

Abstract
Store operated Ca2+ entry (SOCE) is an ancient and ubiquitous Ca2+ signaling pathway discovered decades ago, but the function of SOCE in human physiology is only now being revealed. The relevance of this pathway to striated muscle was solidified with the description of skeletal myopathies that result from mutations in STIM1 and Orai1, the two SOCE components. Here, we consider the evidence for STIM1 and SOCE in cardiac muscle and the sinoatrial node. We highlight recent studies revealing a role for STIM1 in cardiac growth in response to developmental and pathologic cues. We also review the role of STIM1 in the regulation of SOCE and Ca2+ store refilling in a non-Orai dependent manner. Finally, we discuss the importance of this pathway in ventricular cardiomyocytes where SOCE contribute to developmental growth and in pacemaker cells where SOCE likely has a fundamental to generating the cardiac rhythm.
Keywords: Stromal interaction molecule 1 (STIM1), store-operated calcium entry, Store-operated calcium channels, cardiac muscle
Graphical Abstract
Ca2+ signaling in cardiomyocytes, as in all cells, is critical for many cellular functions ranging from muscle contraction to the regulation of gene expression and oxidative metabolism[1]. Coordination of these Ca2+ dependent processes enable muscle cells to adjust cellular output to the imposed workload. The cellular outputs for cardiac muscle depend on the specific cell type. For atrial and ventricular cardiomyocytes, Ca2+ dynamics are essential for the synchronized sarcomeric contraction that occurs in these cells. In contrast, pacemaking cardiomyocytes utilize rhythmic Ca2+ oscillations that drive each heartbeat in the form of a Ca2+ clock[2, 3]. The frequency and amplitude of intracellular Ca2+ transients must be adjustable in order to match demand with alterations in heart rate and the intensity of contraction of the muscle. For example, activity levels or even postural changes require adjustments in heart rate and blood pressure as set by neural or hormonal inputs to cardiomyocytes. While these changes to Ca2+ transients can result in immediate alterations in heart rate and contractility, the muscle cells are also given instructions on how to adjust in the long-term to the altered metabolic demands. For example, greater contractility requires mitochondria to adjust oxidative metabolism in order to maintain bioenergetics and ATP homeostasis[4]. If contractility was increased in the absence of the metabolic adaptation, cardiomyocyte dysfunction would eventually lead to LV systolic dysfunction and ultimately heart failure. Thus enzymes sense the changes in mitochondrial Ca2+ that accompany Ca2+ transients and alter activity for those dehydrogenases involved in the citric acid cycle[5]. Along these same lines, cardiomyocytes exposed to stress alter gene expression profiles to promote greater force production and optimized bioenergetics, which can be adaptive or maladaptive depending of the cellular context. Thus, encoded in Ca2+ transients is not only the information needed to trigger sarcomeric contraction, but also the signals required to activate many facets of the Ca2+-calmodulin signal transduction cascades as well as the regulation of oxidative metabolism[6].
For cardiomyocytes, Ca2+ transients elicited from a single action potential represent the integrated actions of channels and transporters that link membrane depolarization to muscle contraction. In response to depolarization of the sarcolemma via Na channels, Ca2+ enters the myocyte via L-type Ca2+ channels (LTCCs) located in the transverse (T)-tubules and the sarcolemma[7]. This relatively minor Ca2+ influx leads to a much greater release of Ca2+ from the sarcoplasmic reticulum (SR) through the action of ryanodine receptor type-2 (RyR2). Once the concentration of Ca2+ in the myocyte has reached significantly high levels, Ca2+ binds to troponin, leaving actin open to bind with myosin and culminating in sarcomere contraction. Ca2+ release of the RyR2 stores is the key determinant of the amplitude of the Ca2+ transient, but other factors can also alter the transient including Ca2+ extrusion by the Na/Ca2+ exchanger (NCX) and the re-sequestration of Ca2+ into the SR stores by the SR Ca2+ ATPase (SERCA2a)[8]. These are the major pathways regulating the SR-Ca2+ store and therefore the amplitude of the Ca2+ transient that is needed to drive contractility and many recent reviews have addressed these issues directly [9]. But additional factors might contribute to the Ca2+ transients and include the plasma membrane ATPase (PMCA), Ca2+ buffering in the SR (calsequestrin) as well as flux of Ca2+ into and out of other organelles. Diversion of Ca2+ from the cytosol into non-SR organelles including the mitochondria and perhaps nuclei may, in theory, lower the amplitude of the Ca2+ transient, whereas Ca2+ release from endosomes or lysosomes via Two Pore channels (TPC) could facilitate greater Ca2+ release from RyR2 channels via Ca2+ induced Ca2+ release during diastole and regulate autophagy in cardiomyocytes[10–12]. Whether these additional pathways represent primary or indirect mechanism for controlling Ca2+ stores remains to be determined, but these pathways appear to emerge as critical regulators of diastolic Ca2+ and Ca2+ oscillations during pathologic conditions. Ca2+ uptake into organelles may shift the balance of cytosolic and SR Ca2+ levels, prevent Ca2+ dependent inactivation of certain channels (e.g. cav2.2), and make less Ca2+ available to the contractile apparatus. Finally, recent studies have implicated non-voltage dependent Ca2+ entry currents in the regulation of Ca2+ transients under different stress conditions including neurohormal activation, stretch and RyR2 store depletion. Examples of these currents include transient receptor potential channels (TRPC) and store operated Ca2+ entry (SOCE) channels[13–15]. Loss of these control mechanisms as occurs in heart failure or during arrhythmias might alter Ca2+ transients resulting in diminished RyR2 Ca2+ stores and loss of the primary outputs of force production or pacemaking[16, 17]. This review focuses on one of these mechanisms: the sensing of RyR2 store depletion by the S/ER Ca2+ sensor stromal interaction molecule 1 (STIM1) and activation of non-voltage dependent Ca2+ entry through members of the family of Orai channels. The function of SOCE in cardiomyocytes has been a matter of intense debate as it is well established that cardiomyocytes have very efficient mechanisms for refilling SR Ca2+ stores. Nevertheless, reconciling SOCE’s role in cardiac pathophysiology has important implications as it might offer novel therapeutic targets for cardiac arrhythmia and the treatment of heart failure.
The Putney lab was the first to introduce the concept of SOCE in 1986 with a series of experiments in which depletion of internal calcium (Ca2+) stores was shown to control the extent of Ca2+ influx in nonexcitable cells[18]. This mechanism of Ca2+ entry served as a link between extracellular Ca2+ and intracellular Ca2+ stores and thereby meeting the requirements to fulfill the Cell Boundary Theorem for maintaining resting cytosolic Ca2+ levels[19]. When the stores were full, no Ca2+ influx was detected, but when the stores were emptied, Ca2+ entry developed. SOCE is now recognized as a ubiquitous pathway that functions to maintain Ca2+ homeostasis in response to depletion of internal sarco(endo)plasmic (S/ER) Ca2+ stores[18, 20]. Extensive research over the last decade has now defined key aspects of SOCE including a Ca2+ sensor for store depletion and the Ca2+ entry pore. Stromal interaction molecule 1 (STIM1) is a single-pass transmembrane protein that acts as a Ca2+ sensor by activating store-operated calcium channels (SOCCs) following S/ER Ca2+ store depletion [21, 22]. SOCCs are highly selective Ca2+ channels at the plasma membrane comprised of Orai channel multimers [23, 24]. In the presence of adequate S/ER Ca2+ stores, Ca2+ ions bind to an EF-hand motif in the luminal domain of STIM1 to maintain a monomeric form[21, 22]. Depletion of S/ER Ca2+ stores reduces the Ca2+ bound to STIM1 that unfolds and leads to aggregation (puncta formation) and subsequent allosteric activation of SOCCs[25–27]. The function of STIM1 was initially characterized in non-excitable cells; however, evidence from animal models and human mutations suggests a role for STIM1-dependent SOCE in cardiomyocytes. However, much controversy remains as to its role in cardiac growth and failure as well as its participation in cardiac conduction and arrhythmogenesis[28].
STIM1 was initially discovered in a screen to identify surface molecules important for B-cell development[29], however the discovery that STIM1 contributes to Ca2+ signaling occurred decades later in a series of shRNA screens. Here, high throughout screens using SOCE as the readout were performed by several labs and identified STIM1 and Orai1 as key components of the basal SOCE machinery[22–24, 30]. The relevance of SOCE to human pathology quickly emerged when STIM1 and Orai1 mutations were identified in families suffering from severe combined immunodeficiency[31, 32]. These recessive loss of function (LOF) mutations have now been characterized for both STIM1 and Orai1. Additional families with STIM1 or Orai1 mutations solidified the importance of SOCE in T-cell function, but also has raised the importance of SOCE in skeletal muscle function as these patients with LOF mutations develop marked hypotonia, muscle atrophy and weakness[33, 34]. Relevant to this review, no cardiac phenotype has been described in the LOF mutations for STIM1 or Orai1. These patients are critically ill requiring bone marrow transplantation and often succumb to overwhelming infections and sepsis, so a detailed cardiac phenotyping may not be possible; but none of the surviving patients exhibit cardiac dysfunction [34]. Animal models designed to address the role of STIM1 and SOCE in cardiomyocytes are therefore the principle method to define the function of SOCE in cardiac physiology.
