

Abstract
Increasingly, cerebellar syndromes are recognized as affecting multiple systems. Extracerebellar features include peripheral neuropathies affecting proprioception; cranial neuropathies such as auditory and vestibular; and neuronopathies, for example, dorsal root and vestibular. The presence of such features, which in and of themselves may cause ataxia, likely contribute to key disabilities such as gait instability and falls. Based on the evolving available literature and experience, we outline a clinical approach to the diagnosis of adult-onset ataxia where a combination of cerebellar and peripheral or cranial nerve pathology exists. Objective diagnostic modalities including electrophysiology, oculomotor, and vestibular function testing are invaluable in accurately defining an individual's phenotype. Advances in MRI techniques have led to an increased recognition of disease-specific patterns of cerebellar pathology, including those conditions where neuronopathies may be involved. Depending on availability, a stepwise approach to genetic testing is suggested. This is guided by factors such as pattern of inheritance and age at disease onset, and genetic testing may range from specific genetic panels through to whole-exome and whole-genome sequencing. Management is best performed with the involvement of a multidisciplinary team, aiming at minimization of complications such as falls and aspiration pneumonia and maximizing functional status.
The clinical manifestations of cerebellar impairment focus on the central concept of ataxia, which is often summarized as incoordination.1 Increasingly, the far-reaching influence of the cerebellum has resulted in an expanding understanding of the range of clinical expressions of cerebellar disease, which may affect a variable number of domains including, but not limited to, oculomotor, speech, appendicular, gait, and cognition and affect.2 Many clinical features are less than specific for cerebellar disorders, particularly in isolation, and these may include reduced speech intelligibility, motion-induced or spontaneous oscillopsia, and decreased dexterity. Gait disturbance, imbalance, and falls are nonspecific and may be caused by pathology affecting other systems, such as vestibular or somatosensory deficits.3
The noncerebellar manifestations of cerebellar diseases are increasingly recognized and find value in both clinical diagnosis and research progress.3 Previous publications have covered issues such as the existence of peripheral neuropathy in cerebellar diseases, so in this work, we aimed to aid the clinician in approaching patients with cerebellar presentations, particularly regarding extracerebellar pathologies that target ganglia, peripheral, and cranial nerves. Vestibulopathy is an increasingly recognized extracerebellar feature of cerebellar diseases, including the most common sporadic and inherited diseases: idiopathic late-onset cerebellar ataxia (CA)/sporadic late-onset CA, idiopathic cerebellar ataxia with bilateral vestibulopathy (CABV) spinocerebellar ataxia (SCA) 3 and 6, Friedreich ataxia (FRDA), and cerebellar ataxia, neuronopathy, vestibular areflexia syndrome (CANVAS)/RFC1-related disease. Vestibulopathy is not uncommonly believed to be the result of a likely cranial neuropathy (but is generally of uncertain pathology, given the lack of published temporal bone pathology in CAs).
A broad-based unsteady gait may be common to cerebellar, sensory, and vestibular impairment, and therefore, more specific signs are sought to identify or differentiate between these. Oculomotor abnormalities are highly specific, with many reflecting pathology of the cerebellum (and its connections), for example, saccadic visual pursuit, direction-changing nystagmus, and saccadic dysmetria,4 and a lesser number indicating vestibular pathology,5 for example, corrective saccades on performing the head impulse test.6 In addition to oculomotor signs, other more specific localizing abnormalities on examination include cerebellar dysarthria and appendicular ataxia, both seen in cerebellar impairment.7
Cerebellar and vestibular diseases resulting in ataxia may be accompanied by a peripheral neuropathy or neuronopathy.8,9 In addition, peripheral neuropathy or neuronopathy may be associated with vestibular and/or auditory neuropathy.10,11 In individuals with ataxia, identification and characterization of a neuropathy or neuronopathy (cranial, e.g., vestibular or peripheral) is important in defining a person's phenotype as a necessary step in progressing diagnosis and management.
A literature search of PubMed without date of publication limitation was performed using the terms “ataxia” OR “cerebell*” OR “neuropathy” OR “neuronopathy” OR “ganglionopathy” OR “polyneuropathy” OR “vestibulopathy,” with the results manually curated for relevance. If appropriate, primary sources quoted in references were also retrieved and curated. In this study, we focused on the clinical diagnosis of adult-onset ataxia that presents with the combination of cerebellar and peripheral or cranial nerve pathology.
