

Abstract
The American Initiative in Mast Cell Diseases (AIM) held its inaugural investigator conference at Stanford University School of Medicine in May 2019. The overarching goal of this meeting was to establish a Pan-American organization of physicians and scientists with multidisciplinary expertise in mast cell disease. To serve this unmet need, AIM envisions a network where basic, translational, and clinical researchers could establish collaborations with both academia and biopharma to support the development of new diagnostic methods, enhanced understanding of the biology of mast cells in human health and disease, and the testing of novel therapies. In these AIM proceedings, we highlight selected topics relevant to mast cell biology and provide updates regarding the recently described hereditary alpha-tryptasemia. In addition, we discuss the evaluation and treatment of mast cell activation (syndromes), allergy and anaphylaxis in mast cell disorders, and the clinical and biologic heterogeneity of the more indolent forms of mastocytosis. As mast cell disorders are relatively rare, AIM hopes not only to achieve a coordination of scientific efforts in the Americas, but also in Europe by collaborating with the well-established European Competence Network on Mastocytosis (ECNM).
Keywords: Systemic mastocytosis, siglec-8, mast cell activation syndrome, hereditary alpha-tryptasemia, anaphylaxis, European Competence Network on Mastocytosis (ECNM), American Initiative in Mast Cell Diseases (AIM)
Introduction
The evaluation and treatment of patients with mast cell (MC) diseases is undertaken by a cadre of multidisciplinary subspecialists. However, there is no organization in the Americas dedicated to the gathering of specialists to share research findings relevant to this patient population. With the inaugural American Initiative in Mast Cell Diseases (AIM) investigator conference, we sought to launch AIM’s mission of advancing research, education, and treatment concerning mastocytosis and related MC diseases. In this report from the AIM proceedings, we focus on selected presentations related to MC biology, hereditary alpha-tryptasemia (HαT), MC activation syndromes, allergy and anaphylaxis in MC disorders, and the heterogeneity of skin and gastrointestinal manifestations in mastocytosis. Lastly, we highlight a roadmap for future collaborations with the European Competence Network on Mastocytosis (ECNM).
Special Recognition of Dr. K. Frank Austen
A lifetime achievement award was bestowed upon Harvard Emeritus Professor K. Frank Austen, who delivered the keynote lecture on the extrinsic & intrinsic diversity of the major MC subclasses. This work is presented in the next section, entitled: “Characterization of Mast Cell Subset Heterogeneity”. Dr. Austen is a legend in the field of allergy/immunology and the most prominent MC researcher of the last century. He has dedicated his professional life to understanding the physiological and pathological role of MCs and their impact in disease. His contributions, reflected in over 1000 peer-reviewed publications since 1955, have translated into the development of therapies that have impacted patients with asthma, anaphylaxis, and mastocytosis. He has also been a pioneer in understanding the complement and kinin systems in human disease. One of his major research contributions has been elucidation of the pathways of arachidonic acid metabolism to cysteinyl leukotrienes and prostanoids, and the regulation of the action of each of these mediators in allergic inflammation. In 2016, his team described the first transcriptional signature of MCs, observing a unique identity within the mouse immune system with a greater than expected heterogeneity across tissues. As a clinician, he provided the first detailed clinicopathological characterization of human anaphylaxis, and since 1991, has led international collaborations focused on the classification of clonal MC disorders.
Mast Cell Biology
Characterization of Mast Cell Subset Heterogeneity
MCs can be classified into 2 distinct groups: innate and induced. Recognized MC progenitors in the mouse, for both innate and for induced MC subclasses, express the integrin β7, which is essential for their trans-endothelial migration from the blood into peripheral tissues. Innate MCs of adult mice arise in neonatal life from progenitors released in sequence from the extra-embryonic yolk sac and then from the hemogenic endothelium, resulting in profound intrinsic regulation of peripheral phenotypes with minimal heterogeneity.1,2 A third wave of hemogenic endothelium-derived progenitors initially seeds the mucosa,2 whereas adult bone marrow (BM) maintains mucosal populations and provides additional progenitors during a local organ-based Th2 response.3 Adoptive transfer of a mouse BM-derived v-abl immortalized MC line with limited protease expression showed expansion with immunodetection of five proteases for v-abl+ MCs in the liver, and shut down of all proteases in v-abl+ MCs in the small intestine.4 These mouse data suggest that BM-derived MCs have profound tissue-directed secretory protease heterogeneity. A subsequent study of the expansion of mouse BM-derived MCs in type 2 inflammation in the mouse gut with T. spiralis infection and in the lung with ovalbumin sensitization and challenge, allowed comparison with innate MCs. Five proteases were detected in innate MCs for both tissues, while the induced BM-derived MCs in the gut had only mMCP1, those in large bronchi mMCP1, mMCP6 and mMCP7, and those in the trachea additionally demonstrated mMCP4 and CPA.5,6 More recently, expression profiling of submucosal MCs from five mouse tissues and the peritoneal cavity revealed a MC transcriptional signature with minimal heterogeneity distinct from all other immune pathway cells, including basophils.7 Thus, innate and induced mouse MCs are diverse with regard to their derivation from separate progenitors and their tissue-directed heterogeneity-- which is minimal for innate, and profound for induced. Innate are sentinel while induced are ‘after the fact’. The key questions to resolve include: (a) what are the functions of induced BM-derived MCs; and (b) are amplified submucosal MCs in mouse models of inflammatory diseases BM-derived rather than an expansion of the innate? The implications for both questions relate to human target tissue MC expansion in bronchial asthma and rheumatoid synovitis.
As in mice, anatomically distinct compartments in human peripheral tissue contain MCs with distinct protease expression profiles, although human have a greatly reduced protease profile relative to mice. MCs co-expressing tryptase and chymase in conjunction with CPA3 and Cathepsin G (MCTC), thought to be analogous to innate murine MCs, can be found in the stromal compartment of most peripheral tissues. Supporting this concept, transcriptomics analysis identified considerable overlap between the resting transcriptome of murine constitutive skin MCs and human skin MCTC.7 MCs that express tryptase with little to no chymase (MCT), likely analogous to murine induced MCs, reside in the epithelium of mucosal tissues. A bone marrow-derived, circulating committed human MC progenitor has also been identified.8 Like its murine counterpart, this human MC progenitor expresses integrin b7 and is agranular.
Both MCT and MCTC expand during mucosal Th2 inflammation, including asthma and nasal polyposis. However, the transcriptional programs underlying the heterogeneous MC subsets found in barrier tissues, the mechanisms underlying their expansion, and their relationship to the MCT and MCTC observed under homeostatic conditions are poorly understood. To assess this heterogeneity, Dwyer and colleagues flow-sorted MCs based on FcεR1α and CD117 expression from nasal polyps and profiled them using parallel single-cell RNA sequencing. They identified histochemically distinct stromal and intraepithelial MC subsets representing two distinctly polarized ends of a transcriptional gradient, with differential expression of proteases, cytokines, chemokines, and cell surface receptors. Through differential expression analysis and flow cytometric validation, they found that chymase-negative intraepithelial MCs are characterized by robust CD38 expression and low CD117 expression, while chymase-positive stromal MCs have high CD117 expression, lack CD38, and are highly transcriptionally distinct from MCTC in the skin. A third subset was identified which co-expressed CD38 and CD117, and likely gives rise to both polarized MC subsets and contains a population of proliferating MCs. These findings thus suggest a common origin for the expanded human MCT and MCTC found within human mucosal tissue during Th2 inflammation. While murine studies suggest that the expanded MC populations observed during Th2 responses arise from adult bone marrow-derived progenitors,8 human MC progenitors have limited proliferative capacity. Thus, additional studies will be required to characterize the progenitor pool driving MC hyperplasia in nasal polyposis.
Siglec-8: Mast cell Biology and Clinical Targeting
Sialic acid-binding, immunoglobulin-like lectins (Siglecs) are single-pass transmembrane cell surface receptors found primarily on leukocytes that recognize different forms and conformations of sialic acid. Two natural ligands for Siglec-8 are known. Both involve the terminal sugar 6′-sulfated sialyl Lewis X on keratan sulfate.9 In the lower airway, the keratan sulfate is displayed on the glycoprotein aggrecan; on the upper airway, it is displayed on a different glycoprotein, namely DMBT1. So far, the physiologic meaning of these natural ligands remains unknown.
Human MCs express several Siglecs, including CD22 (low levels), CD33, Siglec-5 (low levels), Siglec-6, Siglec-7, Siglec-8 and Siglec-10 (low levels) (Figure 1).10 Among these, Siglec-8, first discovered in 2000, is expressed on MCs, but also on eosinophils. Surface expression of Siglec-8 is maintained on cells within tissues, including BM MCs in SM.11 It is now known that Siglec-8 engagement, either by specific antibodies (via multivalent, specific α2,3-linked sialylated, sulphated artificial or endogenous glycan ligands) or by sulphonamide sialoside analogues,12,13 can result in a number of effects in vitro. These include reduced eosinophil survival and attenuated MC secretion responses. The latter responses have been shown to be a result of downstream signalling events mediated via Siglec-8’s intracellular immunoreceptor tyrosine-based inhibitory motif (ITIM) domain to reduce FcεRI-mediated degranulation and calcium flux responses.14 Siglec-8 also is internalized following ligation, which allows it to function as an endocytic receptor that can be exploited to selectively deliver therapeutic payloads into these cells.15
Figure 1.