Gain of function (GOF) mutations in both STIM1 and Orai1 were recently described in patients presenting with an unusual clinical syndrome that involves tubular aggregate myopathy, hyposplenism and platelet dysfunction as well as hypocalcemia[35–44]. These dominant mutations occur in the hot spots for STIM1 that involve the EF hands located in the S/ER lumen or in the coiled coil CCC domain, both of which are important for Orai1 gating. These patients exhibit muscle weakness with variable penetrance ranging from a positive Gower’s sign (often seen in muscular dystrophies) during early childhood to mild exercise intolerance in adults. Skeletal muscle biopsies from these patients reveal specific myopathic features including aggregation of membranous material seen with Gomori staining. Using specific immunofluorescent antibodies, SERCA pumps are co-localized with STIM1 in these aggregates indicating that the aggregates contain S/ER membranes. Ultrastructural studies with transmission electron microscopy reveal large arrays of SR membrane that occupy portions of the muscle fiber and limit muscle contraction. Functional studies for SOCE in cultured myoblasts reveal augmented SOCE which is consistent with formation of preformed STIM1-Orai1 punctae, although no SOC currents have been characterized. As for a cardiac phenotype, several patients with STIM1 GOF mutations have required implantation of permanent pacemakers for heart rhythm abnormalities implicating STIM1 in cardiac pacemaking[45]. While these studies bring to light the role of SOCE in cardiac and skeletal muscle physiology and offer insight to its role is disease, a greater understanding of the genetic variability of STIM1 and Orai1 will be important. In fact, a recent study of patients undergoing cardiac catheterization found single nucleotide polymorphisms (SNPs) in STIM1 gene correlates with a metabolic defect that includes ER stress and results in an increase in all-cause mortality[46].
The role of STIM1 and SOCE in ventricular cardiomyocytes
Following the description of STIM1 in skeletal muscle and given the similarities of Ca2+ handling in striated muscles, several groups proposed a similar role for SOCE in cardiac muscle [47–49](Table 1). These studies often begin by establishing the presence of STIM1 and Orai channels in the heart but this effort has been somewhat controversial. While the expression of mRNA and protein can be detected in whole heart lysates, the specific cells that express STIM1 and Orai1 remain unclear. Ventricular tissue is composed of many cells including cardiomyocytes, cardiac fibroblasts, and endocardial cells, all of which express STIM and Orai proteins and have SOCE which can complicate expression analysis, often necessitating isolation of primary cells or the use of immunofluorescence of cardiac sections. It is clear from several studies that levels of STIM1 expression are greater in neonatal cardiomyocytes than in adult cells. These issues were first addressed in a careful study of SOCE in neonatal and adult rat myocytes performed in the Hill lab [49]. These studies show that SOCE is robust in neonatal cardiomyocytes, occurring in 100% of cardiomyocytes. In contrast, SOCE occurs in only 10% of adult cardiomyocytes. These findings are consistent with data from our lab in which we used a LacZ reporter for the endogenous STIM1 promoter[50]. We detected STIM1 in the SA node with little expression in the adult ventricle. STIM1 was detected in the smooth muscle cells of the epicardial coronary arteries as has been reported by several groups. Why is STIM1 and SOCE enriched in neonatal but not adult cardiomyocytes? Embryonic and neonatal cardiomyocytes do not have a fully functional SR Ca2+ stores. Over the immediate days and weeks after birth a series of morphogenetic events seek to establish the function Ca2+ store. The T-tubule must invade from the sarcolemma and connect with terminal cisternae, where the RyR2-cav1.2 coupling takes place, SERCA2a is established to create a function uptake mechanism and Ca2+ buffering in the SR is created by calsequestrin isoform shifts[51]. Neonatal cardiomyocytes utilize SOCE to drive cardiac differentiation via Ca2+ dependent gene expression (calcineurin and CamK) and to fill the nascent SR stores with Ca2+[52].
Table 1:
Cardiac phenotype for animal models with altered STIM1 and Orai1 function.
| Species | Design | Phenotype | Reference |
|---|---|---|---|
| rat | Gene silencing STIM1 and Orai1 | Blunted cardiac growth | [85] |
| Rat and mouse | Adenoassociated virus delivery of shRNA for STIM1 | Cardiac atrophy SOC currents | [47, 97] |
| mouse | Tamoxifen inducible cardiac restricted gene knockout | Cardiac atrophy Impaired response to cardiac hypertrophy | [95] |
| mouse | Cardiac restricted gene knockout | Sinoatrial node dysfunction PLB-STIM1 regulation of SR Ca2+ release |
[50, 60] |
| mouse | STIM1-S transgenic mouse line | Cardiac failure Mitochondrial abnormalities | [86] |
| feline | Gene silencing | Reduced SR Ca2+ stores and widened action potential duration | [67] |
| rodent | Gene silencing and overexpression | Blunted hypertrophic response to neurohormonal agonists. | [49] |
| mouse | Cardiac restricted STIM1 deletion | late inset LV dysfunction ER stress | [15, 98] |
Interestingly, the Hill group also described the presence of alternative splice variants STIM1-S and STIM1-L in neonatal cardiomyocytes whereas adult myocytes only express STIM1-S. STIM1-L was originally characterized in skeletal muscle cells and results from a slicing event between exon 11 and 12 where a 106-amino acid peptide containing an actin binding domain is inserted into the C-terminal end of STIM1[53–55]. The role of this spliced variant has been proposed to influence STIM1-oligomerization and Orai1-trapping in membrane domains and seems to accelerate the gating kinetics of SOCE only in stiated muscle (Figure 1A). STIM1-L-Orai1 interaction has only been identified in adult cardiomyocytes subjected to pressure overload; the significance of this complex is unknown in neonatal cardiomyocytes[49]. Alternative forms of STIM proteins have emerged as a novel mechanism to regulate SOCE in cell type specific manner. As described above, STIM1-L can facilitate rapid SOCE by trapping Orai1 channels in designated regions of the cell membrane called S/ER-PM contact sites. These domains might concentrate STIM1 and Orai1 in specialized domains where signal transduction complexes associated with G-protein coupled receptors can regulate Ca2+ entry[54]. Alternatively STIM1-L may activate TRPC channels, a class of non-selective cation channels found to be regulated by STIM1 as SOC channels (Figure 1D)[56]. Interestingly, predictive algorithms for alternative splicing identify as many as 20 alternative transcripts for STIM1. Four variants display a high level of confidence and include not only STIM1-S and STIM1-L but others as well. STIM2 is a second member of the Ca2+ sensor family and may regulate basal Ca2+ levels[21]. STIM2 is also subject to alternative splicing and at least 3 spliced variants have been described. STIM2a is the dominant variant expressed and activates Orai1 channels. STIM2b results from an insertion of eight amino acids into the CAD domain of STIM2. The STIM2b variants can disrupt the STIM2-Orai1 interaction while retaining STIM oligomerization[57]. As a result the STIM2b can inhibit Orai1 Ca2+ currents, reduce SOCE and prevent calcineurin-dependent NFAT translocation into the nucleus[55]. The presence of different STIM species with distinct actions on SOCE add to the complexity of SOCE regulation. The existence of the other variants for STIM proteins remains unknown but does raise the possibility for an array of STIM proteins that might contribute to SOCE and other functions assigned to STIM1. Given the controversies surrounding SOCE in cardiac muscle, it may be important to better define the pattern of expression and function of all spliced variants of STIM in cardiac cells. At present, very little is known about the function of these SOCE components in cardiomyocytes.
Figure 1:

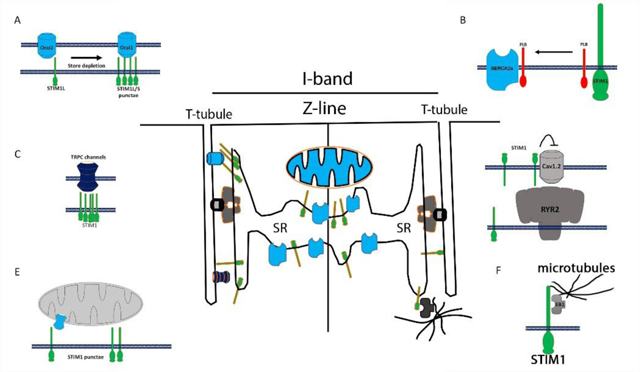
Schematic for STIM1 in cardiomyocytes. A) STIM1 localizes in the SR where it makes connections to different target molecules. B) Canonical SOCE is influenced by the set of spliced variants of STIM proteins. STIM1-L is constitutively associated with Orai1 channels whereas STIM1-S is recruited to sarcolemma in response to Ca2+ store depletion. C) STIM1 was proposed to inhibit cav1.2 in smooth muscle and neurons. Here STIM1 is localized on the cell surface to block voltage activated Ca2+ entry. D) STIM1 spliced forms can bind to and activate transient receptor potential (TRPC) channels. E) EB1 proteins are binding partners with STIM1 and forge a link between STIM1 in the SR membrane to the microtubules network.