Objective Functional Cerebellar Assessment
Objective assessment of cerebellar function (as opposed to structure, i.e., that accomplished by MRI) in most domains is yet to become clinically available; formal testing of vestibular and somatosensory function is readily available and should form an integral part of the clinical assessment of any individual with evidence of CA. The exception to the lack of objective assessment of cerebellar function is oculomotor measurement, which can be performed with a range of widely available video-oculography systems.12 Oculomotor testing is aimed at the identification of commonly seen abnormalities such as saccadic (or broken-up) pursuit, saccadic vestibulo-ocular (VOR) suppression, gaze-evoked nystagmus, dysmetric, latent, or slow saccades to target, and saccadic intrusions.4
Formal Vestibular Function Testing
Diagnosis of a vestibular component aids both the diagnostic process by potentially reducing the number of differential diagnoses and management, for instance, the addition of vestibular rehabilitation to the standard-of-care neurologic rehabilitation.13 Assessment of vestibular function may be performed by a facility that offers formal testing, and the modalities that are available to assess semicircular canal function are the video head impulse test (vHIT), rotational chair, and bithermal caloric irrigation.14 The otolith organs (utricle and saccule) are generally assessed by vestibular-evoked myogenic potentials15 or the subjective visual vertical (or horizontal) test.16 Given the ease of use, brevity of testing, and affordability, many clinicians now have a vHIT unit in their office, and we find this to be a very satisfactory screening test of vestibular involvement in our individuals with CA. It should be noted that the vHIT17 offers several advantages over the clinical HIT (cHIT) including abrogation of false-negative results due to the so-called covert corrective saccades, which are imperceptible to the clinician during the cHIT but clearly recorded by the vHIT.17 In addition, the vHIT offers excellent sensitivity,18 and the system may also be used to objectively test “cerebellar” oculomotor abnormalities such as visual pursuit, VOR suppression, and saccades to target. In addition, these units can be used to identify saccadic (or abnormal) visually enhanced VOR, which confirms the presence of combined bilateral vestibular hypofunction and cerebellar oculomotor dysfunction.19
Vestibular involvement in cerebellar syndromes may be due to a cranial neuropathy/neuronopathy, but regardless of its origin, its assessment is particularly valuable in establishing a patient's phenotype as a means of narrowing the range of differential diagnoses. It is worthwhile noting that although vestibular involvement in CAs is significant, otopathology is available only for very few cerebellar conditions, so whether the etiology is a vestibular neuropathy, vestibular neuronopathy, or end-organ pathology for most CAs is unknown. In the context of cerebellar diseases, vestibular involvement generally manifests as a bilateral hypofunction, affecting the semicircular canals,20 but may also involve the otolith organs, for example, where the vestibular ganglia are involved.21-23 The range of symptoms that accompany bilateral vestibular dysfunction include imbalance and motion-induced oscillopsia. Unfortunately, these symptoms are also seen in cerebellar disease, so they would be compatible with either cerebellar and/or vestibular impairment and, in the case of imbalance, somatosensory impairment as well. Therefore, in addition to electrophysiology, we advise vestibular function testing in all individuals with ataxia.
Neuropathy
When somatosensory loss is present, phenotypic assessment and nerve conduction studies can usually characterize the neuropathy/neuronopathy as axonal or demyelinating, large and/or small fiber, length dependent or nonlength dependent, and symmetric or asymmetric and determine the degree of motor and sensory involvement (Table 1). Impaired vibration and proprioception suggest large-fiber involvement, whereas paraesthesia, neuropathic pain, and burning sensations suggest small-fiber involvement. Our standard nerve conduction study protocol includes at least unilateral median, ulnar, radial, sural, and superficial peroneal antidromic sensory studies along with median, ulnar, fibular, and tibial motor studies. In addition, we find blink reflexes and mechanically activated masseter reflex helpful in assessing neuropathy and neuronopathy because neuropathy affects both tests, but in neuronopathy, the masseter reflex may not be affected because the cell bodies are in the brainstem. Autonomic involvement is suggested by a history of orthostatic dizziness, temperature intolerance, and bladder, gastrointestinal tract, and erectile dysfunction. The presence of any of these symptoms should lead to a careful examination and investigations to determine whether peripheral nerve dysfunction is associated with the overall disorder or whether it may be an incidental disorder, as previously shown in SCA 6.24
Table 1.
Electrophysiologic Findings in Neuropathies/Neuronopathies

Balance depends on the integration of responses to sensory inputs from the vestibular, somatosensory, and visual systems. The vestibular and somatosensory systems react rapidly to acceleration of the head and to postural change and motion especially in the lower limbs, respectively. The visual system reacts later to stabilize the reactions to the earlier vestibular and somatosensory inputs. Therefore, if ataxia or imbalance is present only when visual inputs are removed (such as with eye closure or darkness), then proprioceptive sensory loss and/or vestibular dysfunction are likely.
