mice, exhibits an increased population of microglia and astrocytes expressing the cyclin-dependent kinase inhibitor p16INK4A, which is a marker of senescence. Interestingly, removal of these cells leads to a decrease of hyperphosphorylation of tau and decreased degeneration of cortical and hippocampal neurons [14]. It is possible that depleting the pool of senescent glial cells may similarly be a novel therapeutic approach to alleviate neuropathological progression in AD and PD.
As both activated microglia and senescent microglia produce inflammatory molecules, it is important to distinguish between the functionality and pathogenicity of activated versus senescent microglia. Microglial activation and senescence may both arise from inflammatory insults, as repeated LPS stimulation has been shown to induce senescence in the mouse microglial cell line BV2 [63]. It was previously thought that in the context of neurodegenerative disease, microglia were inappropriately activated, and accordingly, returning them to a homeostatic state would be protective. However, if inflammatory microglial signals in these disease contexts are really from senescent microglia rather than activated microglia, then either eliminating senescent cells or restoring them to a responsive and plastic state would be beneficial. A greater understanding of the activated and senescent microglial phenotypes is imperative for progress in neurodegenerative disease research.
AD microglia
After decades of research focused on neurons, genome-wide association studies (GWAS) have unveiled significant genetic risk for AD in innate immunity/microglia [64–66]. Many of these risk genes appear to be involved in phagocytosis. For example, CD33, TREM2, ABI3, INPP5D, and PLCG2 have all been demonstrated to influence microglia phagocytosis of amyloid-beta or amyloid-beta deposition in the brain [67–70]. In addition to genetics, there have now been several studies that have examined postmortem microglia transcriptomics from aged individuals with or without AD. The microglia from AD patients appear to have an enhanced aging phenotype [71]. Aging microglia have altered expression of genes involved in cell adhesion and actin cytoskeleton dynamics, which suggests a functional decrease in cell motility [72]. Several of the pathways linked to aged microglia are suggestive of a senescent phenotype (Fig. 1). Importantly, genes that were found to vary with aging had very little overlap with genes of aging in murine microglia, emphasizing the importance of studying microglial aging in the human system [73].
Fig. 1.

Pathways enriched in aged microglia. Two hundred seventy-one genes that were enriched in aged microglia were used to identify pathways that are upregulated in aged human microglia compared to microglia from younger individuals [72]. The figure was made in BioRender
Among the genes increased with aging across various studies is IL15. IL-15 is pro-inflammatory and helpful for a productive response against infections. It is also one of the cytokines that are secreted by senescent cells and is considered a part of SASP [74, 75]. Inhibition of IL-15 activity disrupts microglial activation and decreases cytokine and chemokine release [76]. IL-15/interleukin 15 receptor subunit alpha (IL-15RA) signaling may be neuroprotective, as IL-15RA knockout mice have increased motor neuron death after facial nerve axotomy [77]. IL-15 has wide-reaching effects on neural signaling in the brain, as it is thought to be essential in maintaining neurochemical homeostasis and is even thought to have anti-depressive effects on mouse behavior [78]. Additionally, IL-15 holds a very important immunological role in the development of natural killer cells and memory CD8 + T cells.
Another microglial gene that is increased in aging and AD is APOE. APOE has three main isoforms, named APOE e2, e3, and e4. Individuals having the APOE e4 isoform are at higher risk for LOAD [79]. Conversely, the APOE e2 haplotype is protective for AD and is associated with a decrease in the aging microglia phenotype [73]. APOE is a multifunctional protein with an important role in lipid transport [80]. Expression of APOE is upregulated early and implicated in the switch from homeostatic to neurodegenerative disease–associated microglia [81, 82]. While astrocytes are the main cell type for APOE production in the CNS, a recent mouse study suggested that microglial-produced APOE is important for synapse maintenance [83].
PD microglia
Alpha-synuclein aggregates are a hallmark of PD [84], and microglia help clear and degrade misfolded alpha-synuclein [85, 86]. The activation state of microglia modulates the rate of protein degradation, with LPS-activated microglia showing decreased alpha-synuclein degradation and increased cytoplasmic accumulation [86]. And while microglial phagocytosis of extracellular alpha-synuclein can lead to degradation, microglia can also release alpha-synuclein through exosomes. Exosomes from these microglia can transport alpha-synuclein to neurons and induce protein aggregation in the neurons [87]. This mechanism has also been proposed for microglial-mediated transport of tau in AD [88]. These studies indicate a complex role for microglia in both clearing and transferring alpha-synuclein pathology in PD.