Because so few adult cardiomyocytes maintain functional SOCE, it is important to establish whether the Ca2+ entry activated by store depletion corresponds to a SOC current and to determine the identity of the channels that function as the SOC pore. Orai channels have very well defined features, therefore validating the SOCE assays with whole cell current measurements will be important to distinguish the Ca2+ entry from non-specific membrane damage that might occur during the cell isolation process. In addition to store dependence, Orai channels display specific features[58] including: 1) a very positive reversal potential characteristic of a highly selective Ca2+ current, 2) Ca2+ dependent inactivation, 3) monovalence in divalent free conditions, and 4) specific pharmacology (see below). In order to measure these currents in cells with the complicated electrophysiology typically seen in cardiomyocytes, it is necessary to inactivate Na, K and cav2.2 channels using specific solutions, pharmacologic inhibitors and current clamp configurations. To date, traditional Orai currents that meet all of these criteria have only been resolved in SA nodal cells but not in isolated ventricular cardiomyocytes[50]. Thapsigargin is an inhibitor of the SERCA pumps and allows for a slow depletion of SR stores by preventing resequestration of Ca2+ stores following leak of RyR2-stores[59]. Thapsigargin does activate a non-selective current that is permeable to Ca2+ and Na with a reversal potential that is near 0mV indicating a non-selective current in cardiomyocytes[47]. STIM1 does influence this current under some circumstances. However, thapsigargin initiated currents resemble Orai currents only in certain features. The preponderance of evidence does not support Orai1 specifically, and it remains to be determined if these thapsigargin currents require STIM1[60]. Rather the thapsigargin current might represent another STIM1 gated channels such as transient receptor potential channel (TRPC) or another STIM1 target protein such as a transporter or exchanger. It will be important to identify the precise molecular entity that underlies the store operated Ca2+ entry described by several groups in a portion of adult cardiomyocytes. Any future therapeutic strategy designed around SOCE in cardiomyocytes would require a higher level of confidence that Orai channels are the pore for SOCE and therefore the mechanism of action. In contrast, our prior studies using whole cell current recordings and Ca2+ imaging assays from isolated single skeletal muscle fibers and SA nodal myocytes confirmed the presence of STIM1 and Orai1 proteins, and measured a SOC current that displayed all the criteria necessary for Orai channels.
Pharmacologic manipulations by inhibitors of SOC currents provide essential evidence for the molecular identity of Ca2+ currents in a given cell type[61]. Since the initial description of SOCE decades ago, various small molecules have been proposed as SOC inhibitors (Table 2). Several recent reviews can provide key details about the many agents and their specificity, as well provide progress updates on the development of novel SOC inhibitors. For cardiomyocytes, recent studies have utilized lanthanides and pyrazole compounds as blockers of SOCE[47]. Lanthanides are the earliest Orai inhibitors but are not very specific as they can block TRPC channels and L-type Ca2+ channels[62, 63]. Bis(triflourmethyl)pyrozoles were identified by their ability to block NFAT nuclear translocation following T-cell activation[64, 65]. BTP-2 has emerged as the most frequently used inhibitor for SOC channels and are offered as evidence for Orai1 channel involvement in action potential changes and SOCE[48, 66, 67]. Because of several challenges with BTP-2, caution must be applied to interpreting the experiments as proof of Orai channel involvement. First, BTP-2 must be applied extracellularly in order to block SOC currents, indicating that the drug does not alter intracellular events associated with SOCE activation including STIM1 oligomerization or STIM1-Orai1 interaction[68, 69]. Secondly, the mechanism for BTP-2 inhibition of SOCE was called into question by studies showing a specific interaction with TRPM4 channels[70, 71]. Here, TRPM4 channels, which confer Ca2+ activated non-selective currents, can depolarize the cell membrane and thereby limit Ca2+ entry. Given that TRPM4 channels are robustly expressed in cardiomyocytes and underlie human cardiac phenotypes ranging from conduction abnormalities to cardiac hypertrophy[70, 72], it would be important to further understand the mechanism of action for BTP-2 in cardiac electrophysiology[73]. Another pharmacologic agent that effects SOCE is 2-aminoethyldiphenyl borate (2-ABP), and it will block SOC currents with high potency in cardiomyocytes[74, 75]. A curious aspect of 2-ABP is the ability to activate Orai3 channels, independent of STIM1 or store depletion. Orai3 channels are found only in mammals and have been implicated in cardiac hypertrophy based on their expression profile by several investigative groups[76, 77]. It would be interesting to know if 2-ABP can activate Orai3 currents in cardiomyocytes, as they have been shown in non-excitable cells[78]. When looked at as a whole however, the many studies evaluating SOCE in cardiomyocytes under normal conditions and following pathologic insults require careful interpretation based on results with these new pharmacologic agents. Newer Orai channel inhibitors are in development, particularly as anti-inflammatory and immunosuppressant agents. These agents showed improved specificity to inhibit Orai channels but as of yet have not been tested in the cardiomyocytes.
Table 2:
Store-operated Ca2+ entry inhibitors.
Species specific variation in Ca2+ dynamics are well established in mammalian cardiomyocytes and might provide another potential explanation for the differences observed for STIM1 signaling and SOCE in cardiomyocytes. Larger mammals including feline, human and rabbit cardiomyocytes maintain very low resting Ca2+ levels that reflects the relative contributions of the SR Ca2+ stores and NCX1 where the SR is 2–3 fold greater than NCX1 activity [79–81]. In contrast, the SR Ca2+-NCX1 relationship is different for rodent cardiomyocytes where a greater resting Ca2+ levels reflect SR Ca2 that is 10 fold greater than contributions by the NCX1 activity[82]. The impact of this species difference is clear when cardiomyocytes are stimulated at high frequency. High frequency stimulation of larger mammalian cardiomyocytes activates refilling mechanisms and reduces Ca2+ extrusion resulting in greater SR Ca2+ load. In rodents, complex regulation of SR Ca2+-NCX1 relationship and cytosolic Na levels can influence action potential size and shape and reflect different Ca2+ dynamics. High cytosolic Na levels in rodent cardiomyocytes will limit NCX1 extrusion and along with a reduction in fractional Ca2+ release result in a negative force-frequency. We have proposed a role for STIM1 and SOCE in skeletal muscle fibers that are subject to high frequency stimulation in order to refill S/ER Ca2+ stores[83]. SR Ca2+ release from skeletal muscle fibers of STIM1 KO mice is maintained after a single electrical pulse. However Ca2+ transients are not sustained when STIM1 KO fibers are stimulated at high frequency. This failure to refill stores leads to easy fatigue and impaired contractility. Because the SR Ca2+ stores are maintained in maximal state in rodent cardiomyocytes, it is possible that STIM1 and Orai1 are minimized and SOCE sidelined. This provides an important explanation for the differences in SOCE observed in studies with rodents and larger mammals. Interestingly, STIM1 and Orai1 are robustly expressed in SA nodal cells (SAN) of mice and SOCE contributes to cardiac pacemaking[50]. Similar differences in the Ca2+ dynamics between ventricular and SAN cells might also account for the need to maintain a functional SOCE complex. While it is certainly possible that differences in the SOCE complex in cardiomyocytes may be attributed to the variation in Ca2+ dynamics between species, it should be noted that dominant and recessive mutations in the human STIM1 and Orai1 genes have been described and patients exhibit a mild cardiac phenotype that seems to revolve around altered pacemaking and cardiac conduction.
