

Abstract
Rapid flip-flop of phospholipids across the two leaflets of biological membranes is crucial for many aspects of cellular life. The transport proteins that facilitate this process are classified as pump-like flippases and floppases and channel-like scramblases. Unexpectedly, Class A G protein–coupled receptors (GPCRs), a large class of signaling proteins exemplified by the visual receptor rhodopsin and its apoprotein opsin, are constitutively active as scramblases in vitro. In liposomes, opsin scrambles lipids at a unitary rate of >100,000 per second. Atomistic molecular dynamics simulations of opsin in a lipid membrane reveal conformational transitions that expose a polar groove between transmembrane helices 6 and 7. This groove enables transbilayer lipid movement, conceptualized as the swiping of a credit card (lipid) through a card reader (GPCR). Conformational changes that facilitate scrambling are distinct from those associated with GPCR signaling. In this review, we discuss the physiological significance of GPCR scramblase activity and the modes of its regulation in cells.
Keywords: Markov state modeling, membrane transport, molecular dynamics simulations, rhodopsin, scramblase, TMEM16
INTRODUCTION
Phospholipids make up the two leaflets of biological membranes. Within a leaflet, they exhibit considerable intramolecular dynamics and rapid rotational and lateral diffusion. However, in rare cases, they cross the bilayer spontaneously, a process termed flip-flop that entails reorientation of the lipid from one side of the membrane to the other. In a synthetic membrane such as a large unilamellar liposome (56), or in a relatively inert biological membrane such as the envelope of influenza virus (101), flip-flop occurs infrequently—an individual phospholipid moves across the bilayer only approximately once per day—because of the high energy cost associated with moving the phospholipid’s polar headgroup through the oily zone of lipid fatty acyl chains that populate the interior of the membrane. Nevertheless, fast flip-flop is crucial for many aspects of cellular life, where it is needed for cell growth, protein glycosylation in the endoplasmic reticulum (ER), controlled breakdown of the transbilayer lipid asymmetry of the plasma membrane of eukaryotic cells, production of intracellular (IC) transport vesicles, and adaptation of cellular responses to physiological challenges (37, 54, 70, 91, 102, 111). For flip-flop to occur rapidly, on a physiologically relevant timescale, transport proteins are required. Like ion pumps that use metabolic energy to transport ions uphill, against their electrochemical gradient, or ion channels that mediate passive downhill movement of ions, lipid flip-flop is mediated by two classes of proteins: pump-like flippases and floppases and channel-like scramblases (91).
Lipid pump activity was first revealed via observations of the dramatic shape changes that occur when phospholipids are added to red blood cells (24, 106). The cells became spiky as the membrane deformed outward to accommodate excess lipid in the outer leaflet, in accordance with the bilayer couple hypothesis (107). As lipids were transported to the inner leaflet, the cells returned to their discoid shape. Transport required ATP and was specific for the aminophospholipids phosphatidylserine (PS) and phosphatidylethanolamine (PE); shape restoration did not occur when phosphatidylcholine (PC) was tested. The lipid pump associated with this phenomenon was termed aminophospholipid translocase and later identified, through partial purification (116), as a P4 ATPase, the largest subfamily of a superfamily of integral membrane proteins that transport ions and lipids across membranes (62). P4 ATPase flippases are typically out-to-in pumps; i.e., they flip lipids from the exoplasmic leaflet of membranes to the cytoplasmic side. Other lipid pumps, called floppases, catalyze in-to-out transport, moving lipids from the cytoplasmic to the exoplasmic side of the membrane. Floppases are generally ATP binding cassette (ABC) transporters, with the bacterial protein MsbA, important for lipopolysaccharide biosynthesis (79, 126), being among the first to be recognized in this role. Functional studies of flippases and floppases, supported by considerable atomistic structural information, have revealed insights into how they use the energy released from the process of ATP hydrolysis to move phospholipids. The pumps can flip (or flop) approximately 10–100 phospholipids per second, each transport event requiring the hydrolysis of one molecule of ATP to drive large conformational changes. The rate of these events is dictated by the ability of the pumps to turn over ATP.
In contrast to lipid pumps, which belong to either of two major protein families, channel-like lipid scramblases are more diverse, and their activity may be regulated or constitutive. However, despite the absence of a characteristic scramblase sequence motif (but see 71, 72), it is likely that scramblases operate via a universal credit card mechanism (90) whereby a membrane-spanning polar groove in the protein provides a protected pathway for lipid headgroups to traverse the membrane while their hydrophobic tails remain in the bilayer.
The signaling lipid PS, normally sequestered in the cytoplasmic leaflet of the plasma membrane, is externalized by regulated scramblases that are activated during blood clotting (in platelets) and apoptosis (in all cells) (11, 70, 109). Suzuki et al. (113, 114) proposed TMEM16F and XKR8 as the scramblases involved in these events. TMEM16F is activated by increases in IC Ca2+ that occur during cell signaling, whereas XKR8 requires caspase-mediated cleavage to contribute to PS exposure. TMEM16F belongs to the TMEM16/anoctamin family of proteins (10 paralogs in humans), whose founding members (TMEM16A, B) are ion channels lacking scramblase activity. However, other TMEM16 proteins (1, 17), including fungal homologs (15, 64), were demonstrated to have Ca2+-regulated phospholipid scramblase activity upon purification and reconstitution into unilamellar vesicles. Structural studies revealed a membrane-spanning, nanometer-wide polar groove in some of these proteins that becomes exposed to the membrane upon Ca2+ binding to provide the lipid transport pathway implicit in the credit card mechanism (48, 90). Whereas TMEM16 proteins are demonstrably scramblases, the function of XKR proteins is currently less clear, as recent experiments could not demonstrate scramblase activity of intact or caspase-cleaved XKR9 (112).
Constitutively active scramblases are characteristic of biogenic membranes like the ER (90, 91, 102), but their activity has also been documented in other membranes such as the mitochondrial outer membrane (43) and the isolation membrane precursor of autophagosomes (74). The molecular identity of these scramblases is generally not known, with the exceptions of the ER-localized DedA family members TMEM41B and VMP1 (31, 41, 59) and Atg9 (31, 74), which appear to have constitutive phospholipid scramblase activity necessary for processes requiring lipid supply, such as lipid droplet and lipoprotein biogenesis and growth of the autophagosome. At least four constitutively active scramblases are involved in protein glycosylation pathways in the ER—these scramblases have yet to be identified, although their activities are well documented (91, 102, 104, 118). Curiously, bacteriorhodopsin, the light-dependent proton pump of the purple membrane of Halobacterium salinarum, has light-independent phospholipid scramblase activity (119).
This review focuses on the unexpected scramblase activity of rhodopsin-class (Class A) G protein–coupled receptors (GPCRs), the largest subset (4, 28, 40, 50, 77, 125) of an approximately 800-strong family of integral membrane signaling proteins encoded by the human genome. GPCR scramblases appear to be constitutively active when analyzed in vitro, but as discussed below, they are undoubtedly regulated in cells.
BACKGROUND: OPSIN IS A PHOSPHOLIPID SCRAMBLASE
The discovery of the scramblase activity of GPCRs originated with the work of Wu & Hubbell (123), who reported rapid scrambling of phospholipids across the membrane of photoreceptor discs purified from bovine retina. Similar results were later reported by Hessel et al. (38), who showed not only that spin-labeled phospholipids added to the disc exterior could equilibrate with the inner leaflet, but also that the lipid analogs could be completely extracted from discs after they had first been allowed to equilibrate between the two leaflets. This provided direct demonstration of bidirectional lipid transport across disc membranes. Supplementing the assays with ATP did not affect the results. At steady state, the PE and PC analogs were symmetrically distributed across the disc membrane, whereas PS was predominantly located in the outer (cytoplasmic) leaflet. This asymmetry is consistent with a dynamic equilibrium model in which scrambling enables lipids to sample both leaflets of the bilayer, while the strong, protein-based asymmetric charge distribution (positive on the cytoplasmic face on account of the large number of oriented rhodopsin molecules) confines negatively charged PS—but not zwitterionic PC and PE—to the cytoplasmic side. Interestingly, the transbilayer asymmetry of PS changes reversibly in response to light (39) because of changes in transbilayer charge asymmetry associated with light-induced changes in rhodopsin conformation.
To determine the molecular machinery responsible for lipid scrambling in photoreceptor discs, Menon et al. (66) used a fluorescence-based assay (18) to measure scramblase activity in large unilamellar vesicles reconstituted with membrane proteins, i.e., proteoliposomes. They extracted membrane proteins from purified bovine discs using nonionic detergents and reconstituted them into vesicles composed of egg phospholipids, as well as trace quantities (1 of every 500 natural lipids) of nitrobenzoxadiazole (NBD)-modified fluorescent phospholipids as reporters of scramblase activity. Reconstitution was accomplished via three distinct methods, each employing a different nonionic detergent; all yielded the same outcome. The principle of the assay is shown in Figure 1a. Dithionite, a membrane-impermeant reagent, reacts with NBD-phospholipids in the outer leaflet of the vesicles, converting the fluorophore irreversibly to nonfluorescent aminobenzoxadiazole. In large unilamellar vesicles, only approximately half of the NBD-phospholipids react with dithionite, as those in the inner leaflet of the vesicles are protected. In a vesicle with a functional scramblase, lipids are exchanged across the bilayer, causing all fluorophores to be exposed at the outer leaflet, where they are reduced by dithionite. Menon et al. (66) reported three key observations. First, the extent of fluorescence loss in proteoliposomes reconstituted with disc membrane proteins was greater than in protein-free liposomes, indicating that the scramblase activity of intact discs could be recapitulated in synthetic vesicles; second, the amount of disc extract needed to functionalize the vesicles was relatively small, indicating that disc scramblase activity was due to an abundant protein, e.g., opsin, which represents >80% of disc membrane proteins; and finally, pretreatment of the disc extract prior to reconstitution with antiopsin antibodies resulted in loss of activity, whereas vesicles reconstituted with immunopurified opsin were active. Consistent with the latter result, and ruling out the involvement of other disc components, opsin that was expressed in and purified from human embryonic kidney (HEK) cells had scramblase activity.
Figure 1.
