

Abstract
Background:
Given that new therapeutic options for spinocerebellar ataxias are on the horizon, there is a need for markers that reflect disease-related alterations, in particular, in the preataxic stage, in which clinical scales are lacking sensitivity.
Objective:
The objective of this study was to quantify regional brain volumes and upper cervical spinal cord areas in spinocerebellar ataxia type 3 in vivo across the entire time course of the disease.
Methods:
We applied a brain segmentation approach that included a lobular subsegmentation of the cerebellum to magnetic resonance images of 210 ataxic and 48 preataxic spinocerebellar ataxia type 3 mutation carriers and 63 healthy controls. In addition, cervical cord cross-sectional areas were determined at 2 levels.
Results:
The metrics of cervical spinal cord segments C3 and C2, medulla oblongata, pons, and pallidum, and the cerebellar anterior lobe were reduced in preataxic mutation carriers compared with controls. Those of cervical spinal cord segments C2 and C3, medulla oblongata, pons, midbrain, cerebellar lobules crus II and X, cerebellar white matter, and pallidum were reduced in ataxic compared with nonataxic carriers. Of all metrics studied, pontine volume showed the steepest decline across the disease course. It covaried with ataxia severity, CAG repeat length, and age. The multivariate model derived from this analysis explained 46.33% of the variance of pontine volume.
Conclusion:
Regional brain and spinal cord tissue loss in spinocerebellar ataxia type 3 starts before ataxia onset. Pontine volume appears to be the most promising imaging biomarker candidate for interventional trials that aim at slowing the progression of spinocerebellar ataxia type 3.
Keywords: spinocerebellar ataxia, MRI, volumetry, biomarker
Spinocerebellar ataxia type 3/Machado–Joseph disease (SCA3) is worldwide the most common autosomal dominantly inherited ataxia disorder.1 It is caused by unstable expansions of polyglutamine encoding CAG repeats in the ATXN3 gene, resulting in the formation of abnormally elongated disease proteins. Although partial loss of the physiological role of ataxin-3 contributes to the development of SCA3, its pathogenesis is mainly because of newly acquired deleterious actions of elongated ataxin-3.
SCA3 is a multisystem disorder characterized by degeneration of spinocerebellar tracts, dentate nucleus, cerebellar cortex, brain stem nuclei, and basal ganglia.2 The clinical syndrome is characterized by prominent cerebellar ataxia in combination with supranuclear gaze palsy and peripheral neuropathy.3,4 SCA3 takes a progressive course and leads to severe disability and premature death, with a median survival after ataxia onset of 25 years.5–7
Currently, there is no causal treatment for SCA3. However, as the understanding of the molecular mechanisms is rapidly advancing, there are several new treatment approaches. Among the most promising ones are approaches for downregulating or silencing the ATXN3 gene.8 Like in other neurodegenerative diseases, in particular those with a clinically presymptomatic phase, there is a need for markers with known evolution throughout the time course of the disease that have the potential to map disease activity.
Magnetic resonance imaging (MRI) allows the study of structural abnormalities of the brain and spinal cord in vivo. Previous studies in patients with SCA3 showed a pattern of regional brain tissue loss that faithfully reflected the distribution of neurodegenerative changes described in autopsy studies9–13 and revealed that regional volume loss already starts in the preataxia stage.9 Three longitudinal volumetric studies in small numbers of patients with SCA3 suggested that MRI volume is more sensitive to change than clinical scales, which makes MRI volumes promising candidates for biomarkers in clinical trials.14-16 This applies in particular to preventive trials in preataxic mutation carriers, as clinical measures lack sensitivity before ataxia onset. However, further steps toward the validation of MRI regional volumes as biomarkers for SCA3 are hampered by the small numbers of studied MRIs and the time-consuming procedures for segmentation and volumetry.
In this study, we applied an automated method for MRI volumetry, which includes subsegmentation of the cerebellum into its lobules to a large number of existing T1-weighted MRIs of preataxic and ataxic SCA3 mutation carriers that were acquired at 14 centers worldwide. The advantage of the applied method is that it provides individual single-point values that can be used in future longitudinal studies to map individual trajectories.
Materials and Methods
MRI Scans
We collected T1-weighted (T1W) MRIs of 295 SCA3 mutation carriers and 72 healthy controls from 14 sites in 8 countries. There were no restrictions regarding manufacturer, software version, or field strength. Scans with a resolution of greater than 2 mm on at least 1 axis, based on the limited ability to accurately measure small regional brain volumes in scans with large voxel sizes, and scans that were acquired after application of contrast agents were excluded (for details, see Table S2 and Fig. S1). Age, sex, and total Scale for Assessment and Rating of Ataxia (SARA)17 score of all SCA3 participants and healthy controls were available. Information about CAG repeat length was available for SCA3 mutation carriers. Using a SARA cutoff value of 3, SCA3 mutation carriers were divided into preataxic (SARA < 3) and ataxic (SARA ≥ 3) individuals.17,18 This SARA threshold for ataxia was defined as the mean SARA +2 SDs of the healthy control group in the original SARA validation study.17 For complementary analyses, which can be found in the Supplementary data, groups were defined on the basis of subject report, whether and since when gait disturbances were present, here using the terms presymptomatic and symptomatic. Scans with incomplete clinical information were excluded (for details, see Fig. S1). All participants gave their written informed consent.