Ca2+ signaling, important for cardiomyocyte contraction, is the result of a global Ca2+ transient that requires synchronization of many local events[7]. Compartmentalization of the components for Ca2+ cycling such as the RyR2, SERCA2a, LTCC and NCX1 into discrete domains is important to ensure the fidelity of the Ca2+ cycling. When the integrity of these structures is lost, RyR2 channels can be subject to leak, NCX1 can switch between reverse and forward modes, and SERCA can fail to refill stores. This series of events culminates in impaired EC coupling and disorganized Ca2+ transients which impair muscle contraction and trigger cardiac arrhythmias. Common to all these mechanisms is the increase in diastolic Ca2+ that can influence spark frequency and lead to leaky RyR2 stores[84]. Erratic or unregulated SOCE in CM has now joined the ranks of potential pathological mechanisms that impair the Ca2+ cycling in the cardiomyocyte[28]. Because of the local control of Ca2+ cycling, it is important to demonstrate that STIM1 and Orai1 are located in the vicinity of RyR2-LTCC-SERCA-NCX1 complex. What is the function of the Ca2+ sensing domain of STIM1 that resides in the S/ER lumen in cardiomyocytes? Is it localized near components of that apparatus known to control local Ca2+ signals? A consensus seems to exist in the literature that STIM1 is present at the Z-line in both neonatal and adult cardiomyocytes (Figure 1) [47, 67, 85, 86]. However, it has been more difficult to localize Orai channels in cardiac membranes or measure its Ca2+ current (Figure 1A). This localization of STIM1 in the Z-lines would place STIM1 in a sub compartment of the S/ER and suggest that it contributes to refilling of local S/ER stores that may or may not involve Orai1 dependent Ca2+ entry[60]. STIM1 has been shown to regulate Ca2+ signaling in manner independent of Orai channels. For example, STIM1 has previously been shown to interact with and regulate the voltage gated Ca2+ channel cav1.2[87] (Figure 1C). Although this interaction was mapped out in neurons and smooth muscle cells, it has been difficult to demonstrate the relevance of this interaction in STIM1 KO mice[16, 88, 89]. In fact, L-type voltage currents appear to be unchanged in cardiomyocytes as well skeletal and smooth muscle cells[67]. STIM1 localizes to the outer membranes of the nucleus and nucleoplasm, the function of this localization is unknown[90]. One consideration is that STIM1 interacts with microtubules in non-excitable cells through a direct protein-protein interaction via end-binding protein 1 (EB1 proteins). Here, SR-localized STIM1 interacts with EB1, a TIP protein involved microtubule dynamics (Figure 1E). Movement or traffic of STIM1 in the SR membrane or nuclear envelope may be regulated by EB1 via microtubules. STIM1 can also interact with SERCA pumps and influence S/ER Ca2+ loading (Figure 2)[91–93]. In many cell types, STIM1 may influence SERCA activity to shape the dynamics of Ca2+ signaling. In fact, a common thread in many studies that evaluate models of STIM1 overexpression in cardiomyocytes is that increased levels of STIM1 leads to greater Ca2+ store release which results in cell damage and apoptosis[67, 86]. Importantly, deletion of STIM1 from the cardiomyocytes was not associated with a change in SR Ca2+ content, whereas SAN cells from cardiac restricted STIM1 KO mice displayed a reduction in SR Ca2+ stores. It is likely that deletion of STIM1 from the cardiomyocyte may lead to a compensatory effect for other components of the Ca2+ transient.
Figure 2:

Identification of novel STIM1 interacting partners. A) STIM1 domains include the ER luminal domains that contain the SAM and EF hands. These domains function as the Ca2+ sensor of SR store content. Cytosolic oriented STIM1 contains regions critical for Orai channel gained within the CC domains. Additional domains include the ERM and lysine rich regions. B) Design of a screen for STIM1 interacting partners. Biotinylated STIM1 is adheres to a neurtravadin coated plate. Bindings partners from phage display or lysates can be layered into the wells. Detection of the interaction is detected with an ELIZA assay using relevant antibodies. C) Phospholamban was detected as STIM1 partner using the screen. Proposed mechanism is shown for how STIM1 regulates PLN/SERCA2a function in cardiomyocytes.
Our recent work has shed light onto the mechanism by which STIM1 regulates SERCA function[60, 94]. We first described a developmentally controlled relationship between STIM1 and sarcolipin (SLN), an endogenous inhibitor of SERCA. In embryonic muscle, STIM1 exhibits low levels of expression while SLN expression is robust in an effort to keep SR Ca2+ stores low. Post-partum expression of these factors is reversed: STIM1 is robustly expressed while SLN expression is low, only in atrial cells and slow twitch muscle fibers. These results suggest that the primary role for STIM1 in cardiomyocytes is to establish a functional SR Ca2+ store in neonatal mice which may not be required in adult cells. To better understand the function of STIM1, we utilized a phage display library screen to identify novel STIM1 interacting partners (Figure 2A–B). A truncated STIM1 was engineered to contain a biotin tag at the N-terminus which enable us to orient STIM1-c on neutravidin plate (Figure 2A). We were then able to screen a phage library and detect STIM1 interacting proteins using a secondary antibody reaction (Figure 2B). Phospholamban (PLN), a micropeptide that regulates the SERCA pump, was identified to interact directly with STIM1 using this cell free assay. PLN in its monomeric, dephosphylated form can bind SERCA pumps and inhibit the ATPase activity and thereby decreasing Ca2+ store refilling. Adrenergic stimulation of the cardiac membranes leads to inhibition of PLN interaction with SERCA and the associated decrease in store filling. To place the STIM1-PLN interaction into physiologic context, gain and loss of function studies were used to test the idea that Ca2+ load in the cardiomyocyte is determined by a relationship involving STIM1-PLN and SERCA2a (Figure 2C). STIM1 was shown to sequester PLN from SERCA2a and thereby enhance SR Ca2+ loading in the cardiomyocyte. This mechanism may explain our observations that overexpression of STIM1 in cardiomyocytes leads to greater spark frequency in cardiomyocytes. STIM1-induced Ca2+ sparks have been observed in other studies. Our data also provides context for the low levels of STIM1 expression in resting adult cardiomyocytes. Here, STIM1 assembles in large macromolecular complexes at the Z-line where it can influence Ca2+ pumping and promote store refilling via greater SERCA2 efficiency.
The role of STIM1 and SOCE in Cardiac Hypertrophy
Cardiomyocytes respond to pressure overload by activating the fetal gene program which promotes stress resistance. It has been proposed by several laboratories that upregulation of STIM1 and Orai channels during pressure overload is part of the fetal gene program, and enables cells to compensate for depleted SR Ca2+ stores that persist during heart failure[67, 85, 95]. Using gene delivery techniques and loss of function mouse models, recent work suggests that STIM1 upregulation and SOCE are critical for the development of heart failure[47, 96]. SOCE is activated by the neural and hormonal inputs that are long associated with cardiac hypertrophy and failure. Here, STIM1 and SOCE are induced as part of the fetal gene program that is known to be expressed in hypertrophied and the failing heart. STIM1 seems to be a primary event in this process as the transgenic overexpression in the heart leads to cardiac failure[86].
A common theme present in all of the studies discussed above is that STIM1 in cardiomyocytes influences signal transduction pathways associated with cardiac growth. Gain and loss of function studies implicate STIM1 in the signaling events downstream of hypertrophic agonists such as pressure overload, catecholamine excess and TGF-b signaling. Eliminating STIM1-Ca2+ signaling by different techniques (genetic deletion or haploinsufficiency, viral delivery of silencing contracts or pharmacological inhibition) alters the activation state of pathways governed by calcineurin, a Ca2+-calmodulin regulated serine-threonine phosphatase, and the calmodulin dependent kinase CamK[47, 49]. Here transcriptional regulation of nuclear factor activated T-cells (NFAT) family can change the expression profiles for gene associated with cardiac growth and hypertrophy.
Work from the Hulot lab has recently demonstrated that reduction of STIM1 using a cardiotropic adenoassociated virus influenced the response to pressure overload by reducing calcineurin and CamK activity[47]. In a follow up study they examined the long term influence of STIM1 on cardiac function of juvenile and adult mice. They found that long-term silencing of STIM1 led to cardiac atrophy as evidenced by the reduced cardiomyocyte size and cardiac function. An interesting observation from this study is that cardiac dilation and impaired left ventricular (LV) function accompanied the reduced myocyte size[97]. These observations were then validated in adult mice using inducible STIM1 deletion system. Here tamoxifen induced Cre recombinase was used to delete STIM1 from cardiomyocytes. Again, STIM1 deletion for a duration of 40 days led to a dramatic reduction in LV function but not a clear difference in cardiac hypertrophy. These studies therefore suggest that reduction of STIM1 decouples the effects of cardiac hypertrophic growth and LV decompensation[95]. It is well established that LV dysfunction is accompanied by elevated wall stress and increased intracardiac pressures that signal cardiac hypertrophy both in parallel and in series. That each of these models of STIM1 deletion results in LV impairment in the absence of an increase heart weight suggests that either cardiac wall stress is not elevated, or that the reduction in EF are related to changes in heart rate or vascular function. Importantly these studies extended prior studies implicating STIM1 in cell growth pathways linked to AKT signaling and show that STIM1 can regulate mTORC2 complex or by influencing the unfolded protein response linked to ER stress[98]. The Chatham laboratory has proposed that STIM1 functions to mitigate the ER stress in response to developmental cues and hypertrophic agonists [15]. It is easy to see that failure to refill SR Ca2+ stores would impose stress on the S/ER rendering the protein synthetic machinery dysfunctional. Whether specific aspects of ER stress, such as impairment in translation control mechanisms or other mitochondrial dysfunction, occurs in these mice needs to be determined. The notion that STIM1 and SOCE is linked specifically to cardiac growth independent of the typical signaling events linked to wall stress deserves greater attention. Rather surprisingly, when the inducible cardiac STIM1 KO mice were subjected to pressure overload, LV size and cardiac weight were not increased and LV function remained unchanged despite the well-established association of STIM1 induction with cardiac hypertrophy. It would useful to know whether these mice experienced bradycardia, conduction abnormalities or arrhythmias that may account for the reduction in LV function independent of a change in cardiac mass.
The role of SOCE in cardiac pacemaking in SAN cells?