Alpha-synuclein has repeatedly been shown to activate both murine and human microglia [87]. In vitro experiments using the murine microglial BV2 cell line as well as in vivo experiments in mice demonstrate that exosomes carrying alpha-synuclein derived from PD patients enter microglia and induce activation, leading to enhanced microglial cytokine release and NO production [89]. Evidence of microglial activation has been found in the substantia nigra of postmortem PD brains, and in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)–induced mouse model of PD [90, 91]. Activated microglia in the PD brain exhibit increased expression of ICAM-1, a pro-inflammatory intercellular adhesion molecule [92]. These microglia also express the cytokines TNF-alpha and IL-6 [92, 93]. In addition to these pro-inflammatory markers, McGeer et al. described an increase of HLA-DR positive cells in the substantia nigra of patients with PD [94]. HLA-DR, part of the antigen-presenting machinery used to activate T cells, is almost exclusively expressed on innate immune cells, and therefore, in the CNS is specifically expressed in microglia. The human leukocyte antigen (HLA) locus is also genetically associated as discussed below.
Genetics
The advent of unbiased genome-wide association studies has been critical in shifting the focus in neurodegenerative research away from a concentration on only neurons to one that includes other CNS cell types, specifically microglia. Many genes that have been associated with susceptibility to AD are significantly enriched in microglia compared to total brain tissue [73]. Risk genes for PD do not implicate microglia as clearly, as they are expressed in many CNS cell types. However, some of these genes are enriched in microglia, including the HLA region and CTSB. Understanding how one’s genetic background influences microglia behavior in neurodegenerative diseases will lead to a more comprehensive understanding of the disease mechanism, as well as identify potential therapeutic directions.
From work translating genetic associations to functional outcomes, it is becoming clearer that many of the genetic variants associated with LOAD lead to a hypofunctional innate immune system, specifically microglia [95]. For example, the AD-protective allele in the PLCG2 gene leads to a proline to arginine amino acid change, and this mutation results in enhanced immune signaling [96, 97]. The AD-risk alleles in the CD33, IL34, PILRA, and SPI1 loci lead to a dampened immune response [67, 68, 98–100]. Understanding the functional outcomes of these genetic associations has helped to reframe thinking about the approach to targeting the immune system in neurodegenerative disease. The previous thinking that the innate immune system needed to be suppressed is not supported by the genetic findings [20]. We hypothesize that aging, which leads to increased susceptibility to AD and PD, also induces a hypofunctional innate immune system and this, compounded with genetic risk factors, leads to a failure of microglia and the progression of AD.
The genetics of PD are not as clear in implicating innate immunity in the susceptibility to disease. Many of the genetic risk alleles in AD lead to a suppressed immune response, while some of the immune genetic hits in PD modify the pathways of antigen processing and presentation in immune cells. For example, CTSB, which is genetically linked to PD, codes for a lysosomal protease enriched in microglia, which is known to be important in processing antigens, including alpha-synuclein, for presentation by antigen-presenting cells to T cells [101, 102] [103]. Moreover, CTSB has been shown to influence the polarization of T cells through modulation of antigen-presenting cell cytokine production [104]. While we do not know about microglia specifically, the genetic association with PD in the CTSB locus leads to reduced expression levels in multiple tissues [103].
In addition to CTSB, other PD risk genes, LRRK2, GBA, and BAG-3, have all been implicated in antigen presentation as well [105–107]. We have shown that the common genetic association in the LRRK2 locus leads to increased expression of LRRK2 with the risk allele in a model of human microglia [108]. LRRK2 KO mice have innate immune cells that produce more proinflammatory cytokines upon activation and greater T cell proliferation in an antigen presentation assay [105], suggesting that LRRK2 expression may inhibit productive antigen presentation and activation of T cells. Whether the genetic variation changes the spectrum of antigens presented in the CNS of individuals with PD, and how this variation influences T cell phenotypes, is unknown.
The HLA complex region is an immune-specific, genetically associated locus for both AD and PD [109–113]. The proteins encoded in the HLA region present peptides (antigens) to T cells to activate antigen-specific immune responses. HLA is the strongest genetic association for most autoimmune diseases, and the pairing of particular autoimmune risk HLA genes with specific antigens can lead to inappropriate immune activation and destruction of very specific, disease-defining cell types. The HLA association with AD and PD implies that antigen presentation by innate immune cells (including microglia) to T cells is an important part of the susceptibility to both diseases.
One PD-focused study used deep sequencing of the HLA region to identify amino acid changes in the protein coded for by HLA-DRB1 as a genetic risk for PD [114]. HLA-DRB1 codes for part of MHC class II, which is the molecule on antigen-presenting cells responsible for presenting antigens to CD4 + helper T cells. Interestingly, the authors found an interaction between the protective genetic association and a history of smoking, which is a known protective factor for PD [115]. The authors hypothesize that smoking may lead to post-translational modifications of proteins such as alpha-synuclein, which then changes the binding affinity to MHC class II depending on the genetically associated amino acid changes. In a large study dissecting the HLA association with AD, the group found independent MHC class I and class II associations, suggesting that antigen presentation to both helper CD4 + T cells and CD8 + cytotoxic T cells is important to the disease susceptibility [112].