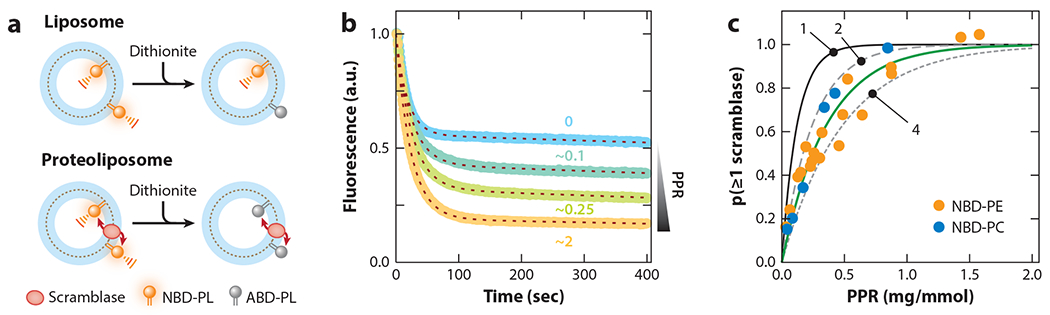
Phospholipid scrambling by opsin in reconstituted vesicles. (a) Scramblase assay Large unilamellar vesicles are reconstituted with a trace amount of fluorescent phospholipid [nitrobenzoxadiazole-phospholipid (NBD-PL)] and with (proteoliposome) or without (liposome) opsin. On adding dithionite to the vesicles, NBD-PL molecules in the outer leaflet are bleached to nonfluorescent aminobenzoxadiazole-phospholipid (ABD-PL). For liposomes (top), dithionite addition lowers fluorescence to approximately 50% of its initial value; for a proteoliposome with a functional scramblase (bottom), fluorescence is eliminated as NBD-PLs in the protected inner leaflet are scrambled to the dithionite-accessible outer leaflet. (b) Representative fluorescence traces corresponding to vesicles reconstituted at different protein–phospholipid ratios (PPRs). The blue trace (PPR = 0) corresponds to protein-free liposomes; the remaining traces correspond to vesicles with increasing PPR (approximately 0.1, 0.25, and 2 mg protein per mmol phospholipid). The dashed lines overlaying each trace represent data fits (mono-exponential decay with a half-time of approximately 15 s, plus a shallow linear component with slope approximately 10−4 s−1). (c) Protein-dependence data indicate that opsin reconstitutes as a dimer or tetramer. The solid, long-dashed, and short-dashed lines correspond to the result expected for the functional reconstitution of opsin as a monomer, dimer, or tetramer, respectively (indicated as 1, 2, and 4, respectively). The green line is the fit to the experimental data [filled circles correspond to assays done with NBD-phosphatidylethanolamine (PE) or NBD-phosphatidylcholine (PC)]. The data in panels b and c are from Goren et al. (32) and Ploier et al. (88).
Figure 1b shows examples of fluorescence time-traces corresponding to scramblase assays on proteoliposomes reconstituted with different amounts of purified opsin. As discussed in more detail in the next section, the extent of fluorescence loss increases with the amount of opsin used, indicating increasing functionalization of individual vesicles in the sample. Evident in Figure 1b is that the rate of fluorescence decay in proteoliposomes is the same as in protein-free liposomes, indicating that scrambling occurs at a rate similar to or faster than the rate at which dithionite bleaches NBD fluorescence. As the membrane of a large unilamellar vesicle has approximately 105 phospholipids, and the half-time (t1/2) of dithionite-mediated fluorescence decay is approximately 10 s, a lower estimate of the scrambling rate is approximately 104 s−1 (32, 66, 81, 88). Recent data using more reactive lipids allow this estimate to be revised upward to 105 s−1 (63). As the number of opsin molecules reconstituted is low (one or a few copies per vesicle on average), this estimate can also be taken as an indicator of the unitary rate of scrambling. Thus, a single opsin scramblase facilitates transbilayer movement of lipids at a rate of >105 s−1, corresponding to at least one flipping event per 10 μs.
The results summarized above were validated with important control experiments. (a) Specific proteolysis showed that opsin was reconstituted symmetrically, with half of the molecules being inserted with the protease-sensitive C terminus facing the vesicle lumen (32). Collisional quenching with iodide ions showed that the fluorescent reporter lipids were symmetrically distributed across the membrane of both protein-free liposomes and proteoliposomes, indicating that the greater extent of fluorescence reduction seen in proteoliposomes was not the result of an asymmetrical transbilayer distribution of the reporter lipid (32, 117). (b) Dithionite was unable to reduce NBD-glucose trapped inside the vesicles, confirming its inability to cross the membrane at an appreciable rate on the timescale of the assay (32). (c) Scramblase activity was demonstrated using an alternate assay in which NBD-phospholipids are extracted from the membrane with fatty-acid-free bovine serum albumin (BSA), measured as a decrease in fluorescence because BSA-bound NBD-phospholipids have a lower quantum yield than their membrane-bound counterparts (18, 66). (d) Reconstitution of high amounts of a bacterial glutamate transporter, an irrelevant trimeric membrane protein of similar monomeric molecular mass as opsin, did not cause lipid scrambling (66). Based on these cumulative data, the authors of this study concluded that opsin is a phospholipid scramblase. To examine whether scramblase activity is a general property of GPCRs, they tested the β1-adrenergic receptor (β1AR) and demonstrated similar activity (66). Subsequent work showed that the β2-adrenergic receptor (β2AR), as well as the adenosine A2A receptor, could also scramble lipids upon reconstitution into large unilamellar vesicles (32), indicating that lipid scrambling is likely a general property of rhodopsin-class (Class A) GPCRs. The question of whether the activation state of GPCRs impacts scramblase activity is discussed below. For the moment, we focus on the scramblase activity of the apoprotein opsin.
LIPID SCRAMBLING BY AN OPSIN MONOMER
Opsin self-associates to form dimers and higher-order multimers in the membrane (22, 44, 53) but purifies as a monomer when solubilized in n-dodecyl-β-D-maltoside (DDM) (29, 45, 53). However, the protein reversibly self-associates when the concentration of DDM is lowered (45, 53) and reconstitutes into vesicles as a dimer or higher-order multimer, as explained below. This behavior makes it difficult to determine the functional oligomeric state of the protein and, more specifically, to address the question of whether an opsin monomer is sufficient to facilitate scrambling. The eventual discovery of scramblase-competent opsin mutants that fail to self-associate during reconstitution (81, 88) made it possible to assign scramblase activity to monomeric opsin.
The typical reconstitution protocol (89) requires incubating opsin with large unilamellar vesicles that are destabilized by treatment with DDM. Subsequent addition of detergent-adsorbing BioBeads results in reconstitution of opsin into the vesicles. As detergent is gradually removed, opsin may directly insert into the vesicle membrane, or it may dimerize or oligomerize before insertion. Clearly, a greater amount of opsin would be needed to functionalize all of the vesicles in the sample if the protein were to dimerize or multimerize prior to reconstitution. The reconstitution process can be described by Poisson statistics (20, 32, 88). If a single reconstitution event directly confers scramblase activity to a given vesicle, then the probability p(≥1 scramblase) that a given vesicle in the ensemble acquires at least one functional scramblase should increase mono-exponentially with the amount of opsin. Thus, for uniformly sized vesicles, p(≥1 scramblase) = 1 – exp(−PPR/α), where α is a fit constant, and PPR is the protein–phospholipid ratio of the sample in units of mg/mmol (for a more detailed analysis that accounts for the size distribution of the vesicles, see 88). At a PPR value of α mg/mmol, there is one functional scramblase per vesicle on average, and approximately 63% of the vesicles in the population possess at least one scramblase. Goren et al. (32) set out to measure α and thereby to determine how many opsin molecules are required to functionalize all of the vesicles in the sample. Taking a fixed amount of phospholipid, i.e., a constant number of vesicles, they varied the amount of opsin used per reconstitution to generate proteoliposome samples over a range of PPR values. These samples were assayed for scramblase activity (Figure 1b), and the fluorescence endpoint data were transformed according to the formula p(≥1 scramblase) = (y – yo)/(ymax – yo), where yo is the percentage reduction in fluorescence obtained with protein-free liposomes (typically 45–50%), and ymax is the maximum percentage reduction observed with proteoliposomes (typically approximately 80–85%)1. Agraph of p(≥1 scramblase) versus PPR is shown in Figure 1c in comparison with predicted graphs for the functional reconstitution of monomers, dimers, and tetramers of opsin. As can be readily seen, the experimental data fall between the predicted traces for the functional reconstitution of dimers and tetramers. Why opsin multimerizes en route to reconstitution is unclear, as recent analyses (53) suggest that self-association of opsins in a detergent micelle is energetically costly and that direct reconstitution of opsin monomers into the vesicle membrane would seem to be preferred.
While testing whether certain point mutations in opsin would result in loss of scramblase activity, Ploier et al. (88) made the serendipitous discovery that these opsins are fully functional as scramblases but reconstitute as monomers. A similar conclusion was reached by Pandey et al. (81), who engineered mutations in transmembrane (TM) helix 4 with the aim of disrupting scramblase activity—in this case, again, scramblase activity was unaffected, but the modified protein reconstituted as a monomer. The p(≥1 scramblase) versus PPR plots for these opsin mutants are steeper than those seen for the wild-type protein and identical to the predicted trace for reconstitution of an opsin monomer shown in Figure 1c. Thus, lipid scrambling can be accomplished by an opsin monomer.
MECHANISM OF SCRAMBLING: LESSONS FROM MOLECULAR DYNAMICS SIMULATIONS
While TMEM16 scramblases possess a structurally evident, polar, membrane-facing groove to facilitate transbilayer lipid transit according to the credit card mechanism (15, 48, 90), there is no analogous feature in the structure of opsin, making it difficult to formulate a specific, experimentally testable hypothesis to suggest how it might scramble lipids. To gain insights into the molecular mechanisms of opsin-mediated lipid scrambling, Morra et al. (68) therefore turned to atomistic molecular dynamics (MD) simulations. The utility of the MD technique has recently expanded significantly to processes of physiological relevance (e.g., protein folding, function-related transitions, or protein–ligand interactions) due to advances in computational hardware and software (23, 46, 65, 69, 84, 127). However, as GPCR-mediated lipid scrambling events are relatively rare, occurring with a frequency estimated at one per approximately 10–100 μs, sufficient sampling of such events with conventional MD approaches still entails a prohibitively high computational cost.
An efficient way to overcome this challenge and to extract quantitative insights into the lipid scrambling process is to use adaptive ensemble MD (AEMD) simulations (5, 49). The AEMD approach leverages the statistical mechanics basis of MD simulations to accumulate very long trajectories from ensemble simulations in which the time propagation of the system is recorded in large numbers of independent replicates, much like in single-molecule experiments. Specifically, within the AEMD framework, the dynamical behavior of a molecular system is sampled in multiple independent unbiased MD trajectories (an ensemble of replicates). These trajectories are analyzed to select one or multiple representative conformations of the system from which the next stage of ensemble MD simulations is initiated (again in multiple replicates). This iterative process continues until satisfactory sampling of mechanistic steps is achieved to enable quantification of kinetic parameters.
To study mechanisms of opsin-mediated lipid scrambling, Morra et al. (68) employed a four-stage AEMD protocol resulting in a cumulative sampling time of >50 μs. In these simulations, opsin was surrounded by a lipid membrane consisting of a 9:1 mixture of POPC and POPG lipids to mimic the experimental conditions of the in vitro scrambling assay (88) (Figure 1b). The simulations revealed gradual transformation of the protein region between TM6 and TM7 into a continuous hydrophilic conduit (Figure 2b) traversed by lipid headgroups from the IC to the extracellular (EC) side (Figure 2a). The translocating lipids engage in polar interactions with specific sets of protein residues at different sites along the TM6–TM7 interface (Figure 2c). At the intracellularly located Site 1, they are involved in ionic interactions with the charged residues E249(6.32) and K311(7.58) [the superscript numbers correspond to the Ballesteros-Weinstein general numbering scheme for GPCR residues (6)]2. Site 2 is located at the level of the bilayer midplane and primarily consists of the polar residues C264(6.47) and S298(7.45), as well as more extracellularly located juxtaposed residues such as Y268(6.51) and K296(7.43). At Site 3, the lipids are seen in geometrically flipped conformation (i.e., the headgroups are at the level of the EC leaflet, whereas the tails face the IC leaflet) and continue interactions with Y268(6.51) and K296(7.43) residues while losing contact with C264(6.47) and S298(7.45).
