

Abstract
In addition to the contribution of cancer cells, the solid tumor microenvironment (TME) has a critical role in determining tumor expansion, antitumor immunity and the response to immunotherapy. Understanding the details of the complex interplay between cancer cells and components of the TME provides an unprecedented opportunity to explore combination therapy for intervening into the immune landscape to improve immunotherapy outcome. One approach is the introduction of multifunctional nanocarriers, capable of delivering drug combinations that provide immunogenic stimuli for improvement of tumor antigen presentation, contemporaneous with the delivery of co-formulated drug or synthetic molecules that provide immune danger signals or interfere in immune escape, immune suppressive and T-cell exclusion pathways. This forward-looking perspective will discuss the use of lipid bilayer encapsulated liposomes and mesoporous silica nanoparticles for combination immunotherapy of the heterogeneous immune landscapes in pancreatic ductal adenocarcinoma (PDAC) and triple negative breast cancer (TNBC). We describe how the combination of remote drug loading and lipid bilayer encapsulation is used for the synthesis of synergistic drug combinations that induce immunogenic cell death, interfere in the PD-1/PD-L1 axis, inhibit the indoleamine-pyrrole 2,3-dioxygenase (IDO-1) immune metabolic pathway, restore spatial access to activated T-cells to the cancer cells, or reduce the impact of immunosuppressive stromal components. We show how an integration of current knowledge and future discovery can be used for a rational approach to nano-enabled cancer immunotherapy.
Keywords: nanocarrier, liposomes, silicasomes, combination therapy, immune landscapes, pancreas cancer, triple negative breast cancer, immunogenic cell death, immune escape, immune suppression, spatial distribution
A. Introduction
A great deal of effort is currently directed to characterizing the tumor microenvironment (TME) of solid cancers, including identification of heterogeneous immune landscapes to initiate custom-designed immunotherapy.1–2 While the introduction of immune checkpoint inhibitors has advanced immunotherapy as one of the cornerstones of cancer treatment, we have come to understand that its success depends on the immunogenic nature of the tumor as well as the makeup of the tumor immune microenvironment (TIME).2–5 Generally speaking, immune inflamed or “hot” tumors are associated with better responses to immune checkpoint inhibitors (ICIs) in cancers such as melanoma, non-small cell lung cancer, head and neck cancer, kidney, liver, and bladder cancer (Figure 1).4–7 In contrast, immunological “cold” tumors exhibit a paucity of T-cell infiltrates (also referred to as “immune desert” landscapes) or present a phenotype where T-cells may be present but excluded from the tumor core, a.k.a. “immune excluded” landscapes (Figure 1).8–9 In addition to spatial exclusion, there are a number of additional reasons why the function of tumor-infiltrating T lymphocytes (TIL) at the tumor site may be constrained from cytotoxic killing, e.g., as a result of the expression of immune checkpoint receptors (e.g., PD-1, CTLA-4, LAG-3, TIM-3 and the adenosine A2A receptor),10–12 or immune metabolic interference by the indoleamine-pyrrole 2,3-dioxygenase or IDO-1) pathway.13–14 It is also important to consider the role of the dysplastic tumor stroma in exerting immune suppressive effects in the TME through the participation of cancer-associated fibroblasts (CAFs), myeloid derived suppressor cells (MDSC), FoxP3 regulatory T-cells and M2 tumor-associated macrophages (TAM).15
Figure 1. The importance of heterogeneous tumor immune microenvironments (TIMES) for immunotherapy.
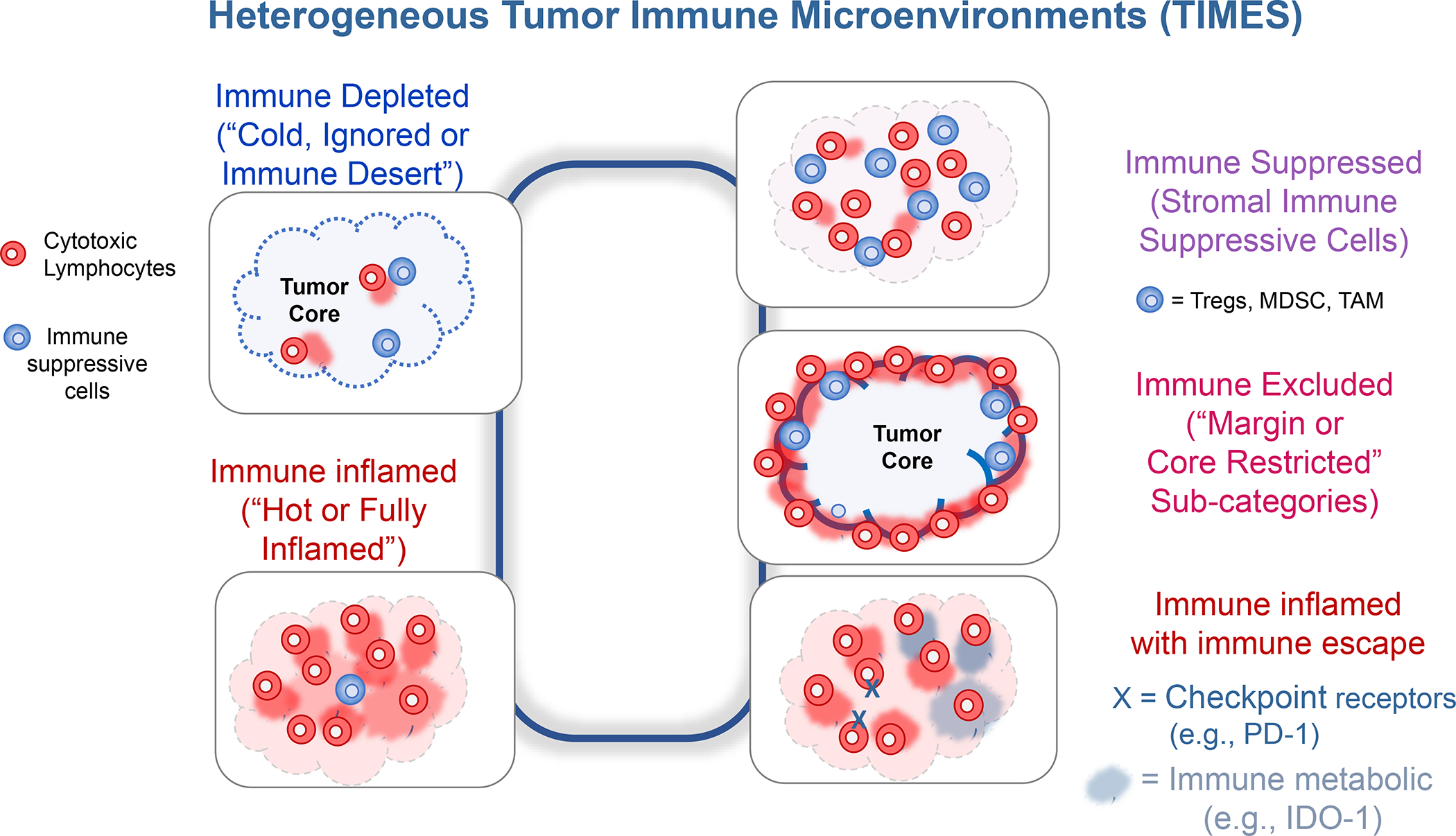
In in spite of the advances by immune checkpoint blocking antibodies for cancer immunotherapy, only 20–30% of patients with responsive cancers mount a robust antitumor immune response, provided that they exhibit an inflamed tumor microenvironment with CTL infiltration. To improve the response rate for these cancers and add to increase the overall number of additional cancers that can be successfully treated with checkpoint blocking antibodies, a number of approaches exist to convert “cold” tumors “hot”, including endogenous and exogenous vaccination approaches. Even when successful at improving CTL recruitment, these attempts may not be enough to achieve cytotoxic killing because of: (i) the immune suppressive effects of the tumor stroma; (ii) recruitment of CD8+ T-cells that are especially excluded from contacting PDAC or TNBC cancer cells; (iii) recruitment of CD8+ T-cells, which are put under constraint by ligation of checkpoint receptors on the immune metabolic effect of the IDO-1 pathway. Thus, in addition to inflamed (“hot”) and immune-depleted (“cold”, “immune desert” or “ignored”) TIMES at the far ends of the spectrum, intermediary categories such as “immune excluded”, “immune suppressed” and “immune escape” landscapes need to be considered for TNBC and PDAC immunotherapy. This requires customized design of treatment combinations to address the challenges in each landscape. Abbreviations: Treg = FoxP3+ regulatory T-cells; MDSC = myeloid derived suppressor cells; TAM = tumor-associated macrophages; IDO-1 = Indoleamine-pyrrole 2,3-dioxygenase.
Against this background, a key question is whether intratumoral heterogeneity can be therapeutically targeted or exploited to improve combination therapy in the era of immune checkpoint blockers? Not only does this require knowledge about the makeup of heterogeneous tumor landscapes, but also provides a rational approach for combining active pharmaceutical ingredients (API) to reprogram “cold” immune landscapes, overcome T-cell exhaustion by checkpoint receptors, circumvent IDO-1 suppression, address the immune suppressive properties of the tumor stroma and overcome T-cell exclusion. While currently these challenges are being addressed by theory, immune phenotyping and modeling, it remains a challenge to replicate the complexity of the TIME outside the human body, leading to the conceptualization of therapeutic combinations beyond the number of study subjects that are available to conduct these studies. This prompted us to consider whether preclinical animal models can be used to therapeutically address the heterogeneous TME of pancreatic ductal adenocarcinoma (PDAC) and triple negative breast cancer (TNBC), including intervention with custom-designed nanocarriers or drug delivery systems (DDS). Following a brief review of heterogeneous TIMEs in human and animal PDAC and TNBC tumors (Subsection B), we will describe our existing experience and ongoing efforts to develop combination therapy with lipid bilayer (LB)-coated nanocarriers, capable of inducing immunogenic cell death responses (Subsection C). These carriers have been selected for the ability to use lipid bilayers for remote drug loading into the aqueous interior of liposomes and silicasomes, as well as allowing drug incorporation in the bilayer (Subsection D). This is accomplishable using natural or synthetic lipid moieties as well as lipid-conjugated prodrugs. Subsection E delineates how these responses can be augmented by additional intervention in stromal immune suppressive pathways, while subsections F and G discuss how these interventions can be integrated and further improved. We will briefly compare in-house chemo-immunotherapy efforts to similar approaches by other investigators for achieving nano-enabled immunotherapy.
B. Heterogeneous immune landscapes
B.1. Why do we focus on PDAC and TNBC landscapes?
The complexity of the tumor microenvironment (TME) resulting from the collective contribution of tumor cells, infiltrating immune cells, fibroblasts, tumor vasculature and extracellular matrix determines the success of solid tumor immunotherapy, including pancreatic, breast, colorectal, lung, melanoma, and head and neck cancers.16–18 This includes the impact on immunotherapy, where a major objective is to improve the response in cancers such as pancreatic ductal adenocarcinoma (PDAC) and triple-negative breast cancer (TNBC). Not only are PDAC and TNBC two of the most formidable cancers that we encounter in oncology, but also present poorly immunogenic landscapes that resist immunotherapy. PDAC is the fourth leading cause of cancer deaths in the United States, with median survival less than 6 months or a 5-year survival rate in the single digit range.19–20 Moreover, this cancer is typically diagnosed at an advanced stage, which precludes surgery and is poorly responsive to chemotherapy or immunotherapy, including treatment with checkpoint blocking antibodies. Similarly, TNBC is considered one of the highest-risk breast cancer subtypes, with a local recurrence rate >70% within 5 years or a 5-year survival rate of 12% for metastatic disease.21 Also, as a result of being negative for the estrogen receptor (ER), progesterone receptor (PR), and the human epidermal growth factor receptor 2 loci (HER2), there is a paucity of targeted therapeutics, with chemotherapy still serving as the primary treatment option.
The variable composition of the TME, comprised of tumor cells, vasculature, extracellular matrix, fibroblasts and infiltrating immune cells contributes to the establishment of heterogeneous immune landscapes, is of major interest for PDAC, TNBC, melanoma, and non-small cell lung cancer. This has given rise to the development of integrated approaches for immune and molecular-directed therapies, making use of new clinicopathologic, genomic/transcriptomic, immunophenotypic and spatial distribution technologies for disease classification and stratification. This discovery is now being used for the development of customized therapy for receptive subgroups to improve ICI response rates as well as prevent unnecessary treatment in patients not likely to respond. Our focus is on addressing heterogeneous immune landscapes in preclinical PDAC and TNBC animal models, making creative use of LB-coated nanocarriers for the delivery of immunogenic stimuli that can be propagated by co-delivered immune modulatory agents. This does not exclude the use of the same carriers to treat other solid cancers that exhibit heterogeneous immune landscapes, including colorectal and renal carcinoma models, as we will be illustrated in our data demonstration, while also citing related studies performed by other investigators.
B.2. Human pancreatic ductal adenocarcinoma landscapes
A paucity of neoantigens, poor recruitment of antigen presenting cells (APC) and interference in cytotoxic T-cell (CTL) killing by multiple immune evasion or immune suppressive pathways lies at the heart of the poor PDAC immune response. This includes an important contribution by the cancer-associated fibroblasts (CAF) that account for 50–80% of the total tumor volume.18 In addition to displaying densely packed collagen plus other extracellular matrix proteins, the stroma engages in the recruitment of regulatory T-cells (Tregs), tumor associated macrophages (TAMs), and MDSCs. This establishes a wide-ranging network of cellular interactions that exert immune suppressive effects in the TME. These cellular interactions are governed by a number of stromal-derived secretory products, including colony-stimulating factor 1 (CSF-1) and a chemokine cascade involving stromal cell-derived factor 1 (SDF-1), a.k.a. CXCL12. The CSF-1 receptor (CSF1R) is predominantly expressed in myeloid cells and is responsible for TAM and MDSC recruitment, while CXCL12 leads to the activation of the CXCR4 receptor 4 (CXCR4), exerting pleiotropic effects in the PDAC stroma.22–23 These include direct and indirect effects on cancer cell growth and migration, tumor angiogenesis and stromal matrix T-cell trapping, preventing CTLs from making contact with cancer cells. 23 This phenomenon is also known as T-cell exclusion, which constitutes a major barrier for CTL killing. It has also been demonstrated that CXCL12/CXCR4 signaling promotes epithelial-to-mesenchymal (EMT) transition by activating the MEK/ERK, PI3K/AKT or Wnt/β-catenin pathway. The expression of CXCL12 in the liver, lungs and bone marrow have the additional role of promoting metastasis of CXCR4-expressing PDAC cells to these sites.22 CXCL12 also exerts important functions remote from the primary tumor site, including regulation of leukocyte release from the bone marrow, allowing recruitment to the primary tumor site.22–23
In addition to spatial separation, the PDAC microenvironment promotes the expression of checkpoint receptors on CTL, which interfere in the function of the T-cell antigen receptors (TCR). This induces a state of CTL “exhaustion” and inability to trigger perforin and granulolysin B release for tumor cell killing. This is particularly likely if there is a source of interferon-γ (IFN-γ) production in the TME, leading to the expression of the programmed death-ligand 1 (PD-L1).24 PD-L1 interacts with programmed death protein 1 (PD-1), a major checkpoint receptor in PDAC. The expression of Cytotoxic T-lymphocyte associated protein 4 (CTLA4) represents another important checkpoint receptor. Not surprising, PD-L1 overexpression is associated with a worse prognosis in PDAC patients.25 An additional source of interference in CD8+ T-cell function and CTL killing comes from the direct and indirect immune suppressive effects of FoxP3+ Tregs and MDSC.18 Tregs may suppress the function of CTLs via direct interaction of receptor–ligand pairs on the Treg and target cells, delivery of suppressive factors such as transforming growth factor-β (TGF-β), cytokine competition or direct cytolysis. The PDAC stroma is also a source of granulocyte colony stimulating factor (G-CSF), which reduces bone marrow supply of conventional CD103+ dendritic cells (cDC) progenitors to the PDAC and BC tumor sites.26 This is in keeping with the poor prognosis of PDAC and BC patients, who express a reduced number circulatory cDC1.26 CD103+ DC are engaged in tumor antigen cross-presentation to the TCR of CTLs.27 Therapeutic intervention to improve the supply of CD103+ DCs constitutes an important therapeutic objective, as will be discussed later. Finally, it is also important to comment on the immune metabolic effects of indole 2,3-dioxygenase (IDO-1), which is overexpressed at the tumor site by DCs, MDSCs, and cancer cells. 13–14, 28 IDO-1 interferes in T-cell activation, in addition to promoting the conversion of naïve T cells to Tregs as well as increasing IL-6 expression that augments MDSC function.28–29
Early attempts to define PDAC subtypes for prognostication purposes were based on molecular discovery by the Cancer Genome Atlas Research Network.30–31 Two major subtypes based on cancer cell characteristics were delineated, namely basal-like (quasi-mesenchymal or squamous) and classical (pancreatic progenitor) phenotypes.32 While the less-differentiated basal phenotype was associated with a poor prognosis, the better differentiated (ductal) features of the classical subtype was were associated with improved outcome. No attempt was made in this category of phenotyping to include immune response features, leading to further attempts to collect molecular disease characteristics (e.g., mutational signaling clusters, chromosomal instability, transcriptomics, metabolomics and epigenomics) (Figure S1), among which the studies of Puleo et al. identified distinct stromal phenotypes, among which the stromal-activated phenotype was characterized by reduced CTL infiltration and the worst prognosis.33–35 These studies were the forerunners for identification of additional PDAC landscapes by employing high-dimensional immune profiling technologies, including multiplex IHC (mIHC), single cell transcriptomics (e.g., single-cell RNA-Seq) and time-of-flight cytometry (CyTOF) (Figure 2).36–39 For instance, Steele et al. analyzed tumor samples by CyTOF, single-cell RNA sequencing and mIHC analysis, leading to the identification a complex cellular network, characteristic of immune-suppressive features.40 Not only did tumors from individual patients demonstrate a spectrum of CD8+ T-cell recruitment densities, but also showed heterogeneous expression of immune checkpoint receptors. While CyTOF and mIHC revealed an inverse correlation in myeloid vs. CD8+ T cell infiltration, cRNA seq analysis showed frequent presence of CD8+ T-cells with exhaustion markers, especially in the advanced stage. mIHC analysis is t conducted by using cyclic or tyramide-based immunofluorescence approaches for providing information on cell composition and spatial distribution (Figure 2).41
Figure 2. High-dimensional immune-profiling and multiplex immunohistochemistry (mIHC) analysis of cancer landscapes.
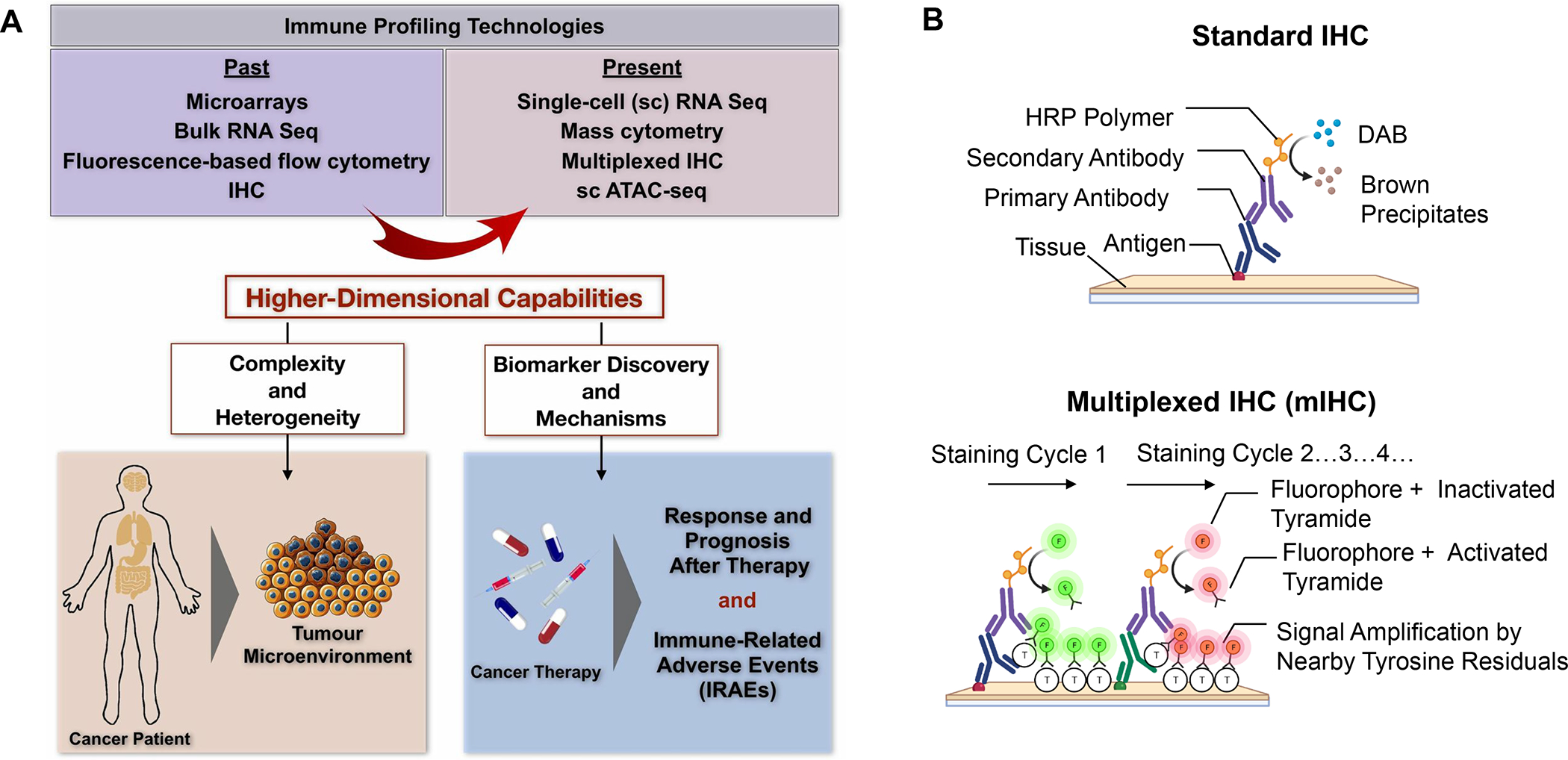
Panel A: mIHC analysis, single cell (sc) transcriptomics (e.g., single-cell RNA-Seq) and cytometry by time of flight (CyTOF) platforms are replacing conventional tools in discovery for understanding complex and heterogeneous tumor microenvironments, including by introducing immune response biomarkers that can be used for chemoimmunotherapy.36–39 ATAC = assay for transposase-accessible chromatin. Reprinted with permission from ref 36 under a Creative Commons Attribution License 4.0 (CC BY). Copyright 2020 The Authors. Panel B: While conventional IHC allows detection of cellular antigens in tissue sections through the employment of enzyme-labeled or fluorochrome-labeled antibodies to identify diverse cell types and spatial location, no more than 4 markers can be used simultaneously as a result of the constraints of chromogenic or fluorescent spectra overlap.36 However, advancements in dye-cycling techniques, where staining, imaging and dye inactivation are done repeatedly, have enabled the detection of multiple different antigens on the same tissue sample by mIHC analysis.37
Additional mIHC analysis of clinical PDAC samples were provided by Carstens et al.,37 Masugi et al.,38 and Seo et al.39 Utilizing tyramide-based signal amplification, Carstens et al. looked at 8 distinct markers (anti-smooth muscle actin, collagen-I, cytokeratin 8, CD3, CD8, CD4 and Foxp3) in the TME of 132 PDAC patients (Figure 3). The predominant T-cell subpopulation was comprised of CTLs, appearing in spectrally unmixed images as a range of different densities in individual patient samples (Figure 3). Patients with the highest CD8+ density had the best survival outcome, particularly when the CTLs were in close proximity to cancer cells.37 While confirming that the association of higher CD8+ densities is associated with the best survival, Masugi et al. specifically called out the prognostic value of high cell density in the tumor center.38 Moreover, this group reported in 240 patient samples that the CTL densities in the tumor center are less than 50% of the densities in the tumor margins, further characterized by the presence of tertiary lymphoid structures, which are also described in human cancer following KRAS peptide immunotherapy.42 However, in spite of differences in the spatial distribution between cores and margins, Masugi et al. did not observe a survival benefit for CTL proximity to the cancer cells. This group also differed from Carstens et al. in observing less CD8+ T-cell infiltration in the aSMA+ tumor center, while the latter group did not observe correlation between anti-smooth muscle actin (aSMA) fibroblast/type I collagen density vs. T-cell infiltration. Masugi et al. also found a link between higher CD8+ densities and PD-L1 expression, which supports the notion that IFN-γ production by cytotoxic T-cells may generate a counterregulatory response.38 Finally, a study by Seo et al. (Figure S33) confirmed less dense CD8+ T-cells infiltration in the juxta-tumoral stroma where cancer cells were present, compared to stromal regions not containing cancer cells.39 These are just a few of the growing number studies using high dimensional immune profiling to elucidate heterogeneous PDAC immune landscapes.
Figure 3. mIHC images of heterogeneous human PDAC immune landscapes.
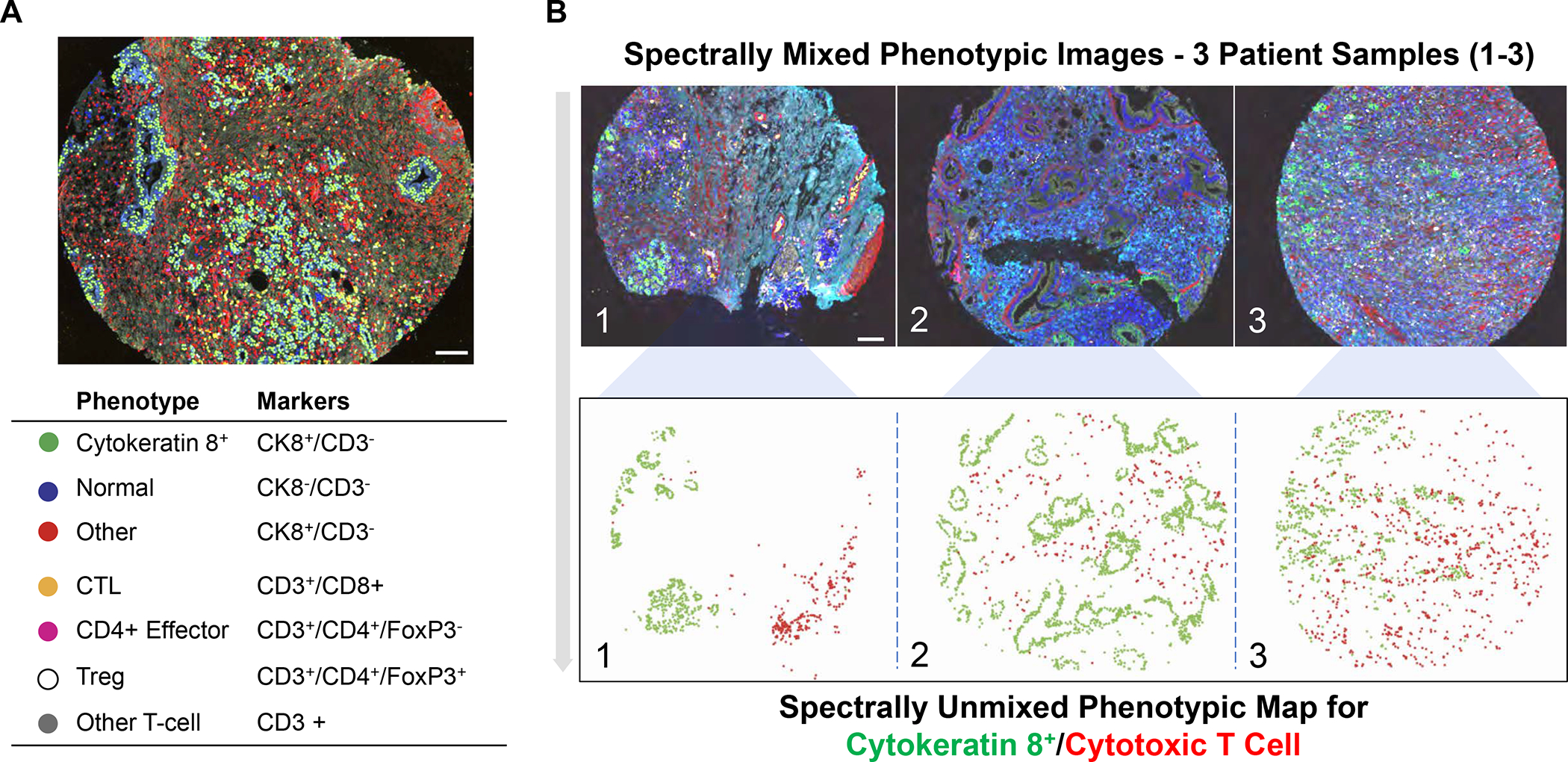
Utilizing tyramide-based signal amplification, Carstens et al. examined 8 distinct markers (anti-smooth muscle actin, collagen-I, cytokeratin 8, CD3, CD8, CD4 and Foxp3) to obtain spectrally mixed and unmixed images of the heterogeneous cell populations and their spatial distribution in paraffin-embedded tumor samples from 132 PDAC patients.37 Panel A: Spectrally mixed image of the cell phenotype map identifying the cell populations defined by the individual markers of the multiplex stain, overlayed on the raw image. Legend: Summary of each defined cell phenotype, color code and associated markers. The scale bar equals 100 nm. Panel B: spectrally mixed (upper panel) and unmixed (lower panel) images three patients (A, B and C) with differing levels of CTL infiltration - patient A showing low infiltration, patient B medium infiltration and patient C high infiltration. The unmixed phenotype map depicts the cytokeratin positive cancer cells (green) and CTLs (red) in the tumor sites. Panel A-B adapted with permission from ref 37 under a Creative Commons Attribution License 4.0 (CC BY). Copyright 2017 The Authors.
Based on the need for linking discovery of heterogeneous landscapes to immunotherapy, Karamitopoulou has suggested subdividing PDAC landscapes into (i) immune-escape, (ii) immune-rich or (iii) mixed microenvironments.31 According to this view, the majority of tumors display an immune-escape phenotype, where the presence of abundant Tregs and M2-macrophages overshadow the paucity of effector CTL, creating tumors with highly suppressive and aggressive phenotypes. These TMEs are characterized by EMT features, extensive tumor budding, somatic gene alterations and microRNA dysregulation. This tumor category corresponds to the poor prognosis seen in squamous,34 quasi-mesenchymal,43 or ‘basal’ subtypes.32 In contrast, the relatively infrequent immune-rich phenotype refers to tumors with a highly cytotoxic immunophenotype, which corresponds to the prognostically favorable classical subtype, which also exhibits low grade tumor budding and prolonged survival.44 This landscape is enriched in effector CD4+ and CD8+ T-cells and M1 macrophages, along with reduced numbers of immunosuppressive Tregs and M2 macrophages. These tumors display a high mutational frequency, increased neoantigen load, improved antigen presentation capabilities and enhanced IFN-γ signaling. Finally, tumors with mixed landscapes display immune-enriched features, which are accompanied by immune exhaustion and unfavorable histopathologic characteristics such as high tumor budding. Immune exhaustion reflects amplification of the JAK/STAT signaling pathway by IFN-γ production in CTL.24, 45 The impact of these evasive mechanisms is to produce the same outcome as in an immune suppressive TME, in spite of higher CTL content. Interestingly, the Karamitopoulou phenotyping does not include an immune exclusion category, in contrast to the TNBC features that we will discuss later.
B.3. Animal model comparison making to human PDAC immune landscapes
Attempts to study heterogeneous human PDAC immune landscapes in preclinical animal studies are limited.46–47 The most frequently studied genetically engineered mouse model is LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx1-Cre (KPC), which uses Cre-Lox to express activated Kras and a mutant p53 version under the control of a pancreatic epithelial cell–specific Pdx1 or Ptf1a promoter.48 These animals develop tumors that mimic several human PDAC characteristics, including a limited number of driver mutations (KRAS, TP53, CDKN2A and SMAD4), also reflected in the human mutational signaling clusters (Figure S1). Overall, there is a low mutational burden and display of neoepitopes in both human and murine PDAC, both of which show the loss of p53 function during stage-wise progression from pancreatic intraepithelial neoplasia (PanIN) to the stage of full-blown desmoplasia.48–50 The TME displays considerable heterogeneity as a result of the dependence on diverse EMT cancer pathways.51–52 An example of mIHC multispectral imaging of stromal features in the KPC model is shown in Figure 4B. Additional overlapping histopathological features with human PDAC include poor vascularity and high metastatic burden.49, 53 While both cancer types show dense infiltration of immunosuppressive TAMs and myeloid cells, the lesser expression of CTL in murine KPC does not mirror the occasional occurrence of an immune-rich subtype in human PDAC.54 However, similar to human PDAC, KPC tumor-bearing mice are mostly refractory to ICIs, including administration of anti-PD-L1/PD-1 and anti-CTLA-4 antibodies.55
Figure 4. Utility of the Kras pancreatic cancer (KPC mouse model) for developing combination immunotherapy.
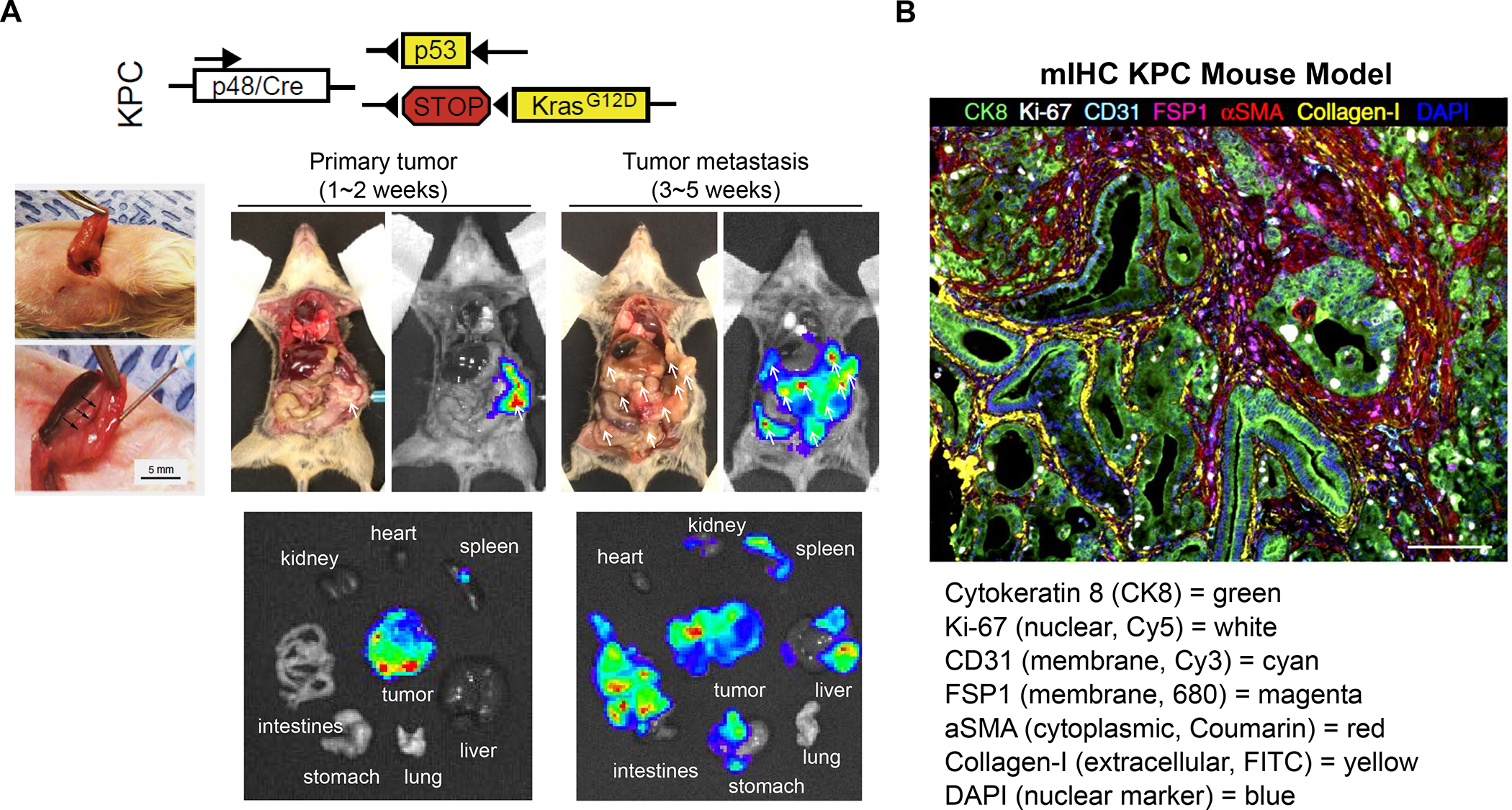
Panel A: The KPC genetic mouse model of pancreas cancer (Pdx1-cre/LSL-Kras G12D/p53R172H) has been widely used because of its fidelity to human PDAC, including activating Kras(G12D) mutations and loss of Trp53, associated desmoplasia, and inflammation.49, 61, 301 The spontaneous model has been instrumental in developing a number of PDAC immunotherapy approaches that are being applied in human studies, even though differences exist for KPC vs. human PDAC immune landscapes, particularly the occurrence of an immune-rich subset in humans vs. the myeloid-dominant TIME of KPC. Noteworthy, the Kras oncogene contributes to immune suppression and immune evasion in this animal model.302 Due to the logistical constraints to breed a sufficient number of animals for accommodating all the treatment combinations that can be studied in one experiment, we developed an orthotopic implant model in immunocompetent B6/129 mice to perform our studies.93 The orthotopic implant procedure involves minor surgery for injecting 2 × 106 KPC-luc cells in the tail of the pancreas (left panel).93 The autopsy and bioluminescence imaging reveal primary tumor growth after 1 to 2 weeks, followed by tumor metastases after 3 to 5 weeks. Macro-metastases are marked by arrows. However, in spite of the utility of the orthotopic KPC model, it is important to note that these tumors lack an autochthonous stroma or the extensive desmoplasia seen human tumors or the spontaneous GEM. Nonetheless, the orthotopic model has proven of considerable benefit in studying chemo-immunotherapy, as we will demonstrate in later sections. Adapted with permission from ref 61. Copyright 2020 Elsevier (upper panel A). Reprinted with permission from ref 93. Copyright 2017 American Society for Clinical Investigation (lower panel A). Panel B: Spectrally unmixed mIHC image obtained from a mouse PDAC tumor, stained with tumor stroma biomarkers, as shown in the figure legend. Adapted with permission from ref 37 under a Creative Commons Attribution License 4.0 (CC BY). Copyright 2017 The Authors.
In spite of the differences in CD8+ T-cell and TAMs densities in human and murine PDAC, the spontaneous KPC animal orthotopic model has been useful for developing new PDAC therapies that could be translated in the clinic.56–57 This includes studies looking at the role of CD40, which is broadly expressed in immune cells and capable of mediating tumor regression by macrophages, independent of T cells.58–59 These studies provided the impetus for clinical trials using CD40 agonists in advanced stage PDAC, including accomplishment of significant tumor shrinkage in a phase 1b clinical trial.59 Immune profiling of these patients demonstrated rapid DC activation as well as reprogramming of the myeloid compartment. Moreover, the impact of CD40 activation was augmented by the release of DC precursors from the bone marrow, using agonists of the Fms-related tyrosine kinase 3(Flt3)/Flt3L pathway.60–61 Another example of KPC-inspired immunotherapy was the elucidation of the role of the CXCL12/CXCR4 axis in directed migration of immune cells under the instruction of several chemokine receptors (CXCR1, CXCR3, CXCR5, CXCR6, CCR2)39, 62 CXCL12 is produced by CAFs and can attract CXCR4-positive inflammatory, vascular, and stromal cells into the tumor mass to support tumor development.63–64 In a later section, we will discuss the impact of blockade of the CXCL12/CXCR4 axis in improving human PDAC immunotherapy responses to ICI’s.65 The TGF-β contribution to immune escape in the PDAC immune landscape was demonstrated by the use of TGFβ inhibitor, galunisertib, in improving impact of ICI treatment in KPC mice, also receiving administration of Gemcitabine.66–67
B.4. Triple negative breast cancer (TNBC) immune landscapes in humans
TNBC represents a subgroup of BC characterized the lack of estrogen receptors, progesterone receptors and human epidermal growth factor receptor 2 (HER2). These tumors represent 15–20% of all BCs, are more prevalent in younger African and Hispanic women and have high rates of distant recurrence, with reduced overall survival.68–69 In spite of its phenotypic characteristics, TNBC is a heterogeneous disease category representing multiple subtypes with marked histopathological, transcriptomic and genomic variability. While a landmark study by Lehmann et al. identified seven TNBC clusters based on histopathological, transcriptomic and therapeutic response differences,70–71 this classification has proven to be of limited prognostic significance.72 This prompted the use of single cell analytics and multi-omics to accomplish delineation of the TNBC type-4 subtypes, which include basal-like 1 and 2, luminal androgen receptor (LAR), and mesenchymal phenotypes.73–76 However, because these phenotypes were still incapable of predicting the response to immunotherapy, Gruosso et al. made use of conventional immune phenotyping of epithelial and stromal compartments to identify TNBC subtypes with gene-based meta-signatures.8 These TIMES were characterized as “Immune desert”, “Fully inflamed”, “Margin-restricted” or “Stroma-restricted” phenotypes (Figure 5). Each subtype represents a significant fraction of human TNBC cases, has prognostic significance and provides therapeutic guidelines (Table 1). Using CD8+ T-cell density in the tumor cores, these landscapes were initially identified as corCD8-HIGH and corCD8-LOW categories, which were further subclassified for CD8+ accumulation in the tumor margins (marCD8hi) (Table 1). Most tumors in the corCD8-LOW category showed some CD8+ T-cell accumulation in the tumor margins (marCD8hi) and were designated as a “margin-restricted” (MR) landscape (Table 1). In contrast, a smaller number of tumor landscapes showed low-density T-cell infiltration everywhere and was designated as an “immune desert” (ID) phenotype. Similarly, the corCD8-HIGH category was subdivided into “fully inflamed” (FI), with high density CD8+ infiltration in the epithelial and stromal compartments, or “stromal-restricted” (SR) if the CD8+ T-cell accumulation occurred in the stroma but not the epithelial compartment. More details of these phenotypes and immunotherapy implications appear in Table 1.
Figure 5. Therapy-naive TNBC tumors are classified into subtypes on the basis of distinct spatial localization of CD8+ T cells.
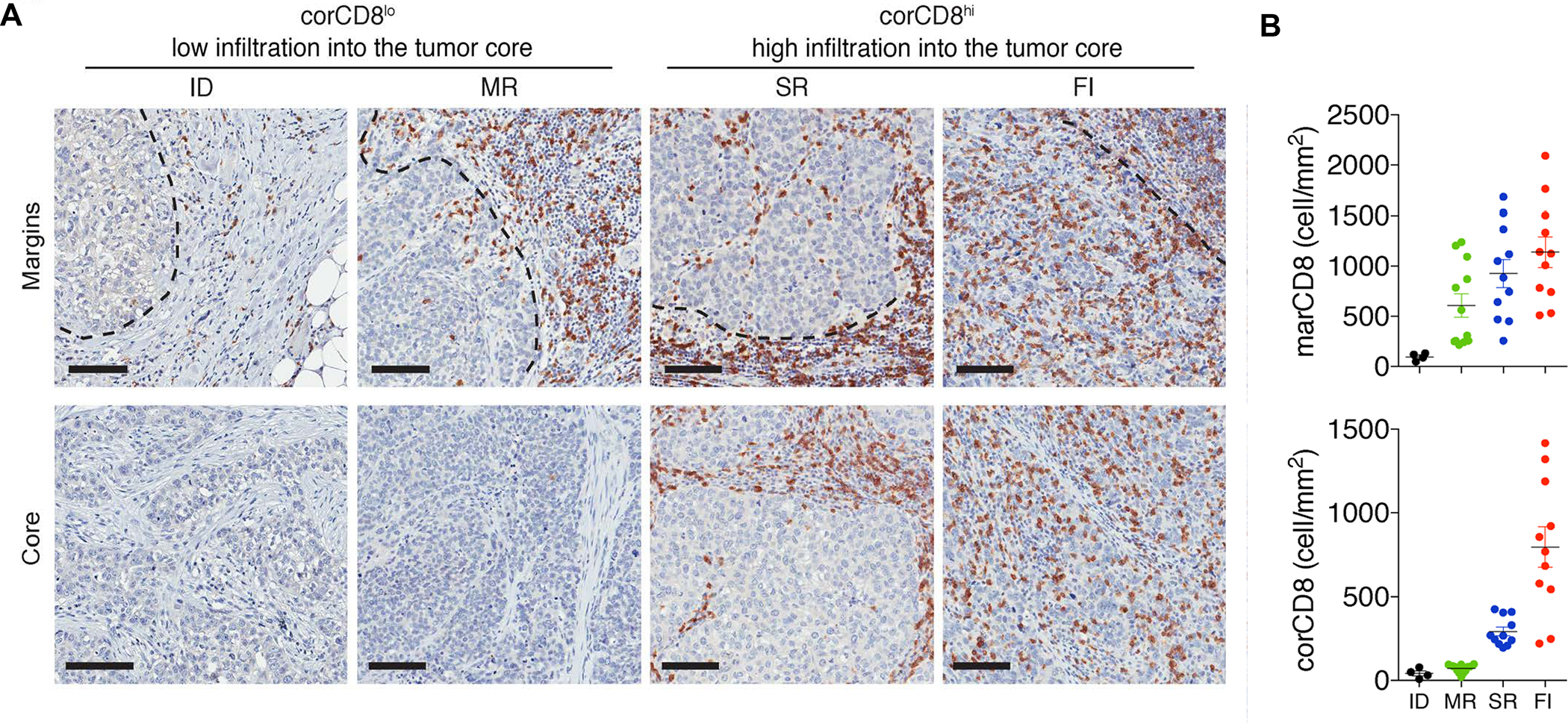
Panel A: Representative images of CD8+ T-cell staining in the vicinity of tumor margins (top panels, dotted lines) and tumor cores (bottom panels), collected from 38 human samples. Scale bars: 100 μm. Panel B: Quantification of CD8+ T cell densities at the tumor margins (marCD8) and in the tumor cores (corCD8). Data represent the mean ± SEM. Tumor phenotypes: ID = immune desert; MR = margin-restricted; SR = stromal-restricted; FI = fully inflamed. Panel A-B reprinted in part with permission from ref 8. Copyright 2019 American Society for Clinical Investigation.
