

Abstract
Total internal reflection fluorescence (TIRF) microscopy (TIRFM) is an elegant optical technique that provides for the excitation of fluorophores in an extremely thin axial region (“optical section”). The method is based on the principle that when excitation light is totally internally reflected in a transparent solid (e.g., coverglass) at its interface with liquid, an electromagnetic field, called the evanescent wave, is generated in the liquid at the solid-liquid interface and is the same frequency as the excitation light. Since the intensity of the evanescent wave exponentially decays with distance from the surface of the solid, only fluorescent molecules within a few hundred nanometers of the solid are efficiently excited. This overview will review the history, optical theory, and hardware configurations used in TIRFM. In addition, it will provide experimental details and methodological considerations for studying receptors at the plasma membrane in neurons.
Keywords: axial resolution, fluorescence microscopy, live cell imaging, receptor trafficking, neurons
INTRODUCTION
TIRFM is a powerful technique for selectively imaging fluorescent molecules (e.g., GFP, membrane dyes, fluorochromes attached to antibodies, etc.), in an aqueous environment, that are very near a solid substance with a high refractive index (e.g., coverglass). Depending on the excitation wavelength and objective numerical aperture, the thickness of the excitation depth, which is called the evanescent field, can be <100 nm from the solid surface. In comparison, the thickness of a confocal image section is ~500 nm. The advantage of such a small illumination volume is three fold: (1) the background is greater than 2000-fold lower than when imaging by normal epifluorescence microscopy (Funatsu et al., 1995), which results in a high signal-to-background ratio; (2) there is virtually no out-of-focus fluorescence collected; (3) cells are exposed to a significantly smaller amount of light (Fig. 1). The following are just a few of the many applications of TIRFM in cellular microscopy.
Figure 1.
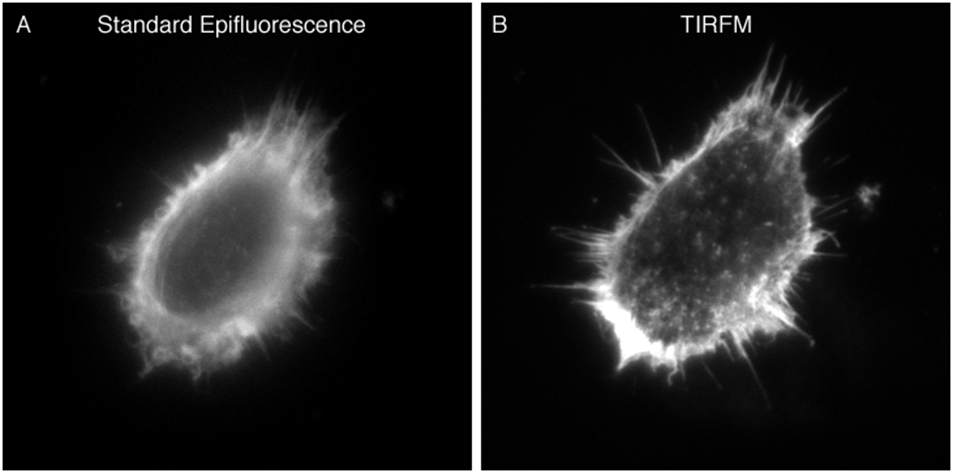
Information revealed by standard epifluoresence (A) and TIRF microscopy (B). HeLa cells were cultured on 18-mm round 1.78 RI coverslips. Post fixation with 4% paraformaldehyde, rhodamine-conjugated phalloidin was used to visualize F-actin. The cell was imaged in HBSS using a 100× 1.65 NA objective and 1.78 RI immersion liquid. Both images were taken at the same plane with the same exposure settings.
Receptors at the PM
TIRFM can be used to measure the kinetics of receptor endocytosis in response to ligand binding (Hellen and Axelrod, 1991; Rao et al., 2022; Riven et al., 2006; Tabor et al., 2016), receptor insertion into the plasma membrane (Yudowski et al., 2006), receptor channel opening and closing (Toglia et al., 2017; Yao and Qin, 2009), receptor clustering (Drenan et al., 2008; Erdelyi et al., 2014; Salavessa et al., 2021; Sungkaworn et al., 2014), the lateral movement of receptors (Fowler et al., 2007; Salavessa et al., 2021), and stochiometry of receptors (Salavessa and Sauvonnet, 2021). For these types of studies, TIRFM is sometimes combined with other advanced microscopy techniques (e.g., fluorescence recovery after photobleaching; see (Hellen and Axelrod, 1991; Riven et al., 2006)). Receptors at the PM can be visualized using fluorescently tagged ligands, antibodies, or small molecules. Alternatively, the receptor of interest can be fused to a fluorescent protein (e.g., GFP).
Exocytosis
Numerous investigators have taken advantage of the utility of TIRFM for studying exocytotic events. By using TIRFM and styryl dyes such as FM1-43 (Zenisek et al., 2002), the loading of vesicles undergoing exocytosis with fluorescent dyes (Pangrsic et al., 2007; Pryazhnikov and Khiroug, 2008), or cargo fused to a fluorescent protein (Toomre et al., 2000), one can observe single exocytic events (Beaumont, 2003; Betz et al., 1996; Pollard and Apps, 2002). When visualizing events of exocytosis, fluorescently loaded vesicles appear bright as they enter the evanescent field. Investigators have been able to study the docking and subsequent fusing of secretory granules to the plasma membrane (Gaus et al., 2021; Mizuno and Izumi, 2022; Urbina and Gupton, 2021). As the vesicles fuse with the PM, the signal rapidly diffuses as the fluorescently labeled contents spill into the extracellular space or enters the PM (Steyer and Almers, 2001). The high signal-to-background ratio in TIRFM makes it possible to automatically detect and distinguish between clustered and dispersed fusion events spatially and temporally (Urbina and Gupton, 2021).
Proteins of the Endocytic Pathway
TIRFM has been extensively used to qualitatively and quantitatively describe the roles different proteins play in exocytosis/endocytosis. For example, TIRFM was used to show that GTPases Rab3A and Rab27A cooperatively regulate docking steps of dense-core vesicles to the PM (Tsuboi and Fukuda, 2006) and to visualize the dissociation of Rab3A from vesicles during exocytosis (Lin et al., 2007). Rab27A and its effector Slp4A on the same granules before membrane fussion (Lam et al., 2010). Others have studied the roles of specific v-SNARE and t-SNARES in vesicle targeting to and fusion with the plasma membrane (Hastoy et al., 2017; Nikolaus et al., 2021; Winkle et al., 2016). The function of clathrin, dynamin, and adaptor proteins have also been extensively studied using TIRFM (Cocucci et al., 2014; Keyel et al., 2004; Merrifield et al., 2002; Saffarian and Kirchhausen, 2008). In such studies, TIRFM has been used to identify the existence of subclasses of clathrin-coated pits and the function of specific adaptor proteins in internalization directed to these subclasses (Pascolutti et al., 2019).
Cell-Substrate Contacts
TIRFM can be used to observe the size, movement, and distance apart of the regions of contact between a cell and the solid substrate (e.g., coverglass) it is bound to (Axelrod, 1981). For these types of experiments, fluorescently labeled molecules (e.g., dextran) can be microinjected into the cell. Alternatively, GFP fusion proteins that localize to focal adhesions can be exogenously expressed. TIRFM has also been used to directly assess mechanisms regulating cell migration (Arts et al., 2021).