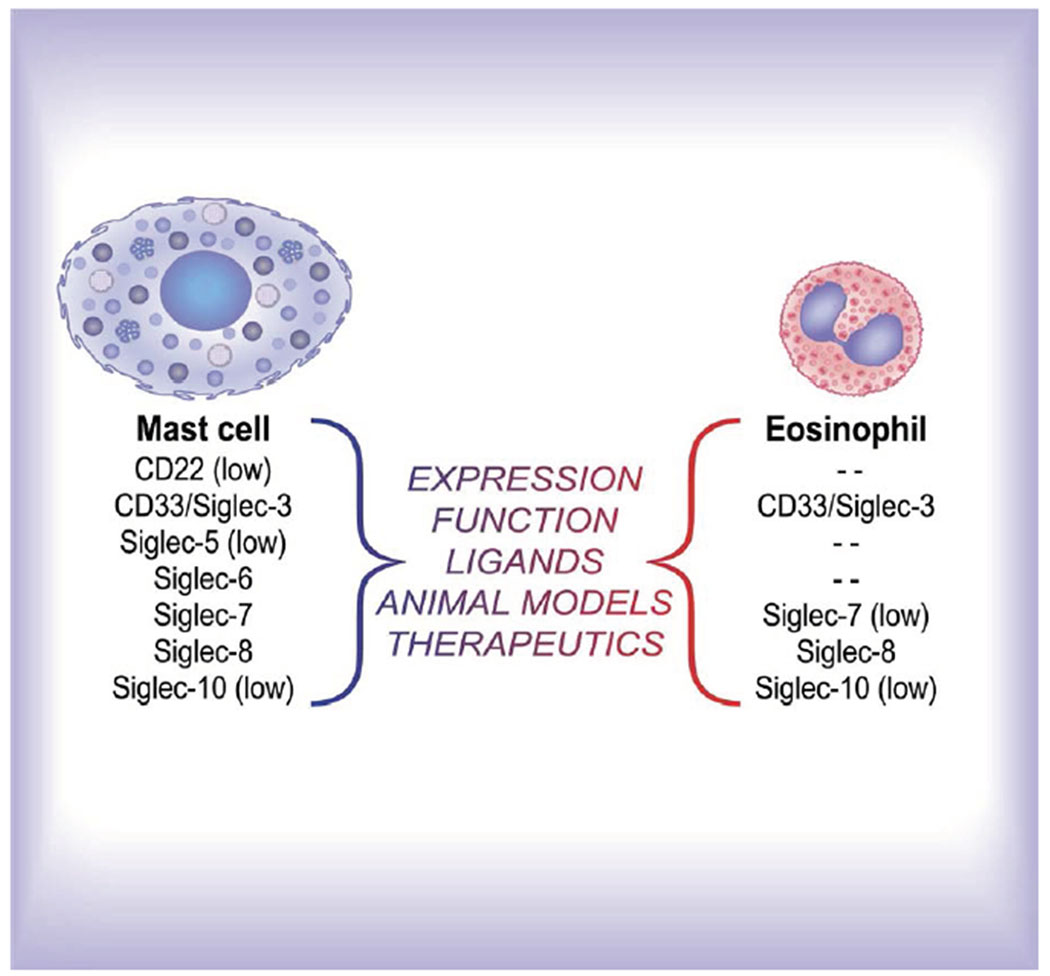
Siglecs on human mast cells and eosinophils. Artwork by Jacqueline Schaffer. This online graphical abstract (https://jlb.onlinelibrary.wiley.com/toc/19383673/0/0) that accompanies a recent publication [21] was reproduced with permission.
Because Siglec-8 is only expressed on human and primate cells and not in lower species, novel knock-in strains of mice have been generated in which Siglec-8 is transgenically expressed on eosinophils and/or MCs.16–19 Together with “humanized” mice, these preclinical models have enabled studies of Siglec-8 function on MC responses in vivo, revealing that anti-Siglec-8 antibody administration induces both protection from anaphylaxis20 and reductions in eosinophils and MCs in a model of eosinophilic gastroenteritis,18 A humanized non-fucosylated IgG1 monoclonal antibody, AK002, has entered clinical trials for the treatment of various diseases involving MCs and/or eosinophils including eosinophilic gastritis and/or gastroenteritis, antihistamine-resistant chronic urticaria, severe allergic conjunctivitis, and indolent systemic mastocytosis (SM).21
Hereditary α-Tryptasemia
An elevated basal serum tryptase (BST) has been traditionally defined as a level of greater than 11.4 ng/mL, and it is seen in approximately 5% of people from Western populations.22,23 This clinical finding can be associated with mastocytosis and other myeloid neoplasms, such as acute myeloid leukemia (AML: about 30%), chronic myeloid leukemia (CML: about 20%) and myeloproliferative neoplasms (MPN, especially those associated with eosinophilia related to fusion tyrosine kinases genes involving PDGFRA, PDGFRB, FGFR1, or JAK2).24–27 Elevated BST is now recognized to be more commonly caused by a genetic trait called hereditary alpha-tryptasemia (HαT). HαT is so-named as it results from increased TPSAB1 copy number encoding wild-type alpha-tryptase. The increased copy number of TPSAB1 leads to its over-expression and elevated levels of pro-tryptases – ostensibly composed predominantly of α-tryptase – in the peripheral blood.28 In the small number of studies to date characterizing individuals with HαT, an association has been observed between increased BST and skin symptoms such as pruritus and urticaria, \as well as with a constellation of comorbidities that may variably include certain connective tissue phenotypes such as joint hypermobility, functional gastrointestinal complaints including irritable bowel syndrome-like complaints and dyspepsia, and symptoms suggestive of autonomic dysfunction.29–31 While these associations require further validation, several studies have demonstrated a similar link between elevated BST and some of these clinical phenotypes.22,32–38 More recently, the prevalence of HαT was found to be uniquely increased in patients with mastocytosis (especially indolent SM), and was associated with an increased risk of severe mediator symptoms/anaphylaxis in these individuals. It is unknown whether the number of germline copies of TPSAB1 may confer a genetic predisposition for the later development of mastocytosis or if alpha tryptase expression level modulates mast cell homeostasis and neoplastic mast cell growth.39,40
Variable expression of these clinical findings has been observed in HαT, with some individuals reporting few or none of the described symptoms. However, all individuals identified with HαT to date have been reported with BST of ≥ 8 ng/mL (median BST in the general population is approximately 5 ng/mL), making this genetic trait fully penetrant. In the absence of severe acute systemic symptoms such as anaphylaxis, serum tryptase is comprised of enzymatically inactive monomeric pro-tryptases, irrespective of whether clonal MC disorders, myeloid disease, or HαT is the culprit.41 Unlike mature enzymatically active tryptases, which are released following MC degranulation, pro-tryptases have no currently known biological function(s).42 Mature tryptases from subjects with HαT, as well as individuals producing TPSAB1-endcoded α-tryptase, have recently been shown to include heterotetrameric αβ-tryptases that increase with greater relative gene composition of α-tryptase.43 Heterotetrameric tryptases, in contrast to α- or β-homotetramers, are able to cleave and directly activate Protease activated receptor-2 (PAR2), as well as augment activation of vibration-dependent EGF-like module-containing mucin-like hormone receptor-like 2 (EMR2).43 These pathways have been associated with gut permeability in vitro, and an increased response to vibratory challenge in vivo, respectively. Thus, such heterotetramers may contribute to some of the symptoms reported with HαT and is distinct from a severe form of familial vibratory urticaria caused by a gain-of-function variant in Adhesion G protein-coupled receptor E2 (ADGRE2) encoding EMR2.44 Whether a particular tryptase genotype may modify clonal or non-clonal MC-associated disorders and related clinical phenotypes is an area of ongoing investigation.
Mast Cell Activation, Allergy, and Anaphylaxis in Mast Cell Diseases
Mast cell activation syndrome (MCAS)
Localized MC activation is common and is presumed to be necessary for normal homeostasis.45 Pathologic MC activation can occur in the context of mastocytosis (clonal), with IgE and non-IgE mediated triggers (secondary) or idiopathically (e.g. idiopathic anaphylaxis). MCAS is a severe systemic manifestation of pathologic MC activation. Based on an initial proposal published in 2010,46 the first international consensus diagnostic criteria for MCAS were coined in 201247 and updated in 2019.48,49 All three criteria (Table 1A) must be met to establish a diagnosis of MCAS, which is divided into three subtypes (Table 1B).
Table 1.