Although in somatosensory disorders, there has been limited investigation of vestibular function, evidence suggests that vestibular and/or auditory neuropathy or neuronopathy is common, but possibly the slow onset of the conditions might obscure the vestibular component.25 When ataxia or imbalance is out of proportion to the objective somatosensory findings, then vestibular involvement should be considered. An ataxic gait is common in individuals with diabetes mellitus and may be associated with vestibulopathies or caused by hyperglycemia and changing glucose levels. Autonomic neuropathy often also contributes. Other neuropathies including Guillain-Barre syndrome (GBS), less commonly chronic inflammatory demyelinating polyradiculoneuropathy (CIDP),26 and inherited neuropathies are associated with cerebellar pathology and vestibular neuropathy. The combination of sensorineural hearing loss (in the case of FRDA, also auditory neuropathy) and vestibular neuropathy is seen in other diseases, including FRDA,27-29 Charcot-Marie Tooth (CMT) disease,30 and Refsum disease.31,32
In demyelinating neuropathies, vestibular dysfunction seems common. Frohman et al.33 in 1996 reported the first case of fluctuating vestibular syndrome paralleling the course and treatment of CIDP. Since then, several authors have confirmed vestibular involvement in CIDP and its variants, as well as GBS.25,26 Therefore, if ataxia is present with somatosensory loss and retained visual function, there is likely to be significant somatosensory loss, with or without vestibular dysfunction, and the latter should be investigated.
Similarly, in CMT, it has been shown that vestibular impairment is common, and it has been suggested that cervical vestibular myogenic potentials might be a more sensitive test than vHIT.34 It is not clear whether the neuropathy in these cases were demyelinating or axonal.
In axonal neuropathies, it is also suggested that vestibular neuropathy may be as common as in demyelinating neuropathies,10 but differing methodologies make this difficult to assess.11 Sensory neuronopathies may be acquired, usually of subacute onset, and may be paraneoplastic, immune mediated, infectious, toxic, idiopathic, or may be inherited, such as CANVAS and FRDA. These are associated with ataxia. Many of these have not been well investigated to assess the mechanism of ataxia, although in the most common phenotype of RFC1-related disease,21,35 CANVAS, the ataxia is also related to the vestibular ganglionopathy and the cerebellar pathology.22,36,37
Somatosensory disorders are a common and integral part of several of the spinocerebellar ataxias (SCAs). Peripheral neuropathy/neuronopathy is frequent in SCA1, SCA2, and SCA3,38 and although there are conflicting reports, it is probably not present in SCA6.24 A length-dependent neuropathy is an integral part of autosomal recessive spastic ataxia of Charlevoix-Saguenay.20 For guidance on the laboratory investigation of peripheral neuropathies, we refer the reader to Lau.39
Magnetic Resonance Imaging
Clinical radiologic assessment using MRI is often used after ataxia symptom presentation, particularly in the absence of an established genetic etiology, to qualitatively identify and grade any atrophy or detect lesions in the posterior fossa.23 However, the value of quantitative assessments of cerebellar, brainstem, and spinal cord macrostructure (i.e., atrophy), microstructure (i.e., diffusion), and even neurochemistry (i.e., spectroscopy) in differential diagnosis and disease monitoring of degenerative ataxias is becoming increasingly clear.40 The need for translation of quantitative research practices to the clinic is particularly salient, given the evidence that the clinical onset of ataxia may often be a late manifestation of well-advanced neuropathology or masked by the peripheral findings in a multisystem disease process. As such, we argue that there should be a low threshold to obtain neuroimaging of the brain and spinal cord in a person presenting with ataxia to evaluate for cerebellar, pontine, and dorsal column or corticospinal tract involvement particularly.
Assessment of precise cerebral and cerebellar anatomical involvement in different disease processes can be obtained via in vivo brain morphometry using MRI combined with postimaging automated software analysis such as voxel-based morphometry41 or feature-based anatomical image parcellation.40 Analogous approaches are also available for diffusion-weighted MRI, including tract-based spatial statistics and probabilistic tractography methods, to identify abnormalities in the integrity of different white matter tracts. These approaches have applications in identifying pathology relative to healthy control participants, as well as differentiating between disease entities with cerebellar involvement.