Image Analysis
The T1W images of each subject were processed with a fully automated image-processing pipeline to obtain volumes of 122 distinct anatomical regions covering the entire brain and 7 compartments containing cerebrospinal fluid. For the cerebellar subsegmentation, a reference database was developed to provide a parcellation scheme including fine-granularity cerebellar subsegmentations on the level of cerebellar lobules into 30 disjoint volumes. These reference data set segmentations were generated from a gold standard segmentation set of 17 T1W images with manual segmentations of the cerebellar lobules provided by J. Diedrichsen (http://www.diedrichsenlab.org). The image-processing and segmentation methods are described in detail in the supplementary data. To enable comparison of subject volume, data adjustment to account for head size was undertaken, using a normalization factor that was estimated from a template registration approach based on Buckner et al.19 In brief, the affine (9-parameter) transformation was computed for the subject T1W to the MNI152 linear T1 template. The magnitude of the scaling factors of the estimated affine matrix was then applied as a normalization factor to estimated volumes. For the final analysis, each 2 hemispheric volumes of hemispheric bilateral volumes were combined, and for the cerebellum the 8 subdivisions of the vermis were combined. In addition, the following compound volumes were analyzed: anterior lobe (cerebellar lobules I–V), superior posterior lobe (cerebellar lobules VI, VIIA [crus I, crus II], and VIIB), inferior posterior lobe (cerebellar lobules VIIIA, VIIIB, and IX), cerebellar gray matter (lobules I–X and vermis) and basal ganglia (pallidum, caudate, putamen). A full list of all volumes is given in Table S1, and an example segmentation is shown in Figure S2.
The upper portion of the cervical spinal cord was depicted on all available MRIs. Analysis of the C3 level was applicable in 256 cases and analysis of the C2 level in 297 of 321 cases. In a semiautomated approach, we used the Spinal Cord Toolbox20 in combination with manual corrections of the automated delineation to compute the cross-sectional area under consideration of an angle correction along the center line. We calculated the mean of the angle-corrected cross-sectional areas for all slices of cervical spinal cord segments C2 and C3 separately. The image processing and segmentation methods are described in detail in the supplementary data. To improve readability, we do not state the term “mean cross-sectional area” each time, but instead we will subsume the values of the mean cross-sectional areas of C2 and C3 and brain regional volumes under the term “metric.”
Statistical Analysis
All analyses were performed using R Software for Statistical Computing, version 3.5.1. R Foundation for Statistical Computing, Vienna.
To investigate group differences between ataxic SCA3 mutation carriers, preataxic mutation carriers, and healthy controls, we used separate linear mixed-effects models (R package lme4) to analyze the relationship of each metric — each brain volume and the mean cross-sectional area of spinal cord levels C2 and C3 — with the covariables age, sex, and group (preataxic, ataxic, healthy control). The latter variables were represented by fixed effects, whereas scanner type was represented by a random effect. Group differences were evaluated using the R package multcomp (function glht). In all metrics that showed a significant group effect, a post hoc multiple comparison via Benjamini–Hochberg correction was applied afterward.21 Given the heterogeneous sample, we chose a strict significance level to reduce the probability of false-positive effects. P < 0.001 after post-hoc Benjamini–Hochberg correction21 was considered significant. In a second analysis, analysis groups were defined by self-report as presymptomatic or symptomatic SCA3 mutation carriers (Supplementary Data).
Only those metrics that showed a significant difference between preataxic SCA3 mutation carriers, either compared with healthy controls or ataxic SCA3 mutation carriers, were considered for the further analyses to meaningfully cover the entire time course of the disease (regional volume loss in relation to disease duration, influencing factors, sample sizes).
To describe the regional changes in relation to disease duration, we z-transformed each metric in relation to healthy controls of the same age and plotted z values against the time scale of estimated disease duration, as described in detail in the supplementary data. We used a uniform time scale for all SCA3 mutation carriers, defined by the predicted time of ataxia onset calculated on the basis of CAG repeat length.22 On this scale, negative values for disease duration indicate the predicted time to ataxia onset and positive values the time from the predicted onset. In a second analysis (Supplementary Data), we used a compound time scale. For presymptomatic carriers, we calculated the time to ataxia onset based on CAG repeat length and present age,22 and in symptomatic carriers, we used the reported time from ataxia onset. The x axis was restricted to (−20 years; 20 years). We applied locally weighted scatterplot smoothing for interpolation to avoid any preassumptions about the curve course, for example, assuming a linear or parabolic curve course.
To identify factors that covary with regional volume loss, we applied linear regression analysis with SARA score, CAG repeat of the longer allele, age, and sex as independent variables. We calculated R2, which indicates the overall proportion of the variance of each metric that is explained by the independent variables and the P value for each independent variable. Here, the P value indicates whether the respective variable contributes to the model in a statistically significant way.
As a measure of effect size, we calculated Cohen’s d values for a presumed 50% reduction of the decrease of SARA and each MRI metric. Calculations were based on the estimated metric slopes of linear models with calculated disease duration as covariate. To allow comparison of the effect sizes of the MRI metrics, we calculated relative values by dividing Cohen’s d of each MRI metric by Cohen’s d of SARA.