Unique among cardiomyocytes, SAN cells utilize Ca2+ less for generating force but rather to control the heart rate. Here, rhythmic and spontaneous Ca2+ release defines the Ca2+ clock, as described by the Lakatta lab[99]. The Ca2+ clock works in parallel with the membrane clock where oscillations in membrane potential can trigger diastolic depolarization by triggering the HCN4 channels activation [100, 101]. Because the expression of STIM1 and Orai1 was so robust in the SAN cells, we considered the SOCE might be an important component of both the membrane and Ca2+ clocks during cardiac pacemaking (Figure 3A)[50]. Notably, Orai1 currents were detected in SAN cells and displayed many of the features typically described for Orai1 channels including inward rectification, Ca2+ selectivity (markedly positive reversal potential) and inhibition by Orai1 blockers. We found that STIM1 and Orai1 punctae are pre-formed in SAN cardiomyocytes. Store depletion did not rearrange STIM1 localization as is described for many non-excitable cells. Deletion of STIM1 from SAN cardiomyocytes resulted in lower resting Ca2+ levels and reduced SR Ca2+ stores. The cardiac specific STIM1 KO mice exhibit spontaneous shifting within the pacemakers and these mice exhibit exaggerated slowing of the heart rate after sustained stimulation. These studies were taken as evidence that STIM1 and SOCE were required for pacemaking. We also found substantial changes in the key components of the Ca2+ clock in STIM1 null SAN cardiomyocytes. Currents assigned to the NCX1 and cav1.2 were altered in response to the loss of SOCE as a form of compensation. We hypothesize that Orai1 mediated Ca2+ entry acts a counter current for the potassium currents that required for the repolarization of the AP for the SAN (Figure 3B). Crosstalk between SOCE and these other currents provide the basis of the idea that STIM1 integrates the membrane and Ca2+ clock systems for cardiac pacemaking (Figure 3C). Modeling of this phenomena was provided and showed the relationship of SOCE, Ca2+ signaling and repolarization events. It will be interested to understand whether STIM1 role in pacemaking is similar to its role in electrically stimulated muscle fibers in order to prevent fatigue. Is solely replenishment of SR Ca2+ stores the main issue or are there other cell functions that are store operated? Changes in gene expression in the SAN confers differential risk of SAN dysfunction and risk of atrial arrhythmias, whether the signaling pathways activated by SOCE can influence profiles in the SAN are unknown.
Figure 3:

STIM1 and SOCE regulate sinus node function. A) STIM1-LacZ is detected in the thick section of the heart of an adult mouse. B-galactosidase staining reveals STIM1 enrichment in the SAN, coronary arteries and aortic valve structures. B) Model of action potential of the SAN. Relevant currents that coordinate diastolic depolarization (green) and repolarization (red) are indicated. Speculative role of SOCE in the SAN AP is shown. C) Model for the role of STIM1 in the SAN to regulate Orai1 directly and cav1.2 and NCX1 indirectly. Because SOC current is an inward Ca2+ current activated by depletion of SR Ca2+ stores it severs to link the membrane and Ca2+ clocks to coordinate diastolic depolarization.
Conclusion
For decades, cardiac electrophysiology and Ca2+ signaling was viewed from a relatively simplistic perspective that revolved around the release and resequestration of Ca2+ by the cav1.2-RyR2-SERCA2a network in order to maintain excitation contraction coupling. Moreover, Ca2+ handling is greatly impaired in the sick and failing cardiomyocytes yet efforts to target this complex therapeutically have been met with only modest success [102, 103]. Given the paucity of new therapies for heart failure, the emergence of SOCE in the cardiomyocyte and the upregulation with cardiac stress is an exciting development in the field and requires more research to refine our understanding of this pathway influences the heart. In particular, STIM1 and/or SOCE represent ideal targets in various clinical situations including arrhythmogenesis, cardiac hypertrophy and failure and sinoatrial node function. It is also important to point out that the development of SOCE inhibitors as immunosuppressant agents and anti-inflammatory agents is the focus of many pharmaceutical firms. As SOCE inhibition can induce SAN dysfunction, it will be important to know if these putative compounds confer side effects such as cardiac arrhythmogenesis and conduction abnormalities that might represent important safety concerns. For now, a greater understanding in cardiomyocytes is needed for the mechanism of action for STIM1 proteins, the identity of the SOC pore in cardiomyocytes as well as a refinement of the pharmacology.
Highlights.
STIM1 is present in neonatal cardiomyocytes and SAN nodal cells.
SOCE is controversial in adult ventricular cardiomyocytes.
STIM1 is localized to the specialized regions of the cardiomyocyte.
STIM1 mutant mice often exhibit defects in cardiac growth.
STIM1 knock out mice develop defects in cardiac pacemaking.
Acknowledgement
This project was supported by Award Number AG045551 (PBR) and DK109911 (PBR). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- Ca2+
Calcium
- SAN
Sinoatrial node
- CPA
Cyclopiazonic acid
- [Ca2+]i
Cytoplasmic calcium concentration
- GoF
Gain of function
- VCM
Ventricular cardiomyocytes
- LoF
Loss of function
- ROCCs
Receptor-operated calcium channels
- S/ER
Sarco(endo)plasmic reticulum
- SCID
Severe-combined immunodeficiency
- STIM1
Stromal interaction molecule 1
- SOCCs
Store-operated calcium channels
- SOCE
Store-operated calcium entry
- TG
Thapsigargin
- VSM
Vascular smooth muscle
- VOCCs
Voltage-operated calcium channels
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Sobie EA, Lederer WJ, Dynamic local changes in sarcoplasmic reticulum calcium: physiological and pathophysiological roles, J Mol Cell Cardiol, 52 (2012) 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wescott AP, Jafri MS, Lederer WJ, Williams GS, Ryanodine receptor sensitivity governs the stability and synchrony of local calcium release during cardiac excitation-contraction coupling, J Mol Cell Cardiol, 92 (2016) 82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Yaniv Y, Sirenko S, Ziman BD, Spurgeon HA, Maltsev VA, Lakatta EG, New evidence for coupled clock regulation of the normal automaticity of sinoatrial nodal pacemaker cells: bradycardic effects of ivabradine are linked to suppression of intracellular Ca(2)(+) cycling, Journal of molecular and cellular cardiology, 62 (2013) 80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Boyman L, Chikando AC, Williams GS, Khairallah RJ, Kettlewell S, Ward CW, Smith GL, Kao JP, Lederer WJ, Calcium movement in cardiac mitochondria, Biophys J, 107 (2014) 1289–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Heineman FW, Balaban RS, Control of mitochondrial respiration in the heart in vivo, Annu Rev Physiol, 52 (1990) 523–542. [DOI] [PubMed] [Google Scholar]
- [6].Colella M, Grisan F, Robert V, Turner JD, Thomas AP, Pozzan T, Ca2+ oscillation frequency decoding in cardiac cell hypertrophy: role of calcineurin/NFAT as Ca2+ signal integrators, Proc Natl Acad Sci U S A, 105 (2008) 2859–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fearnley CJ, Roderick HL, Bootman MD, Calcium signaling in cardiac myocytes, Cold Spring Harb Perspect Biol, 3 (2011) a004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Trafford AW, Diaz ME, Negretti N, Eisner DA, Enhanced Ca2+ current and decreased Ca2+ efflux restore sarcoplasmic reticulum Ca2+ content after depletion, Circ Res, 81 (1997) 477–484. [DOI] [PubMed] [Google Scholar]
- [9].Eisner DA, Ups and downs of calcium in the heart, J Physiol, 596 (2018) 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Garcia-Rua V, Feijoo-Bandin S, Garcia-Vence M, Aragon-Herrera A, Bravo SB, Rodriguez-Penas D, Mosquera-Leal A, Lear PV, Parrington J, Alonso J, Rosello-Lleti E, Portoles M, Rivera M, Gonzalez-Juanatey JR, Lago F, Metabolic alterations derived from absence of Two-Pore Channel 1 at cardiac level, J Biosci, 41 (2016) 643–658. [DOI] [PubMed] [Google Scholar]