Table 1:
Spatially distinct tumor immune microenvironments, Gruosso et al.8
| TIME Category | Special features, meta-signatures and immune characteristics |
|---|---|
| Immune Desert | corCD8-LOW with low margin densities Negative for PD-L1 and IDO-1; Elevated TGF-β signature; Fibrosis with M2 macrophages; Abundant B7-H4; MHC-I may be low Cold” immune status could benefit from ICD-inducing chemo plus modulation of the immune suppressive stromal environment, including TGF-β inhibitors and anti-B7-H4 blocking antibodies |
| Margin Restricted | corCD8-LOW with higher CD8+ densities in the margin Does not appear as a separate category in Hammerl et al.77 Rest of the features similar to immune desert, likely with similar therapeutic indications. |
| Stroma Restricted | corCD8−HIGH with high CD8+ densities in the margin and stroma but with CTL exclusion from the epithelial compartment; Highly expressed CXCL12/CXCR4 axis, reflecting importance of the desmoplastic stroma, presence of TAMs, MDSC and several immune checkpoint receptors Can also exhibit a pro-inflammatory phenotype with an IL-17 signature; The stromal but not epithelial compartment displays PD-L1, IDO-1 and FOXp3+ Tregs; MHC-I may be low Therapeutic focus on stromal immune suppressive mechanisms (e.g., CXCR4), in combination with ICI and chemotherapy. |
| Fully Inflamed | corCD8-HIGH in the margin, stroma and epithelial compartment Pro-inflammatory TIME with abundant CD8+ T-cells; pro-inflammatory macrophages; Robust IFN-γ signature with high PD-L1 and other checkpoint receptors; High IDO-1 expression may recruit Tregs to the epithelial compartment; Normal MHC-I expression Therapy-wise, likely to be ICI responsive, but may involve neoadjuvant chemotherapy and possibly IDO-1 inhibitors. |
An additional comprehensive analysis of TNBC spatial distribution in 681 human tumors was undertaken by Hammerl et al., who used OPAL reagents (Akoya Biosciences) to perform mIHC, coupled with identifying the best differentially expressed genes, using a gene classifier approach.77 This led to the grouping of the immune landscapes as: “Ignored”, “Excluded” and “Inflamed” (Figure S2). Comparatively speaking, these phenotypes are in agreement with the “Immune desert”, “Stromal/margin restricted” and “Fully Inflamed” phenotypes of Gruosso et al., except that the Hammerl study also included patients receiving anti-PD1 treatment in the TONIC trial.78 The TONIC trial collected transcriptomic data from biopsies taken of pre- and post-induction. The salient details of the Hammerl et al. study are summarized in Table 2.
Table 2:
Spatially distinct tumor immune microenvironments, as per Hammerl et al.77
| TIME Category | Spatial features, Meta-signatures, Immune characteristics |
|---|---|
| Ignored (analogous to immune desert) |
Classical immunological “cold” landscape, with sparse CTL infiltration, but may include the presence of CD163+ myeloid cells and M2 macrophages Prominent signatures and gene expression of the WNT and PPARG/RXR pathways. Prominent collagen deposition and production of chemo repellents Not primary anti-PD1 responsive, but TONIC trial data reveals tumor subsets that can be primed with Cisplatin and Doxorubicin Therapeutic focus should be on blockers of the WNT pathway, drugs that target M2 macrophages (e.g., the CSF1R inhibitor, pexidartinib). |
| Excluded (analysis to stromal restricted) |
“Cold” tumor variant with T-cell exclusion, but not distinguishing between margin and stromal spatial distributions Dysplastic stroma, with prominent collagen deposition, Prominent expression of TGF-β and VEGF pathways, which contribute to T-cell evasion. Not primary anti-PD1 responsive, but TONIC trial data reveal tumor subsets can be primed by Cisplatin and Doxorubicin Exhibit a pro-inflammatory phenotype with elevated IL-17 signature; Therapeutic focus on the stromal effects, leading to T cell exclusion, including inhibitors of the TGF-β pathway, inhibitors of VEGF receptor kinases. |
| Inflamed (analogous to fully inflamed) |
Inflamed phenotype, with abundant CD8+ T-cells; widespread distribution, also includes M2 macrophages. High TCR clonality, high DC density, high expression of ICD-associated biomarkers and evidence of CTL cytotoxicity Increased expression of ICI receptors, reflecting a negative feedback loop associated with T-cell immunity (Figure 17B) Phenotype of choice for ICI therapy, including combination of multiple ICIs or the use of CSF1R inhibitors targeting M2 macrophages; Reactivation of type I IFN pathway may help to boost antigen presentation. |
B.5. Animal model comparison making to human TNBC landscapes
In order to perform preclinical studies on heterogeneous immune landscapes that mimic the human findings, we performed orthotopic implants of 4T1, EMT6 and Py8119 cell lines in the mammary pads of syngeneic mice. The characteristics of these TNBC mimicking tumors are described in Table 3. These preclinical models were used to assess the spatial distribution of CD8+ CTLs, using conventional and mIHC analysis under basal growth conditions as well as after the IV injection of a commercial pegylated liposomal Doxorubicin, as outlined in the online Figures, S3–S5. Doxorubicin is frequently used as neoadjuvant therapy in human TNBC and is a robust inducer of immunogenic cell death (ICD), a topic covered in Figure 9.79–80 Figure 6A depicts the spatial distribution of CD8+ T-cells in 4T1, EMT6 and PY8199 tumor landscapes, using conventional IHC staining. The data demonstrate increased CD8+ density with Doxorubicin treatment in all the landscapes, but with differences in the spatial distribution of newly recruited T-cells. Thus, while in untreated animals, 4T1 tumors tend to be fully inflamed, the recruitment of additional CD8+ T-cells during Doxorubicin treatment remained widely distributed, except for stromal-restricted profiles in 30% of cases. The visual impression was confirmed by quantification of CD8+ cell numbers (cells/mm2) in the margins and cores (upper panel). In contrast, the margin-restricted basal landscape in EMT6 tumors maintained the same distribution under basal and treated conditions, except for the Doxorubicin-induced increase in CTL density (middle panel). Also, the fully inflamed phenotype of PY8199 tumors, reverted to a margin- or stromal-restricted distribution pattern upon treatment with Doxorubicin, which increased T-cell density (Fig. 6A, lower panel).
Table 3:
| Model | 4T1 | EMT6 | Py8119 |
|---|---|---|---|
| Mouse Strain | Balb/c | Balb/c | C57BL/6 |
| HR Expression | ER/PR/HER2–negative | ER/PR/HER2–negative | ER/PR/HER2–negative |
| Origin | Spontaneous mammary intraductal tumor from Balb/c | Derived from a transplanted hyperplastic alveolar nodule in Balb/c | Cell clone from transgenic tumor, where a mouse mammary tumor virus promoter drives polyoma middle T antigen (MMTV-PyMY) |
| Tumor Growth, Metastasis and Immunity | Aggressive orthotopic model that exhibits characteristics of human basal-like TNBC, with high metastatic potential, including to lymph nodes, blood, liver, lung, brain, and bone. Relatively high level of tumor inflammation with abundant cytokine and chemokine production, as well as recruitment of activated CTLs, B-cells and PD-1 expression. Granulocytic-MDSC facilitates metastatic spread at pre-metastatic niches. Generally regarded as poorly immunogenic, with resistance to anti-PD1, anti-CTLA4 as well as combination therapy. Metastasis occur after primary treatment resection, with poor immune memory. | Less invasive orthotopic model, with no or limited metastatic potential, generally regarded as moderately immunogenic with partial responsiveness to anti-PD1 and anti-CTLA4 but could response to both. The EMT6 cells express high levels of chemokines and cytokines, MHC-I, and antigen presentation potential, compared to all other orthotopic models. Tumors show less recruitment of granulocytic-MDSC, adoptive transfer of the cells from 4T1 mice increases metastatic potential. No metastasis after tumor resection, good immune memory prevents secondary challenge. | Orthotopic Py8119 tumors do not metastasize (different from the transgenic model) but can establish lung metastasis after IV injection. Generally regarded as a poorly immunogenic model and refractory to anti-PD1, anti-CTLA4 or combination therapy. Successful study of radiation-induced immune responses.291 Oxidized cell lysates delivered in a liposomal spherical nucleic acid (adjuvant), highly immunogenic.292 |
| TME and Spatial Distribution | Fibrotic stroma in which CXCR4 plays a role in T-cell exclusion, Treg recruitment, immune suppression, and metastatic potential. CXCR4 inhibitors synergize with ICI’s in limiting desmoplasia and metastases.199 | CD3+ cells predominantly confined to the invasive margin in untreated tumors.293 T-cell exclusion successfully blocked by anti-TGFβ antibody plus anti-PD-L1.225 | Py8119 tumors express a fibrotic stroma, with high levels of mesenchymal markers and a prominent TGF-β signature. |
| Spatial Distribution (Figure 6) | Fully inflamed phenotype, with increased CTL recruitment during Doxorubicin treatment, where cells remain widely distributed or may become stromal restricted | Margin-localized CTL distribution in untreated and Doxorubicin treated tumors; tumors exhibit denser stroma than 4T1. | The untreated tumor is fully inflamed (with less dense CTL infiltration than 4T1), but assume a margin and stromal restricted distribution with Doxorubicin treatment. |
Figure 9. Immunogenic Cell Death (ICD).
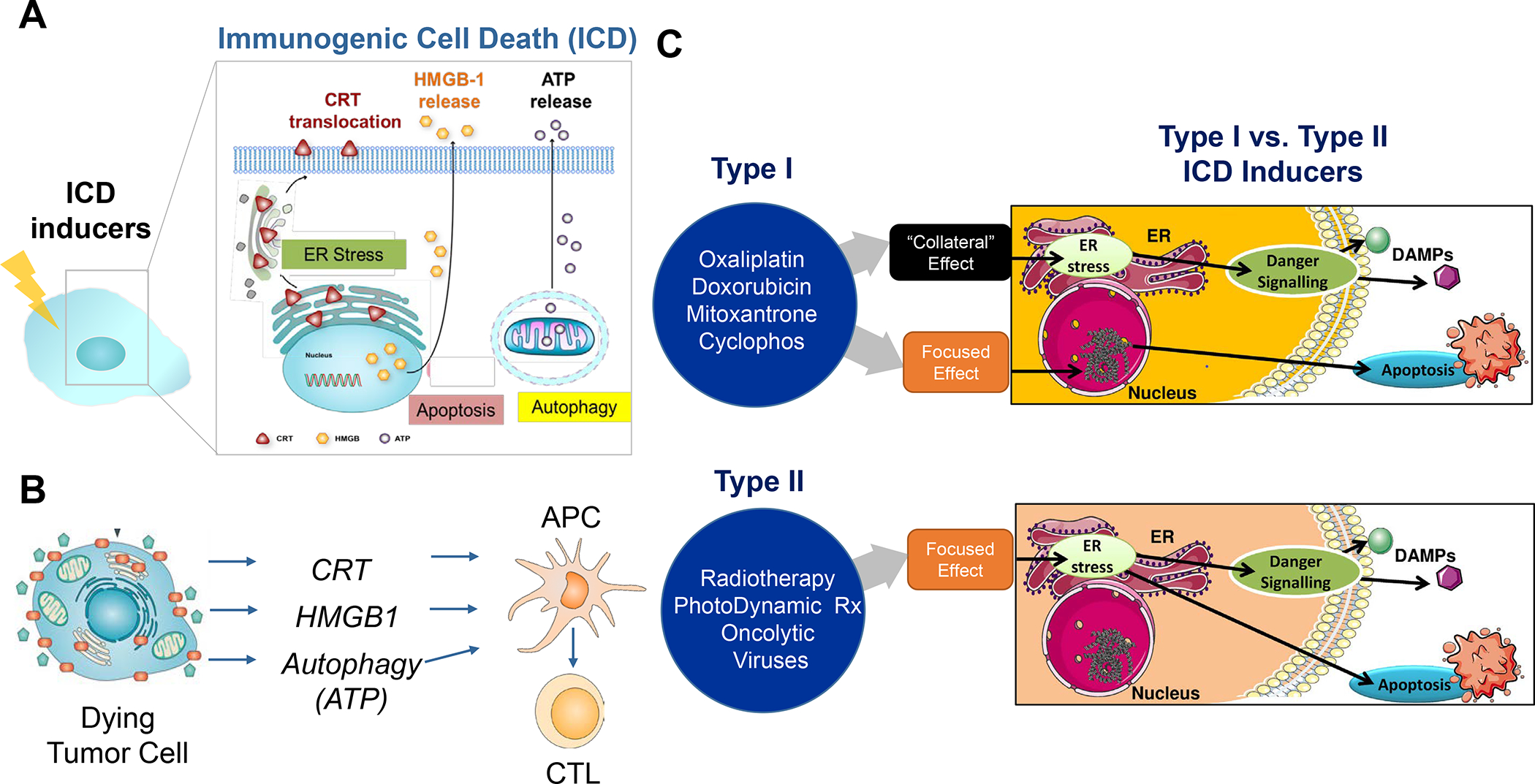
Panel A: ICD is a form of regulated cell death that activates immune responses, and of great interest in converting immune depleted or “cold” tumor microenvironments to immune inflamed or “hot”.79–80, 307 ICD represents a functionally unique response pattern that comprises the induction of organellar and cellular stress, culminating in an apoptosis-like death response that is accompanied by the active secretion or passive release of numerous danger-associated molecular patterns (DAMPs).79, 100, 108–109, 130, 231–232, 258 The principal DAMPs are calreticulin (CRT), heat shock proteins, HMGB1, ATP, and cytokines (type I IFNs and IL-1 family). A number of chemotherapy agents are included in the list of ICD-inducing drugs, including anthracyclines (Doxorubicin, Idarubicin), Mitoxantrone, Bleomycin, Cyclophosphamide, Oxaliplatin, Paclitaxel and Irinotecan. Most of their pharmacologic actions include damage to the cell nucleus and DNA, with collateral effects on cellular stress pathways, including oxidative stress responses, endoplasmic reticulum (ER) stress, mitochondria, autophagy flux, and cell membrane affects, all contributing to DAMPs release. In addition, other small molecule agents such as Bortezomib, cardiac glycosides, Patupilone, Septacidin, Shikonin, Vorinostat, and Wogonin can trigger ICD. Panel B: in order to understand the biology of the immunogenic effect, CRT translocation to the tumor cell surface provides an “eat me” signal, which enhances the uptake of dying tumor cells by APCs, such as dendritic cells (DC). This leads to processing of endogenous tumor antigens, which are displayed to naïve T cells via Type I major histocompatibility complexes on the APC surface. Additional release of adjuvant stimuli, such as HMGB1 from the disintegrating cancer cell nuclei and ATP from autophagic vesicles play a role in DC recruitment and maturation. Panel C: It has been suggested that there are at least two different ICD response pathways: (i) chemotherapeutic agents and drugs that exert their primary effect on the nucleus, with collateral effects on cellular stress and DAMPs release (type I pathway); (ii) irradiation, hypericin-based photodynamic therapy and oncolytic viral stimuli, which primarily promotes cell stress responses, with secondary effects on apoptotic cell death (type II pathway).308. Adapted with permission from ref 308. Copyright 2015 The International Journal of Developmental Biology. All considered, therapeutic use of the ICD pathway is to promote an endogenous tumor vaccination response, which may need to be boosted or propagated to account for the heterogeneity of the immune landscape.
Figure 6. CD8+ T-cells spatial distribution landscapes in 4T1, EMT6 and Py8119 TNBC animal models.
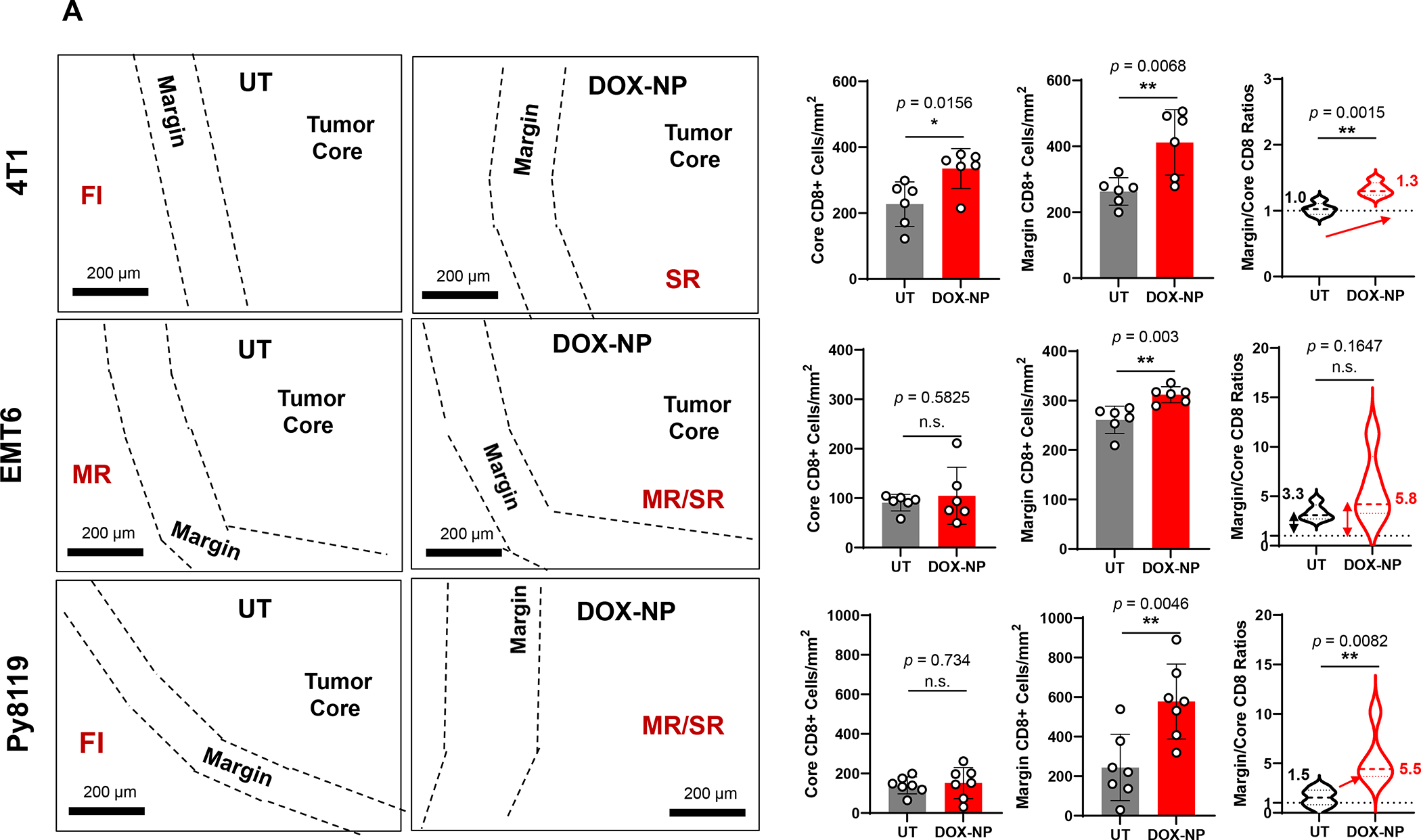
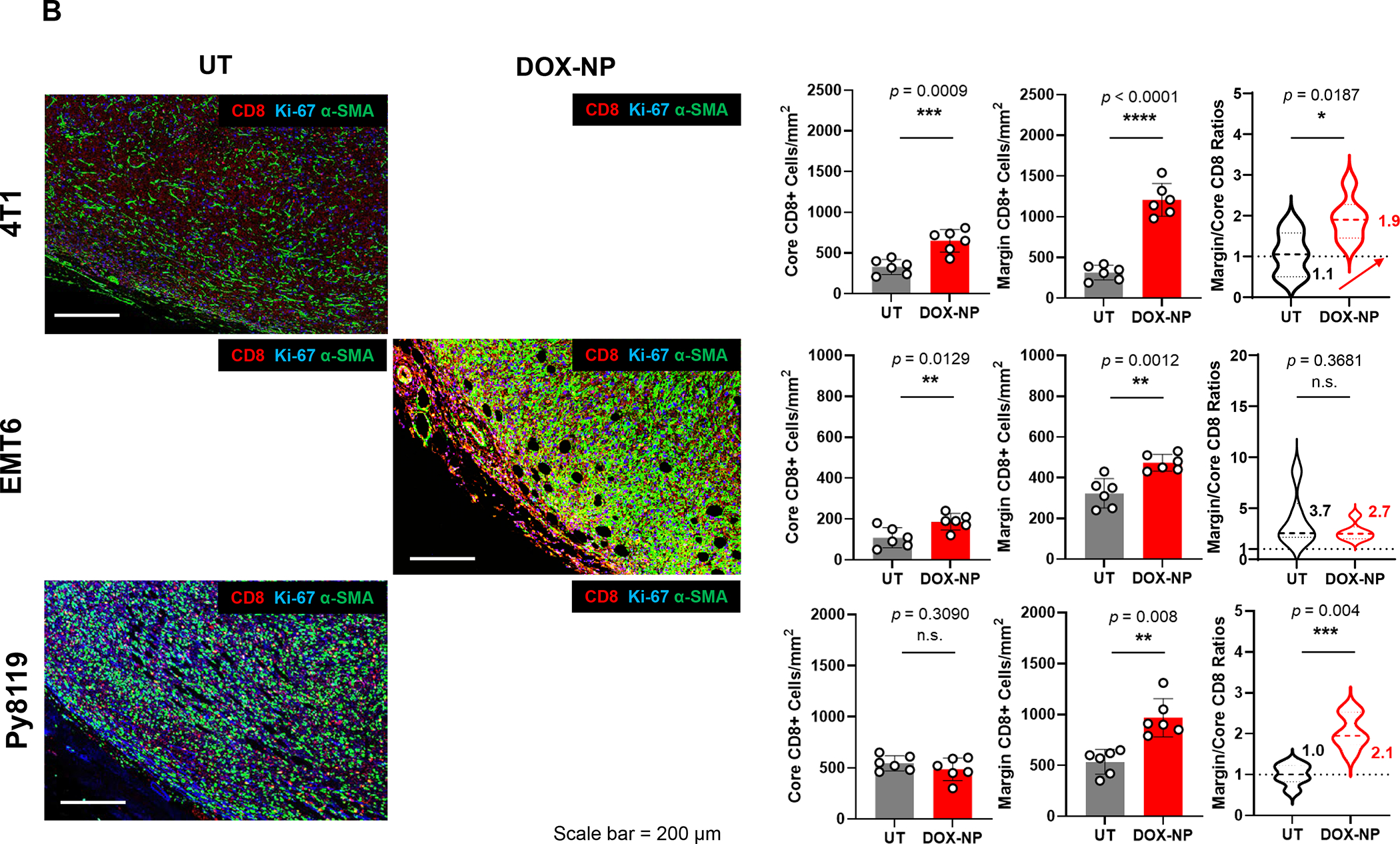
4T1 (Balb/c), EMT6 (Balb/c) and Py8119 (C57BL/6) EC cells were orthotopically implanted in mouse mammary pads on day 0. When the tumors reached 100–150 mm3, animals were IV injected on days 8, 11 and 14 with DOX-NP (5mg/kg; Avanti Polar Lipids) or left untreated (UT). Tumors were collected on day 21 and analyzed by conventional IHC staining (panel A) or multiplex IHC (mIHC) staining (Panel B). Quantitative analysis of CD8+ cells in tumor cores and margins during conventional IHC was performed, using Aperio ImageScope software. For mIHC analysis, tumor sections were stained with primary antibodies: CD8, α-SMA and Ki-67. Quantitative analysis of CD8+ numbers in cores and margins was performed using Akoya InForm Image Analysis software. Doxorubicin treatment induced increased CD8+ T-cell recruitment in all tumor types with both staining methods. Importantly, newly recruited CD8 T-cells tended to be margin- or stroma-restricted in EMT6 and Py8119 tumors, while CTL distribution in 4T1 was across the entire landscape in most tumors with stromal restriction in 30%. The same T-cell distribution was seen with mIHC, where α-SMA staining intensity in the stromal cores, followed the order EMT6 > Py8119 > EMT6. Data are expressed as mean ± SD, n = 6.
Tumor slices from the same animals were used for mIHC analysis, using OPAL reagents (Akoya Biosciences) to obtain spectrally mixed images for CD8, Ki-67, αSMA, Foxp3, CD68/CD163 and DAPI, as described in online Figures S6–S8. Spectral unmixing to focus on CD8, Ki-67 and αSMA confirmed that liposomal Doxorubicin increased the density of CD8+ cells (red fluorescence), which were spatially distributed in similar manner as conventional IHC for each tumor type (Figure 6B). Moreover, α-SMA staining indicated inter-tumor stromal differences, such that the fluorescence staining intensity for EMT6 tumors was > PY8119 > 4T1. All considered, the data in Figure 6 allowed the grouping of the murine TNBC landscapes into categories that partially overlap the human landscapes (Tables 1 and 2). Table 3 summarizes the spatial distribution features together with the distinguishing immunological features of these tumors. While genotyping has been performed on these murine tumors, detailed single-cell RNA-Seq or CyTOF analyses for immunophenotyping are pending.81
It is appropriate to convey that in spite of the ease and reproducibility of the immune landscapes in orthotopic models, these tumors lack an autochthonous stroma and do not accomplish the level of genetic heterogeneity, clonal diversity, response heterogeneity, stepwise tumor progression, and burden of mutational antigens seen in human TNBC or genetically engineered mouse models (GEMM).82–83 In spite of these shortcomings, valuable information could be gleaned from studying TNBC immune responses, following the administration of ICD-inducing drug carriers and co-delivered drugs, as we will discuss.
C. Carriers for the delivery of immunogenic cell death (ICD) stimuli
C.1. Development of a lipid bilayer coated nanocarrier for PDAC chemo-immunotherapy
Recent advances in molecular characterization, genomic sequencing, companion diagnostics, and characterization of immune landscapes in the oncology field have given rise to new cancer therapies.84 Not only are these therapies reshaping immuno-oncology but are increasingly introduced as drug combinations to attain cooperative or synergistic outcomes, as well as improved safety. It is important to observe, however, that only 12% of all combination therapy trials receiving National Institutes of Health (NIH) support ultimately progress to obtaining FDA approval.84–85 Thus, there is an urgent need to improve the combination therapy design, including the choice of agents to combine, appropriate dosing and administration schedules, route and mechanism of delivery, and the consideration of drug-drug interactions and toxicity.86 Similar considerations hold for development of combination therapy to address heterogeneous immune landscapes.
Nanoparticle formulations and drug delivery systems (DDS) have emerged with the promise to introduce new approaches for combination therapy instead of relying on free drugs only.87 This includes our own pioneering approaches for co-formulating two (or more) agents in a macromolecular carrier that enables drug release in precisely balanced ratios and rates to accomplish medicinal effects greater than the sum of the individual agents. Moreover, a variety of different nanocarriers can be used to deliver drugs that differ in solubility characteristics (e.g., hydrophilic vs. hydrophobic), which can be delivered with improved PK and reduced systemic or off-target toxicity.
Based on these considerations, our approach for addressing heterogeneous PDAC and TNBC landscapes over the last decade has been to develop multifunctional nanocarriers that enable: (i) drug co-formulation and co-delivery, including the ability to combine APIs that differ in lipid and aqueous solubility; (ii) drug combinations that trigger anticancer immune responses by delivering ICD stimuli plus API interfering in immune escape and immune suppressive pathways; (iii) protected delivery throughout the drug circulation, accumulation, penetration, internalization, and release (CAPIR) cascade; (iv) harmonized and improved PK; (v) ratiometric design; (vi) improved safety or toxicity reduction. While there are different approaches to drug co-formulation,87 our strategy to intervene in heterogeneous PDAC and TNBC landscapes has been the use of nanocarriers coated with an encapsulating LB, which facilitates remote drug loading in the aqueous interior, while also allowing lipophilic drug incorporation into the bilayer (Figure 7). While a major part of this communication will focus on liposomes and LB-coated mesoporous silica nanoparticles (MSNP) for demonstrating the customized design of nanocarriers to address heterogeneous immune landscapes by co-delivery of ICD-inducing chemotherapeutic agents plus other immune modulatory cargo (Figure 7), we will also include some comparisons to nanocarriers developed by other investigators for accomplishing combination immunotherapy.
Figure 7. Use of LB-coated nanocarriers to deliver drug combinations.
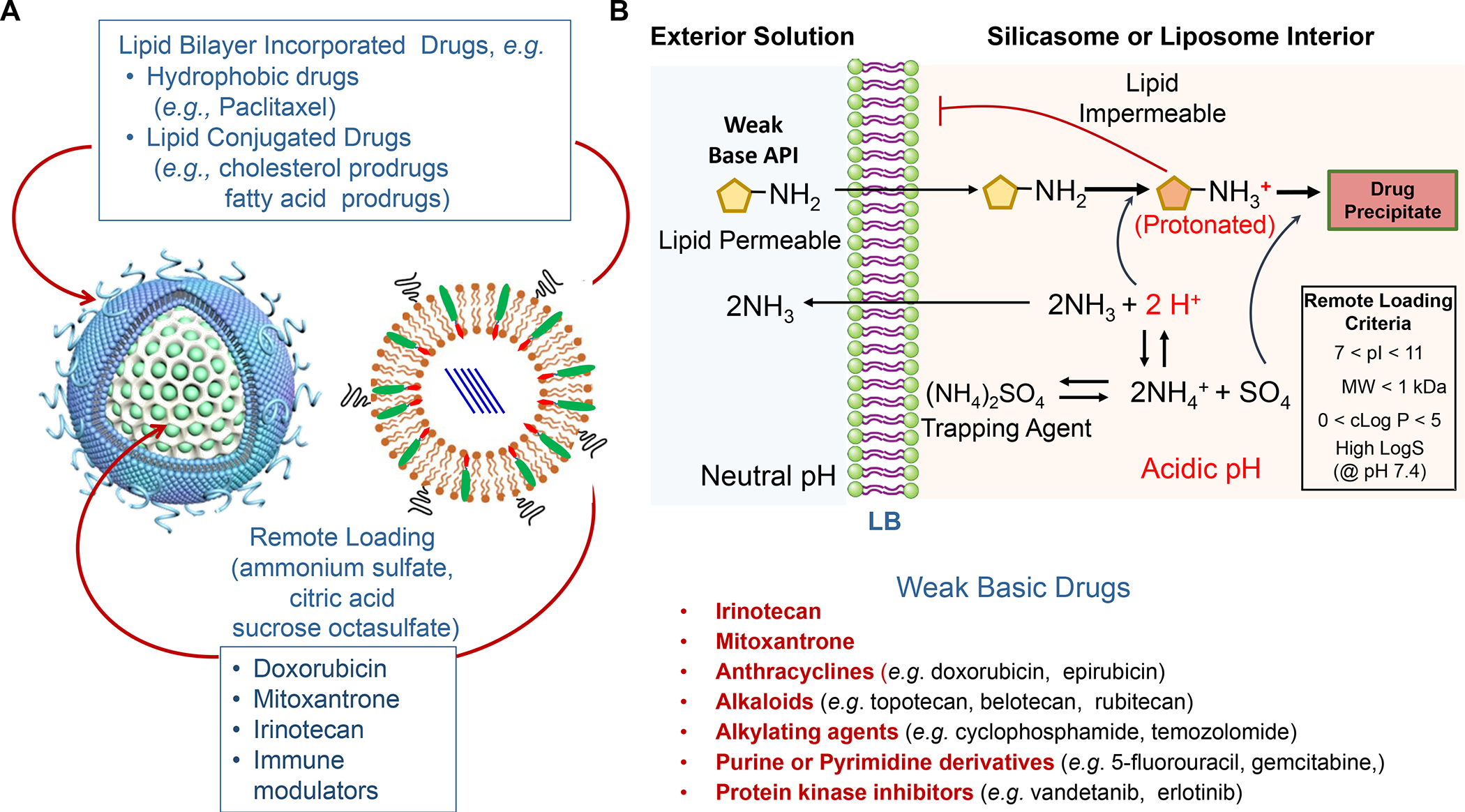
Panel A: Our basic approach to drug-co-formulation in liposomes and silicasomes is to use the hydrophilic interior for remote loading of amphiphilic drugs, while employing the lipophilic environment in the LB to incorporate lipid moieties and prodrugs. The lipid moieties are comprised of natural or synthetic lipid molecules with immune stimulatory effects (Table 4), while prodrugs are prepared by conjugating agents that interfere with immune escape or immune suppressive pathways to a series of lipid molecules (Figures 14–17). Panel B: Drug remote loading is accomplished by using ammonium sulfate, sucralose octasulfate and citrate for generating proton gradients, which allow amphipathic weak-basic molecules (see examples below the schematic) to cross the LB for protonation inside the silicasome pores or lysosomal interior.179 The protonated drug molecule complexes to the cationic group of the trapping agent to yield a drug precipitate, which regulates drug release in the TME in cancer cells. Adapted with permission from ref 179. Copyright 2021 Elsevier.
To transition to nanocarrier use for PDAC immunotherapy, it is worthwhile commenting that an Irinotecan liposome (Onivyde) as well as an albumin-Paclitaxel nanoparticle, a.k.a. nab-Paclitaxel (Abraxane) were introduced to supplement single-agent (Gemcitabine) as well as combination (Irinotecan, oxaliplatin, 5-fluorouracil, leucovorin, a.k.a. FOLFIRINOX) therapy for this disease.88–89 While the principal consideration for developing nab-Paclitaxel was to circumvent the toxicity of the Cremophor EL incipient, Onivyde was approved to improve Irinotecan delivery and treatment efficacy in metastatic PDAC patients failing Gemcitabine therapy. (Onivyde recently also received fast-track approval as first-line therapy for untreated, locally-advanced, and metastatic PDAC, in combination with 5-fluorouracil, leucovorin and oxaliplatin). While the pharmacokinetics (PK) of free Irinotecan was significantly improved through the use of the pegylated liposome, residual toxicity resulted in Onivyde receiving a black box warning (for severe diarrhea and neutropenia). This likely reflects damage to the unilamellar lipid bilayer, resulting from the generation of lipid bilayer loss by plasma proteins and complement, assisted by circulatory shear forces.90 In fact, the leakiness of an unsupported LB was instrumental in our design of a nanocarrier with a supported LB, which offers more stability in addition to allowing Irinotecan remote loading and incorporating lipophilic drugs in the LB.91–92. This culminated in the development of the silicasome platform as the preferred carrier for use in PDAC, as described below.
The Irinotecan-delivering silicasome was developed by encapsulating the proton-generating entrapping agent, triethylammonium sucrose octasulfate (TEA8SOS) in the porous interior, allowing for remote import of weak-basic Irinotecan (pKa = 8.1) (Figure 8A).91 Subsequent comparison of the silicasome carrier to an in-house liposome (with similar features as Onivyde) by intravenous (IV) administration in the orthotopic KPC model, demonstrated improved performance of the silicasome for PK, circulatory half-life, biodistribution and Irinotecan tumor content (Figure 8B). Not only was the silicasome more effective for CTL killing than the liposome at the tumor site, but was also more successful in eliminating metastases. Equally important, the LB-MSNP carrier prevented bone marrow toxicity, intestinal villi blunting and hepatotoxicity seen during free liposomal Irinotecan administration (Figure 8C). We ascribed the toxicity reduction and improved efficacy of LB-coated MSNPs to the increased stability of its supported LB, which also increases loading capacity. The advantages of the LB encapsulated silica nanoparticles in comparison to liposomes is discussed in more detail in Figure 8B. There was no evidence that the bare MSNPs contributed to independent toxicity.
Figure 8. Upscale production of Irinotecan-silicasomes for effective and safe treatment of PDAC.
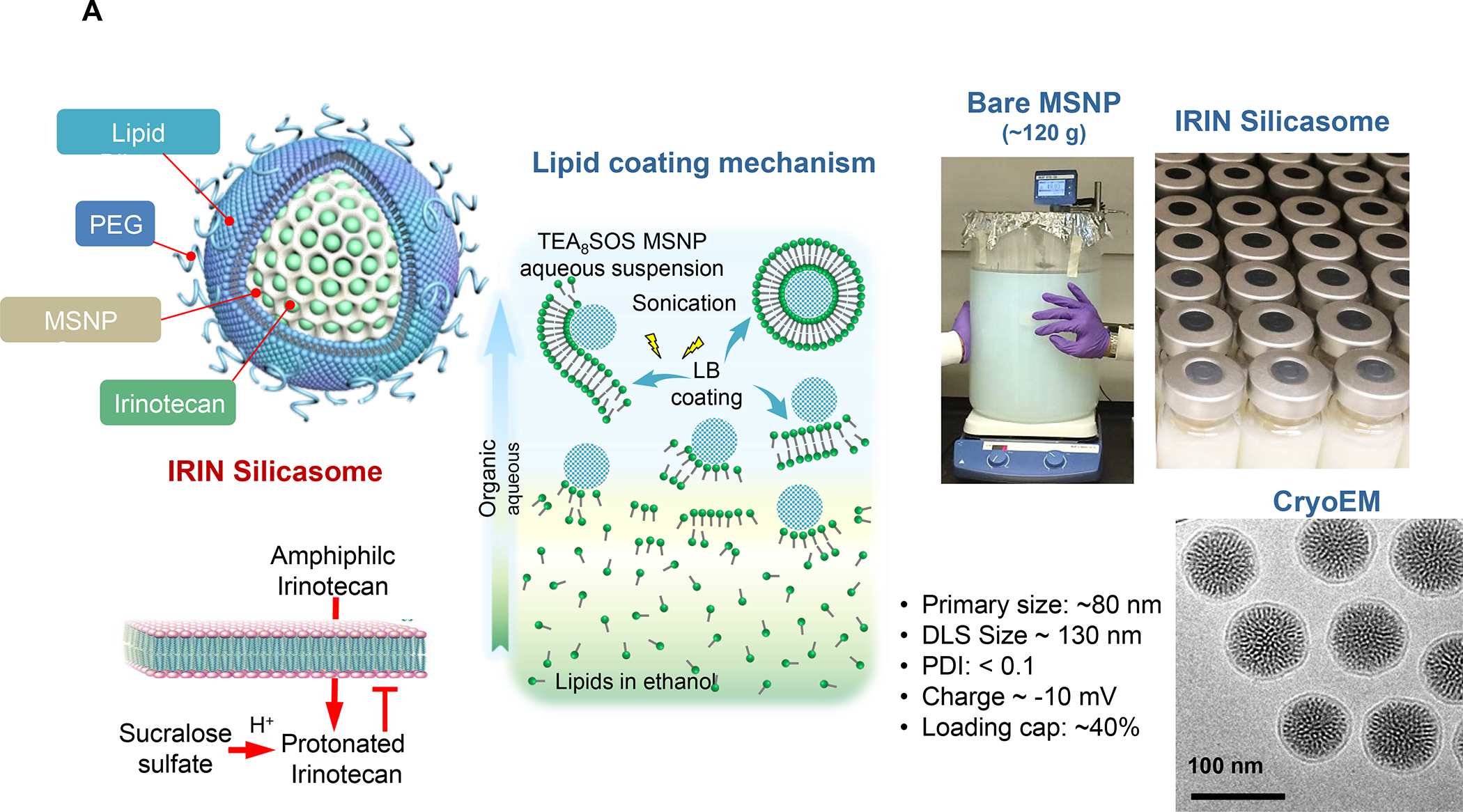
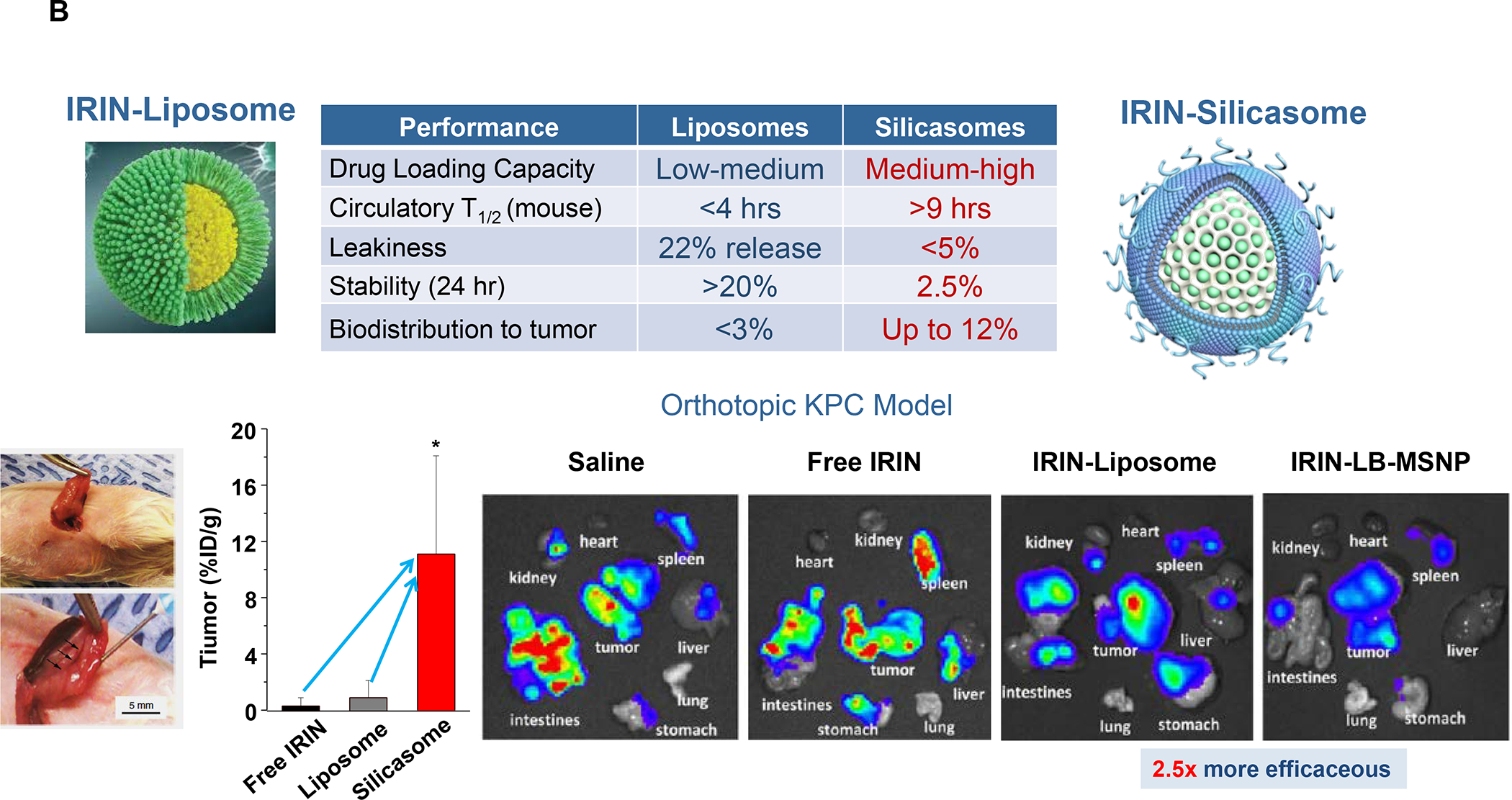
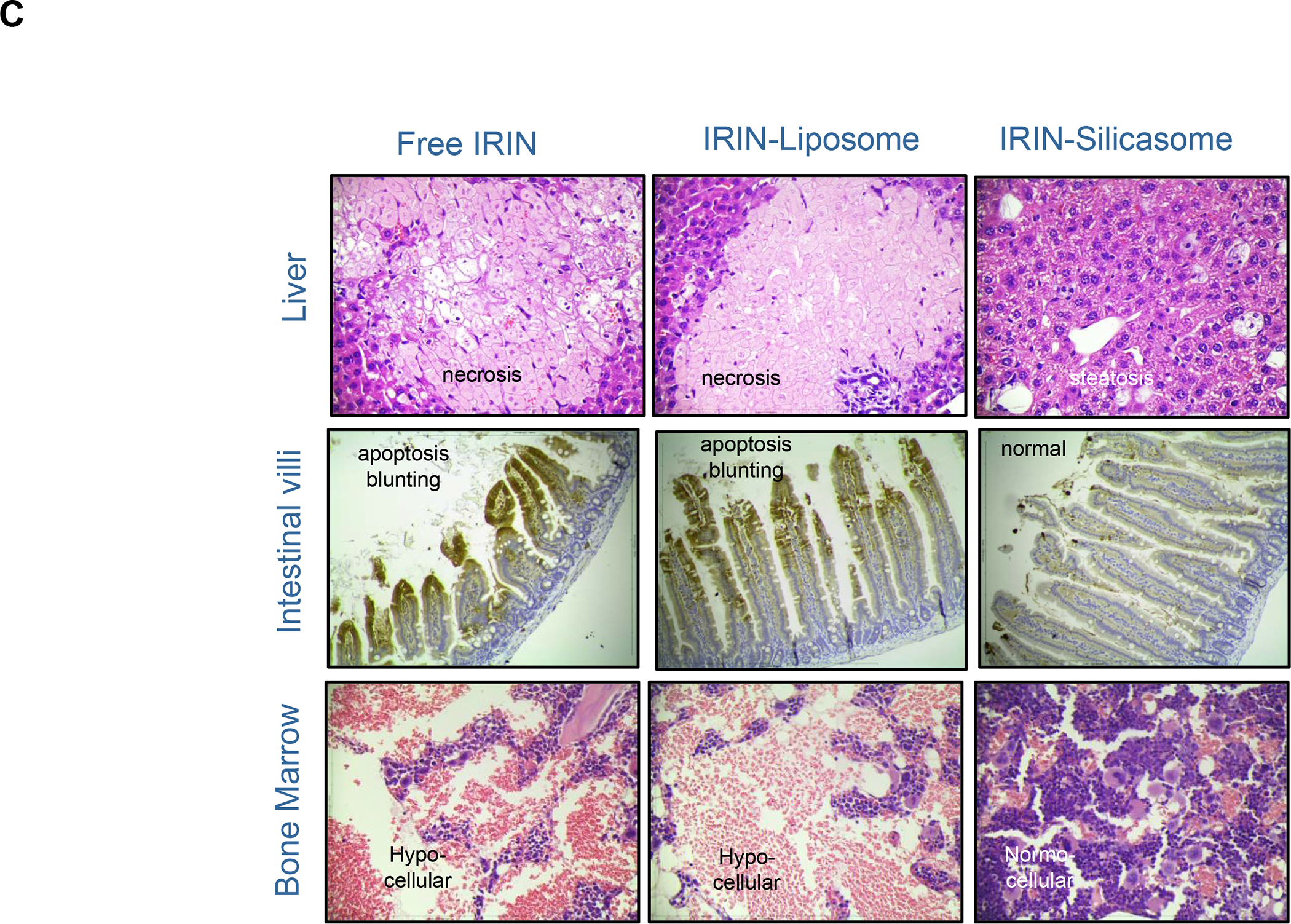
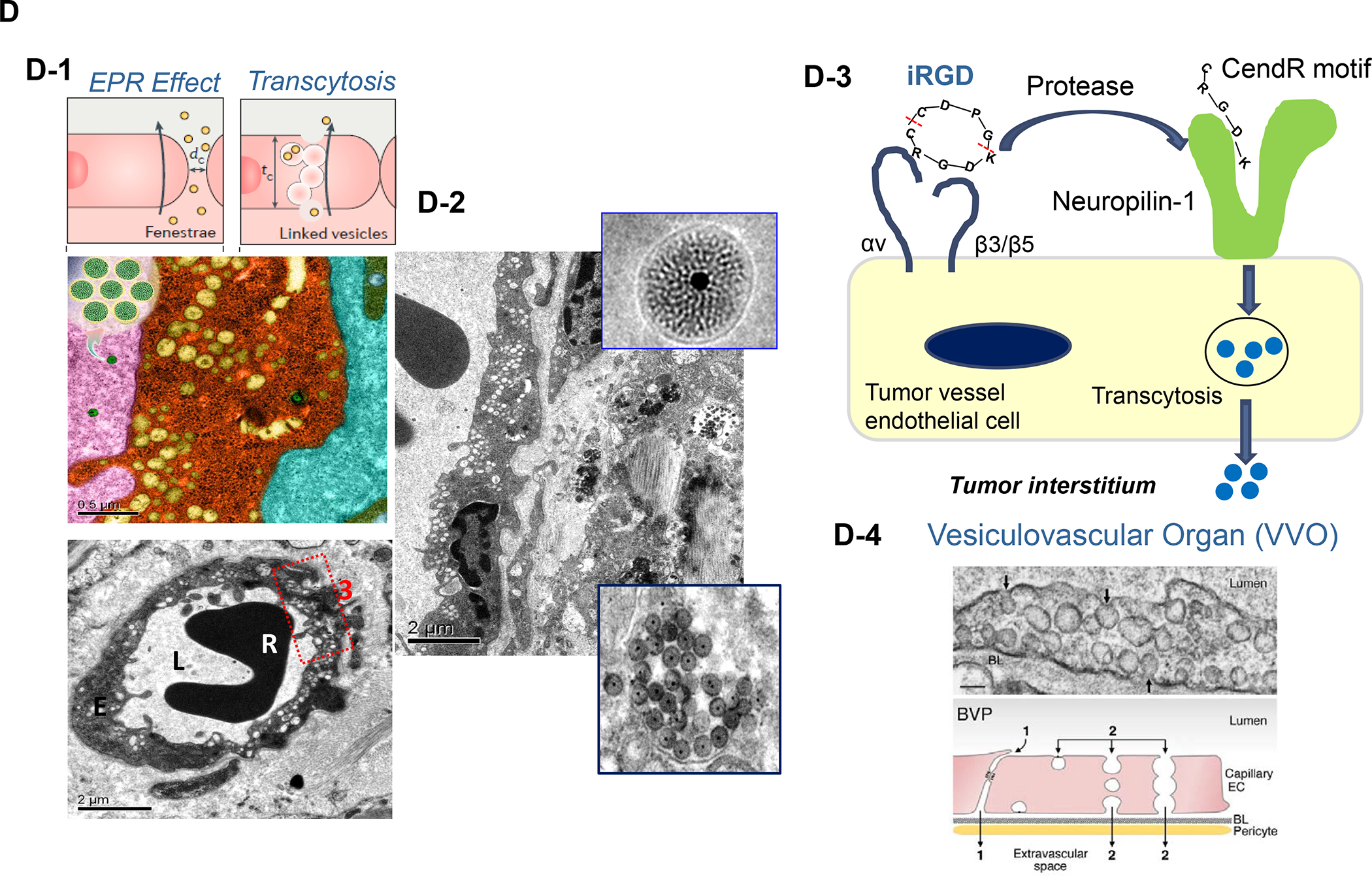
Panel A: We have developed upscale production of an MSNP carrier, where the LB is used for Irinotecan (IRIN) remote loading, following encapsulation of sucralose octasulfate (TEA8SOS) in the particle pores.91–94, 98–99 Large batch production was made possible by using ethanol precipitation for LB coating instead of sonicating a biofilm, which has limitations for coating large particle batches.98 The upscale flow-through sonication procedure involves the direct introduction of an aqueous suspension of MSNP into a concentrated, ethanol suspended lipid solution, followed by controlled energy input in a flow cell sonication device. The coating mechanism is assembly of the suspended lipid monomers onto on the particle surfaces upon introduction into the aqueous environment. This approach is advantageous from the perspective that there is complete and rapid surface coating by the LB (cryoEM visualization) and avoidance of potentially toxic chloroform use. This approach allows the application of LB coating to 120 g MSNP batch sizes. The picture displays the average size dimensions and physicochemical characteristics, including IRIN loading capacity of ~40%. Adapted from ref 98. Copyright 2019 American Chemical Society. Panel B: Improved IRIN delivery and treatment efficacy in an orthotopic KPC model, using a silicasome vs. a liposome.91 The inserted table shows that the increased stability of the supported lipid bilayer improve carrier stability, circulatory half-life, leakiness and drug delivery atthe KPC tumor site, compared to a liposomal equivalent. This includes facilitated drug loading as a result of van der Waal’s forces, hydrogen bonding and electrostatic interactions with the wall of the silicasome pores. Improved drug delivery was accompanied by increased tumor cell killing at the primary and metastatic sites, as shown in the lower panel. Adapted from ref 91. Copyright 2016 American Chemical Society. Panel C: The silicasome carrier does not induce the bone marrow cytopenia, intestinal villi blunting and liver toxicity seen with free or a liposome encapsulated Irinotecan.91 Similar efficacy and safety have also been demonstrated in colon cancer models.98 Adapted from ref 91. Copyright 2016 American Chemical Society. Panel D: Silicasome uptake into the KPC tumor matrix and cancer cells was improved by co-administration of the cyclic iRGD peptide, which promotes transcytosis. Silicasomes were synthesized with an imageable gold nanoparticle core, followed by IV administration of the particles at 50 mg/kg in animals bearing orthotopic KPC tumors.93 Tumor tissues were collected for electron microscopy viewing after 24 hours. D-1 shows conventional and pseudocolor TEM images, demonstrating intravesicular particle transport across the tumor blood vessel wall. The vesicle numbers increased in animals receiving either separate injections or injection of the iRGD-conjugated nanocarrier.93 Adapted with permission from ref 93. Copyright 2017 American Society for Clinical Investigation. D-2 shows TEM images that demonstrate endothelial vesicles and particle localization in the tumor stroma or localized inside cancer cells. Adapted with permission from ref 93. Copyright 2017 American Society for Clinical Investigation. D-3 is a schematic to show the working mechanism of iRGD -induced transcytosis, which involves cyclic peptide binding to overexpressed integrins at the tumor site, peptide cleavage, and release of a CendR motif that activates the tyrosine protein kinase receptor, neurophilin-1.303 Adapted with permission from ref 303. Copyright 2018 Elsevier. This transcytosis mechanism is likely identical to the vesiculovascular organ, delineated by Nagy and Dvorek et al., who performed extensive EM analysis of multiple cancer types in humans (D-4).304–306 Reprinted with permission from ref 305 under the terms of the Creative Commons Attribution 2.0 License. Copyright 2008 The Authors.