Mast Cell Activation Syndrome (MCAS) Diagnostic Criteria
| A. Consensus criteria for MCAS* |
|---|
| Criterion A: Typical clinical signs of severe, recurrent (episodic) systemic MCA are present (often in form of anaphylaxis) (definition of systemic: involving at least 2 organ systems) |
| Criterion B: Involvement of MCs is documented by biochemical studies: preferred marker: increase in serum tryptase level from the individual’s baseline to plus 20% + 2 ng/ml† |
| Criterion C: Response of symptoms to therapy with MC-stabilizing agents, drugs directed against MC mediator production or drugs blocking mediator release or effects of MC-derived mediators‡ |
| B. Recognized Variants of MCAS and Diagnostic Features* | |
|---|---|
| Primary MCAS (Clonal MCAS)** | The KIT D816V mutation is detected and MCs aberrantly display CD25 in most cases (a) with confirmed mastocytosis (CM or SM)† (b) with only 2 minor SM criteria |
| Secondary MCAS | An IgE-mediated allergy, another hypersensitivity reaction, or another immunologic disease that can induce MCA, and thus MCAS, is diagnosed, but no neoplastic MC or KIT D816V is found‡ |
| Idiopathic MCAS | Criteria to diagnose MCAS are met, but no related reactive disease, no IgE-dependent allergy, and no neoplastic/clonal MCs are found‡ |
The consensus criteria for MCAS were first published in Valent et al.47 All 3 MCAS criteria (A + B + C) must be fulfilled to call a condition MCAS.
Other MC-derived markers of MCA (histamine and histamine metabolites, PGD2 metabolites, and heparin) have also been proposed, but are less specific compared with tryptase.
Example: histamine receptor blockers.
From reference 49: Valent P, et al. Proposed Diagnostic Algorithm for Patients with Suspected Mast Cell Activation Syndrome. J Allergy Clin Immunol Pract 2019;7(4): 1125-33 e
The terms clonal MCAS and monoclonal MCAS (MMCAS) can be used synonymously with the term primary MCAS. Whether to add HαT as a form of primary, non-clonal MCAS is under consideration.
Most of the patients suffer from CM or SM. However, in some cases, only 2 minor SM criteria are detected and 1 criteria for SM and CM are not fulfilled.
No KIT mutation at codon 816 is detected, and flow cytometry (if performed) will not detect a clonal population of CD25+MCs.
Serum tryptase, when drawn within 4 hours of symptoms, is the most specific mediator clinically available for testing to confirm MC activation. A formula of 20% of BST plus 2 ng/ml is suggested as a minimal increase to confirm MC activation in a patient with compatible symptoms.50 In the absence of tryptase, urinary metabolites of MC activation products may also be measured (see below). The benefit or harm caused by measuring mediators that are not validated, such as serum heparin and chromogranin,51 also needs to be addressed. The literature regarding MCAS include a multitude of conditions that do not fulfill the outlined criteria, which in turn leads to confusion and frustration among patients and practitioners because of the lack of a definitive diagnosis.49, 52
Utility of Urinary Mast Cell Mediators in the evaluation of SM and MCAS
Currently, selected clinical laboratories in the U.S. can determine urinary levels of the metabolites of three MC mediators: histamine (N-methylhistamine); leukotriene (LT)C4 (LTE4) and prostaglandin (PG) D2 (2,3 dinor-11βPGF2α).53 A urinary sample for the three metabolites can be collected by patients. Because results are expressed “per gm or mg creatinine”, 24-hour urinary collections are no longer necessary. “Spot” urinary samples for these markers are now available at selected centers. Samples mailed to the laboratory by overnight express are stable for at least 7 days, if kept refrigerated.
Significant elevations of urinary 11β-prostaglandin F2α and N-methylhistamine correlate with BM findings in MC disorders;54 the results can be used: 1) to exclude MC-induced symptoms from syndromes with features that symptomatically can overlap (pheochromocytoma , VIPoma and carcinoid syndromes); 2) to verify which mediators are chronically or acutely elevated and thereby allow targeting of treatment; 3) to confirm a diagnosis of suspected MCAS in a patient with low tryptase levels; and 4) to distinguish a MC “event” from a basophil “event” because, although both MC and basophils produce histamine, PGD2 is a product of MC and not basophils. The finding of an elevation of the urinary metabolite of PGD2 supports the diagnosis of MC activation; the specificity of a mildly elevated PGD2 metabolite as the only marker for MCAS has not been studied.
These assays have also shown that elevations of PGD2 are much more common than elevations of histamine in MCAS.55 This may explain the failure of antihistamines in a subset of these patients, whereas treatment with aspirin, an inhibitor of PG synthesis, may be effective in such cases. Available studies confirm increased excretion of LTE4 in SM when compared to a control population, especially in those SM patients with active clinical symptoms.56 The finding of increased excretion of all 3 urinary metabolites correlates with a diagnostic sensitivity for SM of 97% and specificity of 61%.57 Although not formally included in the criteria for SM and MCAS, urinary MC mediator assays can improve diagnosis and target treatment in these disorders.53
Anti-Mediator Therapies in Clonal and Non-Clonal Mast Cell Activation Disorders
Symptoms of MC activation are common in clonal and non-clonal MC activation disorders and are derived from the effects of MC mediators systemically and in targeted tissues. Measurements of tryptase in serum, and urinary histamine, leukotrienes, and PGD2 metabolites, has helped elucidate the clinical expression of MC activation disorders and response to anti-mediator therapies. The Brigham and Women’s Hospital Mastocytosis Center database has captured over 1500 patients, including over 350 patients with non-clonal MC activation disorders, including MCAS.58 Pediatric patients account for 109 patients, of which 17% presented with MCAS. The association of non-clonal MCAS with postural orthostatic tachycardia syndrome (POTS), dysautonomia or Ehlers-Danlos Syndrome (EDS), findings also reported in HαT patients,28–30 were found in 20% of MCAS cases with the majority of patients being female (82.5%), and close to 20% are under the age of 30. MC mediator-related symptoms in such patients affect predominantly the skin (95%), gastrointestinal (90%), respiratory (60%), and skeletal (50%) systems, and can be constitutional (50%), neuropsychiatric (> 30%) or systemic/anaphylactic (30%).58
Avoidance of triggers is critical to reduce MC activation events. These triggers may include specific foods, medications ((non-steroidal anti-inflammatory agents [NSAIDs] in selected patients, vancomycin, quinolones), environmental allergens, and general triggers (temperature changes, lack of sleep, stress). Presence of such triggers provoking symptoms should be evaluated in each patient through a careful history and when indicated, by food/drug testing and challenges.45 Some events may be caused by IgE antibody-independent responsiveness of mast cells to cationic substances, referred to as basic secretagogues. Binding of these molecules to the MRGPRX2 receptor on mast cells can result in systemic pseudo-allergic, or anaphylactoid reactions.59
Pre-medications are generally recommended for radiological, endoscopic procedures and for invasive procedures with contrast dyes, surgery, dental work, and, in sensitive patients, before vaccinations. Anti-histamine H1 and H2 blockers, leukotriene receptor antagonists, and in some cases, low-dose glucocorticoids are recommended based on individual symptom profiles. Skin symptoms are targeted with anti-histamines H1 and H2 blockers, ketotifen, leukotriene receptor antagonists, aspirin and topical cromolyn sodium, which may have anecdotal benefit in cutaneous mastocytosis based on some of the authors’ experience. Gastrointestinal symptoms are targeted with anti-histamine H2 blockers, proton pump inhibitors, sodium cromolyn and ketotifen. Respiratory symptoms are treated with bronchodilators, nebulized cromolyn sodium, with and without glucocorticoids.
Severe hypotension and shock during systemic/anaphylactic events and which can occur in patients with MCAS, are treated with injected epinephrine and patients are recommended to carry at least 2 epinephrine auto-injectors. Recurrent hypotensive events can be treated prophylactically with glucocorticoids with variable success. More recently, anecdotal evidence has shown that omalizumab may be effective in patients with MCAS alone or in mastocytosis with severe MCAS (associated with an IgE-dependent allergy).60–64 Neuropsychiatric symptoms have been treated with anti-histamine H1 blockers, aspirin, anxiolytics, and cromolyn sodium; however, there are limited data regarding their efficacy for these symptoms, and the activity of these agents may reflect their ability to cross the blood-brain barrier and achieve pharmacologically active concentrations. Bone symptoms, including osteopenia, osteoporosis and bone fractures, are treated with calcium, vitamin D, bisphosphonates, denosumab and, if needed, interferon-alfa.65–66 Tyrosine kinase inhibitors (TKIs) currently are reserved for clonal MC activation syndromes, since there is no clinical evidence that activated TKs, including KIT, are involved in non-clonal MCAS. However, in vitro such inhibitors do attenuate mast cell activation. Future dissection of the biology of MC activation disorders, as well as the newly described HαT, will help identify disease-relevant therapeutics, such as anti-tryptase antibodies, which may have utility particularly in the latter condition.67
Allergy and Anaphylaxis in Mast Cell Diseases
Patients with SM have an increased risk of anaphylaxis compared to the general population. Anaphylaxis occurs more often in patients with SM lacking cutaneous involvement and in those with atopic predisposition.68 There is a strong correlation between mastocytosis and Hymenoptera allergy and the most common IgE-mediated triggers for anaphylaxis in subjects with mastocytosis are Hymenoptera venoms (HV) from stings. Idiopathic anaphylaxis is second in frequency followed by pharmacologic agents and foods as triggers.69–71 The prevalence of systemic reactions after Hymenoptera stings in patients with mastocytosis is significantly higher (20-30%) compared to the general population (0.3% - 8.9%); the prevalence of SM in patients with Hymenoptera venom allergy (HVA) (8%) is also higher than in the general population.72 Currently, there are no guidelines to support skin testing for Hymenoptera anaphylaxis in MCAS or mastocytosis. Patients with reactions are candidates for skin testing life-long immunotherapy. A subset of patents with Hymenoptera anaphylaxis have been found to have clonal mast cell disorders, either systemic mastocytosis or monoclonal MCAS, but there is currently no evidence for an increased incidence of Hymenoptera anaphylaxis in MCAS patients.