T cells in AD and PD
Our understanding of microglial diversity in form and function is currently only rudimentary in the human system, especially their antigen-presenting function [116]. Microglia have the machinery to process and present antigens to both CD4 + and CD8 + T cells via MHC class I and II proteins (Fig. 2) [117, 118]. Infiltrating T cells have been described in the hippocampus in AD and the substantia nigra in PD, both primary foci of neurodegeneration in each disease respectively. Interestingly, in addition to T cell infiltration, these regions are also enriched with MHC class I and II expression in AD and PD. As previously described, the HLA region, which encodes MHC proteins, is genetically associated with AD and PD. In this way, microglia T cell interactions are implicated through several lines of evidence in the AD and PD brain [119–121]. A study looking at T cells in the CSF of PD and AD patients found clonally expanded CD8 + T cells in both diseases, suggesting that the T cells are infiltrating in response to a particular antigen [122]. In a study using the 5XFAD AD mouse model, intracerebroventricular injection of amyloid-beta-specific T cells induced MHC class II expression on microglia. These MHC class II–positive microglia have a neuroprotective phenotype with increased plaque-clearing abilities [123]. This highlights the reciprocal signaling between immune cells.
Fig. 2.
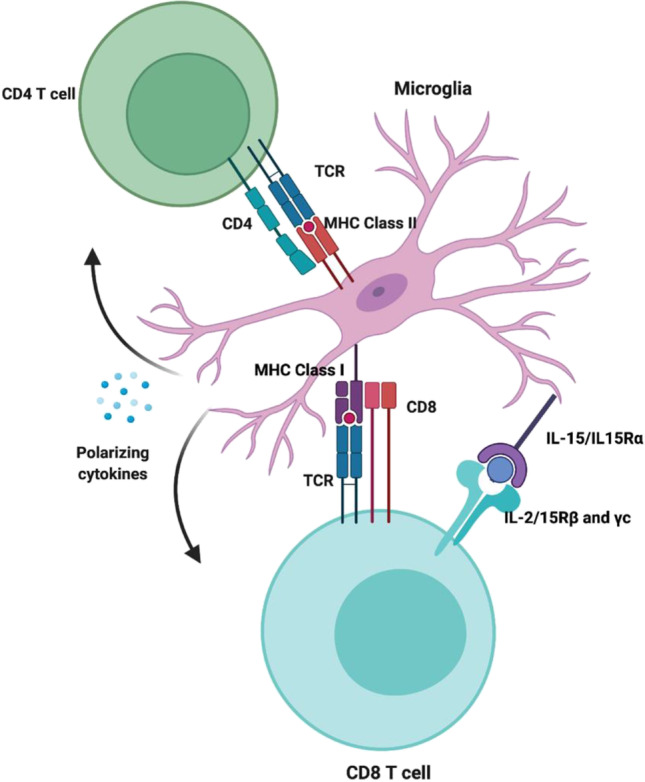
Microglia have antigen-presenting functions. Both CD4 + and CD8 + T cells infiltrate key CNS areas of neurodegeneration in AD and PD. Genetic risk for both diseases has been identified in the region that codes for the MHC proteins, which present antigens to T cells. Microglia also produce cytokines that can support the survival and polarization of T cells. Aged microglia produce more IL-15, an activating T cell cytokine. The figure was made in BioRender
T cells are drastically affected by aging. Atrophy of the thymus with age is a clear example of this phenomenon and leads to a lack of new pluripotent T cells to repopulate the naïve T cell compartment [124]. The diversity of the T-cell receptor (TCR) repertoire found in 20–35-year-olds is reduced by 10–25% in 70–80-year-olds [125]. The phenotype of memory T cells in aging also changes, with an increase of clonality and T cells lacking the expression of necessary co-stimulatory molecules [126].
Peripheral immune aberrations, particularly in lymphocyte subsets, are abundant in PD patients. Specifically, it has been shown that peripheral T cells are diminished in PD patients [127, 128]. In AD, there is an increase in a particular type of CD8 + T cell that has been associated with chronic viral infections, and expansion of this T cell subset was found to be correlated with cognitive decline [122]. These effector memory T cells, which are defined by re-expression of the CD45RA protein, are termed TEMRA. These cells tend to be highly responsive to IL-15, which has been found to be increased in microglia in AD [129]. They have also been associated with a senescent phenotype; however, the senescent population is likely a subset of TEMRA [130]. Interestingly, these TEMRA T cells were found to be reduced in the circulation of individuals newly diagnosed with PD [131]. We have much work to do to understand the nuances of the role of various T cell populations in both AD and PD.
Conclusions
A better understanding of the influence of the aging immune system on neurodegenerative diseases will be helpful in the search for novel therapeutic approaches. In terms of infiltrating T cells, it will be imperative to understand which populations are detrimental and which may be protective in disease contexts. For example, in one study, it was found that higher plasma IL-12p70 and IFNg in cognitively normal individuals was associated with reduced future cognitive decline [132]. IL-12p70 polarizes T cells towards the pro-inflammatory INFγ-producing T cell subset, suggesting that this subset of T cells may be protective. Defining how microglia act as antigen-presenting cells in key areas of neurodegeneration is also important to comprehensively understand their contribution to disease processes. The intersection of aging and genetics is also very likely to be critical to any immune dysregulation. The development of therapies that modify the role of these cells’ contribution to disease pathophysiology or enhance disease resistance will facilitate health and well-being among aging adults and markedly reduce health care expenditures.