- [11].Capel RA, Bolton EL, Lin WK, Aston D, Wang Y, Liu W, Wang X, Burton RA, Bloor-Young D, Shade KT, Ruas M, Parrington J, Churchill GC, Lei M, Galione A, Terrar DA, Two-pore Channels (TPC2s) and Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) at Lysosomal-Sarcoplasmic Reticular Junctions Contribute to Acute and Chronic beta-Adrenoceptor Signaling in the Heart, J Biol Chem, 290 (2015) 30087–30098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Grimm C, Chen CC, Wahl-Schott C, Biel M, Two-Pore Channels: Catalyzers of Endolysosomal Transport and Function, Front Pharmacol, 8 (2017) 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Seth M, Zhang ZS, Mao L, Graham V, Burch J, Stiber J, Tsiokas L, Winn M, Abramowitz J, Rockman HA, Birnbaumer L, Rosenberg P, TRPC1 channels are critical for hypertrophic signaling in the heart, Circulation research, 105 (2009) 1023–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Nakayama H, Wilkin BJ, Bodi I, Molkentin JD, Calcineurin-dependent cardiomyopathy is activated by TRPC in the adult mouse heart, Faseb J, 20 (2006) 1660–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Collins HE, Zhu-Mauldin X, Marchase RB, Chatham JC, STIM1/Orai1-mediated SOCE: current perspectives and potential roles in cardiac function and pathology, Am J Physiol Heart Circ Physiol, 305 (2013) H446–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang YX, STIM1-Ca2+ signaling modulates automaticity of the mouse sinoatrial node, Faseb j, 112 (2015) E5618–5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ohba T, Watanabe H, Murakami M, Sato T, Ono K, Ito H, Essential role of STIM1 in the development of cardiomyocyte hypertrophy, Biochemical and biophysical research communications, 389 (2009) 172–176. [DOI] [PubMed] [Google Scholar]
- [18].Putney JW Jr., A model for receptor-regulated calcium entry, Cell Calcium, 7 (1986) 1–12. [DOI] [PubMed] [Google Scholar]
- [19].Rios E, The cell boundary theorem: a simple law of the control of cytosolic calcium concentration, J Physiol Sci, 60 81–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kwan CY, Putney JW Jr., Uptake and intracellular sequestration of divalent cations in resting and methacholine-stimulated mouse lacrimal acinar cells. Dissociation by Sr2+ and Ba2+ of agonist-stimulated divalent cation entry from the refilling of the agonist-sensitive intracellular pool, J Biol Chem, 265 (1990) 678–684. [PubMed] [Google Scholar]
- [21].Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr., Meyer T, STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx, Curr Biol, 15 (2005) 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD, STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane, Nature, 437 (2005) 902–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A, A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function, Nature, 441 (2006) 179–185. [DOI] [PubMed] [Google Scholar]
- [24].Vig M, Beck A, Billingsley JM, Lis A, Parvez S, Peinelt C, Koomoa DL, Soboloff J, Gill DL, Fleig A, Kinet JP, Penner R, CRACM1 multimers form the ion-selective pore of the CRAC channel, Curr Biol, 16 (2006) 2073–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, Kinet JP, CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry, Science, 312 (2006) 1220–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chen CL, Lin CF, Chiang CW, Jan MS, Lin YS, Lithium inhibits ceramide- and etoposide-induced protein phosphatase 2A methylation, Bcl-2 dephosphorylation, caspase-2 activation, and apoptosis, Molecular pharmacology, 70 (2006) 510–517. [DOI] [PubMed] [Google Scholar]
- [27].Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF, Muallem S, SOAR and the polybasic STIM1 domains gate and regulate Orai channels, Nat Cell Biol, 11 (2009) 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rosenberg P, Socking It to cardiac hypertrophy: STIM1-mediated Ca2+ entry in the cardiomyocyte, Circulation, 124 (2011) 766–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Oritani K, Kincade PW, Identification of stromal cell products that interact with pre-B cells, J Cell Biol, 134 (1996) 771–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G, Stauderman KA, STIM1, an essential and conserved component of store-operated Ca2+ channel function, J Cell Biol, 169 (2005) 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gwack Y, Srikanth S, Oh-Hora M, Hogan PG, Lamperti ED, Yamashita M, Gelinas C, Neems DS, Sasaki Y, Feske S, Prakriya M, Rajewsky K, Rao A, Hair loss and defective T- and B-cell function in mice lacking ORAI1, Mol Cell Biol, 28 (2008) 5209–5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lacruz RS, Feske S, Diseases caused by mutations in ORAI1 and STIM1, Annals of the New York Academy of Sciences, 1356 (2015) 45–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Feske S, ORAI1 and STIM1 deficiency in human and mice: roles of store-operated Ca2+ entry in the immune system and beyond, Immunological reviews, 231 (2009) 189–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Picard C, McCarl CA, Papolos A, Khalil S, Luthy K, Hivroz C, LeDeist F, Rieux-Laucat F, Rechavi G, Rao A, Fischer A, Feske S, STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity, N Engl J Med, 360 (2009) 1971–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bohm J, Chevessier F, De Paula AM, Koch C, Attarian S, Feger C, Hantai D, Laforet P, Ghorab K, Vallat JM, Fardeau M, Figarella-Branger D, Pouget J, Romero NB, Koch M, Ebel C, Levy N, Krahn M, Eymard B, Bartoli M, Laporte J, Constitutive Activation of the Calcium Sensor STIM1 Causes Tubular-Aggregate Myopathy, American journal of human genetics, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bohm J, Chevessier F, Koch C, Peche GA, Mora M, Morandi L, Pasanisi B, Moroni I, Tasca G, Fattori F, Ricci E, Penisson-Besnier I, Nadaj-Pakleza A, Fardeau M, Joshi PR, Deschauer M, Romero NB, Eymard B, Laporte J, Clinical, histological and genetic characterisation of patients with tubular aggregate myopathy caused by mutations in STIM1, J Med Genet, 51 (2014) 824–833. [DOI] [PubMed] [Google Scholar]
- [37].Hedberg C, Niceta M, Fattori F, Lindvall B, Ciolfi A, D’Amico A, Tasca G, Petrini S, Tulinius M, Tartaglia M, Oldfors A, Bertini E, Childhood onset tubular aggregate myopathy associated with de novo STIM1 mutations, Journal of neurology, 261 (2014) 870–876. [DOI] [PubMed] [Google Scholar]
- [38].Misceo D, Holmgren A, Louch WE, Holme PA, Mizobuchi M, Morales RJ, De Paula AM, Stray-Pedersen A, Lyle R, Dalhus B, Christensen G, Stormorken H, Tjonnfjord GE, Frengen E, A dominant STIM1 mutation causes Stormorken syndrome, Human mutation, 35 (2014) 556–564. [DOI] [PubMed] [Google Scholar]
- [39].Morin G, Bruechle NO, Singh AR, Knopp C, Jedraszak G, Elbracht M, Bremond-Gignac D, Hartmann K, Sevestre H, Deutz P, Herent D, Nurnberg P, Romeo B, Konrad K, Mathieu-Dramard M, Oldenburg J, Bourges-Petit E, Shen Y, Zerres K, Ouadid-Ahidouch H, Rochette J, Gain-of-Function mutation in STIM1 (p.R304W) is Associated with Stormorken Syndrome, Human mutation, (2014). [DOI] [PubMed] [Google Scholar]
- [40].Endo Y, Noguchi S, Hara Y, Hayashi YK, Motomura K, Miyatake S, Murakami N, Tanaka S, Yamashita S, Kizu R, Bamba M, Goto Y, Matsumoto N, Nonaka I, Nishino I, Dominant mutations in ORAI1 cause tubular aggregate myopathy with hypocalcemia via constitutive activation of store-operated Ca(2)(+) channels, Hum Mol Genet, 24 (2015) 637–648. [DOI] [PubMed] [Google Scholar]
- [41].Markello T, Chen D, Kwan JY, Horkayne-Szakaly I, Morrison A, Simakova O, Maric I, Lozier J, Cullinane AR, Kilo T, Meister L, Pakzad K, Bone W, Chainani S, Lee E, Links A, Boerkoel C, Fischer R, Toro C, White JG, Gahl WA, Gunay-Aygun M, York platelet syndrome is a CRAC channelopathy due to gain-of-function mutations in STIM1, Mol Genet Metab, 114 (2015) 474–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Okuma H, Saito F, Mitsui J, Hara Y, Hatanaka Y, Ikeda M, Shimizu T, Matsumura K, Shimizu J, Tsuji S, Sonoo M, Tubular aggregate myopathy caused by a novel mutation in the cytoplasmic domain of STIM1, Neurol Genet, 2 (2016) e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bohm J, Bulla M, Urquhart JE, Malfatti E, Williams SG, O’Sullivan J, Szlauer A, Koch C, Baranello G, Mora M, Ripolone M, Violano R, Moggio M, Kingston H, Dawson T, DeGoede CG, Nixon J, Boland A, Deleuze JF, Romero N, Newman WG, Demaurex N, Laporte J, ORAI1 Mutations with Distinct Channel Gating Defects in Tubular Aggregate Myopathy, Hum Mutat, (2017). [DOI] [PubMed] [Google Scholar]
- [44].Noury JB, Bohm J, Peche GA, Guyant-Marechal L, Bedat-Millet AL, Chiche L, Carlier RY, Malfatti E, Romero NB, Stojkovic T, Tubular aggregate myopathy with features of Stormorken disease due to a new STIM1 mutation, Neuromuscul Disord, 27 (2017) 78–82. [DOI] [PubMed] [Google Scholar]