Figure 2.
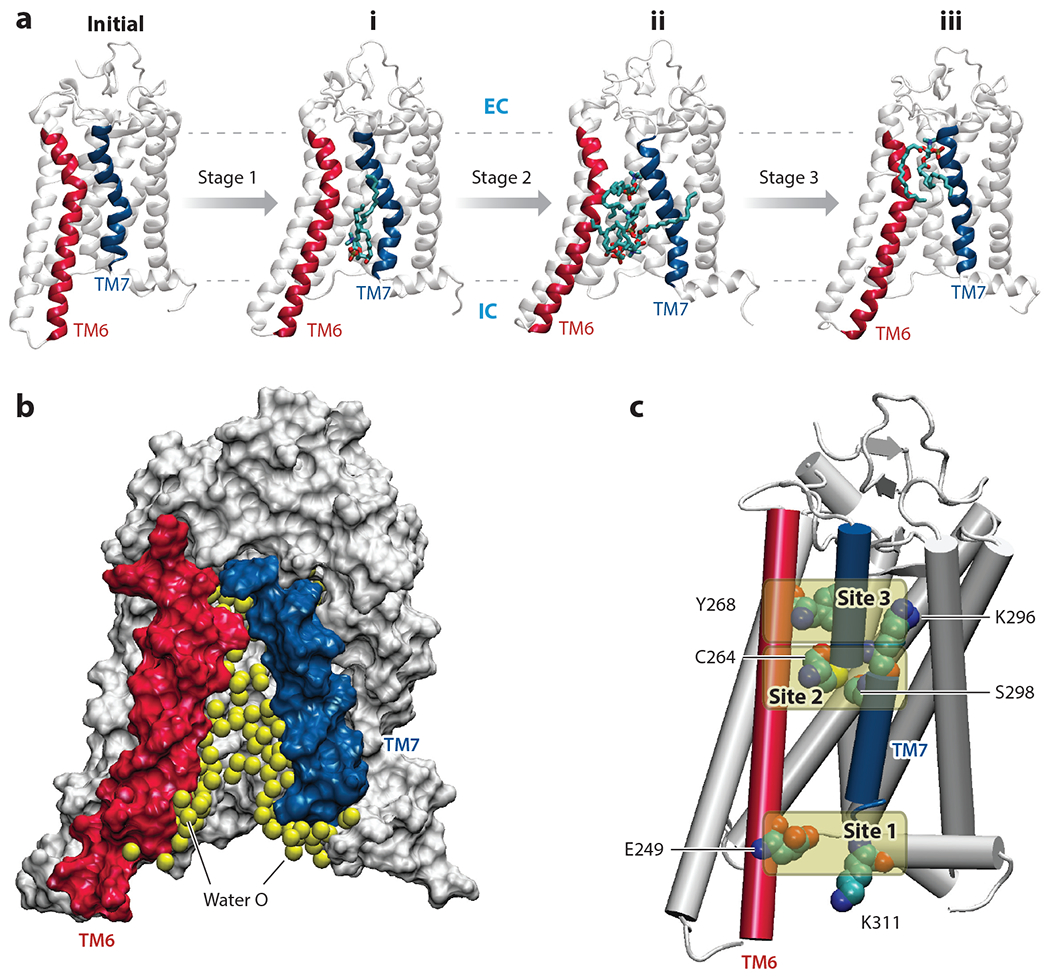
Structural features enabling lipid scrambling. (a) Snapshots illustrating the gradual opening of the lipid pathway in the adaptive ensemble molecular dynamics (AEMD) simulations. Starting from the initial model [Protein Data Bank (PDB) ID 4J4Q] (82), Stage 1 simulations sampled the transient opening of the pathway between the intracellular (IC) ends of transmembrane (TM) 6 and TM7 and the initial lipid insertion (conformation i). When this structure was subjected to subsequent sampling (in Stages 2 and 3), the pathway widened further, allowing multiple lipids to penetrate the pathway (conformation ii) and eventually resulting in complete flip of the most advanced lipid (conformation iii). In the snapshots, TMs 6 and 7 are colored in red and blue, respectively. The penetrating lipids are shown in stick representation. The IC and extracellular (EC) surfaces of the membrane are shown as dashed lines. (b) The hydrophilic lipid translocation pathway is depicted by showing water oxygen atoms (yellow spheres) accumulating inside the region between TMs 6 and 7 during the AEMD simulations. The protein is shown in surface representation, with TMs 6 and 7 shown in red and blue, respectively. (c) Three sites along the TM6–TM7 interface engaged in long-lasting interactions with the headgroups of translocating lipids. Site 1 consists of the E249(6.32)–K311(7.58) pair of residues, Site 2 includes C264(6.47) and S298(7.45), and Site 3 consists of residues Y268(6.51) and K296(7.43) [the superscript numbers correspond to the Ballesteros-Weinstein general numbering scheme for G protein–coupled receptor residues (6)].
Quantitative information about the molecular mechanisms and kinetics of the translocation process can be obtained by performing Markov state model (MSM) analysis of AEMD simulations (8, 55, 75, 76, 80, 86, 87, 92, 110, 120, 122). Morra et al. (68) first carried out dimensionality reduction analysis on the MD trajectories using a time-lagged independent component analysis (tICA) approach to identify the slowest reaction coordinates of the system (67, 73, 85, 98, 99, 105) (see the sidebar titled Dimensionality Reduction Using a Time-Lagged Independent Component Analysis). They then projected the trajectory frames onto the first three tICA vectors, thereby fully capturing the dynamics of the lipid scrambling process (68). The resulting three-dimensional tICA space was discretized into 100 microstates, and MSMs of transitions between the microstates were built (see the sidebar titled Markov State Model Analysis to Study Kinetics of Conformational Transitions). To identify the most probable pathways for opsin-mediated lipid translocation and quantify timescales for the kinetics of the process, Morra et al. (68) then grouped the microstates into 12 macrostates based on their kinetic similarity and applied transition path theory (TPT) analysis (9) (see the sidebar titled Markov State Model Analysis to Study Kinetics of Conformational Transitions).
DIMENSIONALITY REDUCTION USING A TIME-LAGGED INDEPENDENT COMPONENT ANALYSIS.
To study the kinetics of conformational transitions from a set of MD trajectories, the conformational space from the trajectories must be first reduced by a transformation to a space defined by only a few reaction coordinates of interest. Such dimensionality reduction is necessary both for removing redundant (fast) kinetic modes stored in the atomic coordinates of the system and for facilitating clustering of conformations based on kinetic similarity before quantifying kinetics (see the sidebar titled Markov State Model Analysis to Study Kinetics of Conformational Transitions). To perform the transformation, Morra et al. (68) defined as collective variables (CVs) pair-wise distances between every third residue in the TM bundle of opsin (2,016 distances overall) and recorded the time-evolution of these CVs in the trajectories as components of the X(t) vector. The slowest reaction coordinates were then identified in the framework of the tICA approach (67, 73, 85, 98, 99, 105) by solving the generalized eigenvalue problem: CTLV = CVΛ, where Λ and V are the eigenvalue and eigenvector matrices, respectively; CTL(τ) = <X(t)XT(t + τ)> represents a time-lagged covariance matrix (τ being the lag-time); and C = <X(t)XT(t)> is the covariance matrix. The eigenvectors corresponding to the largest eigenvalues define the slowest reaction coordinates.
MARKOV STATE MODEL ANALYSIS TO STUDY KINETICS OF CONFORMATIONAL TRANSITIONS.
In MSM analysis, conformational transitions in a system from MD simulations are represented as a Markov chain (14). Assuming that the transitions between different conformations are sampled at long enough time intervals that each transition is Markovian, the procedure can yield information about long-timescale dynamics by combining the information from short-timescale trajectories. To construct MSMs, Morra et al. (68) used the MSMbuilder package (7). The three-dimensional space of the first three tIC eigenvectors was discretized into microstates and a transition probability matrix (TPM) of transitions between the microstates was built and symmetrized. Multiple TPMs were constructed for different time intervals between transitions (lag-times), and the relaxation timescales of the system were calculated as:
where is the lag-time, λi is the ith eigenvalue of the TPM, and τi is the relaxation timescale corresponding to the ith relaxation mode. The Markovian property of the TPM was established by verifying the independence of τi from .
Using the robust Perron cluster analysis (PCCA+) algorithm (25), the microstates on the tICA space were grouped into 12 macrostates based on their kinetic similarity. A flux matrix was then constructed, and the most probable pathways were identified using a Dijkstra graph theory algorithm (27) by finding the pathways with the highest flux between the starting and final macrostates.
Figure 3 shows the projection of all of the trajectory frames onto the two-dimensional space of the first two time-lagged independent component (tIC) vectors. The landscape is color coded according to the macrostate assignment. The tICA space can be broadly divided into two regions along the tIC1 direction. The region corresponding to tIC1 < 2 contains conformations of the system preceding lipid penetration, and the region with tIC1 > 2 includes states of the system in which the pathway is open and populated with lipids. More specifically, the initial conformation of the system belongs to macrostate 1 (Figure 3), which combines protein structures with an intact IC gate [E249(6.32) and K311(7.58) residues interact] and the TM6–TM7 interface closed to lipids. The system then transitions to macrostate 2 (Figure 3), in which the IC gate breaks, and the sidechain of juxtaposed residue Y306(7.53) on TM7 concomitantly undergoes a conformational switch from pointing toward TM6 to swaying away into the TM bundle toward TM2 (compare macrostates 1 and 2). This conformational change triggers hydration of the IC vestibule between TMs 6 and 7 (the IC ends of TM6 and TM7 move apart by as much as approximately 25 Å in the process), allowing a lipid to penetrate the pathway and gradually travel up toward the middle of the bilayer, where it engages with C264(6.47) and S298(7.45) residues (macrostate 6; see Figure 3). The sustained presence of the lipid headgroup at this site enables further widening of the pathway and penetration of additional lipids (macrostate 7). The flip is complete when the top lipid moves toward the EC end of the pathway, where it engages Y286(6.51) and K296(7.43) (macrostate 8). The requirement for a continuous hydrophilic conduit at the protein–membrane interface to facilitate transbilayer shuttling of lipids (Figure 2b) is generally similar to the scrambling mechanisms found in TMEM16 proteins. Indeed, for both opsin and TMEM16 scramblases, the results from MD data are consistent with the credit card mechanism (10, 47, 51, 52, 57, 90).
Figure 3.