Declarations
Competing interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: EMB is a founder of IMAD Therapeutics. KSH has no conflicts of interest to disclose.
Footnotes
This article is a contribution to the special issue on: Neuroimmune Interactions in Health and Disease - Guest Editors: David Hafler & Lauren Sansing
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Molteni M, Rossetti C. Neurodegenerative diseases: the immunological perspective. J Neuroimmunol. 2017;313:109–115. doi: 10.1016/j.jneuroim.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Doty KR, Guillot-Sestier MV, Town T. The role of the immune system in neurodegenerative disorders: adaptive or maladaptive? Brain Res. 2015;1617:155–173. doi: 10.1016/j.brainres.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Del Giudice G, et al. Fighting against a protean enemy: immunosenescence, vaccines, and healthy aging. NPJ Aging Mech Dis. 2018;4:1. doi: 10.1038/s41514-017-0020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wyss-Coray T. Ageing, neurodegeneration and brain rejuvenation. Nature. 2016;539(7628):180–186. doi: 10.1038/nature20411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Association, A.s., 2015 Alzheimer’s disease facts and figures. Alzheimers Dement, 2015. 11(3): p. 332–84. [DOI] [PubMed]
- 6.Collier TJ, Kanaan NM, Kordower JH. Ageing as a primary risk factor for Parkinson’s disease: evidence from studies of non-human primates. Nat Rev Neurosci. 2011;12(6):359–366. doi: 10.1038/nrn3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bennett DA, et al. Prevalence of parkinsonian signs and associated mortality in a community population of older people. N Engl J Med. 1996;334(2):71–76. doi: 10.1056/NEJM199601113340202. [DOI] [PubMed] [Google Scholar]
- 8.Hely MA, et al. Age at onset: the major determinant of outcome in Parkinson’s disease. Acta Neurol Scand. 1995;92(6):455–463. doi: 10.1111/j.1600-0404.1995.tb00480.x. [DOI] [PubMed] [Google Scholar]
- 9.Jankovic J, Kapadia AS. Functional decline in Parkinson disease. Arch Neurol. 2001;58(10):1611–1615. doi: 10.1001/archneur.58.10.1611. [DOI] [PubMed] [Google Scholar]
- 10.Weinberger B, Grubeck-Loebenstein B. Vaccines for the elderly. Clin Microbiol Infect. 2012;18(Suppl 5):100–108. doi: 10.1111/j.1469-0691.2012.03944.x. [DOI] [PubMed] [Google Scholar]
- 11.Leng J, Goldstein DR. Impact of aging on viral infections. Microbes Infect. 2010;12(14–15):1120–1124. doi: 10.1016/j.micinf.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campisi, J. and F. d'Adda di Fagagna, Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol, 2007. 8(9): p. 729–40. [DOI] [PubMed]
- 13.Ohtani N, et al. The p16INK4a-RB pathway: molecular link between cellular senescence and tumor suppression. J Med Invest. 2004;51(3–4):146–153. doi: 10.2152/jmi.51.146. [DOI] [PubMed] [Google Scholar]
- 14.Bussian TJ, et al. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature. 2018;562(7728):578–582. doi: 10.1038/s41586-018-0543-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dimri GP, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92(20):9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paradis V, et al. Replicative senescence in normal liver, chronic hepatitis C, and hepatocellular carcinomas. Hum Pathol. 2001;32(3):327–332. doi: 10.1053/hupa.2001.22747. [DOI] [PubMed] [Google Scholar]
- 17.Melk A, et al. Cell senescence in rat kidneys in vivo increases with growth and age despite lack of telomere shortening. Kidney Int. 2003;63(6):2134–2143. doi: 10.1046/j.1523-1755.2003.00032.x. [DOI] [PubMed] [Google Scholar]
- 18.Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140(6):871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 19.Mogilenko, D.A., I. Shchukina, and M.N. Artyomov, Immune ageing at single-cell resolution. Nat Rev Immunol, 2021. [DOI] [PMC free article] [PubMed]
- 20.Akiyama H, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21(3):383–421. doi: 10.1016/S0197-4580(00)00124-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greenwood, E.K. and D.R. Brown, Senescent microglia: the key to the ageing brain? Int J Mol Sci, 2021. 22(9). [DOI] [PMC free article] [PubMed]
- 22.Burns A, Iliffe S. Alzheimer’s disease. BMJ. 2009;338:b158. doi: 10.1136/bmj.b158. [DOI] [PubMed] [Google Scholar]
- 23.Beason-Held LL, et al. Changes in brain function occur years before the onset of cognitive impairment. J Neurosci. 2013;33(46):18008–18014. doi: 10.1523/JNEUROSCI.1402-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raskin J, et al. Neurobiology of Alzheimer’s disease: integrated molecular, physiological, anatomical, biomarker, and cognitive dimensions. Curr Alzheimer Res. 2015;12(8):712–722. doi: 10.2174/1567205012666150701103107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014;6(4):37. doi: 10.1186/alzrt269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mullard A. Landmark Alzheimer’s drug approval confounds research community. Nature. 2021;594(7863):309–310. doi: 10.1038/d41586-021-01546-2. [DOI] [PubMed] [Google Scholar]