Long-term Fluorescence Live Cell Imaging
Because of the very thin optical section in TIRFM, which is localized to the cell-substrate contact region, cells tend to survive substantially longer than when imaged by standard epifluorescence techniques (Wang and Axelrod, 1994). In addition to low cell cytotoxicity, since photobleaching occurrs in such a small region, in most experiments there is a continuous flux of new fluorophores into the imaging field. Thus, optimal signal-to-noise ratios can be achieved with the same minimum exposure settings.
THE THEORY BEHIND THE TECHNIQUE
The basic concept of TIRFM in cellular microscopy is simple: an excitation light beam needs to travel at a high incident angle through a glass coverslip upon which cells are cultured. The refractive index (RI; see Table 1 for the refractive indexes of some common reagents) differences between the glass (n2) and intact cell interior (n1) regulate how light is refracted or reflected at the interface as a function of incident angle. At a specific critical angle (θcritical), the beam of light is totally reflected from the glass/water interface, rather than passing through and refracting (see Eq. 1 below; Fig. 2A). The reflection generates a very thin electromagnetic field (the evanescent wave) on the cell side of the coverslip that has an identical frequency to that of the incident light. The depth of the evanescent wave is a function of the incident illumination angle, wavelength, and refractive index differences (see Eq. 2 below) and its intensity exponentially decays with distance from the coverglass (see Eq. 3 below; Fig. 2B). Usually, only fluorophores within 100 to 200 nm of the coverslip are efficiently excited.
Table 1.
Refractive Indices of Commonly Encountered Materials
| Material | Refractive index |
|---|---|
| Water | 1.33 |
| Air | 1.0003 |
| Glass | 1.517 |
| Coverglass | 1.523 |
| Immersion oil | 1.516 |
| Cell cytosol | 1.38 |
| Mount | Variablea |
Mounting media for fluorescence microscopy can vary substantially. To the author's knowledge, none of the standard commercially available mounts are suitable for fixed-sample TIRFM.
Figure 2.

The evanescent wave. (A) The incident angle can be described using a coordinate system arranged to display all three orthogonal directions. The x-y plane represents the interface between the coverglass and the cell cytosol. The plane of incidence is the x-z plane, which is parallel to the excitation light beam. (B) The evanescent field intensity decays exponentially with increasing distance from the interface according to Equation 3. The penetration depth, which is usually between 50 and 300 nm, decreases as the reflection angle grows larger and is dependent on the refractive indices at the interface and the illumination wavelength. Illustrations were adapted from ones provided by Michael W. Davidson, Florida State University.
The following equations are used to calculate the specific critical angle, depth of the evanescent wave, and intensity at depth.
| (Equation 1) |
| (Equation 2) |
Alternatively, d = λ0/(4π n2) × [(sin2θ/sin2θcritical) – 1]−1/2
(Note that d is a decreasing function of the angle of incidence and for θ −θ critical = 0/1° d is of the order of the excitation wavelength λ0.)
| (Equation 3) |
Where Iz = intensity at depth z; I0 = initial intensity (note that at z = 0, Iz = I0). The use of these equations has been simplified by an ImageJ plugin called Calc TIRF (https://imagej.nih.gov/ij/plugins/tirf/index.html ) written by Sebastian Rhode. The plugin calculates max and critical angles, penetration depth, and evanescent field intensity for multiple laser lines simultaneously. Figure 3 shows the output of this plugin for the popular 60x 1.49 N.A. objective.
Figure 3.

TIRFM Calculator. The calculation of max and critical angles, penetration depth, and evanescent field intensity for performing TIRFM has been simplified by an ImageJ plugin called Calc TIRF (https://imagej.nih.gov/ij/plugins/tirf/index.html ) written by Sebastian Rhode. Using this calculator, the max and crtical angles were calculated for the popular 60x 1.49 N.A. objective (inset text A). (A) Penetration depth curves for multiple laser lines and (B) the evanescent field intensity curve is shown.
There are two common methods for producing the evanescent wave for TIRFM. One configuration uses a prism to direct laser light toward the interface between the coverglass and media/cells at an incident angle sufficient to cause total internal reflection. Although the majority of prism-based TIRF microscope designs are centered on inverted microscope bases, upright microscopes can also be used (for review, see (Axelrod, 2001)). The prism-based configuration has been applied to cellular microscopy for nearly 30 years (Axelrod, 1981). The use of TIRF in cellular microscopy increased once an objective-based TIRF (or prism-less TIRF; Fig. 4A) system was introduced to the scientific community (Stout and Axelrod, 1989), and increased even more once a commercial solution became available (Kawano et al., 2000). In objective-based TIRFM, one can easily switch between standard epifluorescence and TIRF by changing the off-axis position of the beam focus at the objective's back focal plane. Several ways have been developed to change the position of the beam. For example, an actuator can be used to change the position of a fiber carrying the laser light in relation to a fluorescence illuminator that attaches to the microscope (Fig. 4A). The following lists the basic characteristics of these two different methods for performing TIRFM.
Figure 4.

TIRFM System calibration. (A) Schematic drawing of a TIRF microscope that uses a micrometer to position a fiber that delivers laser light to the microscope. (B) Schematic drawing of how to use a hemicylindrical glass prism to determine the angle of incidence. (C) Schematic drawing of how to empirically determine the penetration depth of the evanescent wave using 1-μm diameter fluorescent microspheres. Note that in the example 370 equals the measured radius of the microsphere and that a is calculated using the Pythagorean theorem and 500-a = the empirically determined penetration depth. (D) Demonstrates how the objectives numerical aperture and the illumination wavelength affect the penetration depth of the evanescent field. Panel A was provided by Michael W. Davidson, Florida State University.
Characteristics of the Prism Method
Some characteristics of the prism method include: (1) a prism is used to attain critical angle, (2) purer evanescent wave, (3) it is easier to set up than a prism-less system, (4) laser focused to spot size about equal to field of view, (5) access to the sample can be restricted depending on prism position, and (6) there is no commercially available system.
Characteristics of Prism-less Method
Some characteristics of the prism-less method include: (1) the beam must pass through periphery of a high NA objective, (2) higher NA will allow confinement closer to surface, (3) laser focused off-axis at back focal plane of objective, (4) decreased field of illumination, (5) specimen is fully accessible, (6) not as pure an evanescent wave as prism, (7) increased light scattering within objective (decreases signal to noise ratio), and (8) turn-key systems are commercially available.
Although a prism-based system is easier to build than an objective-based one, since the latter is what the average user will find in most core facilities, the rest of this article will focus on performing TIRFM using an objective-based system.
TIRF OBJECTIVES
There is a variety of objectives designed specifically for TIRF imaging available today. TIRF objectives vary substantially in both magnification and NA. Listed are the specifications of a few: 60× 1.45 NA, 60× 1.49, 60× 1.50, 100× 1.50 NA, 100× 1.65 NA, 100× 1.70 NA, and 150× 1.45 NA. The NA of TIRF specific objectives is ≥ 1.45, which allows for an angle of incidence greater than the critical angle. Some objectives also come with compensation collars used to control for varying temperature and coverslip thickness. The objective's specifications will determine the experimental design, and thus the critical angle. For example, below is a comparison between the critical and maximum angles (see Eq. 4) of two different objectives.