Results
Demographic and Clinical Data
Of the 367 collected data sets, 46 had to be excluded (Fig. S1). We analyzed the remaining 321 data sets that comprised data from 210 ataxic SCA3 mutation carriers, 48 preataxic SCA3 mutation carriers, and 63 healthy controls. Demographic data are given in Table 1. Age differed among the 3 subgroups. Ataxic mutation carriers had the highest age (mean ± SD, 46.84 ± 11.24 years) and preataxic mutation carrier the lowest age (mean ± SD, 37.75 ± 9.47 years). Age of the control group was between the 2 groups of mutation carriers (mean ± SD, 42.81 ± 13.65 years). SARA score of the ataxic mutation carriers was on average ± SD, 12.41 ± 5.50). The mean SARA scores of preataxic mutation carriers (mean ± SD, 1.31 ± 0.94) and healthy controls (mean ± SD, 0.22 ± 0.46) was below the cutoff of 3. The number of CAG repeats of the longer allele was higher in ataxic (mean ± SD, 71.10 ± 4.23) than in the preataxic (mean ± SD, 68.29 ± 3.55) mutation carriers.
TABLE 1.
Demographic and characterizing cohort data
| n | Age | Male/female | Age of onseta | Disease duration in yearsa | CAG repeats, longer allele | SARA sum score | |
|---|---|---|---|---|---|---|---|
| Healthy controls | 63 | 42.81 (13.65) | 35/28 | na | na | na | 0.22 (0.46) |
| Preataxic | 48 | 37.75 (9.47) | 18/30 | 39.73 (7.87) | −1.98 (9.83) | 68.29 (3.55) | 1.31 (0.94) |
| Ataxic | 210 | 46.84 (11.24) | 118/92 | 34.12 (9.45) | 12.67 (9.45) | 71.10 (4.23) | 12.41 (5.50) |
Data are expressed as mean and standard deviation for age, age at onset, disease duration, CAG repeat length of the longer allele, and SARA sum score and as number for the group size and the male/female distribution.
Estimated age at onset on the basis of CAG repeat length following the model provided by Tezenas et al (Tezenas du Montcel et al, 2014) and disease duration in years, defined as the actual age minus the estimated age at onset, resulting in negative values for the expected time to onset in preataxic SCA3 mutation carriers and positive values for ataxic SCA3 mutation carriers.
Group Comparisons
To identify regions subject to volume loss before ataxia onset in SCA3, we compared SCA3 preataxic mutation carriers with healthy controls. Volumes of the following brain regions and mean cross-sectional areas of the spinal cord levels were reduced in preataxic mutation carriers compared with healthy controls: cervical spinal cord segments C2 and C3 (P < 0.0001), medulla oblongata (P < 0.0001), pons (P < 0.0001), and pallidum (P < 0.001); see Table 2. None of the cerebellar lobules showed any significant volume loss in preataxic mutation carriers. However, when considering compound volumes, the anterior lobe of preataxic mutation carriers had reduced volume compared with healthy controls (P < 0.001; Table 2).
TABLE 2.
Group differences between preataxic and ataxic SCA3 mutations carriers and healthy controls (HC)
| Metric | Preataxic SCA3 < HC | Ataxic SCA3 < preataxic SCA3 | Ataxic SCA3 < HC |
|---|---|---|---|
| Cervical spinal cord | |||
| Cervical spinal cord, level C3, CSAa | ** | ** | ** |
| Cervical spinal cord, level C2, CSAa | ** | ** | ** |
|
| |||
| Brain stem | |||
| Medulla oblongata | ** | ** | ** |
| Pons | ** | ** | ** |
| Midbrain | * | * | |
|
| |||
| Cerebellum | |||
| Cerebellum white matter | * | * | |
| Cerebellum I–IV | * | ||
| Cerebellum V | * | ||
| Cerebellum VI | * | ||
| Cerebellum crus I | * | ||
| Cerebellum crusII | * | ** | |
| Cerebellum VIIb | * | ||
| Cerebellum IX | * | ||
| Cerebellum X | * | * | |
| Cerebellum vermisb | * | ||
|
| |||
| Cerebrum | |||
| Caudate | ** | ||
| Pallidum | * | * | * |
| Thalamus | * | ||
|
| |||
| Compound volumes | |||
| Cerebellum, anterior lobec | * | * | |
| Cerebellum, superior posterior lobed | * | ||
| Cerebellum, inferior posterior lobee | * | ||
| Cerebellum gray matterf | * | ||
| Basal gangliag | * | ||
| Ventricles | Preataxic SCA3 > HC | Ataxic SCA3 > preataxic SCA3 | Ataxic SCA3 > HC |
| Third ventricle | * | ||
| Fourth ventricle | * | ||
Significance levels are given after post hoc Benjamini-Hochberg correction for multiple comparisons.
P < 0.0001.
P < 0.000.
CSA, mean cross-sectional area.
Entire vermis corresponding to the hemispheric lobules VI–X.
Cerebellar lobules I–V.
Cerebellar lobules VI, crus I, crus II, and VIIB.
Cerebellar lobules VIIIA, VIIIB, and IX.
Cerebellar lobules I–X and vermis.
Palldium, caudate, and putamen.