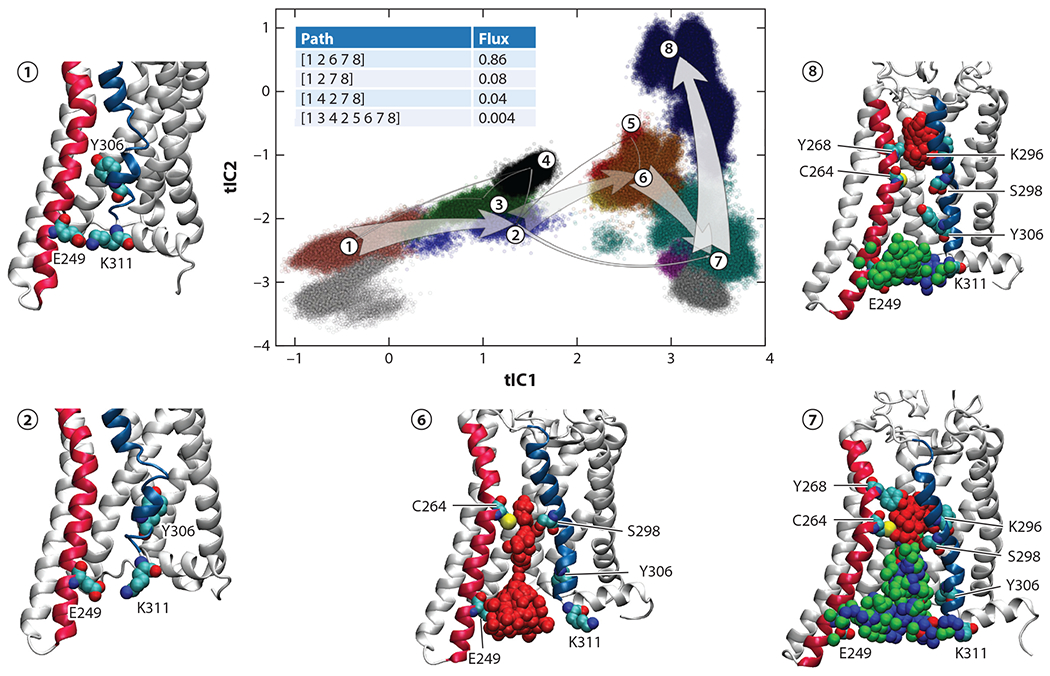
Pathways for opsin-mediated lipid translocation. The top four pathways for lipid translocation identified from TPT analysis are shown on the tIC1 versus tIC2 landscape divided into 12 macrostates (color coded) using the PCCA+ (see the sidebar titled Markov State Model Analysis to Study Kinetics of Conformational Transitions). The thickness of the arrows indicates the relative magnitude of the flux of the pathway. The total flux values for the top pathways are given in the table. The snapshots of the system show representative structures from the macrostates involved in the top pathway. In these snapshots, the relevant region of the protein is shown using the same representation as in Figure 2a; E249(6.32), K311(7.58), Y306(7.53), C264(6.47), S298(7.45), Y268(6.51), and K296(7.43) residues are drawn in space-fill representation [the superscript numbers indicate the Ballesteros-Weinstein number (6) of the residue]. The phosphorus atoms of the three penetrating lipids are shown on the translocation pathway using different colors (red, green, and blue). Figure adapted with permission from Reference 68. Abbreviations: PCCA+, robust Perron cluster analysis; tIC, time-lagged independent component; TPT, transition path theory.
The MSM analysis of the opsin trajectories identified the two slowest relaxation modes of the system, with timescales of 24.5 μs and 8.8 μs, respectively. The first captures the dynamics of the protein that enable initial insertion of the lipid (i.e., dynamics along the tIC1 vector). The second describes the conformational dynamics that accompany lipid translocation through the various sites along the pathway (i.e., dynamics along the tIC2 vector in the regime of tIC1 > 2). Although these timescales are consistent with the experimentally estimated frequency for opsin-mediated lipid scrambling (32, 63, 66, 88), the pathway as described above is incomplete because it does not consider the mechanism and timing of release of the flipped lipid into the EC leaflet (vide infra).
HOW IS THE SCRAMBLED LIPID RELEASED INTO THE EXTRACELLULAR LEAFLET?
To learn how the scrambled lipid is released into the extracellular leaflet, much longer simulations are needed, and these are currently ongoing (G. Khelashvili, A. Razavi, unpublished data). Preliminary analyses reveal that an exit pathway is formed in the region between the extracellular ends of TM5 and TM6 that, interestingly, coincides with the proposed pathways for retinal uptake by opsin (83) and cholesterol ingress to the ligand binding site in the adenosine A2A GPCR (34). Although the simulation data available thus far show only lipids transiting from the IC to the EC face of the membrane, the exit pathway described above may also provide a means for lipids in the EC leaflet to enter the groove.
MSM analyses of the simulations described above suggest that the exit step is rate-limiting in the scrambling process. Thus, while lipid translocation from the IC to the EC side unfolds on timescales of tens of microseconds, the release step is characterized by a timescale of >100 μs, or a scrambling frequency of less than 104 s−1. How can this estimate be reconciled with experimental estimates that opsin-mediated scrambling of NBD-phospholipids occurs at >105 s−1 (vide supra)? The results of Wang et al. (121) offer a possible explanation. These authors compared the standard assay (Figure 1a) with a scramblase assay in which natural phosphatidylinositol (PI), incorporated into reconstituted vesicles as [3H-inositol]PI ([3H]PI), was probed with PI-specific phospholipase C (PI-PLC). Addition of PI-PLC to protein-free liposomes caused rapid (t1/2 < 10 s) hydrolysis of [3H]PI located in the outer leaflet. As inner-leaflet [3H]PI is protected in these liposomes, the extent of hydrolysis plateaued at approximately 50%. The extent of hydrolysis was greater in opsin-proteoliposomes, consistent with scrambling of inner-leaflet PI to the outer leaflet, but occurred biphasically with rapid elimination of [3H]PI in the outer leaflet, followed by slower hydrolysis of scrambled [3H]PI. Indeed, with an endpoint readout taken within 10 min, it was necessary to reconstitute approximately 10 times more opsin to achieve the same extent of scrambling as in a parallel assay with NBD-labeled PI. Thus, NBD-PI may be more rapidly scrambled than natural PI. Alternatively, dithionite may be able to access NBD-PI that has been scrambled but not yet released into the extracellular leaflet, whereas the bulkier PI-PLC probe may require [3H]PI to be released from opsin before it can act. The latter proposal is consistent with lipid release being the slow step in scrambling. Wang et al. (121) reported similar results when assaying a fungal TMEM16 protein, suggesting that a slow release rate may be a general feature of scramblases.
CONFORMATIONAL CHANGES AND DYNAMICS OF SCRAMBLING VERSUS G PROTEIN–COUPLED RECEPTOR ACTIVATION
The TM6–TM7 interface plays a central role in opsin-mediated lipid scrambling, as described above. Conformational changes reposition TM6, and neighboring TM5, away from TM7 to reveal the lipid translocation pathway (Figure 4a,b). Notably, TMs 6 and 5 are also central to the conformational changes that accompany GPCR activation upon G protein binding (125). However, the repositioning of TMs 6 and 5 during scrambling is different from their repositioning during activation, as can be seen by comparing Figure 4c,d and Figure 4a,b. Thus, whereas the TM6 and TM5 helices move away laterally from the TM7/H8 segments for scrambling, the same helices move away from the TM bundle and into the lipid bilayer during activation.
Figure 4.
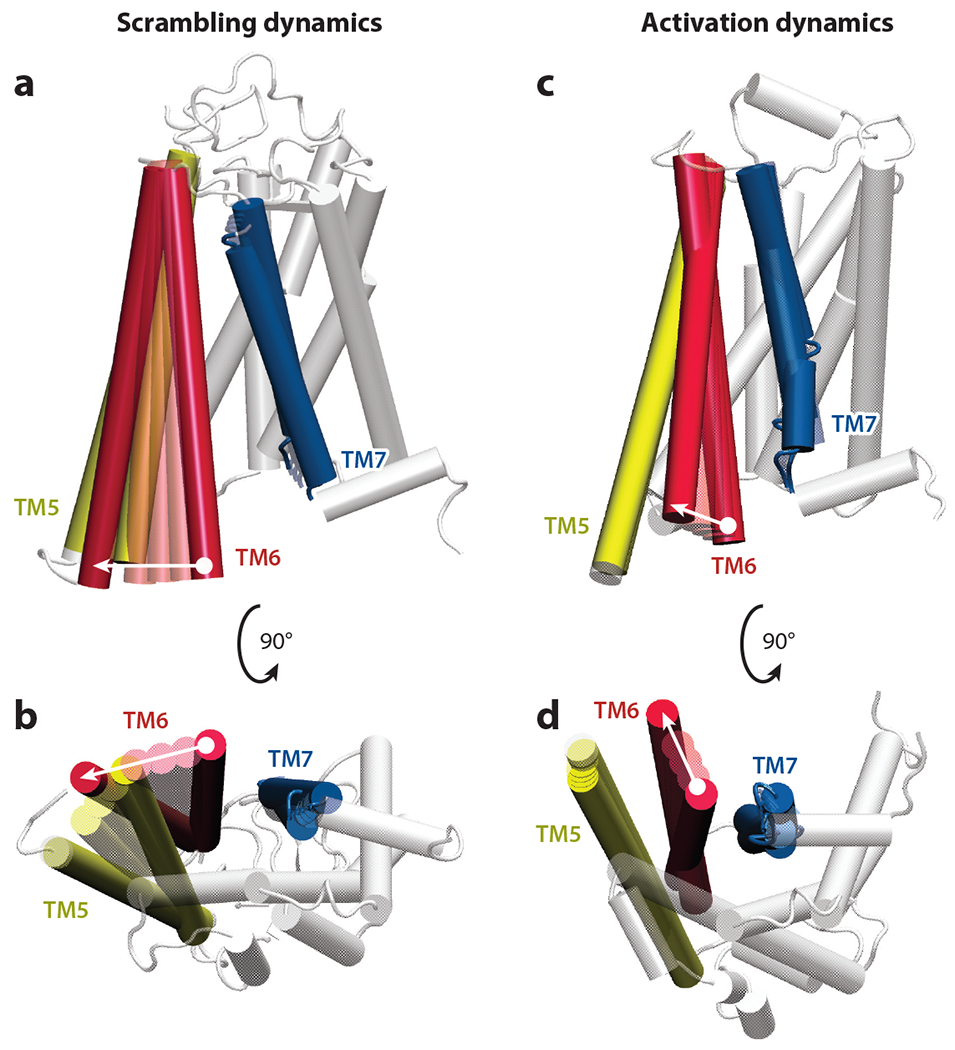
Comparison of conformational changes associated with G protein–coupled receptor (GPCR)-mediated lipid scrambling and GPCR activation dynamics. Membrane and cytoplasmic views of movements of transmembrane (TM) helices during (a,b) lipid scrambling and (c,d) receptor activation are shown. Panels a and b show linear morphing of positions of opsin TMs 5, 6, and 7 during lipid scrambling; panels c and d show linear morphing of positions of β2-adrenergic receptor TMs 5, 6, and 7 during the activation process [between inactive and G protein–bound structures; Protein Data Bank IDs 2RH1 (19) and 3SN6 (97), respectively]. In each panel, the direction of movement in TM6 during the corresponding process is shown by the white arrow. The rest of the protein structure (white) is taken from the target structure used for morphing. Morphing was performed with the VMD plugin.
A more detailed comparative analysis of structural changes in GPCRs during scrambling and activation further highlights differences in the dynamic modes underlying these two processes. By analyzing hundreds of structures of 45 Class A GPCRs, Zhou et al. (125) identified a common activation pathway and associated structural changes. The pathway comprises 34 residue pairs that, upon activation, rearrange in a specific manner; i.e., interactions are formed, broken, or switched. While the major changes were found for residue pairs in well-known activation structural elements such as the DRY (100) and the NPXXY (35) motifs, the structural rearrangements identified by Morra et al. (68) during scrambling, such as breaking of the E249(6.32)–K311(7.58) ionic interaction or widening of the waist of the TM6–TM7 interface [C264(6.47)–S298(7.45) distance increase], were not found, suggesting that the activation and scrambling processes may indeed be decoupled from each other.