- [45].Walter MC, Rossius M, Zitzelsberger M, Vorgerd M, Muller-Felber W, Ertl-Wagner B, Zhang Y, Brinkmeier H, Senderek J, Schoser B, 50 years to diagnosis: Autosomal dominant tubular aggregate myopathy caused by a novel STIM1 mutation, Neuromuscul Disord, 25 (2015) 577–584. [DOI] [PubMed] [Google Scholar]
- [46].Kraus WE, Muoio DM, Stevens R, Craig D, Bain JR, Grass E, Haynes C, Kwee L, Qin X, Slentz DH, Krupp D, Muehlbauer M, Hauser ER, Gregory SG, Newgard CB, Shah SH, Metabolomic Quantitative Trait Loci (mQTL) Mapping Implicates the Ubiquitin Proteasome System in Cardiovascular Disease Pathogenesis, PLoS genetics, 11 (2015) e1005553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hulot JS, Fauconnier J, Ramanujam D, Chaanine A, Aubart F, Sassi Y, Merkle S, Cazorla O, Ouille A, Dupuis M, Hadri L, Jeong D, Muhlstedt S, Schmitt J, Braun A, Benard L, Saliba Y, Laggerbauer B, Nieswandt B, Lacampagne A, Hajjar RJ, Lompre AM, Engelhardt S, Critical role for stromal interaction molecule 1 in cardiac hypertrophy, Circulation, 124 (2011) 796–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Touchberry CD, Elmore CJ, Nguyen TM, Andresen JJ, Zhao X, Orange M, Weisleder N, Brotto M, Claycomb WC, Wacker MJ, Store-operated calcium entry is present in HL-1 cardiomyocytes and contributes to resting calcium, Biochem Biophys Res Commun, 416 (2011) 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Luo X, Hojayev B, Jiang N, Wang ZV, Tandan S, Rakalin A, Rothermel BA, Gillette TG, Hill JA, STIM1-dependent store-operated Ca(2)(+) entry is required for pathological cardiac hypertrophy, J Mol Cell Cardiol, 52 (2012) 136–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zhang H, Sun AY, Kim JJ, Graham V, Finch EA, Nepliouev I, Zhao G, Li T, Lederer WJ, Stiber JA, Pitt GS, Bursac N, Rosenberg PB, STIM1-Ca2+ signaling modulates automaticity of the mouse sinoatrial node, Proceedings of the National Academy of Sciences of the United States of America, 112 (2015) E5618–5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Uehara A, Yasukochi M, Imanaga I, Nishi M, Takeshima H, Store-operated Ca2+ entry uncoupled with ryanodine receptor and junctional membrane complex in heart muscle cells, Cell Calcium, 31 (2002) 89–96. [DOI] [PubMed] [Google Scholar]
- [52].Seth M, Sumbilla C, Mullen SP, Lewis D, Klein MG, Hussain A, Soboloff J, Gill DL, Inesi G, Sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA) gene silencing and remodeling of the Ca2+ signaling mechanism in cardiac myocytes, Proc Natl Acad Sci U S A, 101 (2004) 16683–16688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Darbellay B, Arnaudeau S, Bader CR, Konig S, Bernheim L, STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release, The Journal of cell biology, 194 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sauc S, Bulla M, Nunes P, Orci L, Marchetti A, Antigny F, Bernheim L, Cosson P, Frieden M, Demaurex N, STIM1L traps and gates Orai1 channels without remodeling the cortical ER, J Cell Sci, 128 (2015) 1568–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Miederer AM, Alansary D, Schwar G, Lee PH, Jung M, Helms V, Niemeyer BA, A STIM2 splice variant negatively regulates store-operated calcium entry, Nat Commun, 6 (2015) 6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Horinouchi T, Higashi T, Higa T, Terada K, Mai Y, Aoyagi H, Hatate C, Nepal P, Horiguchi M, Harada T, Miwa S, Different binding property of STIM1 and its novel splice variant STIM1L to Orai1, TRPC3, and TRPC6 channels, Biochemical and biophysical research communications, 428 (2012) 252–258. [DOI] [PubMed] [Google Scholar]
- [57].Soboloff J, Spassova MA, Hewavitharana T, He LP, Xu W, Johnstone LS, Dziadek MA, Gill DL, STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ Entry, Curr Biol, 16 (2006) 1465–1470. [DOI] [PubMed] [Google Scholar]
- [58].Bird GS, DeHaven WI, Smyth JT, Putney JW Jr., Methods for studying store-operated calcium entry, Methods (San Diego, Calif, 46 (2008) 204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Baudet S, Shaoulian R, Bers DM, Effects of thapsigargin and cyclopiazonic acid on twitch force and sarcoplasmic reticulum Ca2+ content of rabbit ventricular muscle, Circ Res, 73 (1993) 813–819. [DOI] [PubMed] [Google Scholar]
- [60].Zhao G, Li T, Brochet DX, Rosenberg PB, Lederer WJ, STIM1 enhances SR Ca2+ content through binding phospholamban in rat ventricular myocytes, Proc Natl Acad Sci U S A, 112 (2015) E4792–4801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Prakriya M, Store-operated Orai channels: structure and function, Curr Top Membr, 71 (2013) 1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kerschbaum HH, Cahalan MD, Single-channel recording of a store-operated Ca2+ channel in Jurkat T lymphocytes, Science, 283 (1999) 836–839. [DOI] [PubMed] [Google Scholar]
- [63].Trebak M, Bird GS, McKay RR, Putney JW Jr., Comparison of human TRPC3 channels in receptor-activated and store-operated modes. Differential sensitivity to channel blockers suggests fundamental differences in channel composition, The Journal of biological chemistry, 277 (2002) 21617–21623. [DOI] [PubMed] [Google Scholar]
- [64].Zitt C, Strauss B, Schwarz EC, Spaeth N, Rast G, Hatzelmann A, Hoth M, Potent inhibition of Ca2+ release-activated Ca2+ channels and T-lymphocyte activation by the pyrazole derivative BTP2, The Journal of biological chemistry, 279 (2004) 12427–12437. [DOI] [PubMed] [Google Scholar]
- [65].Ohga K, Takezawa R, Arakida Y, Shimizu Y, Ishikawa J, Characterization of YM-58483/BTP2, a novel store-operated Ca2+ entry blocker, on T cell-mediated immune responses in vivo, Int Immunopharmacol, 8 (2008) 1787–1792. [DOI] [PubMed] [Google Scholar]
- [66].Sabourin J, Bartoli F, Antigny F, Gomez AM, Benitah JP, Transient Receptor Potential Canonical (TRPC)/Orai1-dependent Store-operated Ca2+ Channels: NEW TARGETS OF ALDOSTERONE IN CARDIOMYOCYTES, J Biol Chem, 291 (2016) 13394–13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Troupes CD, Wallner M, Borghetti G, Zhang C, Mohsin S, von Lewinski D, Berretta RM, Kubo H, Chen X, Soboloff J, Houser S, Role of STIM1 (Stromal Interaction Molecule 1) in Hypertrophy-Related Contractile Dysfunction, Circ Res, 121 (2017) 125–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Jairaman A, Prakriya M, Molecular pharmacology of store-operated CRAC channels, Channels (Austin), 7 (2013) 402–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Shim AH, Tirado-Lee L, Prakriya M, Structural and functional mechanisms of CRAC channel regulation, J Mol Biol, 427 (2015) 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Hof T, Liu H, Salle L, Schott JJ, Ducreux C, Millat G, Chevalier P, Probst V, Guinamard R, Bouvagnet P, TRPM4 non-selective cation channel variants in long QT syndrome, BMC Med Genet, 18 (2017) 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Simard C, Hof T, Keddache Z, Launay P, Guinamard R, The TRPM4 non-selective cation channel contributes to the mammalian atrial action potential, Journal of molecular and cellular cardiology, 59 (2013) 11–19. [DOI] [PubMed] [Google Scholar]
- [72].Kruse M, Schulze-Bahr E, Corfield V, Beckmann A, Stallmeyer B, Kurtbay G, Ohmert I, Schulze-Bahr E, Brink P, Pongs O, Impaired endocytosis of the ion channel TRPM4 is associated with human progressive familial heart block type I, The Journal of clinical investigation, 119 (2009) 2737–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Wolkowicz PE, Huang J, Umeda PK, Sharifov OF, Tabengwa E, Halloran BA, Urthaler F, Grenett HE, Pharmacological evidence for Orai channel activation as a source of cardiac abnormal automaticity, Eur J Pharmacol, 668 (2011) 208–216. [DOI] [PubMed] [Google Scholar]
- [74].Wang P, Umeda PK, Sharifov OF, Halloran BA, Tabengwa E, Grenett HE, Urthaler F, Wolkowicz PE, Evidence that 2-aminoethoxydiphenyl borate provokes fibrillation in perfused rat hearts via voltage-independent calcium channels, Eur J Pharmacol, 681 (2012) 60–67. [DOI] [PubMed] [Google Scholar]
- [75].Peppiatt CM, Collins TJ, Mackenzie L, Conway SJ, Holmes AB, Bootman MD, Berridge MJ, Seo JT, Roderick HL, 2-Aminoethoxydiphenyl borate (2-APB) antagonises inositol 1,4,5-trisphosphate-induced calcium release, inhibits calcium pumps and has a use-dependent and slowly reversible action on store-operated calcium entry channels, Cell Calcium, 34 (2003) 97–108. [DOI] [PubMed] [Google Scholar]
