

Abstract
Cancer accounts for ~9 million deaths a year worldwide, predominantly affecting adults. Adult malignancies are usually examined after extensive clonal evolution and carry many mutations, obscuring the individual contributions of these alterations to oncogenesis. In contrast, pediatric cancers often contain few mutations, most of which cause defects in chromatin-associated proteins. Here, we explore the roles chromatin plays in oncogenesis. We highlight how the developmental regulation of cell proliferation genes and the degradation of chromosome ends are two major bottlenecks in the evolution of malignant cells, and point to a third bottleneck where epigenomic dysfunction triggers expression of tumor suppressor genes, limiting the development of aggressive and metastatic features in tumors. We also identify opportunities for chromatin-based therapies.
Keywords: Cancer, Chromatin, Epigenome, Nucleosome, Tumor Suppressor, Oncogene
Chromatin in cancer cells
Cancer is a disease of metazoans. The growth and patterning of organisms requires coupling cell growth with differentiation in the bodies of multicellular animals. This is accomplished by developmental signalling pathways that coordinate cell division and differentiation. In vertebrates, some cells are set aside during development as pluripotent stem cells that retain the capacity to divide through the lifetime of the organism to maintain the soma. However, a major risk is that cells may lose growth control, and proliferation of somatic clones in the body then drive evolution of cancerous cells into more malignant forms. The hallmarks of cancer are that cells 1) acquire the capacity to proliferate indefinitely, 2) sustain proliferative signalling, 3) inhibit apoptosis, 4) facilitate angiogenesis, 5) evade growth suppressors, and 6) acquire metastatic cellular properties[ [1]. These changes can be complex, and indeed genetic changes in cancer cells are extensive, especially as they are usually examined in very late stages after extensive clonal history and treatment. The role of signalling pathways in development is well appreciated, and its importance was first demonstrated by viral oncogenes that drive aberrant activation of signalling[ [2]. Many of these factors are dominant oncogenic variants of key signalling receptors resulting in constitutive activation. The importance of chromatin in gene regulation is also appreciated, but chromatin also has roles in tumor progression[ [3]. For adult cancers especially, cancer cell genomes are complicated, where it is often difficult to distinguish triggering oncogenic mutations from later selected changes and from hitchhiker mutations[ [4].
In contrast, pediatric cancers are genetically simple[ [5]. There are two distinctive features of these cancers: first, in a number of these cancers as few as one oncogenic mutation have been identified [5]. Second, the initial genetic alterations involve chromatin proteins with general roles in genome function [6]. We discuss the known functions of these proteins here. We identify three critical chromatin-intrinsic bottlenecks to cancer progression: 1) regulatory element nucleosomes restrict expression of proliferation genes, 2) proliferation degrades chromosome ends, and 3) chromatin dysfunction triggers tumor suppressors. While the emphasize cellular features of malignancy, a chromatin-centric view of cancer relates these three bottlenecks to changes in cellular features (Figure 1). Further, this view explains the co-occurrence of sets of mutations observed in many tumors and provides insight into novel therapeutic strategies [7].
Figure 1. Chromatin events associated with the Hallmarks of Cancer.
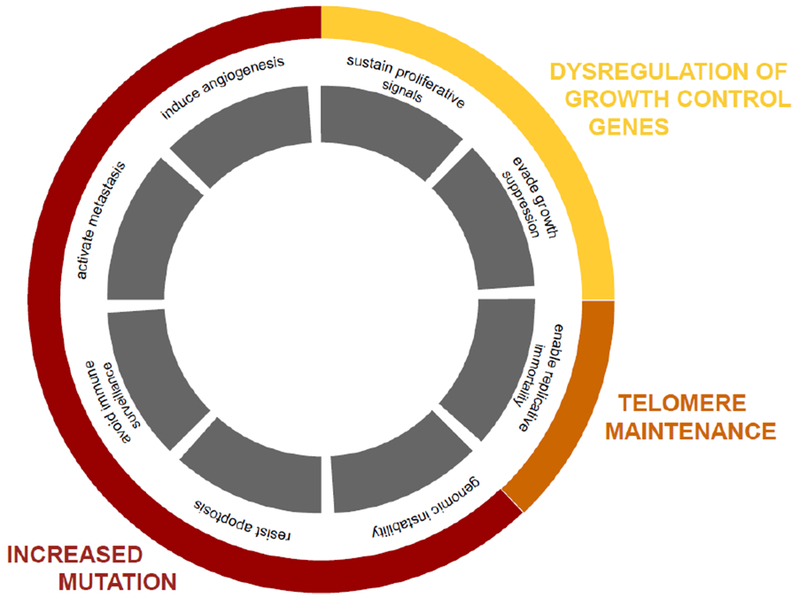
Malignant cells manipulate cellular controls to proliferate without restriction, and manipulate organismal biology to sustain large tumor masses (grey boxes). These features of cancer can be grouped into three classes of chromatin effects: 1) transcription factor- and chromatin-mediated changes in proliferative and tumor suppressor gene expression (yellow), 2) TERT- or recombination-mediated changes to prevent the degradation of linear chromosome ends (orange), and 3) mutagenic evolution of expanding oncogenic cell clones for tumorous properties (red).
The first bottleneck: Nucleosomes restrict expression of proliferation genes
In oncogenesis a somatic clone of cells must start proliferating to become subject to clonal selection and evolution (Figure 2). Proliferation is normally tightly controlled by gene regulation. Chromatin enforces appropriate gene regulation at many levels, by organizing the structure of promoters and regulatory elements, by modulating transcription factor binding at these regulatory elements, and by organizing domains that isolate genes and enhancers. A key aspect in coordinating developmental gene expression is chromatin-mediated repression. Repression is accomplished in several ways, including through DNA methylation of promoters and regulatory elements, through histone modifications that compact chromatin, and through partitioning within the nucleus between active and repressed territories.
Figure 2: Chromatin bottlenecks in tumor clonal evolution.