To define the atrophy pattern of manifest SCA3, we compared ataxic SCA3 mutation carriers and healthy controls. Volumes of the following brain regions and mean crosssectional areas of the spinal cord levels were smaller in ataxic mutation carriers compared with healthy controls: cervical spinal cord segments C2 and C3 (P < 0.0001), medulla oblongata (P < 0.0001), pons (P < 0.0001), midbrain (P < 0.001), cerebellar white matter (P < 0.001), cerebellar lobules I–IV (P < 0.001), V (P < 0.001), VI (P < 0.001), crus I (P < 0.001), crus II (P < 0.0001), VIIB (P < 0.001), IX (P < 0.001), and X (P < 0.001), cerebellar vermis (P < 0.001), thalamus (P < 0.001), caudate (P < 0.0001), and pallidum (P < 0.001); see Table 2. In addition, the volume of the fourth ventricle was larger than in controls (P < 0.001). Comparison of the compound volumes showed reductions of the anterior, superior-posterior, and inferior-posterior lobes (P < 0.001) and cerebellar gray matter (P < 0.001), as well as the basal ganglia (P < 0.001); see Table 2.
Among the regions that showed smaller metrics in ataxic SCA3 mutation carriers compared with healthy controls, a number of regions also showed reduced metrics compared with preataxic mutations carriers, indicating that these regions undergo progressive volume loss in the SCA3 disease course. These regions included cervical spinal cord segments C2 and C3 (P < 0.0001), medulla oblongata (P < 0.0001), pons (P < 0.0001), midbrain (P < 0.001), cerebellar lobules crus II and X (P < 0.001), cerebellar white matter (P < 0.001), and pallidum (P < 0.001); see Table 2. In addition, the volume of the third ventricle was larger than in controls (P < 0.001). Estimates, standard errors, and 95% confidence intervals (CIs) for each group comparison as well as the ANOVA statistics are given in Tables S3 and S4.
When we performed the same analysis in groups defined by self-report as presymptomatic or symptomatic SCA3 mutation carriers, results were generally similar. However, the pallidum and anterior lobe of the cerebellum were not smaller in presymptomatic mutation carriers compared with healthy controls, whereas the superior posterior lobe and total cerebellar gray matter was smaller in presymptomatic compared with symptomatic SCA3 mutation carriers (Table S5).
Regional Volume Loss in Relation to Disease Duration
To study regional volume loss in SCA3 in relation to disease duration, we applied local regression on a time scale defined by the predicted time of ataxia onset, calculated on the basis of CAG repeat length. On this scale, negative values indicate the predicted time to ataxia onset and positive values the time from the predicted onset. For this analysis, we selected those metrics that showed significant alterations in any comparison of preataxic mutation carriers, with either healthy controls or ataxic mutation carriers. Already around 10 to 15 years before ataxia onset, the metrics of cervical spinal cord segments C2 and C3 and the pallidum were reduced by about 1 standard deviation (SD) compared with healthy controls of the same age. Metrics of all regions steadily decreased until a period of 5 to 15 years after ataxia onset. Thereafter, volume loss decelerated, and metrics remained stable or even increased relative to controls, except for the pons and cerebellar lobule X. In the time interval lasting from 5 years before until 5 years after ataxia onset, the metrics of all regions decreased almost linearly. The decline was steepest in the pons and midbrain. At the time of ataxia onset, the metrics of cervical spinal cord segments C2 and C3, the pons, midbrain, and pallidum ranged between about 1 and 2 SD below the control group, whereas the other volumes, in particular the cerebellar volumes, were reduced by less than 1 SD (Fig. 1).
FIG. 1.
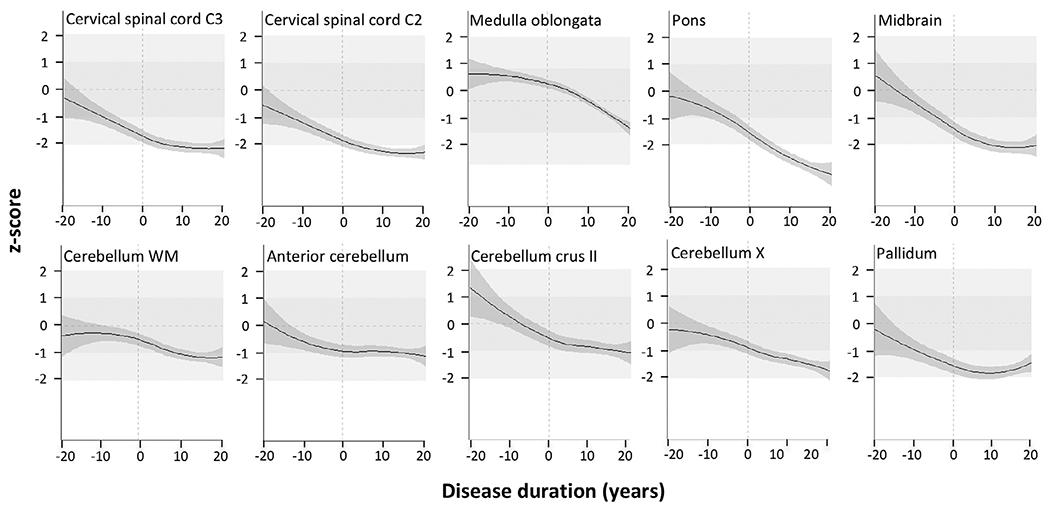
Regional volume loss along the time course of the disease. Each metric, being mean cross-sectional area of cervical spinal cord levels C2 and C3 and volumes of medulla oblongata, pons, midbrain, cerebellar white matter, anterior lobe of the cerebellum, cerebellar lobules crus II and X. and pallidum of SCA3 mutation carriers, was z-transformed in relation to healthy controls of the same age. The x axis represents the estimated disease duration in years. The estimated 95% confidence interval is given in dark gray. For a better orientation, the following reference lines and ranges are given: the vertical dashed line marks the estimated clinical onset; the horizontal dashed line marks the average of the healthy control group, represented by a z score of 0; the medium- and light-gray areas represent the range ± 1 and respective ±2 standard deviations of the healthy control group distribution.