An important finding during the tracking of silicasome biodistribution was the demonstration that these particles gain access to the PDAC microenvironment by a transcytosis process under regulation by the neuropilin-1 (NRP-1) receptor (Figure 8D).93–94 This observation is important in light of the frequently held view that the abnormal permeability of tumor blood vessels constitutes a major pathway for nanoparticle uptake at the tumor site, also described as the enhanced permeability and retention (EPR) effect. This view has been challenged by the growing support for a vesicular transcytosis pathway as an important route for nanoparticle entry into TNBC and PDAC tumor landscapes.91, 95 This thinking is also in keeping with clinical studies showing that the abnormal leakiness of tumor blood vessels is not the only explanation for the improvement of drug delivery by nanocarriers.96 A number of local tumor-related factors such as high interstitial fluid pressure, poor vascular perfusion and irregular vascular distribution negate the impact of abnormal leaking blood vessels at the tumor site. Based on these considerations, Dewhirst and Secomb cautioned that EPR should only be invoked on a case-by-case basis, with actual confirmation of such an effect in the clinical setting.96 Another important outgrowth of the transcytosis concept is that NRP-1–mediated transport across the vascular wall (and possibly also the membrane of certain cancer cells) can be enhanced by the physical attachment or independent administration of a cyclic tumor-penetrating peptide, iRGD (Figure 8D).93 iRGD interacts with tumor-specific αvb3 integrins, which are overexpressed on the tumor vasculature, allowing the CendR motif to bind and activate the non-tyrosine kinase receptor, NRP-1.97 We obtained ultrastructural evidence that iRGD could induce the appearance of grouped vesicles in tumor associated endothelial cells, with the ability to carry silicasomes into the tumor matrix as well as into cancer cells Figure 8D).93 Another important finding was that improved Irinotecan delivery and tumor shrinkage of human PDAC xenografts by iRGD could be correlated to the level of NRP-1 expression on patient-derived PDAC samples.93 Based on promising impact of silicasome therapy in the KPC model, we have developed upscale manufacturing of the Irinotecan-silicasome, with the view to clinical translation (Figure 8A).98
In addition to the cytotoxic effects of chemotherapy, a significant advance has been the discovery that encapsulated Irinotecan delivery induces an immunogenic cell death (ICD) response that can be exploited for chemo-immunotherapy in the KPC model.99 The delivery of an ICD stimulus is analogous to an endogenous vaccination attempt, involving a form of cellular apoptosis that is accompanied by calreticulin (CRT) expression on the dying tumor cell surfaces.100 CRT provides an “eat-me” signal to antigen presenting cells (APC), allowing improved tumor antigen presentation, assisted by the adjuvant effect of high-mobility group box 1 (HMGB1) protein and ATP, which is also released in the process. A brief account of the ICD process appears in Figure 9. While the Irinotecan-induced immunogenic response likely is dependent on the nuclear impact of the topoisomerase I inhibitor, the drug also exerts a collateral effect on lysosomes, as a result of weak base properties (also used for remote drug loading). This alkalinizing affect leads to neutralization of the lysosomal pH soon after drug addition to PDAC cells. Alkalinization disrupts autophagy flux and development of endoplasmic reticulum stress (Figure 10A). Noteworthy, this cellular response is accompanied by increased PD-L1 expression, raising the possibility that the ICD effect may be enhanced by ICIs. This synergy was demonstrated in the KPC model, where treatment with the Irinotecan-silicasome could be seen to induce an ICD response (CRT, HMGB1, perforin and granzyme B expression) that could be augmented by anti-PD1 (Figure 10B). The treatment outcome was associated with improved survival, as demonstrated by Kaplan-Meier analysis (Figure S9).
Figure 10. Use of the Irinotecan-silicasome for PDAC chemo-immunotherapy by an ER stress pathway.

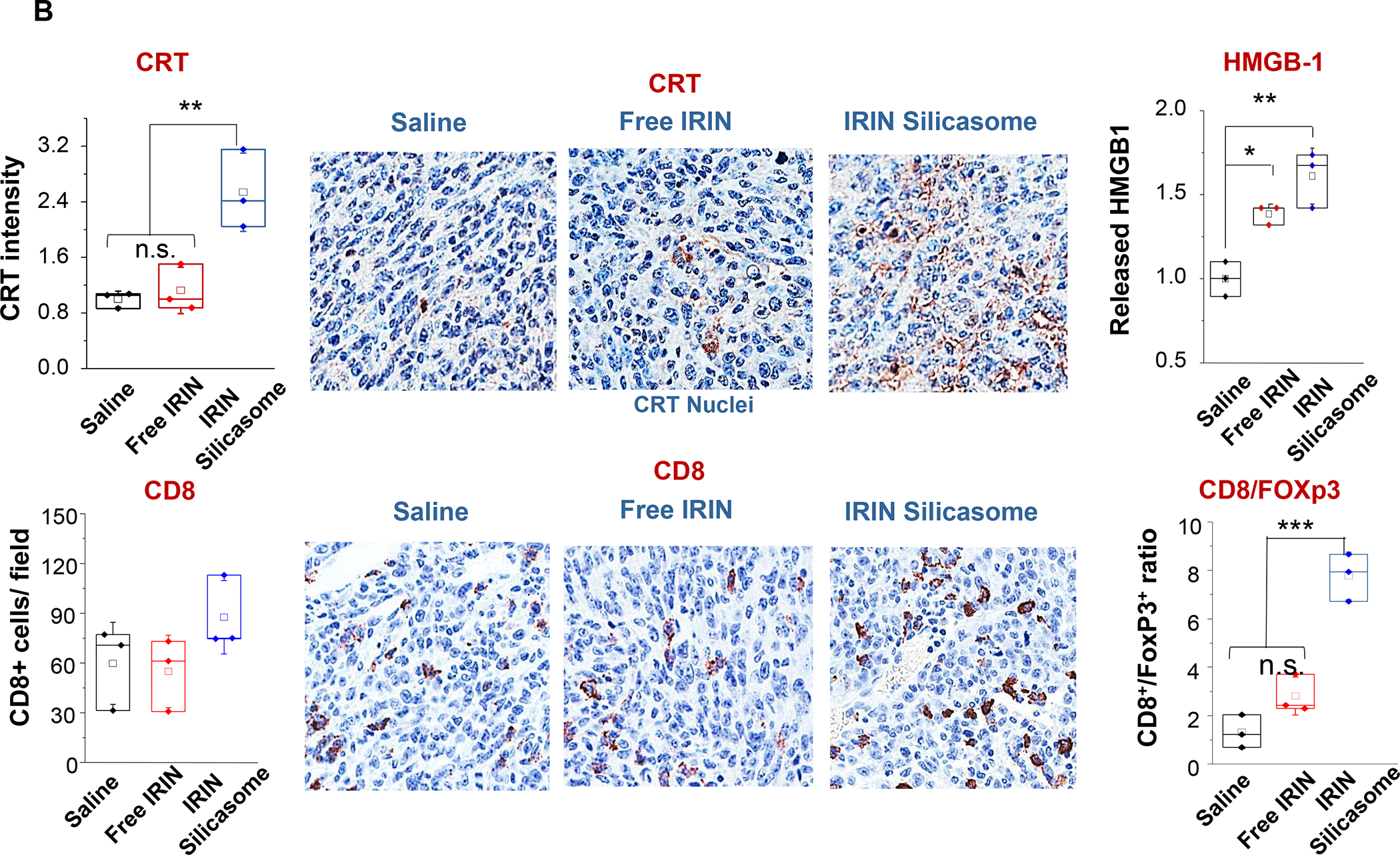
Panel A: In addition to its action as a topoisomerase I inhibitor, Irinotecan (IRIN) induces a robust cell stress response because of its weak basic properties, which induces lysosomal alkalization and interference in autophagy flux. This induces oxidative stress and ER stress. The response is also accompanied by PD-L1 expression in KPC cells. Panel B: The IRIN-silicasome induces an ICD response in orthotopic KPC mice, injected IV on 3 occasions with either free IRIN or the IRIN-silicasome (40 mg/kg), followed by sacrifice 72 h later. IHC analysis, with the assistance of Aperio ImageScope software, was used to determine CRT and HMGB1 release (top) or recruitment of CD8+ and Foxp3+ cells (bottom) to the tumor landscape. Imaging intensity was quantitatively expressed as fold-increase compared to the saline group. Data are expressed as mean ± SEM, n = 3. *p < 0.05; **p < 0.01; ***p < 0.001 (1-way ANOVA followed by a Tukey’s test). Panel A-B reprinted with permission from ref 99 under a Creative Commons Attribution License 4.0 (CC BY). Copyright 2021 The Authors.
In addition to Irinotecan drug loading, silicasomes can be used to deliver other ICD-inducing chemotherapeutic agents, including Gemcitabine and Oxaliplatin (Figure 7). In addition to Irinotecan, the FOLFIRINOX regimen includes Oxaliplatin, a potent chemotherapeutic agent, which leads to dose-limiting toxicity. Oxaliplatin is known to induce an ICD response that is useful for chemo-immunotherapy.101 However, since Pt drugs are coordination compounds, it is not possible to use remote loading or achieve high drug loading capacity in MSNPs, leading us to develop an alternative drug loading procedure (Figure 11A).102 This was accomplished by the using the coordination chemistry features of the Oxaliplatin precursor, cis-dichloro (1,2-diamminocyclohexane) platinum (II) (a.k.a. DACHPtCl2) for attachment to the sidewall localized silanol groups in the pores under basic pH conditions. The carrier could then be coated with an LB, yielding DACHPt-silicasomes, which improved the drug biodistribution and PK to the PDAC site after IV injection. Not only was the drug delivery improved over free Oxaliplatin but also demonstrated an ICD effect in the KPC model. Encapsulated DACHPt resulted in a dramatic improvement of the grade 3–4 neutropenia effects of Oxaliplatin. We also showed that the immunogenic properties of the nanocarrier could be improved by co-administration of anti-PD1 antibody, with survival benefits (Figure 11B).
Figure 11. Synthesis of a DACHPt-silicasome for PDAC chemo-immunotherapy.
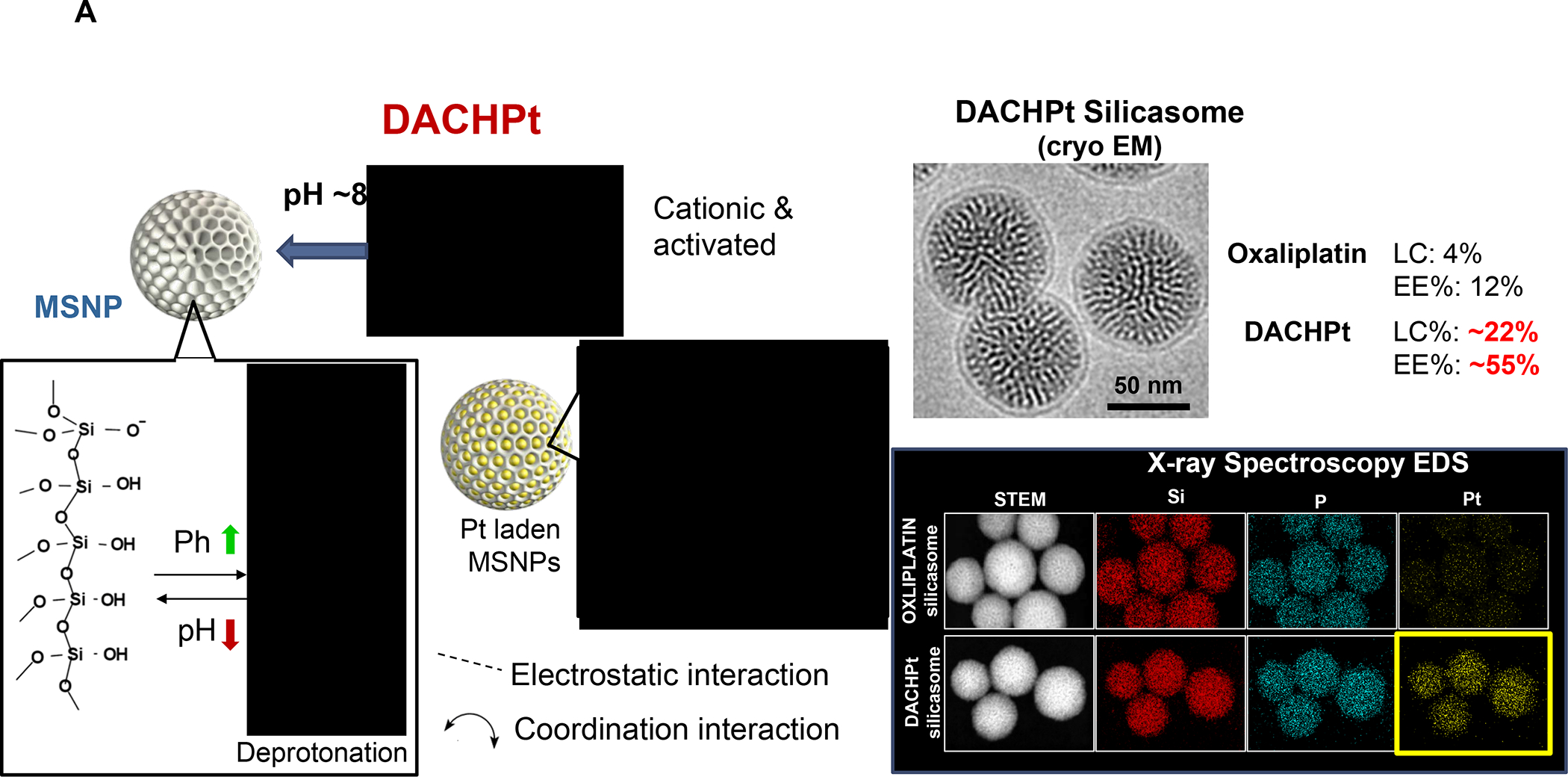
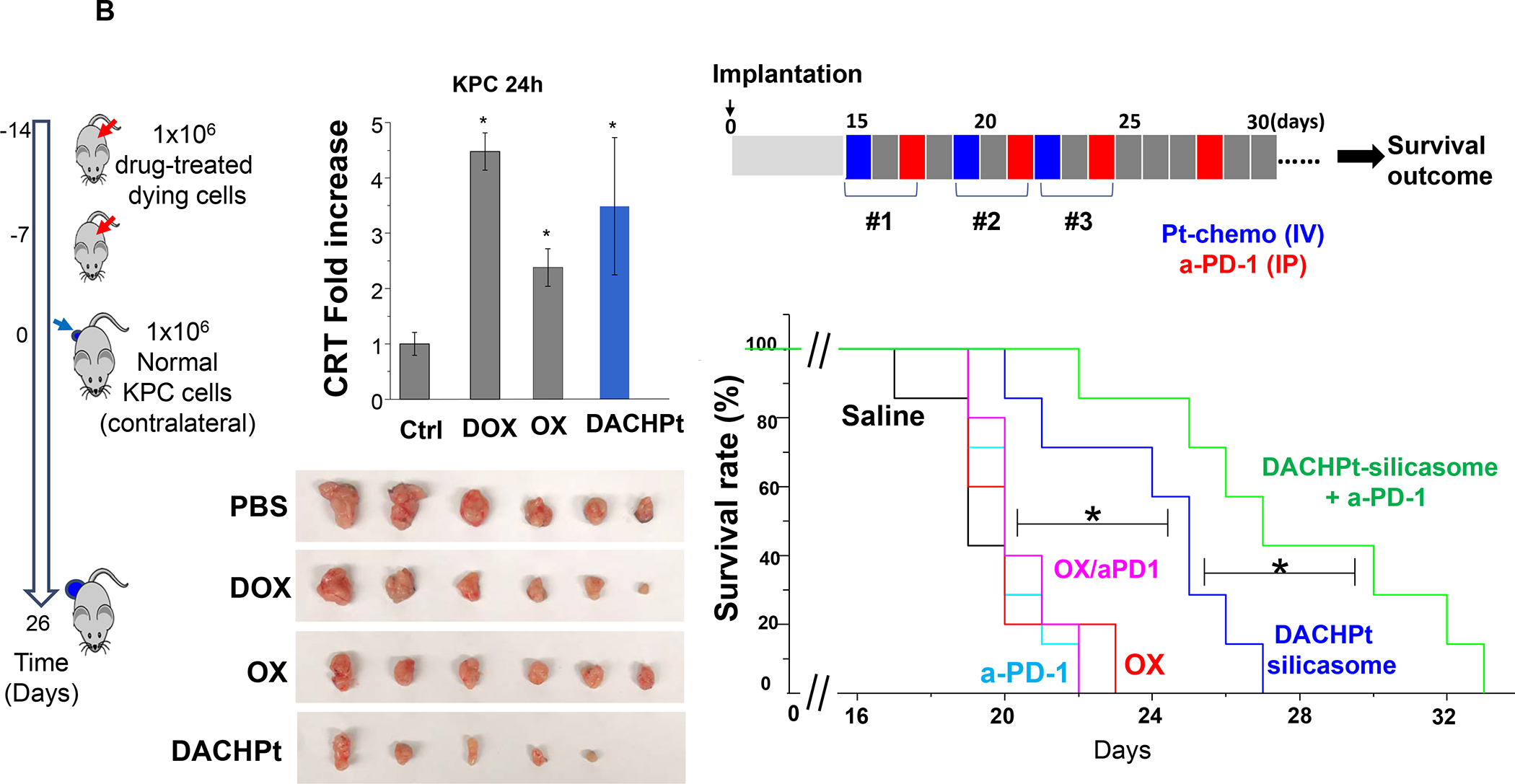
Panel A: The DACHPt carrier is synthesized using the coordination chemistry of the drug under mild alkaline conditions. The panel on the left indicates that pH adjustment to obtain weak alkaline conditions increases the number of silanol groups in the particle pores. This allows electrostatic attachment of cationic DACHPt with achievement 22% loading capacity (compared to 4% for passively loaded Oxaliplatin). The bottom right panel demonstrates X-ray spectroscopy confirmation of the presence of Si, phosphorus (phospholipid) and Pt, also demonstrating that the Pt/Si ratio (w/w) is higher for DACHPt incorporation than Oxaliplatin. Panel B: The panel on the left shows a vaccination response of KPC cells exposed to 20 μm Doxorubicin, 500 μm Oxaliplatin, and 500 μm DACHPt. These agents induce an ICD response, which was confirmed by CRT expression after 24H (upper panel). Data is expressed as mean ± SD, n = 3. *p < 0.05 compared to PBS control. The left and lower panels show the vaccination response in which B6129SF1/J mice received subcutaneous administration of the chemo-treated KPC cells in one flank on two occasions, followed by injecting live KPC cells on the contralateral side. Tumor sizes were assessed at the challenge site on day 26, demonstrating an effective vaccination response to the chemo agents. It was also possible to show the generation of an ICD response in the orthotopic KPC model, with DACHPt being more effective than Oxaliplatin. The panel on the right demonstrates a KPC survival study, where animals treated with DACHPt silicasome at a Pt dose equivalent of 2 mg/kg IV every 3–4 days, in combination with IP administration of 100 μg anti-PD-1 antibody on 4 administrations. Saline, anti-PD-1 only, Oxaliplatin and Oxaliplatin plus anti-PD1 served as controls. Kaplan–Meier plots were used to display animal survival, which was significantly improved by DACHPt silicasome plus-anti-PD1 (n = 5–7, *p< 0.05. Log Rank test). Panel A-B adapted with permission from ref 102. Copyright 2021 John Wiley and Sons.
Other than the in-house use of silicasomes for the delivery of Irinotecan, Oxaliplatin and DACHPt to generate immunogenic effects in a PDAC landscape, amphiphilic diblock copolymer nanoparticles have been used to achieve an ICD outcome with Oxaliplatin and Doxirubicin in pancreatic cancer.103 Moreover, as explained in Figure 9, it is also possible to induce Type II ICD responses with photodynamic therapy, which was demonstrated in a Panc02 pancreas cancer model with a nanocarrier delivering a photosensitizer and a prodrug.104 The carrier was constructed by self-assembly of cyclodextrin-grafted hyaluronic acid, pyropheophorbide a (photosensitizer), and a prodrug. The use of laser excitation in combination with multiple-component nanoparticles was shown to significantly prolong survival in Panc02- bearing mice compared with monotherapy or control groups. An extensive review on the use of nanocarriers for pancreatic treatment, immunotherapy and imaging was recently published by Liu et al. 105
C.2. Use of lipid bilayer coated carriers for TNBC chemo-immunotherapy
Chemotherapy remains the cornerstone of therapy in early and advanced-stage TNBC, which is also the most frequent cancer indication for ICD-inducing chemotherapy.106–108 Consequently, several promising regimens have been established where PD1/PD-L1 inhibitors are combined with agents such as Doxorubicin and Paclitaxel in metastatic, locally-advanced and early stage TNBC.106 One of the best objective responses for metastatic TNBC is reported in the TONIC trial (NCT02499367) where the objective response rate for Doxorubicin plus nivolumab (35%) was superior to the same antibody combined with Cisplatin (23%).78 This is congruent with Doxorubicin being a robust ICD inducer while Cisplatin is not.109 Even more compelling evidence for the utility of chemo-immunotherapy comes from early and advanced-stage disease. For instance, in the early-stage BC phase II I-SPY 2 (NCT01042379) trial, it has been shown that adding pembrolizumab to taxane or anthracycline neoadjuvant therapy could double the pathological complete response (pCR) rate.110 This study was the forerunner of the phase I KEYNOTE-173 (NCT02622074) trial, looking at anti-tumor efficacy of pembrolizumab, combined with Paclitaxel or Doxorubicin, included in various neoadjuvant administration regimens.111 This study demonstrated pCR rates of 60% across all treatment cohorts. Also, the phase III KEYNOTE-522 trial (NCT03036488) demonstrated that neoadjuvant administration of Paclitaxel and carboplatin in combination with pembrolizumab could increase the pCR rates from 51.2 to 64.8% in a locally advanced disease setting.112
Pegylated liposomal Doxorubicin (PLD) is the first-ever nanodrug to be developed for Kaposi’s sarcoma, breast and ovarian cancer, primarily to reduce cardiotoxicity.113 PLD has also been shown to be as effective and less toxic as the free anthracycline in metastatic BC, allowing increased treatment duration as well as accomplishing high-dose drug administration. Currently, a Doxorubicin liposome is being used for its immunogenic effects in combination with ipilimumab and nivolumab in an ongoing ICON (CA209–9FN) chemo-immunotherapy trial in patients with metastatic HR+ breast cancer.114 Liposomes have also been used in preclinical TNBC studies to improve Doxorubicin delivery in 4T1 and MMTV-PyVT tumors, where blood vessel truncation and poor perfusion interfere in intratumoral drug delivery.95, 115–117 While Grasselly et al. have observed a poor response to anti-PD1 or anti-PD L1 antibodies in combination with Doxorubicin and Cyclophosphamide in the 4T1 model,118 a good treatment response was obtained with the same combination in the PyMT mammary tumor model.119 These results agree with our studies in orthotopic 4T1, EMT6 and PY8119 tumor models, showing lack of a combination therapy response in 4T1 tumors, while observing a combined treatment effect in EMT6 and PY8119 tumors. These results also agree with Charles River Laboratories’ immune profiling of syngeneic tumor models, where EMT6 is characterized as modestly-responsive while 4T1 is regarded as refractory. According to Charles River data bank, the response in EMT6 is especially rigorous if anti-PD1 is combined with anti-CTLA-4. This leaves the refractory response in 4T1 to be properly explained in light of the fully inflamed landscape and immunogenic effect of Doxorubicin (Figure 6, Figures S3–S6).118 Additional barriers to address include the CXCL12/CXCR4 cascade, which will be discussed later. Combination treatment with Doxorubicin and anti-PD-1 mAb was effective for generating improved response outcomes in the PyMT model, from which PY8119 cell line was derived.119
While our own studies have not used encapsulated Paclitaxel for TNBC treatment, this ICD-inducing chemo agent is frequently used as first-line therapy in BC patients, where the drug is favored by some oncologists as a result of the underreported toxicity of Doxorubicin.120 Albumin-bound Paclitaxel (Abraxane) was developed to overcome the low Paclitaxel solubility (logP = 3.96), with superior efficacy compared to Taxol in metastatic BC patients.121–122 Noteworthy, the perceived advantage of nab-Paclitaxel in combination with atezolizumab (Tecentriq) in the IMPASSION130 combination immunotherapy trial in TNBC patients did not produce significant results in the follow-up IMpassion131 trial, failing to show a difference in progression-free survival in patients receiving Taxol and Tecentriq vs. people receiving Taxol alone.123–124 Nonetheless, in spite of this outcome, Paclitaxel is a drug that can be considered for incorporation into LB carriers, as we will show later. This requires careful consideration of drug hydrophobicity and its tendency to come out of solution above 3 mol%.125–127 To overcome this problem Zhen et al. have proposed the inclusion of cationic lipids with cis double bonds (e.g., DLin-MC3-DMA, DOTAP or DOPC) in the liposome membrane in place of mono-saturated fatty acids.125
Based on the consideration for liposome use to deliver ICD-inducing drugs, we also developed a liposomal carrier for the anthracenedione, Mitoxantrone (MTO), to develop immunotherapy for TNBC and colon cancer based on an immunogenic response that involves NK cell activation.128–129 Kroemer et al. called attention to this drug as a robust ICD inducer among ~900 anti-cancer compounds screened.130 We constructed a MTO liposome (L-MTO) by using a citric acid buffer as trapping agent for remote drug loading (Figure 12 A). This yielded a 110 nm carrier, which can be seen in the Cryo-EM picture to contain a drug precipitate (Figure 12A). IV administration of the liposome on 4 occasions over 24 days in orthotopic 4T1 tumor-bearing mice showed significant tumor shrinkage, which was accompanied by CRT and HMGB1 expression, in addition to perforin and granzyme B deposition (Figure 12A, Figure S10). Noteworthy, MTO treatment was not accompanied by increased CD8+ T-cell recruitment, showing instead an increased number of NKp46+ cells (Figure 12B). We will discuss the data in Figure 12 related to the combined use of MTO with Indoximod, an IDO-1 inhibitor. These results in 4T1 were corroborated by similar data in the EMT6 and PY8119 breast cancer models (Figure S11 and S12) as well as the CT26 colon cancer model (Figure S13–S15), including the use of 3-color mIHC staining to show that perforin deposition is accompanied by NKp46+ recruitment (Figure S15). While Doxorubicin and Cyclophosphamide also induce NK activation, these chemo agents increase CTL expression (Figure S14). Although the reason for the differential effect of MTO on NKp46+ vs. CD8+ T-cells is unknown, it is important to consider crosstalk between NK cells and DC in promoting antitumor immunity, as conveyed in the legend of Figure S16.131
Figure 12. Mitoxantrone-delivering liposomes induce an ICD response that can be augmented by co-delivery of an IDO-1 inhibitor.
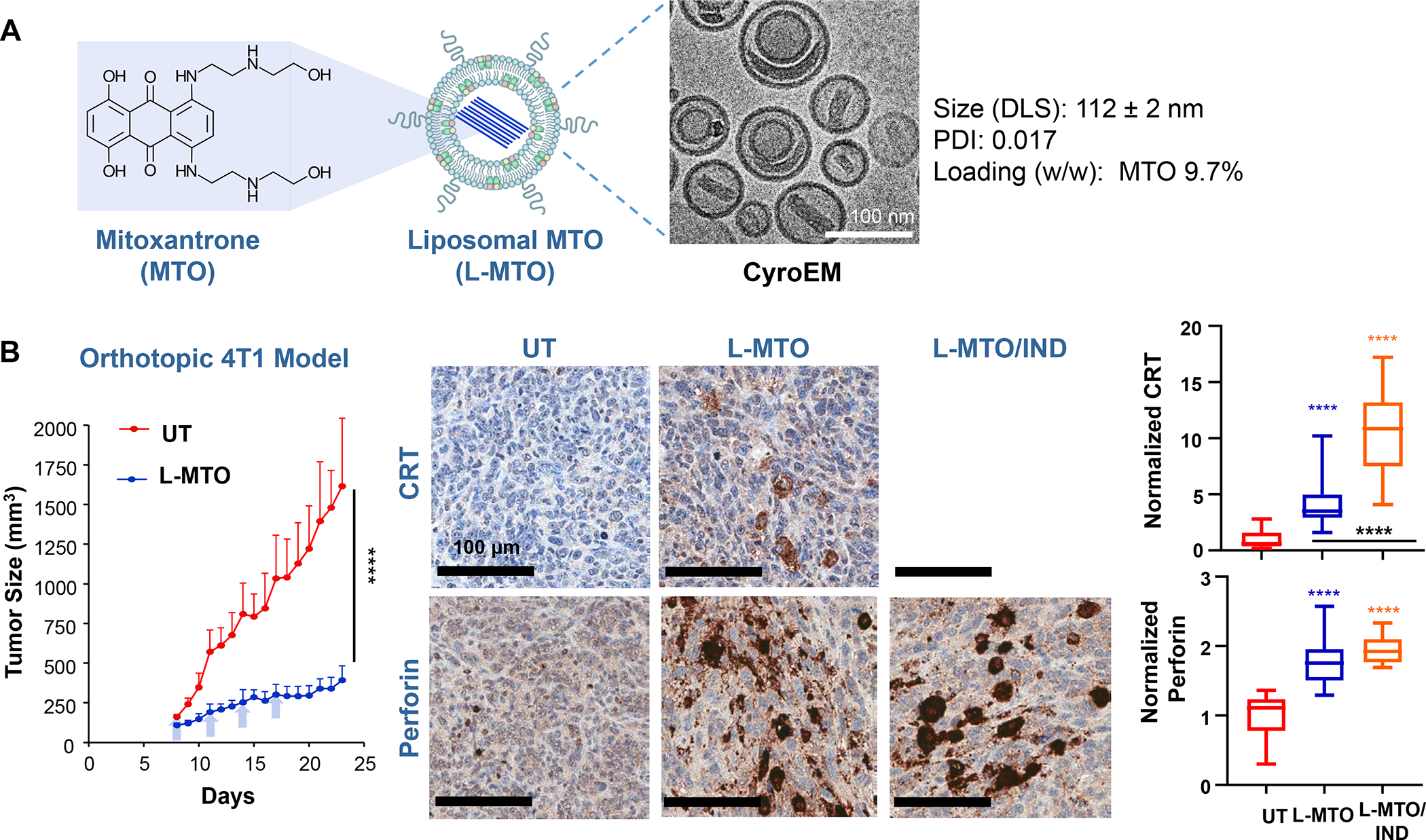
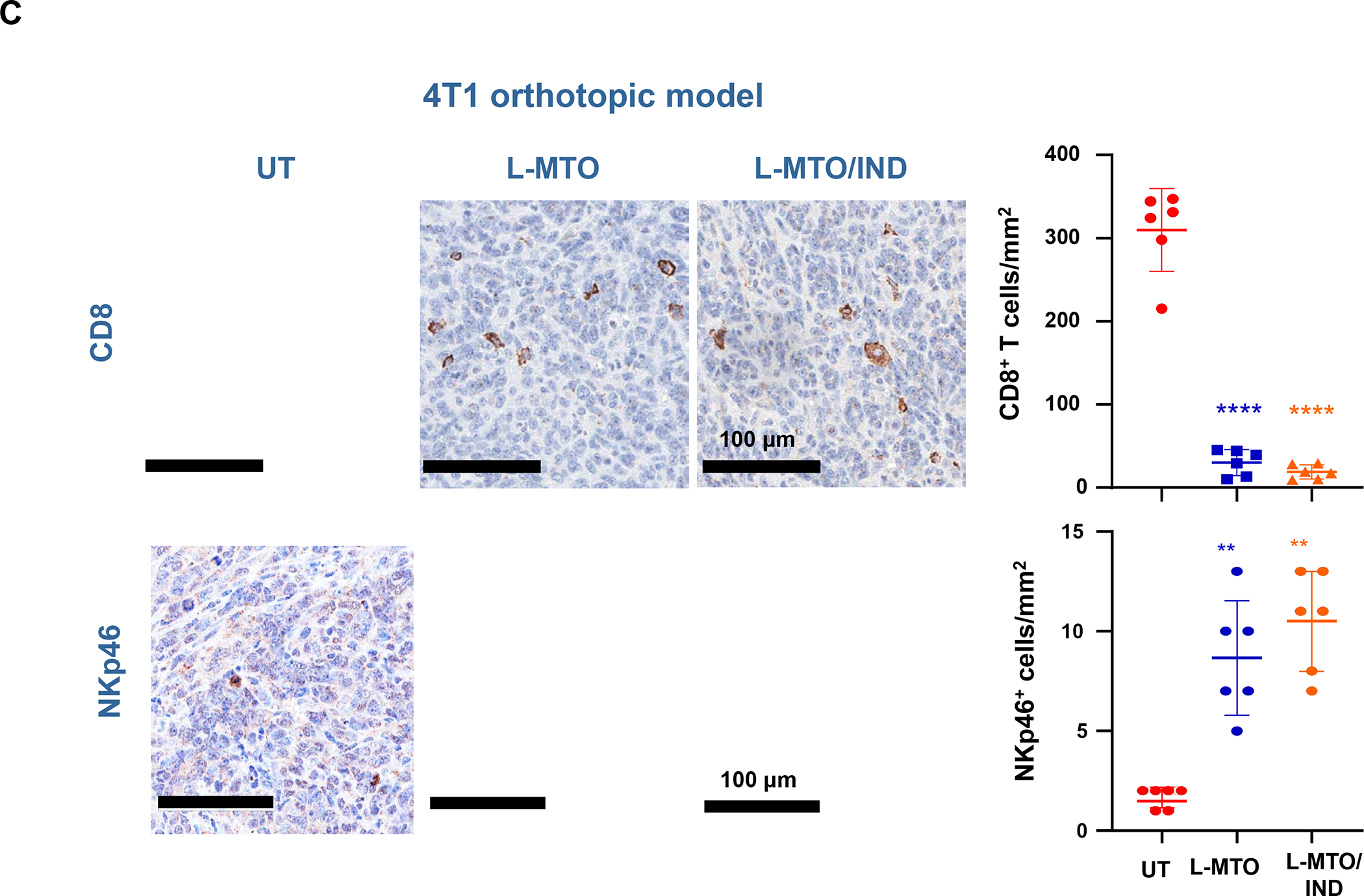
Panel A: Two liposomes were constructed, one containing MTO-only (L-MTO), the second including a combination of MTO plus a prodrug IDO-1 inhibitor. While details about synthesis of the dual delivery liposome are discussed in Figure 18, this passage will provide a side-by-side comparison to maintain interpretation inclusive. The lipid composition of the L-MTO liposome was comprised of DSPC, Chol, CHEMS, and DSPE-PEG2kDa in the molar ratio of 45: 30: 20: 5, while the L-MTO/IND liposome contained DSPC: Chol-IND: CHMS: DSPE-PEG2kDa in the molar ratio of 45: 30: 20: 5. Liposome synthesis was carried out through hydration of a coated biofilm in a round bottom flask, followed by sonication in a citric acid buffer. Free soluble MTO was remotely loaded as described by us, followed by size exclusion chromatography to remove unencapsulated MTO. The purified L-MTO liposomes had an average diameter of ~112 nm with a low polydispersity index at 0.017 and a final drug loading at 9.7% (drug/lipid w/w). Comparable values for the L-MTO/IND liposome were the size of ~100 nm with a polydispersity index at 0.014 and a zeta-potential at −11.7 mV (detailed in Figure 18B). The presence of a drug precipitate in these liposomes is demonstrated in the cryoEM (upper panel). Panel B: L-MTO and L-MTO/IND were administered IV to mice with orthotopic 4T1 tumors to deliver an MTO equivalent dose of 3 mg/kg/injection, with/without the co-delivery of IND at 3 mg/kg/injection. The first injection was on day 8 when tumor size was 100–150 mm3, followed by 3 injections on days 11, 14, and 17. Mice were followed for 23 days. L-MTO administration induced significant tumor shrinkage, further enhanced by IND co-delivery (as explained later in Figure 18C). IHC analysis confirmed significant CRT and perforin expression (similar effects for HMGB1 and granzyme B are shown in Figure S10). Panel C: Different from the immunogenic effects of Doxorubicin and Irinotecan, the robust immunogenic effect of MTO does not include CTL recruitment, resulting instead in the generation of NKp46+ cells, which are particularly relevant for BC immunotherapy (Figure S16). Further data regarding the dual delivery liposome appear in Figure 18. Panel A-C adapted from ref 128. Copyright 2020 American Chemical Society.
The importance of NK cell activation in BC (particularly the HR+ subtype) is their key defense role in achieving immune equilibrium in the early tumor site before immune escape, whereupon the role of cytotoxic T cells become more important (Figure S16).132 MTO was initially approved as a chemotherapeutic agent for the treatment of metastatic BC, acute leukemia, prostate cancer, and non-Hodgkin lymphoma,133 as well as being of benefit in colon cancer treatment.134
In addition to Abraxane and our in-house use of liposomes to deliver Doxorubicin, MTO, or Irinotecan to generate immunogenic effects in a TNBC landscape, additional reports were published for nano-enabled delivery of chemo agents to achieve ICD response outcomes in breast cancer tumor microenvironments. For example, an ICD response in TNBC was achieved by liposomes delivering Doxorubicin alone,135–136 Doxorubicin co-formulated with 5-fluorouridine,137 or using carriers that deliver Gemcitabine138 or cyclophosphamide.139 An alternative strategy for inducing ICD in TNBC was accomplished through the use of liposomal Manganese-Protoporphyrin-enabled sonodynamic therapy (SDT),140 liposomal Porphyrin-enabled photodynamic therapy (PDT),141 or liposomal HSP90 inhibitor e.g., 17-(allylamino)-17-demethoxygeldanamycin (17-AAG).142 The explanation for the use of PDT for the stimulation of Type II ICD responses is explained in Figure 9.
D. Co-formulated drug delivery to propagate ICD stimuli in heterogeneous immune landscapes
D.1. Overall strategies
In addition to relying on free drug combinations to improve immunotherapy outcomes, it is important to consider the utility of our platform of LB carriers, which have been developed to combine the delivery of amphipathic and hydrophobic drugs (Figure 13–16, Tables 4–6). These include natural or synthetic lipid moieties as well as pharmacological agents that can be rendered lipophilic by conjugation to lipid carriers (Figure 13). Other than the advantage of improved PK and drug safety, further payoffs of co-formulated drug delivery include achievement of harmonized P’s as well as cooperative interactions at the same deposition site. The basic design principles for drug co-formulation in liposomes and silicasomes is premised on the use of the aqueous interior for remote import of amphipathic, weak-basic drugs while incorporating the lipophilic drugs into the lipid bilayer (Figure 13). Remote loading of ionizable chemo-drugs (e.g., Doxorubicin, MTO, and Irinotecan) is accomplished by encapsulating protonating agents such as ammonium sulfate, citric acid or sucrose octasulfate in the interior of the LB carriers.143–144 In addition to generating a proton gradient for drug import, the anionic group in the “trapping” agent is used for drug precipitation and the formation of crystals that slowly dissolve. In addition to its role in remote loading, the lipid bilayer is used for incorporating lipophilic or hydrophobic API, including prodrugs and lipid moieties with immune modulatory function. The latter comprise TLR agonists, α-galactosylceramide analogues (promote NK T-cell activation),145 and pro-resolving lipid mediators (impact chronic inflammation, innate immunity and collagen deposition),146 as described in Table 4.138, 147–150
Figure 13. Design of dual-drug nanocarriers to deliver ICD stimuli plus immune modulators interfering in immune escape pathways or delivering adjuvant stimuli.
The schematic illustrates liposome design, making use of remote loading of ICD-inducing chemo drugs, combined with inclusion of immune modulatory lipid moieties and prodrugs into the LB. Natural and synthetic lipid compounds that can be included in the LB are elucidated in Table 3. Prodrug design, with cleavable linkers and utility to interfere in a variety of immune escape pathways, are discussed in Figures 14–17 as well as in online Figures S17–S22. The formation of the lipid bilayer can be accomplished by different techniques, including hydration and sonication of a coated lipid biofilm, microfluidic mixing of ethanol/lipid and aqueous laminar flow channels in a reaction chamber (e.g., NanoAssemblr™). We have also described the use of flow-through sonication in Figure 8A.
Figure 16. Strategies for synthesizing fatty acid-conjugated prodrugs.
Panel A: Fatty acids are versatile design elements for prodrug synthesis. For example, the lipid tails could be saturated or unsaturated, providing different self-assembly properties and membrane rigidity. On the other hand, drugs could be conjugated to the hydrophilic fatty acid head or at the end of the hydrophobic lipid tails, depending on the desired hydrophobicity of the drug/prodrug molecular after the conjugation. Useful building blocks for lipid-head drug conjugations include fatty acid chlorides, fatty acids, fatty aldehyde, and fatty alcohol. Panel B: Thiol/mercapto, amino, and hydroxyl-modified fatty acids are commercially available for synthesizing lipid-tail drug conjugates, yielding cleavable disulfide, imine, and ester bonds for hydrophobic drugs.
Table 4:
Lipid agonists that can be included in the LB of nanocarriers
| Category | Drug | Activity | Reference |
|---|---|---|---|
| TLR4 agonists | MPLA (PHAD®) | Monophosphoryl Lipid A (Synthetic), TLR4 agonist, induces proinflammatory cytokines and antigen-specific effector CD4+ and memory CD8+ T cells. Safe administration to human subjects. Excellent efficacy of Doxorubicin-MPLA liposomes against 4T1 tumors. Additional analogs include: • 3D-PHAD® (Monophosphoryl 3-Deacyl Lipid A): less pyrogenic • 3D(6A)-PHAD® [3D(6-acyl)-PHAD®]: Adjuvant used in GSK’s Adjuvant Systems AS01, AS02, and AS04, including for use in liposomes |
138,147–148 |
| TLR7/8 agonists | MEDI9197; 3M-052 (Telratolimod) A large number of agonists used in preclinical studies are reviewed by Bhagchandani et al, including agonists included in nanoparticles |
Injectable, tissue-retained agonist that forms a tissue depot with gradual, sustained release, allowing local TLR activation without systemic cytokine release; induces local innate immune activation as well as systemic, antigen-specific CD8+ T cell responses which suppress tumor growth; currently under clinical development for use in vaccines and cancer therapy. Design strategies for nanoparticles construction include self-assembly conjugate formulations, conjugation of protein antigens to copolymers or P(Man-TLR7) covalently conjugated onto synthetic polymer scaffolds. |
149–150 |
| α-Galactosyl-ceramide analogues | KRN7000 | Synthetic analog and ligand of the lipid-binding MHC class I-like protein, CD1d. Displays potent antitumor activity in various in vivo models. | 145,294–295 |
| Pro-resolving lipid mediators | LXA4 | Autacoid metabolite of arachidonic acid, interacting with lipoxygenase and acting primarily to dampen inflammation. Downregulates TGF-β1 autocrine signaling, with reversal of mesenchymal phenotypes and metastases in PDAC. | 146,296 |
Table 6.