- 27.Boyle PA, et al. Much of late life cognitive decline is not due to common neurodegenerative pathologies. Ann Neurol. 2013;74(3):478–489. doi: 10.1002/ana.23964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jack CR, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14(4):535–562. doi: 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duyckaerts C, Delatour B, Potier MC. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009;118(1):5–36. doi: 10.1007/s00401-009-0532-1. [DOI] [PubMed] [Google Scholar]
- 30.Alzheimer A, et al. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin Anat. 1995;8(6):429–431. doi: 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- 31.Graeber MB, et al. Rediscovery of the case described by Alois Alzheimer in 1911: historical, histological and molecular genetic analysis. Neurogenetics. 1997;1(1):73–80. doi: 10.1007/s100480050011. [DOI] [PubMed] [Google Scholar]
- 32.Maragakis NJ, Rothstein JD. Mechanisms of disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol. 2006;2(12):679–689. doi: 10.1038/ncpneuro0355. [DOI] [PubMed] [Google Scholar]
- 33.Mosher KI, Wyss-Coray T. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem Pharmacol. 2014;88(4):594–604. doi: 10.1016/j.bcp.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39(6):889–909. doi: 10.1016/S0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 35.Dexter DT, Jenner P. Parkinson disease: from pathology to molecular disease mechanisms. Free Radic Biol Med. 2013;62:132–144. doi: 10.1016/j.freeradbiomed.2013.01.018. [DOI] [PubMed] [Google Scholar]
- 36.Dorsey ER, et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 2007;68(5):384–386. doi: 10.1212/01.wnl.0000247740.47667.03. [DOI] [PubMed] [Google Scholar]
- 37.Hawkes CH. Parkinson’s disease and aging: same or different process? Mov Disord. 2008;23(1):47–53. doi: 10.1002/mds.21766. [DOI] [PubMed] [Google Scholar]
- 38.Olanow CW, Tatton WG. Etiology and pathogenesis of Parkinson’s disease. Annu Rev Neurosci. 1999;22:123–144. doi: 10.1146/annurev.neuro.22.1.123. [DOI] [PubMed] [Google Scholar]
- 39.Polymeropoulos MH, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 40.Singleton AB, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302(5646):841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 41.Spillantini MG, et al. Alpha-synuclein in Lewy bodies. Nature. 1997;388(6645):839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 42.Gibb WR, Lees AJ. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1988;51(6):745–752. doi: 10.1136/jnnp.51.6.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jellinger KA. More frequent Lewy bodies but less frequent Alzheimer-type lesions in multiple system atrophy as compared to age-matched control brains. Acta Neuropathol. 2007;114(3):299–303. doi: 10.1007/s00401-007-0227-4. [DOI] [PubMed] [Google Scholar]
- 44.Cookson MR. alpha-Synuclein and neuronal cell death. Mol Neurodegener. 2009;4:9. doi: 10.1186/1750-1326-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanchez-Guajardo V, et al. Microglia acquire distinct activation profiles depending on the degree of alpha-synuclein neuropathology in a rAAV based model of Parkinson’s disease. PLoS ONE. 2010;5(1):e8784. doi: 10.1371/journal.pone.0008784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.CARLSSON, A., M. LINDQVIST, and T. MAGNUSSON, 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature, 1957. 180(4596): p. 1200. [DOI] [PubMed]
- 47.Carlsson A. A half-century of neurotransmitter research: impact on neurology and psychiatry (Nobel lecture) ChemBioChem. 2001;2(7–8):484–493. doi: 10.1002/1439-7633(20010803)2:7/8<484::AID-CBIC484>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 48.Ikeda M, Kataoka H, Ueno S. Can levodopa prevent cognitive decline in patients with Parkinson’s disease? Am J Neurodegener Dis. 2017;6(2):9–14. [PMC free article] [PubMed] [Google Scholar]
- 49.Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. 2014;41(1):21–35. doi: 10.1016/j.immuni.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alliot F, Godin I, Pessac B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res. 1999;117(2):145–152. doi: 10.1016/S0165-3806(99)00113-3. [DOI] [PubMed] [Google Scholar]
- 51.Askew K, Gomez-Nicola D. A story of birth and death: insights into the formation and dynamics of the microglial population. Brain Behav Immun. 2018;69:9–17. doi: 10.1016/j.bbi.2017.03.009. [DOI] [PubMed] [Google Scholar]
- 52.Ginhoux F, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ginhoux F, et al. Origin and differentiation of microglia. Front Cell Neurosci. 2013;7:45. doi: 10.3389/fncel.2013.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hammond TR, Robinton D, Stevens B. Microglia and the brain: complementary partners in development and disease. Annu Rev Cell Dev Biol. 2018;34:523–544. doi: 10.1146/annurev-cellbio-100616-060509. [DOI] [PubMed] [Google Scholar]