The initial association between HVA and SM was described in patients with urticaria pigmentosa (UP), but more recently HVA has been described with increased frequency in patients with SM without skin involvement. Anaphylactic reactions in patients with SM typically present without angioedema or hives and instead with cardiovascular symptoms, such as hypotension and collapse. Patients may not report MC activation symptoms between acute episodes and severe reactions to HV may be the sole manifestation and presenting symptom of mastocytosis. Among those presenting in this manner, BST is lower than median levels in SM generally, and can be normal. This is likely related to a lower MC burden, since these patients have fewer aggregates in the BM.
The prevalence of mastocytosis among patients with drug hypersensitivity is lower than in patients with HVA, but the severity of reactions, including anaphylaxis, is similar. SM patients with HVA and documented by skin testing or serum IgE testing should be treated with life-long venom immunotherapy (VIT), which confers increased protection in the majority (86%) of restung patients.73 Patients afflicted with SM who also have severe systemic reactions and anaphylaxis should carry at least two epinephrine auto-injectors. Omalizumab has been found to be safe and effective in preventing recurrent unprovoked anaphylaxis.60–63,74
Clinical Heterogeneity of Mastocytosis
Skin Disease in Pediatric and Adult Systemic Mastocytosis
Cutaneous lesions in mastocytosis are highly heterogeneous with localized and disseminated forms. CM is divided into three variants 1) maculopapular cutaneous mastocytosis (MPCM), also known as UP; 2) diffuse CM (DCM); and 3) mastocytoma of the skin. The classification of CM has been based on macroscopic features of skin lesions, their distribution, or the onset of the disease.75–80 The typical maculopapular cutaneous lesions (urticaria pigmentosa, UP) should be subdivided into two variants, namely a monomorphic variant with small maculopapular lesions, typically seen in adult patients, and a polymorphic variant with large irregularly shaped lesions, typically observed in pediatric patients (Table 2).75 Clinical observations suggest that the monomorphic variant, if it develops in children, often persists into adulthood, whereas the polymorphic variant usually resolves around adolescence. Overall, more than 80% of all patients with mastocytosis exhibit characteristic brown or red skin lesions.80 The Darier’s sign, defined by swelling and reddening upon mechanical stroking or rubbing of the lesions, is usually demonstrable.81
Table 2.
Characteristics of Skin Lesions in Adult vs. Childhood-onset Mastocytosis75
| Adulthood-onset mastocytosis | Childhood-onset mastocytosis | |
|---|---|---|
| Most frequent category of mastocytosis | ISM | Cutaneous mastocytosis |
| Typical course of the disease | Chronic | Temporary |
| Frequency of anaphylaxis (%) | 50 | <10 |
| Typical tryptase level (ug/L) | >20 | <20 |
| Typica location of KIT mutation | Exon 17, most frequently KIT D816V | Exon 8, 9, 11, or 17 or absent |
| Most frequent type of cutaneous lesions | Maculopapular | Maculopapular |
| Typical morphology of maculopapular lesions | Monomorphic | Polymorphic |
| Typical size of maculopapular lesions | Small | Large |
| Typical distribution of maculopapular lesions | Thigh, trunk | Trunk, head, extremities |
From Reference 75: Hartmann K, et al. Cutaneous manifestations in patients with mastocytosis: Consensus report of the European Competence Network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European Academy of Allergology and Clinical Immunology. J Allergy Clin Immunol 2016;137:35-45.
Mastocytomas and DCM occur almost exclusively in the pediatric population. In adult-onset mastocytosis, cutaneous lesions are usually associated with SM, most often ISM.82–84 By contrast, in the majority of pediatric patients, CM is found without histologically evident involvement of other organs.85–88 Patients with well-differentiated SM (WDSM) mainly have a pediatric-onset disease89 with persistent skin manifestations (polymorphic MPCM and DCM) typically seen in pediatric patients. These patients have increased MCs in the bone marrow that lack the characteristic markers (CD2, CD25) of the clonal MCs frequently seen in ISM. The course of mastocytosis in adults is usually chronic, whereas children often show a self-limited course with spontaneous resolution around puberty.90–92 Studies have shown that adult and pediatric patients differ with respect to mutations in the KIT gene.93–96 While more than 80% of adult patients with mastocytosis carry the KIT D816V missense variant in exon 17, patients with childhood-onset mastocytosis may not have detectable KIT mutations or express other KIT varjants affecting exons 8, 9, 11 or 17.95
Large polymorphic lesions in children generally have a favorable prognosis for disease resolution.97 A recent report documented the similarities of cutaneous mastocytosis in patients of different racial and ethnic groups. Although the lesions in these patients may demonstrate darker pigmentation, they are not so dissimilar that they would lead to a misdiagnosis.98
Therapy is targeted to avoid known triggers, such as physical and environmental insults, as well as patient-specific triggers, including foods, drugs or venom. Additional management includes employing local skin care with emollients, glucocorticoids and antibiotic ointments when indicated, and psoralens and ultra-violet light (PUVA) on a limited basis in adults.
Gastrointestinal Involvement in Mast Cell Disease
The gastrointestinal (GI) tract is a major compartment where MCs reside and are thought to carry out various functions such as homeostasis and pathogen defense.99 However, MCs may become inappropriately activated in the setting of allergy, recent infection, inflammation, and as part of a primary MC disorder, sometimes with signs of MCAS. At the mucosal surface, MCs may affect the epithelial barrier and secretion, which may result in heartburn, diarrhea, and abdominal bloat and cramps. On the serosal side, MCs may interact with the autonomic nervous system and may contribute to abdominal pain and cramping, as well as alterations in GI motility.
The study of patients with MCAS may serve as a model to determine how MCs interact with other immune and stromal cells within the GI compartment in certain pathogenic states, such as in post-infectious irritable bowel syndrome. When MCs in patients with MCAS have been activated, the aforementioned GI symptoms may be associated with MC mediator-related symptoms in other organs, which may be worsened by predictable triggers. These symptoms are typically treatable with a combination of medications that block MC activation and/or MC mediators, dietary changes (e.g. avoidance of dyes, chemicals, preservatives, alcohol), and stress reduction.100 Diagnostic studies, such as endoscopy and radiology examinations, are primarily used to exclude other GI disorders, because the majority of patients with MCAS will have normal-appearing GI tracts and normal numbers and appearance of MCs as seen on histology. With some exceptions, the MCs in MCAS are dispersed throughout the lamina propria of the mucosal layer without clustering or reaching significantly elevated numbers. Indeed, the numbers of MCs observed in the small intestine and colon are not used to diagnose MCAS per current diagnostic guidelines.
Patients with ISM may present with similar GI symptoms with or without signs of MCAS, but endoscopy and biopsy may be used to detect the clonal MCs and diagnostic features may be identified, such as clustering or sheets of >15 MCs per high power field in the mucosa, typically just below the surface epithelium. Clonal MCs will usually stain positive for CD25.101 Patients with smoldering SM (SSM) or aggressive SM (ASM) may experience more persistent diarrhea, associated with infiltration of clonal MCs beneath the epithelial surface, leading to malabsorption in cases of severe advanced SM. Oral budesonide or systemic glucocorticoids have been used to decrease the MC burden and to relieve diarrhea in this setting, and may also correct malabsorption; however, further cytoreduction is usually required for GI-related organ damage in advanced SM. Further studies are needed to phenotype the populations of MCs in the GI tract of patients with various MC disorders in order to personalize treatments.
Lessons Learned from the ECNM Experience: A Road Map for AIM
The ECNM was established as a multidisciplinary cooperative initiative in 2002.102,103 During the last 18 years, the ECNM contributed to the development of new markers, definitions, and standards in the field of mastocytosis, and has supported the World Health Organization (WHO) and international community in establishing classifications of MC disorders.104–108 Members of the ECNM organized annual meetings, as well as several working conferences and workshops.103 In addition, the ECNM supported the development and conduct of interventional and observational cohort studies. The ECNM structure consists of reference centers, which typically have a specific unitary focus on MC disorders (e.g. pathology or allergy/immunology), and centers of excellence that have multidisciplinary expertise in MC disorders. The major strategic goals of the ECNM are aligned with, and provide a roadmap for AIM. These are: 1) to provide new essential information about the disease to patients and physicians; and 2) to encourage academic and biopharma collaborations to develop and implement tools for prognostication and treatment of various MC disorders.102,103
A series of multi-center studies on diagnostic criteria for mastocytosis published between 1990 and 2000 served as the basis for 2001 WHO classification.104,108 The prognostic significance of the diagnostic criteria and of the WHO classification was confirmed in several different validation studies.83,106 In addition, a number of new potential prognostic markers were identified.101 However, because of the rarity of advanced systemic mastocytosis subtypes, the study cohorts were too small to reach definitive conclusions. To address open issues in these rare and complex diseases, the ECNM initiated a multi-center registry, with the aim to: 1) create a web-based collection of data from mastocytosis patients that is regularly updated by the participants; 2) perform prospective evaluations of prognostic factors in mastocytosis patients; 3) analyze the course of disease and treatment responses in different patient cohorts; and 4) establish multi-parametric scores in mastocytosis.