Critical angle:
60× 1.45 NA objective:
[calculated using n2 = 1.52 (RI of coverglass and immersion liquid) and n1 = 1.38 (cell cytosol)]
100× 1.65 NA objective:
[calculated using n2 = 1.78 (RI coverglass and immersion liquid) and n1 = 1.38 (cell cytosol)]
Maximum angle of incident light that can be generated by the objective can be calculated using Equation 4 (shown below) as follows:
A comparison of the two objectives using the above equations reveals that when aligning a system that uses a 60× 1.45 NA objective to produce the evanescent wave there are only 7.32° for which total internal reflection can occur. In contrast, using the 100× 1.65 NA objective one has 17.14° to work with. Thus, as objective NA decreases the ability to align a system for TIRF imaging becomes more difficult. To the author's knowledge, the smallest NA objective used for objective based TIRFM has been a 60× 1.4 NA objective, which only has ~1.9° of freedom.
As mentioned above, the depth of the evanescent wave is a function of the incident illumination angle, wavelength, and refractive index differences. Considering Equation 2, as the angle of incidence increases, penetration depth decreases. Choosing an angle of incidence for which internal reflection will occur for both the 60× 1.45 and 100× 1.65 objectives, their performance regarding penetration depth can be compared (Fig. 4D). For example, using an angle of incidence of 66.5°, a wavelength of incident light (λ0) of 488 nm, and n2 = 1.38 (cytosol), the penetration depth of the evanescent wave for the 60× 1.45 NA objective would be ~197.6 nm, while that using the 100× 1.65 NA objective would be ~44.5 nm. The >4× difference in penetration depth between these two objectives under the same conditions would result in drastic differences in the excitation of fluorophores (see below). Thus, it is crucial that when designing TIRFM experiments one fully considers all hardware and experimental parameters. In addition, although 60× 1.45 and 60× 1.49 objectives have similar NAs, comparing Figures 3 and 4 show how this small difference changes performance.
EMPIRICALLY DETERMINING INCIDENT ANGLE/PENETRATION DEPTH
When using TIRFM for performing qualitative measures, just knowing that the system is in TIRF imaging mode is sometimes good enough. For these types of experiments, one can use 0.1 μm TetraSpeck fluorescent microspheres (Invitrogen) to confirm the microscope is in TIRF imaging mode. First, set up the microscope with a live cell chamber (see below). Then, add 100 μl of water containing 5 μl of the TetraSpeck solution. When visualizing, some of the beads will be sitting directly on the coverglass and not moving, while others will appear and disappear rather rapidly. In TIRF mode, there will be a very high signal to background ratio and all you will see are events at the coverglass. In epifluorescence mode, there will be a substantial amount of blur from out of focus beads and beads at, and in relatively close proximity to, the coverglass will always be visible.
When performing quantitative fluorescence TIRFM it is important to know that your system is performing as expected. Although the software that accompanies several commercially available turnkey systems reports on incident angle and penetration depth, do not take for granted that the values you see are correct, particularly since the penetration depth is most likely being calculated using Equation 2. To determine the angle of incidence you will need a hemicylindrical glass prism (BK-7, which has a RI of 1.52; note this prism will not work for the 100× 1.65 NA objective). Add immersion oil to the top of the objective, then place a highly polished coverslip on the stage, and carefully focus on the upper surface of the coverslip. The prism needs to be centered (flat side down) directly over the objective, contacting the coverslip through a layer of immersion oil (Fig. 4B). Since the RI of the coverslip and prism are the same, the light will not be reflected, and it will pass through the prism. From this point forward, laser safety goggles should be worn. First adjust the beam so that laser light is shining directly above on the room ceiling (note: you will probably need to reposition some of the microscope hardware to see the beam on the ceiling). Mark the position on the micrometer and then start very slowly walking the beam across the ceiling and eventually onto the wall. At a specific position, the beam will disappear because you have exceeded θmaximum and the internal aperture of the objective is blocking the laser light from exiting the objective. Mark the position on the micrometer. Move the beam back (decreasing the angle of incidence) until you clearly see the laser light on the wall again. To confirm that total internal reflection is occurring, move the objective to an area of the coverslip that is not under the prism. Here, almost no light will emerge if in TIRF mode. There are two different ways to determine the angle of incidence. One method is to mark where the light leaves the prism and measure the radians starting from the bottom edge of the prism. The radians can then be directly converted into degrees. Alternatively, others have projected the light leaving the prism on a screen that rests perpendicularly on a surface level with the top of the objective (Zenisek et al., 2002). By measuring the distance from the center of the prism to the screen, and the distance from the bottom of the screen to where the light from the prism hits, one can calculate the incident light angle (Fig. 4B). You can build a jig so that the screen is always positioned in the same place in relation to the objective/prism. By doing this, you can place marks on the screen for specific incidence angles and then when setting up the system, position the beam to the mark you want for the given experiment. To calibrate a system, the incident angle at different positions of the fiber, or mirror in some systems, will need to be measured (also see (Niederauer et al., 2018)).
As stated above, software packages usually determine the penetration depth of the evanescent wave using Equation 2. However, in that equation d is only the depth at which the intensity of the evanescent wave is 37% of the initial intensity. Thus, for all quantitative studies, the experimental depth should be determined. There are several different ways to empirically determine the evanescent wave penetration depth (Keyel et al., 2004; Niederauer et al., 2018; Saffarian and Kirchhausen, 2008; Sarkar et al., 2004). The author usually directs new users towards the method used by Keyel, Watkins, and Traub for measuring penetration depth (Keyel et al., 2004). Briefly, image 1.0-μm TetraSpeck fluorescence microspheres by TIRFM as described above for the 0.1-μm diameter microspheres. Image several beads that are clearly resting on the coverslip. Since the diameter of the 1.0-μm microspheres is substantially greater than the evanescent wave penetration depth, only a portion of the bead (<1/2) will be imaged if the system is in TIRF mode. The next step is to determine the average measured diameter of the bead, followed by the use of the Pythagorean theorem to determine the experimental penetration depth. For example, using a 60× 1.45 NA objective with an incident light angle of 68.2°, the calculated penetration depth for an excitation wavelength of 488 nm in aqueous medium (RI = 1.33) is 82.3 nm. Using the method above to empirically determine the penetration depth, 1-μm fluorescence microspheres had an average diameter of 740 nm (Fig. 4C). Thus, the measured evanescent wave penetration depth was 164 nm, which is twice the calculated depth. It is important to note that both the calculated and empirically determined depth of penetration would be different if the fluorescence microspheres were within a cell. To get beads into cells, one can use particle bombardment or microinjection. Depending on wavelength and objective, under these conditions the author has found anywhere from 1.4 to 2.12 times difference between calculated (Fig. 4D) and empirically determined penetration depths. Others have used cellular structures to help extrapolate measurements using beads in aqueous solution to experimental conditions (Keyel et al., 2004). Also, important to mention here is that the physical limits inherent in the optical system and the diffraction limit of light make it impossible to empirically determine the exact penetration depth of the evanescent wave. With that said, it is clear by the differences in calculated and empirically determined penetration depths that it is imperative to perform some form of experimental measure for the accurate analysis and interpretation of data.
TIRF IMAGING OF PLASMA MEMBRANE RECEPTORS IN NEURONS
Neurons are highly polarized cells that have three distinctive compartments: somatic, axonal, and dendritic. To define the unique features of each compartment, it is usually necessary to use microscopy to perform an equivalent of biochemical analysis. The following describes two examples of using TIRFM to study the endocytic pathway in primary hippocampal neurons.
Materials
The microscope used in the examples below is an Olympus IX81 inverted microscope equipped with a TIRFM illuminator [multispectral (488, 568, and 635) single fiber], 60× 1.45 NA objective, Hamamatsu C4742-98 CCD camera, and high-precision prior motorized XY stage, and is controlled by SlideBook 4.2 software (Intelligent Imaging Innovations).