When we performed the same analysis on a compound time scale that used the calculated time to expected ataxia onset in presymptomatic mutation carriers and the reported time since ataxia onset in symptomatic mutation carriers, the results were generally similar (Fig. S4).
Factors Determining Regional Volume Loss
To identify factors that covary with regional tissue loss in SCA3, we performed a linear regression analysis with SARA sum score, CAG repeat length of the longer allele, age, and sex as potential determining factors. Again, we selected those metrics for analyses that showed significant alterations in any comparison of preataxic mutation carriers, either with healthy controls or with ataxic mutation carriers. SARA sum score contributed highly significantly to the models of cervical spinal cord segments C2 and C3, the medulla oblongata, pons, and midbrain. Furthermore, CAG repeat length influenced pons, midbrain, and pallidum, age on pons, midbrain anterior cerebellum, and cerebellar crus II, and sex on medulla oblongata and midbrain (each P < 0.001). The multivariate models derived from this analysis explained between 5.82% and 46.33% of the variance of the respective metrics. The proportion of the explained variance was highest for the pons (46.33%) and midbrain (33.49%) and lowest for the cerebellar regions (5.82%–16.60%); see Table 3. Estimates, standardized beta, standard error, and 95% confidence intervals are given in Table S6.
TABLE 3.
Linear regression analysis of potential factors determining regional volume loss in SCA3 including SARA sum score, CAG repeat length of the longer allele, age, and sex as independent variables and the respective metric as dependent variable
| Metric | Cervical spinal cord level C3 | Cervical spinal cord level C2 | Medulla oblongata | Pons | Midbrain | Cerebellum white matter | Cerebellum, anterior lobe | Cerebellum crus II | Cerebellum X | Pallidum | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Adjusted R2 x 100a | 23.35 | 22.15 | 17.83 | 46.33 | 33.49 | 5.82 | 11.13 | 16.60 | 6.44 | 22.58 | |
| p b | SARA | c | c | c | c | c | |||||
| CAG repeat of longer allele | c | c | c | ||||||||
| Age | c | c | c | c | |||||||
| Sex | c | c | |||||||||
Explained portion of the metrics variance, given by the adjusted R2 multiplied with 100.
P value for each independent variable SARA sum score, CAG repeat length of the longer allele, age, and sex, indicating whether the respective variable statistically significantly contributes to the model.
P < 0.001.
Calculation of Effect Sizes
Based on the estimated slopes of linear models of regional volume changes in relation to disease duration, we calculated Cohen’s d values as a measure of effect size for each metric, We report the effects sizes of each metric relative to the effect size of SARA to allow a comparison. Cohen’s d of pontine volume was larger than that of SARA (1.03 times the Cohen’s d of SARA). Cohen’s d values of cervical spinal cord segment C3 (0.81 times the Cohen’s d of SARA) and C2 (0.68 times the Cohen’s d of SARA), medulla oblongata (0.61 times the Cohen’s d of SARA), midbrain (0.97 times the Cohen’s d of SARA), and pallidum (0.97 times the Cohen’s d of SARA) were in the same magnitude, but smaller than that of SARA (Table 4). For all cerebellar regions, Cohen’s d values were less than 0.5 times the Cohen’s d of SARA (Table 4).
TABLE 4.
Calculation of effect sizes relative to the effect size of SARA
| Metric | Slope | Standard error of slope | Relative Cohen’s da |
|---|---|---|---|
| Cervical spinal cord, level C3 | −0.53 | 0.08 | 0.81 |
| Cervical spinal cord, level C2 | −0.43 | 0.08 | 0.68 |
| Medulla oblongata | −24.50 | 4.55 | 0.61 |
| Pons | −210.01 | 23.11 | 1.03 |
| Midbrain | −72.76 | 8.46 | 0.97 |
| Cerebellum white matter | −226.05 | 68.67 | 0.35 |
| Cerebellum, anterior lobe | −39.51 | 16.78 | 0.26 |
| Cerebellum crus II | −104.17 | 33.15 | 0.35 |
| Cerebellum X | −5.93 | 1.50 | 0.45 |
| Pallidum | −35.86 | 4.17 | 0.97 |
Linear models with the estimated disease duration were the basis for the estimated slope of all respective metrics as well as for the SARA sum score. As a measure of effect size, we calculated Cohen’s d values for a presumed 50% reduction of the decrease of SARA and each MRI metric.
Cohen’s d is given in relation to SARA: relative Cohen’s d = Cohen’s d of the metric/Cohen’s d of SARA.