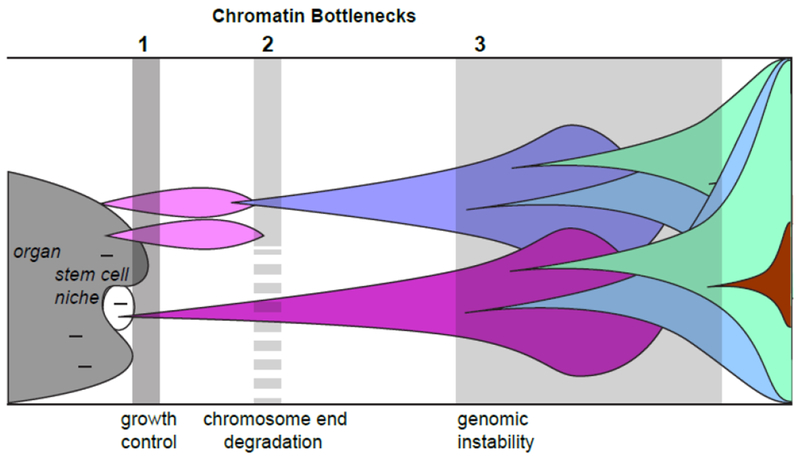
Many mutations (dashes) can occur in cells of organs (dark grey) or in stem cell compartments (white). Most chromatin mutations are lethal (the first bottleneck), but occasional subtle mutations may survive and relieve growth control of the tissue. Such clones can expand (pink), but will extinguish as chromosome ends degrade (the second bottleneck). Cells that reactivate TERT or engage in telomeric recombination can bypass this bottleneck and continue to expand (purple). Note that this bottleneck may be more permeable (dashed barrier) to malignant stem cells where TERT is already expressed. As malignant clones expand, mutations that alter cellular and tumor properties such as more rapid growth, altered metabolism, metastasis, and drug resistance (once treatment begins) will be favored by selection. Increased mutation rates and genome instability will promote evolution of malignant properties, but is limited by epigenomic surveillance and DNA damage responses (the third bottleneck). Cells that mutate or silence surveillance tumor suppressors can evolve aggressive phenotypes rapidly.
At the most fundamental level, histones package the DNA of eukaryotes into nucleosomes, and this chromatin structure limits access to polymerases and regulatory transcription factors [8]. Thus, eukaryotic polymerases require accessory factors to transcribe chromatin, and must manipulate nucleosomes to expose transcription factor binding sites at promoters and enhancers. Eukaryotes devote considerable energy to move nucleosomes around these sites using ATP-dependent chromatin remodelers. These multi-protein complexes are recruited to target sites in cells by transcription factors, and facilitate binding of additional regulatory cofactors. The role of nucleosome positioning in cancer is highlighted by numerous studies showing that chromatin remodelers are among the most frequently mutated genes in cancers [9]. An exploration of malignant rhabdoid tumors (MRT) and atypical teratoid/rhabdoid tumors (ATRT), aggressive pediatric cancers of the kidney and the central nervous system, respectively, demonstrates the contribution of aberrant nucleosome dynamics to cancer. Each of these malignancies has biallelic mutations in SMARCB1, an ATPase-containing subunit of the Brahma-Associated Factor (BAF) chromatin remodelling complex present in canonical BAF (cBAF) and the related polybromo-associated BAF (PBAF) complexes, but absent from the recently-discovered noncanonical BAF (ncBAF) complex [10–13]. Nucleosome remodelling by BAF complexes is critical for differentiation, as they act to remove and reposition nucleosomes to allow binding of transcriptional activators and repressors near developmentally restricted regulatory elements [14]. Cells in SMARCB1-deficient tumors have increased nucleosome occupancy over these elements, which fail to activate appropriately in response to differentiation cues [15]. Therefore, stem cell-like enhancer-promoter interactions can inappropriately persist, as evidenced by the activation of the super-enhancers of SPRY1 and SOX2 genes in rhabdoid tumors. Importantly, reintroduction of SMARCB1 in malignant rhabdoid tumors restores BAF activity at differentiation-promoting enhancers, terminating the stem cell regulatory network. Given that this tumor has no other identified driver mutations, failure to remodel nucleosomes at these regulatory elements is central to tumorigenesis.
It is quite remarkable that loss of function of widely-expressed chromatin proteins such as SMARCB1, ARID1A or H3.3 promote oncogenesis in developmentally-restricted tissue contexts. This is best illustrated by patients with Rhabdoid Tumor Predisposition Syndrome, in which patients with SMARCB1 germline mutations develop rhabdoid tumors in infancy [16]. Why mutations in general chromatin factors have age- and tissue-specific oncogenic potential remains a major question in the field of cancer epigenomics. We suggest that mutations in chromatin factors may be oncogenic in two ways. First, chromatin regulation is redundant, where multiple remodelling complexes are broadly targeted to regulatory elements, and act together with transcription factors. For many genes, no one remodeler is essential, but certain genes may be more or less dependent on specific remodeler complexes. Rhabdoid tumors suggest that regulation of pluripotency genes is particularly dependent on cBAF and PBAF, while promoters and CTCF sites that depend on ncBAF may be relatively insensitive to reduced cBAF or PBAF activity [14, 15]. The finding of different BAF complexes’ abilities to remodel subsets of regulatory elements also presents a therapeutic opportunity, as targeting ncBAF may hinder tumorigenesis in rhabdoid tumors [17]. Recent analyses of chromatin accessibility across cancers may identify additional relationships between remodeler deficiency and pathological regulatory element activity that drive oncogenesis, and may also provide new therapeutic strategies.
Second, because certain aspects of chromatin regulation underlie essential cellular and organismal processes, only subtle mutations that largely preserve functions of chromatin factor complexes will survive to allow further evolution of a malignancy (Figure 2) [18]. Pediatric cancers are rare, consistent with the idea that chromatin mutations are usually lethal, with only certain mutations surviving to be involved in malignancy.