For maintaining cell viability during imaging, the glass coverslips on which neurons were grown (see below) were placed in an imaging chamber (model RC-41LP) that was inserted into a micro-incubator chamber (model DH-40i). A perfusion valve controller (model VC-6) was used to exchange medium throughout the experiment and temperature control was performed using a dual-channel heater controller (model TC-344B; stage and objective) and an in-line solution heater (SH-27B), all from Warner Instruments.
An expression vector for vesicular stomatitis viral G protein fused to green fluorescent protein (VSVG-GFP; Dr. M. Aridor, University of Pittsburgh) was used.
The examples use a high-affinity NPY-Y1 receptor agonist Leu 31, Pro 34 NPY1-36 covalently labeled with Alexa 488 (NPY-488; Tamas Bartfai; The Scripps Research Institute).
The rats used in the examples are Sprague Dawley E18 timed pregnant rats (Harlan). The imaging solution (HEPES-buffered Tyrode's solution) used was composed of: 119 mM NaCl, 5 mM KCl, 25 mM HEPES buffer, 2 mM CaCl2, 2 mM MgCl2, 6 g/liter glucose (adjust pH to 7.4 with NaOH).
Culturing Primary Rat Hippocampal Neurons
For both studies, neurons were cultured at low density from embryonic day (E) 18 rats as previously described (Aridor et al., 2004; Fish and Krucker, 2008; MacLaurin et al., 2007). A detailed protocol of the method has been previously published (Goslin and Banker, 1991). Briefly, hippocampal neurons were plated onto poly-l-lysine-coated glass coverslips (no. 1.5 18-mm round) that are inverted over a monolayer of glial cells after a 2-hr incubation. To achieve low-density cultures, cells were plated at a density of 2700 cells per cm2. Paraffin dots attached to the coverslips were used to keep the neurons separated from the cells making up the glial feeder. Neuron cultures were fed every few days by replacing 1/2 the medium with fresh medium. Most neurons, >90%, developed the characteristic mature morphology of spiny neurons between 16 to 21 days in vitro (DIV).
Microscope System Alignment and Set up
In all examples listed below, the same basic microscope set up routine and alignment were performed before TIRF imaging began. Thus, the step-by-step set of instructions listed here are for all examples.
1. Prior to starting the experiment determine the angle of incidence and penetration depths of the illumination wavelengths that will be used following the instructions under the section entitled Empirically determining incident angle/penetration depth.
2. Preheat all hardware components (objective, imaging chamber, micro-incubator chamber) to 38.5°C ~1 hr prior to starting the experiment. Since in the system used here the imaging chamber, which will hold the coverslip, and micro-incubator are two separate components, the micro-incubator is inserted into the stage and the objective/micro-incubator is heated using a dual-channel heater controller. The imaging chamber is placed in the incubator that is housing the neurons.
3. For long-term experiments or those requiring the medium to be exchanged, set up a perfusion device. For Example 2 below, a gravity-fed perfusion system equipped with a valve controller was used to exchange the medium. An in-line solution heater, which was placed just before the imaging chamber, was used to heat the solution to 37° C.
4. Approximately 10 min before moving the neurons to the microscope, insert a coverslip on which the neurons are grown into the imaging chamber, cover with imaging solution, and return to the incubator.
5. Add immersion oil to the top of the objective, place the imaging chamber in the micro-incubator, and carefully focus on the upper surface of the coverslip. It is essential that the coverslip bottom be thoroughly cleaned before the imaging chamber is transferred to the microscope.
Example 1: Visualizing VSVG-GFP transport to the PM
In this example, TIRFM was used to visualize the fusion of VSVG-GFP exocytic vesicles with the PM. [*Copy Editor: I think that a reference to Figure 5 was crossed out accidentally. Please query author if “(Figure 5)” should be added at the end of the first sentence.] For the expression of exogenous vesicular stomatitis viral G protein fused to green fluorescent protein (VSVG-GFP), plasmid DNA was transported into neurons by electroporation prior to plating using a Nucleofector II device (Amaxa) and the manufacturer's rat neuron kit (VPG-1003). VSVG-GFP trafficking was imaged in cultures at 7 days in vitro. The images were captured at 11.32 frames per sec with 2 × 2 binning. Using a 60× 1.45 NA objective, illumination wavelength of 488 nm, and a 70° angle of incidence, the empirically determined evanescent field penetration depth was 191 nm. When visualizing an exocytosis event (the arrival of a VSVG-GFP vesicle at the PM), the vesicle first becomes brighter as it enters the evanescent field. As the vesicle fuses with the PM, the signal rapidly diffuses as VSVG-GFP enters the PM (Fig. 5).
Figure 5.
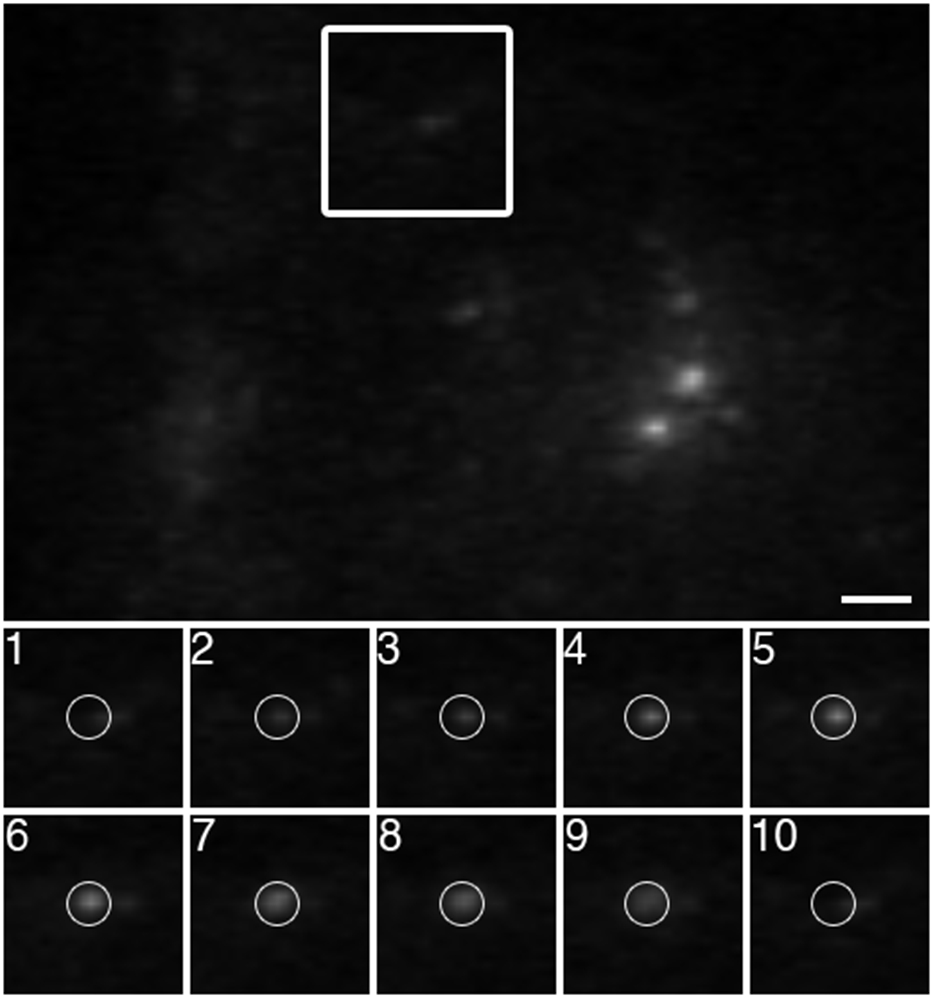
Visualizing the arrival of VSVG-GFP to the PM. VSVG-GFP plasmid DNA was transported into neurons by electroporation prior to plating. At 7 days in vitro, the trafficking of VSVG-GFP vesicles to the PM was imaged by TIRFM using a 60× 1.45 NA objective at 11.32 frames per sec. The panels labeled 1 through 10 are sequential frames. As a VSVG-GFP vesicle nears the PM, it becomes brighter. Upon fusing with the PM, the signal rapidly diffuses. Bar = 2 μm.