Discussion
In this cross-sectional MRI study, we assessed regional tissue loss in a large cohort of preataxic and ataxic SCA3 mutation carriers. We applied a refined and optimized brain segmentation approach that allowed reliable subsegmentation of the cerebellum into the cerebellar lobules. For the spinal cord, we relied on the assessment of the mean cross-sectional area of the upper cervical levels. The advantage of the applied methodology is that the availability of individual single-point metrics allows consideration of brain regional volumes as outcome measures in future interventional trials. To study the change of metrics in relation to disease duration, we used a uniform time scale defined by the predicted age of ataxia onset for all mutation carriers according to a previously published model.18,22 To check whether this methodological approach treating all mutation carriers consistently distorts the disease course, we repeated the analysis using a compound time scale in which ataxia onset was calculated only in presymptomatic individuals, whereas in symptomatic individuals we used the reported age at onset. Both approaches led to similar results.
Our results in SCA3 mutation carriers reflect the neurodegeneration pattern observed in autopsy studies very well23-25 and confirm previous MRI studies in smaller numbers of patients.9,14,16,26,27 We will discuss several aspects of this in more detail in the following section. Even though our data do not allow defining a clear temporal order of involvement of affected regions across the entire disease range, the inclusion of SCA3 mutation carriers from the earliest preataxia to late symptomatic disease stages allowed us to detect regional volume changes occurring before the onset of ataxia. In the preataxic stage, volume loss of cerebellar regions was less pronounced than that of the affected extracerebellar regions including the spinal cord, brain stem, and pallidum, suggesting prominent involvement of extracerebellar regions at the onset of neurodegeneration. This observation is in line with the known pattern of SCA3 — in contrast to many other SCAs, in which the cerebellar cortex is severely affected, cerebellar involvement in SCA3 is mild and variable, and the Purkinje cell layer of the cerebellar cortex has even been reported to be completely spared in some autopsy studies.23-25 The rather mild cerebellar volume loss and given that age was the only factor that covaried with cerebellar volume suggest that cerebellar degeneration in SCA3 may represent a secondary, age-related phenomenon. This is corroborated by evidence of an age-dependent decline of cerebellar volume in healthy individuals,28,29 whereas parts of the brain stem such as the pons appear to be less affected by age-related volume loss.30,31 Based on evidence from neuropathological studies and the results of previous voxel-based morphometric as well as diffusion MRI studies showing prominent white matter loss in patients with SCA3,9,14,24,27,32 it is likely that the anatomical substrate of the volume changes of cervical spinal cord segments C2 and C3 and the medulla oblongata reflects pathology of afferent spinal pathways, in particular the spinocerebellar tracts. In addition, considering the neuropathological evidence of substantial neuronal loss in the dentate nucleus,24 degeneration of the dentato-rubro-thalamo-cortical tract would also affect the thalamic and midbrain volumes.
Anatomically the pons is connected with the cerebellum via the middle cerebellar peduncles. As in other polyglutamine diseases, the expanded disease protein accumulates in neuronal nuclei forming ubiquitin-positive neuronal intranuclear inclusions (NIIs).33,34 The distribution of NIIs does not fully match that of the neurodegeneration, but in SCA3, NIIs are abundant in pontine nuclei,35 and atrophy of the pons is a common feature in the autopsy cases.24 The pons volume showed the most continuous decline and steepest decrease around ataxia onset of all further studied metrics in our study. Furthermore, a model with ataxia severity, CAG repeat length, age, and sex could only explain a small proportion of the variance in cerebellar volume, whereas the pontine volume showed the highest proportion, with almost 50% of its variance explained by a model including these factors. This is remarkable, in particular, given the very heterogeneous sample of MRIs included in this study.
Calculation of effect size has 2 major limitations. First, our calculations were based on cross-sectional data, which do not allow an accurate estimation of between-subject variability in the longitudinal rate of change. However, as we calculated effect size for all metrics and SARA in the same way, we believe that the data are useful for comparison between metrics. Consequently, we provided effect sizes of the MRI metrics in relation to the effect size of SARA. Second, we assumed a linear progression throughout the whole time course of the disease, although the evolution of almost all metrics was nonlinear. The effect size of pontine volume marginally exceeded that of SARA. The effect size of all other metrics was smaller than that of SARA, although those of cervical spinal cord segment C2 and C3, pons, midbrain, and pallidum were of the same magnitude as that of SARA. The current values are not comparable to effect sizes calculated on the basis of longitudinal data.14,16,36
We suggest that extracerebellar regions represent the core sites of the disease process in SCA3. This suggestion is based on the observed time course of volume changes and the close relation of the metrics of extracerebellar regions with clinical disease severity and CAG repeat length. Our observation that the effect size of the pontine volume was marginally higher than that of SARA and that a relevant proportion of pontine volume variance was explained with a model including ataxia severity, CAG repeat length, and age suggests that pontine volume is a promising biomarker candidate for interventional studies that aim to slow the progression of SCA3. However, longitudinal studies including of preataxic SCA3 mutation carriers are inevitable to adequately test the validity of such imaging biomarker candidates.