- [76].Saliba Y, Keck M, Marchand A, Atassi F, Ouille A, Cazorla O, Trebak M, Pavoine C, Lacampagne A, Hulot JS, Fares N, Fauconnier J, Lompre AM, Emergence of Orai3 activity during cardiac hypertrophy, Cardiovascular research, 105 (2015) 248–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Zhang X, Zhang W, Gonzalez-Cobos JC, Jardin I, Romanin C, Matrougui K, Trebak M, Complex role of STIM1 in the activation of store-independent Orai1/3 channels, J Gen Physiol, 143 (2014) 345–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Shuttleworth TJ, Orai3--the ‘exceptional’ Orai?, J Physiol, 590 (2012) 241–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Bers DM, Cardiac excitation-contraction coupling, Nature, 415 (2002) 198–205. [DOI] [PubMed] [Google Scholar]
- [80].Kohlhaas M, Zhang T, Seidler T, Zibrova D, Dybkova N, Steen A, Wagner S, Chen L, Brown JH, Bers DM, Maier LS, Increased sarcoplasmic reticulum calcium leak but unaltered contractility by acute CaMKII overexpression in isolated rabbit cardiac myocytes, Circ Res, 98 (2006) 235–244. [DOI] [PubMed] [Google Scholar]
- [81].Piacentino V 3rd, Weber CR, Chen X, Weisser-Thomas J, Margulies KB, Bers DM, Houser SR, Cellular basis of abnormal calcium transients of failing human ventricular myocytes, Circ Res, 92 (2003) 651–658. [DOI] [PubMed] [Google Scholar]
- [82].Bers DM, Cardiac Na/Ca exchange function in rabbit, mouse and man: what’s the difference?, J Mol Cell Cardiol, 34 (2002) 369–373. [DOI] [PubMed] [Google Scholar]
- [83].Li T, Finch EA, Graham V, Zhang ZS, Ding JD, Burch J, Oh-hora M, Rosenberg P, STIM1-Ca(2+) signaling is required for the hypertrophic growth of skeletal muscle in mice, Molecular and cellular biology, 32 (2012) 3009–3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Marx SO, Marks AR, Regulation of the ryanodine receptor in heart failure, Basic Res Cardiol, 97 Suppl 1 (2002) I49–51. [DOI] [PubMed] [Google Scholar]
- [85].Voelkers M, Salz M, Herzog N, Frank D, Dolatabadi N, Frey N, Gude N, Friedrich O, Koch WJ, Katus HA, Sussman MA, Most P, Orai1 and Stim1 regulate normal and hypertrophic growth in cardiomyocytes, Journal of molecular and cellular cardiology, 48 (2010) 1329–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Correll RN, Goonasekera SA, van Berlo JH, Burr AR, Accornero F, Zhang H, Makarewich CA, York AJ, Sargent MA, Chen X, Houser SR, Molkentin JD, STIM1 elevation in the heart results in aberrant Ca(2)(+) handling and cardiomyopathy, J Mol Cell Cardiol, 87 (2015) 38–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Wang Y, Deng X, Mancarella S, Hendron E, Eguchi S, Soboloff J, Tang XD, Gill DL, The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels, Science (New York, N.Y, 330 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Kassan M, Ait-Aissa K, Radwan E, Mali V, Haddox S, Gabani M, Zhang W, Belmadani S, Irani K, Trebak M, Matrougui K, Essential Role of Smooth Muscle STIM1 in Hypertension and Cardiovascular Dysfunction, Arterioscler Thromb Vasc Biol, 36 (2016) 1900–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Park CY, Shcheglovitov A, Dolmetsch R, The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels, Science (New York, N.Y, 330 (2010) 101–105. [DOI] [PubMed] [Google Scholar]
- [90].Lee SH, Hadipour-Lakmehsari S, Miyake T, Gramolini AO, Three-dimensional imaging reveals endo(sarco)plasmic reticulum-containing invaginations within the nucleoplasm of muscle, Am J Physiol Cell Physiol, 314 (2018) C257–C267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Jousset H, Frieden M, Demaurex N, STIM1 Knockdown Reveals That Store-operated Ca2+ Channels Located Close to Sarco/Endoplasmic Ca2+ ATPases (SERCA) Pumps Silently Refill the Endoplasmic Reticulum, J Biol Chem, 282 (2007) 11456–11464. [DOI] [PubMed] [Google Scholar]
- [92].Lopez JJ, Jardin I, Bobe R, Pariente JA, Enouf J, Salido GM, Rosado JA, STIM1 regulates acidic Ca2+ store refilling by interaction with SERCA3 in human platelets, Biochem Pharmacol, 75 (2008) 2157–2164. [DOI] [PubMed] [Google Scholar]
- [93].Sampieri A, Zepeda A, Asanov A, Vaca L, Visualizing the store-operated channel complex assembly in real time: identification of SERCA2 as a new member, Cell calcium, 45 (2009) 439–446. [DOI] [PubMed] [Google Scholar]
- [94].Seth M, Li T, Graham V, Burch J, Finch E, Stiber JA, Rosenberg PB, Dynamic regulation of sarcoplasmic reticulum Ca(2+) stores by stromal interaction molecule 1 and sarcolipin during muscle differentiation, Dev Dyn, 241 (2012) 639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Parks C, Alam MA, Sullivan R, Mancarella S, STIM1-dependent Ca(2+) microdomains are required for myofilament remodeling and signaling in the heart, Sci Rep, 6 (2016) 25372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Zhu-Mauldin X, Marsh SA, Zou L, Marchase RB, Chatham JC, Modification of STIM1 by O-linked N-acetylglucosamine (O-GlcNAc) attenuates store-operated calcium entry in neonatal cardiomyocytes, The Journal of biological chemistry, 287 (2012) 39094–39106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Benard L, Oh JG, Cacheux M, Lee A, Nonnenmacher M, Matasic DS, Kohlbrenner E, Kho C, Pavoine C, Hajjar RJ, Hulot JS, Cardiac Stim1 Silencing Impairs Adaptive Hypertrophy and Promotes Heart Failure Through Inactivation of mTORC2/Akt Signaling, Circulation, 133 (2016) 1458–1471; discussion 1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Collins HE, He L, Zou L, Qu J, Zhou L, Litovsky SH, Yang Q, Young ME, Marchase RB, Chatham JC, Stromal interaction molecule 1 is essential for normal cardiac homeostasis through modulation of ER and mitochondrial function, Am J Physiol Heart Circ Physiol, 306 (2014) H1231–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Lakatta EG, Maltsev VA, Vinogradova TM, A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker, Circulation research, 106 659–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Monfredi O, Maltseva LA, Spurgeon HA, Boyett MR, Lakatta EG, Maltsev VA, Beat-to-Beat Variation in Periodicity of Local Calcium Releases Contributes to Intrinsic Variations of Spontaneous Cycle Length in Isolated Single Sinoatrial Node Cells, PLoS ONE, 8 (2013) e67247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Li Y, Sirenko S, Riordon DR, Yang D, Spurgeon H, Lakatta EG, Vinogradova TM, CaMKII-dependent phosphorylation regulates basal cardiac pacemaker function via modulation of local Ca2+ releases, Am J Physiol Heart Circ Physiol, 311 (2016) H532–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Desai AS, Barnard D, Bouchard A, Jaski B, Lyon AR, Pogoda JM, Rudy JJ, Zsebo KM, Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebocontrolled, phase 2b trial, Lancet, 387 (2016) 1178–1186. [DOI] [PubMed] [Google Scholar]
- [103].McCauley MD, Wehrens XH, Targeting ryanodine receptors for anti-arrhythmic therapy, Acta Pharmacol Sin, 32 (2011) 749–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Goto J, Suzuki AZ, Ozaki S, Matsumoto N, Nakamura T, Ebisui E, Fleig A, Penner R, Mikoshiba K, Two novel 2-aminoethyl diphenylborinate (2-APB) analogues differentially activate and inhibit store-operated Ca(2+) entry via STIM proteins, Cell Calcium, 47 (2010) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Chung SC, McDonald TV, Gardner P, Inhibition by SK&F 96365 of Ca2+ current, IL-2 production and activation in T lymphocytes, Br J Pharmacol, 113 (1994) 861–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Ishikawa J, Ohga K, Yoshino T, Takezawa R, Ichikawa A, Kubota H, Yamada T, A pyrazole derivative YM -58483, potently inhibits store-operated sustained Ca2+ influx and IL-2 production in T lymphocytes, J Immunol, 170 (2003) 4441–4449. [DOI] [PubMed] [Google Scholar]
- [107].Li J, McKeown L, Ojelabi O, Stacey M, Foster R, O’Regan D, Porter KE, Beech DJ, Nanomolar potency and selectivity of a Ca(2)(+) release-activated Ca(2)(+) channel inhibitor against store-operated Ca(2)(+) entry and migration of vascular smooth muscle cells, Br J Pharmacol, 164 (2011) 382–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Chen G, Panicker S, Lau KY, Apparsundaram S, Patel VA, Chen SL, Soto R, Jung JK, Ravindran P, Okuhara D, Bohnert G, Che Q, Rao PE, Allard JD, Badi L, Bitter HM, Nunn PA, Narula SK, DeMartino JA, Characterization of a novel CRAC inhibitor that potently blocks human T cell activation and effector functions, Mol Immunol, 54 (2013) 355–367. [DOI] [PubMed] [Google Scholar]