Example 2: Imaging NPY-Y1 receptor at the PM
In this example, a fluorescently labeled high-affinity Y1r agonist Leu31, Pro 34 NPY1-36 (NPY-488) was used to detect the arrival of new receptors at the PM (Fig. 6). An advantage to using NPY-488 is that once the labeled agonist binds Y1r, the complex is stable and when endocytosed it is degraded (Bernet et al., 1994). Therefore, the appearance of new NPY-488/Y1r complexes at the PM represents the arrival of new receptors. The empirically determined penetration depth of the evanescent field was 192 nm. Since the optical section is very thin and located at the interface between the coverslip and neuron, imaging could be performed in the presence of NPY-488 in the culture medium without free NPY-488 interfering with bound NPY-488. In this experiment, NPY-488 was added to the culture medium 10 min after step 5 above. Although we have previously found that the Y1 receptors at the PM in cultured hippocampal neurons are functional as high-affinity binding sites (1.65 picomoles/mg protein), we added a large excess of NPY-488 to the medium (10−6 M) to make sure all NPY receptors at the PM would be bound to NPY-488. Again, having a substantial amount of free fluorescently labeled ligand in the medium does not interfere with our imaging. Imaging was performed at 2 frames/min without camera binning. Shortly after adding NPY-488 to the medium, the presence of NPY-Y1 receptors on the PM of some hippocampal neurons was revealed by NPY-488 receptor binding (fluorescent puncta in Fig. 6A; note the high signal-to-background ratio). Using time-lapse imaging, the removal of NPY-Y1rs from the PM and the arrival of new receptors can be easily followed (Fig. 6B).
Figure 6.
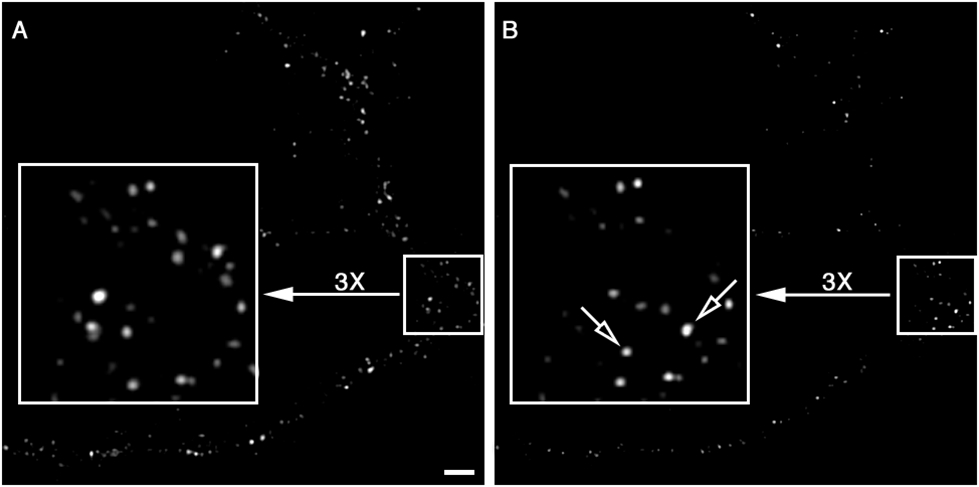
Visualization of Y1r arrival at the PM by TIRFM. (A) Taken shortly after NPY-488 was added to the culture medium. The signal-to-background ratio is not affected by the presence of free fluorescently labeled agonist in the medium. (B) Captured 10 min after the image in A. Note the loss of 488 fluorescence along the dendritic branches and in some cases (open arrows) the detection of new receptors at the PM. Bar = 2 μm.
MULTI-WAVELENGTH TIRFM
There is an increasing demand for multi-wavelength TIRFM. Multi-wavelength TIRFM has been used to study the interaction between antithrombin with Corline Heparin Surfaces in real time (Klinth et al., 2006), to visualize the splicing of single pre-mRNA molecules (Crawford et al., 2008), transcription mechanisms (Friedman and Gelles, 2015), and actin (Chin et al., 2016), to name a few ways. All the major microscopy companies now offer turn-key multi-wavelength TIRFM solutions and investigators have built novel systems (e.g., see (Sobhy et al., 2011)). Depending on hardware, multi-wavelength TIRFM is usually performed in three general ways: (1) different channels are collected sequentially either using the same or different incident angles for each wavelength; (2) different channels are collected simultaneously using the same angle of incidence; (3) multi-angle illumination combined with simultaneous capture. In configuration 1, an actuator is needed to change the angle of incidence during imaging. In methods 2 and 3, the systems need to be able to pass multiple excitation wavelengths simultaneously. In configuration 2, this is accomplished by using either a single multiline laser, also known as a mixed-gas laser (e.g., Argon/Krypton ion laser), or a laser combiner that houses several single line lasers that can exit the combiner simultaneously. In configuration 3, each wavelength remains independent from the others until it passes a unique set of hardware used to adjust the angle of incidence. In configurations 2 and 3, there also needs to be a mechanism for the simultaneous capture of the different emitted wavelengths. This can be accomplished using matched cameras connected to the microscope using a multi-camera adaptor. Alternatively, simultaneous multi-wavelength imaging can be performed on a single camera using devices like the DV2 and Quad-view from Mag Biosystems. These devices connect to the camera port and use a beamsplitter(s) to split the emission light from the microscope into independent channels that are simultaneously projected onto different quadrants of the same CCD.
There is some debate over if it is necessary to use wavelength-specific angles of incidence when performing multi-wavelength TIRFM. The answer can be reached by performing a simple experiment, but first it is important to understand how excitation wavelength affects evanescent wave penetration depth, which can easily be visualized by performing a few calculations using Equation 12.18.2 [d = λ0/4π (n22 sin2θ – n12)−1/2]. The calculations shown below are based on a 60× 1.45 NA objective for which θcritical = 65.22° and θmaximum = 72.54° (using n2 = 1.52 and n1 = 1.38).
If d = 120.66
and λ0 = 488 then θ = 68.79°
and λ0 = 568 then θ = 70.18°
and λ0 = 647 then θ = 71.86°
If d = 162.13
and λ0 = 488 then θ = 67.14°
and λ0 = 568 then θ = 67.86°
and λ0 = 647 then θ = 68.69°
If θ = 68.79°
and λ0 = 488 then d = 120.66°
and λ0 = 568 then d = 140.44°
and λ0 = 647 then d = 159.97°
There are a few important things to notice about the above calculations. First, as the wavelength increases the penetration depth increases. Second, the shorter the wavelength the less the incident angle needs to change for the same change in penetration depth. Now for a simple experiment that demonstrates how differences in penetration depth affect experimental outcome.