It should be possible, in principle, to uncover the distinct conformational changes associated with activation and scrambling using techniques such as single-molecule Förster resonance energy transfer (smFRET) (94). In one such study, Gregorio et al. (33) examined distance changes between the IC ends of the TM4 and TM6 helices of β2AR during activation. By incorporating fluorophores at positions 4.40 and 6.28, they could monitor the ligand- and G protein–induced displacements of TM6 away from the protein bundle, as seen in Figure 4c,d. Remarkably, analysis of opsin trajectories from MD simulations (68) revealed that the average distance between these residues was the same in the closed and open groove conformations associated with scrambling. To use smFRET to track conformational transitions related to scrambling, it would be necessary to label the protein at sites other than those used by Gregorio et al. (33). For example, smFRET measurements between the IC ends of TM5 and TM7, or between the IC end of TM6 and the EC end of TM1, would be predicted to be insensitive to activation dynamics while revealing changes associated with scrambling.
To test experimentally if the conformational changes associated with activation and scrambling are in any way correlated, Goren et al. (32) compared the scramblase activity of opsin under different activation states. Thus, when opsin is covalently attached to the inverse agonist 11-cis retinal, the resulting rhodopsin molecule is conformationally silent, unable to couple to the G protein transducin (Gt). Consequently, unlike other GPCRs, inverse agonist-bound rhodopsin has essentially no basal signaling activity. Only upon light-induced isomerization of the retinal ligand does rhodopsin transition to the conformationally flexible Meta II state that can bind Gt to initiate signaling. Eventually, the all-trans retinal agonist dissociates, producing a flexible apo opsin molecule that resets to rhodopsin upon acquisition of 11-cis-retinal. Goren et al. (32) measured scramblase activity of opsin, rhodopsin, and a constitutively active Meta II mimic carrying the M257Y mutation (26, 36). For the latter construct, the apoprotein (M257Y-opsin) as well as the corresponding rhodopsin with bound all-trans retinal were considered. The scramblase activity of M257Y-opsin and M257Y-rhodopsin was indistinguishable from that of wild-type opsin. Rhodopsin was also scrambling competent, an observation that was subsequently reproduced by Shihoya et al. (108). Thus, within the time resolution of the assay, all experimentally accessible conformational variants of the opsin–rhodopsin system appear to be equivalently active as phospholipid scramblases upon reconstitution into vesicles. The latter observation suggests a key point: Rhodopsin can explore the conformational space necessary for scrambling even as it is silent with respect to activation.
IS THE SCRAMBLING MECHANISM CONSERVED ACROSS CLASS A G PROTEIN–COUPLED RECEPTORS?
Several residues implicated in the scrambling mechanism in opsin are conserved in Class A GPCRs. Indeed, the Y7.53 residue (Y306 in opsin), which undergoes a conformational switch to open the pathway at the IC side, is highly conserved across the family (89% among all human members; Figure 5a). There is also an interesting conservation pattern for the charged residues at positions 6.32 and 7.58, which comprise Site 1 (Figure 5b). In opsin, the ionic interaction between these residues (E249 and K311, respectively) breaks, enabling a lipid headgroup to enter the pathway. Position 6.32 is generally occupied by a charged residue (81%), often a basic residue (Lys or Arg) (76%; see Figure 5a). Position 7.58, situated at the entrance to the helix 8 segment, is generally occupied by a polar residue (76%), but only 55% of the sequences contain a charged residue at this position; in 37% of cases, 7.58 is a basic residue, and in 18% of cases, it is anionic. Thus, in some GPCRs, both the 6.32 and 7.58 positions are occupied by basic residues, which would be incompatible for ionic interactions. However, we find that the adjacent 7.59 position is an anionic residue in 24% of sequences (Figure 5a; e.g., see β2AR in Figure 5c), creating a dynamic mechanism for opening the lipid pathway on the IC side similar to that observed in opsin. It is noteworthy that, in those GPCRs in which a residue in the 6.32 position does not have a bonding partner in the TM7–helix 8 region (e.g., the opioid receptors; Figure 5a), the IC side of the TM6–TM7 interface could be intrinsically more prone to lipid headgroup insertion, thus facilitating opening of the pathway.
Figure 5.
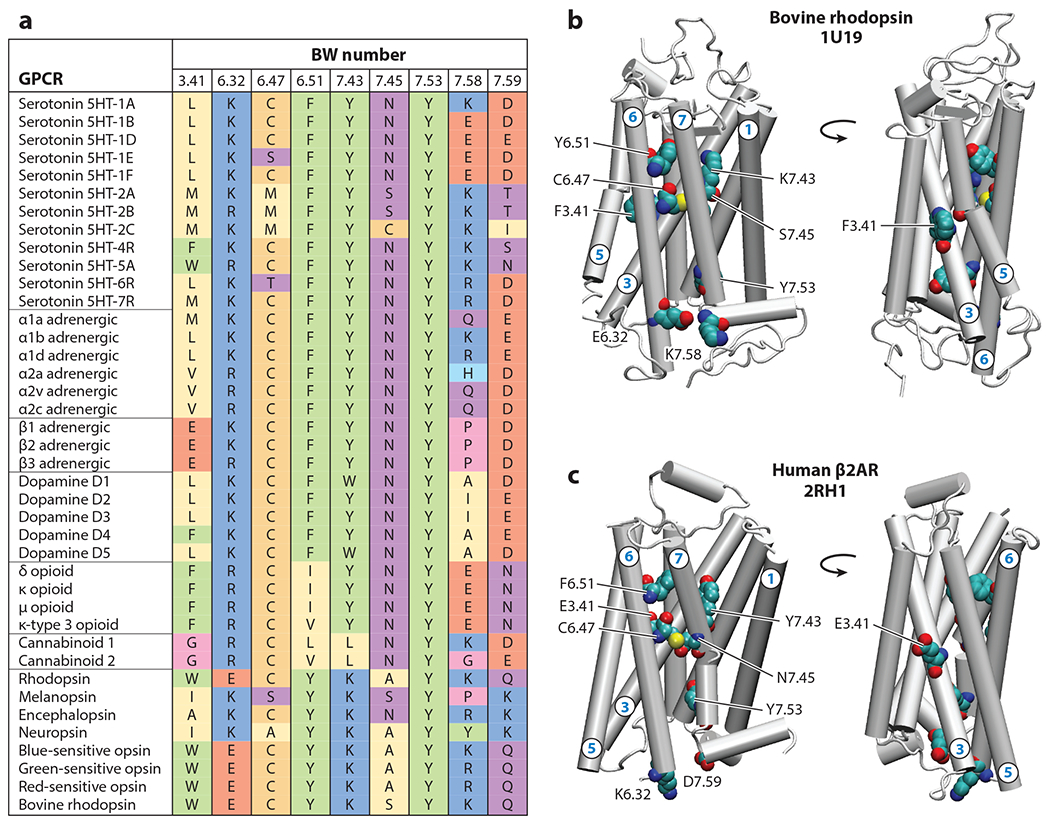
Structural motifs implicated in opsin-mediated lipid scrambling are conserved within Class A G protein–coupled receptors (GPCRs). (a) Sequence alignment at selected positions of major families of human Class A GPCRs. For comparison, the results for bovine rhodopsin are also included. The selected residue positions are given by the Ballesteros-Weinstein (BW) generic residue numbering for GPCRs (6). The amino acids are color coded to represent their type: Red indicates negatively charged, blue indicates positively charged, purple indicates polar, yellow indicates hydrophobic, and green indicates aromatic. Other colors represent special cases: Cys (gold), Pro (cyan), and Gly (pink). (b) Two views of the bovine rhodopsin structure [Protein Data Bank (PDB) ID 1U19] (78) illustrating positions of the key residues shown in the alignment table. Relevant helices are numbered. (c) The same views as in panel b but for human β2-adrenergic receptor (β2AR) structure (PDB ID 2RH1) (19).
The Site 2 region forms a constriction in the middle of the membrane that must open to allow passage of lipid headgroups. This constriction is created by sidechains at positions 6.47 and 7.45, which in opsin are occupied by Cys and Ser, respectively. Our conservation analysis (Figure 5a) reveals that 70% of sequences contain Cys at the 6.47 position, while in 90% of cases, 7.45 is a polar residue, with Asn being the most common (66%). Thus, the physicochemical properties of amino acids at Site 2 appear to be well-conserved among Class A GPCRs.
Lastly, the extracellularly located Site 3, which is crucial for coordinating the headgroup of the scrambled lipid, comprises residues 6.51 and 7.43, which in opsin are the polar residues Tyr and Lys, respectively. Conservation analysis shows that the 6.51 position has some preference for aromatic residues (65%), with Tyr being the most likely amino acid (35%), followed by Phe (30%). In nearly half of the sequences, the position is occupied by a residue capable of forming a hydrogen bond. Position 7.43 is occupied by Lys in all opsin members of the family, as it forms a critical Schiff base linkage with retinal. Overall, in 56% of cases, the position is occupied by a polar residue, with Tyr being the most prevalent (31%), suggesting that Site 3 in GPCRs is likely to consist of amino acids capable of forming hydrogen bonds with the lipid headgroup to coordinate its dynamics. Together, these analyses support the mechanistic role of the TM6–TM7 interface in lipid scrambling by all Class A GPCRs.
In support of these conclusions, previous MD simulations of the cannabinoid CB2 GPCR (42) showed that its lipid-like ligand sn-2-arachidonoylglycerol (2-AG) enters the receptor through an opening between TMs 6 and 7. Furthermore, as 2-AG moves toward its binding pocket, its polar headgroup is coordinated by residues at positions 6.47 and 6.51, which are important for the lipid translocation pathway in opsin.
While the importance of the TM6–TM7 interface in the lipid scrambling mechanism seems clear, intriguingly, our conservation analysis also identified an alternative region that, in some GPCRs, could potentially serve as a lipid translocation pathway. Thus, we found that the membrane-exposed position 3.41 in the middle of the TM3 helix, usually occupied by hydrophobic or noncharged polar residues, is occupied by anionic Glu in the adrenergic receptors (Figures 5a,c). Given its location and exposure to lipids, it is possible that this residue in the adrenergic GPCRs could attract water molecules and, with them, create the necessary hydrophilic environment for lipid translocation. Ongoing MD simulations of β1AR and β2AR (G. Khelashvili, unpublished data) show increased solvation around E3.41 and sustained presence of lipid head-groups near this residue. Enhanced conformational sampling of these systems in the future should address whether this position is important for lipid scrambling in this specific subfamily of GPCRs.