Quantitative research undertaken to date has largely focused on the inherited ataxias. In the SCAs, for example, degeneration is preferential in cerebellar lobules VII-IX in SCA2, additionally involves lobule VI and the medial anterior lobe in SCA3, while affecting lobules IX-X in SCA7. The disproportionate involvement of motor pathways in SCA3 correlates with the more severe motor phenotype. In FRDA, atrophy is preferentially localized to the cerebellar anterior lobe and superior cerebellar peduncle, although with increasing corticospinal and cerebral involvement with disease progression.42
In idiopathic progressive ataxias, research has principally focused on multiple system atrophy (MSA). Gray and white matter abnormalities in the posterior fossa appear to be restricted to the cerebellar variant of MSA (relative to the parkinsonian variant). Faber et al. also demonstrated that cerebellar degeneration occurs in individuals with both MSA–cerebellar subtype and sporadic late-onset CA, but that the brainstem and corticospinal tracts are affected only in those with MSA–cerebellar subtype.43 In individuals with CANVAS, case series have identified prominent involvement of cerebellar atrophy involving anterior and dorsal vermis, as well as crus I3. A recent quantitative analysis of a cohort of individuals with the RFC1 gene expansion also reported cerebellar degeneration largely weighted to the vermis, anterior lobe (lobules I–IV), and superior aspects of the posterior lobe (lobules V, VI and crus I). Subcortical structures were also strongly implicated, alongside a relevant sparing of the cerebral cortex.44
Neurodegenerative disorders causing chronic ataxia require different MRI acquisition protocols than those used in the diagnosis of other common neurologic disorders, such as stroke and epilepsy, although typically are broadly similar or equivalent to those used for other neurodegenerative and movement disorders. A major distinction for ataxias, however, is the additional value of spinal cord measures. Qualitative grading and quantitative measurements of cerebellar atrophy are also aided by higher-resolution images acquired at high field strengths (3T). Objective automated imaging analysis and MRI will continue to play an important role in the clinical evaluation of individuals with ataxia.
Genetic Testing
The first step in genetic analysis of ataxia is to draw a 3-generation pedigree. This will allow analysis of pattern of inheritance including autosomal dominant, autosomal recessive, X-linked, and mitochondrial. If the pedigree is consistent with autosomal-dominant, adult-onset ataxia, then testing for the common SCAs is the first genetic test. This will often include testing for the trinucleotide repeat expansions that underlie SCA 1, 2, 3, 6, and 7. This currently requires a stand-alone test because such expansions cannot be identified by genomic sequencing in most diagnostic laboratories. If no cause is found, then genomic sequencing should be considered to identify missense and nonsense pathogenic variants, as well as insertions, small deletions, and duplications as the cause. This can be whole-genome sequencing, whole-exome sequencing, or panel testing.
Ataxia in a young person should prompt testing for FRDA and ataxia telangiectasia (alpha fetoprotein is almost always elevated in the latter). Again, if no cause is found, then genomic testing should be considered. In an individual who has an onset of ataxia in adulthood where the family history is consistent with recessive inheritance or the individual is the only one in the family with the condition, in addition to testing for FRDA, testing for the biallelic pentanucleotide repeat expansion in RFC1 should take place. Testing by chromosomal microarray for copy number variants will occasionally diagnose the cause for ataxia such as SCA15, which is often due to a deletion of ITPR1. Testing for the CGG repeat expansion in FMR1 will enable the diagnosis of fragile X tremor ataxia syndrome (FXTAS).
Whole-genome sequencing can identify most genetic causes of ataxia including repeat expansions, point mutations, and copy number variants.45 The mitochondrial genome can be interrogated at the same time as the nuclear genome. Mitochondrial genome sequencing can identify several causes of ataxia with neuropathy such as neuropathy, ataxia, and retinitis pigmentosa, most commonly due to heteroplasmic point mutations in MT-ATP6. While whole-genome sequencing is currently relatively expensive, in the future, it is likely that this will be the first step in the genetic investigation of ataxia, removing the need for multiple individual tests.
For more detailed information on the genetic testing approach to ataxia, the reader is directed to Witek et al.46 For the general investigation of the etiology of CA, we refer the reader to previously published work.47 In the context of CA with a neuropathy or neuronopathy, after an objective investigation with electrophysiology and oculomotor and vestibular function testing, establishment of the individual's phenotype will guide genetic testing.
Multidisciplinary Management of Ataxia
Pharmaceutical intervention for the ataxias is constantly evolving and will not be a subject of this section. The foundation of successful management of ataxia lies with the multidisciplinary team, focused on management of the motor and nonmotor aspects of CA. Moreover, being familiar with clinical management guidelines is an essential component of evidence-based management of individuals with CA.48
We include the following in our routine management of individuals with the spectrum of diseases covered by this article in an effort to mitigate complications and aid the maintenance of functional status. In the first instance, it is crucial individuals with ataxia have access to multidisciplinary care including neurology, physiotherapy, occupational therapy, speech pathology, social work, audiology, and psychology (Table 2). In the context of there often being no “cure” for these conditions, regular review, instigation, and maintenance of appropriate medical and allied health management are the single most important interventions available to ensure a person with CA can maintain health and well-being for as long as possible. Without such intervention, the rate of progression and associated dependence and debility (including associated care costs) is significantly greater.49
Table 2.