Supplementary Material
Funding agencies:
This publication is an outcome of ESMI, an EU Joint Programme - Neurodegenerative Disease Research (JPND) project (see www.jpnd.eu). The project is supported under the aegis of JPND through the following funding organizations: Germany, Federal Ministry of Education and Research (BMBF; funding codes 01ED1602A/B); Netherlands, The Netherlands Organisation for Health Research and Development; Portugal, Foundation for Science and Technology and Regional Fund for Science and Technology of the Azores; United Kingdom, Medical Research Council. This project has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement 643417. At the US sites this work was in part supported by the National Ataxia Foundation and the National Institute of Neurological Disorders and Stroke (NINDS) grant R01 NS080816. The Center for Magnetic Resonance Research is supported by the National Institute of Biomedical Imaging and Bioengineering (NIBIB) grant P41 EB027061, and the Institutional Center Cores for Advanced Neuroimaging award P30 NS076408 and S10 OD017974 grant.
Relevant conflicts of interest/financial disclosures:
M.C.F. was supported by grants from Brazilian governmental agencies (CNPq and FAPESP). B.v.W. received funding from ZonMW, EU Joint Programme — Neurodegenerative Disease Research (JPND) project, was supported under the aegis of JPND through funding from the Netherlands Organisation for Health Research and Development. A.D. received BIOSCA NCT01470729 sponsorship from Assistance Publique — Hopitaux de Paris. P.G. received support from the EU Joint Programme — Neurodegenerative Disease Research (JPND) project and was supported under the aegis of JPND through funding from the Medical Research Council. H.G.-M. received a JPND grant rom the Medical Research Council and support from CureSCA3 and Fathers Foundation. L.S. received support from the EU Joint Programme - Neurodegenerative Disease Research (JPND) project and support under the aegis of JPND through the Federal Ministry of Education and Research (BMBF; funding codes 01ED1602A/B). G.O. — the study was in part funded by the National Ataxia Foundation. The Center for Magnetic Resonance Research is supported by the National Institute of Biomedical Imaging and Bioengineering (NIBIB) grant P41 EB027061 and the Institutional Center Cores for Advanced Neuroimaging award P30 NS076408. R.J. is a paid employee of IXICO Plc, contracted to undertake this work. The involvement of R.W. was funded by IXICO. T.K. received support from the EU Joint Programme — Neurodegenerative Disease Research (JPND) project and under the aegis of JPND through the Federal Ministry of Education and Research (BMBF; funding codes 01ED1602A/B).
Footnotes
Supporting Data
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site.
References
- 1.Ruano L, Melo C, Silva MC, Coutinho P. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology 2014;42(3):174–183. [DOI] [PubMed] [Google Scholar]
- 2.Rub U, Schols L, Paulson H, et al. Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog Neurobiol 2013;104:38–66. [DOI] [PubMed] [Google Scholar]
- 3.Maciel P, Gaspar C, DeStefano AL, et al. Correlation between CAG repeat length and clinical features in Machado-Joseph disease. Am J Hum Genet 1995;57(1):54–61. [PMC free article] [PubMed] [Google Scholar]
- 4.Schmitz-Hubsch T, Coudert M, Bauer P, et al. Spinocerebellar ataxia types 1, 2, 3, and 6: disease severity and nonataxia symptoms. Neurology 2008;71(13):982–989. [DOI] [PubMed] [Google Scholar]
- 5.Jacobi H, du Montcel ST, Bauer P, et al. Long-term disease progression in spinocerebellar ataxia types 1, 2, 3, and 6: a longitudinal cohort study. Lancet Neurol 2015;14(11):1101–1108. [DOI] [PubMed] [Google Scholar]
- 6.Diallo A, Jacobi H, Cook A, et al. Prediction of survival with longterm disease progression in Most common spinocerebellar ataxia. Mov Disord 2019;34(8):1220–1227. [DOI] [PubMed] [Google Scholar]
- 7.Jardim LB, Hauser L, Kieling C, et al. Progression rate of neurological deficits in a 10-year cohort of SCA3 patients. Cerebellum 2010;9(3):419–428. [DOI] [PubMed] [Google Scholar]
- 8.McLoughlin HS, Moore LR, Chopra R, et al. Oligonucleotide therapy mitigates disease in spinocerebellar ataxia type 3 mice. Ann Neurol 2018;84(1):64–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rezende TJR, de Paiva JLR, Martinez ARM, et al. Structural signature of SCA3: from presymptomatic to late disease stages. Ann Neurol 2018;84(3):401–408. [DOI] [PubMed] [Google Scholar]
- 10.Fahl CN, Branco LM, Bergo FP, D’Abreu A, Lopes-Cendes I, Franca MC Jr. Spinal cord damage in Machado-Joseph disease. Cerebellum 2015;14(2):128–132. [DOI] [PubMed] [Google Scholar]
- 11.Guimaraes RP, D’Abreu A, Yasuda CL, et al. A multimodal evaluation of microstructural white matter damage in spinocerebellar ataxia type 3. Mov Disord 2013;28(8):1125–1132. [DOI] [PubMed] [Google Scholar]