OUT-OF-THE-GROOVE SCRAMBLING
Opsin scrambles NBD analogs of all common glycerophospholipids, both zwitterionic (PC, PE) and anionic (PI, PS), as well as the ceramide-based phospholipid sphingomyelin (32, 66). Modeled as spheres, the headgroups of these lipids have a diameter of approximately 1 nm, and thus they are readily accommodated within the waist of the TM6–TM7 groove as it dilates to approximately 1 nm between C264(6.47) and S298(7.45) during lipid transit. Unexpectedly, opsin also scrambles large lipids, namely natural glycosylphosphatidylinositol (66) and synthetic PEG2000-PE (63), that have headgroups with diameters two to three times larger than those of common phospholipids. This phenomenon might be explained by membrane packing defects, or membrane thinning promoted by protein–lipid interactions at the groove, creating an energetically favorable environment to facilitate scrambling (63). Indeed, because of lipids penetrating toward Site 2 (Figure 2c), the membrane thins considerably in the vicinity of the TM6–TM7 helices, going from a thickness (phosphorus-to-phosphorus distance between lipids in the opposing leaflets) of approximately 3.5 nm to one of approximately 1.6 nm. Such a dramatic extent of thinning would bring the two bilayer leaflets within approximately 2 nm of each other, a length scale comparable to the dimensions of the large-headed lipids noted above. These lipids may therefore flip-flop between the leaflets with minimal interactions with the protein groove per se. Thus, the groove may not only provide a lipid transit pathway according to the credit card mechanism, but also enable an out-of-the-groove pathway. A fungal TMEM16 protein is also able to scramble phospholipids as large as PEG5000-PE with a headgroup diameter of approximately 4 nm, indicating that an out-of-the-groove mechanism may be quite general (63). Remarkably, despite its overall lack of substrate specificity, opsin cannot scramble the isoprenoid-based glycolipid Man5GlcNAc2-PP-dolichol (66), a key intermediate in the assembly of the oligosaccharide precursor necessary for protein N-glycosylation in the ER (102, 104). The structural features of this lipid—a large headgroup, diphosphate linkage, and long (approximately C100) isoprenoid tail—suggest that, while it would not be accommodated within the TM6–TM7 groove, even an out-of-the-groove mechanism may not sufficiently lower the energy barrier to enable it to be scrambled.
QUESTIONS AND FUTURE DIRECTIONS
The scramblase activity of Class A GPCRs, suggested originally in work on photoreceptor discs (38, 123) and evinced in vitro (30, 66), is unexpected and may be viewed as a moonlighting function of proteins otherwise well-known as signaling receptors. It would be interesting to learn whether GPCRs outside the Class A group also moonlight as scramblases. The MD simulations of opsin detailed above suggest a credible hypothesis about how scrambling is facilitated, but experimental validation is clearly necessary, and this is an important task for the future. In this section, we present and discuss two key open questions about the biology of GPCR-mediated lipid scrambling: What is the physiological significance of GPCR-mediated lipid scrambling, and how is it regulated in cells?
Function
There appears to be a clear-cut role for opsin’s scramblase function in the context of the retina. Retinal photoreceptor disc membranes possess two lipid pumps. One is an ABC transporter, ABCA4, that functions unconventionally as a flippase (rather than a floppase; see the Introduction) to transport both PE and N-retinylidene-PE (NRPE) to the cytoplasmic side of the disc (60, 95, 96). On reaching the cytoplasmic side, NRPE dissociates to release all-trans retinal, which enters the visual cycle, leaving behind PE. The inability to transport NRPE is a cause of age-related macular degeneration. The second disc lipid pump is Atp8a2, a P4-ATPase flippase specific for PS and PE (2, 21). By acting as flippases, both ABCA4 and Atp8a2 expand the cytoplasmic leaflet at the expense of the luminal leaflet, introducing bilayer stress. Ernst & Menon (30) suggested that opsin’s scramblase activity would be critical in continuously resetting the number density of lipids in the two leaflets, thereby preventing disc membrane distortion and enabling the key transport of NRPE to continue.
The biological significance of lipid scrambling by other Class A GPCRs is less clear and presents an intriguing challenge for the future. The predominant steady state pool of these proteins is found in the plasma membrane, with a portion—depending on activation state—in endocytic compartments. Mammalian cells express many different GPCRs; for example, HEK293 cells have at least 75 endogenous GPCRs (3), the cumulative copy number probably exceeding 105 receptors per cell. Yet these cells have an asymmetric plasma membrane with PS sequestered in the inner leaflet. Thus, the scramblase activity of plasma membrane–localized GPCRs may be suppressed, for example, by the high cholesterol content of these membranes (vide infra), making it possible for resident flippases to correct any loss of asymmetry. However, GPCRs are translated on membrane-bound ribosomes and integrated into the ER membrane prior to being exported to the plasma membrane. It is therefore possible that they provide or contribute significantly to the constitutive phospholipid scramblase activity that is necessary for ER membrane biogenesis.
Regulation
The apparent silencing of GPCR scramblase activity in the plasma membrane suggests that the unique characteristics of this membrane, such as high cholesterol content, greater thickness, predominance of phospholipids with saturated acyl chains, and presence of specific phosphoinositides, may be inhibitory, singly or in combination. Cholesterol likely plays a role, as it is known to interact with GPCRs (16, 19, 58, 61, 93, 103, 115, 124), and through its effects on lipid bilayer properties (e.g., lipid tail ordering, bending rigidity), it may suppress the protein conformational dynamics necessary for revealing the TM6–TM7 lipid pathway. Interestingly, whereas rhodopsin molecules in cholesterol-poor disc membranes can execute the light-induced conformational changes necessary for Gt binding and downstream signaling, the same molecules in the enveloping cholesterol-rich plasma membrane of the rod outer segment are inactive until cholesterol is eliminated by treating the isolated membranes with cholesterol oxidase (12, 13). These data suggest that cholesterol constrains the ability of rhodopsin to undergo conformational changes necessary for activation, and that it may similarly constrain TM movements needed for scrambling (30). Indeed, ongoing simulations (G. Khelashvili, A. Razavi, G. Morra, unpublished data) reveal specific modes of cholesterol localization near the TM6–TM7 interface that stabilize the closed conformation of the translocation pathway, thereby inhibiting lipid scrambling. These computational predictions await experimental tests.
ACKNOWLEDGMENTS
We thank Giulia Morra and Asghar Razavi for their contributions to the work described in this review and for many insightful discussions. We also thank Joshua Levitz for comments on the manuscript. This work was supported by National Institutes of Health grants R01 EY027969 and R21 EY028314. G.K. is also supported by the HRH Prince Alwaleed Bin Talal Bin Abdulaziz Alsaud Institute of Computational Biomedicine at Weill Cornell Medical College through gratefully acknowledged support from the 1923 Fund.
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
Glossary
- Flippase
out-to-in lipid pump that uses the energy released from the process of ATP hydrolysis to move lipids uphill from the exoplasmic side of the membrane to the cytoplasmic side; flippases are usually P4-ATPases
- Floppase
in-to-out lipid pump that uses the energy released from the process of ATP hydrolysis to move lipids uphill from the cytoplasmic side of the membrane to the exoplasmic side; floppases are usually ABC transporters
- Scramblase
a lipid channel that catalyzes bidirectional (ATP-independent) movement of lipids from one side of the membrane to the other; some scramblases are constitutively active, whereas others are regulated, for example, by Ca2+ ions
- PS
phosphatidylserine
- PE
phosphatidylethanolamine
- PC
phosphatidylcholine
- Proteoliposomes
large unilamellar vesicles reconstituted with membrane proteins
- DDM
n-dodecyl-β-D-maltoside; a nonionic detergent with 0.1% (w/v) = 1.96 mM
- TM
transmembrane
- MD
molecular dynamics
- AEMD
adaptive ensemble MD
- MSM
Markov state model
- tICA
time-lagged independent component analysis
- TPT
transition path theory
- PI-PLC
phosphatidylinositol-specific phospholipase C
Footnotes
Even when sufficient opsin is used to populate all vesicles, the extent of fluorescence reduction observed was only approximately 80–85% (in Figure 1b, the trace corresponding to the highest value of PPR), indicating that approximately 30-40% of the vesicles are refractory to reconstitution. The reasons for this are not clear, but the phenomenon of incomplete reconstitution has been widely noted.
In the Ballesteros-Weinstein numbering scheme for GPCR residues, the most conserved residue in each TM is assigned the number 50, and then a pair of numbers (A1.A2) is used to describe amino acid residues in TMs. A1 is the TM number, and A2 denotes the position of the amino acid relative to the most conserved residue in the TM, with numbers decreasing toward the N terminus and increasing toward the C terminus.