- 55.Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10(11):1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 56.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 57.Schafer DP, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Streit WJ, et al. Dystrophic microglia in the aging human brain. Glia. 2004;45(2):208–212. doi: 10.1002/glia.10319. [DOI] [PubMed] [Google Scholar]
- 59.Streit WJ, et al. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2009;118(4):475–485. doi: 10.1007/s00401-009-0556-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shahidehpour RK, et al. Dystrophic microglia are associated with neurodegenerative disease and not healthy aging in the human brain. Neurobiol Aging. 2021;99:19–27. doi: 10.1016/j.neurobiolaging.2020.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Caldeira C, et al. Key aging-associated alterations in primary microglia response to beta-amyloid stimulation. Front Aging Neurosci. 2017;9:277. doi: 10.3389/fnagi.2017.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hu Y, et al. Replicative senescence dictates the emergence of disease-associated microglia and contributes to Aβ pathology. Cell Rep. 2021;35(10):109228. doi: 10.1016/j.celrep.2021.109228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu HM, et al. Repeated lipopolysaccharide stimulation induces cellular senescence in BV2 cells. NeuroImmunoModulation. 2012;19(2):131–136. doi: 10.1159/000330254. [DOI] [PubMed] [Google Scholar]
- 64.Lambert JC, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 65.Hollingworth P, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43(5):429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Naj AC, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43(5):436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bradshaw EM, et al. CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat Neurosci. 2013;16(7):848–850. doi: 10.1038/nn.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Griciuc A, et al. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78(4):631–643. doi: 10.1016/j.neuron.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Salih, D.A., et al., Genetic variability in response to amyloid beta deposition influences Alzheimer’s disease risk. Brain Commun, 2019. 1(1): p. fcz022. [DOI] [PMC free article] [PubMed]
- 70.Yeh FL, et al. TREM2 Binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron. 2016;91(2):328–340. doi: 10.1016/j.neuron.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 71.Srinivasan K, et al. Alzheimer’s patient microglia exhibit enhanced aging and unique transcriptional activation. Cell Rep. 2020;31(13):107843. doi: 10.1016/j.celrep.2020.107843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Galatro TF, et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat Neurosci. 2017;20(8):1162–1171. doi: 10.1038/nn.4597. [DOI] [PubMed] [Google Scholar]
- 73.Olah M, et al. A transcriptomic atlas of aged human microglia. Nat Commun. 2018;9(1):539. doi: 10.1038/s41467-018-02926-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Coppé JP, et al. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Perera PY, et al. The role of interleukin-15 in inflammation and immune responses to infection: implications for its therapeutic use. Microbes Infect. 2012;14(3):247–261. doi: 10.1016/j.micinf.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gomez-Nicola D, Valle-Argos B, Nieto-Sampedro M. Blockade of IL-15 activity inhibits microglial activation through the NFkappaB, p38, and ERK1/2 pathways, reducing cytokine and chemokine release. Glia. 2010;58(3):264–276. doi: 10.1002/glia.20920. [DOI] [PubMed] [Google Scholar]
- 77.Huang Z, Ha GK, Petitto JM. IL-15 and IL-15R alpha gene deletion: effects on T lymphocyte trafficking and the microglial and neuronal responses to facial nerve axotomy. Neurosci Lett. 2007;417(2):160–164. doi: 10.1016/j.neulet.2007.01.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu X, et al. Interleukin-15 affects serotonin system and exerts antidepressive effects through IL15Rα receptor. Psychoneuroendocrinology. 2011;36(2):266–278. doi: 10.1016/j.psyneuen.2010.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Saunders AM, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43(8):1467–1472. doi: 10.1212/WNL.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 80.LaDu MJ, et al. Apolipoprotein E and apolipoprotein E receptors modulate A beta-induced glial neuroinflammatory responses. Neurochem Int. 2001;39(5–6):427–434. doi: 10.1016/S0197-0186(01)00050-X. [DOI] [PubMed] [Google Scholar]
- 81.Krasemann S, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47(3):566–581.e9. doi: 10.1016/j.immuni.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Keren-Shaul H, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169(7):1276–1290.e17. doi: 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 83.Henningfield CM, et al. Microglia-specific ApoE knock-out does not alter Alzheimer’s disease plaque pathogenesis or gene expression. Glia. 2022;70(2):287–302. doi: 10.1002/glia.24105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dickson, D.W., Parkinson’s disease and parkinsonism: neuropathology. Cold Spring Harb Perspect Med, 2012. 2(8). [DOI] [PMC free article] [PubMed]