This concept is evident in recent literature demonstrating the role of H3 variants in cancer. Flumans have ~60 histone genes to produce the massive amount of protein needed to package the entire genome into chromatin. Chromatin packaging is essential in all eukaryotes, and thus it was surprising that monoallelic recurrent substitution mutations in H3.1 and H3.3 histones occur in several malignancies including diffuse midline gliomas (DMGs), sarcomas, head and neck squamous carcinomas, giant cell bone tumor, chondroblastomas and acute myeloid leukemias [19, 20]. These substitutions lie in a region of the histone tail that is critical for chromatin-mediated silencing through modifications of the H3K27 residue and for transcriptional regulation through modifications of the H3K36 residue. This region is required for docking of multiple histone modifying enzymes and effector proteins, including Polycomb Repressive Complexes (PRCs). H3K27M is a dominant mutation, as only a fraction of the H3 histone pool carries the substitution. The substitution increases binding affinity of the H3 tail to the EZH2 histone methyltransferase subunit of PRC2 and reduces the in vitro activity of the enzyme [19, 21, 22]. Exogenous expression of H3.3K27M, an H3 variant that is deposited in a replication-independent manner, leads to loss of the H3K27me3 modification at some loci while gain at other loci, resulting in highly variable phenotypes depending on cellular and tissue-specific context [23, 24]. Although the H3K27M mutation is likely an early event in tumorigenesis [25], is not oncogenic on its own; a second genetic hit must occur for tumorigenesis, and this second hit commonly results in constitutive activation of ACVR1 or PDGFRA signalling receptors [24, 26].
Tumorigenesis in mouse models with the H3K27M mutation is limited to an early window in embryogenesis, implying that a developmentally restricted cell population is prone to oncogenesis with this combination of mutations [24]. Diffuse Midline Glioma (DMG) precursors may be a transient oligodendrocyte progenitor stem cell population in the ventral pons [27]. PRC-mediated repression is required for cellular differentiation, and H3K27M mutant stem cells have a decreased competence for differentiation [28]. However, they maintain sufficient residual PRC activity to silence tumor suppressor genes such as CDKN2A [22]. Consistent with the subtle inhibition of PRC2 activity by the H3K27M “oncohistone”, PRC2 subunit mutations are not seen in these malignancies, and further EZH2 inhibition in H3K27M-mutant DMGs leads to growth arrest through derepression of the CDKN2A locus [29, 30]. These data demonstrate that H3.3K27M-containing nucleosomes inhibit PRC2, but some loci maintain repression. The differential sensitivity of loci to H3K27M inhibition, the developmental and tissue-specific restriction of H3 mutations in cancers, and the interaction with secondary mutations such as ACVR1, PDGFRA, and RUNX1 remain unknown.
Additional levels of chromatin control mediated by regulatory elements may also be important in oncogenesis. In mammals, DNA modification in the form of 5-methylcytosine of CpG dinucleotides is critical for gene expression, transposon silencing, and X-chromosome inactivation. DNA methylation affects gene expression because 1) methylation of factor binding sites blocks factor binding, and 2) methylated DNA binds Methylated Cytosine binding Proteins (MeCPs and MBDs), recruiting histone modifying enzymes, and forming repressive chromatin. DNA methylation is removed by the Ten-Eleven Translocation (TET) enzymes, and many of these are translocation partners with MLL in acute leukemias. Inactivating TET-2 mutations are found in pre-malignant clones in patients with MDS and AML, which carry a high risk of transformation [31].
Long-range chromosome interactions organize chromatin within the nucleus, and this is in part regulated by the DNA-binding chromatin factor CTCF. CTCF binds at specific DNA sequences and recruits cohesin, which together link binding sites, mediate chromatin looping, and isolate active gene regions from inactive ones. CTCF binding sites are among the most frequently mutated sites in cancer, and cohesin mutations, most frequently in STAG2, have been identified in several malignancies, with well-characterized roles in acute myeloid leukemia [3, 32]. A clear example of chromatin effects on CTCF binding comes from IDH1/2 mutations in adult gliomas and secondary glioblastomas, with striking effects on two major epigenetic systems in cells [33]. In these tumors, the oncometabolite 2-hydroxyglutarate accumulates in the tumor environment and inhibits alpha-ketoglutarate-dependent enzymes, including the Jumonji domain-containing (JmJC) histone demethylases and Ten-Eleven Translocation (TET) DNA demethylases. Inhibition of TET2 results in aberrant methylation of a CTCF binding site near the PDGFRA promoter, which is important in gliomagenesis. Upregulation of PDGFRA occurs when disruption of a Topological Associated Domain (TAD) boundary activates the PDGFRA promoter by a neighboring enhancer. Interestingly, a recent study also showed this to be the case for BAF-mutant cancers, as ncBAF localizes to CTCF sites even in SMARCB1-deficient tumors to promote oncogenesis [17], further demonstrating the importance of insulators in cancer.
The second bottleneck: Proliferation degrades chromosome ends
The expansion of many cell lineages is limited by an inherent problem in DNA replication at the ends of linear eukaryotic chromosomes. Replicative polymerases synthesize DNA in a 5’-to-3’ direction using an RNA primer, and removal of the distal-most RNA primer results in a daughter strand that is shorter than the parental template. This shorter strand cannot be extended by conventional DNA polymerases. Instead, germline and somatic stem cells in mammals use the TERT telomerase reverse transcriptase to specifically extend their telomeric DNA, allowing these cells to divide indefinitely. Without extension, chromosome ends grow shorter with each cell division, eventually deleting telomeric protein binding sites, eliciting DNA damage responses and cell cycle arrest [34]. Thus oncogenic cells must extend telomeres de novo to proliferate, and a major way they do this is by expressing TERT [35]. Neuroblastoma, a pediatric tumor of the sympathetic nervous system, provides the most informative example that the replication-associated shortening of chromosome ends is a bottleneck for oncogenesis. Neuroblastoma is one of the most common pediatric solid tumors, affecting ~800 children per year. It is the most common cancer in infants under 12 months, and is a genetically heterogeneous disease [6, 36]. A subset of infantile patients presents with metastatic disease in liver, bone marrow and skin, but these tumors surprisingly spontaneously regress without treatment. These tumors appear to fail to reactivate TERT, resulting in a malignancy that extinguishes itself due to inability to maintain telomere length [37, 38].