LITERATURE CITED
- 1.Alvadia C, Lim NK, Clerico Mosina V, Oostergetel GT, Dutzler R, Paulino C. 2019. Cryo-EM structures and functional characterization of the murine lipid scramblase TMEM16F. eLife 8:e44365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andersen JP, Vestergaard AL, Mikkelsen SA, Mogensen LS, Chalat M, Molday RS. 2016. P4-ATPases as phospholipid flippases: structure, function, and enigmas. Front. Physiol 7:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atwood BK, Lopez J, Wager-Miller J, Mackie K, Straiker A. 2011. Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC Genom. 12:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Audet M, Stevens RC. 2019. Emerging structural biology of lipid G protein-coupled receptors. Protein Sci. 28:292–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balasubramanian V, Jensen T, Turilli M, Kasson P, Shirts M, Jha S. 2018. Adaptive ensemble biomolecular applications at scale. arXiv 1804.04736 [Google Scholar]
- 6.Ballesteros JA, Weinstein H. 1995. Integrated methods for the construction of three dimensional models and computational probing of structure-function relations in G-protein coupled receptors. Methods Neurosci. 25:366–428 [Google Scholar]
- 7.Beauchamp KA, Bowman GR, Lane TJ, Maibaum L, Haque IS, Pande VS. 2011. MSMBuilder2: modeling conformational dynamics at the picosecond to millisecond scale. J. Chem. Theory Comput 7:3412–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beauchamp KA, McGibbon R, Lin YS, Pande VS. 2012. Simple few-state models reveal hidden complexity in protein folding. PNAS 109:17807–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berezhkovskii A, Hummer G, Szabo A. 2009. Reactive flux and folding pathways in network models of coarse-grained protein dynamics. J. Chem. Phys 130:205102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bethel NP, Grabe M. 2016. Atomistic insight into lipid translocation by a TMEM16 scramblase. PNAS 113:14049–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bevers EM, Williamson PL. 2016. Getting to the outer leaflet: physiology of phosphatidylserine exposure at the plasma membrane. Physiol. Rev 96:605–45 [DOI] [PubMed] [Google Scholar]
- 12.Boesze-Battaglia K, Albert AD. 1990. Cholesterol modulation of photoreceptor function in bovine retinal rod outer segments. J. Biol. Chem 265:20727–30 [PubMed] [Google Scholar]
- 13.Boesze-Battaglia K, Fliesler SJ, Albert AD. 1990. Relationship of cholesterol content to spatial distribution and age of disc membranes in retinal rod outer segments. J. Biol. Chem 265:18867–70 [PMC free article] [PubMed] [Google Scholar]
- 14.Bowman GR, Pande VS,Noe F. 2014. An Introduction to Markov State Models and Their Application to Long Timescale Molecular Simulation. Berlin: Springer [Google Scholar]
- 15.Brunner JD, Lim NK, Schenck S, Duerst A, Dutzler R. 2014. X-ray structure of a calcium-activated TMEM16 lipid scramblase. Nature 516:207–12 [DOI] [PubMed] [Google Scholar]
- 16.Burger K, Gimpl G, Fahrenholz F. 2000. Regulation of receptor function by cholesterol. Cell Mol. Life Sci 57:1577–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bushell SR, Pike ACW, Falzone ME, Rorsman NJG, Ta CM, et al. 2019. The structural basis of lipid scrambling and inactivation in the endoplasmic reticulum scramblase TMEM16K. Nat. Commun 10:3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang QL, Gummadi SN, Menon AK. 2004. Chemical modification identifies two populations of glycerophospholipid flippase in rat liver ER. Biochemistry 43:10710–18 [DOI] [PubMed] [Google Scholar]
- 19.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, et al. 2007. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318:1258–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cliff L, Chadda R, Robertson JL. 2020. Occupancy distributions of membrane proteins in heterogeneous liposome populations. Biochim. Biophys. Acta Biomembr 1862:183033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coleman JA, Kwok MC, Molday RS. 2009. Localization, purification, and functional reconstitution of the P4-ATPase Atp8a2, a phosphatidylserine flippase in photoreceptor disc membranes. J. Biol. Chem 284:32670–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Comar WD, Schubert SM, Jastrzebska B, Palczewski K, Smith AW. 2014. Time-resolved fluorescence spectroscopy measures clustering and mobility of a G protein-coupled receptor opsin in live cell membranes. J. Am. Chem. Soc 136:8342–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corradi V, Sejdiu BI, Mesa-Galloso H, Abdizadeh H, Noskov SY, et al. 2019. Emerging diversity in lipid-protein interactions. Chem. Rev 119:5775–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daleke DL, Huestis WH. 1985. Incorporation and translocation of aminophospholipids in human erythrocytes. Biochemistry 24:5406–16 [DOI] [PubMed] [Google Scholar]
- 25.Deuflhard P, Weber M. 2005. Robust Perron cluster analysis in conformation dynamics. Linear Algebra Appl. 398:161–84 [Google Scholar]
- 26.Deupi X, Edwards P, Singhal A, Nickle B, Oprian D, et al. 2012. Stabilized G protein binding site in the structure of constitutively active metarhodopsin-II. PNAS 109:119–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dijkstra EW. 1959. A note on two problems in connexion with graphs. Numer. Math 1:269–71 [Google Scholar]
- 28.Erlandson SC,McMahon C, Kruse AC. 2018. Structural basis for G protein-coupled receptor signaling. Annu. Rev. Biophys 47:1–18 [DOI] [PubMed] [Google Scholar]
- 29.Ernst OP, Gramse V, Kolbe M, Hofmann KP, Heck M. 2007. Monomeric G protein-coupled receptor rhodopsin in solution activates its G protein transducin at the diffusion limit. PNAS 104:10859–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ernst OP, Menon AK. 2015. Phospholipid scrambling by rhodopsin. Photochem. Photobiol. Sci 14:1922–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghanbarpour A, Valverde DP, Melia TJ, Reinisch KM. 2021.A model for a partnership of lipid transfer proteins and scramblases in membrane expansion and organelle biogenesis. PNAS 118:e2101562118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goren MA, Morizumi T, Menon I, Joseph JS, Dittman JS, et al. 2014. Constitutive phospholipid scramblase activity of a G protein-coupled receptor. Nat. Commun 5:5115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gregorio GG, Masureel M, Hilger D, Terry DS, Juette M, et al. 2017. Single-molecule analysis of ligand efficacy in β2-AR–G-protein activation. Nature 547:68–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guixa-Gonzalez R, Albasanz JL, Rodriguez-Espigares I, Pastor M, Sanz F, et al. 2017. Membrane cholesterol access into a G-protein-coupled receptor. Nat. Commun 8:14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han DS, Wang SX, Weinstein H. 2008. Active state-like conformational elements in the beta2-AR and a photoactivated intermediate of rhodopsin identified by dynamic properties of GPCRs. Biochemistry 47:7317–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han M, Smith SO, Sakmar TP. 1998. Constitutive activation of opsin by mutation of methionine 257 on transmembrane helix 6. Biochemistry 37:8253–61 [DOI] [PubMed] [Google Scholar]
- 37.Hankins HM, Baldridge RD, Xu P, Graham TR. 2015. Role of flippases, scramblases and transfer proteins in phosphatidylserine subcellular distribution. Traffic 16:35–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hessel E, Herrmann A, Muller P, Schnetkamp PP, Hofmann KP 2000. The transbilayer distribution of phospholipids in disc membranes is a dynamic equilibrium evidence for rapid flip and flop movement. Eur. J. Biochem 267:1473–83 [DOI] [PubMed] [Google Scholar]
- 39.Hessel E, Muller P, Herrmann A, Hofmann KP. 2001. Light-induced reorganization of phospholipids in rod disc membranes. J. Biol. Chem 276:2538–43 [DOI] [PubMed] [Google Scholar]
- 40.Hilger D, Masureel M, Kobilka BK. 2018. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol 25:4–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang D, Xu B, Liu L, Wu L, Zhu Y, et al. 2021. TMEM41B acts as an ER scramblase required for lipoprotein biogenesis and lipid homeostasis. Cell Metab. 33:1655–70.e8 [DOI] [PubMed] [Google Scholar]
- 42.Hurst DP, Grossfield A, Lynch DL, Feller S, Romo TD, et al. 2010. A lipid pathway for ligand binding is necessary for a cannabinoid G protein-coupled receptor. J. Biol. Chem 285:17954–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Janssen MJ, Koorengevel MC, de Kruijff B, de Kroon AI. 1999. Transbilayer movement of phosphatidylcholine in the mitochondrial outer membrane of Saccharomyces cerevisiae is rapid and bidirectional. Biochim. Biophys. Acta 1421:64–76 [DOI] [PubMed] [Google Scholar]
- 44.Jastrzebska B, Chen Y, Orban T, Jin H, Hofmann L, Palczewski K. 2015. Disruption of rhodopsin dimerization with synthetic peptides targeting an interaction interface. J. Biol. Chem 290:25728–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jastrzebska B, Maeda T, Zhu L, Fotiadis D, Filipek S, et al. 2004. Functional characterization of rhodopsin monomers and dimers in detergents. J. Biol. Chem 279:54663–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang T, Wen PC, Trebesch N, Zhao Z, Pant S, et al. 2020. Computational dissection of membrane transport at a microscopic level. Trends Biochem. Sci 45:202–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang T, Yu K, Hartzell HC, Tajkhorshid E. 2017. Lipids and ions traverse the membrane by the same physical pathway in the nhTMEM16 scramblase. eLife 6:e28671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kalienkova V, Clerico Mosina V, Paulino C. 2021. The groovy TMEM16 family:molecular mechanisms of lipid scrambling and ion conduction. J. Mol. Biol 433:166941. [DOI] [PubMed] [Google Scholar]
- 49.Kasson PM, Jha S. 2018. Adaptive ensemble simulations of biomolecules. Curr. Opin. Struct. Biol 52:87–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Katritch V, Cherezov V, Stevens RC. 2013. Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol 53:531–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khelashvili G, Cheng X, Falzone ME, Doktorova M, Accardi A, Weinstein H. 2020. Membrane lipids are both the substrates and a mechanistically responsive environment of TMEM16 scramblase proteins. J. Comput. Chem 41:538–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khelashvili G, Falzone ME, Cheng X, Lee BC, Accardi A, Weinstein H. 2019. Dynamic modulation of the lipid translocation groove generates a conductive ion channel in Ca2+-bound nhTMEM16. Nat. Commun 10:4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Khelashvili G, Pillai AN, Lee J, Pandey K, Payne AM, et al. 2021. Unusual mode of dimerization of retinitis pigmentosa-associated F220C rhodopsin. Sci. Rep 11:10536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kobayashi T, Menon AK. 2018. Transbilayer lipid asymmetry. Curr. Biol 28:R386–91 [DOI] [PubMed] [Google Scholar]
- 55.Kohlhoff KJ, Shukla D, Lawrenz M, Bowman GR, Konerding DE, et al. 2014. Cloud-based simulations on Google Exacycle reveal ligand modulation of GPCR activation pathways. Nat. Chem 6:15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kornberg RD, McConnell HM. 1971.Inside-outside transitions of phospholipids in vesicle membranes. Biochemistry 10:1111–20 [DOI] [PubMed] [Google Scholar]
- 57.Lee BC, Khelashvili G, Falzone M, Menon AK, Weinstein H, Accardi A. 2018. Gating mechanism of the extracellular entry to the lipid pathway in a TMEM16 scramblase. Nat. Commun 9:3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee JY, Lyman E. 2012. Predictions for cholesterol interaction sites on the A2A adenosine receptor. J. Am. Chem. Soc 134:16512–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li YE, Wang Y, Du X, Zhang T, Mak HY, et al. 2021. TMEM41B and VMP1 are scramblases and regulate the distribution of cholesterol and phosphatidylserine. J. Cell. Biol 220:e202103105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu F, Lee J, Chen J. 2021. Molecular structures of the eukaryotic retinal importer ABCA4. eLife 10:e63524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu W, Chun E, Thompson AA, Chubukov P, Xu F, et al. 2012. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337:232–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lyons JA, Timcenko M, Dieudonne T, Lenoir G, Nissen P. 2020. P4-ATPases: how an old dog learnt new tricks—structure and mechanism of lipid flippases. Curr. Opin. Struct. Biol 63:65–73 [DOI] [PubMed] [Google Scholar]