The Role of Members of a Multidisciplinary Team in Nonmotor and Motor Issues Related to Cerebellar Ataxia
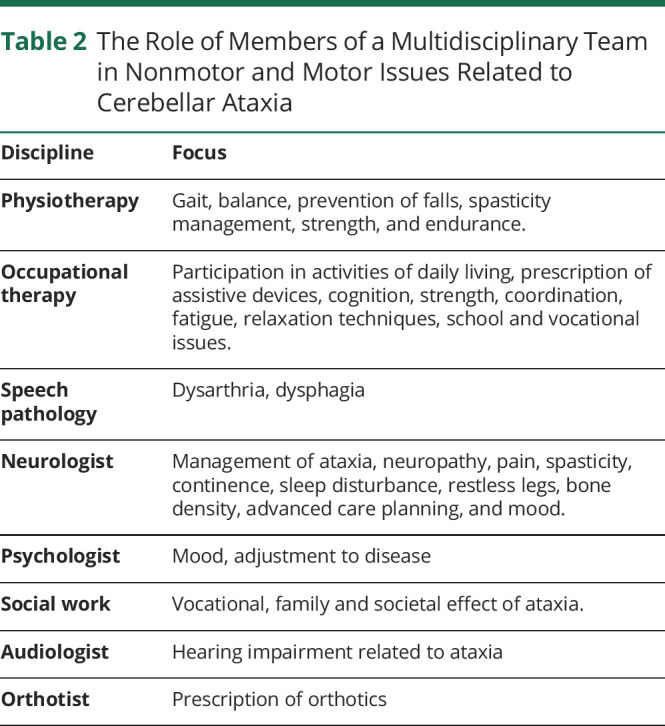
Intensive neurorehabilitation has demonstrated efficacy in improving gait, coordination, functional status, health, and well-being in individuals with CA. In particular, regular access to periods of intensive neurorehabilitation supplemented by a targeted home-based activity program is important in maintaining function.50
Speech therapy aims to optimize verbal communication where cerebellar dysarthria is present,51 and given the risk of aspiration pneumonia, management of dysphagia is a management priority.52 The role of occupational therapy differs depending on the individual's circumstances, however, should comprise at a minimum an assessment of function in daily activities including participation in personal care, domestic, school, vocational, and community-based activities.53 The occupational therapist is crucial to identify the effect of changes in cognition and fatigue on functional performance. If necessary, prescription of assistive devices and/or advice regarding accessibility and safety in the home and community should be provided. Physiotherapy finds multiple applications including functional mobility and fall prevention.54 Spasticity, pain, and spasms are best managed by a multidisciplinary team that can offer physical therapy, focal and/or systemic medications, and application of orthoses.
More recently, noninvasive brain stimulation (NIBS) techniques have been the focus of multiple studies exploring the benefit of NIBS as a stand-alone and/or adjunct to traditional intervention for ataxia such as physiotherapy.55 In addition, a person with ataxia should undergo dual energy X-ray absoptiometry bone densitometry and bone health treatment as required in an effort to mitigate the potentially serious sequelae of falls.56 Finally, individuals with ataxia may have an increased risk of sleep disordered breathing that may necessitate regular surveillance and intervention as indicated.57,58
It seems likely that the expanding diversity in phenotypes of known CAs will continue. Similarly, new cerebellar diseases and genes will be discovered. These factors place an ever-increasing demand on detailed, objective assessment and characterization of our patients as a necessary step toward greater diagnostic acuity and, in a wider context, disease and treatment discovery.
Glossary
- CA
cerebellar ataxia
- CABV
cerebellar ataxia with bilateral vestibulopathy
- CANVAS
cerebellar ataxia, neuronopathy, vestibular areflexia syndrome
- CIDP
chronic inflammatory demyelinating polyradiculoneuropathy
- CMT
Charcot-Marie-Tooth
- FRDA
Friedreich ataxia
- GBS
Guillain-Barre Syndrome
- FXTAS
fragile X tremor ataxia syndrome
- MSA
multiple system atrophy
- NIBS
noninvasive brain stimulation
- SCA
spinocerebellar ataxia
- vHIT
video head impulse test
- VOR
vestibulo-ocular reflex
Contributor Information
Leslie J. Roberts, Email: lesroberts1@bigpond.com.