Unfortunately, other subtypes of neuroblastoma often progress to lethal disease. These tumors show recurrent mutations implicating at least four different mechanisms that escape the telomeric bottleneck [39]. One recurrent mutation is amplification of the MYCN locus, in which N-Myc directly activates the TERT promoter [40, 41]. In other cases, a chromosome rearrangement brings a strong enhancer in proximity to the TERT promoter [42]. Both of these mutational classes reactivate telomerase expression, resulting in replicative immortality.
A second class of mutations in lethal neuroblastomas do not upregulate telomerase. This class includes recurrent mutations in the ATRX chromatin remodeler or DAXX histone chaperone complex that are mutually exclusive with MYCN alterations. ATRX or DAXX-deficient neuroblastomas extend chromosome ends by the Alternative Lengthening of Telomeres (ALT) mechanism, where telomeric repeats at deprotected chromosome ends participate in homologous recombination, lengthening chromosomes [43]. The ATRX chromatin remodeling complex normally assembles nucleosomes at telomeres using the histone H3.3 variant, which may be necessary to restore nucleosomes displaced by aberrant G-quadruplex DNA structures, replication fork stalling, and transcription at telomeres. Without ATRX, telomeric DNA becomes exposed and recombinogenic [44, 45]. While this suffices to circumvent the telomeric bottleneck, ALT is less common in neuroblastomas than telomerase-mediated telomere extension because telomeric recombination also results in prolonged DNA damage response activation, genomic damage (chromothripsis), and sensitivity to replication stress [39].
Recently, a third subset of neuroblastoma and a subset of melanoma patients were found to lack TERT activation or ALT [46, 47]. These subtypes were referred to as an “Ever Shorter Telomeres (EST)” phenotype. Telomeres in EST tumors have long repeat arrays, perhaps resulting from transient activation of TERT, or from pre-oncogenic cells that naturally have long telomeres. While chromosome ends do eventually degrade in these tumors, their extended lifespan can result in clinically significant tumor burdens [46]. Interestingly, a recent analysis of over 400 pre-treatment neuroblastomas demonstrated that patients can be risk-stratified according to telomere maintenance mechanism status, thereby relating a chromatin bottleneck to prognosis [48].
Stem cells may be uniquely sensitive to oncogenesis because they express pluripotency factors, including the Myc transcription factor, a potent oncogene [3, 49]. As stem cells must divide throughout a lifetime, they also express telomerase to maintain chromosome ends; thus the telomeric bottleneck to oncogenesis is relieved in these cells [50]. Why then are stem cell-derived cancers not more common? Stem cells express a collection of master transcription factors that activate each other (aka the stem cell circuit) to maintain the pluripotent state [51]. Shut down of these genes is coordinated with inter-cellular signalling that stimulates differentiation. Thus, oncogenic expansion of stem cells requires mutations that together 1) hyperactivate a signalling receptor and 2) prevent repression of the stem cell circuit. This explains the oncogenic combination of H3K27M mutations with hyperactivating signalling receptor mutations.
A similar scenario may apply in Wilms tumor [52]. These are pediatric renal tumors that display stem cell epigenomic features, and appear to derive from renal blastemal stem cells. Wilms tumor cells have reduced differentiation capabilities into stromal and epithelial lineages derived from renal blastemal cells, suggesting that they are unable to shut off stem cell epigenomic features although they can aberrantly express some markers associated with differentiation [52]. More generally, the developmental specificity of mutations for specific cancers will depend on the particular genes and transcription factors that activate stem cell programs in those cells.
The third bottleneck: Chromatin dysfunction activates genomic sensors
Certain cell types like stem cells express TERT, making them particularly susceptible to oncogenic transformation [53]. As stem cell cancers are relatively rare, mechanisms must be present in these precarious cells to protect against uncontrolled proliferation. Such protection may result from coupling repression of pluripotency genes to activation of proliferation genes (Figure 3A). Polycomb-mediated gene silencing is required in differentiating cells to silence pluripotency genes [51, 54]. Thus, one way to maintain pluripotency gene and TERT expression is to cripple this silencing system. However, at least one critical DNA damage response gene appears to respond to changes in epigenomic silencing. The CDKN2A locus encodes both p16 (INK4A) which inhibits cell division and p14 (ARF) which stimulates apoptosis in response to DNA damage [55, 56]. The locus is encompassed in a silenced domain with bivalent promoters, where active histone modifications poise the genes for induction upon loss of PRC activity [52]. This provides a mechanism to sense global changes in Polycomb silencing, thereby monitoring the epigenome. For example, adult T cell acute lymphoblastic leukemia (T-ALL) is caused by loss of the EZH2 histone methyltransferase complex in precursor cells [57]. However, global reduction of Polycomb-mediated silencing derepresses the CDKN2A locus, and triggers growth arrest and cell death in most T-ALL precursor cells. In some cells, deletion of the CDKN2A locus ablates these checkpoints, and these are the cells that proliferate as a tumor. In this way, CDKN2A is a sensitive barometer for Polycomb-mediated silencing, eliminating cells that lose this epigenetic silencing system.
Figure 3. Chromatin mechanisms that limit cell proliferation.