ECNM registry projects are approved by local ethics committees of participating centers. The first patients were enrolled in 2011, and by 2019, 32 centers from 15 European countries and one US center (Stanford, CA) had joined the registry and accrued 3,830 patients with >8,000 follow up examinations. Moreover, data on symptomatic and cytoreductive therapies have been captured. Since 2015, 29 ECNM registry projects have been initiated in various centers. Four of these projects have been published.109–112 Figure 2 highlights the distribution of mastocytosis subtypes comprising the registry.
Figure 2.
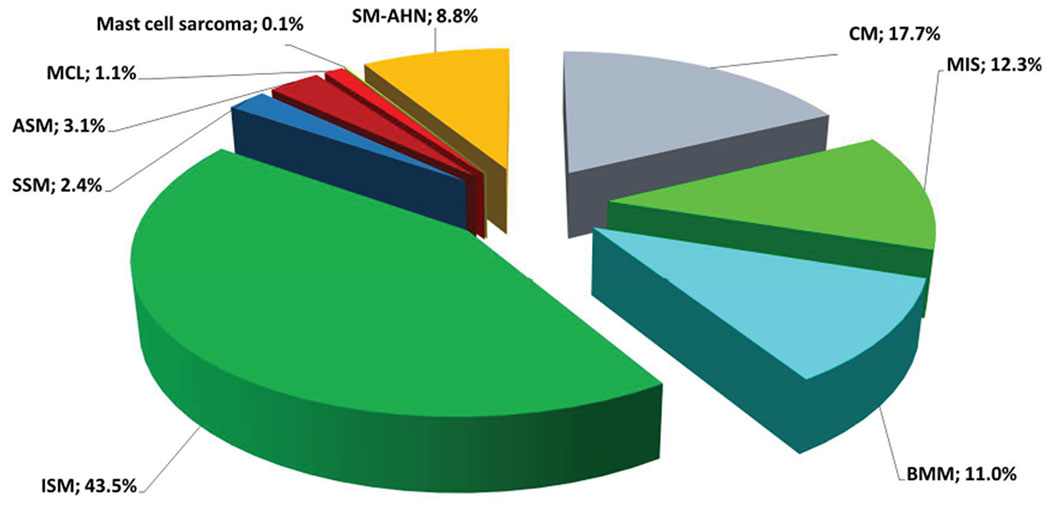
Distribution of mastocytosis subtypes from the ECNM Registry. The total number of patients included in the graph is 2,361 from 23 centers. The date of data cut off is 11/2017. Permission from Dr. Wolfgang Sperr and members of the ECNM who contribute to the registry. ASM, aggressive systemic mastocytosis; BMM, bone marrow mastocytosis; CM, cutaneous mastocytosis; ECNM, European Competence Network on Mastocytosis; ISM, indolent systemic mastocytosis; MIS, mastocytosis in the skin; MCL, mast cell leukemia; SM-AHN: systemic mastocytosis with an associated hematologic neoplasm; SSM: smoldering systemic mastocytosis.
In order to address new developments in the field, several patient- and disease-related parameters, including co-morbidities and genetic data, are included in the registry. Moreover, the ECNM consortium has a plan to link the ECNM registry data with a robust biobank system. Overall, the analyses and projects arising on the basis of the ECNM registry should improve prognostication and individualized management of patients with mastocytosis. A major goal will be to merge ECNM and AIM efforts to establish and maintain a common ECNM/AIM registry. In turn, observations gleaned from these combined efforts will further inform opportunities for inter-organizational collaborations as well as for productive engagement with patient-centered groups.
Acknowledgements:
Research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number R13TR002722 (to J.G.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. J.G. expresses gratitude to the Charles and Ann Johnson Foundation and staff of the Mastocytosis Center at Stanford for their support. MC, JL and DDM are supported in part, by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases, NIH. Dr Dwyer is supported by the Asthma and Allergic Diseases Cooperative Research Centers Opportunity Fund Award U19AI07053 from the National Institutes of Health. PV has been supported by the Austrian Science Fund (FWF) grants F4701-B20, F4704-B20, and P32470-B.
Conflict-of-Interest (COI) Disclosures:
Jason Gotlib has served as a Chair of the Study Steering Committee (SSC) for the global trial of midostaurin in advanced systemic mastocytosis (AdvSM) (Novartis), Chair of the response adjudication committee (RAC) for studies of avapritinib in advSM (Blueprint Medicines), and SSC Co-Chair for the phase II trial of ripretinib in advSM (Deciphera Pharmaceuticals). He has received funding for the conduct of these trials, and has received honoraria and reimbursement of travel expenses from Novartis, Blueprint Medicines, and Deciphera Pharmaceuticals. Tracy George has served on the SSC for clinical trials in SM for Novartis and Blueprint Medicines, and she has received consulting fees and reimbursement of travel expenses from Novartis and Blueprint Medicines. Dr. Bochner receives remuneration for serving on the scientific advisory board of Allakos, Inc. and owns stock in Allakos. He receives publication-related royalty payments from Elsevier and UpToDate®. He is a co-inventor on existing Siglec-8–related patents and thus may be entitled to a share of royalties received by Johns Hopkins University during development and potential sales of such products. Dr. Bochner is also a co-founder of Allakos, which makes him subject to certain restrictions under University policy. The terms of this arrangement are being managed by Johns Hopkins University and Northwestern University in accordance with their conflict-of-interest policies.
Matthew J. Hamilton serves on a Scientific Advsory Board for Allakos. Lawrence B. Schwartz is a consultant for Deciphera Pharmaceuticals, Blueprint Medicines, Allakos, and Genentech; participated in clinical trials sponsored by Deciphera Pharmaceuticals, and Blueprint Medicines; and, as inventor of the commercial tryptase assay, receives funds from VCU collected as royalties from Thermo Fisher. Celalettin Ustin has served as a consultant and received honoraria from Incyte, Inc, and Jazz Pharmaceuticals. Hans-Peter Horny serves on a scientific advisory board and Study Steering Committee for Blueprint Medicines and Deciphera Pharmaceuticals, and has served as a consultant and received honoraria from Blueprint Medicines, Deciphera Pharmaceuticals, and Novartis. Alberto Orfao has received consultancy honoraria from Novartis. Michael Deininger is a paid consultant and/or member of the scientific advisory board for Fusion Pharma, Takeda, Novartis, Incyte, Sangama, SPARC, Pfizer and DisperSol. Her serves on the SMC for the Optic (Takeda), Explorer (Blueprint), Pathfinder (Blueprint) and BFORE (Pfizer) clinical trials. Deepti Radia received funding for the conduct of the global trial of midostaurin in AdvSM (Novartis), and for studies of avapritinib in advSM (Blueprint Medicines). She has also served on the RAC for studies of avapritinib in advSM (Blueprint Medicines). Mohamad Jawhar received consultancy honoraria from Novartis and Blueprint, and research support from Novartis. Hanneke Kluin-Nelemans received institutional support from Novartis. Michel Arock has served as a consultant and received honoraria from Blueprint Medicines and Novartis and has received a research grant from Deciphera Pharmaceuticals. Wolfgang Sperr received honoraria from Thermo Fisher, AbbVie, Novartis, Pfizer, Incyte, Deciphera Pharmaceuticals, Jazz Pharmaceuticals, Teva and Celgene. Peter Valent received consultancy honoraria from Novartis, Incyte, Blueprint, Deciphera Pharmaceuticals, and Thermo Fisher, and research support from Novartis, Blueprint and Deciphera Pharmaceuticals. Mariana Castells is a consultant for Blueprint Medicines and one of the PIs of the PIONEER clinical trial for indolent SM. Cem Akin has received research support from Blueprint Medicines and has served as a consultant for Blueprint Medicines and Novartis, including SSC for clinical trials in SM.
Abbreviations
- AIM
American Initiative in Mast Cell Diseases
- BM
Bone marrow
- CM
Cutaneous mastocytosis
- DCM
Diffuse cutaneous mastocytosis
- ECNM
European Competence Network on Mastocytosis
- HαT
Hereditary alpha-tryptasemia
- ISM
Indolent systemic mastocytosis
- MC
Mast cells
- MCA
Mast cell activation
- MCAS
Mast cell activation syndrome
- MPCM
Maculopapular cutaneous mastocytosis
- SM
Systemic mastocytosis
- WHO
World Health Organization
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Melody Carter, Frank Austen, Daniel Dwyer, Jonathan J. Lyons, Joseph Butterfield, Patrizia Bonnadonna, Catherine Weiler, Stephen Galli, Hannke Oude Elberink, Anne Maitland, Theoharis Theoharides, and Dean D. Metcalfe report no COI.