Michael McVeigh, Email: 11mmcv@gmail.com.
Linda Seiderer, Email: linda.seiderer@svha.org.au.
Ian H. Harding, Email: ian.harding@monash.edu.
Louise A. Corben, Email: louise.corben@mcri.edu.au.
Martin Delatycki, Email: martin.delatycki@vcgs.org.au.
Study Funding
No targeted funding reported.
Disclosure
The authors report no relevant disclosures. Go to Neurology.org/NG for full disclosures.
References
- 1.Ashizawa T, Xia G. Ataxia. Continuum. 2016;22(4 Movement Disorders):1208–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koziol LF, Budding D, Andreasen N, et al. . Consensus paper: the cerebellum's role in movement and cognition. Cerebellum. 2014;13(1):151-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schniepp R, Möhwald K, Wuehr M. Gait ataxia in humans: vestibular and cerebellar control of dynamic stability. J Neurol. 2017;264(suppl 1):87-92. [DOI] [PubMed] [Google Scholar]
- 4.Kheradmand A, Zee DS. Cerebellum and ocular motor control. Front Neurol. 2011;2:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schubert MC, Minor LB. Vestibulo-ocular physiology underlying vestibular hypofunction, Phys Ther. 2004;84(4):373-385. [PubMed] [Google Scholar]
- 6.Halmagyi GM, Curthoys IS. A clinical sign of canal paresis. Arch Neurol. 1988;45(7):737-739. [DOI] [PubMed] [Google Scholar]
- 7.Bodranghien F, Bastian A, Casali C, et al. . Consensus paper: revisiting the symptoms and signs of cerebellar syndrome. Cerebellum. 2016;15(3):369-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liang L, Chen T, Wu Y. The electrophysiology of spinocerebellar ataxias. Neurophysiol Clin. 2016;46(1):27-34. [DOI] [PubMed] [Google Scholar]
- 9.Szmulewicz DJ, McLean CA, Rodriguez ML, et al. Dorsal root ganglionopathy is responsible for the sensory impairment in CANVAS. Neurology. 2014;82(16):1410-1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palla A, Schmid-Priscoveanu A, Studer A, Hess K, Straumann D. Deficient high-acceleration vestibular function in patients with polyneuropathy. Neurology. 2009;72(23):2009-2013. [DOI] [PubMed] [Google Scholar]
- 11.Buetti B, Luxon LM. Vestibular involvement in peripheral neuropathy: a review. Int J Audiol. 2014;53(6):353-359. [DOI] [PubMed] [Google Scholar]
- 12.Haslwanter T, Clarke AH. Eye movement measurement: electro-oculography and video-oculography. In: Edders SD, Zee DS editors. Handbook of Clinical Neurophysiology: Elsevier; 2010:61-79. [Google Scholar]
- 13.Hassannia F, Misale P, Sulway S, et al. . Effectiveness of vestibular rehabilitation therapy in patients with idiopathic cerebellar ataxia with bilateral vestibulopathy (iCABV). J Vestib Res. Epub 2022 May 6. [DOI] [PubMed] [Google Scholar]
- 14.Curthoys IS. The interpretation of clinical tests of peripheral vestibular function. Laryngoscope. 2012;122(6):1342-1352. [DOI] [PubMed] [Google Scholar]
- 15.Colebatch JG, Halmagyi GM. Vestibular evoked potentials in human neck muscles before and after unilateral vestibular deafferentation. Neurology. 1992;(8): 1635-1635. [DOI] [PubMed] [Google Scholar]
- 16.Friedmann G. The judgement of the visual vertical and horizontal with peripheral and central vestibular lesions. Brain. 1970;93(2):313-328. [DOI] [PubMed] [Google Scholar]
- 17.MacDougall HG, Weber KP, McGarvie LA, Halmagyi GM, Curthoys IS. The video head impulse test: diagnostic accuracy in peripheral vestibulopathy. Neurology. 2009;73(14):1134-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halmagyi GM, Chen L, MacDougall HG, Weber KP, McGarvie LA and Curthoys IS. The video head impulse test. Front Neurol. 2017;8:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szmulewicz DJ. Combined central and peripheral degenerative vestibular disorders: CANVAS, idiopathic cerebellar ataxia with bilateral vestibulopathy (CABV) and other differential diagnoses of the CABV phenotype. Curr Otorhinolaryngol Rep. 2017;5:167-174. [Google Scholar]
- 20.Bouchard JP, Barbeau A, Bouchard R, Bouchard RW. Electromyography and nerve conduction studies in Friedreich's ataxia and autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Can J Neurol Sci. 1979;6(2):185-189. [DOI] [PubMed] [Google Scholar]