- 85.Brück D, et al. Glia and alpha-synuclein in neurodegeneration: a complex interaction. Neurobiol Dis. 2016;85:262–274. doi: 10.1016/j.nbd.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee HJ, et al. Clearance and deposition of extracellular alpha-synuclein aggregates in microglia. Biochem Biophys Res Commun. 2008;372(3):423–428. doi: 10.1016/j.bbrc.2008.05.045. [DOI] [PubMed] [Google Scholar]
- 87.Guo M, et al. Microglial exosomes facilitate α-synuclein transmission in Parkinson’s disease. Brain. 2020;143(5):1476–1497. doi: 10.1093/brain/awaa090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Asai H, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015;18(11):1584–1593. doi: 10.1038/nn.4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xia Y, et al. Microglia as modulators of exosomal alpha-synuclein transmission. Cell Death Dis. 2019;10(3):174. doi: 10.1038/s41419-019-1404-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Banati RB, Daniel SE, Blunt SB. Glial pathology but absence of apoptotic nigral neurons in long-standing Parkinson’s disease. Mov Disord. 1998;13(2):221–227. doi: 10.1002/mds.870130205. [DOI] [PubMed] [Google Scholar]
- 91.Członkowska A, et al. Microglial reaction in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induced Parkinson’s disease mice model. Neurodegeneration. 1996;5(2):137–143. doi: 10.1006/neur.1996.0020. [DOI] [PubMed] [Google Scholar]
- 92.Imamura K, et al. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 2003;106(6):518–526. doi: 10.1007/s00401-003-0766-2. [DOI] [PubMed] [Google Scholar]
- 93.Hunot S, et al. Nitric oxide synthase and neuronal vulnerability in Parkinson’s disease. Neuroscience. 1996;72(2):355–363. doi: 10.1016/0306-4522(95)00578-1. [DOI] [PubMed] [Google Scholar]
- 94.McGeer PL, et al. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38(8):1285–1291. doi: 10.1212/WNL.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 95.Chatila, Z.K. and E.M. Bradshaw, Alzheimer’s disease genetics: a dampened microglial response? Neuroscientist, 2021: p. 10738584211024531. [DOI] [PubMed]
- 96.Magno L, et al. Alzheimer’s disease phospholipase C-gamma-2 (PLCG2) protective variant is a functional hypermorph. Alzheimers Res Ther. 2019;11(1):16. doi: 10.1186/s13195-019-0469-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Takalo M, et al. The Alzheimer’s disease-associated protective Plcγ2-P522R variant promotes immune functions. Mol Neurodegener. 2020;15(1):52. doi: 10.1186/s13024-020-00402-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.de Rojas I, et al. Common variants in Alzheimer’s disease and risk stratification by polygenic risk scores. Nat Commun. 2021;12(1):3417. doi: 10.1038/s41467-021-22491-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rathore N, et al. Paired Immunoglobulin-like Type 2 Receptor Alpha G78R variant alters ligand binding and confers protection to Alzheimer’s disease. PLoS Genet. 2018;14(11):e1007427. doi: 10.1371/journal.pgen.1007427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Huang, K.L., et al., A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat Neurosci, 2017. 20(8): p. 1052–1061. [DOI] [PMC free article] [PubMed]
- 101.Zhang T, et al. Lysosomal cathepsin B plays an important role in antigen processing, while cathepsin D is involved in degradation of the invariant chain inovalbumin-immunized mice. Immunology. 2000;100(1):13–20. doi: 10.1046/j.1365-2567.2000.00000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.McGlinchey RP, Lee JC. Cysteine cathepsins are essential in lysosomal degradation of α-synuclein. Proc Natl Acad Sci U S A. 2015;112(30):9322–9327. doi: 10.1073/pnas.1500937112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chang D, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet. 2017;49(10):1511–1516. doi: 10.1038/ng.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gonzalez-Leal IJ, et al. Cathepsin B in antigen-presenting cells controls mediators of the Th1 immune response during Leishmania major infection. PLoS Negl Trop Dis. 2014;8(9):e3194. doi: 10.1371/journal.pntd.0003194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kubo, M., et al., Leucine-rich repeat kinase 2 controls inflammatory cytokines production through NF-κB phosphorylation and antigen presentation in bone marrow-derived dendritic cells. Int J Mol Sci, 2020. 21(5). [DOI] [PMC free article] [PubMed]
- 106.Liu J, et al. Gaucher disease gene GBA functions in immune regulation. Proc Natl Acad Sci U S A. 2012;109(25):10018–10023. doi: 10.1073/pnas.1200941109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wenger T, et al. Autophagy inhibition promotes defective neosynthesized proteins storage in ALIS, and induces redirection toward proteasome processing and MHCI-restricted presentation. Autophagy. 2012;8(3):350–363. doi: 10.4161/auto.18806. [DOI] [PubMed] [Google Scholar]