References
- 1.Gentek R, Ghigo C, Hoeffel G, Bulle MJ, Msallam R, Gautier G, et al. Hemogenic endothelial fate mapping reveals dual developmental origin of mast cells. Immunity 2018;48:1160–71. [DOI] [PubMed] [Google Scholar]
- 2.Li Z, Liu S, Xu J, Zhang X, Han D, Liu J, et al. Adult connective tissue-resident mast cells originate from late erythro-myeloid progenitors. Immunity 2018;49:640–53. [DOI] [PubMed] [Google Scholar]
- 3.Gurish MF, Tao H, Abonia JP, Arya A, Friend DS, Parker CM, et al. Intestinal mast cell progenitors require CD49dbeta7 (alpha4beta7 integrin) for tissue-specific homing. J Exp Med 2001;194:1243–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gurish MF, Pear WS, Stevens RL, Scott ML, Sokol K, Ghildyal N, et al. Tissue-regulated differentiation and maturation of a v-abl-immortalized mast cell-committed progenitor. Immunity 1995;3:175–86. [DOI] [PubMed] [Google Scholar]
- 5.Gurish MF, Austen KF. Developmental origin and functional specialization of mast cell subsets. Immunity 2012;7:25–33. [DOI] [PubMed] [Google Scholar]
- 6.Xing W, Austen KF, Gurish MF, Jones TG. Protease phenotype of constitutive connective tissue and of induced mucosal mast cells in mice is regulated by the tissue. Proc Natl Acad Sci. USA 2011;108:14210–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dwyer DF, Barrett NA, Austen KF, Immuological Genome Project Consortium. Expression profiling of constitutive mast cells reveals a unique identity within the immune system. Nature Immunology 2016;17:878–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dahlin JS, Malinovschi A, Öhrvik H, Sandelin M, Janson C, Alving K, et al. Lin- CD34hi CD117int/hi FcεRI+ cells in human blood constitute a rare population of mast cell progenitors. Blood 2016;127:383–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Youngblood BA, Leung J, Falahati R, Williams J, Schanin J, Brock EC, et al. Discovery, function, and therapeutic targeting of siglec-8. Cells 2020;10:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bochner BS. “Siglec”ting the allergic response for therapeutic targeting. Glycobiology 2016; 26:546–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hudson SA, Herrmann H, Du J, Cox P, Haddad El-B, Butler B, et al. Developmental, malignancy-related, and cross-species analysis of eosinophil, mast cell, and basophil Siglec-8 expression. J Clin Immunol 2011;31:1045–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez-Gil A, Porell RN, Fernandes SM, Wei Y, Yu H, Carroll DJ, et al. Editor’s Choice: Sialylated keratan sulfate proteoglycans are Siglec-8 ligands in human airways. Glycobiology 2018;28:786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nycholat CM, Duan S, Knuplez E, Worth C, Elich M, Yao A, et al. A sulfonamide sialoside analogue for targeting Siglec-8 and -F on immune cells. J Am Chem Soc 2019;141:14032–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yokoi H, Choi OH, Hubbard W, Lee HS, Canning BJ, Lee HH, et al. Inhibition of FcεRI dependent mediator release and calcium flux from human mast cells by sialic acid-binding immunoglobulin-like lectin 8 engagement. J Allergy Clin Immunol 2008;121:499–505. [DOI] [PubMed] [Google Scholar]
- 15.O’Sullivan JA, Carroll DJ, Cao Y, Salicru AN, Bochner BS. Leveraging Siglec-8 endocytic mechanisms to kill human eosinophils and malignant mast cells. J Allergy Clin Immunol 2018; 141:1774–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Sullivan JA, Wei Y, Carroll DJ, Moreno-Vinasco L, Cao Y, Zhang F, et al. Frontline science: characterization of a novel mouse strain expressing human Siglec-8 only on eosinophils. J Leukoc Biol 2018;104:11–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei Y, Chhiba KD, Zhang F, Ye X, Wang L, Zhang L, et al. Mast cell-specific expression of human siglec-8 in conditional knock-in mice. Int J Mol Sci 2018;20:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Youngblood BA, Brock EC, Leung J, Falahati R, Bochner BS, Rasmussen HS, et al. Siglec 8 antibody reduces eosinophil and mast cell infiltration in a transgenic mouse model of eosinophilic gastroenteritis. JCI Insight 2019;4:e126219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knuplez E, Krier-Burris R, Cao Y, Marsche G, O’Sullivan J, Bochner BS. Frontline Science: Superior mouse eosinophil depletion in vivo targeting transgenic Siglec-8 instead of endogenous Siglec-F: Mechanisms and pitfalls. J Leukoc Biol 2020;108:43–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Youngblood BA, Brock EC, Leung J, Falahati R, Bryce PJ, Bright J, et al. AK002, a humanized sialic acid-binding immunoglobulin-like lectin-8 antibody that induces antibody dependent cell-mediated cytotoxicity against human eosinophils and inhibits mast cell-mediated anaphylaxis in mice. Int Arch Allergy Immunol 2019;180:91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Sullivan JA, Chang AT, Youngblood BA, Bochner BS. Eosinophil and mast cell Siglecs: From biology to drug target. J Leukoc Biol 2020;108:73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fellinger C, Hemmer W, Wohrl S, Sesztak-Greinecker G, Jarisch R, Wantke F. Clinical characteristics and risk profile of patients with elevated baseline serum tryptase. Allergol Immunopathol (Madr) 2014;42:544–52. [DOI] [PubMed] [Google Scholar]
- 23.Gonzalez-Quintela A, Vizcaino L, Gude F, Rey J, Meijide L, Fernandez-Merino C, et al. Factors influencing serum total tryptase concentrations in a general adult population. Clin Chem Lab Med 2010;48:701–6. [DOI] [PubMed] [Google Scholar]
- 24.Schwartz LB, Metcalfe DD, Miller JS, Earl H, Sullivan T. Tryptase levels as an indicator of mast-cell activation in systemic anaphylaxis and mastocytosis. N Engl J Med 1987;316:1622–26. [DOI] [PubMed] [Google Scholar]
- 25.Sperr WR, El-Samahi A, Kundi M, Girschikofsky M, Winkler S, Lutz D, et al. Elevated tryptase levels selectively cluster in myeloid neoplasms: a novel diagnostic approach and screen marker in clinical haematology. Eur J Clin Invest 2009;39:914–23. [DOI] [PubMed] [Google Scholar]
- 26.Sperr WR, Jordan JH, Baghestanian M, Kiener HP, Samorapoompichit P, Semper H, et al. Expression of mast cell tryptase by myeloblasts in a group of patients with acute myeloid leukemia. Blood 2001;98:2200–9. [DOI] [PubMed] [Google Scholar]
- 27.Sperr WR, Pfeiffer T, Hoermann G, Herndlhofer S, Sillaber C, Mannhalter C, et al. Serum-tryptase at diagnosis: a novel biomarker improving prognostication in Ph(+) CML. Am J Cancer Res 2014;5:354–62. [PMC free article] [PubMed] [Google Scholar]
- 28.Lyons JJ. Hereditary alpha tryptasemia: genotyping and associated clinical features. Immunol Allergy Clin North Am 2018;38:483–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lyons JJ, Sun G, Stone KD, Nelson C, Wisch L, O’Brien M, et al. Mendelian inheritance of elevated serum tryptase associated with atopy and connective tissue abnormalities. J Allergy Clin Immunol 2014;133:1471–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lyons JJ, Yu X, Hughes JD, Le QT, Jamil A, Bai Y, et al. Elevated basal serum tryptase identifies a multisystem disorder associated with increased TPSAB1 copy number. Nat Genet 2016;48:1564–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sabato V, Chovanec J, Faber M, Milner JD, Ebo D, Lyons JJ. First identification of an inherited TPSAB1 quintuplication in a patient with clonal mast cell disease. J Clin Immunol 2018;38:457–59. [DOI] [PubMed] [Google Scholar]
- 32.Aniceto V, Dias MM, Melo JML, Trevisan-Neto O, Aragon DC, Maia LSM, et al. Serum baseline tryptase level as a marker for the severity of anaphylaxis. Int Arch Allergy Immunol 2019;179:201–8. [DOI] [PubMed] [Google Scholar]
- 33.Haeberli G, Bronnimann M, Hunziker T, Muller U. Elevated basal serum tryptase and hymenoptera venom allergy: relation to severity of sting reactions and to safety and efficacy of venom immunotherapy. Clin Exp Allergy 2003;33:1216–20. [DOI] [PubMed] [Google Scholar]
- 34.Kucharewicz I, Bodzenta-Lukaszyk A, Szymanski W, Mroczko B, Szmitkowski M. Basal serum tryptase level correlates with severity of hymenoptera sting and age. J Investig Allergol Clin Immunol 2007;17:65–9. [PubMed] [Google Scholar]
- 35.Kutty GR, Downs-Kelly E, Crispin HT, Peterson KA. Elevated tryptase in EoE Is an independent phenomenon associated with extra-esophageal symptoms. Dig Dis Sci 2019;64:152–57. [DOI] [PubMed] [Google Scholar]