- 21.Rafehi H, Szmulewicz DJ, Bennett MF, et al. . Bioinformatics-based identification of expanded repeats: a non-reference intronic pentamer expansion in RFC1 causes CANVAS. Am J Hum Genet. 2019;105(1):151-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szmulewicz DJ, Merchant SN, Halmagyi GM. Cerebellar ataxia with neuropathy and bilateral vestibular areflexia syndrome: a histopathologic case report. Otol Neurotol. 2011;32(8):e63-e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deistung A, Stefanescu MR, Ernst TM, et al. Structural and functional magnetic resonance imaging of the cerebellum: considerations for assessing cerebellar ataxias. Cerebellum. 2016;15(1):21-25. [DOI] [PubMed] [Google Scholar]
- 24.Zhang W, Jasinarachchi M, Seiderer L, Szmulewicz DJ, Roberts LJ. The electrophysiological findings in spinocerebellar ataxia type 6: evidence from 24 patients. J Clin Neurophysiol. Epub 2021 Apr 30. [DOI] [PubMed] [Google Scholar]
- 25.Blanquet M, Petersen JA, Palla A, et al. Vestibulo-cochlear function in inflammatory neuropathies. Clin Neurophysiol. 2018;129(4):863-873. [DOI] [PubMed] [Google Scholar]
- 26.Akdal G, Tanrıverdizade T, Şengün İ, et al. . Vestibular impairment in chronic inflammatory demyelinating polyneuropathy. J Neurol. 2018;265(2):381-387. [DOI] [PubMed] [Google Scholar]
- 27.Rance R, Fava R, Baldock H, et al. . Speech perception ability in individuals with Friedreich ataxia, Brain, 2008;131(8):2002-2012. [DOI] [PubMed] [Google Scholar]
- 28.Ell J, Prasher D, Rudge P. Neuro-otological abnormalities in Friedreich's ataxia. J Neurol Neurosurg Psychiatry. 1984;47(1):26-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fahey MC, Cremer PD, Aw ST, et al. . Vestibular, saccadic and fixation abnormalities in genetically confirmed Friedreich ataxia. Brain. 2008;131(Pt 4):1035-1045. [DOI] [PubMed] [Google Scholar]
- 30.Nadol JB Jr, Hedley-Whyte ET, Amr SS, O Apos Malley JT, Kamakura T. Histopathology of the inner ear in charcot-marie-tooth syndrome caused by a missense variant (p.Thr65AL) in the MPZ gene. Audiol Neurootol. 2018;23(6):326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oysu C, Aslan I, Basaran B, Baserer N, The site of the hearing loss in Refsum's disease. Int J Pediatr Otorhinolaryngol 2001;61(2):129-134. [DOI] [PubMed] [Google Scholar]
- 32.Taylor RL, Jankelowitz SK, Young AS, Sullivan D, Halmagyi GM, Welgampola MS. Reversible vestibular neuropathy in adult Refsum disease. Neurology. 2018;8;90(19):890-892. [DOI] [PubMed] [Google Scholar]
- 33.Frohman EM, Tusa R, Mark AS, Cornblath DR. Vestibular dysfunction in chronic inflammatory demyelinating polyneuropathy. Ann Neurol. 1996;39(4):529-535. [DOI] [PubMed] [Google Scholar]
- 34.Poretti A, Palla A, Tarnutzer AA, et al. Vestibular impairment in patients with Charcot-Marie-tooth disease. Neurology. 2013;80(23):2099-2105. [DOI] [PubMed] [Google Scholar]
- 35.Cortese A, Simone R, Sullivan R, et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat Genet. 2019;51(4):649-658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ishai R, Seyyedi M, Chancellor AM, et al. . The pathology of the vestibular system in CANVAS. Otol Neurotol. 2021;42(3):e332-e340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szmulewicz DJ, Roberts L, McLean CA, MacDougall HG, Halmagyi GM, Storey E. Proposed diagnostic criteria for cerebellar ataxia with neuropathy and vestibular areflexia syndrome (CANVAS). Neurol Clin Pract. 2016;6(1):61-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yadav R, Pal PK, Krishna N, Amar BR, Jain S, Purushottam M. Electrophysiological evaluation of spinocerebellar ataxias 1, 2 and 3. J Neurol Sci. 2012;15;(1-2):142-145. [DOI] [PubMed] [Google Scholar]
- 39.Lau KHV. Laboratory evaluation of peripheral neuropathy. Semin Neuroll. 2019;39:531-541. [DOI] [PubMed] [Google Scholar]