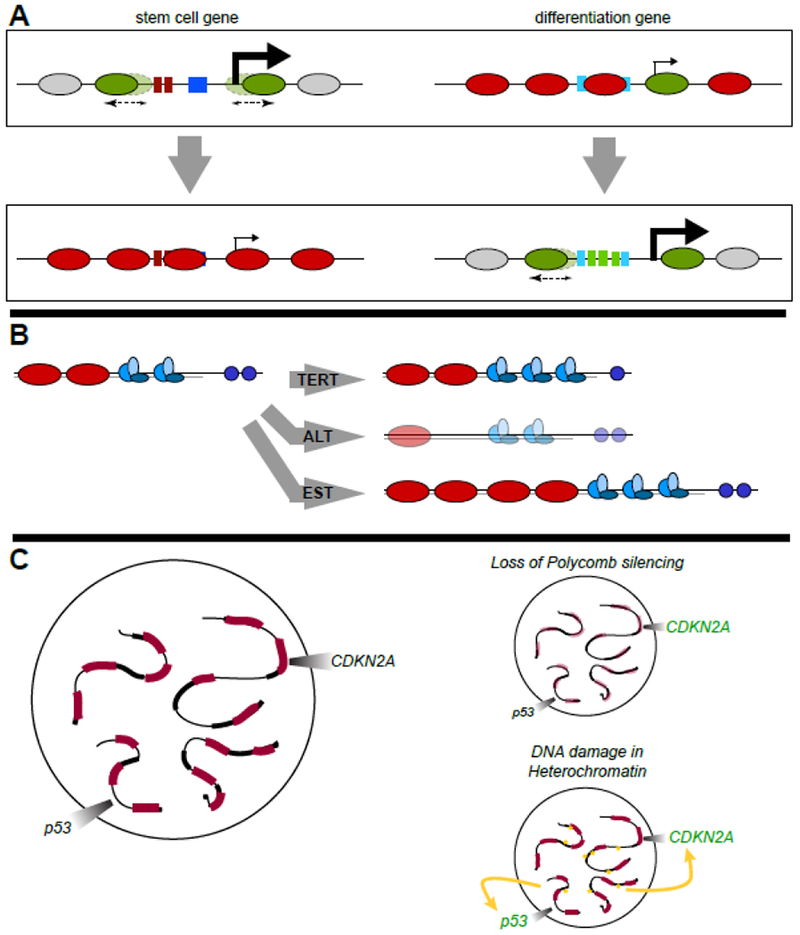
(A) Chromatin structure of promoters control gene expression. Transcription factor binding sites (boxes) at the promoters of pluripotency and differentiation genes are packaged in nucleosomes (ovals). Nucleosomes are marked with active histone modifications (green) or with repressive modifications (red), and moved by chromatin remodelers (dashed arrows) to block or expose factor binding sites and silence or promote gene expression, respectively. In stem cells (top) chromatin remodelers maintain access at promoters of pluripotency factors, while differentiation genes are silent. As cells differentiate tissue-specific transcription factors activate differentiation genes, and pluripotency promoters are occluded by nucleosomes and repressed by Polycomb-mediated silencing. Chromatin defects that inhibit efficient silencing of pluripotency genes may stall cells in an undifferentiated state that is prone to uncontrolled proliferation upon additional mutation.
(B) Telomeres are the specialized ends of linear chromosomes. Nucleosomes (red ovals) package subtelomeric DNA, while the more distal double-stranded telomere repeat sequences are bound by shelterin protein complexes (blue) and the single-stranded DNA of the chromosome end is bound by the hPOT1 capping protein (purple). In most somatic cells the chromosome end shortens after incomplete DNA replication in each S phase of the cell cycle, eventually deleting hPOT1 and shelterin binding sites and activating DNA damage responses. In germline and stem cells, TERT telomerase maintains the length of the chromosome end by extending the shorter bottom strand after every S phase. Malignant cells may extend chromosome ends by at least three ways (grey arrows): 1) by reactivating TERT and synthesizing new telomeric DNA; 2) by ALT, where loss of nucleosomes packaging telomeric regions permits ectopic recombination and extends chromosome ends; and 3) by EST, where cells with extremely long telomeres are not extended but are long enough to support clonal expansion.
(C) Epigenomic surveillance by tumor suppressors. Extensive chromosomal regions within a nucleus are packaged in silenced chromatin (red), including the tumor suppressor CDKN2A locus. Other regions contain repetitive DNA sequences (thick black lines). Loss of chromatin proteins can derepress CDKN2A, thus eliminating cells with defective epigenomic silencing. DNA damage induced during DNA replication often occurs in heterochromatic repeats but triggers CDKN2A and p53 expression, eliminating damaged cells.
The concept that the CDKN2A locus is a sensor of gene silencing is also evident in Malignant Peripheral Nerve Sheath Tumor (MPNST), which is associated with neurofibromatosis type 1 [58]. MPNSTs are strongly associated with only three co-occurring mutations: in the GTPase accessory factor NF1, in components of the EZH2 methyltransferase complex, and in CDKN2A [59]. NF1 is a potent inhibitor of the Ras signalling pathway and NF1 mutations prolong Ras activation [60]. Mutations in the PRC2 component SUZ12 promote tumorigenesis in NF1-deficient cells, because PRC2 normally dampens Ras activation of downstream gene targets, but this loss also derepresses the CDKN2A locus. Thus, MPNSTs can only proliferate with a combination of the three mutations that bypasses the three chromatin bottlenecks to cancer.
Compared to adult malignancies, the CDKN2A locus is less frequently deleted in most pediatric tumors [6]. In two of these pediatric tumors – DMGs and Wilms Tumor – Polycomb silencing is maintained at the CDKN2A locus, in spite of global reductions in H3K27me3. The global reduction may be due to the stem cell origin of these tumors, but the critical effect is that chromatin-mediated silencing is equivalent to genetic deletion of the CDKN2A locus. Thus, stem cells in developing tissues with subtle mutations in chromatin remodelers are likely able to maintain enough chromatin regulation to silence tumor suppressors while derepressing other loci important for uncontrolled growth.