- 36.Sahiner UM, Yavuz ST, Buyuktiryaki B, Cavkaytar O, Yilmaz EA, Tuncer A, et al. Serum basal tryptase may be a good marker for predicting the risk of anaphylaxis in children with food allergy. Allergy 2014;69:265–68. [DOI] [PubMed] [Google Scholar]
- 37.Siles R, Xu M, Hsieh FH. The utility of serum tryptase as a marker in chronic spontaneous urticaria. Acta Derm Venereol 2013;93:354–55. [DOI] [PubMed] [Google Scholar]
- 38.van der Linden PW, Hack CE, Poortman J, Vivié-Kipp YC, Struyvenberg A, van der Zwan JK. Insect-sting challenge in 138 patients: relation between clinical severity of anaphylaxis and mast cell activation. J Allergy Clin Immunol 1992;90:110–18. [DOI] [PubMed] [Google Scholar]
- 39.Greiner G, Sprinzl B, Gorska A, Ratzinger F, Gurbisz M, Witzender N, et al. Hereditary alpha tryptasemia is a valid genetic biomarker for severe mediator-related symptoms in mastocytosis. Blood 2020;137:238–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lyons JJ, Chovanec J, O’Connell MP, Liu Y, Selb Julij, Zanotti R, et al. Heritable risk for severe anaphylaxis associated with increased a-tryptase-encoding germline copy number at TPSAB1. J Allergy Clin Immunol 2021;147:622–32. [DOI] [PubMed] [Google Scholar]
- 41.Schwartz LB, Min HK, Ren S, Xia HZ, Hu J, Zhao W, et al. Tryptase precursors are preferentially and spontaneously released, whereas mature tryptase is retained by HMC-1 cells, Mono-Mac-6 cells, and human skin-derived mast cells. J Immunol 2003;170:5667–73. [DOI] [PubMed] [Google Scholar]
- 42.Caughey GH. Tryptase genetics and anaphylaxis. J Allergy Clin Immunol 2006;117:1411–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Le QT, Lyons JJ, Naranjo AN, Olivera A, Lazarus RA, Metcalfe DD, et al. Impact of naturally forming human alpha/beta-tryptase heterotetramers in the pathogenesis of hereditary alpha-tryptasemia. J Exp Med 2019;216:2348–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boyden SE, Desai A, Cruse G, Young ML, Bolan HC, Scott LM, et al. Vibratory urticaria associated with a missense variant in ADGRE2. New Engl J Med 2016;374:656–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Akin C Mast cell activation syndromes. J Allergy Clin Immunol 2017;140:349–55. [DOI] [PubMed] [Google Scholar]
- 46.Akin C, Valent P, Metcalfe DD. Mast cell activation syndrome: Proposed diagnostic criteria. J Allergy Clin Immunol 2010;126:1099–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Valent P, Akin C, Arock M, Brockow K, Butterfield JH, Carter MC, et al. Definitions, criteria and global classification of mast cell disorders with special reference to mast cell activation syndromes: a consensus proposal. Int Arch Allergy Immunol 2012;157:215–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weiler CR, Austen KF, Akin C, Barkoff MS, Bernstein JA, Bonadonna P, et al. AAAAI Mast Cell Disorders Committee Work Group Report: Mast cell activation syndrome (MCAS) diagnosis and management. J Allergy Clin Immunol 2019;144:883–96. [DOI] [PubMed] [Google Scholar]
- 49.Valent P, Akin C, Bonadonna P, Hartmann K, Brockow K, Niedoszytko M, et al. Proposed diagnostic algorithm for patients with suspected mast cell activation syndrome. J Allergy Clin Immunol Pract 2019;7:1125–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Valent P, Bonadonna P, Hartmann K, Broesby-Olsen S, Brockow K, Butterfield JH, et al. Why the 20% + 2 tryptase formula is a diagnostic gold standard for severe systemic mast cell activation and mast cell activation syndrome. Int Arch Allergy Immunol 2019;180:44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hanjra P, Lee C-C R, Maric I, Carter M, Olicera A, Metcalfe DD, et al. Chromogranin A is not a biomarker of mastocytosis. J Allergy Clin Immunol Pract. 2018;6:687–689.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khokhar D, Akin C. Mast cell activation: when the whole is greater than the sum of its parts. Med Clin North Am. 2020;104:177–87. [DOI] [PubMed] [Google Scholar]
- 53.Butterfield J, Weiler CR. The utility of measuring urinary metabolites of mast cell mediators in systemic mastocytosis and mast cell activation syndrome. J Allergy Clin Immunol Pract 2020;8:2533–41. [DOI] [PubMed] [Google Scholar]
- 54.Divekar R, Butterfield J. Urinary 11beta-PGF2alpha and N-methyl histamine correlate with bone marrow biopsy findings in mast cell disorders. Allergy 2015;70:1230–38. [DOI] [PubMed] [Google Scholar]
- 55.Ravi A, Butterfield J, Weiler CR. Mast cell activation syndrome: improved identification by combined determinations of serum tryptase and 24-hour urine 11 β-prostaglandin 2α. J Allergy Clin Immunol Pract 2014; 2:775–78. [DOI] [PubMed] [Google Scholar]
- 56.Butterfield JH. Increased leukotriene E4 excretion in systemic mastocytosis. Prostaglandins Other Lipid Mediat 2010;92:73–6. [DOI] [PubMed] [Google Scholar]
- 57.Lueke AJ, Meeusen JW, Donato LJ, Gray AV, Butterfield JH, Saenger AK. Analytical and clinical validation of an LC-MS/MS method for urine leukotriene E4: A marker of systemic mastocytosis. Clin Biochem 2016;49:979–82. [DOI] [PubMed] [Google Scholar]
- 58.Castells M, Butterfield J. Mast cell activation syndrome and mastocytosis: initial treatment options and long-term management. J Allergy Clin Immunol Pract 2019;4:1097–106. [DOI] [PubMed] [Google Scholar]
- 59.McNeil BD, Pundir P, Meeker S, Han L, Undem BJ, Kulka M, et al. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature. 2015;519:237–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Slapnicar C, Trinkaus M, Hicks L, Vadas P. Efficacy of omalizumab in indolent systemic mastocytosis. Case Rep Hematol 2019;2019:3787586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jendoubi F, Gaudenzio N, Gallini A, Negretto M, Paul C, Bulai Livideanu CB. Omalizumab in the treatment of adult patients with mastocytosis: a systematic review. Clin Exp Allergy 2020;50:654–61. [DOI] [PubMed] [Google Scholar]
- 62.Distler M, Maul JT, Steiner UC, Jandus P, Kolios AGA, Murer C, et al. Efficacy of omalizumab in mastocytosis: allusive indication obtained from a prospective, double-blind, multicenter study (XOLMA study). Dermatology 2020;236:529–39. [DOI] [PubMed] [Google Scholar]
- 63.Lemal R, Fouquet G, Terriou L, Vaes M, Livideanu CB, Frenzel L, et al. Omalizumab Therapy for Mast Cell-Mediator Symptoms in Patients with ISM, CM, MMAS, and MCAS. J Allergy Clin Immunol Pract 2019;7:2387–2395.e3. [DOI] [PubMed] [Google Scholar]
- 64.Smiljkovic D, Kiss R, Lupinek C, Hoermann G, Greiner G, Witzeneder N, et al. Microarray-Based Detection of Allergen-Reactive IgE in Patients with Mastocytosis. J Allergy Clin Immunol Pract 2020:8:2761–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rossini M, Zanotti R, Orsolini G, Tripi G, Viapiana O, Idolazzi L, et al. Prevalence, pathogenesis, and treatment options for mastocytosis-related osteoporosis. Osteoporos Int 2016;27:2411–21. [DOI] [PubMed] [Google Scholar]
- 66.Orsolini G, Gavioli I, Tripi G, Vippiana O, Gatti D, Idolazzi L, et al. Denosumab for the treatment of mastocytosis-related osteoporosis: a case series. Calcif Tissue Int 2017;100:595–98. [DOI] [PubMed] [Google Scholar]
- 67.Maun HR, Jackman JK, Choy DF, Loyet KM, Staton TL, Jia G, et al. An allosteric anti-tryptase antibody for the treatment of mast cell-mediated severe asthma. Cell 2019;179:417–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gulen T, Hagglund H, Dahlen B, Nilsson G. High prevalence of anaphylaxis in patients with systemic mastocytosis: a single-centre experience. Clin Exp Allergy 2014;44:121–29. [DOI] [PubMed] [Google Scholar]
- 69.Greenhawt M, Akin C. Mastocytosis and allergy. Curr Opin Allergy Clin Immunol 2007;7:387–92. [DOI] [PubMed] [Google Scholar]
- 70.González de Olano D, de la Hoz Caballer B, Núñez López R, Sánchez Muñoz L, Cuevas Agustín M, Diéguez MC, et al. Prevalence of allergy and anaphylactic symptoms in 210 adult and pediatric patients with mastocytosis in Spain: a study of the Spanish network on mastocytosis (REMA). Clin Exp Allergy 2007;7:1547–55. [DOI] [PubMed] [Google Scholar]
- 71.Bonadonna P, Lombardo C. Drug allergy in mastocytosis. Immunol Allergy Clin North Am 2014;34:397–405. [DOI] [PubMed] [Google Scholar]
- 72.Bonadonna P, Scaffidi L. Hymenoptera anaphylaxis as a clonal mast cell disorder. Immunol Allergy Clin North Am 2018;38:455–68. [DOI] [PubMed] [Google Scholar]
- 73.Bonadonna P, Zanotti R, Pagani M, et al. Anaphylactic reactions after discontinuation of hymenoptera venom immunotherapy: a clonal mast cell disorder should be suspected. J Allergy Clin Immunol Pract 2018;6:1368–72. [DOI] [PubMed] [Google Scholar]