Summary of experimentation with LB-enabled nanocarriers for Immunotherapy.
| Interior Drug Loading | Liposomes | Silicasomes | Cancer type | LB-assisted Drug Co-formulation |
|---|---|---|---|---|
| DoxorubicinA | + | + | BC | IDO-1 prodrug inhibitors - Indoximod - Linrodostat GSK3 prodrug inhibitor - TWS119 PD-1 prodrug inhibitor -BMS-8 TLR agonists - 3M-052 |
| MitoxantroneA | + | + | BC, Colon, Renal | |
| IrinotecanA | + | + | PDAC, BC, Colon | |
| OxaliplatinB | − | + | PDAC | |
| DACH-PtB | − | + | PDAC, Colon | |
| Paclitaxel | + | + | PDAC, BC | |
| GemcitabineA | + | + | PDAC, BC | |
| CXCR4 inhibitors: AMD3100A AMD3465A AMD11070A | + | + | PDAC, BC | |
| GSK3 inhibitor: AZD1080A | + | + | PDAC |
= remote loading with a proton gradient
= Other loading method, e.g., passive loading for Oxaliplatin or silica sidewall complexation for DACH-Pt
The synthesis of lipid-conjugated prodrugs offers a robust approach to endow ICD-inducing carriers with cleavable release of drugs that propagate immunogenic effects by interfering in immune escape or immune suppressive pathways. These include prodrugs targeting the IDO-1, PD-1/PD-L1 axis and other checkpoint pathways (Figure 13). Not only does the lipid conjugation improve the PK of drug delivery, but protects the conjugated drug from hydrolysis and enzymatic cleavage.128, 151–152 Moreover, the inclusion of an enzyme-cleavable linker (e.g., for esterase) provides a means of drug release in the TME (Figure 13). Prodrug synthesis is accomplished by using medicinal chemistry criteria to locate drug groups that can be covalently linked to the chosen lipid species, while ensuring bilayer compatibility and stability. In addition, the aim of prodrug design is to accomplish amphiphilicity (proper polarity arrangements) with cleavable drug-linkage chemistry (Figure S17A). Attention is also required for the 3D geometry of the amphiphilic prodrugs and the size of ingredient hydrophobic vs. hydrophilic domains. This is explained by the synthesis of a prodrug, where IDO-1 inhibitor, Indoximod, was conjugated to cholesterol (Figure S17A). The synthesis is illustrative of the use of the critical packing parameter, Cpp, according to the formula Cpp = V/(A0 x LC), where V is the hydrophobic tail volume, A0 is the optimal surface area of the hydrophilic headgroups, and LC is the critical chain length of the hydrophobic tail (Figure S17B). The calculated theoretical Cpp predicts the overall geometry of the self-assembled aggregates, including spherical micelles (Cpp < 0.35), flexible bilayers/vesicles (Cpp = 0.6~0.85), flexible bilayers (Cpp = 0.85~0.95), planar bilayers (Cpp = 0.95~1.15), for inverted micelles (Cpp > 1.15)153–154 For optimal liposomal bilayer compatibility, Cpp should be in the range of 0.5~1.0. Importantly, the protonation status of the drugs at physiological pH should also be considered if it significantly changes the hydrophobicity/polarity of the drug, impacting the Cpp (Figure S17B). Depending on the physicochemical properties of the prodrug, formulations could be further designed to ensure optimal lipid bilayer compatibility by fine-tuning surface zeta potential (Figure S17C). Collectively this determines the pharmacokinetic, pharmacodynamic, drug release and stability that ultimately governs therapeutic efficacy (Figure S17D).
A variety of lipid structures can be used for prodrug synthesis, including phospholipids, cholesterol, fatty acids, and glycerides (Figure 14). For ester-conjugated prodrugs, Drug-OH could be conjugated to lipid-COOH, e.g., fatty acids, cholesteryl hemisuccinate (CHEMS) or 1,2-dioleoyl-sn-glycero-3-succinate (DGS) derivatives, e.g., dipalmitoyl-sn-glycerol-3-succinate (16:0 DGS), or di-oleoyl-sn-glycerol-3-succinate (18:1 DGS). This is achieved through standard esterification reactions such as the Steglich esterification, which pre-activates the lipid-COOH using carbodiimide-derivatives with 4-dimethylaminopridine (DMAP) as the catalyst.155 The alternative is Fisher esterification. Similarly, lipid-acid chlorides could be used to synthesize ester-conjugated prodrugs. Alternatively, ester bonds could be formed between Drug-OH and Lipid-OH via converting an OH into an alkyl tosylate (a good leaving group), to be substituted with lipids/drug containing a nucleophile, e.g., hydroxide ion (-O− salt+) to form an ester. Other strategies for synthesizing acid/enzyme cleavable lipid-drug conjugates via ester, amide, ether, disulfide, imine, carbamate conjugations, etc., are summarized in Figure 14.
Figure 14. General strategies to synthesize lipid-drug conjugates.
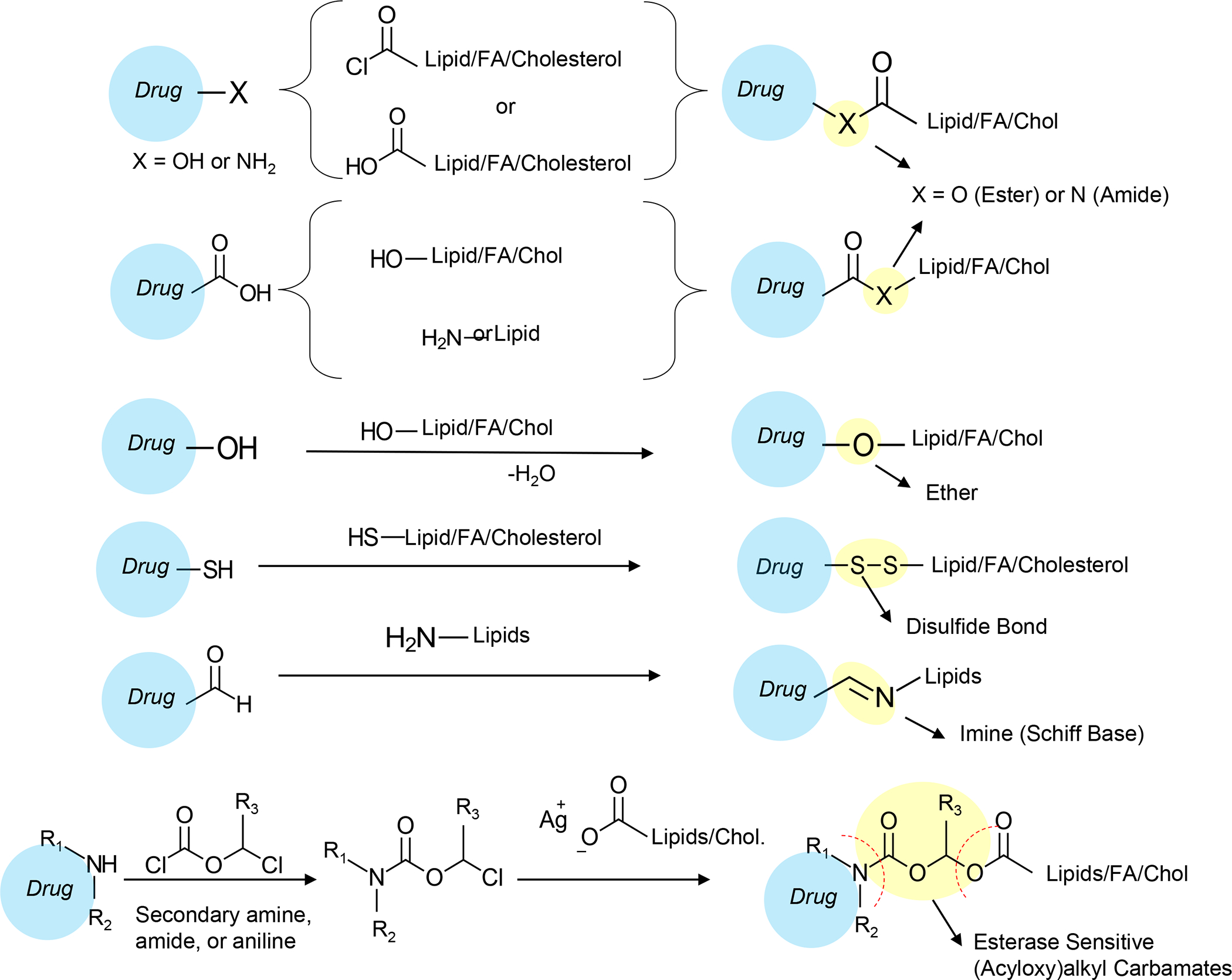
Cholesterol derivatives (detailed in Fig. 15), phospholipids, and fatty acids (detailed in Figure 16), collectively provide a wide selection of chemical building blocks for synthesizing designer prodrug conjugates, most preferably through the formation of ester, amide, ether, disulfide, imine, and carbamate conjugations. These linkers are acid/redox-sensitive or subject to enzymatic (e.g., esterase or protease) cleavage for drug release at target sites. The various drug lipidation options open new gateways to nano-enabled drug/gene delivery through lipid-bilayer drug anchoring/embedding, improved pharmacokinetics for tumor delivery, reduced toxicity due to systematic exposure, and additional drug combinations through remote drug loading.
In the design of cholesterol-conjugated (cholesteryl) prodrugs, useful building blocks such as cholesterol/cholesteryl chloride, cholesteryl mercaptan, cholesteryl chloroformate, and cholesteryl hemisuccinate (CHEMS) can be used to conjugate to both hydrophilic and hydrophobic drugs, using the chemistries described earlier (Figure 15). In designing fatty acid-linked drug conjugates, drugs could be conjugated to the hydrophilic fatty acid head or at the end of the hydrophobic lipid tails, depending on the hydrophobicity of the drug molecule. Useful building blocks for lipid-head drug conjugations include fatty acid chlorides, fatty acids, fatty aldehydes, and fatty alcohols (Figure 16A). Thiol/mercapto, amino, and hydroxyl-modified fatty acids are also commercially available for synthesizing lipid-tail drug conjugates, yielding cleavable disulfide, imine, and ester bonds for hydrophobic drugs (Figure 16B).
Figure 15. Strategies for synthesizing cholesteryl-conjugated prodrugs.
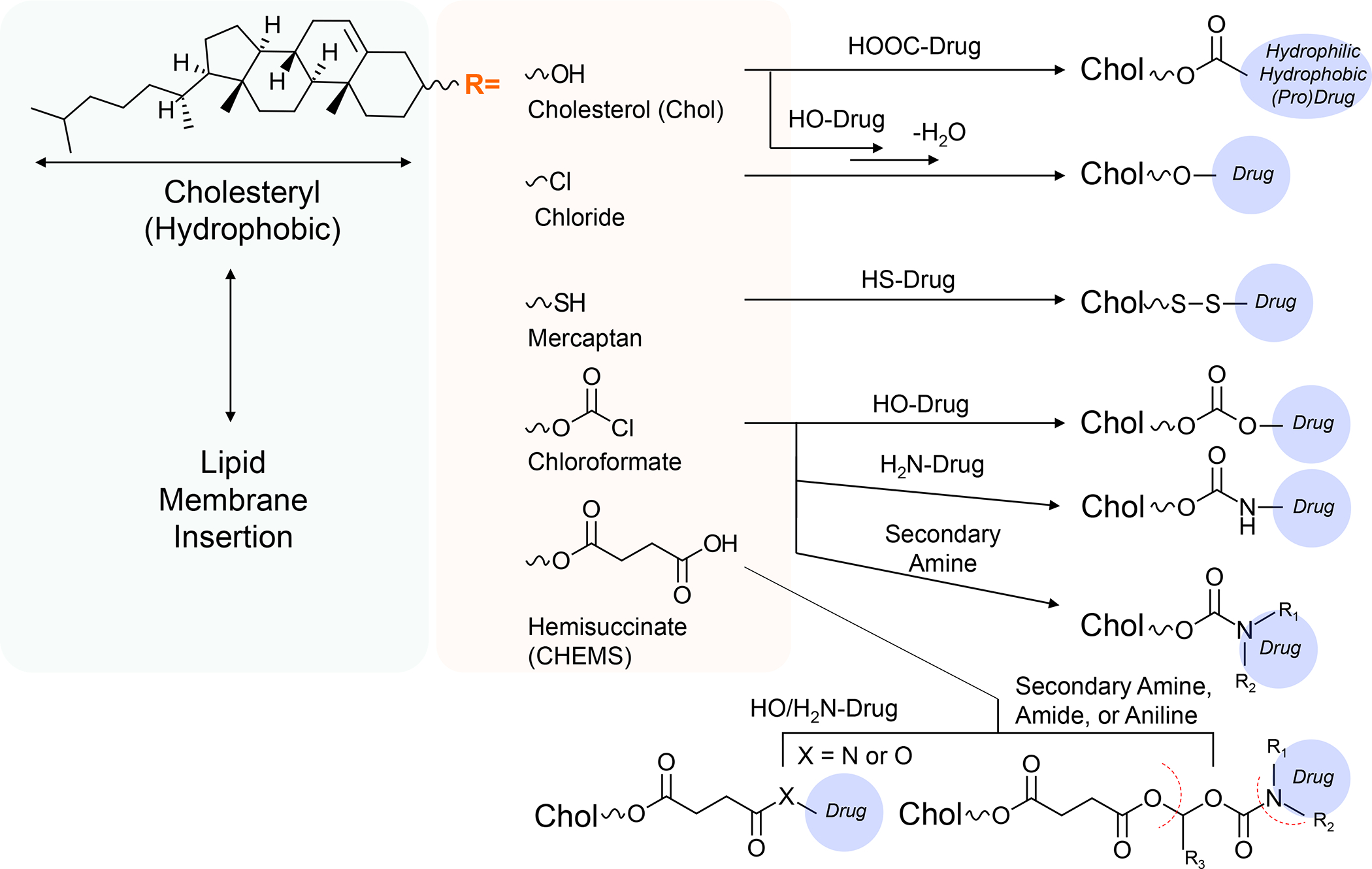
Cholesterol-conjugated (cholesteryl) prodrugs provide a different type of anchor for LB incorporation. Compared to lipids, cholesterol increases lipid bilayer rigidity and eliminates bilayer phase transition in a concentration-dependent way, thereby increasing liposomal stability and slowing drug release (if desired). Useful cholesteryl building blocks such as cholesterol/cholesteryl chloride, cholesteryl mercaptan, cholesteryl chloroformate, and cholesteryl hemisuccinate (CHEMS) are commercially available for conjugation to hydrophilic and hydrophobic drugs by reactions detailed in Figure 14.
Compared to fatty acids, phospholipid-prodrug conjugates provide a significantly higher level of modular designs and complexity to obtain lipid-conjugated prodrugs. As shown in Figure S18, the general structure of phospholipids often includes a glycerol backbone. The three hydroxyls of the glycerol backbone are linked to: (i) single hydrophobic lipid tails (a.k.a. lysolipid) or two hydrophobic lipid tails via ester linkages, and (ii) a hydrophilic head group through a phosphodiester bond (hence named as phospholipids, or iii) directly conjugated to succinate as glycerol-3-succinate (GS). The most common phosphodiester conjugated head groups include phosphatidylcholine (PC), phosphor-(1’-rac-glycerol) (PG), phosphoethanolamine (PE), phosphor-L-serine (PS). A variety of head groups provide avenues for conjugation to hydrophilic drugs. An important consideration is the hydrophilicity of the drug after linkage as the conjugation sites also double up as drug solvation sites. Additional design strategies, making use of the glycerol backbone, as well as controlling the rigidity of the lipid membrane are discussed online in Figures S19–S20.
D.2. Use of IDO-1 prodrugs to augment ICD-initiated immune responses
The first example of a prodrug strategy to augment immunity by ICD-inducing liposomes makes use of an inhibitor of the indoleamine 2,3-dioxygenase (IDO-1) pathway. IDO-1 is a heme-containing enzyme, which acts as the first and rate-limiting enzyme in the kynurenine (KYN) pathway, where it is responsible for tryptophan (TRP) catabolism, leading to kynurenine (KYN) excess (Figure 17, left panel).156–157 Through its immune metabolic activity, IDO-1 acts as an immune checkpoint molecule that leads to robust immune suppression through actions such as: (i) negative regulation of the mTOR/P-S6 kinase pathway (T-cell activation); (ii) stimulation of the aryl hydrocarbon receptor (AhR) pathway, which promotes further IDO-1 expression as well as Treg generation; and (iii) activation of a serine/threonine-protein kinase, GCN2 (general control nonderepressible 2) kinase, which increases IL-6 production and development of Tregs and MDSC. Moreover, IL-6 also engages in an auto-feedback loop that augments IDO-1 overexpression. IDO-1 overexpression by dendritic cells, macrophages, and cancer cells plays a role in loco-regional interference in T-cell activation in a variety human cancers such as TNBC, pancreas, lung, prostate, colorectal, cervical, gastric, ovarial and head and neck cancers. IDO-1 is also expressed at the site of ICD induction by newly recruited CD8+ CTLs that produce the cytokine, IFN-γ. IFN-γ, in turn, induces transcriptional activation of the PD-L1 and IDO-1 promotors, generating a counterregulatory response that allows cooperation between these checkpoint pathways (Figure 17, right panel).128, 158–160
Figure 17. The role of the indoleamine 2,3-dioxygenase (IDO-1) metabolic pathway in immune interference, including in the counter-regulatory IFN-γ response pathway.
Panel A: Schematic to explain the role of IDO-1 in immune metabolic TME regulation by converting tryptophan to kynurenine. The kynurenine excess and tryptophan depletion interfere in the mTOR pathway and P−S6 kinase activity but enhance the activation of a kinase, “general control nonderepressible 2” (GNC2), as well as the transcriptional activity of the aryl hydrocarbon receptor (AhR). The overall impact is decreased CTL activity, T-cell anergy and increased Treg production. In addition, the activation of increased IL-6 production by AhR is responsible for enhanced IDO-1 production. Panel B: Mechanistic explanation of the counter-regulatory effect of ICD-induced IFN-γ release, which leads to upregulated IDO-1 and PD-L1 expression and Treg generation. This also explains cooperation between the metabolic and receptor-mediated checkpoint pathways towards immune escape in immune landscapes with an IFN-γ genomic footprint. Panel A-B adapted from ref 128. Copyright 2020 American Chemical Society.
In the previously covered section dealing with Figure 12 and S10–S12, we have demonstrated that Doxorubicin- and MTO-delivering liposomes trigger ICD responses in different TNBC landscapes. To investigate the impact of the non-competitive IDO-1 inhibitor, 1-methyl-D tryptophan (a.k.a. Indoximod) in the same tumor environment, we developed an Indoximod (IND) prodrug for incorporation into a dual-delivery liposome, L-MTO/IND. Guided by the medicinal chemistry criteria discussed in Figure S17, amine and carboxyl groups on IND were identified as potential conjugation sites for cholesterol linkage (Figure 18A).128 Conjugation to the -COOH group was accomplished by initially protecting the drug amine by tert-butoxycarbonyl (tert-Boc), allowing Steglich esterification to the -COOH group to cholesterol. The acid-sensitive tert-Boc group was subsequently removed by trifluoroacetic acid (TFA). This generated a Chol-INDNH3+/TFA− salt from which TFA was removed by an ion-exchange resin to yield Chol-IND-NH2. High-resolution mass spectroscopy (MS) and 1H-NMR analysis confirmed the prodrug structure (Figure 18A). Subsequent construction of the L-MTO/IND was accomplished by lipid film hydration, which also took into consideration the prodrug’s ability to generate a cationic charge.128 The charge was neutralized by including cholesteryl hemisuccinate (CHEMS), which contains a carboxyl group (pKa ≈ 5.8) (Figures 15 and 18B). We have already covered the synthesis of the single-drug L-MTO carrier in the TNBC experimentation, as described in Figure 12.128 A similar study was performed on the CT26 colon cancer model to compare L-MTO/IND with the MTO-only liposomes (Figures S13–S15). Noteworthy, both liposomes induced ICD markers (CRT and HMGB-1) in TNBC and the colon TME, where immune activation led to the deposition of perforin and granzyme B (Figures S10–S15). As noted in Figure 12, MTO induced a NK instead of a CD8+ T-cell response (Figures 18C and S15). Noteworthy, the reduction in tumor size in EMT6 and 4T1 was significantly enhanced by IND-Chol co-delivery (Figure 18C). The same outcome was generated in CT26, where perforin release by NKp46+ cells (instead of CTLs) were responsible for improved survival, as demonstrated by Kaplan-Meier analysis (Figures S13–S15).
Figure 18. Co-delivery of the IDO-inhibiting cholesteryl-Indoximod prodrug synergizes with mitoxantrone in augmenting chemo-immunotherapy in animal TNBC tumor models.
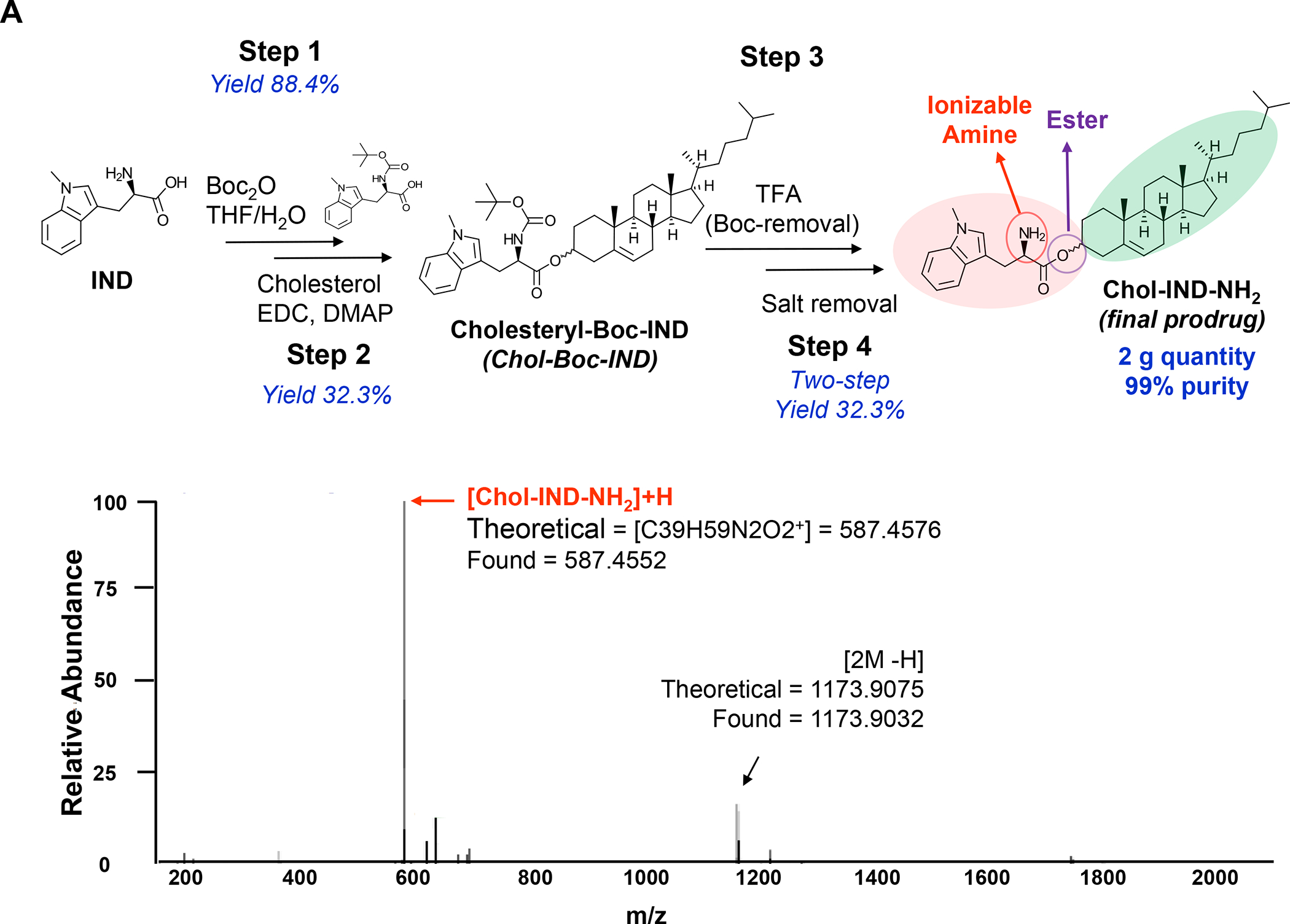
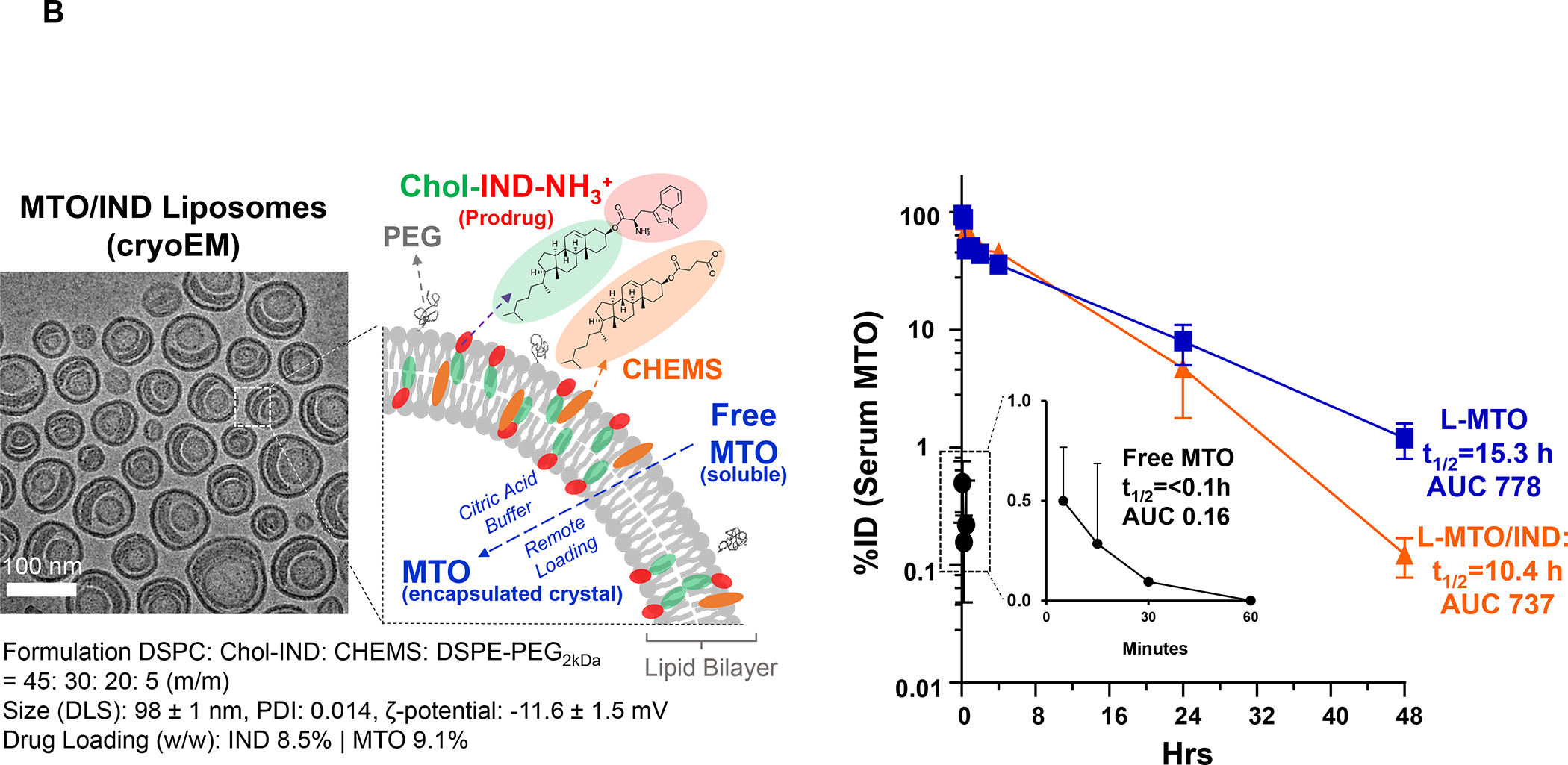
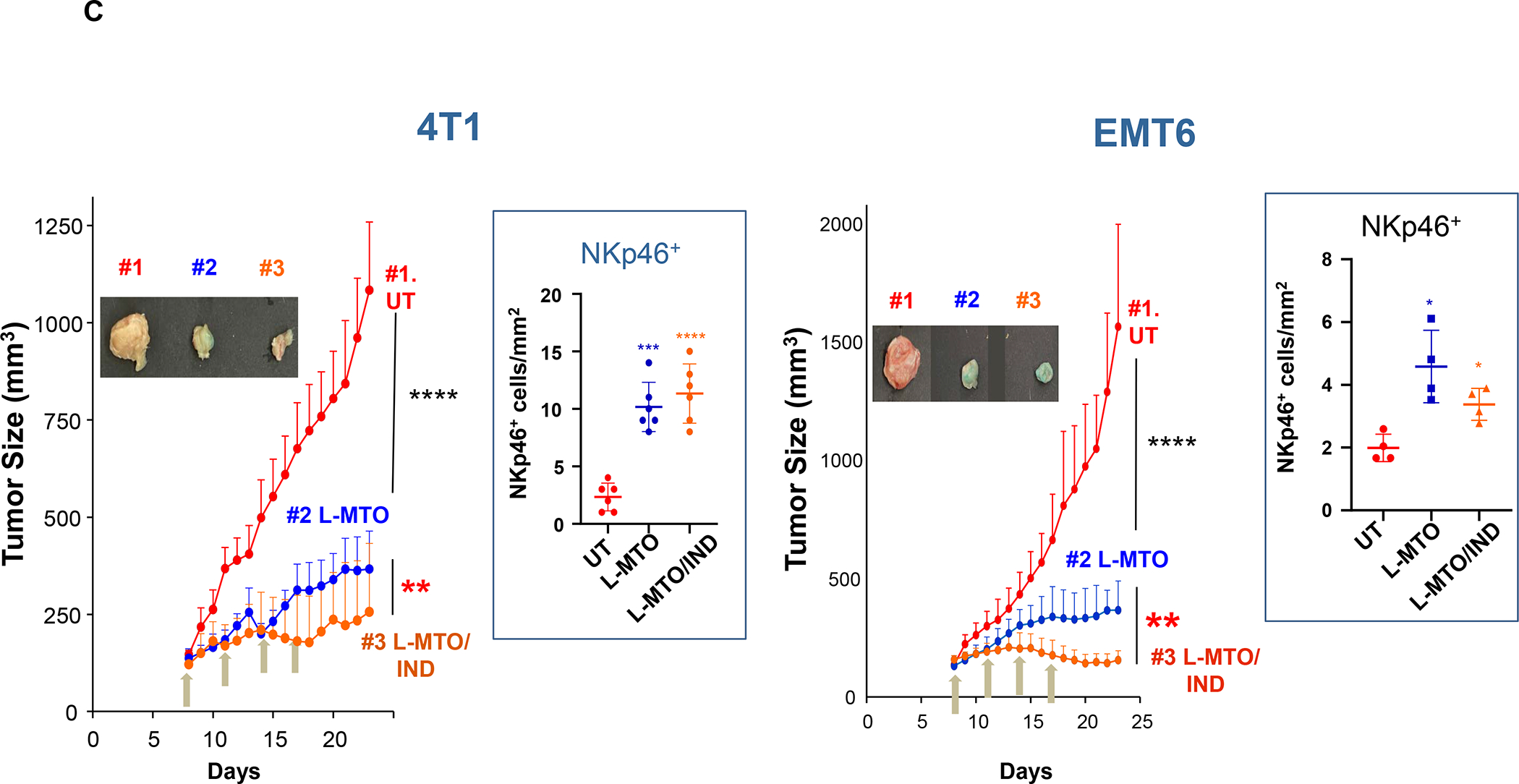
The dual-delivery L-MTO/IND data presented here supplements the data discussion in Figure 12 to show how the liposome was constructed, in addition to discussing the impact on another TNBC model. Panel A: A cholesteryl-ester-conjugated prodrug using the non-competitive IDO-1 inhibitor, 1-methyl-D tryptophan (a.k.a. Indoximod/IND) was used for prodrug development, involving 4 steps: (i) Boc-protection of IND, yielding Boc-IND; (ii) conjugation of Boc-IND to cholesterol by Steglich esterification; (iii) removal of the Boc group; (iv) desalting to yield CholIND-NH2. This streamlined approach is scalable and capable of providing highly purified gramscale quantities. Prodrug structure was confirmed by mass spectrometry and NMR.128 Panel B: The prodrug was incorporated into a liposomal carrier, making use of cholesteryl hemisuccinate (CHEMS) to neutralize the cationic charge of the ionized Chol-IND-NH3+ prodrug at physiological pH. This yielded L-MTO/IND liposomes with the physicochemical characteristics and cryoEM imaging features depicted in the lower panel. Both the L-MTO and L-MTO/IND liposomes showed significant improvement in PK and circulatory half-life, compared to free MTO (right panel). Panel C: L-MTO/IND showed significant improvement in tumor growth in both the 4T1 and EMT6 TNBC models, compared to L-MTO. Moreover, the ICD effect was mediated by NK cells instead of CTLs. Highly efficient liposomal drug delivery at both tumor sites was reflected by the blue coloration of the tumor tissue due to MTO. Panel A-C adapted from ref 128. Copyright 2020 American Chemical Society.
In addition to our in-house studies, Lan et al. developed a dual-functional immunostimulatory polymeric prodrug carrier (PEG2k-Fmoc-1-MT) for simultaneously delivering 1-methyl tryptophan plus Doxorubicin to trigger a BC chemo-immunotherapy response.161 Not only were the prodrug micelles more effective for apoptosis and growth inhibition in 4T1 cells, but also enhanced kynurenine production in effector CD4+ and CD8+ T cells. This was accompanied by an increased drug delivery and accumulation of DOX and 1-MT at the tumor site, in addition to decreased tryptophan/kynurenine ratios in the blood and tumor tissue. There was also evidence for generation of an ICD response, characterized by increased recruitment of effector CD4+ and CD8+ T-cells, increased IFN-γ release and reduced Treg expression. The overall impact was enhanced tumor cell killing and an improved anti-cancer immune response. Wan et al. also described a dual-functional immuno-stimulatory polymeric prodrug carrier, capable of indoximod (IND) and Doxorubicin co-delivery.162 The carrier was comprised of hydrophilic blocks of poly (oligo (ethylene glycol) methacrylate) (POEG) that contained Doxorubicin, which interacted with hydrophobic blocks conjugated to Indoximod. This allowed a synergistic immune response between the ICD-induced infiltration of IFN-γ producing CD8+ T-cells, with the immune metabolic effect of Indoximod, including its ability to reduce the number of Tregs. The Dox/POEG-b-PVBIND micelles led to significantly improved tumor regression in an orthotopic murine breast cancer model compared to single drug carriers, confirming the efficacy of dual drug delivery.
While Indoximod was historically the first reported drug to interfere in the IDO-1 pathway (Ki = 34 μM), it does not function as a direct enzyme inhibitor, serving instead as a tryptophan mimetic that regulates amino acid transport, with modulation of downstream signaling effects.163 While effective in combination with chemotherapy and checkpoint blocking antibodies, D-1-methyl tryptophan is poorly soluble with unfavorable PK, prompting the synthesis of additional prodrugs (including NLG802) to increase drug exposure at the cancer site.164 Thus, additional effort has been placed into developing new and improved competitive inhibitors, based on the structure-activity relationships at work in the heme iron-binding pocket of the enzyme. 165 This culminated in the synthesis of Epicadostat, Linrodostat mesylate (BMS-986205) and Navoximod (NLG-919) (Figure S21 and S22). While the competitive inhibitor, Epacadostat (IC50 = 10 nM) showed synergy with checkpoint blocking antibodies in clinical trials, drug failure in a phase III study (in spite of a promising phase II study) became a turning point for IDO-1 inhibitor development, with several pharmaceutical companies abandoning clinical trials. However, dosimetry differences between the phase II and III studies suggest that variances in drug distribution could be responsible for the divergent outcomes, with the implication that small molecule IDO-1 inhibitors should not be written off. Linrodostat mesylate (BMS-986205), an orally available IDO1 inhibitor with IC50 = 1.1 nM, is a direct IDO-1 inhibitor. Over 20 clinical studies have been performed with BMS-986205 or are ongoing,165 including data that the combination this drug with checkpoint blocking antibodies or chemotherapy is effective and safe.166
In our planning to develop a BMS-986205 prodrug through a commercial supplier, medicinal chemistry criteria was used to assess available drug groups for conjugation to lipid moieties.167 This identified the amide group that could be conjugated to carboxy-terminal lipid building blocks, including saturated and unsaturated fatty acids, DGS-derivatives and CHEMS (Figure 19). To date, BMS-986205 has been successfully conjugated to stearic acid for incorporation into a nanocarrier lipid bilayer. In addition to BMS-986205, Navoximod (NLG-919) is a potent (EC50 = 75 nM) non-competitive IDO-1 inhibitor that can be considered for prodrug design, as shown in Figure S22. This includes the use of a hydroxyl group on NLG919 for synthesis of ether or ester-conjugated prodrugs linked to hydroxyl-terminated fatty alcohol or cholesterol. Another possibility is to use carboxyl-terminated lipid building blocks (fatty acids, DGS-derivatives, and CHEMS).
Figure 19. Design of a series of Linrodostat prodrugs to boost nano-enabled chemoimmunotherapy.
The competitive IDO-1 inhibitor, Linrodostat (BMS-986205), does not provide access to ester, ether or amine bonding for prodrug design. However, it is possible to accomplish bio-cleavable prodrugs by establishing an (acyloxy)alkyl carbamate site.309 Linrodostat contains an amide that can be reacted with chloromethyl chloroformate to generate a chloromethyl linker, which reacts with the silver salts carboxylic acid groups associated with fatty acids, CHEMS, and DGS derivatives. This yields AgCl precipitates and the formation of (acyloxy)alkyl carbamate-conjugated prodrugs.167
In addition to our in-house data, Shen et al. developed a bifunctional liposome comprising an Oxaliplatin prodrug conjugated to a phospholipid plus alkylated NLG919.168 This allowed oxaliplatin release from the bifunctional liposome, capable of inducing ICD in CT26 cancer cells along with decreased conversion of tryptophan to immunosuppressive kynurenine by NLG919. Dual drug delivery was also accompanied by increased pharmacokinetics in vivo, in addition to superior anti-tumor immunity in subcutaneous and orthotopic CT26 tumors, characterized by increased recruitment of cytokine producing CD8+ T-cells and Treg downregulation.
D.3. Prodrugs or remote loaded drugs for interfering in the PD-1/PD-L1 axis
While the use of blocking antibodies has proven highly effective for intervening in the immune suppressive effects of the PD-1/PD-L1 axis, antibody treatment has limitations, including high production cost, long half-life, poor tumor penetration and triggering of immunoglobulin-related side effects. Small molecule inhibitors (SMI) of the PD-1/PD-L1 axis have therefore been considered as alternatives or complementary therapies, making the development of small molecule or peptide-based inhibitors an active area of investigation.169 Our interest in these compounds is their use for remote loading or as prodrugs that can be incorporated into our LB nanocarriers. PD-L1 (a.k.a. B7-H1 or CD274) is one of two ligands that interact with PD-1 in nonlymphoid organs, where these ligands are expressed in response to IFN-γ production (Figure 17B). Ligand binding to PD-1 delivers an inhibitory signal to the T-cell antigen receptor (TCR), which is responsible for triggering CTL killing (Figure 20). The search for SMI to disrupt PD-1/PD-L1 interactions was accelerated by the elucidation of the crystal structure and clarification of the mechanism of action of antibodies interfering in these receptor binding activities.170 This has sparked design of macrocyclic peptides and non-peptide organic inhibitors for performing the same function as antibodies.171–174 One example includes a series of inhibitors developed by Bristol-Myers Squibb (BMS), e.g., BMS-202, BMS-1001, and BMS-1166, which displays a scaffold structure where a substituted biphenyl group is connected to an aromatic ring by a benzyl ether bond (Figure S23). This interaction leads to PD-L1 dimerization, which interferes in receptor binding (Figure 21). Among these compounds, BMS-202 has been tested in vivo, including in 4T1 and B16-F10 subcutaneous models.175–176 Noteworthy, for the 4T1 experimentation, hydrophobic BMS-202 was encapsulated in a nanocarrier to improve poor biodistribution, leading to a significant slowing in tumor growth compared to free drug or anti-PD-L1 mAb.175 Moreover, inclusion of a photosensitizer, chlorin e6 (Ce6), in the same carrier augmented the antitumor immune responses based on the dye’s photodynamic properties, allowing the induction of an ICD response. Nanoparticles with photodynamic properties constitute another example of the use of particle physicochemical properties to induce ICD.177–178
Figure 20. Interference in PD-1 expression by small molecule GSK3 inhibitors.
Panel A: Schematic depicting the PD-1/PD-L1 signaling axis, which is responsible for suppressing CTL killing through interference in T-cell antigen receptor (TCR) signal transduction. PD-1 is expressed on “exhausted” T-cells, leading to the recruitment of SHP2 phosphatase, which interferes in recruitment of signaling components that bind to the tyrosine-based motifs in post-TCR complex. This prevents the release of cytolytic granules. Constitutionally active GSK3 is responsible for preventing the transcriptional activation of the T-bet promoter (Tbx21). Panel B: Introduction of a GSK3 inhibitor allows restoration of T-bet expression and interference in activation of the PD-1 promoter (Pdcd1) complex. The disappearance of PD-1 from the cell surface restores TCR signal transduction, allowing tumor cell killing by CTLs. In this sense, the transcriptional suppression of PD-1 provides the same outcome as anti-PD-1 blocking antibodies. Panel A-B reprinted with permission from ref 179. Copyright 2021 Elsevier.
Figure 21. Use of a reporter gene assay to illustrate BMS-8 interference in PD-1/PD-L1 interactions, as well as the approach of constructing a prodrug.
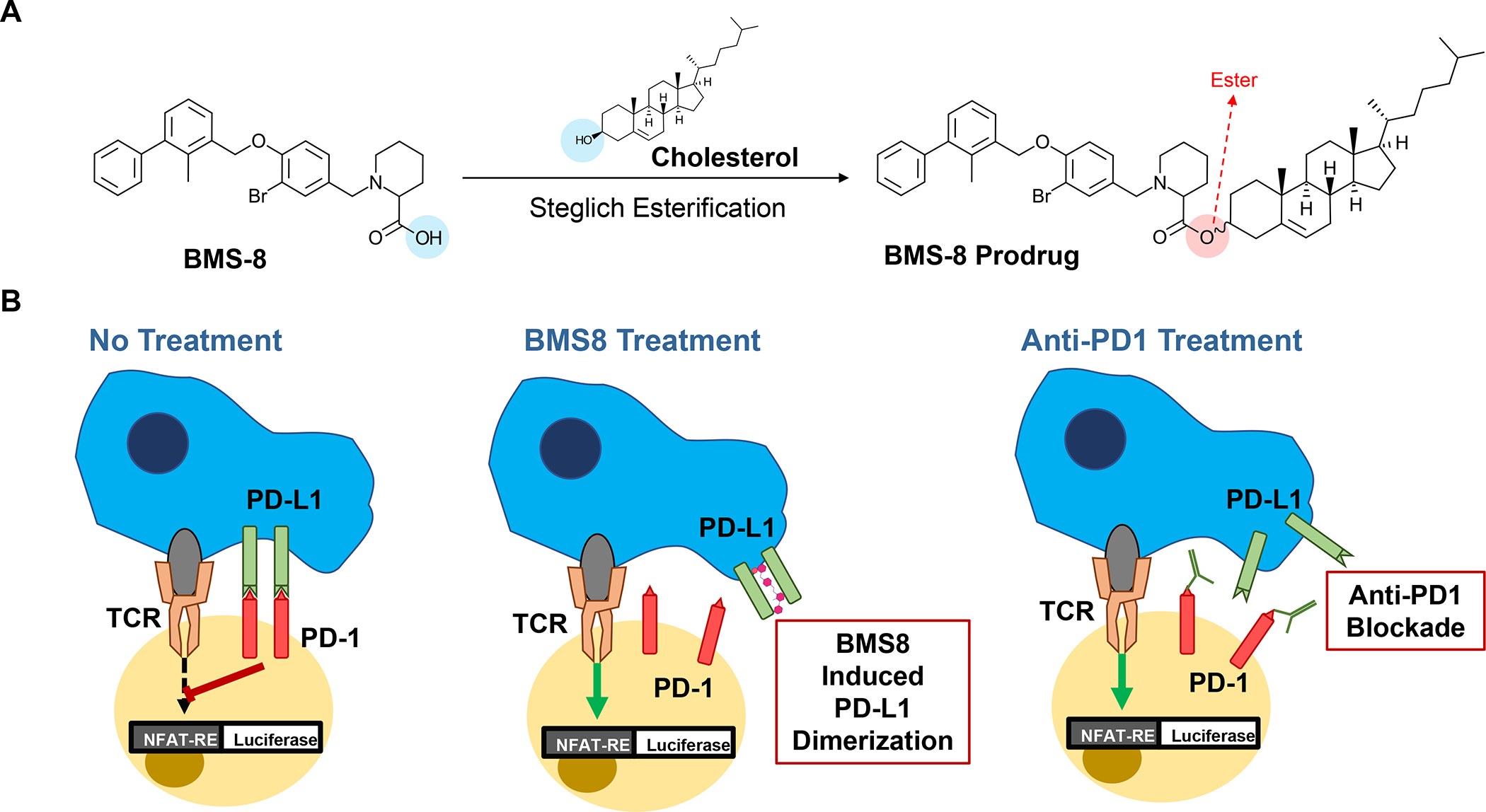
BMS-8 was chosen from among a short list of SMI (Figure S23), capable of disrupting PD-1/PD-L1 interactions, to develop a prodrug for LB incorporation. Panel A: BMS-8 exhibits a carboxy group that can be used for cholesterol conjugation, using Steglich esterification, as described in Figure S25. Panel B: BMS-8 exhibits a core/scaffold structure that induces PD-L1 dimerization, which prevents its ability to bind to PD-1. The blocking action can be demonstrated by the Promega PD-1 Cell Based Assay, which utilizes a Jurkat cell line, stably transfected with a nuclear factor of activated T-cell (NFAT) luciferase reporter plus a copy of cell surface expressed PD-1. TCR ligation in the absence of PD-1 engagement induces NFAT-luc activity. However, the TCR signal is blocked by antigen presenting (aAPC) CHO-K1 cells, stably transfected with PD-L1 (Figure 20). The addition of SMI inhibitors of the PD-1/PD-L1 axis restores TCR signal transduction, providing a quantitative readout to assess the avidity binding interference as by BMS inhibitors or antibodies, as shown in Figure S24.