- 12.Hernandez-Castillo CR, King M, Diedrichsen J, Fernandez-Ruiz J. Unique degeneration signatures in the cerebellar cortex for spinocerebellar ataxias 2, 3, and 7. Neuroimage 2018;20:931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klaes A, Reckziegel E, Franca MC Jr, et al. MR imaging in spinocerebellar ataxias: a systematic review. Am J Neuroradiol 2016;37(8):1405–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adanyeguh IM, Perlbarg V, Henry PG, et al. Autosomal dominant cerebellar ataxias: imaging biomarkers with high effect sizes. Neuroimage 2018;19:858–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reetz K, Rodriguez-Labrada R, Dogan I, et al. Brain atrophy measures in preclinical and manifest spinocerebellar ataxia type 2. Ann Clin Transl Neurol 2018;5(2):128–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Piccinin CC, Rezende TJR, de Paiva JLR, et al. A 5-year longitudinal clinical and magnetic resonance imaging study in spinocerebellar ataxia type 3. Mov Disord 2020;35(9):1679–1684. [DOI] [PubMed] [Google Scholar]
- 17.Schmitz-Hubsch T, du Montcel ST, Baliko L, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 2006;66(11):1717–1720. [DOI] [PubMed] [Google Scholar]
- 18.Jacobi H, du Montcel ST, Romanzetti S, et al. Conversion of individuals at risk for spinocerebellar ataxia types 1, 2, 3, and 6 to manifest ataxia (RISCA): a longitudinal cohort study. Lancet Neurol 2020;19(9):738–747. [DOI] [PubMed] [Google Scholar]
- 19.Buckner RL, Head D, Parker J, et al. A unified approach for morphometric and functional data analysis in young, old, and demented adults using automated atlas-based head size normalization: reliability and validation against manual measurement of total intracranial volume. Neuroimage 2004;23(2):724–738. [DOI] [PubMed] [Google Scholar]
- 20.De Leener B, Levy S, Dupont SM, et al. SCT: spinal cord toolbox, an open-source software for processing spinal cord MRI data. Neuroimage 2017;145((Pt A)):24–43. [DOI] [PubMed] [Google Scholar]
- 21.Benjamini YHY. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc, Series B 1995;57:289–300. [Google Scholar]
- 22.Tezenas du Montcel S, Durr A, Rakowicz M, et al. Prediction of the age at onset in spinocerebellar ataxia type 1, 2, 3 and 6. J Med Genet 2014;51(7):479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seidel K, Siswanto S, Brunt ER, den Dunnen W, Korf HW, Rub U. Brain pathology of spinocerebellar ataxias. Acta Neuropathol 2012;124(1):1–21. [DOI] [PubMed] [Google Scholar]
- 24.Koeppen AH. The neuropathology of spinocerebellar ataxia type 3/Machado-Joseph disease. Adv Exp Med Biol 2018;1049:233–241. [DOI] [PubMed] [Google Scholar]
- 25.Durr A, Stevanin G, Cancel G, et al. Spinocerebellar ataxia 3 and Machado-Joseph disease: clinical, molecular, and neuropathological features. Ann Neurol 1996;39(4):490–499. [DOI] [PubMed] [Google Scholar]
- 26.Klockgether T, Skalej M, Wedekind D, et al. Autosomal dominant cerebellar ataxia type I. MRI-based volumetry of posterior fossa structures and basal ganglia in spinocerebellar ataxia types 1, 2 and 3. Brain 1998;121(Pt 9):1687–1693. [DOI] [PubMed] [Google Scholar]
- 27.Schulz JB, Borkert J, Wolf S, et al. Visualization, quantification and correlation of brain atrophy with clinical symptoms in spinocerebellar ataxia types 1, 3 and 6.Neuroimage 2010;49(1):158–168. [DOI] [PubMed] [Google Scholar]
- 28.Bernard JA, Leopold DR, Calhoun VD, Mittal VA. Regional cerebellar volume and cognitive function from adolescence to late middle age. Hum Brain Mapp 2015;36(3):1102–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han S, An Y, Carass A, Prince JL, Resnick SM. Longitudinal analysis of regional cerebellum volumes during normal aging. Neuroimage 2020;220:117062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lambert C, Chowdhury R, Fitzgerald TH, et al. Characterizing aging in the human brainstem using quantitative multimodal MRI analysis. Front Hum Neurosci 2013;7:462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luft AR, Skalej M, Schulz JB, et al. Patterns of age-related shrinkage in cerebellum and brainstem observed in vivo using three-dimensional MRI volumetry. Cereb Cortex 1999;9(7):712–721. [DOI] [PubMed] [Google Scholar]
- 32.Lukas C, Schols L, Bellenberg B, et al. Dissociation of grey and white matter reduction in spinocerebellar ataxia type 3 and 6: a voxel-based morphometry study. Neurosci Lett 2006;408(3):230–235. [DOI] [PubMed] [Google Scholar]
- 33.Paulson HL, Perez MK, Trottier Y, et al. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron 1997;19(2):333–344. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt T, Landwehrmeyer GB, Schmitt I, et al. An isoform of ataxin-3 accumulates in the nucleus of neuronal cells in affected brain regions of SCA3 patients. Brain Pathol 1998;8(4):669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seidel K, Siswanto S, Fredrich M, et al. On the distribution of intranuclear and cytoplasmic aggregates in the brainstem of patients with spinocerebellar ataxia type 2 and 3. Brain Pathol 2017;27(3):345–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reetz K, Costa AS, Mirzazade S, et al. Genotype-specific patterns of atrophy progression are more sensitive than clinical decline in SCA1, SCA3 and SCA6. Brain 2013;136(Pt 3):905–917. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.