- 74.Broesby-Olsen S, Vestergaard H, Mortz CG, Jensen B, Havelund T, Hermann AP, et al. Mastocytosis Centre Odense University Hospital (MastOUH). Omalizumab prevents anaphylaxis and improves symptoms in systemic mastocytosis: Efficacy and safety observations. Allergy 2018;73:230–38. [DOI] [PubMed] [Google Scholar]
- 75.Hartmann K, Escribano L, Grattan C, Brockow K, Carter MC, Álvarez-Twose I, et al. Cutaneous manifestations in patients with mastocytosis: Consensus report of the European Competence Network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European Academy of Allergology and Clinical Immunology. J Allergy Clin Immunol 2016;137:35–45. [DOI] [PubMed] [Google Scholar]
- 76.Wolff K, Komar M, Petzelbauer P. Clinical and histopathological aspects of cutaneous mastocytosis. Leuk Res 2001; 25:519–28. [DOI] [PubMed] [Google Scholar]
- 77.Metcalfe DD. Classification and diagnosis of mastocytosis: current status. J Invest Dermatol 1991;96:2S–4S; discussion S, 60S-5S. [PubMed] [Google Scholar]
- 78.Soter NA. The skin in mastocytosis. J Invest Dermatol 1991; 96:32S–8S. [PubMed] [Google Scholar]
- 79.Sagher F, Even-Paz Z. Incidence of Mastocytosis. In: Sagher, Even-Paz Z, editors. Mastocytosis and the mast cell: Karger, Basel, New York; 1967. p. 14–7. [Google Scholar]
- 80.Akin C, Metcalfe DD. Systemic mastocytosis. Annu Rev Med 2004;55:419–32. [DOI] [PubMed] [Google Scholar]
- 81.Galen BT, Rose MG. Darier’s sign in mastocytosis. Blood 2014;123:1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Berezowska S, Flaig MJ, Rueff F, Walz C, Haferlach T, Krokowski M, et al. Adult-onset mastocytosis in the skin is highly suggestive of systemic mastocytosis. Mod Pathol 2014;27:19–29. [DOI] [PubMed] [Google Scholar]
- 83.Lim KH, Tefferi A, Lasho TL, Finke C, Patnaik M, Butterfield JH, et al. Systemic mastocytosis in 342 consecutive adults: survival studies and prognostic factors. Blood 2009; 113:5727–36. [DOI] [PubMed] [Google Scholar]
- 84.Lanternier F, Cohen-Akenine A, Palmerini F, Feger F, Yang Y, Zermati Y, et al. Phenotypic and genotypic characteristics of mastocytosis according to the age of onset. PLoS One 2008; 3:e1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Brockow K, Akin C, Huber M, Metcalfe DD. Assessment of the extent of cutaneous involvement in children and adults with mastocytosis: relationship to symptomatology, tryptase levels, and bone marrow pathology. J Am Acad Dermatol 2003;48:508–16. [DOI] [PubMed] [Google Scholar]
- 86.Hartmann K, Metcalfe DD. Pediatric mastocytosis. Hematol Oncol Clin North Am 2000; 14:625–40. [DOI] [PubMed] [Google Scholar]
- 87.Lange M, Nedoszytko B, Gorska A, Zawrocki A, Sobjanek M, Kozlowski D. Mastocytosis in children and adults: clinical disease heterogeneity. Arch Med Sci 2012;8:533–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Torrelo A, Álvarez-Twose I, Escribano L. Childhood mastocytosis. Curr Opin Pediatr 2012;24:480–6. [DOI] [PubMed] [Google Scholar]
- 89.Álvarez-Twose I, Jara-Acevedo M, Morgado JM, García-Montero A, Sánchez-Muñoz L, Teodosio C, et al. Clinical, immunophenotypic, and molecular characteristics of well differentiated systemic mastocytosis. J Allergy Clin Immunol 2016;137:168–78. [DOI] [PubMed] [Google Scholar]
- 90.Carter MC, Clayton ST, Komarow HD, Brittain EH, Scott LM, Cantave D, et al. Assessment of clinical findings, tryptase levels, and bone marrow histopathology in the management of pediatric mastocytosis. J Allergy Clin Immunol 2015;136:1673–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ben-Amitai D, Metzker A, Cohen HA. Pediatric cutaneous mastocytosis: a review of 180 patients. Isr Med Assoc J 2005;7:320–2. [PubMed] [Google Scholar]
- 92.Caplan RM. The natural course of urticaria pigmentosa. Analysis and follow-up of 112 cases. Arch Dermatol 1963;87:146–57. [DOI] [PubMed] [Google Scholar]
- 93.Ma D, Stence AA, Bossler AB, Hackman JR, Bellizzi AM. Identification of KIT activating mutations in paediatric solitary mastocytoma. Histopathology 2014. 64:218–25. [DOI] [PubMed] [Google Scholar]
- 94.Soucie E, Brenet F, Dubreuil P. Molecular basis of mast cell disease. Mol Immunol 2015; 63:55–60. [DOI] [PubMed] [Google Scholar]
- 95.Bodemer C, Hermine O, Palmerini F, Yang Y, Grandpeix-Guyodo C, Leventhal PS, et al. Pediatric mastocytosis is a clonal disease associated with D816V and other activating c-KIT mutations. J Invest Dermatol 2010;130:804–15. [DOI] [PubMed] [Google Scholar]
- 96.Yanagihori H, Oyama N, Nakamura K, Kaneko F. c-kit Mutations in patients with childhood-onset mastocytosis and genotype-phenotype correlation. J Mol Diagn 2005;7:252–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wiechers T, Rabenhorst A, Schick T, Preussner LM, Forster A, Valent P, et al. Large maculopapular cutaneous lesions are associated with favorable outcome in childhood-onset mastocytosis. J Allergy Clin Immunol 2015;136:1581–90. [DOI] [PubMed] [Google Scholar]
- 98.Kirshenbaum AS, Abuhay H, Bolan H, Metcalfe DD, Carter MC. Maculopapular cutaneous mastocytosis in a diverse population. J Allergy Clin Immunol Pract 2019;7:2845–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang L, Song J, Hou X. Mast cells and irritable bowel syndrome: from the bench to the bedside. J Neurogastroenterol Motil 2016;22:181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hamilton MJ, Hornick JL, Akin C, Castells MC, Greenberger NJ. Mast cell activation syndrome: a newly recognized disorder with systemic clinical manifestation. J Allergy Clin Immunol 2011;128:147–152. [DOI] [PubMed] [Google Scholar]
- 101.Doyle LA, Sepehr GJ, Hamilton MJ, Akin C, Castells MC, Hornick JL. A clinicopathologic study of 24 cases of systemic mastocytosis involving the gastrointestinal tract and assessment of mucosal mast cell density in irritable bowel syndrome and aymptomatic patients. Am J Surg Pathol 2014; 38:832–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Valent P, Arock M, Bischoff SC, Bühring H-J, Brockow K, Escribano L, et al. The European Competence Network on Mastocytosis (ECNM). Wien Klin Wochenschr 2004;116:647–51. [DOI] [PubMed] [Google Scholar]
- 103.Valent P, Arock M, Bonadonna, Brockow K, Broesby-Olsen S, Escribano L, et al. European Competence Network on Mastocytosis (ECNM): 10-year jubilee, update, and future perspectives. Wien Klin Wochenschr 2012;124:807–14. [DOI] [PubMed] [Google Scholar]
- 104.Valent P, Horny HP, Escribano L, Longley BJ, Li CY, Schwartz LB, et al. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res 2001;25:603–25. [DOI] [PubMed] [Google Scholar]
- 105.Valent P, Akin C, Escribano L, Födinger M, Hartmann K, Castells M, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest 2007;37:435–53. [DOI] [PubMed] [Google Scholar]
- 106.Horny HP, Akin C, Arber D, Peterson LA, Tefferi A, Metcalfe DD, et al. Mastocytosis. In: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Eds: Swerdlow SH, Campo, Harris NL, Jaffe ES, Pileri SA, Stein, et al. IARC Press Lyon, France, vol 3, pp 61–69, 2017. [Google Scholar]
- 107.Valent P, Akin C, Metcalfe DD. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood 2017;129:1420–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Valent P, Akin C, Hartmann K, Nilsson G, Reiter A, Hermine O, et al. Advances in the classification and treatment of mastocytosis: current status and outlook toward the future. Cancer Res 2017;77:1261–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Valent P, Oude Elberink JNG, Gorska A, Lange M, Zanotti R, van Anrooij B, et al. The Data Registry of the European Competence Network on Mastocytosis (ECNM): set up, projects, and perspectives. J Allergy Clin Immunol Pract 2019;7:81–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sperr WR, Kundi M, Alvarez-Twose I, van Anrooij B, Oude Elberink JNG, Gorska A, et al. Proposed international prognostic scoring system for mastocytosis (IPSM): A retrospective validation study. Lancet Haematol 2019. 6:e638–e649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kluin-Nelemans HC, Reiter A, Illerhaus A, van Anrooij B, Hartmann K, Span LFR, et al. Prognostic impact of eosinophils in mastocytosis: analysis of 2350 patients collected in the ECNM registry. Leukemia 2020; 34:1090–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Trizuljak J, Sperr WR, Nekvindova L, Elberink HO, Gleixner KV, Gorska A, et al. Clinical features and survival of patient with indolent systemic mastocytosis defined by the updated WHO classification. Allergy 2020;75:1927–38. [DOI] [PMC free article] [PubMed] [Google Scholar]