For our studies, BMS-8 was selected for conjugation to cholesterol, an option that was not available for other compounds in this category (Figure 21). An in vitro reporter gene assay confirmed that BMS-8 interferes the expression of a luciferase reporter gene, stably transfected into the Jurkat T-cell line (Figure 21 and Figure S24). The BMS-8 alcohol group was used for Steglich esterification to the cholesterol carboxylic group (Figure 21). Full synthesis details of this prodrug appear online, including confirmation of its 1H-NMR structure (Figures S25–S26). Different from Chol-IND, the BMS8 prodrug (BMS8-PD) does not carry a cationic charge and could easily be incorporated into silicasome and liposome carriers, coated with a DSPC:Cholesterol:BMS8 Prodrug: PE-PEG2000 bilayer at molar ratios equal to 60:15: 20: 5 (Figure S27). These could also be developed as dual drug carriers by importing ICD-inducing chemo agents into the silicasome, using sucralose sulfate, or into liposomes, using ammonium sulfate (Figure S27). A subcutaneous KPC animal model was used to assess the efficacy of the silicasome carrier, as detailed in Figure 22. This demonstrated effective tumor shrinking in response to free Irinotecan, an outcome that was significantly enhanced by co-administration of anti-PD1, BMS8, BMS8-PD or treatment with the dual delivery (Irinotecan/BMS8-PD) silicasome (Figure 22). Noteworthy, however, there was no improvement in the effect of the encapsulated over a combination of the free drugs, both of which resulted in a significant increase in apoptotic cell death and tumor growth inhibition (Figure S28). Looking at the spatial distribution of CD8+ T-cells, demonstrated enhanced recruitment of CTL by anti-PD1, BMS8 and the dual delivery silicasome, above and beyond the Irinotecan contribution (Figure S29). This included evidence of an increase in CTL density in the tumor cores and margins. Although these data suggest little additional benefit by encapsulated delivery of the PD-1 blocking prodrug, experimentation in a more rigorous orthotopic or genetically engineered model may be necessary to evaluate if similar success can be achieved as described for the BMS-202 carrier.
Figure 22. Demonstration of the efficacy of free and encapsulated BMS-8 in a subcutaneous KPC model.
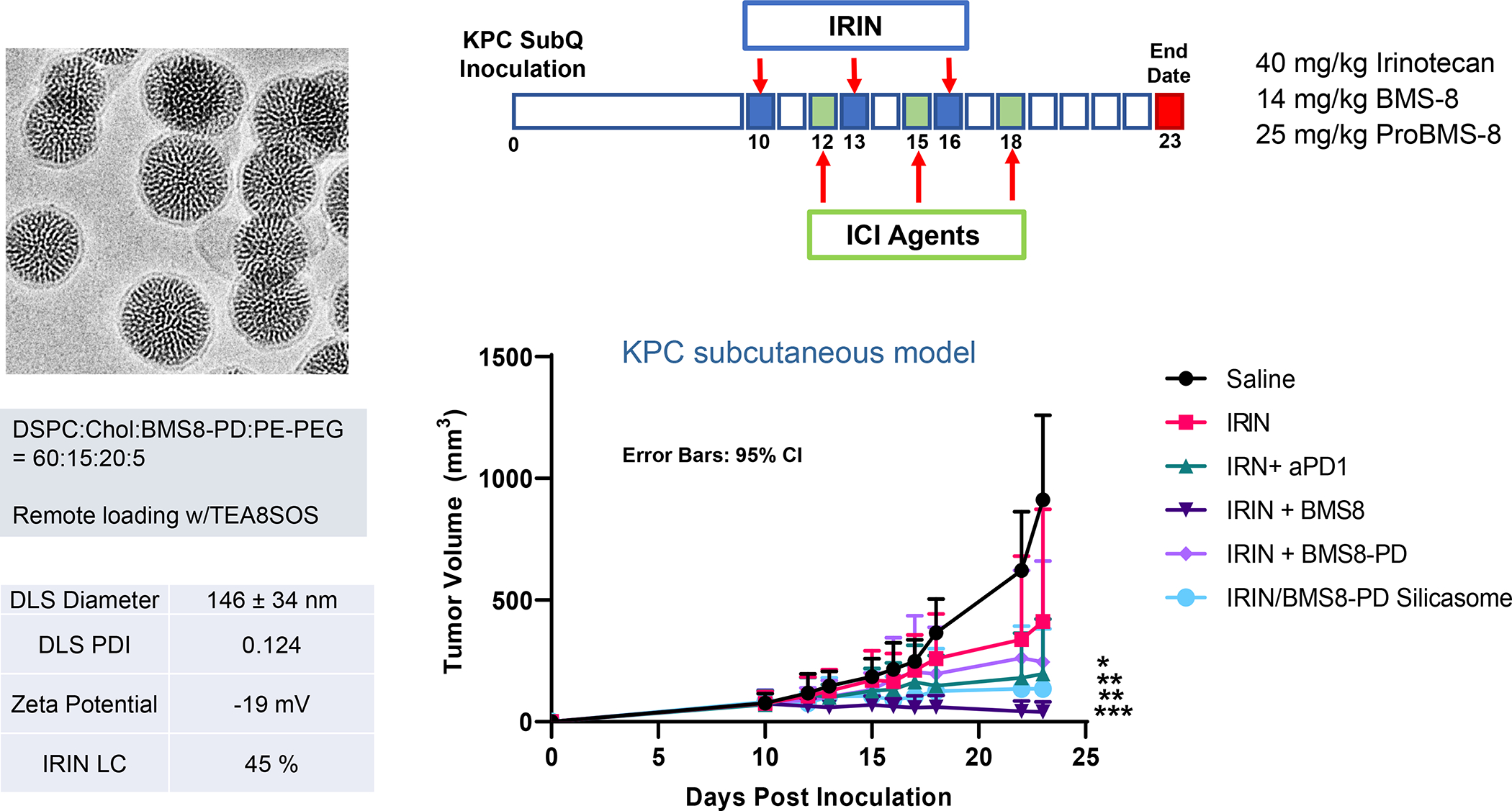
Mice received subcutaneous implementation with KPC tumor cells on days zero, followed by tracking tumor volumes for 23 days (upper panel). Animals received IV Irinotecan (IRIN) injection at a dose of 40 mg/Kg on days 10, 13 and 16, followed by the administration of BMS-8, BMS-8 prodrug (PD), anti-PD-1 antibody, or a dual-delivery IRIN/BMS-8PD silicasome on days 12, 15 and 18 (doses appear in the upper panel). The silicasome was synthesized by prodrug incorporation into the LB and remote loading (sucralose sulfate). Carrier physicochemical characterization is shown in the panel on the left. Animals were sacrificed on day 23 to collect tumor tissues for assessment of cytotoxic killing (Figure S28) and CTL recruitment (Figure S28). Tumor size plotting (lower panel) shows effective tumor shrinking in response to free IRIN, an outcome that was significantly enhanced by anti-PD1, BMS8, and BMS8-PD, as well as the dual-delivery (Irinotecan/BMS8-PD) silicasome. There was no improvement in tumor reduction of encapsulated over free BMS-8 in spite of a statistically significant increase (p = 0.047) in the BMS-8 tumor drug content by encapsulated (1.24 μg/g) vs. free drug (1.10 μg/g) delivery.
D.4. Remote loading of GSK3 inhibitors and GSK3 prodrugs to interfere in the PD-1/PD-L1 axis
In addition to employing SMI as avidity blockers to disrupt the PD-1/PD-L1 interaction, it is possible to overcome PD-1 inhibitory effects at TCR level by preventing the surface expression of PD-1 on CTLs (Figure 20).179–180 This is accomplishable by small-molecule inhibition of the signaling hub protein kinase, glycogen synthase kinase-3 (GSK3), which suppresses expression of the T-cell transcription factor, T-bet, involved in PD-1 expression.180 Thus, while constitutionally active GSK3 prevents transcriptional activation of the T-bet promoter (Tbx21), introduction of a GSK3 inhibitor (e.g., the SMI, SB415286) allows T-bet expression, which interferes in the activation of the PD-1 promoter (Pdcd1) (Figure 20). This is followed by the disappearance of PD-1 from the T-cell surface, leading to the restoration of TCR signaling, cellular withdrawal from an exhausted immune state and enhanced cytotoxic killing.180 These findings suggest that appropriately selected GSK-3 inhibitors could be implemented in place of receptor blocking antibodies for cancer immunotherapy, as demonstrated for SB415286 in melanoma and lymphoblastic lymphoma murine models.180 Moreover, Ugolkov et al., demonstrated, overcoming chemoresistance in human BC therapy with the GSK-3 inhibitors, 9-ING-41 and 9-ING-87.181 However, while the effect of 9-ING-41 was augmented by Irinotecan, it was not resolved whether this is due to an immunogenic effect since GSK3 inhibitors (GSK3i) also interfere in tumor cell growth. An important consideration in the use of these inhibitors is avoidance of off-target toxicity due to the pleiotropic action of GSK3.179, 182
We used medicinal chemistry criteria to identify GSK3i candidates for remote import across the carrier LB, in addition to identifying chemical groups for prodrug synthesis. In the first example, the selected criteria included use of molecular weight, partition coefficient, isoelectric point and solubility coefficient to predict the remote loading potential of weak-basic GSK3i compounds (Figure S30).179, 183 We identified 4 compounds out of 17 commercially available GSK3i for successful proton gradient import, among which the best loading capacity (~9%) was achieved with the compound, AZD1080 (Figure S30). AZD1080 has proven as effective as checkpoint blocking antibodies in animal studies.184 Essentially the same design steps were used to construct representative silicasome and liposome carriers. The assessment of the AZD1080-silicasome was initially carried out in the MC38 colon cancer model, previously assessed by Charles River Laboratories as a highly sensitive animal model for ICI screening.179 IV administration of silicasome-encapsulated AZD1080 significantly improved biodistribution and drug delivery to the MC38 tumor site (Figure 23B). The improved PK was accompanied by cytotoxic killing of tumor cells by CD8+ T-cells, including a subset that showed reduced PD-1 expression in the TME (Figure 23D). Further experimentation in syngeneic KPC, CT26 (colorectal) and lung cancer (LLC) models confirmed silicasome efficacy, demonstrating that encapsulated AZT1080 is similar to or more efficacious than anti-PD-1 (Figure S31)179
Figure 23. Effective delivery of a GSK3 inhibitor and tumor growth inhibition, using a remote loaded silicasome carrier179.
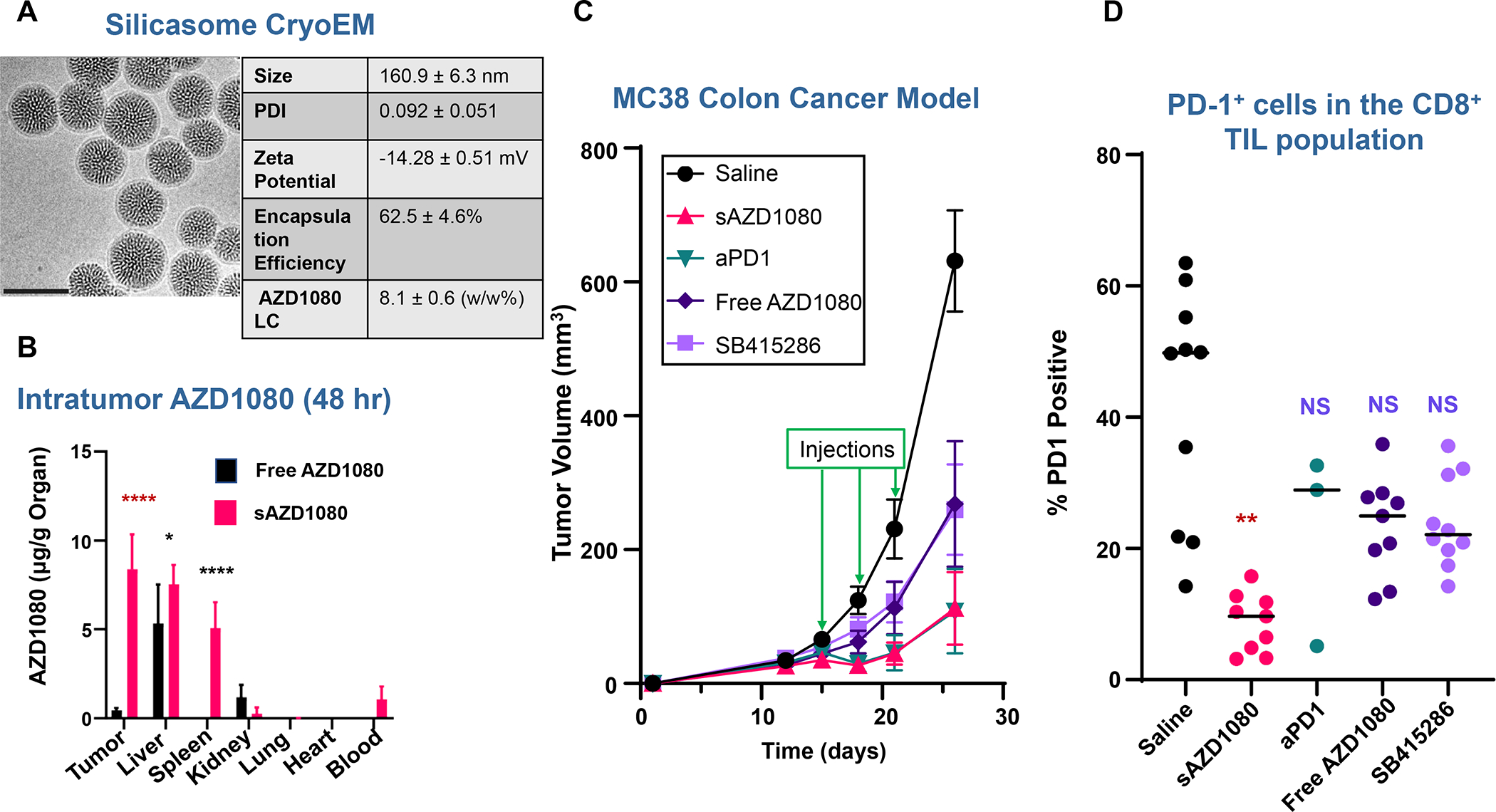
Figure S29 describes the use of medicinal chemistry criteria to accomplish the selection and remote loading of the GSK3 inhibitor, AZD1080, in a silicasome, with the characteristics shown in panel A. After growing subcutaneous MC38 tumors to ~300 mm3, mice were injected IV with 5 mg/kg encapsulated or free AZD1080. Animals were sacrificed after 24 and 48 hours, and blood, tumors, livers, spleens, kidneys, lungs, and hearts were harvested for HPLC quantification of tissue AZD1080 concentration (panel B), as described by Allen et al.179 Significance was assessed by 1-way ANOVA, * p < 0.05, **** p < 0.0001, n = 4. Panel C: Following the establishment of subcutaneous MC38 growth, treatment with free AZD1080, sAZD1080, anti-PD-1 antibody, SB415286, or saline commenced 10 days after inoculation (n = 9 animals/group). The silicasome was injected IV to deliver AZD1080 at a dose of 5 mg/kg. The controls were animals receiving IP doses of 5, 8 and 4 mg/kg, respectively, of free AZD1080, SB415286 (another GSK3i) and anti-PD-1. Treatment was repeated every 3 days for a total of 3 administrations. Mice were sacrificed 4 days after the final treatment. Panel D: Flow cytometry analysis to determine PD-1 staining intensity on tumor-infiltrating CD8+ T-cells, demonstrating a significant decrease in staining intensity in tumor tissue from animals treated with the AZD1080-silicasome vs. other treatments. The data confirmed interference in PD-1 expression by the encapsulated drug (** p < 0.01, NS = not significant). Comparable outcomes were achieved in subcutaneous pancreatic cancer (KPC), CD26 (colon cancer) and lung cancer (LLC) tumors, shown in Figure S30. Panel A-D adapted with permission from ref 179. Copyright 2021 Elsevier.
Not only do the above results confirm the promise of an encapsulated GSK3i to augment cancer immunotherapy, but also open the possibility of combination chemo-immunotherapy. In addition to using remote drug loading, commercial GSK3i compounds can be designed as lipid-conjugated prodrugs. This is illustrated by the conceptualized design of a TWS119 prodrug, conjugated to cholesterol or a fatty acid by an ester bond (Figure S32). TWS119 is a 4,6-disubstituted pyrrolopyrimidine, which impacts proliferation, survival and cytolytic activity of γδ T-cells following GSK3 inhibition; this leads to downstream activation of the canonical wnt/β-catenin and the mammalian target of rapamycin (mTOR) pathways.185 The cytolytic effects have been associated with shrinking of HCT116 colon tumors in mice.185 Moreover, TWS119 improved the generation of T memory stem cell-like (T-SCM) cells in the mouse or human CD8+ T-cell compartment, with evidence of increased persistence, proliferation, and anti-tumor activity.186
D.5. Prodrugs to interfere in the adenosine A2A receptor immune checkpoint pathway
Blocking of receptors other than the PD-1/PD-L1 axis can be used to augment ICD responses. This includes a class of drugs that blocks adenosine at the level he adenosine A2A receptor (A2AR), also expressed on the surface of CD8+ tumor-infiltrating CTL, MDSC, and NK cells.187–188 While historically, the structure-activity relationships of A2AR binding interactions have been used in the treatment of neurological disease,189–190 there is growing interest in blocking the A2AR cascade in the immuno-oncology space.191–193 This is particularly relevant from a chemotherapy perspective, where ICD is responsible for ATP release at the solid tumor site. Additional overexpression of the ecto-nucleotidase, CD39, or the 5′-nucleotidase, CD73, at the same site could contribute to the generation of additional adenosine catabolites by ATP dephosphorylation. 187 The accompanying activation of overexpressed A2AR by these catabolites could diminish the protective effect of T-cells, NK cells and DCs, while also boosting the inhibitory effects of Tregs and MDSCs.187 This explains why tumors obtained from A2AR-antagonist-treated mice show dense infiltration of CD8+ T-cells and NK cells, while exhibiting few Tregs.
A number of A2AR antagonists have emerged for cancer immunotherapy, including SMI with xanthine or non-xanthine structures for launching immunotherapy studies in solid tumors in animals and clinical trials in humans.187–188 This includes the use of the non-xanthine antagonist, ZM241385, to suppress tumor growth and metastases in TNBC or colon cancer preclinical models.194 ZM241385 has low bioavailability during oral administration.195 Our use of medicinal chemistry criteria to identify A2AR antagonists that could be delivered as lipid-conjugated prodrugs, demonstrated a phenolic hydroxyl group on ZM241385 for conjugation to several lipid moieties (Figure 24)196 This includes the availability of ester or ether linkages to terminal hydroxyls on fatty alcohol or cholesterol, as well as conjugation to the carboxyl-terminal groups on fatty acids, DGS-derivatives, and CHEMS. Not only does bilayer incorporation of these prodrugs offer to improve the unfavorable drug PK seen in oral administration,195 but also allows development of co-formulated carriers. There is indeed strong rationale to consider combining A2AR antagonists with chemotherapy, radiation therapy or PD-1/PDL-1 and CTLA-4 checkpoint blockade.187 This is underscored by the synergy between A2R antagonists and anti-PD-1, including the observation that PD-1 blockade enhances A2AR expression in tumor-infiltrating CTLs or that A2AR engagement of CTL responses promotes PD-1 expression.197 Moreover, chemo and radiation therapy elevate the expression of CD39 and CD73, explaining why concurrent treatment with an anti-CD73 mAb or pharmacologic blockade of CD39 activity enhance tumor control and survival in mice treated with ICD inducing chemo.134, 198
Figure 24. Prodrug design of the A2AR Inhibitor, ZM241385, four nanocarrier delivery.
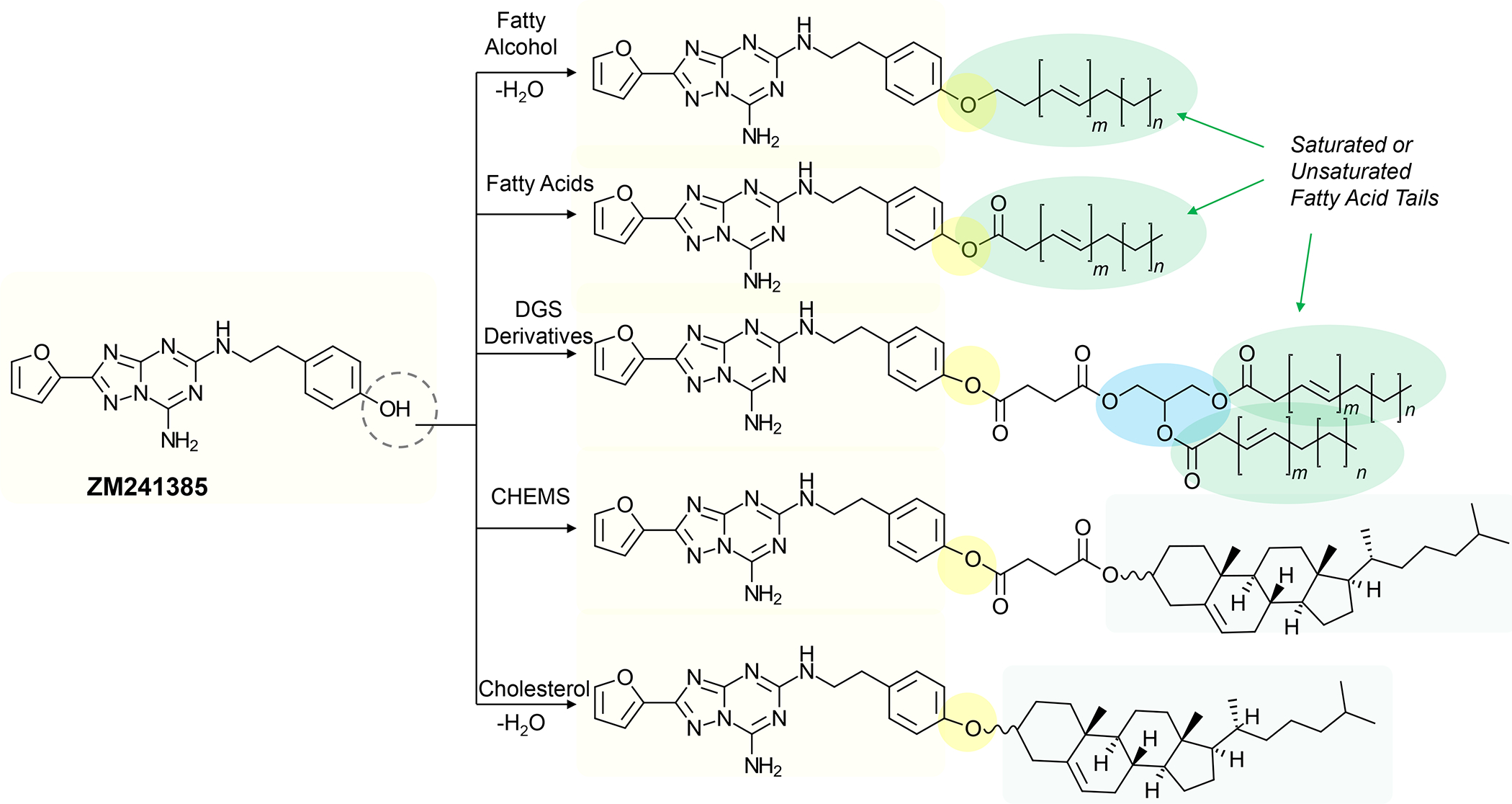
The A2A receptor provides another avenue of immune escape in the solid tumor immune landscape and can be inhibited by the non-xanthine antagonist ZM241385. We identified the phenolic hydroxyl group on this compound for prodrug design. This enables synthesis of ester or ether drug-linkages to fatty alcohols or cholesterol, as well as the possibility to conjugate the drug to fatty acid, DGS or CHEMS carboxyl-terminal groups. Not only does bilayer incorporation of these drug conjugates offer improvement of the unfavorable drug PK,195 but also allows the development of co-formulated multi-drug carriers.
E. Augmentation of cancer immunotherapy by nanocarriers impacting T-cell spatial distribution and the immune suppressive effect of the tumor stroma
E.1. Interference in the CXCL12/CXCR4 axis
There is growing evidence from preclinical PDAC and TNBC studies that, in addition to the poor cancer immunogenicity, important stromal cell types such as TAFs, MDSCs and TAM) restrain CTL efficacy, in addition to preventing cellular contact with cancer cells.18 Among the networks of cellular activation pathways that control the balance between immune attack and immune evasion, the CXCL12/CXCR4 axis is of particular importance for cellular communication in the TME, with influences on tumor vascularization, Treg recruitment, spatial distribution of T-cells, cancer cell proliferation and metastasis.22–23 Consequently, CXCL12/CXCR4 overexpression correlates with poor prognosis, including for BC and PDAC.199 It is relevant, therefore, to consider the impact of the CXCL12/CXCR4 axis on the immune suppressive effects of stromal cells in immunotherapy design. This includes the use of SMI such as Plerixafor (AMD3100) and Mavorixafor (AMD11070) or the synthetic peptide, BL-8040, to interfere in CXCL12 binding to CXCR4, with the potential of preventing T-cell exclusion, reducing Treg and MDSC recruitment, and metastasis inhibition.65, 199 A number of preclinical studies support the use of CXCR4 antagonists for sensitization to chemotherapy and ICIs in solid cancers. This includes the work of Chen et al., who used Plexifor with success in metastatic BC models (including 4T1) to decrease desmoplasia, limit metastatic spread, improve blood perfusion, increase CTL recruitment, and to decrease immunosuppression.199 Similarly, the efficacy of BL-8040, was documented in numerous preclinical studies (e.g., melanoma, breast and lung cancers), where CXCR4 blockade could also be seen to mobilize T-cells and NK cells from the bone marrow and lymph nodes to the tumor site.65, 200 BL-8040 was also used to show improved survival in combination with checkpoint blocking antibodies in a phase II clinical trial in patients with metastatic PDAC (Figure S33A).65 The response was characterized by increased CD8+ T-cell infiltration, while MDSC and Treg numbers are suppressed. Seo et al. also demonstrated that CXCR4 blockade with AMD3100 could improve CTL migration to the juxta-tumoral PDAC compartment in a time sequence multicolor fluorescence study in tumor cell slices (Figure S33B).39 In addition, Biasci et al. conducted an experimental study in which AMD3100 was administered in combination with PD-1/PD-L1 inhibitors in PDAC and colorectal patients with microsatellite stability.201 Transcriptional analysis in paired biopsy samples from metastatic tumor sites, demonstrated an integrated immune response, including improved spatial distancing between cancer cells and CTLs.
While CXCR4 antagonists are clinically approved for mobilizing hematopoietic bone marrow precursors, their impact overcoming T-cell exclusion and recruitment of immunosuppressive cells in solid tumors has resulted in implementation of new combination immunotherapies that include the use of nano-DDS. One approach has been the attachment of AMD3100 to pegylated nab-Paclitaxel nanoparticles, leading to improved outcomes in ovarial cancer.202 In contrast, our approach has been to determine if the weak basic properties of AMD3100, AMD3465 and AMD11070 could be used to achieve remote loading in LB carriers, using TEA8SOS or ammonium sulfate trapping agents (Figure 25A). The best loading capacity (LC) in liposomes, was for AMD11070 (LC =17%), prompting construction of an AMD11070-silicasome (Figure 25A). The first animal study with the AMD11070-liposome was to assess the impact of combination therapy with a liposome delivering Doxorubicin in orthotopic 4T1 and EMT6 tumor models. Following same-day IV administration of both carriers and the dosing schedule shown in Figure 25B, tumor shrinkage could be obtained in 4T1 tumors using free AMD11070 alone, liposomal L-AMD11070, DOX-NP®, DOX-NP® plus free AMD11070, and DOX-NP® plus L-AMD11070. The latter treatment combination resulted in the most significant tumor reduction, with evidence of improved cytotoxic killing (Figure 25B). While both free and encapsulated AMD11070 failed to show a significant effect on CD8+ spatial distribution during mIHC analysis (Figure S34), both treatments had a significant impact on lung metastases, in keeping with the important role of CXCR4 in this disease setting (Figure 25B). Similar experimentation in EMT6 demonstrated that co-administration of encapsulated Doxorubicin and AMD11070 resulted in significant tumor shrinkage, albeit that there was no statistical difference with respect to DOX-NP® plus free AMD070 (Figures 25C and S35). Nonetheless, encapsulated AMD11070 was effective in allowing more abundant CTL recruitment to the tumor core (Figure 25C). This was accompanied by increased perforin deposition at the tumor site and increased cytotoxic killing (not shown).
Figure 25. Nano-formulated CXCR4 inhibitors suppress tumor metastases and intervene in CTL exclusion in orthotopic TNBC and PDAC tumor models.
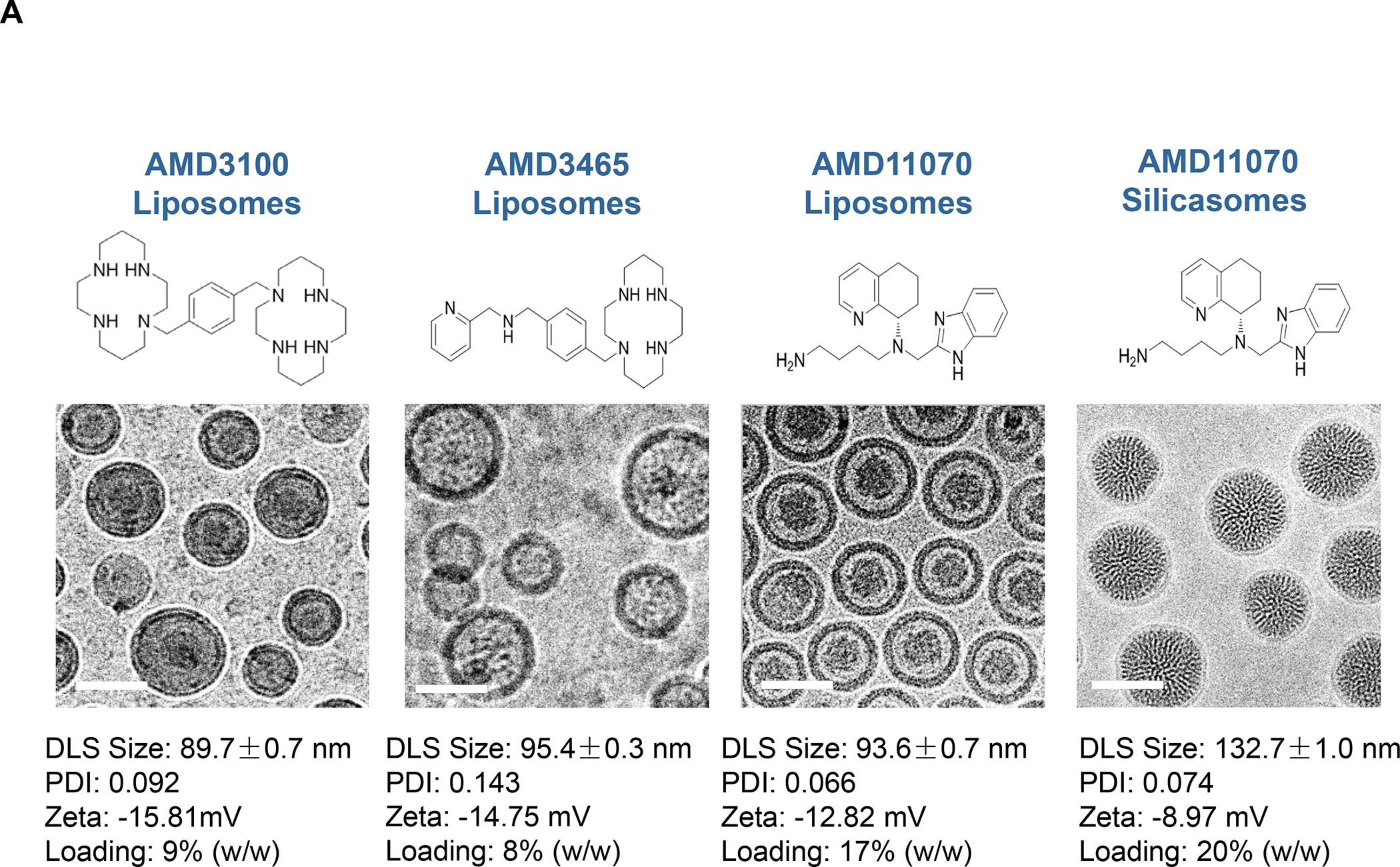
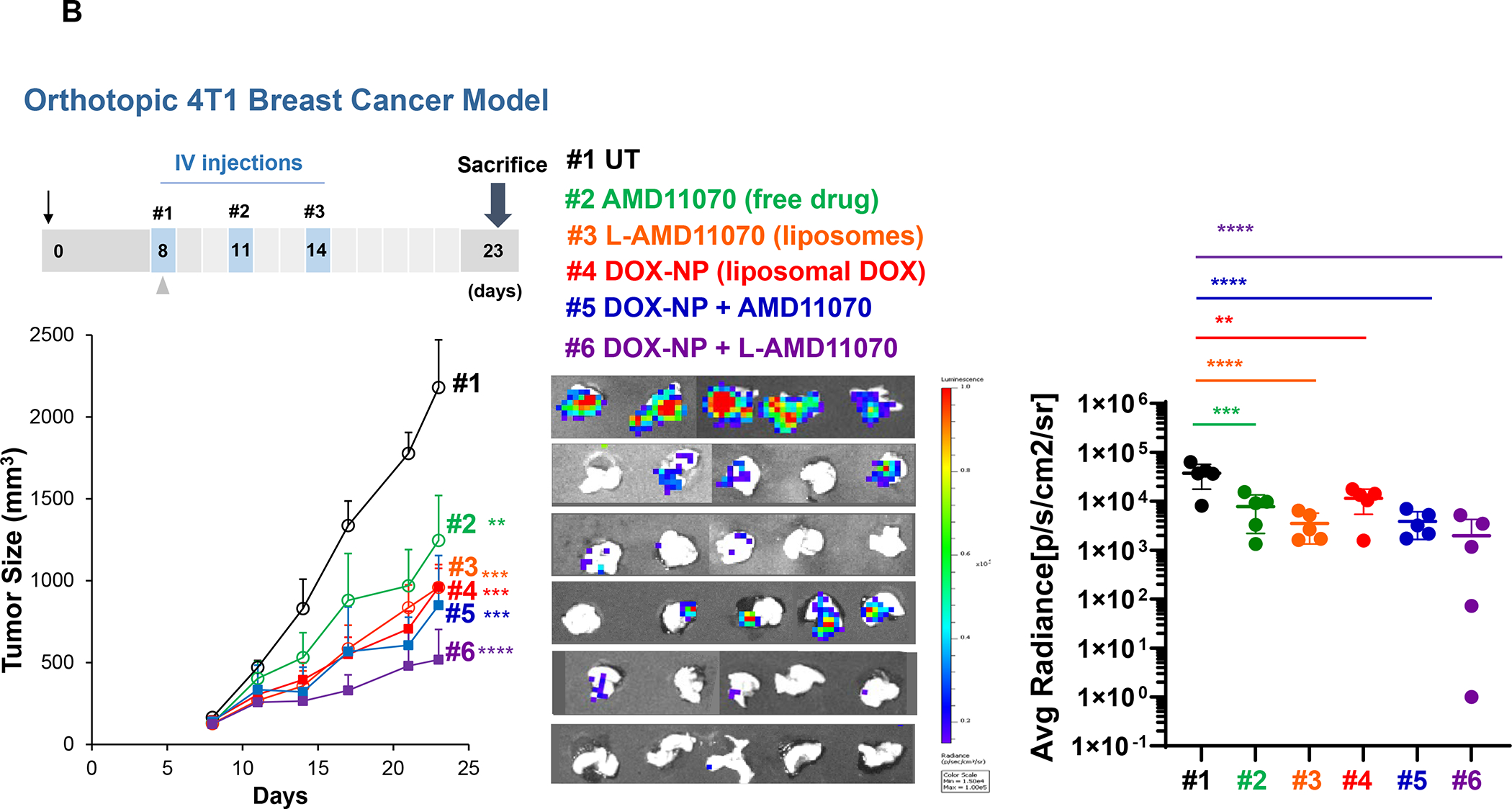
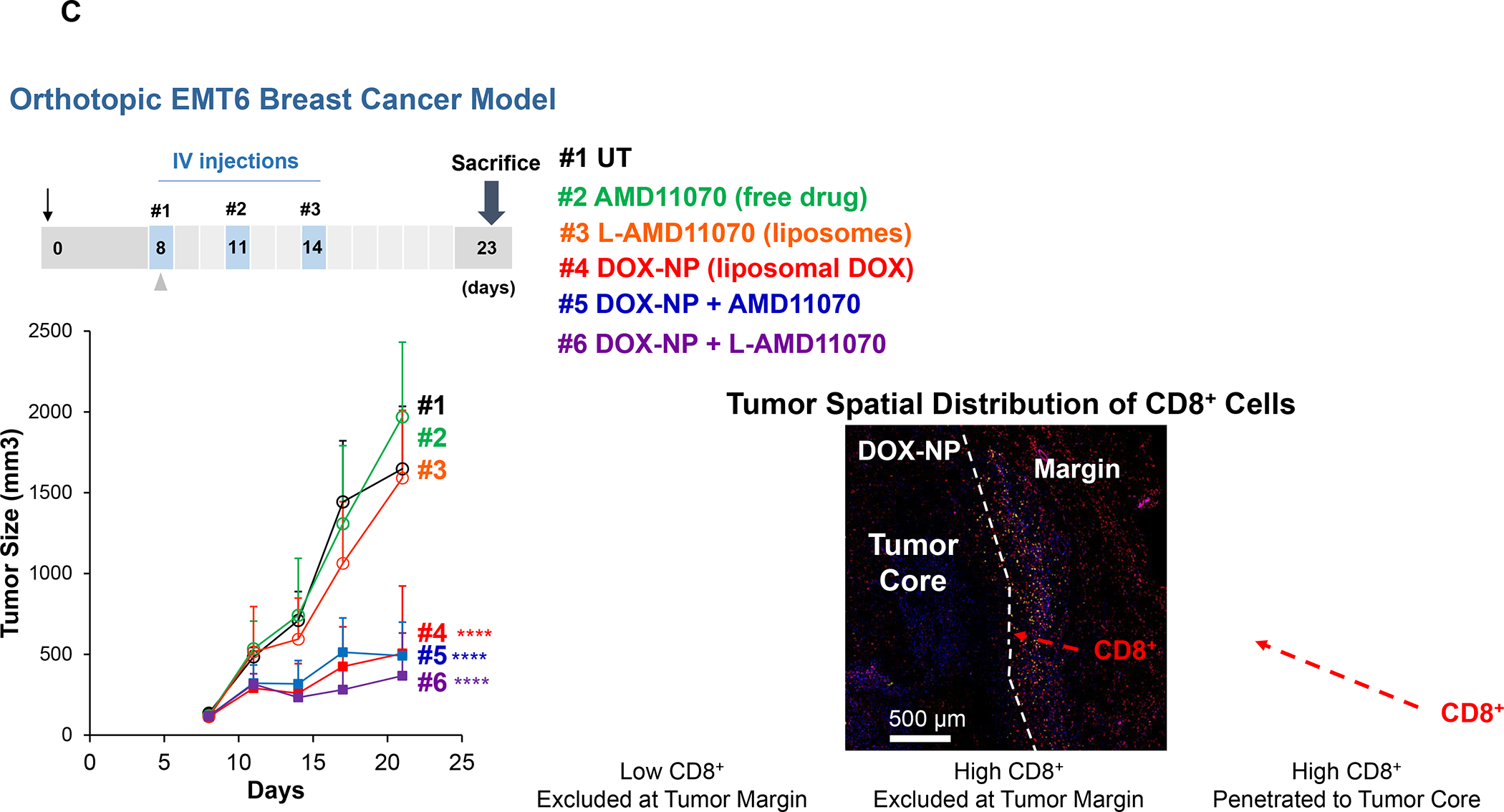
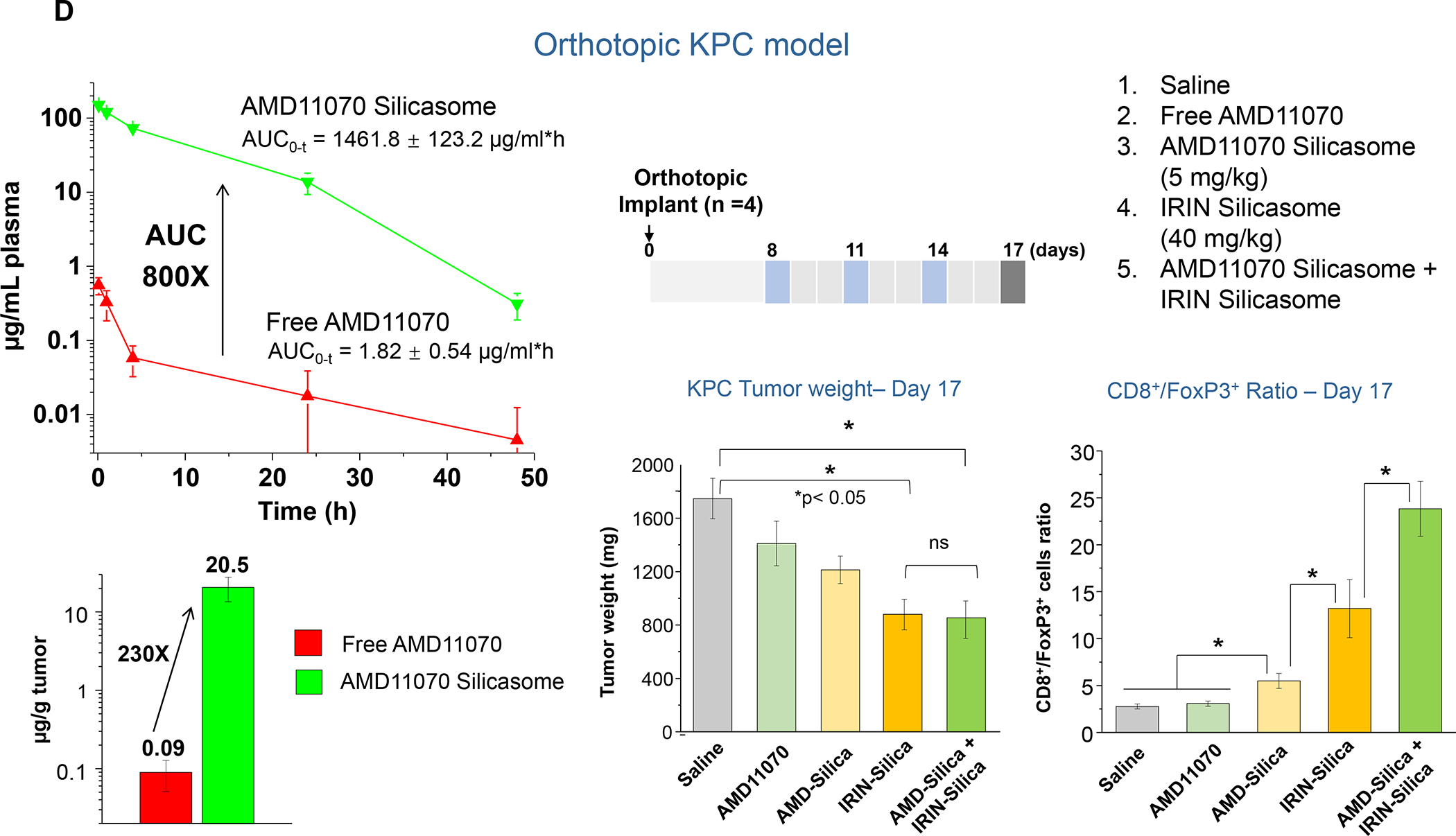
Panel A: We utilized the weak-basic properties of a selected series of CXCR4 inhibitors (AMD3100, AMD3465, and AMD11070) to assess drug loading capacities across liposomal and silicasome LB. This was accomplished using triethylammonium sucrose octasulfate (TEA8SOS) or ammonium sulfate to achieve loading capacities of 8~20% in liposomes. CryoEM images of the liposomes used for TEA8SOS loading and accompanying physicochemical characteristics are shown. AMD11070 was chosen to construct a silicasome, using the same trapping agent. For lipid coating, 40 mg/mL of the purified, bare MSNPs were used to soak in 80 mM TEA8SOS, before the addition of a 50% (w/v) lipids mixture (DSPC/Chol/DSPE-PEG2000, in the molar ratio 3:2:0.15. Panel B: Orthotopic 4T1 tumors were established as described in Figures 6 and 18. These animals develop a high rate of lung metastasis. Liposomal Doxorubicin (DOX-NP) induces significant 4T1 shrinkage (bottom left panel), with evidence of an immunogenic response as shown in Figure S4–S6. Free AMD11070 alone also leads to tumor shrinkage, which was significantly enhanced when combined with DOX-NP. In addition, combination therapy with DOX-NP plus liposomal AMD11070 provided additional tumor size reduction, in addition to accomplishing the highest density of CTL recruitment (Figure S34). IVIS imaging of explanted animal lungs demonstrated significant reduction in metastases under all conditions where AMD11070 was used. Panel C: Similar analysis was carried out in the orthotopic EMT6 model, which is characterized by extensive CD8+ exclusion from the tumor core, even under basal growth conditions (Figure 6). In this setting, neither free nor L-AMD11070 was able to interfere in tumor growth. However, combination of free or encapsulated AMD11070 with DOX-NP contributed to growth inhibition, which did not differ significantly between free and encapsulated drugs. However, spatial analysis of the tumor landscape, demonstrated that the increased recruitment of CD8+ T-cells during co-administration of DOX-NP, showed a significant shift in cell distribution to the tumor core. These changes were also accompanied by a reduction in Treg recruitment to the tumor landscape. Panel: D: AMD11070-silicasomes were used to perform a PK study in 10–12-week-old female B6/129SF1/J mice bearing KPC tumors (left panel). The animals received IV injection of free AMD11070 or AMD11070-silicasome at a drug dose of 5 mg/kg, followed by collection of blood samples at 5 min, 1, 4, 24, and 48 hrs. After separation of the plasma fraction, the drug was extracted in an acidic methanol solution (0.1 mol/L phosphoric acid/methanol, 1:4 v/v). Drug content in the tumor tissue was obtained from KPC tumor bearing animals 24hr after drug administration. The PK data were analyzed by PKSolver software, using a one-compartment model. We also investigated treatment impact on tumor weight and the CD8/Treg ratio in orthotopic KPC tumor bearing mice, as shown in the right panel. Animals received 3 IV administrations on days 8, 11 and 14, using the formulations shown in the legend, or were left untreated. Animals were sacrificed on day 17. Orthotopic tumors were collected, weighed, and prepared for sectioning to perform mIHC analysis, as described in Figure 6. While tumor growth inhibition by IRIN was not increased by co-delivery of the AMD11070-silicasome, this treatment resulted in a significant increase in the CD8/FoxP3 ratio in combination treatment of Irinotecan silicasome. * p< 0.05 compared to saline.
The AMD11070-silicasome was used to investigate the impact on drug biodistribution and immunogenic effects in the murine orthotopic KPC model. A PK study was performed, as described in Figure 25D (left side panel). The animals received IV injection of free AMD11070 or the AMD11070-silicasome at dose equivalents of 5 mg/kg, followed by collecting blood samples at 5 min, 1, 4, 24, and 48 hrs. The data demonstrate a significant prolongation of the circulatory half-life by drug encapsulation, leading to an 800-fold increase in the area under the curve (AUC) and a 230-fold increase in drug delivery at the tumor site. In subsequent experimentation in the KPC orthotopic model, mice were randomly assigned to 5 groups (n = 4): saline, free AMD11070, AMD11070 silicasome, IRIN-silicasome, IRIN-silicasome + AMD11070 silicasome (right panel). This was followed by IV administration of a 40 mg/kg Irinotecan dose equivalent or 5 mg/kg AMD070 dose equivalent on 4 occasions, as shown in Figure 25D. Animals were sacrificed on day 17 before tumor collection and immunophenotyping. While there was no additional tumor shrinkage during co-delivery of the AMD11070 silicasome over the short observation period, there was a significant increase in the CD8/FoxP3 ratio (Figure S35 and S36). Long-term animal survival studies are ongoing.