- 63.Malvezzi M, Andra KK, Pandey K, Lee BC, Falzone ME, et al. 2018. Out-of-the-groove transport of lipids by TMEM16 and GPCR scramblases. PNAS 115:E7033–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Malvezzi M, Chalat M, Janjusevic R, Picollo A, Terashima H, et al. 2013. Ca2+-dependent phospholipid scrambling by a reconstituted TMEM16 ion channel. Nat. Commun 4:2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marrink SJ, Corradi V, Souza PCT, Ingolfsson HI, Tieleman DP, Sansom MSP. 2019. Computational modeling of realistic cell membranes. Chem. Rev 119:6184–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Menon I, Huber T, Sanyal S, Banerjee S, Barre P, et al. 2011. Opsin is a phospholipid flippase. Curr. Biol 21:149–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Molgedey L, Schuster HG. 1994. Separation of a mixture of independent signals using time delayed correlations. Phys. Rev. Lett 72:3634–37 [DOI] [PubMed] [Google Scholar]
- 68.Morra G, Razavi AM, Pandey K, Weinstein H, Menon AK, Khelashvili G. 2018. Mechanisms of lipid scrambling by the G protein-coupled receptor opsin. Structure 26:356–67.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Muller MP, Jiang T, Sun C, Lihan M, Pant S, et al. 2019. Characterization of lipid-protein interactions and lipid-mediated modulation of membrane protein function through molecular simulation. Chem. Rev 119:6086–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nagata S, Sakuragi T, Segawa K. 2020. Flippase and scramblase for phosphatidylserine exposure. Curr Opin. Immunol 62:31–38 [DOI] [PubMed] [Google Scholar]
- 71.Nakao H, Ikeda K, Ishihama Y, Nakano M. 2016. Membrane-spanning sequences in endoplasmic reticulum proteins promote phospholipid flip-flop. Biophys. J 110:2689–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nakao H, Sugimoto Y, Ikeda K, Saito H, Nakano M. 2020. Structural feature of lipid scrambling model transmembrane peptides: same-side positioning of hydrophilic residues and their deeper position. J. Phys. Chem. Lett 11:1662–67 [DOI] [PubMed] [Google Scholar]
- 73.Naritomi Y, Fuchigami S. 2011. Slow dynamics in protein fluctuations revealed by time-structure based independent component analysis: the case of domain motions. J. Chem. Phys 134:065101. [DOI] [PubMed] [Google Scholar]
- 74.Noda NN. 2021. Atg2 and Atg9: intermembrane and interleaflet lipid transporters driving autophagy. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1866:158956. [DOI] [PubMed] [Google Scholar]
- 75.Noe F, Clementi C. 2017. Collective variables for the study of long-time kinetics from molecular trajectories: theory and methods. Curr. Opin. Struct. Biol 43:141–47 [DOI] [PubMed] [Google Scholar]
- 76.Noe F, Fischer S. 2008. Transition networks for modeling the kinetics of conformational change in macromolecules. Curr. Opin. Struct. Biol 18:154–62 [DOI] [PubMed] [Google Scholar]
- 77.Odoemelam CS, Percival B, Wallis H, Chang M-W, Ahmad Z, et al. 2020. G-protein coupled receptors: structure and function in drug discovery. RSC Adv. 10:36337–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. 2004. The retinal conformation and its environment in rhodopsin in light of a new 2.2 Å crystal structure. J. Mol. Biol 342:571–83 [DOI] [PubMed] [Google Scholar]
- 79.Padayatti PS, Lee SC, Stanfield RL, Wen PC, Tajkhorshid E, et al. 2019. Structural insights into the lipid A transport pathway in MsbA. Structure 27:1114–23.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pande VS, Beauchamp K, Bowman GR. 2010. Everything you wanted to know about Markov state models but were afraid to ask. Methods 52:99–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pandey K, Ploier B, Goren MA, Levitz J, Khelashvili G, Menon AK. 2017. An engineered opsin monomer scrambles phospholipids. Sci. Rep 7:16741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Park JH, Morizumi T, Li Y, Hong JE, Pai EF, et al. 2013. Opsin, a structural model for olfactory receptors? Angew. Chem. Int. Ed. Engl 52:11021–24 [DOI] [PubMed] [Google Scholar]
- 83.Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. 2008. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature 454:183–87 [DOI] [PubMed] [Google Scholar]
- 84.Perez A, Martinez-Rosell G, De Fabritiis G. 2018. Simulations meet machine learning in structural biology. Curr. Opin. Struct. Biol 49:139–44 [DOI] [PubMed] [Google Scholar]
- 85.Perez-Hernandez G, Paul F, Giorgino T, De Fabritiis G, Noe F. 2013. Identification of slow molecular order parameters for Markov model construction. J. Chem. Phys 139:015102. [DOI] [PubMed] [Google Scholar]
- 86.Pinamonti G, Zhao J, Condon DE, Paul F, Noe F, et al. 2017. Predicting the kinetics of RNA oligonucleotides using Markov state models. J. Chem. Theory Comput 13:926–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Plattner N, Doerr S, De Fabritiis G, Noe F. 2017. Complete protein–protein association kinetics in atomic detail revealed by molecular dynamics simulations and Markov modelling. Nat. Chem 9:1005–11 [DOI] [PubMed] [Google Scholar]
- 88.Ploier B, Caro LN, Morizumi T, Pandey K, Pearring JN, et al. 2016. Dimerization deficiency of enigmatic retinitis pigmentosa-linked rhodopsin mutants. Nat. Commun 7:12832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ploier B, Menon AK. 2016. A fluorescence-based assay of phospholipid scramblase activity. J. Vis. Exp 20:54635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pomorski T, Menon AK. 2006. Lipid flippases and their biological functions. CellMol. Life Sci 63:2908–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pomorski TG, Menon AK. 2016. Lipid somersaults: uncovering the mechanisms of protein-mediated lipid flipping. Prog. Lipid Res 64:69–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Prinz JH, Wu H, Sarich M, Keller B, Senne M, et al. 2011. Markov models of molecular kinetics: generation and validation. J. Chem. Phys 134:174105. [DOI] [PubMed] [Google Scholar]
- 93.Pucadyil TJ, Chattopadhyay A. 2006. Role of cholesterol in the function and organization of G-protein coupled receptors. Prog. Lipid Res 45:295–333 [DOI] [PubMed] [Google Scholar]
- 94.Quast RB, Margeat E. 2019. StudyingGPCR conformational dynamics by single molecule fluorescence. Mol. Cell Endocrinol 493:110469. [DOI] [PubMed] [Google Scholar]
- 95.Quazi F, Lenevich S, Molday RS. 2012. ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat. Commun 3:925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Quazi F, Molday RS. 2014. ATP-binding cassette transporter ABCA4 and chemical isomerization protect photoreceptor cells from the toxic accumulation of excess 11-cis-retinal. PNAS 111:5024–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, et al. 2011. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477:549–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Razavi AM, Khelashvili G, Weinstein H. 2017. A Markov state-based quantitative kinetic model of sodium release from the dopamine transporter. Sci. Rep 7:40076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Razavi AM, Khelashvili G, Weinstein H. 2018. How structural elements evolving from bacterial to human SLC6 transporters enabled new functional properties. BMC Biol 16:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Roth CB, Hanson MA, Stevens RC. 2008. Stabilization of the human beta2-adrenergic receptor TM4-TM3-TM5 helix interface by mutagenesis of Glu1223.41, a critical residue in GPCR structure. J. Mol. Biol 376:1305–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rothman JE, Tsai DK, Dawidowicz EA, Lenard J. 1976. Transbilayer phospholipid asymmetry and its maintenance in the membrane of influenza virus. Biochemistry 15:2361–70 [DOI] [PubMed] [Google Scholar]
- 102.Sanyal S, Menon AK. 2009. Flipping lipids: why an’ what’s the reason for? ACS Chem. Biol 4:895–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sarkar P, Chattopadhyay A. 2020. Cholesterol interaction motifs in G protein-coupled receptors: slippery hot spots? Wiley Interdiscip. Rev. Syst. Biol. Med 12:e1481. [DOI] [PubMed] [Google Scholar]
- 104.Schenk B, Fernandez F, Waechter CJ. 2001. The ins(ide) and out(side) of dolichyl phosphate biosynthesis and recycling in the endoplasmic reticulum. Glycobiology 11:61R–70R [DOI] [PubMed] [Google Scholar]
- 105.Schwantes CR, Pande VS. 2013. Improvements in Markov state model construction reveal many non-native interactions in the folding of NTL9. J. Chem. Theory Comput 9:2000–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Seigneuret M, Devaux PF. 1984. ATP-dependent asymmetric distribution of spin-labeled phospholipids in the erythrocyte membrane: relation to shape changes. PNAS 81:3751–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sheetz MP, Singer SJ. 1974. Biological membranes as bilayer couples: a molecular mechanism of drug-erythrocyte interactions. PNAS 71:4457–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shihoya W, Inoue K, Singh M, Konno M, Hososhima S, et al. 2019. Crystal structure of heliorhodopsin. Nature 574:132–36 [DOI] [PubMed] [Google Scholar]
- 109.Shin HW, Takatsu H. 2020. Phosphatidylserine exposure in living cells. Crit. Rev. Biochem. Mol. Biol 55:166–78 [DOI] [PubMed] [Google Scholar]
- 110.Shukla D, Meng Y, Roux B, Pande VS. 2014. Activation pathway of Src kinase reveals intermediate states as targets for drug design. Nat. Commun 5:3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shukla S, Baumgart T. 2021. Enzymatic trans-bilayer lipid transport: mechanisms, efficiencies, slippage, and membrane curvature. Biochim. Biophys. Acta Biomembr 1863:183534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Straub MS, Alvadia C, Sawicka M, Dutzler R. 2021. Cryo-EM structures of the caspase-activated protein XKR9 involved in apoptotic lipid scrambling. eLife 10:e69800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Suzuki J, Denning DP, Imanishi E, Horvitz HR, Nagata S. 2013. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 341:403–6 [DOI] [PubMed] [Google Scholar]
- 114.Suzuki J, Umeda M, Sims PJ, Nagata S. 2010. Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468:834–38 [DOI] [PubMed] [Google Scholar]
- 115.Taghon GJ, Rowe JB, Kapolka NJ, Isom DG. 2021. Predictable cholesterol binding sites in GPCRs lack consensus motifs. Structure 29:499–506.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tang X, Halleck MS, Schlegel RA, Williamson P. 1996.A subfamily of P-type ATPases with aminophospholipid transporting activity. Science 272:1495–97 [DOI] [PubMed] [Google Scholar]
- 117.Vehring S, Pakkiri L, Schroer A, Alder-Baerens N, Herrmann A, et al. 2007. Flip-flop of fluorescently labeled phospholipids in proteoliposomes reconstituted with Saccharomyces cerevisiae microsomal proteins. Eukaryot. Cell 6:1625–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Verchere A, Cowton A, Jenni A, Rauch M, Haner R, et al. 2021. Complexity of the eukaryotic dolichol-linked oligosaccharide scramblase suggested by activity correlation profiling mass spectrometry. Sci. Rep 11:1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Verchere A, Ou WL, Ploier B,Morizumi T, Goren MA, et al. 2017. Light-independent phospholipid scramblase activity of bacteriorhodopsin from Halobacterium salinarum. Sci. Rep 7:9522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Voelz VA, Bowman GR, Beauchamp K, Pande VS. 2010. Molecular simulation of ab initio protein folding for a millisecond folder NTL9(1–39). J. Am. Chem. Soc 132:1526–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang L, Iwasaki Y, Andra KK, Pandey K, Menon AK, Butikofer P. 2018. Scrambling of natural and fluorescently tagged phosphatidylinositol by reconstituted G protein-coupled receptor and TMEM16 scramblases. J. Biol. Chem 293:18318–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wieczorek M, Abualrous ET, Sticht J, Alvaro-Benito M, Stolzenberg S, et al. 2017. Major histocompatibility complex (MHC) class I and MHC class II proteins: conformational plasticity in antigen presentation. Front. Immunol 8:292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wu G, Hubbell WL. 1993. Phospholipid asymmetry and transmembrane diffusion in photoreceptor disc membranes. Biochemistry 32:879–88 [DOI] [PubMed] [Google Scholar]
- 124.Yeliseev A, Iyer MR, Joseph TT, Coffey NJ, Cinar R, et al. 2021. Cholesterol as a modulator of cannabinoid receptor CB2 signaling. Sci. Rep 11:3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhou Q, Yang D, Wu M, Guo Y, Guo W, et al. 2019. Common activation mechanism of class A GPCRs. eLife 8:e50279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhou Z, White KA, Polissi A, Georgopoulos C, Raetz CR. 1998. Function of Escherichia coli MsbA, an essential ABC family transporter, in lipid A and phospholipid biosynthesis. J. Biol. Chem 273:12466–75 [DOI] [PubMed] [Google Scholar]
- 127.Zuckerman DM, Chong LT. 2017. Weighted ensemble simulation: review of methodology, applications, and software. Annu. Rev. Biophys 46:43–57 [DOI] [PMC free article] [PubMed] [Google Scholar]