- 108.Ryan, K.J., et al., A human microglia-like cellular model for assessing the effects of neurodegenerative disease gene variants. Sci Transl Med, 2017. 9(421). [DOI] [PMC free article] [PubMed]
- 109.Hamza TH, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet. 2010;42(9):781–785. doi: 10.1038/ng.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wissemann WT, et al. Association of Parkinson disease with structural and regulatory variants in the HLA region. Am J Hum Genet. 2013;93(5):984–993. doi: 10.1016/j.ajhg.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nalls MA, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet. 2014;46(9):989–993. doi: 10.1038/ng.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Steele NZ, et al. Fine-mapping of the human leukocyte antigen locus as a risk factor for Alzheimer disease: a case-control study. PLoS Med. 2017;14(3):e1002272. doi: 10.1371/journal.pmed.1002272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jansen IE, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51(3):404–413. doi: 10.1038/s41588-018-0311-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hollenbach JA, et al. A specific amino acid motif of. Proc Natl Acad Sci U S A. 2019;116(15):7419–7424. doi: 10.1073/pnas.1821778116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Quik M. Smoking, nicotine and Parkinson’s disease. Trends Neurosci. 2004;27(9):561–568. doi: 10.1016/j.tins.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 116.Mastroeni D, et al. Laser-captured microglia in the Alzheimer’s and Parkinson’s brain reveal unique regional expression profiles and suggest a potential role for hepatitis B in the Alzheimer’s brain. Neurobiol Aging. 2018;63:12–21. doi: 10.1016/j.neurobiolaging.2017.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shaked I, et al. Early activation of microglia as antigen-presenting cells correlates with T cell-mediated protection and repair of the injured central nervous system. J Neuroimmunol. 2004;146(1–2):84–93. doi: 10.1016/j.jneuroim.2003.10.049. [DOI] [PubMed] [Google Scholar]
- 118.Das R, Chinnathambi S. Microglial priming of antigen presentation and adaptive stimulation in Alzheimer’s disease. Cell Mol Life Sci. 2019;76(19):3681–3694. doi: 10.1007/s00018-019-03132-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.McGeer PL, et al. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett. 1987;79(1–2):195–200. doi: 10.1016/0304-3940(87)90696-3. [DOI] [PubMed] [Google Scholar]
- 120.Brochard V, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest. 2009;119(1):182–192. doi: 10.1172/JCI36470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dressman, D. and W. Elyaman, T cells: a growing universe of roles in neurodegenerative diseases. Neuroscientist, 2021: p. 10738584211024907. [DOI] [PubMed]
- 122.Gate D, et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature. 2020;577(7790):399–404. doi: 10.1038/s41586-019-1895-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mittal, K., et al., CD4 T cells induce a subset of MHCII-expressing microglia that attenuates Alzheimer pathology. iScience, 2019. 16: p. 298–311. [DOI] [PMC free article] [PubMed]
- 124.Palmer DB. The effect of age on thymic function. Front Immunol. 2013;4:316. doi: 10.3389/fimmu.2013.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Qi Q, et al. Diversity and clonal selection in the human T-cell repertoire. Proc Natl Acad Sci U S A. 2014;111(36):13139–13144. doi: 10.1073/pnas.1409155111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Herndler-Brandstetter D, et al. The impact of aging on memory T cell phenotype and function in the human bone marrow. J Leukoc Biol. 2012;91(2):197–205. doi: 10.1189/jlb.0611299. [DOI] [PubMed] [Google Scholar]
- 127.Bhatia D, et al. T-cell dysregulation is associated with disease severity in Parkinson’s disease. J Neuroinflammation. 2021;18(1):250. doi: 10.1186/s12974-021-02296-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fiszer U, et al. Parkinson’s disease and immunological abnormalities: increase of HLA-DR expression on monocytes in cerebrospinal fluid and of CD45RO+ T cells in peripheral blood. Acta Neurol Scand. 1994;90(3):160–166. doi: 10.1111/j.1600-0404.1994.tb02699.x. [DOI] [PubMed] [Google Scholar]
- 129.Pangrazzi L, et al. Increased IL-15 production and accumulation of highly differentiated CD8. Front Immunol. 2017;8:715. doi: 10.3389/fimmu.2017.00715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Verma K, et al. Human CD8+ CD57- TEMRA cells: too young to be called “old”. PLoS ONE. 2017;12(5):e0177405. doi: 10.1371/journal.pone.0177405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kouli A, et al. T lymphocyte senescence is attenuated in Parkinson’s disease. J Neuroinflammation. 2021;18(1):228. doi: 10.1186/s12974-021-02287-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yang, H.S., et al., Plasma IL-12/IFN-γ axis predicts cognitive trajectories in cognitively unimpaired older adults. Alzheimers Dement, 2021. [DOI] [PMC free article] [PubMed]