To complement above results, dual-delivery carriers can be used to combine CXCR4 antagonists with other therapeutic agents. This includes LB-coated carriers with remote loading of AMD11070 in addition to the encapsulation of IDO-1 or ICI prodrugs in the lipid membrane. CXCR4 antagonists could also be anchored to PEG that is incorporated in a LB of nanocarriers that are also used for loading ICD-inducing drugs or GSK3 inhibitors.202–203 This topological arrangement also allows the conjugated drug to target CXCR4 expressing tumor or metastatic sites.204 Moreover, PEG can also be used to synthesize self-assembling dual drug carriers to improve anticancer therapy.205
E.2. Complementary therapies to reprogram immune suppressive stromal cells
Other than the use of CXCR4 antagonists, additional mechanisms exist to reprogram immunosuppressive cells in the PDAC stroma.18 These include CD40 ligation, interference in TGFβ activity and STING targeting. CD40 provides an integrative therapeutic approach based on its involvement in T-cell-dependent as well as innate immune response pathways in mice and humans.206–207 CD40 plays an important role in “licensing” dendritic cells for improved antigen presentation, while also inducing stromal depleting macrophage activity. These properties have allowed anti-CD40 mAb to improve the objective response rate in metastatic PDAC clinical trials, in combination with Gemcitabine and anti-PD-1.207 Chemotherapy addition prior to anti-CD40 administration provides a robust vaccine-like effect in murine PDAC tumors,207–208 where a combination of ICI, chemotherapy and anti-CD40 augments tumor regression and survival.55 In fact, the most effective response to anti-CD40 immunotherapy in the KPC model is when the antibodies are combined with Gemcitabine, nab-Paclitaxel, anti-PD-1 and anti-CTLA-4 mAb.55
The integrative effects of CD40 in cancer immunotherapy allow the development of further combination approaches that take into consideration communication of the tumor stroma with myelopoetic cells in the bone marrow.26 In addition to the TME effects, inflammatory cytokine production (e.g., granulocyte-CSF) by the desmoplastic TNBC or PDAC stroma is also involved in recruiting MDSC and TAM precursors from the bone marrow.209 The bone marrow also supplies conventional dendritic cell (DC) progenitors, which develop into APC at the tumor site.26 This supply source is negatively impacted by stromal cytokines, leading to DC deprivation and poor survival.26 These findings are instrumental in combining anti-CD40 with the Fms-related tyrosine kinase 3 ligand (Flt3L), with activation of the cDC precursors that are released from the bone marrow.26, 61, 210 Consequently, Flt3L administration during early tumorigenesis resulted in robust cDC infiltration at the PDAC site, with significant improvement in disease control. We further envisage that the combination of ICD-inducing carriers with anti-CD40 and CXCR4 antagonists will improve immune suppressive effects of the PDAC and TNBC stroma.
The “stimulator of interferon genes” (STING) is a cytosolic protein, which is abundantly expressed in tumor cells and stroma in human PDAC, where it plays an important role in type I interferon (IFN) production by activating the IRF3/NF-kB signaling pathway.211–212 This introduces the possibility to use STING agonists for the activation of APCs and the programming of MDSCs in a cancer setting, including as an adjuvant during vaccination by antigenic peptides.213–214 However, in spite of pre-clinical efficacy, clinical translation of STING agonists has yielded disappointing results. Poor PK and the requirement of the STING agonist cyclic dinucleotides (CDN) to cross the cell membrane are major barriers, necessitating intra-tumoral dosing.215–216 In this context, nanocarriers are particularly impactful for robust STING activation at 1/100th the extracellular dose of free CDN’s.217 While the physicochemical properties of polymer nanoparticles are well suited for CDN delivery, this task is not easy to perform in LB carriers due to the strong negative charge and degradability of CDNs.217 However, our extensive experience with poly lactic-co-glycolic acid (PLGA) nanoparticles offers a potential use advantage from the perspective of expanding nano-enabled immunotherapy to include attention to liver metastases. This is based on the success that we achieved with PLGA nanoparticles in targeting tolerogenic APC in the liver, including liver sinusoidal endothelial cells (LSECs).218–220 Based on their role as tolerogenic APC, antigen delivery LSECs are capable of generating regulatory T-cells, which suppresses allergic and anaphylactic immune responses to food allergens.218 We postulate that the tolerogenic effect of liver APCs could be reversed by STING delivering nanoparticles to reprogram their involvement in switching to a protective immune state against metastatic pancreas, bowel, breast and lung cancer cells.221 Previous attempts to reprogram the protective effect of the liver for cancer metastases include the use of an engineered CXCL12 trap, which includes the TLR5 agonist, Entolimod, capable of restoring the immune response outcome.221–222 In addition, Yu et al. developed a lipid nanoparticle incorporating the cationic defense peptide, melittin, for LSEC targeting to block metastatic spread in animal hepatic and BC models.223 To achieve similar outcomes, we have developed technology for encapsulating the anionic STING agonist, amidobenzimidazole (diABZI), PLGA nanoparticles for targeting tolerogenic APC in the liver (Figure S37). This particle has robust type I IFN inducing properties and can be successfully targeted to the liver by a peptide ligand (apoB peptide) binding to LSEC scavenger receptors.218–220 Future studies will evaluate its effect on metastatic tumor spread in KPC in and TNBC models, using the achieved outcome of mellitin-delivering nanoparticles as guideline.
TGFβ is a pleiotropic cytokine that is capable of generating tumor-promoting, as well as tumor suppressive effects.224 While capable of suppressing epithelial cell proliferation at an early tumor stage, TGFβ exerts immune suppressive effects at the more advanced stage of tumor growth, with extensive desmoplasia.224 This involves Treg generation as well as interference in effector T-cell function by checkpoint receptor expression.66, 224–226 It is therefore not surprising that interfering in TGFβ activity can enhance the action of ICIs in preclinical studies, including PDAC.225 The principle strategies to interfere in TGF-β function include compounds that block TGF-β binding to its receptors, inhibition of intracellular signaling, or blocking of angiotensin II type I receptor. Our own studies have demonstrated that delivery of a TGF-β receptor kinase inhibitor, LY364947, complexed to the MSNP surface, exerts a robust effect on the PDAC stroma, with improved vascular access to the tumor site.227 This will be discussed later (Figure 27). Blockade of the angiotensin II type I receptor by losartan leads to reduced TGFβ production in fibroblasts and is effective in preclinical PDAC models as well as in patients undergoing neoadjuvant FOLFIRINOX therapy.228–229 Losartan encapsulation in liposomes improves the response to Paclitaxel in BC by inhibiting collagen deposition.230
Figure 27. Two-wave of therapy to improve KPC chemo delivery by stromal-vascular engineering.
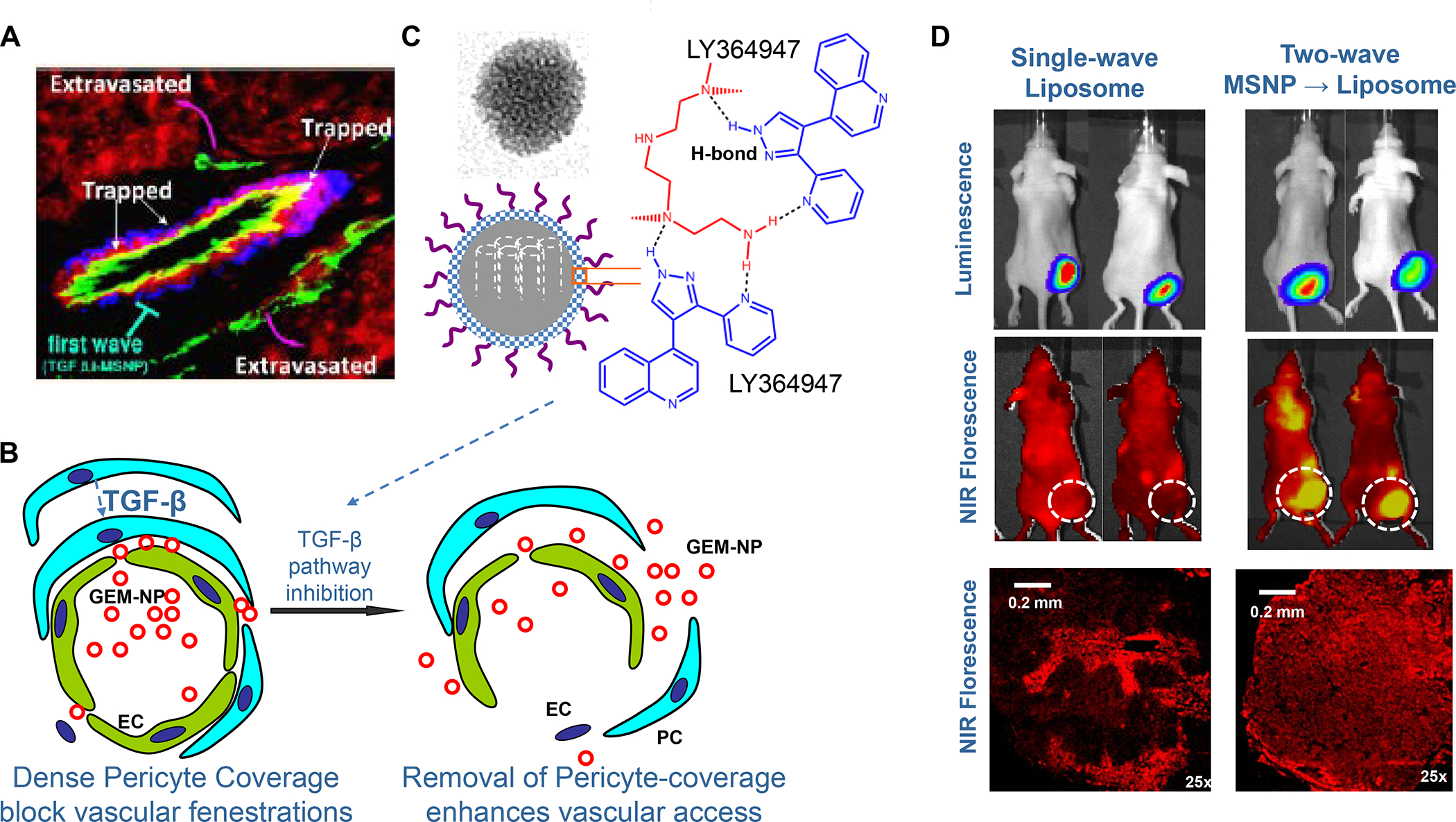
The dysplastic PDAC stroma plays a role in limiting vascular access to the tumor site through the recruitment of stromal pericytes to the abluminal endothelial cell surface. This recruitment is mediated by a TGF-β signaling pathway (panels A and B). Panel A demonstrates that the dense PDAC stroma blocks vascular access of IV-injected NIR-labeled liposomes, which becomes entrapped between the endothelial cells (CD31 green fluorescent marker) and the pericytes (NG2 blue fluorescent marker). The schematic below that illustrates these relationships. Pericytes adhere to endothelial cells as a consequence of TGF-β production in the stroma, which engages TGF-β type I receptor kinase activity. LY364947 is a SMI of this kinase, and can be delivered to the PDAC site by H-bonding to the surface MSNPs, decorated with a PEG/polyethylenimine polymer (Panel C). The drug is released in the TME by the acidic stromal pH, which interferes in drug binding to the decorated particle surface (Panel B). Panel D: In vivo experimentation using the LY364947-MSNP as a first wave carrier, injected IV, to increase uptake of the NIR-labeled, GEM-delivering liposomes injected 60 minutes later. The IVIS imaging in the upper panel demonstrates subcutaneous KPC growth of luciferase transfected tumor cells. The middle panel shows fluorescence imaging of the same tumors injected with the NIR-labeled liposomes, with and without prior LY364947-MSNP administration. The increased fluorescence intensity at the tumor site during two-way therapy is as a result of increased lysosomal release into the tumor site, as shown in the explanted tumors following animal sacrifice. Panel A-D adapted from ref 227. Copyright 2013 American Chemical Society.
F. Integration of nano-DDS in PDAC and TNBC combination treatment strategies
F.1. Combination drug use challenges for cancer immunotherapy
The combination of chemotherapy with ICI’s or other immune modulators are continuing to introduce new immunotherapy approaches in the oncology landscape.231 However, while most oncologists are familiar with the use of ICD-inducing chemo agents (e.g., Doxorubicin, Epirubicin, Idarubicin, Mitoxantrone, Bortezomib, Cyclophosphamide or Oxaliplatin) in combination with immunotherapy, there are still significant challenges to overcome. The first barrier is that the approval of chemo drugs for cytotoxic killing does not cover off-target immune effects. Most of these drugs have been developed in preclinical xenograft models using data extrapolation from patient studies, based on maximum tolerated dose (MTD).231–232 As a result, most of ICD-inducing chemo agents are used in dosing and treatment regimens aimed at tumor killing with the lowest incidence of side effects in normal tissues. Unfortunately, this approach does not consider the increased sensitivity of the bone marrow and secondary lymphoid organs, which frequently leads to lymphopenia and neutropenia, which adversely impact the anti-tumor immune response. These restrictions can be overcome by using immunocompetent preclinical models and introduction of dosing schedules that are less harmful.
In theory, when two or more therapeutics are administrated contemporaneously, their combined effects may amount to lesser (antagonistic), equal (additive) or increased (synergy) responses compared to individual drug effects.233–234 However, it is difficult to differentiate between synergistic and additive drug interactions in humans, where combination therapy frequently leads to drug/drug interactions and toxicity. It is important, therefore, to consider appropriate use of software tools and approaches that enable ratiometric combination of therapeutics, including in nanocarriers.92, 233–234
An important consideration for combination therapy is differences in the solubility, PK and biodistribution potential of individual drugs, with impacts on treatment efficacy and safety. The traditional approaches for construction of combination chemotherapy often fail to consider differences in drug PK,235 which can be specifically addressed by nanocarriers.236 High interstitial fluid pressure also limits small molecule diffusion in densely packed TMEs, with possible impacts on the drug spatial distribution. Treatment delivery to heterogeneous landscapes should consider the role of stimulus-responsive nanocarriers that can perform on-demand delivery. The inclusion of cholesteryl hemisuccinate (CHEMS) in some of our carriers exemplifies this approach.128 CHEMS is an ionizable lipid that is widely used in acid-sensitive delivery systems. While negatively charged at neutral pH, lipid charge reverts to neutral under more acidic conditions, causing bilayer disruption. We have also illustrated how MSNPs can be endowed with pH-sensitive nanovalves, esterase-sensitive pore stoppers and heat-sensitive release from iron oxide embedded MSNPs being placed under an oscillating external magnetic field.237–239
F.2. Benefits of co-encapsulated drug delivery for immunotherapy
Compared to the administration of free drugs, encapsulated drugs provide special advantages, such as protection against degradation, prolonged circulation time, improved PK, sustained drug release, ability to deliver insoluble drugs and limiting drug toxicity. Moreover, the advantage of DDS extends to drug co-formulation, with the promise of achieving harmonized PK, ratiometric design, overcoming multiple drug resistance mechanisms and allowing release control with respect to time, sequence and delivery locality.240–243 Our focus on LB-coated nanocarriers is particularly germane to improve immunotherapy. Not only does the LB approach enable high drug loading capacities, but also allows incorporation of lipid moieties and prodrugs with a diverse range of functions to boost immune responses. This is exemplified by our studies on the L-MTO/IND, discussed in Figure 18. This allows the drug synergy at the site of IFN-γ release (Figure 17B). In addition to LB-coated carriers, multiple DDS have been developed to improve the PK of chemo agents in diverse preclinical settings, making use of chemo-immunotherapy plus co-delivery of immune modulatory agents.244–246
The design of co-delivery systems, should consider the importance of ratiometric loading and drug release to improve the efficacy of combination therapy.87, 234, 247–248 While it is known that drug ratio is important at the cancer cell level, the impact of ratios in vivo is poorly investigated, with decisions often based on MTD. Mayer et al. systematically examined chemotherapeutic agents with unique pharmacological actions to determine ratiometric design.234 This included the demonstration of optimal molar ratios for Irinotecan/floxuridine (1:1), cytarabine/daunorubicin (5:1) and cisplatin/daunorubicin (10:1), as compared to combinations leading to additive or antagonistic outcomes. Moreover, these ratios remained fixed in the blood or the tumor site after liposomal delivery, allowing the development of Vyxeos, a liposome that combines daunorubicin and cytarabine in a 1:5 molar ratio.249–250 Vyxeos received FDA approval for treatment of acute myeloid leukemia (AML), resulting in significant improvement in survival outcome and compliance, compared to the traditional treatment and dosing regimens.
Other than preventing dangerous drug-drug interactions, ratiometric design allows synergy to be achieved at sublethal chemo doses. This is illustrated by our efforts to develop a more effective and safer treatment approach for Gemcitabine delivery to the PDAC site. The rationale was that the administration of commercial albumin-bound Paclitaxel (PTX) (Abraxane) improves the therapeutic efficacy of Gemcitabine (GEM) by inhibiting PDAC stromal abundance and expression of GEM-inactivating enzyme, cytidine deaminase (CDA).92, 251–252 This led to the development of a ratiometric-designed silicasome carrier, capable of delivering an optimal GEM/PTX ratio to increase the content of activated GEM (dFdCTP) at the tumor site (Figure 26).92 High GEM loading (40 wt %) was achieved by LB encapsulation, which also allowed hydrophobic PTX to be incorporated into the coated bilayer. This enabled ratiometric testing of the drug combination, assisted by CompuSyn software (Figure S38). This allowed us to select a PTX dose below the cytotoxic threshold, yet still effective for delivering a pro-oxidative, pharmacodynamic effect that suppresses stromal volume and CDA expression. The ultimate outcome was ratiometric design of a dual-delivery silicasome that is more effective in treating orthotopic tumors than free GEM, encapsulated GEM only or free GEM plus Abraxane (Figure 26). To achieve the same level of tumor reduction, the Abraxane dose had to be increased 12-fold.92 High-performance liquid chromatography analysis of tumor-associated GEM metabolites confirmed that the ratiometric designed silicasome could enhance the tumor drug content of the active GEM metabolite 13-fold over free GEM.
Figure 26. Use of a ratiometric designed silicasome carrier for improved chemotherapy in a human PDAC xenograft.
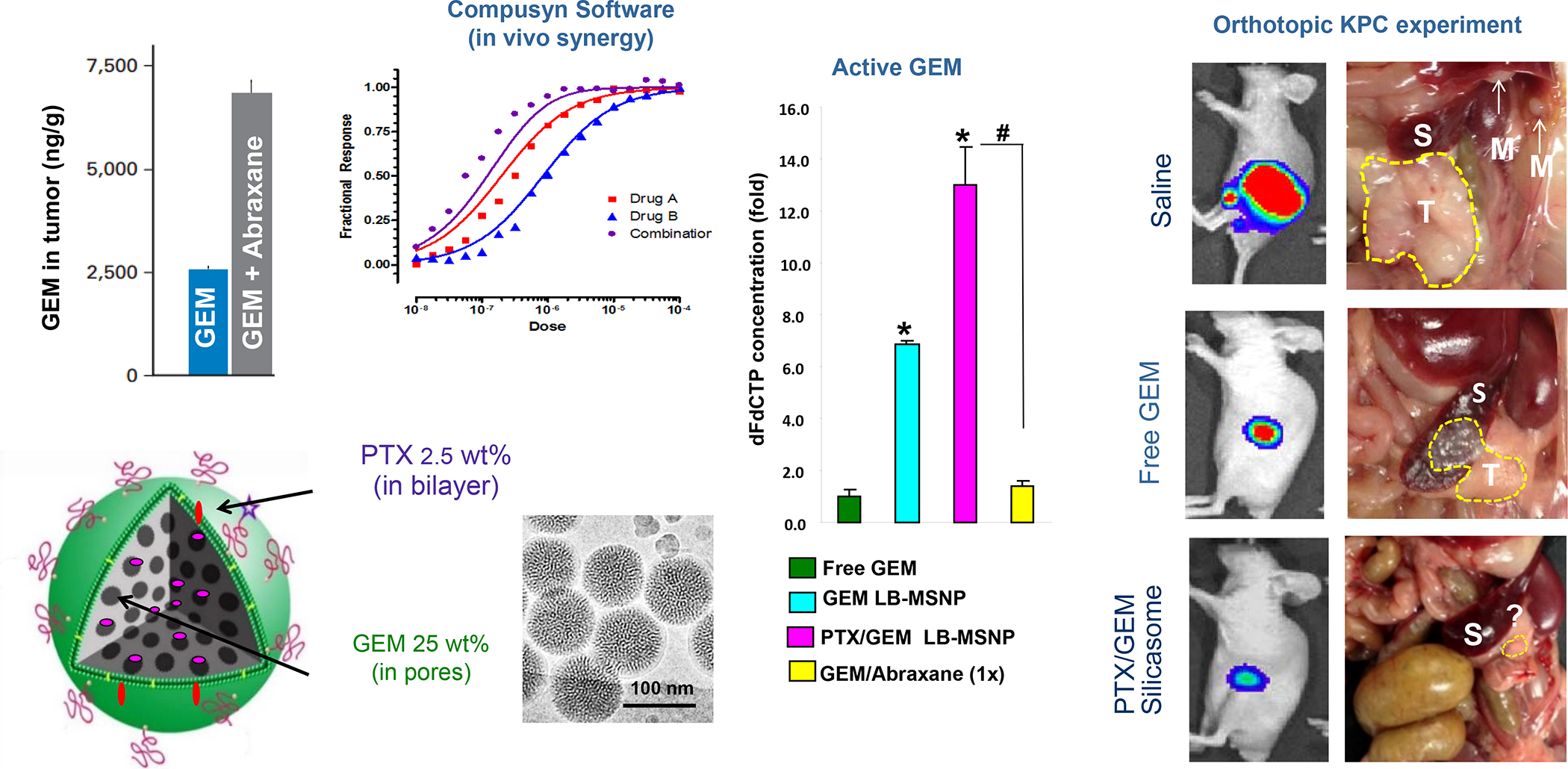
The dysplastic PDAC stroma contributes to chemo resistance, including inactivation of the first-line drug Gemcitabine (GEM) by the stromal enzyme, cytidine deaminase (CDA). Since it has been shown that Paclitaxel (PTX) can decrease GEM uptake through oxidative stress-mediated stromal depletion, we opted for a silicasome carrier that can deliver PTX plus GEM. This was accomplished by ratiometric design of a nanocarrier that incorporates GEM in the porous interior, with a sublethal amount of hydrophobic PTX incorporated in the LB. Ratiometric design, as described in Figure S38, yielded a dual-delivery carrier that incorporates 25 wt% GEM with 2.5 wt% PTX. The 10:1 ratiometric delivery was responsible for decreasing the stromal abundance, while increasing uptake of activated GEM 13-fold. We also demonstrated that the sublethal PTX dose could achieve the stromal response by generating oxidative stress instead of cell killing. To demonstrate the in vivo efficacy, mice carrying subcutaneous PANC-1 human xenografts received IV injection of PTX/GEM-loaded silicasomes. Drug co-delivery provided more effective tumor shrinkage than the GEM-only carrier, free GEM, or free GEM plus Abraxane. Comparable tumor shrinkage required coadministration of 12 times the amount of free Abraxane. Adapted from ref 92. Copyright 2015 American Chemical Society.
Nanoparticle platforms for combination drug delivery holds considerable advantages to overcoming anti-tumor multidrug resistance (MDR), including for breast/ovarian or prostate cancer.235.227, 253–255 This includes the option of using the same particle for co-delivery of nucleic acids that knock down drug metabolic pathways, which interfere with the action of the codelivered chemotherapeutic agent.227, 256–257 For instance, Meng et al. developed MSNPs to deliver Doxorubicin plus a siRNA construct that knocks down the P-glycoprotein (Pgp) drug exporter in DC cells, in vitro and in vivo.227 This led to significant improvement in the cellular and tumor Doxorubicin concentration compared to free or encapsulated drug without siRNA.
F.3. Rational integration of nano-DDS for PDAC and TNBC chemo-immunotherapy
In spite of the paradigm shift of checkpoint therapy, only 10–38% of cancer patients respond to ICIs, prompting development of multiple combination strategies to improve treatment outcome. This established an urgent need to select the best combinations for testing in clinical settings, often limited by the number of available subjects to test out all the possible drug combinations. Throughout this communication, we have argued that switching the immune landscape of “cold” or non-inflamed tumors to “hot” is a key consideration that can be accomplished by ICD-inducing nanocarriers. Moreover, we have illustrated how the immunogenic response can be propagated by co-formulation or co-delivering drugs interfering in immune escape, immune exclusion, and immune suppressive pathways, resulting in improved immunotherapy responses in PDAC and TNBC landscapes. BC, in particular, provides the best historical example of how chemotherapy can be effectively combined with checkpoint blocking antibodies and other immunomodulators that can be considered for a nano-DDS approach.231 A prime example of how to improve delivery of drug combinations by nano-DDS is the ICON chemo-immunotherapy phase IIb trial (CA209–9FN) in HR+ metastatic BC, which makes use of a pegylated Doxorubicin-delivering liposome plus Cyclophosphamide to deliver an ICD stimulus.114 The chemotherapy delivery is intended to synergize with co-administered checkpoint blocking antibodies, ipilimumab (anti-CTLA-4) and nivolumab (anti-PD-1). Encapsulated Doxorubicin was introduced to minimize cardiac toxicity, as well as to extend the duration of therapy to be compatible with longer-lasting ICI effects. Importantly, the study utilizes a semi-metronomic dosing regimen to limit drug toxicity and leukopenia associated with an every 3–4 week Doxorubicin dosing schedule. Instead, lower dose administration every two weeks prevents interference in the effector immune response. While the outcome of the trial is awaited, data from recently conducted TNBC trials support the notion that anthracyclines are superior to taxanes in rendering “cold” tumors “hot”, at the expense of more toxicity. For instance, the TONIC trial attempted to determine which chemotherapeutics work best if applied prior to PD1-blockade.78 Therapy induction with Doxorubicin yielded the highest response rate to nivolumab, compared to other chemo agents or irradiation. This study also provided biological evidence of the immunogenic effects of Doxorubicin at the tumor site. The Keynote 522 study provided further evidence that neoadjuvant chemotherapy can increase the response outcome in the treatment group receiving pembrolizumab (anti-PD1) co-administration.112
We envisage that the success of chemo-immunotherapy in patient care will be augmented by nano-DDS in combination with SMI of the PD1/PD-L1 and A2AR checkpoint pathways, the IDO-1 immune metabolic pathway, GSK3, and the CCL12/CXCR4 axis. In addition, it is possible to combine ICD stimuli with TLR agonists, CD40 agonists, Flt3L agonists, blockers of the WNT pathway, antibodies that target M2 macrophages (e.g., CSF1R inhibitors) or augment the contribution of NK cells. When applied to the recently acquired knowledge of heterogeneous immune landscapes, purposeful use of these interventions could reprogram the immune desert (ignored), stromal/margin-restricted (excluded) and inflamed TNBC landscapes (Tables 1 and 2). Examples of custom-designed nano-DDS strategies for TNBC immunotherapy are provided in Table 5. It is also important to consider the contribution of NK activation, as discussed in Figure S16. This can be accomplished by MTO delivery and further augmented by the NKG2-Atargeting mAb, monalizumab, or recombinant interleukin 15 (IL-15).258 Moreover, CXCR4 antagonists and A2AR blockade can augment NK activity.
Table 5:
Customized, nano-enabled combination therapy for TNBC
| TIME Category | Primary and complementary strategies |
|---|---|
| Ignored or Immune Desert Landscape. | • The primary strategy is to change the “cold” immune status by encapsulated delivery of ICD inducing chemo: Doxorubicin, Mitoxantrone or Paclitaxel • Depending on the level of success, the consolidation strategy would be to propagate the immune response by combination strategies that make use of antibody blockade or co-formulation of the nanocarrier with prodrugs or SMI that interfere in PD-1/PD-L1, GSK3, and A2AR and B7-H4* checkpoint pathways • Key supplementary treatment to augment the outcome should address stromal fibrosis and presence of immune suppressive stromal cell types, using carriers that deliver AMD11070, with possible assistance by losartan or a CSF1R inhibitor, e.g., pexidartinib**, which interferes in M2 macrophage activity • Additional consideration could be the inclusion WNT pathway blockers, e.g. WNT794 |
| Excluded or Stroma-restricted Landscape | • The primary strategy is to change the “cold” immune status by ICD therapy as described above • Consolidation strategy as described above, except that B7-H4 may not be upregulated in this setting • Key supplementary treatment could target the highly expressed CXCL12/CXCR4 axis, as well as the stromal therapies listed above. • Additional considerations could include inhibitors of VEGF receptor kinases |
| Inflamed or Fully inflamed Landscape | • The primary strategy is single or combination ICI therapy, but should consider metronomic chemotherapy to maintain the activated immune status, • The consolidation strategy, based on the role of the IFN-γ mediated JAK–STAT axis feedback loop, is to provide co-formulated drugs interfering in the PD-1/PD-L1, A2AR and IDO-1 pathways, possibly from the beginning. • Key supplementary treatment includes the use of type I IFN stimuli to boost and maintain antigen presentation, as well as use of a CSF1R inhibitor to target M2 macrophages that are often also overexpressed in the TME. • Additional consideration should also be given to intervening in the IFN-γ mediated JAK–STAT axis |
B7-H4 vs. PD-L1: Expression of these checkpoint receptors are mutually exclusive in BC, which could help to explain why only a subset respond to anti-PD1 treatment.297 mAb MIH43 increases the sensitivity of TNBC cell lines to Doxorubicin, Paclitaxel or carboplatin.298
Pexidartinib is a tyrosine kinase inhibitor, which targets the CSF1R, KIT proto-oncogene receptor tyrosine kinase (KIT), and FMS-like tyrosine kinase 3 (FLT3). While primarily approved for the treatment of tenosynovial giant cell tumors, CSF1R inhibition also regulates the anti-tumor activity of M2 macrophages in the solid tumor setting.299 The drug is a is a pyrrolopyridine compound which can be orally administered and loaded into a liposome. It has also been shown that an amphiphile CSF1R inhibitor could be incorporated through the use of co-lipids into a liposomal membrane for effective use in an experimental breast cancer model.300
To assist the implementation of these treatment strategies, it is important to consider compendium diagnostics and gene classifier tools to characterize and follow the TIME changes during immunotherapy.77 In addition to mIHC, a number of commercial multiplexed cancer immune analysis platforms have emerged to assist immunophenotyping. Combes et al. have recently introduced an approach for identifying and classifying recurrent immune features across 12 different tumor types, using computational clustering of flow cytometry and transcriptomic data cell collection in sub-compartments.259 Their comprehensive characterization of the TME across many human solid cancer types show the capability of using common and reproducible immune cellular networks and archetypes that can be used to develop chemoimmunotherapy, using publicly available website tools.
The COMBAT clinical trial in PDAC has demonstrated that effective chemoimmunotherapy can be introduced by a liposomal Irinotecan carrier in combination with Leucovorin and Fluorouracil (NAPOLI-1 regimen), pembrolizumab (anti-PD1) and the CXCR4 antagonist, BL-8040.65, 260 This treatment regimen introduced an increase in CXCR4+ T-cells and the activation of CD4+ and CD8+ T-cells, along with a decreased density of granulocytic MDSCs in the TME. 65 This agrees with our approach of using silicasome carriers to combine the delivery of Irinotecan plus AMD11070 in the orthotopic KPC setting (Figures 25D and S36). Based on the clinical success of Onivyde, we propose that the Irinotecan-silicasome will improve chemo-immunotherapy efficacy by combination therapy with IDO-1, PD-1, GSK3, and/or A2AR inhibitors. Another approach is to co-deliver the ICD stimulus with lipophilic TLR agonists, incorporated into the lipid bilayer. It is also possible to achieve combination therapy by co-administration of silicasomes that are independently prepared to deliver drug combinations that are injected sequentially on the same day, as we will discuss later (Figure 27). Alternatively, we could construct silicasome carriers that deliver DACH-Pt plus immunomodulators incorporated into the lipid bilayer. As for TNBC, the immune suppressive effects of the stroma can be targeted by silicasome carriers that deliver CXCR4 inhibitors, losartan and/or CSF1R inhibitors. Yet another variation of nano-DDS delivery, is to combine the carriers with checkpoint blocking antibodies, anti-CD40 mAb and/or the administration of Flt3L agonists. Table 6 briefly summarizes illustrative combination therapies in experimentation with liposomes and silicasomes for PDAC, TNBC and occasionally other cancer types. Some of the milestones achieved with co-formulated drug delivery appears throughout this review.
G. Further strategies to enhance nano-enabled immunotherapy
We have referred to the adverse impact that MTD chemo dose schedules may have on achieving immunogenic responses due to bone marrow toxicity.131, 261 In addition, high-dose chemotherapy contributes to poor regrowth of tumor vasculature and selection of drug-resistant tumors cell clones in the drug-free recovery interval.262 Thus, for chemo-immunotherapy to be effective, the cytotoxic dose needs to be lower than the threshold for systemic toxicity, as addressed by the metronomic dosing schedule in the ICON trial (CA209–9FN).114 Metronomic dosing refers to more frequent (even daily) administration of lower chemo doses than is typically used in standard therapy, with the advantage that toxicity reduction can sustain the chemo-immunotherapy response of ICD-inducing drugs. For the metronomic regimen to be effective, it is necessary to adjust the dose below the threshold for bone marrow toxicity, as well as amending the duration of the drug-free interval to achieve optimal balance of tumor cell killing vs. sustaining the immune response (Figure S39).131 This consideration is readily testable with LB carriers in preclinical TNBC and PDAC models. Another approach is co-administration of Cyclophosphamide, which is frequently used for metronomic effects in clinical trials.263 Cyclophosphamide elicits a sustained immune response when administered on an intermittent 6-day repeating metronomic schedule.264 A number of clinical trials have demonstrated the immune stimulatory potential of low-dose Cyclophosphamide, which could be remotely loaded in LB-coated carriers (Figure 7).
Drug delivery by LB-coated carriers can be improved if combined with vascular normalization approaches that improve poor perfusion in the tumor vascular tree.93, 227, 242 Figure 8D outlines our approach for improving silicasome delivery at the orthotopic PDAC site by cyclic iRGD peptides that bind to overexpressed αvβ3 and αvβ5 integrins at the vascular site.93 Another approach is to increase vascular access by reducing pericyte coverage of PDAC vasculature.227 Pericytes constitute an important stromal component, which is recruited to the vascular endothelium by a TGF-β triggered signaling pathway. To decrease the obstruction of vascular fenestrations, with the intent of improving carrier uptake, we constructed a MSNP carrier that assembles a TGF-β receptor kinase inhibitor, LY364947, on the particle surface through electrostatic binding to a surface coated polyethylenimine/PEG co-polymer (Figure 27). IV administration of these particles into orthotopic KPC bearing mice resulted in a dramatic improvement in the tumor uptake of second wave liposome therapy, which delivers Gemcitabine. The improved liposomal biodistribution was associated with significant tumor shrinkage compared to monotherapy. This approach illustrates the potential utility of therapeutic waves of therapy, which can be developed in multiple ways, including delivery of the ICD stimulus prior to inhibitors of the PD1/PD-L1, A2AR, IDO-1 or CXCR4 pathways. Hypothetically, the second wave of therapy could involve a number of drug combinations that target one or more barriers in the TIME.
A further approach to vascular normalization is the use of dexamethasone to reduce interstitial fluid pressure, as demonstrated for increased Docetaxel uptake at the BC tumor site.265–267 Vascular normalization can also be accomplished with VEGFA blocking antibodies or inhibitors, with the caveat that this strategy may be better suited for nano-formulations < 40 nm in size.268 Nonetheless, the VEGFA antibody, bevacizumab, has been demonstrated to improve immunotherapy outcomes and survival in clinical trials for hepatocellular or non-small cell lung cancers in combination with anti–PD-L1 (atezolizumab) and chemotherapy.269–270 This dovetails with the demonstration that checkpoint blockers also contribute to vascular normalization. Meprkis et al. have developed a mathematical framework for predicting how vascular and stromal normalization strategies can be combined to improve immunotherapy efficacy.271
While a lot of emphasis is placed on DDS delivery to the primary cancer site, it is important to consider that beyond the TME, the tumor-bearing state also impacts the peripheral immune compartment in cancer patients.272 This reflects the importance of the bone marrow, secondary lymphoid tissues and regional lymph nodes in the cancer-immunity cycle. Accordingly, it is important to consider the functional characteristics of the immune system in tumor-bearing subjects when planning cancer immunotherapy, including the contribution of dendritic cells recruited from the bone marrow, and the impact of their reduced recruitment may have on proliferation and differentiation of CD8+ T-cells in PDAC and TNBC landscapes. We have already referred to the utility of FLT3L as an adjunct therapy to release DC precursors from the bone marrow,60 including the possibility to use anti-CD40 mAb for licensing of newly recruited APC.
It is also important to consider that a significant number of the cells being targeted by immunotherapy are located in secondary lymphoid organs and regional lymph nodes.273–274 Even though tumor-draining lymph nodes are traditionally viewed as important sites of immune activation due to the collection of tumor antigens and antigen-specific T-cells, it is often overlooked that lymph nodes also collect immune suppressive cytokines, Tregs and metastatic cancer cells, which could promote an immune tolerant rather than an immune defense environment. Thus, the delivery of immune stimuli to regional lymph nodes is an important area of focus for immunotherapy.273–274 From a nano-DDS perspective, it is important to consider that physicochemical properties that promote particle accumulation at the primary tumor site (e.g., stealth surfaces leading to longer circulatory half-lives and limited cellular uptake) do not automatically favor lymph node accumulation.274–276 A number of strategies can be implemented to improve lymph node targeting by LB-coated carriers, including inclusion of TLR7 (e.g., Telratolimod) or TLR9 agonists (e.g., CpG) in the LB.277–278 We are currently focusing on the impact of TLR7 agonist inclusion in silicasome and liposome bilayers in animal models. Another approach is the use of a lipophilic prodrug strategy to improve particle uptake by intestinal lymph nodes, including the consideration that longer lipid chains are more effective for lymphatic transport.274, 277 For this purpose, Porter et al. developed a range of lipophilic prodrugs with oil-water partition coefficients (LogDs) >5 and high (>50 mg/g) triglyceride content to promote lymphatic transport.279–280 This design feature is easily accommodated in our prodrug synthesis strategies (Figures 14–16). A complementary strategy is to make use of cationic lipid nanoparticles (LNP), as described below.
While our multifunctional liposome and silicasome particle platforms have been instrumental in devising combination immunotherapy strategies, this does not exclude the use of other particle platforms that can be used as adjunct carriers to promote the use of an endogenous vaccination approach. This includes the use of LNP that can be endowed with mRNA constructs that encode antigens and epitopes, and can be adapted for delivery to the primary tumor site, lymph nodes and spleen in combination with immune adjuvants.273, 281 LNPs are recognized as effective delivery systems for nucleic acids, which are incorporated by ionizable lipids, with the assistance of helper phospholipids, cholesterol, and PEGylated lipids.273, 281 This technology is ideal for generating antigen-specific T-cells that recognize major tumor specific antigens and major antigenic epitopes. This strategy could be particularly effective to boost antigen specific responses to TSA at the level of lymph nodes and spleen, thereby contributing to the cancer immunity cycle when the activated T-cells are returned to the primary tumor site. This holds the promise of improving the robustness of the T-cell response in the primary cancer site, where the endogenous vaccination response contribute to supply of neoantigens. PDAC provides an excellent example for the use of peptide-based vaccines to generate immune responses against tumor-specific antigens (TSA), including KRAS, telomerase, and gastrin.42, 282 Kras is mutated in >90% of PDAC patients, where the mutations occur at 3 major sites (G12, G13 and Q61), with G12D contributing to 45% of all Kras mutations.283 While peptides representing these mutational sites have been used successfully to generate CTL responses in human PDAC tumors, a recently expressed expert opinion strongly advocates that vaccination responses can benefit from combination therapy.284 This strategy could be employed for TSA and tumor-associated antigens (e.g., Muc1, mesothelin, and CEA) in PDAC and TNBC.42, 285
Our silicasome platform should also consider the contribution of physical design properties on the chemoimmunotherapy response. This includes consideration of particle morphology, as previously demonstrated by the use of different MSNP aspect ratios to affect particle uptake by GTPase-dependent uptake mechanisms in cancer cells.286 Since the importance of particle morphology, size, shape, and surface functionalization on cellular uptake, adjuvant effects and the immune reactivity have been covered extensively in the literature, we will not further labor the details here except to comment that these design features provide additional design features to impact nano-enabled vaccination approaches in future.287–288
Supplementary Material
Acknowledgment
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Numbers 5R01CA247666, U01CA198846 and 1R01CA247666–01A1. We thank the Translational Pathology Core Laboratory (TPCL) at UCLA Jonsson Comprehensive Cancer Center for IHC/mIHC staining, the Electron Imaging Center for Nanomachines the use of EM instruments at, the Molecular Instrumentation Center for NMRs and Mass Spectrometry, the CNSI Advanced Light Microscopy/Spectroscopy Shared Facility for confocal fluorescent microscopy. We want to acknowledge the research contributions of Dr. Sean Allen to the data in Figures 21–23 and S24–S29, in addition to the cited published contributions.179. We also want to acknowledge the collaborative contributions or Prof. Huan Meng, Prof. Jeffrey Zink, Prof. Tim Donahue, and many others.
Abbreviation Definition
- A2AR
Adenosine A2A Receptor
- AhR
Aryl Hydrocarbon Receptor
- AKT
Ak Strain Transforming Factor
- AML
Acute Myeloid Leukemia
- APC
Antigen Presenting Cell
- API
Active Pharmaceutical Ingredients
- ATP
Adenosine Triphosphate
- AUC
Area Under the Curve
- αSMA
α-Smooth Muscle Actin
- BC
Breast Cancer
- CAFs
Cancer-associated Fibroblasts
- CAPIR
Circulation, Accumulation, Penetration, Internalization, and Release
- CCR
C-C Motif Chemokine Receptor
- CD
Cluster of Differentiation
- CDA
Cytidine Deaminase
- cDC
Conventional (CD103+) Dendritic Cells
- CDN
Cyclic Dinucleotides
- CDKN2A
Cyclin-dependent Kinase Inhibitor 2A
- Ce6
Chlorin e6
- CHEMS
Cholesteryl Hemisuccinate
- Chol
Cholesterol
- Cpp
Critical Packing Parameters
- CRT
Calreticulin
- cRNA
Complementary RNA
- CSF-1
Colony-stimulating Factor 1
- CSF1R
Colony-stimulating Factor 1 Receptor
- CTLs
Cytotoxic T Lymphocytes
- CTLA-4
Cytotoxic T-lymphocyte-associated protein 4
- CXCL
C-X-C Motif Chemokine Ligand
- CXCR
C-X-C Chemokine Receptor
- CyTOF
Cytometry by Time of Flight
- DACH-Pt
Dichloro(1,2-Diaminocyclohexane)Platinum(II)
- DC
Dendritic Cell
- DDS
Drug Delivery System
- DGS
1,2-dioleoyl-sn-glycero-3-succinate (18:1) 1,2-dipalmitoyl-sn-glycero-3-succinate (16:0)
- DOX
Doxorubicin
- EC50
Half Maximal Effective Concentration
- EMT
Epithelial-to-Mesenchymal Transition
- EPR
Enhanced Permeation and Retention
- ER
Estrogen Receptor
- ERK
Extracellular Signal-Regulated Kinase
- FoxP3
Forkhead box P3 protein
- FI
Fully Inflamed
- FLT3
Fms Related Receptor Tyrosine Kinase 3
- FLT3L
FMS-like Tyrosine Kinase 3 Ligand
- GEMM
Genetically Engineered Mouse Models
- GEM
Gemcitabine
- GS
Glycerol-3-succinate 3
- GSK3
Glycogen Synthase Kinase-3
- HER2
Human Epidermal Growth Factor Receptor 2
- HMGB1
High mobility group box 1
- IC50
Half Maximal Inhibitory Concentration
- ICD
Immunogenic Cell Death
- ICI
Immune Checkpoint Inhibitors
- ID
Immune Desert
- IDO-1
Indoleamine-pyrrole 2,3-dioxygenase
- IFN-γ
Interferon Gamma
- IHC/mIHC
Immunohistochemistry/multiplex IHC
- IL-
Interleukin
- IND
Indoximod (1-methyl-D-tryptophan)
- iRGD
A cyclic peptide (sequence: CRGDKGPDC)
- IRIN
Irinotecan
- IV
Intravenous
- JAK
Janus Kinases
- KRAS
Kirsten Rat Sarcoma Viral Oncogene Homolog
- KPC
KrasG12D/+; LSL-Trp53R172H/+; Pdx1-Cre
- KYN
Kynurenine
- LAG-3
Lymphocyte-activation gene 3
- LB
Lipid Bilayer
- LC
Loading Capacity
- LLC
Louis Lung Carcinoma
- LNP
Lipid Nanoparticles
- LSECs
Liver Sinusoidal Endothelial Cells
- MDSCs
Myeloid-derived Suppressor Cells
- MDR
Multidrug Resistance
- MEK
Mitogen-activated ERK Kinase
- MMTV-PyVT (PyMT)
Mouse Mammary Tumor Virus-Polyoma Virus Middle Tumor-antigen MSNP Mesoporous Silica Nanoparticle
- MR
Margin Restricted
- MS
Mass Spectroscopy
- MTD
Maximum Tolerated Dose
- MTO
Mitoxantrone
- mTOR
Mammalian Target of Rapamycin
- mTORC1
Mammalian Target of Rapamycin Complex 1
- NK
Natural Killer Cells
- NMR
Nuclear Magnetic Resonance
- NRP-1
Neuropilin-1
- OXA
Oxaliplatin
- PC
Phosphatidylcholine
- pCR
Pathological Complete Response
- PD-1
Programmed cell Death protein 1
- PDT
Photodynamic Therapy
- PDAC
Pancreatic Ductal Adenocarcinoma
- PD-L1
Programmed Death-ligand 1
- PE
Phosphoethanolamine
- PEG
Polyethylene Glycol
- PG
Phosphor-(1’-rac-glycerol)
- Pgp
P-glycoprotein
- PI3K
Phosphatidylinositol 3-kinase
- PK
Pharmacokinetics
- PLD
Pegylated liposomal Doxorubicin
- PLGA
Poly Lactic-co-glycolic Acid
- PR
Progesterone Receptor
- PS
Phosphor-L-serine
- PTX
Paclitaxel
- SDF-1
Stromal Cell-derived Factor 1
- Seq
Sequencing
- SMAD4
Mothers Against Decapentaplegic Homolog 4
- SMI
Small Molecule Inhibitors
- SR
Stromal Restricted
- STAT
Signal Transducer and Activator of Transcription Protein
- STING
Stimulator of Interferon Genes
- TAM
Tumor-associated Macrophages
- TCR
T-cell Antigen Receptor
- TEA8SOS
triethylammonium Sucrose Octasulfate
- TGFβ
Transforming growth factor beta
- TME
Tumor Microenvironment
- TIL
Tumor-infiltrating T Lymphocytes
- TIM-3
T-cell Immunoglobulin and Mucin Domain-containing Protein 3
- TIME
Tumor Immune Microenvironment
- TLR
Toll-like Receptor
- TNBC
Triple-negative Breast Cancer
- TP53
Tumor Protein p53
- Treg
Regulatory T-cell
- TRP
Tryptophan
- TSA
Tumor Specific Antigen
- VEGFA
Vascular Endothelial Growth Factor A
Footnotes
Competing Financial Interests
Andre E. Nel is co-founder and equity holder in Westwood Biosciences Inc. and NAMMI Therapeutics. Nel also serves on the Board for Westwood Biosciences Inc. The remaining authors declare no conflicts of interest.