Experimental steps
An Olympus IX81 microscope equipped with a TIRF illuminator, laser combiner with 488-, 568-, and 635-nm lasers, and 60× 1.45 NA objective was used in this experiment.
1. Set up the microscope to empirically measure the angle of incidence using the 488-nm laser line. In our experiment, the determined angle of incidence = 68.1°. Thus, the calculated evanescent wave penetration depth for 488 nm = 82.78 nm, and for 635 nm = 107.72 nm (using n1 = 1.33 and n2 = 1.52).
2. Following the procedures above, empirically determine the evanescent wave penetration depth. An experimental penetration depth was found for 488 nm = 161.2 nm and for 635 = 201.8 nm.
3. Dry down 10 μl of a 488 nm/625 nm fluorescence microsphere mixture (1:1 solution of 0.1-μm fluorescent microspheres in water) onto a microscope coverglass bottom chamber (e.g., Warner Instrument DH-35i culture dish chamber) and empirically determine the 488 nm/625 nm microsphere ratio of the mixture (the author had a ratio of 1/0.96).
4. Change the coverglass, add 200 μl of the microsphere mixture, and quantify the number of microspheres in each channel. Per imaging field, the author had an average ratio of 1 (488 nm)/1.22 (625 nm).
5. Change the incident angle for one wavelength so that the penetration depths are the same. Since in this experiment the incident angle can not increase enough so that the 635-nm line has a calculated penetration depth of 82.78 (it would need to be 74.31°, but the maximum angle for the experimental conditions is 72.54°), change the incident angle such that the evanescent wave produced by the 488-nm excitation line equals that of the one produced by the 635-nm line. Solving for θ in Equation 12.18.2 [θ = sin−1 (λ0/d4π)2 + (n12/n22)−1/2], estimate the new angle of incidence. The author used a new angle of 65.04°, which is between θcritical and θmaximum (61.05° and 72.54°, respectively).
(Note: For this step, it is necessary that you have a calibrated system that allows you to accurately change the incident angle.)
6. Perform a new estimate of the number of 488-nm microspheres/field. Using similar evanescent wave penetration depths, the new ratio was 1 (488 nm)/1.03 (625 nm).
7. Optional: empirically determine the 488 nm evanescent wave penetration depth at the new angle of incidence. The new measured depth was 199.8 nm.
Author's Recommendations
Clearly, for this experiment using the same angle of incidence resulted in a misleading finding. The question is why. TIRF can be viewed as a probability curve: increase evanescent wave penetration = increase probability at any given imaging site of exciting a molecule. Thus, in experiments where the goal is to quantitate the number of different objects within the evanescent field using different fluorescent probes, it is important that the penetration depths of the different illumination lines be matched. For experiments in which simultaneous imaging is required, configuration 3 (multi-angle illumination combined with simultaneous capture) should be used. However, since the penetration depth for each line in a multi-angle illumination system should be empirically determined at the onset of each experiment, which is time consuming, for experiments that do not require simultaneous imaging, the author recommends using a system in which different channels are collected sequentially using different incident angles for each wavelength. If the system is calibrated, one can use Equation 2 and an actuator to achieve very similar evanescent field penetration depths without using empirical measures to set the angle of incidence for each line. The use of an actuator provides high accuracy and the capability to switch rapidly (~50 msec) between wavelengths. Importantly, the author has found that although the calculated and empirically determined penetration depths are always different, when using angles of incidence for different illumination wavelengths that have the same calculated penetration depth, the empirically determined penetration depths are also the same. Thus, even without empirically determining the penetration depths, one can be relatively assured that when using calculated angles of incidence the same penetration depth across illumination lines can be achieved.
It is important to point out that the above TIRFM imaging was performed in water rather than within the cell cytosol. Thus, θcritical = 61.05°, rather than 65.22° (that for imaging in cytosol). Very small changes in n1 have a big effect on differences in penetration depth when performing multi-wavelength TIRFM. For example, the calculated evanescent wave penetration depths for 488 nm and 635 nm are 82.78 nm and 107.72 nm, respectively, at an angle of incidence = 68.1° using n1 = 1.33 and n2 = 1.52. However, they are 133.53 and 173.76, respectively, if using n1 = 1.38. Thus, in an aqueous environment there is a difference in evanescent penetration depth of 24.94 nm (calculated) between the two wavelengths, while in cytosol the difference jumps to 40 nm. These and other considerations covered in this unit are important to keep in mind when designing and performing TIRFM experiments and analyzing/reviewing TIRFM data.
FINAL EXPERIMENTAL SUGGESTIONS
1. The coverglass (total internal reflection surface) should be thoroughly cleaned.
2. Depending on the design of the system, photobleaching within the evanescent field may be greater than under epifluorescence conditions. Several precautions should be taken to reduce photobleaching. For example, minimize the amount of light fluorophores are exposed to by reducing light intensity and exposure time without compromising image quality. This can be done in part by maximizing photon detection and capture using an ideal detector(s) (i.e., sensitive, high quantum efficiency, and low read noise), and carefully selecting fluorophores. In addition, reagents like Trolux (6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid; Sigma Aldrich, cat. no. 238813), DABCO (1,4-diazabicyclo[2.2.2]octane), nPG (n-propyl gallate), ascorbic acid, and OxyFluor (Oxyrase) all possess antioxidant activity and can be added to the medium to increase photostability. If studying protein and lipid mobility in the PM of red blood cells, perform deoxygenation (Corbett et al., 1994).
3. Since objective-based TIRFM uses high magnification/NA objectives, there is a high propensity for focus drift, which can be caused by subtle temperature fluctuations and air circulation, to wreak havoc on time-lapse experiments performed at physiological temperatures and experiments in which the temperature needs to be changed. Thus, for these types of experiments, the author highly recommends using some form of microscope autofocus mechanism, which is available from nearly all the major microscope manufacturers and several third-party suppliers.
CONCLUDING REMARKS
This overview has touched on the history, optical theory, and hardware configurations of TIRFM. In addition, it provides useful tools for aligning a TIRF microscope and practical experimental protocols. Since the purpose of this unit was to provide useful information to both beginner and advanced TIRF microscopists, advanced methodologies were not covered. Thus, it is important to point out that advanced approaches for using TIRF microscopy to study biological problems are continuously being developed. For more than 30 years, TIRF imaging has been combined with fluorescence photobleaching recovery (Burghardt and Axelrod, 1981) and fluorescence correlation spectroscopy (Thompson and Axelrod, 1983). In addition, atomic force microscopy and TIRFM have been combined to study protein unfolding, and differential evanescence nanometry and standing wave TIRF have been used to perform superresolution fluorescence microscopy (Chung et al., 2006; Chung et al., 2007; Saffarian and Kirchhausen, 2008).
TIRFM purists often prefer using a prism-based approach because the illumination across the field is more even and cleaner than TIRFM using an objective. The difference in illumination between the two techniques is in part due to light scattering in the objective. We previously worked to circumvent this problem by building a system that placed an upright microscope equipped with a spinning disc confocal above an inverted objective based TIRFM system. (https://www.photonics.com/Articles/Combined_Upright_Inverted_Microscope_Offers_New/a25257). The idea was that by using a second objective to collect emitted photons we would illuminate [*Copy editor: I believe this should be “eliminate” – please query.] some of the objective based noise. Although this system accomplished our goals, it is a difficult approach for cleaning up the TIRF signal. Recently, two companies (3i and GATACA Systems) have developed approaches to achieve both cleaner and even illumination. 3i’s Vector2 TIRF (https://www.intelligent-imaging.com/tirf) spins the laser beam using galvo mirrors around the periphery of the back focal plane at high-speed. This approach averages out artifacts so that images are evenly illuminated and have minimum noise. 3i has combined VectorTIRF with spinning disc confocal, photoactivation, and ablation systems to build custom, flexible imaging microscopes.