Epigenomic status of other chromosome regions may also impinge on oncogenesis. Almost 50% of the human genome is repetitive DNA sequences that are not included in genome assemblies; this includes copies of transposons and retrotransposons that are targeted for silencing by DNA methylation and by chromatin silencing marked by methylation of the histone H3K9 residue. While heterochromatin is gene-poor, there are two risks to repetitive sequences. First, most DNA damage occurs in heterochromatin simply because of the sheer amount of DNA included in these regions. Second, heterochromatic DNA damage can lead to chromosomal rearrangements and genome instability, because of ectopic recombination between scattered repeat sequences. DNA damage in heterochromatin is surveyed by both p53 and gene products of the CDKN2A locus. DNA damage in heterochromatin thus acts as a second barometer for the status of the epigenome, as DNA damage near tumor suppressor genes will often co-occur with many more damage events in heterochromatin that can be easily sensed. Thus eliminating either p53 or CDKN2A pathways during oncogenesis will increase tolerance to all DNA damage, with a concomitant increase in genome instability. Cancers appear to suffer losses in heterochromatic silencing as well, as multiple transposons and retrotransposons are derepressed, further mutating the genome. For example, the BRCA1 protein localizes to heterochromatin in normal cells, where part of its contribution to tumor suppression is by repressing transcription of repetitive satellite sequences [61]. While these are non-coding transcripts, they can on their own induce DNA damage, inhibit checkpoints, and potentially lead to genomic instability [61, 62]. A substantial fraction of heterochromatin sequences may be the remnants of genomic conflicts by selfish genetic elements. Such genomic conflicts are often cryptic, since host suppressor genes continually restrain those elements. But recent work has shown that massive genetic catastrophes including widespread transposon mobilization occurs when suppressor genes are lost. This way of thinking leads to the idea that epigenomic changes deep in heterochromatin might have dramatic consequences for genome stability, emphasizing that derepression of heterochromatin may contribute to tumor progression.
Concluding Remarks
Aberrant chromatin regulation has been observed in a wide variety of cancers, but the precise contributions to tumor suppression and oncogenesis are only now beginning to be understood. The rarity of pediatric cancer and the recurrent mutations in chromatin-regulating genes allows us to identify three major bottlenecks to cancer that are intrinisic to chromatin. While more common adult malignancies contain mutations in signalling genes that activate developmental or proliferative programs (eg. BRAF and RAS) these must be accompanied by multiple secondary mutations to escape chromatin bottlenecks; these include reactivation of TERT, mutations in chromatin remodelers, and disruption of CTCF binding sites. While we have outlined chromatin bottlenecks as a sequence of genetic changes, some of these may occur simultaneously by pleiotropic actions of oncogenes. For example, the Myc oncogene may stimulate proliferation, reactivate telomerase, and induce genomic instability, affecting all three chromatin bottlenecks. We note also that the three bottlenecks do not apply equally to malignant cells. Tumors deriving from a stem cell population initiate as TERT-positive cells, obviating the need for mutations to bypass the chromosome end degradation bottleneck. However, these three chromatin bottlenecks can inform efforts to develop interventional therapies for all tumors.
Agents that target chromatin of promoters and regulatory elements offer the opportunity to interfere with the drivers of uncontrolled proliferation in tumors. HDAC inhibitors have been used to induce differentiation with some success in pre-clinical models [63, 64], and their efficacy may be due to improved repression of oncogenes or enhancement of differentiation genes [65, 66]. Therapies that target defective long-range chromatin interactions are already available. For example, DNA hypomethylating agents can restore CTCF binding and downregulate PDGFRA in pre-clinical models of IDH-1 mutant gliomas, suggesting a therapeutic opportunity for humans [67, 68]. IDH-1/2 inhibitors may act by enhancing DNA and histone methylation levels and restoring normal chromatin structure. Similarly, PARP inhibitors are synthetically lethal with cohesin mutations in pre-clinical models [69], and clinical trials are in development.
For the second bottleneck, drug inhibition of the telomerase protein complex is well-established as an attractive target, although few effective therapies currently exist [70–72]. However, the prolonged DNA damage response activation in ALT-positive malignant cells suggests that components of DNA repair pathways might be exploited. This is supported by the sensitivity of ALT-positive cells to Ataxia-Telangiectasia and Rad3-related kinase (ATR) inhibitors [73], although not all ALT-positive cells are sensitive [74]. The recent realization that the EST phenotype occurs in cells without telomere lengthening suggests that these tumors might be treated by repeated resection, reducing tumor burden until telomere lengths are critically short [46, 47]. Perturbing telomere maintenance mechanisms is likely to be a useful adjuvant to current approaches to treating malignancies.
Finally, articulation of epigenomic surveillance where tumor suppressors sense chromatin status suggests another front to attack malignancies. Drugs that modulate histone modifications have proven efficacious in preclinical models of both liquid and solid malignancies [29, 75, 76]. Modulating histone methylations that direct Polycomb-mediated silencing may be an important route for targeting malignancies. For example, inhibiting the EZH2 histone methyltransferase in tumor cells with low EZH2 activity could activate CDKN2A and eliminate tumor cells. Indeed, treatment of malignant rhabdoid tumors with EZH2 inhibitors has proven efficacious in preclinical models, with clinical trials now underway [76]. Interestingly, in malignancies with mutations in the SMARCA4 chromatin remodeler, EZH2 inhibition activates the SMARCA2 chromatin remodeler, leading to growth arrest [75]. This indicates that interactions between histone modifications and chromatin remodelers may be exploited as a therapeutic strategy [75]. However, caution must be used as well, as other stem cell populations may be exquisitely sensitive to EZH2 inhibition, in particular precursors in T-cell development. This is evidenced by inactivating EZH2 mutations in T-ALL [77] and the recent identification of a secondary T-cell lymphoma in a patient receiving an EZH2 inhibitor for a CNS malignancy. It will be exciting to see if elucidation of the tissue-specificity of mutations in chromatin regulating genes will lead to new therapeutic strategies and windows for high-risk malignancies (see outstanding questions).
Highlights:
The Hallmarks of Cancer correspond to specific changes in chromatin control that normally limit cell proliferation.
Mutational burdens in many pediatric cancers are unusually simple but include defects in core chromatin regulators. While most mutations in chromatin regulators are expected to be lethal events, rare subtle mutations may be viable and oncogenic.