Supporting Information Available
Additional figures and results, as described in the text, are available free of charge via the Internet at http://pubs.acs.org
References
- 1.Allott EH; Dean K; Robson T; Meaney C, Characterizing and Exploiting Tumor Microenvironments to Optimize Treatment Outcomes. Cancers (Basel) 2021, 13 (22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Picard E; Verschoor CP; Ma GW; Pawelec G, Relationships Between Immune Landscapes, Genetic Subtypes and Responses to Immunotherapy in Colorectal Cancer. Front. Immunol. 2020, 11, 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang J; Zhang C, Regulation of cancer-immunity cycle and tumor microenvironment by nanobiomaterials to enhance tumor immunotherapy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020, 12, e1612. [DOI] [PubMed] [Google Scholar]
- 4.Rodallec A; Sicard G; Fanciullino R; Benzekry S; Lacarelle B; Milano G; Ciccolini J, Turning Cold Tumors into Hot Tumors: Harnessing the Potential of Tumor Immunity Using Nanoparticles. Expert. Opin. Drug Metab. Toxicol. 2018, 14, 1139–1147. [DOI] [PubMed] [Google Scholar]
- 5.Petitprez F; Meylan M; de Reynies A; Sautes-Fridman C; Fridman WH, The Tumor Microenvironment in the Response to Immune Checkpoint Blockade Therapies. Front. Immunol. 2020, 11, 784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sevenich L, Turning “Cold” Into “Hot” Tumors-Opportunities and Challenges for Radio-Immunotherapy Against Primary and Metastatic Brain Cancers. Front Oncol 2019, 9, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Michel LL; von Au A; Mavratzas A; Smetanay K; Schutz F; Schneeweiss A, Immune Checkpoint Blockade in Patients with Triple-Negative Breast Cancer. Target Oncol. 2020, 15, 415–428. [DOI] [PubMed] [Google Scholar]
- 8.Gruosso T; Gigoux M; Manem VSK; Bertos N; Zuo D; Perlitch I; Saleh SMI; Zhao H; Souleimanova M; Johnson RM; Monette A; Ramos VM; Hallett MT; Stagg J; Lapointe R; Omeroglu A; Meterissian S; Buisseret L; Van den Eynden G et al. Spatially Distinct Tumor Immune Microenvironments Stratify Triple-Negative Breast Cancers. J. Clin. Invest. 2019, 129, 1785–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hegde PS; Karanikas V; Evers S, The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [DOI] [PubMed] [Google Scholar]
- 10.Leone RD; Emens LA, Targeting Adenosine for Cancer Immunotherapy. J. Immunother. Cancer 2018, 6, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pardoll DM, The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khair DO; Bax HJ; Mele S; Crescioli S; Pellizzari G; Khiabany A; Nakamura M; Harris RJ; French E; Hoffmann RM; Williams IP; Cheung A; Thair B; Beales CT; Touizer E; Signell AW; Tasnova NL; Spicer JF; Josephs DH; Geh JL; MacKenzie Ross A; Healy C; Papa S; Lacy KE; Karagiannis SN, Combining Immune Checkpoint Inhibitors: Established and Emerging Targets and Strategies to Improve Outcomes in Melanoma. Front. Immunol. 2019, 10:453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mellor AL; Keskin DB; Johnson T; Chandler P; Munn DH, Cells Expressing Indoleamine 2,3-Dioxygenase Inhibit T Cell Responses. J. Immunol. 2002, 168, 3771–3776. [DOI] [PubMed] [Google Scholar]
- 14.Munn DH; Mellor AL, IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends in Immunology 2016, 37 (3), 193–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vannucci L, Stroma as an Active Player in the Development of the Tumor Microenvironment. Cancer Microenvironment 2015, 8 (3), 159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oliver AJ; Lau PKH; Unsworth AS; Loi S; Darcy PK; Kershaw MH; Slaney CY, Tissue-Dependent Tumor Microenvironments and Their Impact on Immunotherapy Responses. Front. Immunol. 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vitale I; Shema E; Loi S; Galluzzi L, Intratumoral Heterogeneity in Cancer Progression and Response to Immunotherapy. Nat. Med. 2021, 27 (2), 212–224. [DOI] [PubMed] [Google Scholar]
- 18.Ho WJ; Jaffee EM; Zheng L, The Tumour Microenvironment in Pancreatic cancer — Clinical Challenges and Opportunities. Nat. Rev. Clin. Oncol. 2020, 17 (9), 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korc M, Pancreatic Cancer-associated Stroma Production. Am. J. Surg. 2007, 194, S84–S86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stark AP; Sacks GD; Rochefort MM; Donahue TR; Reber HA; Tomlinson JS; Dawson DW; Eibl G; Hines OJ, Long-term Survival in Patients with Pancreatic Ductal Adenocarcinoma. Surgery 2016, 159, 1520–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berger ER; Park T; Saridakis A; Golshan M; Greenup RA; Ahuja N, Immunotherapy Treatment for Triple Negative Breast Cancer. Pharmaceuticals 2021, 14, 763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo F; Wang Y; Liu J; Mok SC; Xue F; Zhang W, CXCL12/CXCR4: a Symbiotic Bridge Linking Cancer Cells and Their Stromal Neighbors in Oncogenic Communication Networks. Oncogene 2016, 35, 816–826. [DOI] [PubMed] [Google Scholar]
- 23.Joyce JA; Fearon DT, T Cell Exclusion, Immune Privilege, and the Tumor Microenvironment. Science 2015, 348, 74–80. [DOI] [PubMed] [Google Scholar]
- 24.Kalbasi A; Ribas A, Tumour-intrinsic Resistance to Immune Checkpoint Blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X; Li X; Wei X; Jiang H; Lan C; Yang S; Wang H; Yang Y; Tian C; Xu Z; Zhang J; Hao J; Ren H, PD-L1 is a Direct Target of Cancer-FOXP3 in Pancreatic Ductal Adenocarcinoma (PDAC), and Combined Immunotherapy with Antibodies Against PD-L1 and CCL5 is Effective in the Treatment of PDAC. Signal Transduct. Target. Ther. 2020, 5, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meyer MA; Baer JM; Knolhoff BL; Nywening TM; Panni RZ; Su X; Weilbaecher KN; Hawkins WG; Ma C; Fields RC; Linehan DC; Challen GA; Faccio R; Aft RL; DeNardo DG, Breast and Pancreatic Cancer Interrupt IRF8-dependent Dendritic Cell Development to Overcome Immune Surveillance. Nat. Commun. 2018, 9, 1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hildner K; Edelson BT; Purtha WE; Diamond M; Matsushita H; Kohyama M; Calderon B; Schraml BU; Unanue ER; Diamond MS; Schreiber RD; Murphy TL; Murphy KM, Batf3 Deficiency Reveals a Critical Role for CD8alpha+ Dendritic Cells in Cytotoxic T Cell Immunity. Science 2008, 322, 1097–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Munn DH; Mellor AL, Indoleamine 2,3 Dioxygenase and Metabolic Control of Immune Responses. Trends in Immunol. 2013, 34, 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith C; Chang MY; Parker KH; Beury DW; DuHadaway JB; Flick HE; Boulden J; Sutanto-Ward E; Soler AP; Laury-Kleintop LD; Mandik-Nayak L; Metz R; Ostrand-Rosenberg S; Prendergast GC; Muller AJ, IDO Is a Nodal Pathogenic Driver of Lung Cancer and Metastasis Development. Cancer Discov. 2012, 2, 722–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Regel I; Mayerle J; Ujjwal Mukund M, Current Strategies and Future Perspectives for Precision Medicine in Pancreatic Cancer. Cancers 2020, 12, 1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karamitopoulou E, Tumour Microenvironment of Pancreatic Cancer: Immune Landscape is Dictated by Molecular and Histopathological Features. Br. J. Cancer 2019, 121, 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moffitt RA; Marayati R; Flate EL; Volmar KE; Loeza SGH; Hoadley KA; Rashid NU; Williams LA; Eaton SC; Chung AH; Smyla JK; Anderson JM; Kim HJ; Bentrem DJ; Talamonti MS; Iacobuzio-Donahue CA; Hollingsworth MA; Yeh JJ, Virtual Microdissection Identifies Distinct Tumor- and Stroma-specific Subtypes of Pancreatic Ductal Adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Puleo F; Nicolle R; Blum Y; Cros J; Marisa L; Demetter P; Quertinmont E; Svrcek M; Elarouci N; Iovanna J; Franchimont D; Verset L; Galdon MG; Devière J; de Reyniès A; Laurent-Puig P; Van Laethem J-L; Bachet J-B; Maréchal R, Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018, 155, 1999–2013.e3. [DOI] [PubMed] [Google Scholar]
- 34.Bailey P; Chang DK; Nones K; Johns AL; Patch A-M; Gingras M-C; Miller DK; Christ AN; Bruxner TJC; Quinn MC; Nourse C; Murtaugh LC; Harliwong I; Idrisoglu S; Manning S; Nourbakhsh E; Wani S; Fink L; Holmes O; Chin V et al. , Australian Pancreatic Cancer Genome, I., Genomic Analyses Identify Molecular Subtypes of Pancreatic Cancer. Nature 2016, 531, 47–52. [DOI] [PubMed] [Google Scholar]
- 35.Waddell N; Pajic M; Patch A-M; Chang DK; Kassahn KS; Bailey P; Johns AL; Miller D; Nones K; Quek K; Quinn MCJ; Robertson AJ; Fadlullah MZH; Bruxner TJC; Christ AN; Harliwong I; Idrisoglu S; Manning S; Nourse C; Nourbakhsh E et al. , Whole Genomes Redefine the Mutational Landscape of Pancreatic Cancer. Nature 2015, 518, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chuah S; Chew V, High-dimensional Immune-profiling in Cancer: Implications for Immunotherapy. Journal for ImmunoTherapy of Cancer 2020, 8, e000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carstens JL; Correa de Sampaio P; Yang D; Barua S; Wang H; Rao A; Allison JP; LeBleu VS; Kalluri R, Spatial Computation of Intratumoral T cells Correlates with Survival of Patients with Pancreatic Cancer. Nat. Commun. 2017, 8, 15095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Masugi Y; Abe T; Ueno A; Fujii-Nishimura Y; Ojima H; Endo Y; Fujita Y; Kitago M; Shinoda M; Kitagawa Y; Sakamoto M, Characterization of Spatial Distribution of Tumor-infiltrating CD8+ T Cells Refines Their Prognostic Utility for Pancreatic Cancer Survival. Modern Pathology 2019, 32 (10), 1495–1507. [DOI] [PubMed] [Google Scholar]
- 39.Seo YD; Jiang X; Sullivan KM; Jalikis FG; Smythe KS; Abbasi A; Vignali M; Park JO; Daniel SK; Pollack SM; Kim TS; Yeung R; Crispe IN; Pierce RH; Robins H; Pillarisetty VG, Mobilization of CD8+ T Cells via CXCR4 Blockade Facilitates PD-1 Checkpoint Therapy in Human Pancreatic Cancer. Clin. Cancer Res. 2019, 25, 3934–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steele NG; Carpenter ES; Kemp SB; Sirihorachai VR; The S; Delrosario L; Lazarus J; Amir E.-a. D.; Gunchick V; Espinoza C; Bell S; Harris L; Lima F; Irizarry-Negron V; Paglia D; Macchia J; Chu AKY; Schofield H; Wamsteker E-J et al. , Multimodal Mapping of the Tumor and Peripheral Blood Immune Landscape in Human Pancreatic Cancer. Nat. Cancer 2020, 1, 1097–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tan WCC; Nerurkar SN; Cai HY; Ng HHM; Wu D; Wee YTF; Lim JCT; Yeong J; Lim TKH, Overview of Multiplex Immunohistochemistry/Immunofluorescence Techniques in the Era of Cancer Immunotherapy. Cancer Communications 2020, 40, 135–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu AA; Jaffee E; Lee V, Current Status of Immunotherapies for Treating Pancreatic Cancer. Curr. Oncol. Rep. 2019, 21, 60. [DOI] [PubMed] [Google Scholar]
- 43.Collisson EA; Sadanandam A; Olson P; Gibb WJ; Truitt M; Gu S; Cooc J; Weinkle J; Kim GE; Jakkula L; Feiler HS; Ko AH; Olshen AB; Danenberg KL; Tempero MA; Spellman PT; Hanahan D; Gray JW, Subtypes of Pancreatic Ductal Adenocarcinoma and Their Differing Responses to Therapy. Nat. Med. 2011, 17, 500–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wartenberg M; Cibin S; Zlobec I; Vassella E; Eppenberger-Castori S; Terracciano L; Eichmann MD; Worni M; Gloor B; Perren A; Karamitopoulou E, Integrated Genomic and Immunophenotypic Classification of Pancreatic Cancer Reveals Three Distinct Subtypes with Prognostic/Predictive Significance. Clin. Cancer Res. 2018, 24, 4444–4454. [DOI] [PubMed] [Google Scholar]
- 45.Spranger S; Gajewski TF, Impact of Oncogenic Pathways on Evasion of Antitumour Immune Responses. Nat. Rev. Cancer 2018, 18, 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pham TND; Shields MA; Spaulding C; Principe DR; Li B; Underwood PW; Trevino JG; Bentrem DJ; Munshi HG, Preclinical Models of Pancreatic Ductal Adenocarcinoma and Their Utility in Immunotherapy Studies. Cancers 2021, 13 (3), 440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kong K; Guo M; Liu Y; Zheng J, Progress in Animal Models of Pancreatic Ductal Adenocarcinoma. J. Cancer 2020, 11, 1555–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muzumdar MD; Dorans KJ; Chung KM; Robbins R; Tammela T; Gocheva V; Li CM-C; Jacks T, Clonal Dynamics Following p53 Loss of Heterozygosity in Kras-driven Cancers. Nat. Commun. 2016, 7, 12685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hingorani SR; Wang L; Multani AS; Combs C; Deramaudt TB; Hruban RH; Rustgi AK; Chang S; Tuveson DA, Trp53R172H and KrasG12D Cooperate to Promote Chromosomal Instability and Widely Metastatic Pancreatic Ductal Adenocarcinoma in Mice. Cancer Cell 2005, 7, 469–483. [DOI] [PubMed] [Google Scholar]
- 50.Li J; Pan J; Zhu X; Su Y; Bao L; Qiu S; Zou C; Cai Y; Wu J; Tham IWK, Recombinant Adenovirus-p53 (Gendicine) Sensitizes a Pancreatic Carcinoma Cell Line to Radiation. Chin. J. Cancer Res. 2013, 25, 715–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aiello NM; Maddipati R; Norgard RJ; Balli D; Li J; Yuan S; Yamazoe T; Black T; Sahmoud A; Furth EE; Bar-Sagi D; Stanger BZ, EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev.Cell 2018, 45, 681–695.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rhim Andrew D.; Mirek Emily T.; Aiello Nicole M.; Maitra A; Bailey Jennifer M.; McAllister F; Reichert M; Beatty Gregory L.; Rustgi Anil K.; Vonderheide Robert H.; Leach Steven D.; Stanger Ben Z., EMT and Dissemination Precede Pancreatic Tumor Formation. Cell 2012, 148, 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Olive KP; Tuveson DA, The Use of Targeted Mouse Models for Preclinical Testing of Novel Cancer Therapeutics. Clin. Cancer Res. 2006, 12, 5277–5287. [DOI] [PubMed] [Google Scholar]
- 54.Lee JW; Komar CA; Bengsch F; Graham K; Beatty GL, Genetically Engineered Mouse Models of Pancreatic Cancer: The KPC Model (LSL-KrasG12D/+;LSL-Trp53R172H/+;Pdx-1-Cre), Its Variants, and Their Application in Immuno-oncology Drug Discovery. Curr. Protoc. Pharmacol. 2016, 73, 14.39.1–14.39.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Winograd R; Byrne KT; Evans RA; Odorizzi PM; Meyer ARL; Bajor DL; Clendenin C; Stanger BZ; Furth EE; Wherry EJ; Vonderheide RH, Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol. Res. 2015, 3, 399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li J; Byrne KT; Yan F; Yamazoe T; Chen Z; Baslan T; Richman LP; Lin JH; Sun YH; Rech AJ; Balli D; Hay CA; Sela Y; Merrell AJ; Liudahl SM; Gordon N; Norgard RJ; Yuan S; Yu S; Chao T; Ye S et al. , Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018, 49, 178–193.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shibuya KC; Goel VK; Xiong W; Sham JG; Pollack SM; Leahy AM; Whiting SH; Yeh MM; Yee C; Riddell SR; Pillarisetty VG, Pancreatic Ductal Adenocarcinoma Contains an Effector and Regulatory Immune Cell Infiltrate that Is Altered by Multimodal Neoadjuvant Treatment. PLOS ONE 2014, 9, e96565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Elgueta R; Benson MJ; De Vries VC; Wasiuk A; Guo Y; Noelle RJ, Molecular Mechanism and Function of CD40/CD40L Engagement in the Immune System. Immunological Reviews 2009, 229, 152–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beatty GL; Chiorean EG; Fishman MP; Saboury B; Teitelbaum UR; Sun W; Huhn RD; Song W; Li D; Sharp LL; Torigian DA; O’Dwyer PJ; Vonderheide RH, CD40 Agonists Alter Tumor Stroma and Show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science 2011, 331, 1612–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cueto FJ; Sancho D, The Flt3L/Flt3 Axis in Dendritic Cell Biology and Cancer Immunotherapy. Cancers 2021, 13, 1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hegde S; Krisnawan VE; Herzog BH; Zuo C; Breden MA; Knolhoff BL; Hogg GD; Tang JP; Baer JM; Mpoy C; Lee KB; Alexander KA; Rogers BE; Murphy KM; Hawkins WG; Fields RC; DeSelm CJ; Schwarz JK; DeNardo DG, Dendritic Cell Paucity Leads to Dysfunctional Immune Surveillance in Pancreatic Cancer. Cancer Cell 2020, 37, 289–307.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Feig C; Jones JO; Kraman M; Wells RJB; Deonarine A; Chan DS; Connell CM; Roberts EW; Zhao Q; Caballero OL; Teichmann SA; Janowitz T; Jodrell DI; Tuveson DA; Fearon DT, Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 20212–20217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Orimo A; Gupta PB; Sgroi DC; Arenzana-Seisdedos F; Delaunay T; Naeem R; Carey VJ; Richardson AL; Weinberg RA, Stromal Fibroblasts Present in Invasive Human Breast Carcinomas Promote Tumor Growth and Angiogenesis through Elevated SDF-1/CXCL12 Secretion. Cell 2005, 121, 335–348. [DOI] [PubMed] [Google Scholar]
- 64.Burger JA; Kipps TJ, CXCR4: a Key Receptor in the Crosstalk Between Tumor Cells and Their Microenvironment. Blood 2006, 107, 1761–1767. [DOI] [PubMed] [Google Scholar]
- 65.Bockorny B; Semenisty V; Macarulla T; Borazanci E; Wolpin BM; Stemmer SM; Golan T; Geva R; Borad MJ; Pedersen KS; Park JO; Ramirez RA; Abad DG; Feliu J; Muñoz A; Ponz-Sarvise M; Peled A; Lustig TM; Bohana-Kashtan O; Shaw SM et al. , BL-8040, a CXCR4 Antagonist, in Combination with Pembrolizumab and Chemotherapy for Pancreatic Cancer: the COMBAT trial. Nat. Med. 2020, 26, 878–885. [DOI] [PubMed] [Google Scholar]
- 66.Principe DR; Park A; Dorman MJ; Kumar S; Viswakarma N; Rubin J; Torres C; McKinney R; Munshi HG; Grippo PJ; Rana A, TGFβ Blockade Augments PD-1 Inhibition to Promote T-Cell–Mediated Regression of Pancreatic Cancer. Mol. Cancer Ther. 2019, 18, 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Principe DR; Narbutis M; Kumar S; Park A; Viswakarma N; Dorman MJ; Kamath SD; Grippo PJ; Fishel ML; Hwang RF; Thummuri D; Underwood PW; Munshi HG; Trevino JG; Rana A, Long-Term Gemcitabine Treatment Reshapes the Pancreatic Tumor Microenvironment and Sensitizes Murine Carcinoma to Combination Immunotherapy. Cancer Res. 2020, 80, 3101–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Prakash O; Hossain F; Danos D; Lassak A; Scribner R; Miele L, Racial Disparities in Triple Negative Breast Cancer: A Review of the Role of Biologic and Non-biologic Factors. Front. Public Health 2020, 8: 576964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scott LC; Mobley LR; Kuo T-M; Il’yasova D, Update on triple-negative breast cancer disparities for the United States: A population-based study from the United States Cancer Statistics database, 2010 through 2014. Cancer 2019, 125, 3412–3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lehmann BD; Bauer JA; Chen X; Sanders ME; Chakravarthy AB; Shyr Y; Pietenpol JA, Identification of Human Triple-negative Breast Cancer Subtypes and Preclinical Models for Selection of Targeted Therapies. J. Clin. Investig. 2011, 121, 2750–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Masuda H; Baggerly KA; Wang Y; Zhang Y; Gonzalez-Angulo AM; Meric-Bernstam F; Valero V; Lehmann BD; Pietenpol JA; Hortobagyi GN; Symmans WF; Ueno NT, Differential Response to Neoadjuvant Chemotherapy Among 7 Triple-Negative Breast Cancer Molecular Subtypes. Clin. Cancer Res. 2013, 19 (19), 5533–5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marra A; Trapani D; Viale G; Criscitiello C; Curigliano G, Practical Classification of Triple-negative Breast Cancer: Intratumoral Heterogeneity, Mechanisms of Drug Resistance, and Novel Therapies. npj Breast Cancer 2020, 6, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Burstein MD; Tsimelzon A; Poage GM; Covington KR; Contreras A; Fuqua SAW; Savage MI; Osborne CK; Hilsenbeck SG; Chang JC; Mills GB; Lau CC; Brown PH, Comprehensive Genomic Analysis Identifies Novel Subtypes and Targets of Triple-Negative Breast Cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Karaayvaz M; Cristea S; Gillespie SM; Patel AP; Mylvaganam R; Luo CC; Specht MC; Bernstein BE; Michor F; Ellisen LW, Unravelling Subclonal Heterogeneity and Aggressive Disease States in TNBC Through Single-cell RNA-seq. Nat. Commun. 2018, 9, 3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jiang Y-Z; Ma D; Suo C; Shi J; Xue M; Hu X; Xiao Y; Yu K-D; Liu Y-R; Yu Y; Zheng Y; Li X; Zhang C; Hu P; Zhang J; Hua Q; Zhang J; Hou W; Ren L; Bao D et al. , Genomic and Transcriptomic Landscape of Triple-Negative Breast Cancers: Subtypes and Treatment Strategies. Cancer Cell 2019, 35, 428–440.e5. [DOI] [PubMed] [Google Scholar]
- 76.Kim C; Gao R; Sei E; Brandt R; Hartman J; Hatschek T; Crosetto N; Foukakis T; Navin NE, Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hammerl D; Martens JWM; Timmermans M; Smid M; Trapman-Jansen AM; Foekens R; Isaeva OI; Voorwerk L; Balcioglu HE; Wijers R; Nederlof I; Salgado R; Horlings H; Kok M; Debets R, Spatial Immunophenotypes Predict Response to Anti-PD1 Treatment and Capture Distinct Paths of T cell Evasion in Triple Negative Breast Cancer. Nat. Commun. 2021, 12, 5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Voorwerk L; Slagter M; Horlings HM; Sikorska K; van de Vijver KK; de Maaker M; Nederlof I; Kluin RJC; Warren S; Ong S; Wiersma TG; Russell NS; Lalezari F; Schouten PC; Bakker NAM; Ketelaars SLC; Peters D; Lange CAH; van Werkhoven E; van Tinteren H et al. , Immune Induction Strategies in Metastatic Triple-negative Breast Cancer to Enhance the Sensitivity to PD-1 Blockade: the TONIC Trial. Nat. Med. 2019, 25, 920–928. [DOI] [PubMed] [Google Scholar]
- 79.Fucikova J; Kepp O; Kasikova L; Petroni G; Yamazaki T; Liu P; Zhao L; Spisek R; Kroemer G; Galluzzi L, Detection of Immunogenic Cell Death and Its Relevance for Cancer Therapy. Cell Death Dis. 2020, 11, 1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Janicka M; Gubernator J, Use of Nanotechnology for Improved Pharmacokinetics and Activity of Immunogenic Cell Death Inducers Used in Cancer Chemotherapy. Expert Opin. Drug Deliv. 2017, 14, 1059–1075. [DOI] [PubMed] [Google Scholar]
- 81.Zhong W; Myers JS; Wang F; Wang K; Lucas J; Rosfjord E; Lucas J; Hooper AT; Yang S; Lemon LA; Guffroy M; May C; Bienkowska JR; Rejto PA, Comparison of the Molecular and Cellular Phenotypes of Common Mouse Syngeneic Models with Human Tumors. BMC Genomics 2020, 21, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Olson B; Li Y; Lin Y; Liu ET; Patnaik A, Mouse Models for Cancer Immunotherapy Research. Cancer Discov. 2018, 8, 1358–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Holen I; Speirs V; Morrissey B; Blyth K, In Vivo Models in Breast Cancer Research: Progress, Challenges and Future Directions. Disease Models & Mechanisms 2017, 10, 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Paller CJ; Huang EP; Luechtefeld T; Massett HA; Williams CC; Zhao J; Gravell AE; Tamashiro T; Reeves SA; Rosner GL; Carducci MA; Rubinstein L; Ivy SP, Factors Affecting Combination Trial Success (FACTS): Investigator Survey Results on Early-Phase Combination Trials. Front. Med. 2019, 6:!22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Maitland ML; Hudoba C; Snider KL; Ratain MJ, Analysis of the Yield of Phase II Combination Therapy Trials in Medical Oncology. Clin. Cancer Res. 2010, 16, 5296–5302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Riviere MK; Le Tourneau C; Paoletti X; Dubois F; Zohar S, Designs of Drug-combination Phase I Trials in Oncology: a Systematic Review of the Literature. Ann. Oncol. 2015, 26, 669–674. [DOI] [PubMed] [Google Scholar]
- 87.Ma L; Kohli M; Smith A, Nanoparticles for Combination Drug Therapy. ACS Nano 2013, 7, 9518–9525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Frampton JE, Liposomal Irinotecan: A Review in Metastatic Pancreatic Adenocarcinoma. Drugs 2020, 80, 1007–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Caputo D; Pozzi D; Farolfi T; Passa R; Coppola R; Caracciolo G, Nanotechnology and Pancreatic Cancer Management: State of the Art and Further Perspectives. World J. Gastrointest. Oncol. 2021, 13, 231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ashley CE; Carnes EC; Phillips GK; Padilla D; Durfee PN; Brown PA; Hanna TN; Liu J; Phillips B; Carter MB; Carroll NJ; Jiang X; Dunphy DR; Willman CL; Petsev DN; Evans DG; Parikh AN; Chackerian B; Wharton W; Peabody DS et al. , The Targeted Delivery of Multicomponent Cargos to Cancer Cells by Nanoporous Particle-supported Lipid Bilayers. Nat. Mater. 2011, 10, 389–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu X; Situ A; Kang Y; Villabroza KR; Liao Y; Chang CH; Donahue T; Nel AE; Meng H, Irinotecan Delivery by Lipid-Coated Mesoporous Silica Nanoparticles Shows Improved Efficacy and Safety over Liposomes for Pancreatic Cancer. ACS Nano 2016, 10, 2702–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Meng H; Wang M; Liu H; Liu X; Situ A; Wu B; Ji Z; Chang CH; Nel AE, Use of a Lipid-Coated Mesoporous Silica Nanoparticle Platform for Synergistic Gemcitabine and Paclitaxel Delivery to Human Pancreatic Cancer in Mice. ACS Nano 2015, 9, 3540–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu X; Lin P; Perrett I; Lin J; Liao Y-P; Chang CH; Jiang J; Wu N; Donahue T; Wainberg Z; Nel AE; Meng H, Tumor-penetrating Peptide Enhances Transcytosis of Silicasome-based Chemotherapy for Pancreatic Cancer. J. Clin. Investig. 2017, 127, 2007–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu X; Jiang J; Meng H, Transcytosis - An Effective Targeting Strategy That is Complementary to “EPR effect” for Pancreatic Cancer Nano Drug Delivery. Theranostics 2019, 9, 8018–8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sindhwani S; Syed AM; Ngai J; Kingston BR; Maiorino L; Rothschild J; MacMillan P; Zhang Y; Rajesh NU; Hoang T; Wu JLY; Wilhelm S; Zilman A; Gadde S; Sulaiman A; Ouyang B; Lin Z; Wang L; Egeblad M; Chan WCW, The Entry of Nanoparticles into Solid Tumours. Nat. Mater. 2020, 19, 566–575. [DOI] [PubMed] [Google Scholar]
- 96.Dewhirst MW; Secomb TW, Transport of Drugs from Blood Vessels to Tumour Tissue. Nat. Rev. Cancer 2017, 17, 738–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ruoslahti E, Access Granted: iRGD Helps Silicasome-encased Drugs Breach the Tumor Barrier. J. Clin. Investig. 2017, 127, 1622–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu X; Jiang J; Chan R; Ji Y; Lu J; Liao Y-P; Okene M; Lin J; Lin P; Chang CH; Wang X; Tang I; Zheng E; Qiu W; Wainberg ZA; Nel AE; Meng H, Improved Efficacy and Reduced Toxicity Using a Custom-Designed Irinotecan-Delivering Silicasome for Orthotopic Colon Cancer. ACS Nano 2019, 13, 38–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu X; Jiang J; Liao Y-P; Tang I; Zheng E; Qiu W; Lin M; Wang X; Ji Y; Mei K-C; Liu Q; Chang CH; Wainberg ZA; Nel AE; Meng H, Combination Chemo-Immunotherapy for Pancreatic Cancer Using the Immunogenic Effects of an Irinotecan Silicasome Nanocarrier Plus Anti-PD-1. Advanced Science 2021, 8, 2002147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Obeid M; Tesniere A; Ghiringhelli F; Fimia GM; Apetoh L; Perfettini J-L; Castedo M; Mignot G; Panaretakis T; Casares N; Métivier D; Larochette N; van Endert P; Ciccosanti F; Piacentini M; Zitvogel L; Kroemer G, Calreticulin Exposure Dictates the Immunogenicity of Cancer Cell Death. Nat. Med. 2007, 13, 54–61. [DOI] [PubMed] [Google Scholar]
- 101.Hato SV; Khong A; de Vries IJM; Lesterhuis WJ, Molecular Pathways: The Immunogenic Effects of Platinum-Based Chemotherapeutics. Clin. Cancer Res. 2014, 20, 2831–2837. [DOI] [PubMed] [Google Scholar]
- 102.Liu X; Jiang J; Chang CH; Liao Y-P; Lodico JJ; Tang I; Zheng E; Qiu W; Lin M; Wang X; Ji Y; Mei K-C; Nel AE; Meng H, Development of Facile and Versatile Platinum Drug Delivering Silicasome Nanocarriers for Efficient Pancreatic Cancer Chemo-Immunotherapy. Small 2021, 17, 2005993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhao X; Yang K; Zhao R; Ji T; Wang X; Yang X; Zhang Y; Cheng K; Liu S; Hao J; Ren H; Leong KW; Nie G, Inducing Enhanced Immunogenic Cell Death with Nanocarrier-based Drug Delivery Systems for Pancreatic Cancer Therapy. Biomaterials 2016, 102, 187–197. [DOI] [PubMed] [Google Scholar]
- 104.Sun F; Zhu Q; Li T; Saeed M; Xu Z; Zhong F; Song R; Huai M; Zheng M; Xie C; Xu L; Yu H, Regulating Glucose Metabolism with Prodrug Nanoparticles for Promoting Photoimmunotherapy of Pancreatic Cancer. Adv. Sci. 2021, 8, 2002746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liu L; Kshirsagar PG; Gautam SK; Gulati M; Wafa EI; Christiansen JC; White BM; Mallapragada SK; Wannemuehler MJ; Kumar S; Solheim JC; Batra SK; Salem AK; Narasimhan B; Jain M, Nanocarriers for Pancreatic Cancer Imaging, Treatments, and Immunotherapies. Theranostics 2022, 12, 1030–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Thomas R; Al-Khadairi G; Decock J, Immune Checkpoint Inhibitors in Triple Negative Breast Cancer Treatment: Promising Future Prospects. Front. Oncol. 2021, 10:600573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Emens LA; Middleton G, The Interplay of Immunotherapy and Chemotherapy: Harnessing Potential Synergies. Cancer Immunology Research 2015, 3, 436–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zitvogel L; Apetoh L; Ghiringhelli F; Kroemer G, Immunological Aspects of Cancer Chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [DOI] [PubMed] [Google Scholar]
- 109.Bezu L; Gomes-da-Silva LC; Dewitte H; Breckpot K; Fucikova J; Spisek R; Galluzzi L; Kepp O; Kroemer G, Combinatorial Strategies for the Induction of Immunogenic Cell Death. Front. Immunol. 2015, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nanda R; Liu MC; Yau C; Shatsky R; Pusztai L; Wallace A; Chien AJ; Forero-Torres A; Ellis E; Han H; Clark A; Albain K; Boughey JC; Jaskowiak NT; Elias A; Isaacs C; Kemmer K; Helsten T; Majure M; Stringer-Reasor E et al. , Effect of Pembrolizumab Plus Neoadjuvant Chemotherapy on Pathologic Complete Response in Women With Early-Stage Breast Cancer: An Analysis of the Ongoing Phase 2 Adaptively Randomized I-SPY2 Trial. JAMA Oncol. 2020, 6, 676–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schmid P; Salgado R; Park YH; Muñoz-Couselo E; Kim SB; Sohn J; Im SA; Foukakis T; Kuemmel S; Dent R; Yin L; Wang A; Tryfonidis K; Karantza V; Cortés J; Loi S, Pembrolizumab Plus Chemotherapy as Neoadjuvant Treatment of High-risk, Early-stage Triple-negative Breast Cancer: Results From the Phase 1b Open-label, Multicohort KEYNOTE-173 Study. Ann. Oncol. 2020, 31, 569–581. [DOI] [PubMed] [Google Scholar]
- 112.Schmid P; Cortes J; Pusztai L; McArthur H; Kümmel S; Bergh J; Denkert C; Park YH; Hui R; Harbeck N; Takahashi M; Foukakis T; Fasching PA; Cardoso F; Untch M; Jia L; Karantza V; Zhao J; Aktan G; Dent R; O’Shaughnessy J, Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [DOI] [PubMed] [Google Scholar]
- 113.Zhao M; Ding X.-f.; Shen J.-y.; Zhang X.-p.; Ding X.-w.; Xu B, Use of Liposomal Doxorubicin for Adjuvant Chemotherapy of Breast Cancer in Clinical Practice. J. Zhejiang Univ. Sci. B 2017, 18, 15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kyte JA; Andresen NK; Russnes HG; Fretland SØ; Falk RS; Lingjærde OC; Naume B, ICON: a Randomized Phase IIb Study Evaluating Immunogenic Chemotherapy Combined with Ipilimumab and Nivolumab in Patients with Metastatic Hormone Receptor Positive Breast Cancer. J. Transl. Med. 2020, 18, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Goel S; Gupta N; Walcott BP; Snuderl M; Kesler CT; Kirkpatrick ND; Heishi T; Huang Y; Martin JD; Ager E; Samuel R; Wang S; Yazbek J; Vakoc BJ; Peterson RT; Padera TP; Duda DG; Fukumura D; Jain RK, Effects of Vascular-Endothelial Protein Tyrosine Phosphatase Inhibition on Breast Cancer Vasculature and Metastatic Progression. J. Natl. Cancer Inst. 2013, 105, 1188–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nagy JA; Chang SH; Dvorak AM; Dvorak HF, Why are Tumour Blood Vessels Abnormal and Why is it Important to Know? British Journal of Cancer 2009, 100, 865–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Klein D, The Tumor Vascular Endothelium as Decision Maker in Cancer Therapy. Front. Oncol. 2018, 8:367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Grasselly C; Denis M; Bourguignon A; Talhi N; Mathe D; Tourette A; Serre L; Jordheim LP; Matera EL; Dumontet C, The Antitumor Activity of Combinations of Cytotoxic Chemotherapy and Immune Checkpoint Inhibitors Is Model-Dependent. Front. Immunol. 2018, 9:2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sirait-Fischer E; Olesch C; Fink AF; Berkefeld M; Huard A; Schmid T; Takeda K; Brüne B; Weigert A, Immune Checkpoint Blockade Improves Chemotherapy in the PyMT Mammary Carcinoma Mouse Model. Front. Oncol. 2020, 10:1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hurvitz SA; McAndrew NP; Bardia A; Press MF; Pegram M; Crown JP; Fasching PA; Ejlertsen B; Yang EH; Glaspy JA; Slamon DJ, A Careful Reassessment of Anthracycline Use in Curable Breast Cancer. npj Breast Cancer 2021, 7 (1), 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gradishar WJ; Tjulandin S; Davidson N; Shaw H; Desai N; Bhar P; Hawkins M; O’Shaughnessy J, Phase III Trial of Nanoparticle Albumin-Bound Paclitaxel Compared With Polyethylated Castor Oil–Based Paclitaxel in Women With Breast Cancer. J. Clin. Oncol. 2005, 23, 7794–7803. [DOI] [PubMed] [Google Scholar]
- 122.Yuan H; Guo H; Luan X; He M; Li F; Burnett J; Truchan N; Sun D, Albumin Nanoparticle of Paclitaxel (Abraxane) Decreases while Taxol Increases Breast Cancer Stem Cells in Treatment of Triple Negative Breast Cancer. Mol. Pharmaceutics 2020, 17, 2275–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schmid P; Rugo HS; Adams S; Schneeweiss A; Barrios CH; Iwata H; Diéras V; Henschel V; Molinero L; Chui SY; Maiya V; Husain A; Winer EP; Loi S; Emens LA, Atezolizumab Plus nab-Paclitaxel as First-line Treatment for Unresectable, Locally Advanced or Metastatic Triple-negative Breast Cancer (IMpassion130): Updated Efficacy Results From a Randomised, Double-blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2020, 21, 44–59. [DOI] [PubMed] [Google Scholar]
- 124.Miles D; Gligorov J; André F; Cameron D; Schneeweiss A; Barrios C; Xu B; Wardley A; Kaen D; Andrade L; Semiglazov V; Reinisch M; Patel S; Patre M; Morales L; Patel SL; Kaul M; Barata T; O’Shaughnessy J; Zhang Q et al. , Primary Results from IMpassion131, a Double-blind, Placebo-controlled, Randomised Phase III Trial of First-line Paclitaxel With or Without Atezolizumab for Unresectable Locally Advanced/Metastatic Triple-negative Breast Cancer. Ann. Oncol. 2021, 32 (8), 994–1004. [DOI] [PubMed] [Google Scholar]
- 125.Zhen Y; Ewert KK; Fisher WS; Steffes VM; Li Y; Safinya CR, Paclitaxel Loading in Cationic liposome Vectors is Enhanced by Replacement of Oleoyl with Linoleoyl Tails with Distinct Lipid Shapes. Sci. Rep. 2021, 11, 7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Koudelka Š; Turánek J, Liposomal Paclitaxel Formulations. J. Controlled Release 2012, 163, 322–334. [DOI] [PubMed] [Google Scholar]
- 127.Fasol U; Frost A; Büchert M; Arends J; Fiedler U; Scharr D; Scheuenpflug J; Mross K, Vascular and Pharmacokinetic Effects of EndoTAG-1 in Patients with Advanced Cancer and Liver Metastasis. Ann. Oncol. 2012, 23, 1030–1036. [DOI] [PubMed] [Google Scholar]
- 128.Mei K-C; Liao Y-P; Jiang J; Chiang M; Khazaieli M; Liu X; Wang X; Liu Q; Chang CH; Zhang X; Li J; Ji Y; Melano B; Telesca D; Xia T; Meng H; Nel AE, Liposomal Delivery of Mitoxantrone and a Cholesteryl Indoximod Prodrug Provides Effective Chemo-immunotherapy in Multiple Solid Tumors. ACS Nano 2020, 14, 13343–13366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sconocchia G; Eppenberger S; Spagnoli GC; Tornillo L; Droeser R; Caratelli S; Ferrelli F; Coppola A; Arriga R; Lauro D; Iezzi G; Terracciano L; Ferrone S, NK Cells and T cells Cooperate During the Clinical Course of Colorectal Cancer. OncoImmunology 2014, 3, e952197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sukkurwala AQ; Adjemian S; Senovilla L; Michaud M; Spaggiari S; Vacchelli E; Baracco EE; Galluzzi L; Zitvogel L; Kepp O; Kroemer G, Screening of Novel Immunogenic Cell Death Inducers Within the NCI Mechanistic Diversity Set. OncoImmunology 2014, 3, e28473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wu J; Waxman DJ, Immunogenic Chemotherapy: Dose and Schedule Dependence and Combination with Immunotherapy. Cancer Lett. 2018, 419, 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yamazaki T; Buqué A; Ames TD; Galluzzi L, PT-112 Induces Immunogenic Cell Death and Synergizes with Immune Checkpoint Blockers in Mouse Tumor Models. OncoImmunology 2020, 9, 1721810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Heidemann E; Stoeger H; Souchon R; Hirschmann WD; Bodenstein H; Oberhoff C; Fischer JT; Schulze M; Clemens M; Andreesen R; Mahlke M; König M; Scharl A; Fehnle K; Kaufmann M, Is First-line Single-agent Mitoxantrone in the Treatment of High-risk Metastatic Breast Cancer Patients as Effective as Combination Chemotherapy? No Difference in Survival But Higher Quality of Life Were Found in a Multicenter Randomized Trial. Ann. Oncol. 2002, 13, 1717–1729. [DOI] [PubMed] [Google Scholar]
- 134.Michaud M; Martins I; Sukkurwala AQ; Adjemian S; Ma Y; Pellegatti P; Shen S; Kepp O; Scoazec M; Mignot G; Rello-Varona S; Tailler M; Menger L; Vacchelli E; Galluzzi L; Ghiringhelli F; Virgilio F d.; Zitvogel, L.; Kroemer, G., Autophagy-Dependent Anticancer Immune Responses Induced by Chemotherapeutic Agents in Mice. Science 2011, 334, 1573–1577. [DOI] [PubMed] [Google Scholar]
- 135.Liu J; Zhao Z; Qiu N; Zhou Q; Wang G; Jiang H; Piao Y; Zhou Z; Tang J; Shen Y, Co-delivery of IOX1 and Doxorubicin for Antibody-independent Cancer Chemoimmunotherapy. Nat. Commun. 2021, 12 (1), 2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wu S; Liu D; Li W; Song B; Chen C; Chen D; Hu H, Enhancing TNBC Chemoimmunotherapy via Combination Reprogramming Tumor Immune Microenvironment with Immunogenic Cell Death. Int. J. Pharm. 2021, 598, 120333. [DOI] [PubMed] [Google Scholar]
- 137.Wu D; Vogus D; Krishnan V; Broto M; Pusuluri A; Zhao Z; Kapate N; Mitragotri S, Optimized 5-Fluorouridine Prodrug for Co-Loading with Doxorubicin in Clinically Relevant Liposomes. Pharmaceutics 2021, 13, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wu D; Zhao Z; Kim J; Razmi A; Wang LL-W; Kapate N; Gao Y; Peng K; Ukidve A; Mitragotri S, Gemcitabine and Doxorubicin in Immunostimulatory Monophosphoryl Lipid A Liposomes for Treating Breast Cancer. Bioeng. Transl. Med. 2021, 6, e10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kyte JA; Røssevold A; Falk RS; Naume B, ALICE: a Randomized Placebo-controlled Phase II Study Evaluating Atezolizumab Combined with Immunogenic Chemotherapy in Patients with Metastatic Triple-negative Breast Cancer. J. Transl. Med. 2020, 18, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chen H; Liu L; Ma A; Yin T; Chen Z; Liang R; Qiu Y; Zheng M; Cai L, Noninvasively Immunogenic Sonodynamic Therapy with Manganese Protoporphyrin Liposomes Against Triple-negative Breast Cancer. Biomaterials 2021, 269, 120639. [DOI] [PubMed] [Google Scholar]
- 141.Liu D; Chen B; Mo Y; Wang Z; Qi T; Zhang Q; Wang Y, Redox-Activated Porphyrin-Based Liposome Remote-Loaded with Indoleamine 2,3-Dioxygenase (IDO) Inhibitor for Synergistic Photoimmunotherapy through Induction of Immunogenic Cell Death and Blockage of IDO Pathway. Nano Lett. 2019, 19, 6964–6976. [DOI] [PubMed] [Google Scholar]
- 142.Liu Y; Qiu N; Shen L; Liu Q; Zhang J; Cheng Y-Y; Lee K-H; Huang L, Nanocarrier-mediated Immunogenic Chemotherapy for Triple Negative Breast Cancer. J. Controlled Release 2020, 323, 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gubernator J, Active Methods of Drug Loading into Liposomes: Recent Strategies for Stable Drug Entrapment and Increased In Vivo Activity. Expert. Opin. Drug Deliv. 2011, 8, 565–580. [DOI] [PubMed] [Google Scholar]
- 144.Yang W; Yang Z; Fu J; Guo M; Sun B; Wei W; Liu D; Liu H, The Influence of Trapping Agents on the Antitumor Efficacy of Irinotecan Liposomes: Head-to-head Comparison of Ammonium Sulfate, Sulfobutylether-β-cyclodextrin and Sucrose Octasulfate. Biomater. Sci. 2019, 7, 419–428. [DOI] [PubMed] [Google Scholar]
- 145.Fujii S.-i.; Shimizu K; Smith C; Bonifaz L; Steinman RM, Activation of Natural Killer T Cells by α-Galactosylceramide Rapidly Induces the Full Maturation of Dendritic Cells In Vivo and Thereby Acts as an Adjuvant for Combined CD4 and CD8 T Cell Immunity to a Coadministered Protein. J. Exp. Med. 2003, 198, 267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Serhan CN; Chiang N; Van Dyke TE, Resolving Inflammation: Dual Anti-inflammatory and Pro-resolution Lipid Mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wang L; Wang Z; Qin Y; Liang W, Delivered Antigen Peptides to Resident CD8α+ DCs in Lymph node by Micelle-based Vaccine Augment Antigen-specific CD8+ Effector T Cell Response. Eur. J. Pharm. Biopharm. 2020, 147, 76–86. [DOI] [PubMed] [Google Scholar]
- 148.Chatzikleanthous D; O’Hagan DT; Adamo R, Lipid-Based Nanoparticles for Delivery of Vaccine Adjuvants and Antigens: Toward Multicomponent Vaccines. Mol. Pharmaceutics 2021, 18, 2867–2888. [DOI] [PubMed] [Google Scholar]