GATACA Systems’ iLas 2 platform (https://www.gataca-systems.com/products/gataca-products/ilas-2/) is a similar multi-application system that offers full control over laser illumination, which allows for azimuthal averaging TIRF (artifact reduction) and 3D TIRF. The TIRF imaging can also be combined with FRAP. The iLas 2 platform provides a highly uniform TIRF field and can be used for simultaneous multichannel TIRF with different or similar penetration depths of the unique laser lines.
Acknowledgement
This work was supported by the National Institute of Mental Health (Grant MH119701).
Footnotes
Conflict of Interest
The author declares no conflict of interest.
Data Availability Statement
Data available on request from the authors.
LITERATURE CITED
- Aridor M, Guzik AK, Bielli A, Fish KN, 2004. Endoplasmic reticulum export site formation and function in dendrites. J Neurosci 24: 3770–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arts JJG, Mahlandt EK, Schimmel L, Gronloh MLB, van der Niet S, Klein B, Fernandez-Borja M, van Geemen D, Huveneers S, van Rijssel J, Goedhart J, van Buul JD, 2021. Endothelial Focal Adhesions Are Functional Obstacles for Leukocytes During Basolateral Crawling. Front Immunol 12: 667213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod D, 1981. Cell-substrate contacts illuminated by total internal reflection fluorescence. J Cell Biol 89: 141–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod D, 2001. Total internal reflection fluorescence microscopy in cell biology. Traffic 2: 764–74. [DOI] [PubMed] [Google Scholar]
- Beaumont V, 2003. Visualizing membrane trafficking using total internal reflection fluorescence microscopy. Biochem Soc Trans 31: 819–23. [DOI] [PubMed] [Google Scholar]
- Bernet F, Maubert E, Bernard J, Montel V, Dupouy JP, 1994. In vitro steroidogenic effects of neuropeptide Y (NPY1-36), Y1 and Y2 receptor agonists (Leu31-Pro34 NPY, NPY18-36) and peptide YY (PYY) on rat adrenal capsule/zona glomerulosa. Regul Pept 52: 187–93. [DOI] [PubMed] [Google Scholar]
- Betz WJ, Mao F, Smith CB, 1996. Imaging exocytosis and endocytosis. Curr Opin Neurobiol 6: 365–71. [DOI] [PubMed] [Google Scholar]
- Burghardt TP, Axelrod D, 1981. Total internal reflection/fluorescence photobleaching recovery study of serum albumin adsorption dynamics. Biophys J 33: 455–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin SM, Jansen S, Goode BL, 2016. TIRF microscopy analysis of human Cof1, Cof2, and ADF effects on actin filament severing and turnover. J Mol Biol 428: 1604–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung E, Kim D, So PT, 2006. Extended resolution wide-field optical imaging: objective-launched standing-wave total internal reflection fluorescence microscopy. Opt Lett 31: 945–7. [DOI] [PubMed] [Google Scholar]
- Chung E, Kim D, Cui Y, Kim YH, So PT, 2007. Two-dimensional standing wave total internal reflection fluorescence microscopy: superresolution imaging of single molecular and biological specimens. Biophys J 93: 1747–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocucci E, Gaudin R, Kirchhausen T, 2014. Dynamin recruitment and membrane scission at the neck of a clathrin-coated pit. Mol Biol Cell 25: 3595–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett JD, Cho MR, Golan DE, 1994. Deoxygenation affects fluorescence photobleaching recovery measurements of red cell membrane protein lateral mobility. Biophys J 66: 25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford DJ, Hoskins AA, Friedman LJ, Gelles J, Moore MJ, 2008. Visualizing the splicing of single pre-mRNA molecules in whole cell extract. RNA 14: 170–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenan RM, Nashmi R, Imoukhuede P, Just H, McKinney S, Lester HA, 2008. Subcellular trafficking, pentameric assembly, and subunit stoichiometry of neuronal nicotinic acetylcholine receptors containing fluorescently labeled alpha6 and beta3 subunits. Mol Pharmacol 73: 27–41. [DOI] [PubMed] [Google Scholar]
- Erdelyi M, Simon J, Barnard EA, Kaminski CF, 2014. Analyzing receptor assemblies in the cell membrane using fluorescence anisotropy imaging with TIRF microscopy. PLoS One 9: e100526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish KN, Krucker T, 2008. Functional consequences of hippocampal neuronal ectopia in the apolipoprotein E receptor-2 knockout mouse. Neurobiol Dis 32: 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler CE, Aryal P, Suen KF, Slesinger PA, 2007. Evidence for association of GABA(B) receptors with Kir3 channels and regulators of G protein signalling (RGS4) proteins. J Physiol 580: 51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman LJ, Gelles J, 2015. Multi-wavelength single-molecule fluorescence analysis of transcription mechanisms. Methods 86: 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funatsu T, Harada Y, Tokunaga M, Saito K, Yanagida T, 1995. Imaging of single fluorescent molecules and individual ATP turnovers by single myosin molecules in aqueous solution. Nature 374: 555–9. [DOI] [PubMed] [Google Scholar]
- Gaus B, Bruning D, Hatlapatka K, Rustenbeck I, 2021. Changes in granule mobility and age contribute to changes in insulin secretion after desensitization or rest. BMJ Open Diabetes Res Care 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goslin K, Banker G, 1991. Rat hippocampal neurons in low density culture. In: Culturing nerve cells. Vol., Banker G, Goslin K, ed.^eds. MIT, Cambridge, MA, pp. 251–281. [Google Scholar]
- Hastoy B, Scotti PA, Milochau A, Fezoua-Boubegtiten Z, Rodas J, Megret R, Desbat B, Laguerre M, Castano S, Perrais D, Rorsman P, Oda R, Lang J, 2017. A Central Small Amino Acid in the VAMP2 Transmembrane Domain Regulates the Fusion Pore in Exocytosis. Sci Rep 7: 2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellen E, Axelrod D, 1991. Kinetics of epidermal growth factor/receptor binding on cells measured by total internal reflection/fluorescence recovery after photobleaching. J. Fluorescence 1: 113–128. [DOI] [PubMed] [Google Scholar]
- Kawano Y, Abe C, Kaneda T, Aono Y, Abe K, Tamura K, Terakawa S, 2000. High-numerical-aperture objective lenses and optical system improved objective type total internal reflection fluorescence microscopy. Proc SPIE 4098: 142. [Google Scholar]
- Keyel PA, Watkins SC, Traub LM, 2004. Endocytic adaptor molecules reveal an endosomal population of clathrin by total internal reflection fluorescence microscopy. J Biol Chem 279: 13190–204. [DOI] [PubMed] [Google Scholar]
- Klinth JE, Larsson R, Andersson PO, Ekdahl KN, 2006. A novel application of multi-wavelength TIRF spectroscopy for real time monitoring of antithrombin interactions with immobilized heparin. Biosens Bioelectron 21: 1973–80. [DOI] [PubMed] [Google Scholar]