Chromatin mutations interact with cellular signalling at specific childhood developmental stages to induce oncogenic cells.
Chromosome end degradation limits uncontrolled proliferation, but multiple chromatin defects can circumvent this barrier. These define distinct sets of co-occuring oncogenic mutations.
Some tumor suppressors are repressed by chromatin-mediated silencing and act as sensors to maintain epigenomic silencing. Bypassing these sensors defines additional sets of recurring oncogenic mutations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hanahan D and Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144 (5), 646–74. [DOI] [PubMed] [Google Scholar]
- 2.Mesri EA et al. (2014) Human viral oncogenesis: a cancer hallmarks analysis. Cell Host Microbe 15 (3), 266–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yao X et al. (2016) Epigenomic Consequences of Coding and Noncoding Driver Mutations. Trends Cancer 2 (10), 585–605. [DOI] [PubMed] [Google Scholar]
- 4.Vogelstein B et al. (2013) Cancer genome landscapes. Science 339 (6127), 1546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lawrence MS et al. (2013) Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499 (7457), 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grobner SN et al. (2018) The landscape of genomic alterations across childhood cancers. Nature 555 (7696), 321–327. [DOI] [PubMed] [Google Scholar]
- 7.Polak P et al. (2015) Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature 518 (7539), 360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teves SS et al. (2014) Transcribing through the nucleosome. Trends Biochem Sci 39 (12), 577–86. [DOI] [PubMed] [Google Scholar]
- 9.Kadoch C and Crabtree GR (2015) Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci Adv 1 (5), e1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fruhwald MC et al. (2016) Atypical teratoid/rhabdoid tumors-current concepts, advances in biology, and potential future therapies. Neuro Oncol 18 (6), 764–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim KH and Roberts CW (2014) Mechanisms by which SMARCB1 loss drives rhabdoid tumor growth. Cancer Genet 207 (9), 365–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johann PD et al. (2016) Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 29 (3), 379–393. [DOI] [PubMed] [Google Scholar]
- 13.Mashtalir N et al. (2018) Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 175 (5), 1272–1288 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakayama RT et al. (2017) SMARCB1 is required for widespread BAF complex-mediated activation of enhancers and bivalent promoters. Nat Genet 49 (11), 1613–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X et al. (2017) SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat Genet 49 (2), 289–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agaimy A and Foulkes WD (2018) Hereditary SWI/SNF complex deficiency syndromes. Semin Diagn Pathol 35 (3), 193–198. [DOI] [PubMed] [Google Scholar]
- 17.Michel BC et al. (2018) A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat Cell Biol 20 (12), 1410–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aloia L et al. (2013) Polycomb complexes in stem cells and embryonic development. Development 140 (12), 2525–34. [DOI] [PubMed] [Google Scholar]
- 19.Funato K and Tabar V (2018) Histone Mutations in Cancer. Annual Reviews of Cancer Biology 2, 337–351. [Google Scholar]
- 20.Lehnertz B et al. (2017) H3(K27M/I) mutations promote context-dependent transformation in acute myeloid leukemia with RUNX1 alterations. Blood 130 (20), 2204–2214. [DOI] [PubMed] [Google Scholar]
- 21.Williams RT and Sherr CJ (2008) The INK4-ARF (CDKN2A/B) locus in hematopoiesis and BCR-ABL-induced leukemias. Cold Spring Harb Symp Quant Biol 73, 461–7. [DOI] [PubMed] [Google Scholar]
- 22.Stafford JM et al. (2018) Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci Adv 4 (10), eaau5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herz HM et al. (2014) Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science 345 (6200), 1065–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pathania M et al. (2017) H3.3(K27M) Cooperates with Trp53 Loss and PDGFRA Gain in Mouse Embryonic Neural Progenitor Cells to Induce Invasive High-Grade Gliomas. Cancer Cell 32 (5), 684–700 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nikbakht H et al. (2016) Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat Commun 7, 11185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mackay A et al. (2017) Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 32 (4), 520–537 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones C and Baker SJ (2014) Unique genetic and epigenetic mechanisms driving paediatric diffuse high-grade glioma. Nat Rev Cancer 14 (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Funato K et al. (2014) Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 346 (6216), 1529–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mohammad F et al. (2017) EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med 23 (4), 483–492. [DOI] [PubMed] [Google Scholar]
- 30.Piunti A et al. (2017) Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat Med 23 (4), 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bowman RL et al. (2018) Clonal Hematopoiesis and Evolution to Hematopoietic Malignancies. Cell Stem Cell 22 (2), 157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katainen R et al. (2015) CTCF/cohesin-binding sites are frequently mutated in cancer. Nat Genet 47 (7), 818–21. [DOI] [PubMed] [Google Scholar]
- 33.Flavahan WA et al. (2016) Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529 (7584), 110–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olovnikov AM (1996) Telomeres, telomerase, and aging: origin of the theory. Exp Gerontol 31 (4), 443–8. [DOI] [PubMed] [Google Scholar]
- 35.Maciejowski J and De Lange T (2017) Telomeres in Cancer: tumor suppression and genome instability. Nat Rev Mol Cell Bio 18, 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pugh TJ et al. (2013) The genetic landscape of high-risk neuroblastoma. Nat Genet 45 (3), 279–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brodeur GM and Bagatell R (2014) Mechanisms of neuroblastoma regression. Nat Rev Clin Oncol 11 (12), 704–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hiyama E et al. (1995) Correlating telomerase activity levels with human neuroblastoma outcomes. Nat Med 1 (3), 249–55. [DOI] [PubMed] [Google Scholar]
- 39.Hertwig F et al. (2016) Telomere maintenance is pivotal for high-risk neuroblastoma. Cell Cycle 15 (3), 311–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nikiforov MA et al. (2002) TRRAP-dependent and TRRAP-independent transcriptional activation by Myc family oncoproteins. Mol Cell Biol 22 (14), 5054–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rickman DS et al. (2018) The Expanding World of N-MYC-Driven Tumors. Cancer Discov 8 (2), 150–163. [DOI] [PubMed] [Google Scholar]