- 149.Singh M; Khong H; Dai Z; Huang X-F; Wargo JA; Cooper ZA; Vasilakos JP; Hwu P; Overwijk WW, Effective Innate and Adaptive Antimelanoma Immunity through Localized TLR7/8 Activation. J. Immunol. 2014, 193, 4722–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bhagchandani S; Johnson JA; Irvine DJ, Evolution of Toll-like receptor 7/8 agonist therapeutics and their delivery approaches: From antiviral formulations to vaccine adjuvants. Adv. Drug Delivery Rev. 2021, 175, 113803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zaro JL, Lipid-Based Drug Carriers for Prodrugs to Enhance Drug Delivery. AAPS J. 2015, 17, 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lambert DM, Rationale and Applications of Lipids as Prodrug Carriers. Eur. J. Pharm. Sci. 2000, 11, S15–S27. [DOI] [PubMed] [Google Scholar]
- 153.Lombardo D; Kiselev MA; Magazù S; Calandra P, Amphiphiles Self-Assembly: Basic Concepts and Future Perspectives of Supramolecular Approaches. Adv. Condens. Matter. Phys. 2015, 2015, 151683. [Google Scholar]
- 154.Kulkarni CV, Lipid Crystallization: From Self-assembly to Hierarchical and Biological Ordering. Nanoscale 2012, 4, 5779–5791. [DOI] [PubMed] [Google Scholar]
- 155.Neises B; Steglich W, Simple Method for the Esterification of Carboxylic Acids. Angew. Chem. Int. Ed. 1978, 17, 522–524. [Google Scholar]
- 156.Prendergast GC; Smith C; Thomas S; Mandik-Nayak L; Laury-Kleintop L; Metz R; Muller AJ, Indoleamine 2,3-Dioxygenase Pathways of Pathogenic Inflammation and Immune Escape in Cancer. Cancer Immunol. Immunother. 2014, 63, 721–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Labadie BW; Bao R; Luke JJ, Reimagining IDO Pathway Inhibition in Cancer Immunotherapy via Downstream Focus on the Tryptophan–Kynurenine–Aryl Hydrocarbon Axis. Clin. Cancer Res. 2019, 25, 1462–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Castro F; Cardoso AP; Gonçalves RM; Serre K; Oliveira MJ, Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Mojic M; Takeda K; Hayakawa Y, The Dark Side of IFN-γ: Its Role in Promoting Cancer Immunoevasion. International Journal of Molecular Sciences 2018, 19 (1), 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ribas A; Wolchok JD, Cancer Immunotherapy Using Checkpoint Blockade. Science 2018, 359, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lan Y; Liang Q; Sun Y; Cao A; Liu L; Yu S; Zhou L; Liu J; Zhu R; Liu Y, Codelivered Chemotherapeutic Doxorubicin via a Dual-Functional Immunostimulatory Polymeric Prodrug for Breast Cancer Immunochemotherapy. ACS Appl. Mater. Interfaces 2020, 12, 31904–31921. [DOI] [PubMed] [Google Scholar]
- 162.Wan Z; Sun J; Xu J; Moharil P; Chen J; Xu J; Zhu J; Li J; Huang Y; Xu P; Ma X; Xie W; Lu B; Li S, Dual Functional Immunostimulatory Polymeric Prodrug Carrier with Pendent Indoximod for Enhanced Cancer Immunochemotherapy. Acta Biomater. 2019, 90, 300–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Fox E; Oliver T; Rowe M; Thomas S; Zakharia Y; Gilman PB; Muller AJ; Prendergast GC, Indoximod: An Immunometabolic Adjuvant That Empowers T Cell Activity in Cancer. Front. Oncol. 2018, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kumar S; Jaipuri FA; Waldo JP; Potturi H; Marcinowicz A; Adams J; Van Allen C; Zhuang H; Vahanian N; Link C; Brincks EL; Mautino MR, Discovery of Indoximod Prodrugs and Characterization of Clinical Candidate NLG802. Eur. J. Med. Chem. 2020, 198, 112373. [DOI] [PubMed] [Google Scholar]
- 165.Tang K; Wu Y-H; Song Y; Yu B, Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitors in Clinical Trials for Cancer Immunotherapy. Journal of Hematology & Oncology 2021, 14, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Sonpavde G; Necchi A; Gupta S; Steinberg GD; Gschwend JE; Heijden MSVD; Garzon N; Ibrahim M; Raybold B; Liaw D; Rutstein M; Galsky MD, ENERGIZE: a Phase III Study of Neoadjuvant Chemotherapy Alone or With Nivolumab With/Without Linrodostat Mesylate for Muscle-invasive Bladder Cancer. Future Oncol. 2020, 16, 4359–4368. [DOI] [PubMed] [Google Scholar]
- 167.Simplício AL; Clancy JM; Gilmer JF, Prodrugs for Amines. Molecules 2008, 13, 519–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Shen F; Feng L; Zhu Y; Tao D; Xu J; Peng R; Liu Z, Oxaliplatin-/NLG919 Prodrugs-constructed Liposomes for Effective Chemoimmunotherapy of Colorectal Cancer. Biomaterials 2020, 255, 120190. [DOI] [PubMed] [Google Scholar]
- 169.Guzik K; Tomala M; Muszak D; Konieczny M; Hec A; Błaszkiewicz U; Pustuła M; Butera R; Dömling A; Holak TA, Development of the Inhibitors That Target the PD-1/PD-L1 Interaction—A Brief Look at Progress on Small Molecules, Peptides and Macrocycles. Molecules 2019, 24, 2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zak KM; Kitel R; Przetocka S; Golik P; Guzik K; Musielak B; Dömling A; Dubin G; Holak TA, Structure of the Complex of Human Programmed Death 1, PD-1, and Its Ligand PD-L1. Structure 2015, 23, 2341–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Weinmann H, Cancer Immunotherapy: Selected Targets and Small-Molecule Modulators. ChemMedChem 2016, 11, 450–466. [DOI] [PubMed] [Google Scholar]
- 172.Zarganes-Tzitzikas T; Konstantinidou M; Gao Y; Krzemien D; Zak K; Dubin G; Holak TA; Dömling A, Inhibitors of Programmed Cell Death 1 (PD-1): a Patent Review (2010–2015). Expert. Opin. Ther. Pat. 2016, 26, 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Konstantinidou M; Zarganes-Tzitzikas T; Magiera-Mularz K; Holak TA; Dömling A, Immune Checkpoint PD-1/PD-L1: Is There Life Beyond Antibodies? Angew. Chem. Int. Ed. 2018, 57, 4840–4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wang T; Wu X; Guo C; Zhang K; Xu J; Li Z; Jiang S, Development of Inhibitors of the Programmed Cell Death-1/Programmed Cell Death-Ligand 1 Signaling Pathway. J. Med. Chem. 2019, 62, 1715–1730. [DOI] [PubMed] [Google Scholar]
- 175.Zhang R; Zhu Z; Lv H; Li F; Sun S; Li J; Lee C-S, Immune Checkpoint Blockade Mediated by a Small-Molecule Nanoinhibitor Targeting the PD-1/PD-L1 Pathway Synergizes with Photodynamic Therapy to Elicit Antitumor Immunity and Antimetastatic Effects on Breast Cancer. Small 2019, 15, 1903881. [DOI] [PubMed] [Google Scholar]
- 176.Hu Z; Yu P; Du G; Wang W; Zhu H; Li N; Zhao H; Dong Z; Ye L; Tian J, PCC0208025 (BMS202), a Small Molecule Inhibitor of PD-L1, Produces an Antitumor Effect in B16-F10 Melanoma-bearing Mice. PLOS ONE 2020, 15, e0228339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Duan X; Chan C; Lin W, Nanoparticle-Mediated Immunogenic Cell Death Enables and Potentiates Cancer Immunotherapy. Angew. Chem. Int. Ed. 2019, 58, 670–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Gao J; Wang W.-q.; Pei Q; Lord MS; Yu H.-j., Engineering Nanomedicines Through Boosting Immunogenic Cell Death for Improved Cancer Immunotherapy. Acta Pharmacol. Sin. 2020, 41, 986–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Allen SD; Liu X; Jiang J; Liao Y-P; Chang CH; Nel AE; Meng H, Immune Checkpoint Inhibition in Syngeneic Mouse Cancer Models by a Silicasome Nanocarrier Delivering a GSK3 Inhibitor. Biomaterials 2021, 269, 120635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Taylor A; Rothstein D; Rudd CE, Small-Molecule Inhibition of PD-1 Transcription Is an Effective Alternative to Antibody Blockade in Cancer Therapy. Cancer Res. 2018, 78, 706–717. [DOI] [PubMed] [Google Scholar]
- 181.Ugolkov A; Gaisina I; Zhang J-S; Billadeau DD; White K; Kozikowski A; Jain S; Cristofanilli M; Giles F; O’Halloran T; Cryns VL; Mazar AP, GSK-3 Inhibition Overcomes Chemoresistance in Human Breast Cancer. Cancer Lett. 2016, 380, 384–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Eldar-Finkelman H; Martinez A, GSK-3 Inhibitors: Preclinical and Clinical Focus on CNS. Front. Mol. Neurosci. 2011, 4:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Cern A; Barenholz Y; Tropsha A; Goldblum A, Computer-aided Design of Liposomal Drugs: In Silico Prediction and Experimental Validation of Drug Candidates for Liposomal Remote Loading. J. Controlled Release 2014, 173, 125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Georgievska B; Sandin J; Doherty J; Mörtberg A; Neelissen J; Andersson A; Gruber S; Nilsson Y; Schött P; Arvidsson PI; Hellberg S; Osswald G; Berg S; Fälting J; Bhat RV, AZD1080, a Novel GSK3 Inhibitor, Rescues Synaptic Plasticity Deficits in Rodent Brain and Exhibits Peripheral Target Engagement in Humans. J. Neurochem. 2013, 125, 446–456. [DOI] [PubMed] [Google Scholar]
- 185.Chen Y.-q.; Zheng L; Aldarouish M; Zhou Z.-h.; Pan N; Liu J.-q.; Chen F.-x.; Wang L.-x., Wnt Pathway Activator TWS119 Enhances the Proliferation and Cytolytic Activity of Human γδT Cells Against Colon Cancer. Exp. Cell Res. 2018, 362 (1), 63–71. [DOI] [PubMed] [Google Scholar]
- 186.Gattinoni L; Lugli E; Ji Y; Pos Z; Paulos CM; Quigley MF; Almeida JR; Gostick E; Yu Z; Carpenito C; Wang E; Douek DC; Price DA; June CH; Marincola FM; Roederer M; Restifo NP, A Human Memory T cell Subset with Stem cell–like Properties. Nat. Med. 2011, 17, 1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Vigano S; Alatzoglou D; Irving M; Ménétrier-Caux C; Caux C; Romero P; Coukos G, Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front. Immunol. 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Yu F; Zhu C; Xie Q; Wang Y, Adenosine A2A Receptor Antagonists for Cancer Immunotherapy. J. Med. Chem. 2020, 63, 12196–12212. [DOI] [PubMed] [Google Scholar]
- 189.Baraldi PG; Tabrizi MA; Gessi S; Borea PA, Adenosine Receptor Antagonists: Translating Medicinal Chemistry and Pharmacology into Clinical Utility. Chem. Rev. 2008, 108, 238–263. [DOI] [PubMed] [Google Scholar]
- 190.Jazayeri A; Andrews SP; Marshall FH, Structurally Enabled Discovery of Adenosine A2A Receptor Antagonists. Chem. Rev. 2017, 117, 21–37. [DOI] [PubMed] [Google Scholar]
- 191.Hatfield SM; Sitkovsky M, A2A Adenosine Receptor Antagonists to Weaken the Hypoxia-HIF-1α Driven Immunosuppression and Improve Immunotherapies of Cancer. Curr. Opin. Pharmacol. 2016, 29, 90–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Congreve M; de Graaf C; Swain NA; Tate CG, Impact of GPCR Structures on Drug Discovery. Cell 2020, 181, 81–91. [DOI] [PubMed] [Google Scholar]
- 193.Merighi S; Battistello E; Giacomelli L; Varani K; Vincenzi F; Borea PA; Gessi S, Targeting A3 and A2A Adenosine Receptors in the Fight Against Cancer. Expert. Opin. Ther. Targets 2019, 23, 669–678. [DOI] [PubMed] [Google Scholar]
- 194.Beavis PA; Divisekera U; Paget C; Chow MT; John LB; Devaud C; Dwyer K; Stagg J; Smyth MJ; Darcy PK, Blockade of A2A Receptors Potently Suppresses the Metastasis of CD73+ Tumors. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 14711–14716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Byeon J-J; Park M-H; Shin S-H; Park Y; Lee B. i.; Choi J.-m.; Kim N; Park S.-j.; Park M.-j.; Lim J.-h.; Na Y-G; Shin YG, In Vitro, In Silico, and In Vivo Assessments of Pharmacokinetic Properties of ZM241385. Molecules 2020, 25, 1106/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Palmer TM; Poucher SM; Jacobson KA; Stiles GL, 125I-4-(2-[7-amino-2-[2furyl][1,2,4]triazolo[2,3-a][1,3,5] triazin-5-yl-amino]ethyl)phenol, a High Affinity Antagonist Radioligand Selective for The A2a Adenosine Receptor. Mol. Pharmacol. 1995, 48, 970–974. [PMC free article] [PubMed] [Google Scholar]
- 197.Beavis PA; Milenkovski N; Henderson MA; John LB; Allard B; Loi S; Kershaw MH; Stagg J; Darcy PK, Adenosine Receptor 2A Blockade Increases the Efficacy of Anti–PD-1 through Enhanced Antitumor T-cell Responses. Cancer Immunol. Res. 2015, 3, 506–517. [DOI] [PubMed] [Google Scholar]
- 198.Loi S; Pommey S; Haibe-Kains B; Beavis PA; Darcy PK; Smyth MJ; Stagg J, CD73 Promotes Anthracycline Resistance and Poor Prognosis in Triple Negative Breast Cancer. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 11091–11096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Chen IX; Chauhan VP; Posada J; Ng MR; Wu MW; Adstamongkonkul P; Huang P; Lindeman N; Langer R; Jain RK, Blocking CXCR4 Alleviates Desmoplasia, Increases T-lymphocyte Infiltration, and Improves Immunotherapy in Metastatic Breast Cancer. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 4558–4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Tamamura H; Hori A; Kanzaki N; Hiramatsu K; Mizumoto M; Nakashima H; Yamamoto N; Otaka A; Fujii N, T140 Analogs as CXCR4 Antagonists Identified as Anti-metastatic Agents in the Treatment of Breast Cancer. FEBS Letters 2003, 550 (1–3), 79–83. [DOI] [PubMed] [Google Scholar]
- 201.Biasci D; Smoragiewicz M; Connell CM; Wang Z; Gao Y; Thaventhiran JED; Basu B; Magiera L; Johnson TI; Bax L; Gopinathan A; Isherwood C; Gallagher FA; Pawula M; Hudecova I; Gale D; Rosenfeld N; Barmpounakis P; Popa EC; Brais R et al. , CXCR4 Inhibition in Human Pancreatic and Colorectal Cancers induces an Integrated Immune Response. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 28960–28970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Xue J; Li R; Gao D; Chen F; Xie H, CXCL12/CXCR4 Axis-Targeted Dual-Functional Nano-Drug Delivery System Against Ovarian Cancer. Int. J. Nanomedicine 2020, 15, 5701–5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Zheng N; Liu W; Li B; Nie H; Liu J; Cheng Y; Wang J; Dong H; Jia L, Co-delivery of Sorafenib and Metapristone Encapsulated by CXCR4-targeted PLGA-PEG Nanoparticles Overcomes Hepatocellular Carcinoma Resistance to Sorafenib. J. Exp. Clin. Cancer Res. 2019, 38, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Zhang F; Gong S; Wu J; Li H; Oupicky D; Sun M, CXCR4-Targeted and Redox Responsive Dextrin Nanogel for Metastatic Breast Cancer Therapy. Biomacromolecules 2017, 18, 1793–1802. [DOI] [PubMed] [Google Scholar]
- 205.Li Y; Zhang T; Liu Q; He J, PEG-Derivatized Dual-Functional Nanomicelles for Improved Cancer Therapy. Frontiers in Pharmacology 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Vonderheide RH; Bajor DL; Winograd R; Evans RA; Bayne LJ; Beatty GL, CD40 Immunotherapy for Pancreatic Cancer. Cancer Immunology, Immunotherapy 2013, 62, 949–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Vonderheide RH, CD40 Agonist Antibodies in Cancer Immunotherapy. Annu. Rev. Med. 2020, 71, 47–58. [DOI] [PubMed] [Google Scholar]
- 208.Nowak AK; Robinson BWS; Lake RA, Synergy between Chemotherapy and Immunotherapy in the Treatment of Established Murine Solid Tumors. Cancer Res. 2003, 63, 4490–4496. [PubMed] [Google Scholar]
- 209.Gabrilovich DI; Nagaraj S, Myeloid-derived Suppressor Cells as Regulators of The Immune System. Nat. Rev. Immunol. 2009, 9, 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Hammerich L; Marron TU; Upadhyay R; Svensson-Arvelund J; Dhainaut M; Hussein S; Zhan Y; Ostrowski D; Yellin M; Marsh H; Salazar AM; Rahman AH; Brown BD; Merad M; Brody JD, Systemic Clinical Tumor Regressions and Potentiation of PD1 Blockade with In Situ Vaccination. Nat. Med. 2019, 25, 814–824. [DOI] [PubMed] [Google Scholar]
- 211.Musella M; Galassi C; Manduca N; Sistigu A, The Yin and Yang of Type I IFNs in Cancer Promotion and Immune Activation. Biology 2021, 10, 856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Ager CR; Boda A; Rajapakshe K; Lea ST; Di Francesco ME; Jayaprakash P; Slay RB; Morrow B; Prasad R; Dean MA; Duffy CR; Coarfa C; Jones P; Curran MA, High Potency STING Agonists Engage Unique Myeloid Pathways to Reverse Pancreatic Cancer Immune Privilege. J. Immunother. Cancer 2021, 9, e003246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Fu J; Kanne DB; Leong M; Glickman LH; McWhirter SM; Lemmens E; Mechette K; Leong JJ; Lauer P; Liu W; Sivick KE; Zeng Q; Soares KC; Zheng L; Portnoy DA; Woodward JJ; Pardoll DM; Dubensky TW; Kim Y, STING Agonist Formulated Cancer Vaccines Can Cure Established Tumors Resistant to PD-1 Blockade. Sci. Transl. Med. 2015, 7, 283ra52–283ra52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Kinkead HL; Hopkins A; Lutz E; Wu AA; Yarchoan M; Cruz K; Woolman S; Vithayathil T; Glickman LH; Ndubaku CO; McWhirter SM; Dubensky TW Jr.; Armstrong TD; Jaffee EM; Zaidi N, Combining STING-based Neoantigen-targeted Vaccine with Checkpoint Modulators Enhances Antitumor Immunity in Murine Pancreatic Cancer. JCI Insight 2018, 3 (20). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Motedayen Aval L; Pease JE; Sharma R; Pinato DJ, Challenges and Opportunities in the Clinical Development of STING Agonists for Cancer Immunotherapy. J. Clin. Med. 2020, 9, 3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Shae D; Becker KW; Christov P; Yun DS; Lytton-Jean AKR; Sevimli S; Ascano M; Kelley M; Johnson DB; Balko JM; Wilson JT, Endosomolytic Polymersomes Increase the Activity of Cyclic Dinucleotide STING Agonists to Enhance Cancer Immunotherapy. Nat. Nanotechnol. 2019, 14, 269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Junkins RD; Gallovic MD; Johnson BM; Collier MA; Watkins-Schulz R; Cheng N; David CN; McGee CE; Sempowski GD; Shterev I; McKinnon K; Bachelder EM; Ainslie KM; Ting JPY, A Robust Microparticle Platform for a STING-Targeted Adjuvant that Enhances Both Humoral and Cellular Immunity During Vaccination. J. Controlled Release 2018, 270, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Liu Q; Wang X; Liao Y-P; Chang CH; Li J; Xia T; Nel AE, Use of a Liver-targeting Nanoparticle Platform to Intervene in Peanut-induced Anaphylaxis Through Delivery of an Ara h2 T-cell Epitope. Nano Today 2022, 42, 101370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Liu Q; Wang X; Liu X; Liao Y-P; Chang CH; Mei K-C; Jiang J; Tseng S; Gochman G; Huang M; Thatcher Z; Li J; Allen SD; Lucido L; Xia T; Nel AE, Antigen- and Epitope-Delivering Nanoparticles Targeting Liver Induce Comparable Immunotolerance in Allergic Airway Disease and Anaphylaxis as Nanoparticle-Delivering Pharmaceuticals. ACS Nano 2021, 15, 1608–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Liu Q; Wang X; Liu X; Kumar S; Gochman G; Ji Y; Liao Y-P; Chang CH; Situ W; Lu J; Jiang J; Mei K-C; Meng H; Xia T; Nel AE, Use of Polymeric Nanoparticle Platform Targeting the Liver To Induce Treg-Mediated Antigen-Specific Immune Tolerance in a Pulmonary Allergen Sensitization Model. ACS Nano 2019, 13, 4778–4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Burdelya LG; Brackett CM; Kojouharov B; Gitlin II; Leonova KI; Gleiberman AS; Aygun-Sunar S; Veith J; Johnson C; Haderski GJ; Stanhope-Baker P; Allamaneni S; Skitzki J; Zeng M; Martsen E; Medvedev A; Scheblyakov D; Artemicheva NM; Logunov DY; Gintsburg AL et al. , Central Role of Liver in Anticancer and Radioprotective Activities of Toll-like Receptor 5 Agonist. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, E1857–E1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Goodwin TJ; Zhou Y; Musetti SN; Liu R; Huang L, Local and Transient Gene Expression Primes the Liver to Resist Cancer Metastasis. Sci. Transl. Med. 2016, 8, 364ra153–364ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Yu X; Chen L; Liu J; Dai B; Xu G; Shen G; Luo Q; Zhang Z, Immune Modulation of Liver Sinusoidal Endothelial Cells by Melittin Nanoparticles Suppresses Liver Metastasis. Nat. Commun. 2019, 10, 574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Principe DR; Doll JA; Bauer J; Jung B; Munshi HG; Bartholin L; Pasche B; Lee C; Grippo PJ, TGF-β: Duality of Function Between Tumor Prevention and Carcinogenesis. J. Natl. Cancer Inst. 2014, 106, djt369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Mariathasan S; Turley SJ; Nickles D; Castiglioni A; Yuen K; Wang Y; Kadel Iii EE; Koeppen H; Astarita JL; Cubas R; Jhunjhunwala S; Banchereau R; Yang Y; Guan Y; Chalouni C; Ziai J; Şenbabaoğlu Y; Santoro S; Sheinson D; Hung J et al. , TGFβ Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature 2018, 554, 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Soares KC; Rucki AA; Kim V; Foley K; Solt S; Wolfgang CL; Jaffee EM; Zheng L, TGF-β Blockade Depletes T Regulatory Cells From Metastatic Pancreatic Tumors in a Vaccine Dependent Manner. Oncotarget 2015, 6, 43005–43015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Meng H; Zhao Y; Dong J; Xue M; Lin Y-S; Ji Z; Mai WX; Zhang H; Chang CH; Brinker CJ; Zink JI; Nel AE, Two-Wave Nanotherapy To Target the Stroma and Optimize Gemcitabine Delivery To a Human Pancreatic Cancer Model in Mice. ACS Nano 2013, 7, 10048–10065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Chauhan VP; Martin JD; Liu H; Lacorre DA; Jain SR; Kozin SV; Stylianopoulos T; Mousa AS; Han X; Adstamongkonkul P; Popović Z; Huang P; Bawendi MG; Boucher Y; Jain RK, Angiotensin Inhibition Enhances Drug Delivery and Potentiates Chemotherapy by Decompressing Tumour Blood Vessels. Nat. Commun. 2013, 4, 2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Murphy JE; Wo JY; Ryan DP; Clark JW; Jiang W; Yeap BY; Drapek LC; Ly L; Baglini CV; Blaszkowsky LS; Ferrone CR; Parikh AR; Weekes CD; Nipp RD; Kwak EL; Allen JN; Corcoran RB; Ting DT; Faris JE; Zhu AX; Goyal L; Berger DL; Qadan M; Lillemoe KD; Talele N; Jain RK; DeLaney TF; Duda DG; Boucher Y; Fernández-Del Castillo C; Hong TS, Total Neoadjuvant Therapy With FOLFIRINOX in Combination With Losartan Followed by Chemoradiotherapy for Locally Advanced Pancreatic Cancer: A Phase 2 Clinical Trial. JAMA Oncology 2019, 5, 1020–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Xia T; He Q; Shi K; Wang Y; Yu Q; Zhang L; Zhang Q; Gao H; Ma L; Liu J, Losartan Loaded Liposomes Improve the Antitumor Efficacy of Liposomal Paclitaxel Modified with pH Sensitive Peptides by Inhibition of Collagen in Breast Cancer. Pharm. Dev. Technol. 2018, 23, 13–21. [DOI] [PubMed] [Google Scholar]
- 231.Vanmeerbeek I; Sprooten J; De Ruysscher D; Tejpar S; Vandenberghe P; Fucikova J; Spisek R; Zitvogel L; Kroemer G; Galluzzi L; Garg AD, Trial Watch: Chemotherapy-induced Immunogenic Cell Death in Immuno-oncology. OncoImmunology 2020, 9, 1703449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Pol J; Vacchelli E; Aranda F; Castoldi F; Eggermont A; Cremer I; Sautès-Fridman C; Fucikova J; Galon J; Spisek R; Tartour E; Zitvogel L; Kroemer G; Galluzzi L, Trial Watch: Immunogenic Cell Death Inducers for Anticancer Chemotherapy. OncoImmunology 2015, 4, e1008866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Zhang RX; Wong HL; Xue HY; Eoh JY; Wu XY, Nanomedicine of Synergistic Drug Combinations for Cancer Therapy – Strategies and Perspectives. J. Controlled Release 2016, 240, 489–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Mayer LD; Harasym TO; Tardi PG; Harasym NL; Shew CR; Johnstone SA; Ramsay EC; Bally MB; Janoff AS, Ratiometric Dosing of Anticancer Drug Combinations: Controlling Drug Ratios After Systemic Administration Regulates Therapeutic Activity in Tumor-bearing Mice. Mol. Cancer Ther. 2006, 5, 1854–1863. [DOI] [PubMed] [Google Scholar]
- 235.Hu Q; Sun W; Wang C; Gu Z, Recent Advances of Cocktail Chemotherapy by Combination Drug Delivery Systems. Adv. Drug Delivery Rev. 2016, 98, 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.MacEwan SR; Callahan DJ; Chilkoti A, Stimulus-responsive Macromolecules and Nanoparticles for Cancer Drug Delivery. Nanomedicine 2010, 5, 793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Meng H; Xue M; Xia T; Zhao Y-L; Tamanoi F; Stoddart JF; Zink JI; Nel AE, Autonomous in Vitro Anticancer Drug Release from Mesoporous Silica Nanoparticles by pH-Sensitive Nanovalves. Journal of the American Chemical Society 2010, 132, 12690–12697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Liong M; Lu J; Kovochich M; Xia T; Ruehm SG; Nel AE; Tamanoi F; Zink JI, Multifunctional Inorganic Nanoparticles for Imaging, Targeting, and Drug Delivery. ACS Nano 2008, 2 (5), 889–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Lin F-C; Zink JI, Probing the Local Nanoscale Heating Mechanism of a Magnetic Core in Mesoporous Silica Drug-Delivery Nanoparticles Using Fluorescence Depolarization. Journal of the American Chemical Society 2020, 142, 5212–5220. [DOI] [PubMed] [Google Scholar]
- 240.Nezhadi S; Dorkoosh FA, Co-delivery Systems: Hope for Clinical Application? Drug Deliv. Transl. 2021. [DOI] [PubMed] [Google Scholar]
- 241.Meng H; Mai WX; Zhang H; Xue M; Xia T; Lin S; Wang X; Zhao Y; Ji Z; Zink JI; Nel AE, Codelivery of an Optimal Drug/siRNA Combination Using Mesoporous Silica Nanoparticles To Overcome Drug Resistance in Breast Cancer in Vitro and in Vivo. ACS Nano 2013, 7, 994–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Shim G; Kim M-G; Kim D; Park JY; Oh Y-K, Nanoformulation-based Sequential Combination Cancer Therapy. Adv. Drug Delivery Rev. 2017, 115, 57–81. [DOI] [PubMed] [Google Scholar]
- 243.Mohammad IS; Teng C; Chaurasiya B; Yin L; Wu C; He W, Drug-delivering-drug Approach-based Codelivery of Paclitaxel and Disulfiram for Treating Multidrug-resistant Cancer. Int. J. Pharm. 2019, 557, 304–313. [DOI] [PubMed] [Google Scholar]
- 244.Wan X; Beaudoin JJ; Vinod N; Min Y; Makita N; Bludau H; Jordan R; Wang A; Sokolsky M; Kabanov AV, Co-delivery of Paclitaxel and Cisplatin in Poly(2-oxazoline) Polymeric Micelles: Implications for Drug Loading, Release, Pharmacokinetics and Outcome of Ovarian and Breast Cancer Treatments. Biomaterials 2019, 192, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Qiu N; Liu Y; Liu Q; Chen Y; Shen L; Hu M; Zhou X; Shen Y; Gao J; Huang L, Celastrol Nanoemulsion Induces Immunogenicity and Downregulates PD-L1 to Boost Abscopal Effect in Melanoma Therapy. Biomaterials 2021, 269, 120604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Liu Q; Chen F; Hou L; Shen L; Zhang X; Wang D; Huang L, Nanocarrier-Mediated Chemo-Immunotherapy Arrested Cancer Progression and Induced Tumor Dormancy in Desmoplastic Melanoma. ACS Nano 2018, 12, 7812–7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Mayer LD; Janoff AS, Optimizing Combination Chemotherapy by Controlling Drug Ratios. Mol. Interv. 2007, 7, 216–223. [DOI] [PubMed] [Google Scholar]
- 248.Lammers T; Storm G; Kiessling F, Nanomedicine Formulations for Combination Therapies. Nano Rev 2010, 1, 10.3402/nano.v1i0.5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Feldman EJ; Lancet JE; Kolitz JE; Ritchie EK; Roboz GJ; List AF; Allen SL; Asatiani E; Mayer LD; Swenson C; Louie AC, First-In-Man Study of CPX-351: A Liposomal Carrier Containing Cytarabine and Daunorubicin in a Fixed 5:1 Molar Ratio for the Treatment of Relapsed and Refractory Acute Myeloid Leukemia. J. Clin. Oncol. 2011, 29, 979–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Krauss AC; Gao X; Li L; Manning ML; Patel P; Fu W; Janoria KG; Gieser G; Bateman DA; Przepiorka D; Shen YL; Shord SS; Sheth CM; Banerjee A; Liu J; Goldberg KB; Farrell AT; Blumenthal GM; Pazdur R, FDA Approval Summary: (Daunorubicin and Cytarabine) Liposome for Injection for the Treatment of Adults with High-Risk Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 2685–2690. [DOI] [PubMed] [Google Scholar]
- 251.Von Hoff DD; Ervin T; Arena FP; Chiorean EG; Infante J; Moore M; Seay T; Tjulandin SA; Ma WW; Saleh MN; Harris M; Reni M; Dowden S; Laheru D; Bahary N; Ramanathan RK; Tabernero J; Hidalgo M; Goldstein D; Van Cutsem E; Wei X; Iglesias J; Renschler MF, Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Frese KK; Neesse A; Cook N; Bapiro TE; Lolkema MP; Jodrell DI; Tuveson DA, nab-Paclitaxel Potentiates Gemcitabine Activity by Reducing Cytidine Deaminase Levels in a Mouse Model of Pancreatic Cancer. Cancer Discov. 2012, 2, 260–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Wang H; Huang Y, Combination Therapy Based on Nano Codelivery for Overcoming Cancer Drug Resistance. Medicine in Drug Discovery 2020, 6, 100024. [Google Scholar]
- 254.Zhang J; Du Z; Pan S; Shi M; Li J; Yang C; Hu H; Qiao M; Chen D; Zhao X, Overcoming Multidrug Resistance by Codelivery of MDR1-Targeting siRNA and Doxorubicin Using EphA10-Mediated pH-Sensitive Lipoplexes: In Vitro and In Vivo Evaluation. ACS Appl. Mater. Interfaces 2018, 10, 21590–21600. [DOI] [PubMed] [Google Scholar]
- 255.Alimoradi H; Greish K; Barzegar-Fallah A; Alshaibani L; Pittalà V, Nitric Oxide-releasing Nanoparticles Improve Doxorubicin Anticancer Activity. Int. J. Nanomed. 2018, 13, 7771–7787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Li J; Wang Y; Zhu Y; Oupický D, Recent Advances in Delivery of Drug–nucleic acid Combinations for Cancer Treatment. J. Controlled Release 2013, 172 (2), 589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Xiong X-B; Lavasanifar A, Traceable Multifunctional Micellar Nanocarriers for Cancer-Targeted Co-delivery of MDR-1 siRNA and Doxorubicin. ACS Nano 2011, 5 (6), 5202–5213. [DOI] [PubMed] [Google Scholar]
- 258.Buque A; Bloy N; Petroni G; Kroemer G; Galluzzi L, NK cells Beat T Cells at Early Breast Cancer Control. OncoImmunology 2020, 9 (1), 1806010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Combes AJ; Samad B; Tsui J; Chew NW; Yan P; Reeder GC; Kushnoor D; Shen A; Davidson B; Barczak AJ; Adkisson M; Edwards A; Naser M; Barry KC; Courau T; Hammoudi T; Argüello RJ; Rao AA; Olshen AB; Spitzer M et al. , Discovering Dominant Tumor Immune Archetypes in a Pan-cancer Census. Cell 2022, 185, 184–203.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Wang-Gillam A; Li C-P; Bodoky G; Dean A; Shan Y-S; Jameson G; Macarulla T; Lee K-H; Cunningham D; Blanc JF; Hubner RA; Chiu C-F; Schwartsmann G; Siveke JT; Braiteh F; Moyo V; Belanger B; Dhindsa N; Bayever E; Von Hoff DD et al. , Nanoliposomal Irinotecan with Fluorouracil and Folinic Acid in Metastatic Pancreatic Cancer After Previous Gemcitabine-based Therapy (NAPOLI-1): a Global, Randomised, Open-label, Phase 3 Trial. The Lancet 2016, 387, 545–557. [DOI] [PubMed] [Google Scholar]
- 261.Pasquier E; Kavallaris M; André N, Metronomic Chemotherapy: New Rationale for New Directions. Nat. Rev. Clin. Oncol. 2010, 7, 455–465. [DOI] [PubMed] [Google Scholar]
- 262.Holohan C; Van Schaeybroeck S; Longley DB; Johnston PG, Cancer Drug Resistance: An Evolving Paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [DOI] [PubMed] [Google Scholar]
- 263.Lien K; Georgsdottir S; Sivanathan L; Chan K; Emmenegger U, Low-dose Metronomic Chemotherapy: A Systematic Literature Analysis. Eur. J. Cancer 2013, 49, 3387–3395. [DOI] [PubMed] [Google Scholar]
- 264.Doloff JC; Waxman DJ, VEGF Receptor Inhibitors Block the Ability of Metronomically Dosed Cyclophosphamide to Activate Innate Immunity–Induced Tumor Regression. Cancer Research 2012, 72, 1103–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Popilski H; Abtew E; Schwendeman S; Domb A; Stepensky D, Efficacy of Paclitaxel/Dexamethasone Intra-tumoral Delivery in Treating Orthotopic Mouse Breast Cancer. J. Controlled Release 2018, 279, 1–7. [DOI] [PubMed] [Google Scholar]
- 266.Martin JD; Panagi M; Wang C; Khan TT; Martin MR; Voutouri C; Toh K; Papageorgis P; Mpekris F; Polydorou C; Ishii G; Takahashi S; Gotohda N; Suzuki T; Wilhelm ME; Melo VA; Quader S; Norimatsu J; Lanning RM; Kojima M; Stuber MD; Stylianopoulos T; Kataoka K; Cabral H, Dexamethasone Increases Cisplatin-Loaded Nanocarrier Delivery and Efficacy in Metastatic Breast Cancer by Normalizing the Tumor Microenvironment. ACS Nano 2019, 13, 6396–6408. [DOI] [PubMed] [Google Scholar]
- 267.Zhang L; Su H; Liu Y; Pang N; Li J; Qi X-R, Enhancing Solid Tumor Therapy with Sequential Delivery of Dexamethasone and Docetaxel Engineered in a Single Carrier to Overcome Stromal Resistance to Drug Delivery. J. Controlled Release 2019, 294, 1–16. [DOI] [PubMed] [Google Scholar]
- 268.Chauhan VP; Stylianopoulos T; Martin JD; Popović Z; Chen O; Kamoun WS; Bawendi MG; Fukumura D; Jain RK, Normalization of Tumour Blood Vessels Improves the Delivery of Nanomedicines in a Size-dependent Manner. Nat. Nanotechnol. 2012, 7, 383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Jain RK, Delivery of Molecular and Cellular Medicine to Solid Tumors. Adv. Drug Delivery Rev. 2012, 64, 353–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Socinski MA; Jotte RM; Cappuzzo F; Orlandi F; Stroyakovskiy D; Nogami N; Rodríguez-Abreu D; Moro-Sibilot D; Thomas CA; Barlesi F; Finley G; Kelsch C; Lee A; Coleman S; Deng Y; Shen Y; Kowanetz M; Lopez-Chavez A; Sandler A; Reck M, Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [DOI] [PubMed] [Google Scholar]
- 271.Mpekris F; Voutouri C; Baish JW; Duda DG; Munn LL; Stylianopoulos T; Jain RK, Combining Microenvironment Normalization Strategies to Improve Cancer Immunotherapy. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 3728–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Hiam-Galvez KJ; Allen BM; Spitzer MH, Systemic Immunity in Cancer. Nat. Rev. Cancer 2021, 21, 345–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Schudel A; Francis DM; Thomas SN, Material Design for Lymph Node Drug Delivery. Nat. Rev. Mater. 2019, 4, 415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Feeney OM; Gracia G; Brundel DHS; Trevaskis NL; Cao E; Kaminskas LM; Porter CJH, Lymph-directed Immunotherapy – Harnessing Endogenous Lymphatic Distribution Pathways for Enhanced Therapeutic Outcomes in Cancer. Adv. Drug Delivery Rev. 2020, 160, 115–135. [DOI] [PubMed] [Google Scholar]
- 275.Irvine DJ; Swartz MA; Szeto GL, Engineering Synthetic Vaccines Using Cues from Natural Immunity. Nature Materials 2013, 12, 978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Thomas SN; Vokali E; Lund AW; Hubbell JA; Swartz MA, Targeting the Tumor-draining Lymph Node with Adjuvanted Nanoparticles Reshapes the Anti-tumor Immune Response. Biomaterials 2014, 35, 814–824. [DOI] [PubMed] [Google Scholar]
- 277.Choo EF; Boggs J; Zhu C; Lubach JW; Catron ND; Jenkins G; Souers AJ; Voorman R, The Role of Lymphatic Transport on the Systemic Bioavailability of the Bcl-2 Protein Family Inhibitors Navitoclax (ABT-263) and ABT-199. Drug Metabolism and Disposition 2014, 42, 207–212. [DOI] [PubMed] [Google Scholar]
- 278.Liu H; Moynihan KD; Zheng Y; Szeto GL; Li AV; Huang B; Van Egeren DS; Park C; Irvine DJ, Structure-based Programming of Lymph-node Targeting in Molecular Vaccines. Nature 2014, 507, 519–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Hu L; Quach T; Han S; Lim SF; Yadav P; Senyschyn D; Trevaskis NL; Simpson JS; Porter CJH, Glyceride-Mimetic Prodrugs Incorporating Self-Immolative Spacers Promote Lymphatic Transport, Avoid First-Pass Metabolism, and Enhance Oral Bioavailability. Angew. Chem. Int. Ed. 2016, 55, 13700–13705. [DOI] [PubMed] [Google Scholar]
- 280.Han S; Hu L; Quach T; Simpson JS; Trevaskis NL; Porter CJH, Profiling the Role of Deacylation-Reacylation in the Lymphatic Transport of a Triglyceride-Mimetic Prodrug. Pharm. Res. 2015, 32, 1830–1844. [DOI] [PubMed] [Google Scholar]
- 281.Nakamura T; Harashima H, Dawn of Lipid Nanoparticles in Lymph Node Targeting: Potential in Cancer Immunotherapy. Adv. Drug Delivery Rev. 2020, 167, 78–88. [DOI] [PubMed] [Google Scholar]
- 282.Salman B; Zhou D; Jaffee EM; Edil BH; Zheng L, Vaccine Therapy for Pancreatic Cancer. OncoImmunology 2013, 2, e26662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Bryant KL; Mancias JD; Kimmelman AC; Der CJ, KRAS: Feeding Pancreatic Cancer Proliferation. Trends in Biochemical Sciences 2014, 39 (2), 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Zhang Y; Ma J-A; Zhang H-X; Jiang Y-N; Luo W-H, Cancer Vaccines: Targeting KRAS-driven Cancers. Expert Rev. Vaccines 2020, 19, 163–173. [DOI] [PubMed] [Google Scholar]
- 285.Toh U; Sakurai S; Saku S; Takao Y; Okabe M; Iwakuma N; Shichijo S; Yamada A; Itoh K; Akagi Y, Early Phase II Study of Mixed 19-peptide Vaccine Monotherapy for Refractory Triple-negative Breast Cancer. Cancer Sci. 2020, 111, 2760–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Meng H; Yang S; Li Z; Xia T; Chen J; Ji Z; Zhang H; Wang X; Lin S; Huang C; Zhou ZH; Zink JI; Nel AE, Aspect Ratio Determines the Quantity of Mesoporous Silica Nanoparticle Uptake by a Small GTPase-Dependent Macropinocytosis Mechanism. ACS Nano 2011, 5, 4434–4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Dobrovolskaia MA; McNeil SE, Immunological Properties of Engineered Nanomaterials. Nat. Nanotechnol. 2007, 2, 469–478. [DOI] [PubMed] [Google Scholar]
- 288.Zhu J; Sevencan C; Zhang M; McCoy RSA; Ding X; Ye J; Xie J; Ariga K; Feng J; Bay BH; Leong DT, Increasing the Potential Interacting Area of Nanomedicine Enhances Its Homotypic Cancer Targeting Efficacy. ACS Nano 2020, 14, 3259–3271. [DOI] [PubMed] [Google Scholar]
- 289.Fantozzi A; Christofori G, Mouse Models of Breast Cancer Metastasis. Breast Cancer Res. 2006, 8, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Rockwell SC; Kallman RF; Fajardo LF, Characteristics of a Serially Transplanted Mouse Mammary Tumor and Its Tissue-Culture-Adapted Derivative. J. Natl. Cancer Inst. 1972, 49, 735–749. [PubMed] [Google Scholar]
- 291.Aguilera TA; Rafat M; Castellini L; Shehade H; Kariolis MS; Hui AB-Y; Stehr H; von Eyben R; Jiang D; Ellies LG; Koong AC; Diehn M; Rankin EB; Graves EE; Giaccia AJ, Reprogramming the Immunological Microenvironment Through Radiation and Targeting Axl. Nat. Commun. 2016, 7, 13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Callmann CE; Cole LE; Kusmierz CD; Huang Z; Horiuchi D; Mirkin CA, Tumor Cell Lysate-loaded Immunostimulatory Spherical Nucleic Acids as Therapeutics for Triple-negative Breast Cancer. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 17543–17550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Yu JW; Bhattacharya S; Yanamandra N; Kilian D; Shi H; Yadavilli S; Katlinskaya Y; Kaczynski H; Conner M; Benson W; Hahn A; Seestaller-Wehr L; Bi M; Vitali NJ; Tsvetkov L; Halsey W; Hughes A; Traini C; Zhou H; Jing J; Lee T; Figueroa DJ; Brett S; Hopson CB; Smothers JF; Hoos A; Srinivasan R, Tumor-immune Pofiling of Mrine Sngeneic Tmor Mdels as a Famework to Gide Mchanistic Sudies and Pedict Terapy Rsponse in Dstinct Tmor Mcroenvironments. PLOS ONE 2018, 13, e0206223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Kobayashi E; Motoki K; Uchida T; Fukushima H; Koezuka Y, KRN7000, A Novel Immunomodulator, and Its Antitumor Activities. Oncol. Res. 1995, 7, 529–534. [PubMed] [Google Scholar]
- 295.Morita M; Motoki K; Akimoto K; Natori T; Sakai T; Sawa E; Yamaji K; Koezuka Y; Kobayashi E; Fukushima H, Structure-Activity Relationship of .alpha.-Galactosylceramides against B16-Bearing Mice. J. Med. Chem. 1995, 38, 2176–2187. [DOI] [PubMed] [Google Scholar]
- 296.Zong L; Chen K; Jiang Z; Chen X; Sun L; Ma J; Zhou C; Xu Q; Duan W; Han L; Lei J; Li X; Ma Q; Wang Z, Lipoxin A4 Reverses Mesenchymal Phenotypes to Attenuate Invasion and Metastasis via the Inhibition of Autocrine TGF-β1 Signaling in Pancreatic Cancer. J. Exp. Clin. Cancer Res. 2017, 36, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Altan M; Kidwell KM; Pelekanou V; Carvajal-Hausdorf DE; Schalper KA; Toki MI; Thomas DG; Sabel MS; Hayes DF; Rimm DL, Association of B7-H4, PD-L1, and Tumor Infiltrating Lymphocytes with Outcomes in Breast Cancer. npj Breast Cancer 2018, 4, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Wang L; Yang C; Liu X.-b.; Wang L; Kang F.-b., B7-H4 Overexpression Contributes to Poor Prognosis and Drug-resistance in Triple-negative Breast Cancer. Cancer Cell Int. 2018, 18, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Benner B; Good L; Quiroga D; Schultz TE; Kassem M; Carson WE; Cherian MA; Sardesai S; Wesolowski R, Pexidartinib, a Novel Small Molecule CSF-1R Inhibitor in Use for Tenosynovial Giant Cell Tumor: A Systematic Review of Pre-Clinical and Clinical Development. Drug. Des. Devel. Ther. 2020, 14, 1693–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Ramesh A; Kumar S; Nandi D; Kulkarni A, CSF1R- and SHP2-Inhibitor-Loaded Nanoparticles Enhance Cytotoxic Activity and Phagocytosis in Tumor-Associated Macrophages. Adv. Mater. 2019, 31, 1904364. [DOI] [PubMed] [Google Scholar]
- 301.Morton JP; Timpson P; Karim SA; Ridgway RA; Athineos D; Doyle B; Jamieson NB; Oien KA; Lowy AM; Brunton VG; Frame MC; Evans TRJ; Sansom OJ, Mutant p53 Drives Metastasis and Overcomes Growth Arrest/Senescence in Pancreatic Cancer. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Ischenko I; D’Amico S; Rao M; Li J; Hayman MJ; Powers S; Petrenko O; Reich NC, KRAS Drives Immune Evasion in a Genetic Model of Pancreatic Cancer. Nat. Commun. 2021, 12, 1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Meng H; Nel AE, Use of Nano Engineered Approaches to Overcome the Stromal Barrier in Pancreatic Cancer. Adv. Drug Delivery Rev. 2018, 130, 50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Dvorak AM; Feng D, The Vesiculo–Vacuolar Organelle (VVO): A New Endothelial Cell Permeability Organelle. Journal of Histochemistry & Cytochemistry 2001, 49, 419–431. [DOI] [PubMed] [Google Scholar]
- 305.Nagy JA; Benjamin L; Zeng H; Dvorak AM; Dvorak HF, Vascular Permeability, Vascular Hyperpermeability and Angiogenesis. Angiogenesis 2008, 11, 109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Nagy JA; Dvorak AM; Dvorak HF, Vascular Hyperpermeability, Angiogenesis, and Stroma Generation. Cold Spring Harbor Perspectives in Medicine 2012, 2, a006544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Kroemer G; Galluzzi L; Kepp O; Zitvogel L, Immunogenic Cell Death in Cancer Therapy. Annu.l Rev. Immunol. 2013, 31, 51–72. [DOI] [PubMed] [Google Scholar]
- 308.Garg AD; Dudek-Peric AM; Romano E; Agostinis P, Immunogenic Cell Death. Int. J. Dev. Biol. 2015, 59, 131–40. [DOI] [PubMed] [Google Scholar]
- 309.Alexander J; Cargill R; Michelson SR; Schwam H, (Acyloxy)alkyl Carbamates as Novel Bioreversible Prodrugs for Amines: Increased Permeation Through Biological Membranes. J. Med. Chem. 1988, 31, 318–322. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.