- Lam AD, Ismail S, Wu R, Yizhar O, Passmore DR, Ernst SA, Stuenkel EL, 2010. Mapping dynamic protein interactions to insulin secretory granule behavior with TIRF-FRET. Biophys J 99: 1311–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CC, Huang CC, Lin KH, Cheng KH, Yang DM, Tsai YS, Ong RY, Huang YN, Kao LS, 2007. Visualization of Rab3A dissociation during exocytosis: a study by total internal reflection microscopy. J Cell Physiol 211: 316–26. [DOI] [PubMed] [Google Scholar]
- MacLaurin SA, Krucker T, Fish KN, 2007. Hippocampal dendritic arbor growth in vitro: regulation by Reelin-Disabled-1 signaling. Brain Res 1172: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrifield CJ, Feldman ME, Wan L, Almers W, 2002. Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nat Cell Biol 4: 691–8. [DOI] [PubMed] [Google Scholar]
- Mizuno K, Izumi T, 2022. Munc13b stimulus-dependently accumulates on granuphilin-mediated, docked granules prior to fusion. Cell Struct Funct. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederauer C, Blumhardt P, Mucksch J, Heymann M, Lambacher A, Schwille P, 2018. Direct characterization of the evanescent field in objective-type total internal reflection fluorescence microscopy. Opt Express 26: 20492–20506. [DOI] [PubMed] [Google Scholar]
- Nikolaus J, Hancock K, Tsemperouli M, Baddeley D, Karatekin E, 2021. Optimal Detection of Fusion Pore Dynamics Using Polarized Total Internal Reflection Fluorescence Microscopy. Front Mol Biosci 8: 740408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangrsic T, Potokar M, Stenovec M, Kreft M, Fabbretti E, Nistri A, Pryazhnikov E, Khiroug L, Giniatullin R, Zorec R, 2007. Exocytotic release of ATP from cultured astrocytes. J Biol Chem 282: 28749–58. [DOI] [PubMed] [Google Scholar]
- Pascolutti R, Algisi V, Conte A, Raimondi A, Pasham M, Upadhyayula S, Gaudin R, Maritzen T, Barbieri E, Caldieri G, Tordonato C, Confalonieri S, Freddi S, Malabarba MG, Maspero E, Polo S, Tacchetti C, Haucke V, Kirchhausen T, Di Fiore PP, Sigismund S, 2019. Molecularly Distinct Clathrin-Coated Pits Differentially Impact EGFR Fate and Signaling. Cell Rep 27: 3049–3061 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard HB, Apps DK, 2002. New technologies in exocytosis and ion movement. Ann N Y Acad Sci 971: 617–9. [DOI] [PubMed] [Google Scholar]
- Pryazhnikov E, Khiroug L, 2008. Sub-micromolar increase in [Ca(2+)](i) triggers delayed exocytosis of ATP in cultured astrocytes. Glia 56: 38–49. [DOI] [PubMed] [Google Scholar]
- Rao TC, Nawara TJ, Mattheyses AL, 2022. Live-Cell Total Internal Reflection Fluorescence (TIRF) Microscopy to Investigate Protein Internalization Dynamics. Methods Mol Biol 2438: 45–58. [DOI] [PubMed] [Google Scholar]
- Riven I, Iwanir S, Reuveny E, 2006. GIRK channel activation involves a local rearrangement of a preformed G protein channel complex. Neuron 51: 561–73. [DOI] [PubMed] [Google Scholar]
- Saffarian S, Kirchhausen T, 2008. Differential evanescence nanometry: live-cell fluorescence measurements with 10-nm axial resolution on the plasma membrane. Biophys J 94: 2333–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salavessa L, Lagache T, Malarde V, Grassart A, Olivo-Marin JC, Canette A, Trichet M, Sansonetti PJ, Sauvonnet N, 2021. Cytokine receptor cluster size impacts its endocytosis and signaling. Proc Natl Acad Sci U S A 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salavessa L, Sauvonnet N, 2021. Stoichiometry of Receptors at the Plasma Membrane During Their Endocytosis Using Total Internal Reflection Fluorescent (TIRF) Microscopy Live Imaging and Single-Molecule Tracking. Methods Mol Biol 2233: 3–17. [DOI] [PubMed] [Google Scholar]
- Sarkar A, Robertson RB, Fernandez JM, 2004. Simultaneous atomic force microscope and fluorescence measurements of protein unfolding using a calibrated evanescent wave. Proc Natl Acad Sci U S A 101: 12882–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobhy MA, Elshenawy MM, Takahashi M, Whitman BH, Walter NG, Hamdan SM, 2011. Versatile single-molecule multi-color excitation and detection fluorescence setup for studying biomolecular dynamics. Rev Sci Instrum 82: 113702. [DOI] [PubMed] [Google Scholar]
- Steyer JA, Almers W, 2001. A real-time view of life within 100 nm of the plasma membrane. Nat Rev Mol Cell Biol 2: 268–75. [DOI] [PubMed] [Google Scholar]
- Stout AL, Axelrod D, 1989. Evanescent field excitation of fluorescence by epi-illumination microscopy. Appl. Opt 28: 5237–5242. [DOI] [PubMed] [Google Scholar]
- Sungkaworn T, Rieken F, Lohse MJ, Calebiro D, 2014. High-resolution spatiotemporal analysis of receptor dynamics by single-molecule fluorescence microscopy. J Vis Exp: e51784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabor A, Weisenburger S, Banerjee A, Purkayastha N, Kaindl JM, Hubner H, Wei L, Gromer TW, Kornhuber J, Tschammer N, Birdsall NJ, Mashanov GI, Sandoghdar V, Gmeiner P, 2016. Visualization and ligand-induced modulation of dopamine receptor dimerization at the single molecule level. Sci Rep 6: 33233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson NL, Axelrod D, 1983. Immunoglobulin surface-binding kinetics studied by total internal reflection with fluorescence correlation spectroscopy. Biophys J 43: 103–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toglia P, Ullah G, Pearson JE, 2017. Analyzing optical imaging of Ca(2+) signals via TIRF microscopy: The limits on resolution due to chemical rates and depth of the channels. Cell Calcium 67: 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toomre D, Steyer JA, Keller P, Almers W, Simons K, 2000. Fusion of constitutive membrane traffic with the cell surface observed by evanescent wave microscopy. J Cell Biol 149: 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi T, Fukuda M, 2006. Rab3A and Rab27A cooperatively regulate the docking step of dense-core vesicle exocytosis in PC12 cells. J Cell Sci 119: 2196–203. [DOI] [PubMed] [Google Scholar]
- Urbina F, Gupton SL, 2021. Automated Detection and Analysis of Exocytosis. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MD, Axelrod D, 1994. Time-lapse total internal reflection fluorescence video of acetylcholine receptor cluster formation on myotubes. Dev Dyn 201: 29–40. [DOI] [PubMed] [Google Scholar]
- Winkle CC, Hanlin CC, Gupton SL, 2016. Utilizing Combined Methodologies to Define the Role of Plasma Membrane Delivery During Axon Branching and Neuronal Morphogenesis. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Qin F, 2009. Interaction with phosphoinositides confers adaptation onto the TRPV1 pain receptor. PLoS Biol 7: e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudowski GA, Puthenveedu MA, von Zastrow M, 2006. Distinct modes of regulated receptor insertion to the somatodendritic plasma membrane. Nat Neurosci 9: 622–7. [DOI] [PubMed] [Google Scholar]
- Zenisek D, Steyer JA, Feldman ME, Almers W, 2002. A membrane marker leaves synaptic vesicles in milliseconds after exocytosis in retinal bipolar cells. Neuron 35: 1085–97. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data available on request from the authors.