- 40.Öz G, Harding IH, Krahe J. Reetz K. MR imaging and spectroscopy in degenerative ataxias: towards multi-modal, multi-site, multi-stage monitoring of neurodegeneration. Curr Opin Neurol. 2020;33(4):451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kerestes R, Han S, Balachander S, et al. A standardized pipeline for examining human cerebellar grey matter morphometry using structural magnetic resonance imaging. J Vis Exp. 2022;4(180). [DOI] [PubMed] [Google Scholar]
- 42.Harding IH, Chopra S, Arrigoni F, et al. . Brain structure and degeneration staging in Friedreich ataxia: magnetic resonance imaging volumetrics from the ENIGMA‐ataxia working group. Ann Neurol. 2021;90(4):570-583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Faber J, Giordano I, Jiang X, et al. . Prominent white matter involvement in multiple system Atrophy of cerebellar type. Mov Disord. 2020;35(5):816–824. [DOI] [PubMed] [Google Scholar]
- 44.Matos PCA, Rezende TJ, Schmitt GS, et al. . Brain Structural signature of RFC1–related disorder. Mov Disord. 2021;36(11):2634-2641. [DOI] [PubMed] [Google Scholar]
- 45.Tankard RM, Bennett MF, Degorski P, Delatycki MB, Lockhart PJ, Bahlo M. Detecting expansions of tandem repeats in cohorts sequenced with short-read sequencing data. Am J Hum Genet. 2018;103(6):858-873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Witek N, Hawkins J, Hall D. Genetic ataxias: update on classification and diagnostic approaches. Curr Neurol Neurosci Rep. 2021;21:13. [DOI] [PubMed] [Google Scholar]
- 47.de Silva RN, Vallortigara J, Greenfield J, Hunt B, Giunti P, Hadjivassiliou M. Diagnosis and management of progressive ataxia in adults. Pract Neurol. 2019;19(3):196-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Silva R, Greenfield J, Cook A, et al. . Guidelines on the diagnosis and management of the progressive ataxias. Orphanet J Rare Dis. 2019;14(1):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fonteyn EM, Keus SH, Verstappen CC, Schöls L, de Groot IJ, van de Warrenburg BP. The effectiveness of allied health care in patients with ataxia: a systematic review. J Neurol. 2014;261:251-258. [DOI] [PubMed] [Google Scholar]
- 50.Ilg W, Brötz D, Burkard S, et al. . Long-term effects of coordinative training in degenerative cerebellar disease. Mov Disord. 2010;25:2239-2246. [DOI] [PubMed] [Google Scholar]
- 51.Vogel AP, Stoll LH, Oettinger Aet al. . Speech treatment improves dysarthria in multisystemic ataxia: a rater-blinded, controlled pilot-study in ARSACS. J Neurol. 2019;266;260-1266. [DOI] [PubMed] [Google Scholar]
- 52.Ilg W, Branscheidt M, Butala A, et al. Consensus paper: neurophysiological assessments of ataxias in daily practice. Cerebellum. 2018;17(5):628-653. [DOI] [PubMed] [Google Scholar]
- 53.Silva RC, Saute JA, Silva AC, Coutinho AC, Saraiva-Pereira ML, Jardim LB. Occupational therapy in spinocerebellar ataxia type 3: an open-label trial. Braz J Med Biol Res. 2010;43(6):537–542. [DOI] [PubMed] [Google Scholar]
- 54.Ilg W, Bastian AJ, Boesch S, et al. . Consensus paper: management of degenerative cerebellar disorders. Cerebellum 2014;13:248-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manto M, Argyropoulos GPD, Bocci T, et al. . Consensus paper: novel directions and next steps of non-invasive brain stimulation of the cerebellum in health and disease. 2021. [DOI] [PubMed] [Google Scholar]
- 56.Farias AM, Appenzeller S, França MC, et al. Assessment of bone mineral density of patients with spinocerebellar ataxia type 3. J Mov Disord. 2019;12(1):43-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.LaGrappe D, Massey L, Kruavit A. Sleep disorders among aboriginal Australians with machado-joseph disease: quantitative results from a multiple methods study to assess the experience of people living with the disease and their caregivers. Neurobiol Sleep Circadian Rhythms. 2022;12:100075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corben LA, Ho M, Copland J, Tai G, Delatycki MB. Increased prevalence of sleep disordered breathing in Friedreich ataxia. Neurology. 2013;81(1):40-45. [DOI] [PubMed] [Google Scholar]