- 42.Peifer M et al. (2015) Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 526 (7575), 700–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dilley RL and Greenberg RA (2015) ALTernative Telomere Maintenance and Cancer. Trends Cancer 1 (2), 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Voon HPJ et al. (2016) Compromised Telomeric Heterochromatin Promotes ALTernative Lengthening of Telomeres. Trends Cancer 2 (3), 114–116. [DOI] [PubMed] [Google Scholar]
- 45.Clynes D et al. (2015) Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat Commun 6, 7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dagg RA et al. (2017) Extensive Proliferation of Human Cancer Cells with Ever-Shorter Telomeres. Cell Rep 19 (12), 2544–2556. [DOI] [PubMed] [Google Scholar]
- 47.Viceconte N et al. (2017) Highly Aggressive Metastatic Melanoma Cells Unable to Maintain Telomere Length. Cell Rep 19 (12), 2529–2543. [DOI] [PubMed] [Google Scholar]
- 48.Ackermann SC,M; Hero B; Welte A; Kahlert Y; Roderwieser A; Bartenhagen C; Walter E; Gecht J; Kerschke L; Volland R; Menon R; Heuckmann JM; Gartlgruber M; Hartlieb S; Henrich K; Okonechnikov K; Altmuller J; Nurnberg P,; Lefever S; deWilde B; Sand F; Ikram F; Rosswog C; Fischer J; Thiessen J; Hertwig F; Singhi AD; Thorsten S; Vogel W; Perner S; Krug B; Schmidt M; Rhmann S; Achter V; Lang U; Vokuhl C; Ortmann M; Buttner R; Eggert A; Speleman F; O’Sullivan RJ; Thomas RK; Berthold F; Vandesompele J; Schramm A; Westermann F; Schulte JH; Peifer M; Fischer M (2018) A Mechanistic Classification of Clinical Phenotypes in Neuroblastoma. Science 362 (6419), 1165–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ohm JE et al. (2007) A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet 39 (2), 237–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gunes C and Rudolph KL (2013) The role of telomeres in stem cells and cancer. Cell 152 (3), 390–3. [DOI] [PubMed] [Google Scholar]
- 51.Laugesen A and Helin K (2014) Chromatin repressive complexes in stem cells, development, and cancer. Cell Stem Cell 14 (6), 735–51. [DOI] [PubMed] [Google Scholar]
- 52.Aiden AP et al. (2010) Wilms tumor chromatin profiles highlight stem cell properties and a renal developmental network. Cell Stem Cell 6 (6), 591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wainwright EN and Scaffidi P (2017) Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity. Trends Cancer 3 (5), 372–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Niwa H (2018) The principles that govern transcription factor network functions in stem cells. Development 145 (6). [DOI] [PubMed] [Google Scholar]
- 55.Bracken AP et al. (2007) The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev 21 (5), 525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dietrich N et al. (2007) Bypass of senescence by the polycomb group protein CBX8 through direct binding to the INK4A-ARF locus. EMBO J 26 (6), 1637–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ntziachristos P et al. (2012) Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med 18 (2), 298–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morgan MA and Shilatifard A (2015) Chromatin signatures of cancer. Genes Dev 29 (3), 238–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brohl AS et al. (2017) The genomic landscape of malignant peripheral nerve sheath tumors: diverse drivers of Ras pathway activation. Sci Rep 7 (1), 14992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.De Raedt T et al. (2014) PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 514 (7521), 247–51. [DOI] [PubMed] [Google Scholar]
- 61.Zhu O et al. (2011) BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature 477 (7363), 179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhu O et al. (2018) Heterochromatin-Encoded Satellite RNAs Induce Breast Cancer. Mol Cell 70 (5), 842–853 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kerl K et al. (2013) The histone deacetylase inhibitor SAHA acts in synergism with fenretinide and doxorubicin to control growth of rhabdoid tumor cells. BMC Cancer 13, 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nagaraja S et al. (2017) Transcriptional Dependencies in Diffuse Intrinsic Pontine Glioma. Cancer Cell 31 (5), 635–652 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang L et al. (2018) Resetting the epigenetic balance of Polycomb and COMPASS function at enhancers for cancer therapy. Nat Med 24 (6), 758–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lasko LM et al. (2017) Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 550 (7674), 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.DiNardo CD et al. (2018) Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N Engl J Med 378 (25), 2386–2398. [DOI] [PubMed] [Google Scholar]
- 68.Stein EM et al. (2017) Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 130 (6), 722–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu Y et al. (2018) Somatic mutation of the cohesin complex subunit confers therapeutic vulnerabilities in cancer. J Clin Invest 128 (7), 2951–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Buseman CM et al. (2012) Is telomerase a viable target in cancer? Mutat Res 730 (1-2), 90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Feldser DM and Greider CW (2007) Short telomeres limit tumor progression in vivo by inducing senescence. Cancer Cell 11 (5), 461–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Greenberg RA et al. (1999) Short dysfunctional telomeres impair tumorigenesis in the INK4a(delta2/3) cancer-prone mouse. Cell 97 (4), 515–25. [DOI] [PubMed] [Google Scholar]
- 73.Flynn RL et al. (2015) Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 347 (6219), 273–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Deeg KI et al. (2016) Cancer Cells with Alternative Lengthening of Telomeres Do Not Display a General Hypersensitivity to ATR Inhibition. Front Oncol 6, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Januario T et al. (2017) PRC2-mediated repression of SMARCA2 predicts EZH2 inhibitor activity in SWI/SNF mutant tumors. Proc Natl Acad Sci U S A 114 (46), 12249–12254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Knutson SK et al. (2013) Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 110 (19), 7922–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Simon C et al. (2012) A key role for EZH2 and associated genes in mouse and human adult T-cell acute leukemia. Genes Dev 26 (7), 651–6. [DOI] [PMC free article] [PubMed] [Google Scholar]